Cathode properties of a controlled crystallinity nano-Li1.2Cr0.4Mn0.4O2 cathode for lithium ion batteries†
Received
16th May 2024
, Accepted 30th September 2024
First published on 8th October 2024
Abstract
The milled-Li1.2Cr0.4Mn0.4O2 (milled-LCMO) cathode, a promising material for next-generation Li ion batteries, is prepared by dry ball-milling of layered rocksalt-type Li1.2Cr0.4Mn0.4O2 (layered-LCMO) obtained by solid-state synthesis. Despite undergoing ball-milling treatment, resulting in separation into Cr-rich and Mn-rich phases along with Li2O, milled-LCMO still exhibited a reversible capacity of 277 mA h g−1 at a rate of 16 mA g−1. However, it was also revealed that its cyclability was poor due to the contribution of oxygen redox in the charging process. On the other hand, layered-LCMO exhibited better cyclability because charge and discharge reactions proceeded only through the Cr redox. The thermally treated Li1.2Cr0.4Mn0.4O2 was prepared as a cathode material that combines the favorable properties of these two materials. In fact, each thermally treated sample showed a larger reversible capacity than the layered-LCMO obtained by the solid-phase method, and the cyclability recovered as the heat treatment temperature increased.
Introduction
Layered 3d-transition metal oxides having a basic composition of LiMO2 (M = Co, Ni, Mn, etc.) for cathode materials have been the subject of intensive research to advance the energy density of lithium ion batteries (LIBs). To date, many compounds have been reported in which the 3d-transition metal portion of LiMO2 is replaced with Co, Ni, Mn, and other elements.1 However, the actual storage capacities of LIBs are limited to 138–160 mA h g−1, and the suitable insertion/extraction reaction for LiMO2 (M = Co, Ni, Mn, etc.) proceeds at no more than 4.4 V for the charging voltage. To improve the energy density of LiMO2, the Li2MnO3–LiMO2 solid solution system has drawn attention as a cathode material for LIBs. Li2MnO3–LiMO2 solid solution systems (M = Mn, Co, and Ni), such as Li1.2Mn0.54Ni0.13Co0.13O2, Li1.2Ni0.6Mn0.2O2, Li1.17Mn0.51Ni0.15Co0.15Al0.025O2, etc., exhibited large capacities of over 200 mA h g−1 by utilizing both transition metals and oxygen redox.2 However, there are concerns about the cycle stability and safety of these materials due to the utilization of oxygen redox. On the other hand, layered Li1.2Cr0.4Mn0.4O2, which forms a Li2MnO3–LiCrO2 solid solution system, showed a large capacity of 180 mA h g−1, corresponding to a 0.56-electron reaction. Because this reaction proceeds only by Cr3+/Cr6+ redox, this cathode material has exhibited relatively good cyclability.3 However, it has an irreversible capacity of 80 mA h g−1 in the first cycle, and further improvements in reversible capacity are needed in order to advance toward the theoretical capacity of 323 mA h g−1. To increase the rechargeable capacity of Li1.2Cr0.4Mn0.4O2, we focused on reports that the rechargeable capacity of LiCrO2 can be improved by tailoring the size of LiCrO2 domains.4 Therefore, nano-Li1.2Cr0.4Mn0.4O2 (milled-LCMO) was prepared using the dry ball-milling method from layered Li1.2Cr0.4Mn0.4O2 (layered-LCMO), and its electrochemical properties were evaluated. In addition, to investigate the influence of domain size on cathode properties, the obtained milled-LCMO was re-sintered at 500–800 °C, and its cathode properties were also evaluated. Moreover, its local structures were investigated using X-ray diffraction (XRD) and neutron diffraction (ND). The detailed surface spatial/chemical structures were examined with spectral imaging using a scanning/transmission electron microscope (S/TEM) equipped with an electron energy-loss spectrometer (EELS). The charge compensation during the charge–discharge reaction was also investigated using X-ray absorption near-edge structure (XANES) measurement.
Results and discussion
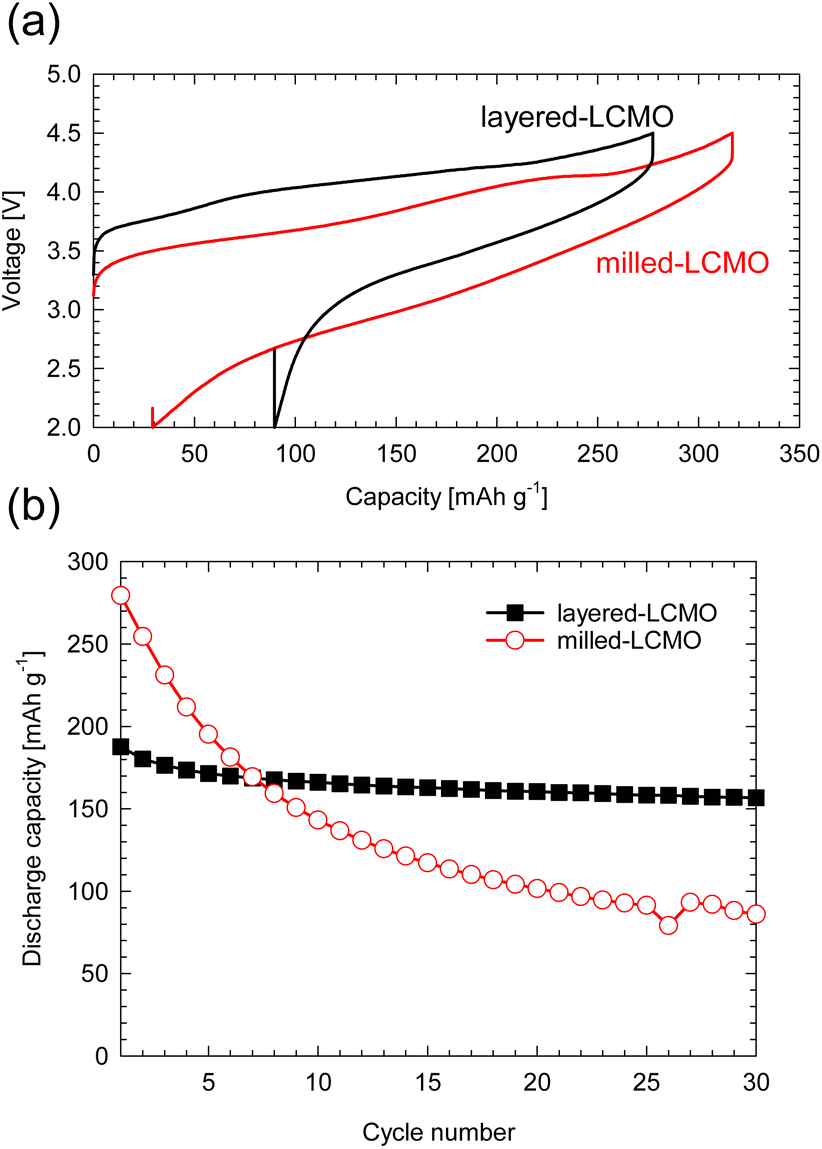
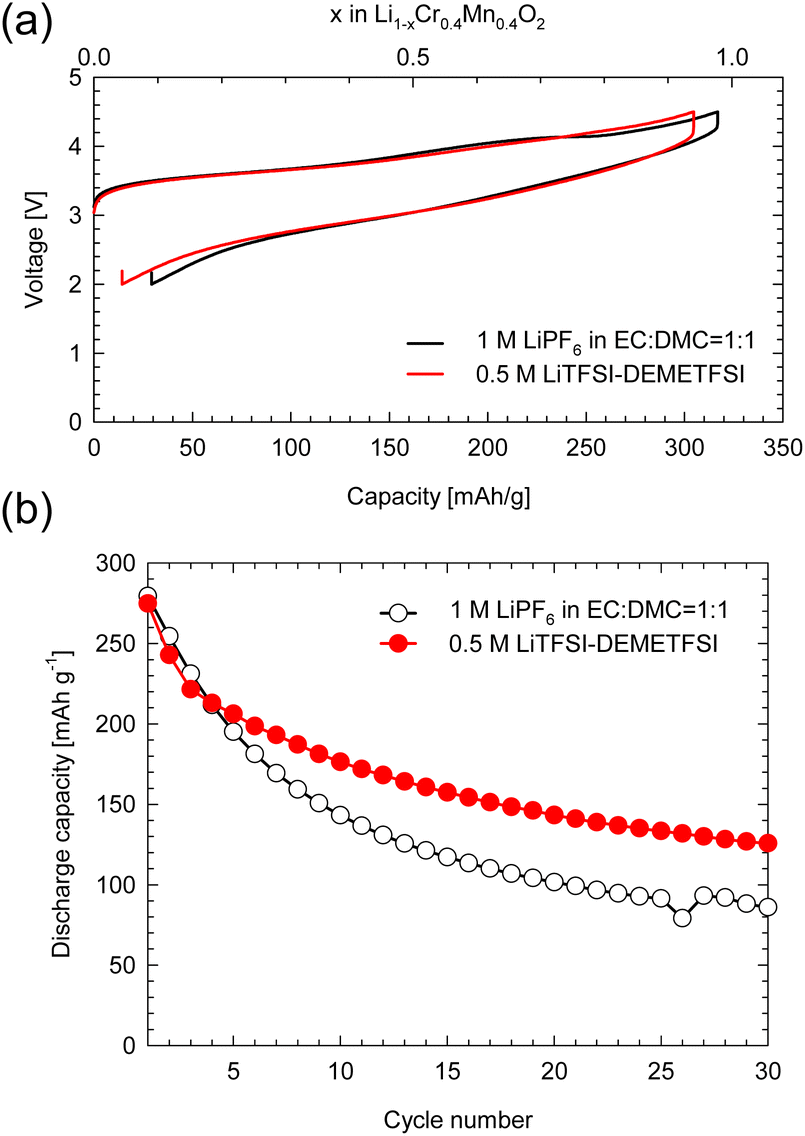
Fig. S1 (a)† shows the XRD profiles of layered-LCMO prepared by the solid-state synthesis method and milled-LCMO prepared by the dry ball-milling method. The observed diffraction peaks for layered-LCMO were indexed as a trigonal system with the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The lattice constant for layered-LCMO was a = b = 2.87 Å and c = 14.31 Å. On the other hand, the (0 0 3) peak was eliminated by the ball-milling treatment, while the intensity of a halo peak, which is broader peak at around 15–25°, increased, and the broad diffraction peaks are observed at 38°, 44° and 64°. This result suggested that the crystal system was changed from trigonal to cubic, which is indexed as the space group Fmm. The lattice constant of the disordered rocksalt phase remained the same, with a value of a = 4.10 Å. Fig. S1(b) and (c)† show SEM images of the obtained samples to confirm the particle size and the particle morphology. The obtained layered-LCMO powders had a primary particle size of 1 μm, and the primary particle size for milled-LCMO was 100–200 nm. To clarify the effect of milling treatment on the electronic structure around the Cr ion and Mn ion, we measured the Cr K-edge and Mn K-edge XANES spectra in milled-LCMO and layered-LCMO. Fig. S2(a)† shows the Cr K-edge XANES spectra of layered-LCMO and milled-LCMO. The main peak positions corresponding to 1s → 4p dipole-allowed transitions remained unchanged between layered-LCMO and milled-LCMO. In addition, the pre-edge feature for milled-LCMO agreed with that for layered-LCMO. These results suggested that the oxidation state of chromium was Cr3+. On the other hand, the Mn K-edge edge position in layered-LCMO was consistent with that of MnO2, while the K-edge position in milled-LCMO shifted to a lower energy level than that in layered-LCMO (Fig. S2(b)†). In other words, this result suggested that the oxidation number of manganese in milled-LCMO was reduced to +3 from +4 by the ball-milling treatment. In the case of the disordered rocksalt structure, where the cations of Li, Cr, and Mn occupy the 4b site as shown Fig. S2(c),† it is considered that the oxidation number of chromium is +3 and that of manganese is +4. In addition, it is considered that the cations of Li, Cr, and Mn exist uniformly within the particles. To investigate the dispersion states of Li, Cr, and Mn ions within the milled-LCMO particles, STEM-EELS measurement was performed. Fig. S3(a–e)† show ADF-STEM images and EELS elemental distributions (O K-edge, Li K-edge, Cr L2,3-edge, and Mn L2,3-edge) of milled-LCMO. Each composition was calculated using the following formula: X comp. = CX/(CO + CCr + CMn), where CX denotes the projected concentration of element X and X = O, Cr, and Mn, respectively. In addition, the Li concentration was calculated as CLi normalized by using the sample thickness. Milled-LCMO shows heterogeneous Cr and Mn compositions. In addition, the Li K-edge exhibits a small pre-peak at around 58 eV in addition to the main peak at 62 eV, as shown in Fig. S3(f).† This characteristic pre-peak at around 58 eV agreed with that of Li2O.5 It is thus presumed that milled-LCMO is partially decomposed into several different phases (Cr-rich Li–Cr–Mn–O, Mn-rich Li–Cr–Mn–O, and Li2O). To compare the cathode properties of layered-LCMO and milled-LCMO, the charge–discharge measurement was performed at a rate of 16 mA g−1 in the voltage range of 2.0–4.5 V. Here, 0.05 C represents a current density at which the discharge for the theoretical capacity, considering a 1-electron reaction of Li1.2Cr0.4Mn0.4O2 finishes within 1 h. Fig. 1 summarizes (a) the charge–discharge profiles and (b) cyclability of the obtained samples. Layered-LCMO and milled-LCMO showed initial charge/discharge capacities of 277 mA h g−1/188 mA h g−1 and 320 mA h g−1/277 mA h g−1, respectively. The irreversible capacities were 89 mA h g−1 (layered-LCMO) and 44 mA h g−1 (milled-LCMO), respectively. The cell voltage for the layered-LCMO increased gradually, with inflection points observed at around 3.8 V and 4.2 V during the initial charge process. On the other hand, the cell voltage for milled-LCMO also increased through inflection points at around 3.6 V and 4.2 V during the initial charge process. The average charging voltages were 4.1 V (layered-LCMO) and 3.8 V (milled-LCMO). On the other hand, the average discharging voltages were 3.5 V (layered-LCMO) and 3.0 V (milled-LCMO). These results suggested that the average operating voltage of milled-LCMO was lower than that of layered-LCMO. It considered to contribute for increasing the rechargeable capacity and lower operating voltage of milled-LCMO, which was formed multiple phases and the reduced oxidation state of Mn to +3 from +4. Fig. 1b summarizes the cyclability of layered-LCMO and milled-LCMO. After 30 cycles, the capacity retention for layered-LCMO was 84%, while milled-LCMO exhibited a significantly lower capacity retention of 31%. It is known that the dissolution of V ions, which have properties similar to those of Cr ions, affects the cyclability of vanadium systems such as Li1.2Nb0.2V0.6O2.6 Therefore, the dissolution of Cr ions may contribute to the poor cyclability. To investigate the dissolution of Cr ions during the charge–discharge process, a battery evaluation was performed using a highly viscous ionic liquid electrolyte with high viscosity and reduced solubility of Cr and Mn ions. Fig. 2 shows (a) charge–discharge profiles and (b) cyclability of milled-LCMO using 1 M LiPF6 in EC:DMC electrolyte and 0.5 M LiTFSI-DEMETFSI electrolyte at a rate of 16 mA g−1 in the voltage range of 2.0–4.5 V. The test temperature was 25 °C. The charge–discharge capacity and overpotential hardly changed even when a highly viscous electrolyte was used. However, the capacity retention after 30 cycles was improved to 45% by using an ionic liquid electrolyte. Regarding the elution of Mn and Cr ions from electrodes, the Mn and Cr concentrations in the electrolyte were measured using ICP-AES. It was revealed that 0.05% Cr ions and 0.06% Mn ions were eluted from the electrodes in 1 M LiPF6 in EC:DMC electrolyte. On the other hand, Cr and Mn ions were not detected in 0.5 M LiTFSI-DEMETDSI electrolyte. The elution of Cr ions and Mn ions is one of the causes of cycle deterioration, but these results suggested that there are other factors that cause cycle deterioration. Based on the above results, to achieve both the large reversible capacity and better cyclability of milled-LCMO, we aimed to prepare a cathode material including Mn3+ that has the advantages of both milled-LCMO and layered-LCMO. Therefore, milled-LCMO was resintered in an Ar atmosphere to control the conditions of the several phases formed by milling treatment.
m. The lattice constant for layered-LCMO was a = b = 2.87 Å and c = 14.31 Å. On the other hand, the (0 0 3) peak was eliminated by the ball-milling treatment, while the intensity of a halo peak, which is broader peak at around 15–25°, increased, and the broad diffraction peaks are observed at 38°, 44° and 64°. This result suggested that the crystal system was changed from trigonal to cubic, which is indexed as the space group Fmm. The lattice constant of the disordered rocksalt phase remained the same, with a value of a = 4.10 Å. Fig. S1(b) and (c)† show SEM images of the obtained samples to confirm the particle size and the particle morphology. The obtained layered-LCMO powders had a primary particle size of 1 μm, and the primary particle size for milled-LCMO was 100–200 nm. To clarify the effect of milling treatment on the electronic structure around the Cr ion and Mn ion, we measured the Cr K-edge and Mn K-edge XANES spectra in milled-LCMO and layered-LCMO. Fig. S2(a)† shows the Cr K-edge XANES spectra of layered-LCMO and milled-LCMO. The main peak positions corresponding to 1s → 4p dipole-allowed transitions remained unchanged between layered-LCMO and milled-LCMO. In addition, the pre-edge feature for milled-LCMO agreed with that for layered-LCMO. These results suggested that the oxidation state of chromium was Cr3+. On the other hand, the Mn K-edge edge position in layered-LCMO was consistent with that of MnO2, while the K-edge position in milled-LCMO shifted to a lower energy level than that in layered-LCMO (Fig. S2(b)†). In other words, this result suggested that the oxidation number of manganese in milled-LCMO was reduced to +3 from +4 by the ball-milling treatment. In the case of the disordered rocksalt structure, where the cations of Li, Cr, and Mn occupy the 4b site as shown Fig. S2(c),† it is considered that the oxidation number of chromium is +3 and that of manganese is +4. In addition, it is considered that the cations of Li, Cr, and Mn exist uniformly within the particles. To investigate the dispersion states of Li, Cr, and Mn ions within the milled-LCMO particles, STEM-EELS measurement was performed. Fig. S3(a–e)† show ADF-STEM images and EELS elemental distributions (O K-edge, Li K-edge, Cr L2,3-edge, and Mn L2,3-edge) of milled-LCMO. Each composition was calculated using the following formula: X comp. = CX/(CO + CCr + CMn), where CX denotes the projected concentration of element X and X = O, Cr, and Mn, respectively. In addition, the Li concentration was calculated as CLi normalized by using the sample thickness. Milled-LCMO shows heterogeneous Cr and Mn compositions. In addition, the Li K-edge exhibits a small pre-peak at around 58 eV in addition to the main peak at 62 eV, as shown in Fig. S3(f).† This characteristic pre-peak at around 58 eV agreed with that of Li2O.5 It is thus presumed that milled-LCMO is partially decomposed into several different phases (Cr-rich Li–Cr–Mn–O, Mn-rich Li–Cr–Mn–O, and Li2O). To compare the cathode properties of layered-LCMO and milled-LCMO, the charge–discharge measurement was performed at a rate of 16 mA g−1 in the voltage range of 2.0–4.5 V. Here, 0.05 C represents a current density at which the discharge for the theoretical capacity, considering a 1-electron reaction of Li1.2Cr0.4Mn0.4O2 finishes within 1 h. Fig. 1 summarizes (a) the charge–discharge profiles and (b) cyclability of the obtained samples. Layered-LCMO and milled-LCMO showed initial charge/discharge capacities of 277 mA h g−1/188 mA h g−1 and 320 mA h g−1/277 mA h g−1, respectively. The irreversible capacities were 89 mA h g−1 (layered-LCMO) and 44 mA h g−1 (milled-LCMO), respectively. The cell voltage for the layered-LCMO increased gradually, with inflection points observed at around 3.8 V and 4.2 V during the initial charge process. On the other hand, the cell voltage for milled-LCMO also increased through inflection points at around 3.6 V and 4.2 V during the initial charge process. The average charging voltages were 4.1 V (layered-LCMO) and 3.8 V (milled-LCMO). On the other hand, the average discharging voltages were 3.5 V (layered-LCMO) and 3.0 V (milled-LCMO). These results suggested that the average operating voltage of milled-LCMO was lower than that of layered-LCMO. It considered to contribute for increasing the rechargeable capacity and lower operating voltage of milled-LCMO, which was formed multiple phases and the reduced oxidation state of Mn to +3 from +4. Fig. 1b summarizes the cyclability of layered-LCMO and milled-LCMO. After 30 cycles, the capacity retention for layered-LCMO was 84%, while milled-LCMO exhibited a significantly lower capacity retention of 31%. It is known that the dissolution of V ions, which have properties similar to those of Cr ions, affects the cyclability of vanadium systems such as Li1.2Nb0.2V0.6O2.6 Therefore, the dissolution of Cr ions may contribute to the poor cyclability. To investigate the dissolution of Cr ions during the charge–discharge process, a battery evaluation was performed using a highly viscous ionic liquid electrolyte with high viscosity and reduced solubility of Cr and Mn ions. Fig. 2 shows (a) charge–discharge profiles and (b) cyclability of milled-LCMO using 1 M LiPF6 in EC:DMC electrolyte and 0.5 M LiTFSI-DEMETFSI electrolyte at a rate of 16 mA g−1 in the voltage range of 2.0–4.5 V. The test temperature was 25 °C. The charge–discharge capacity and overpotential hardly changed even when a highly viscous electrolyte was used. However, the capacity retention after 30 cycles was improved to 45% by using an ionic liquid electrolyte. Regarding the elution of Mn and Cr ions from electrodes, the Mn and Cr concentrations in the electrolyte were measured using ICP-AES. It was revealed that 0.05% Cr ions and 0.06% Mn ions were eluted from the electrodes in 1 M LiPF6 in EC:DMC electrolyte. On the other hand, Cr and Mn ions were not detected in 0.5 M LiTFSI-DEMETDSI electrolyte. The elution of Cr ions and Mn ions is one of the causes of cycle deterioration, but these results suggested that there are other factors that cause cycle deterioration. Based on the above results, to achieve both the large reversible capacity and better cyclability of milled-LCMO, we aimed to prepare a cathode material including Mn3+ that has the advantages of both milled-LCMO and layered-LCMO. Therefore, milled-LCMO was resintered in an Ar atmosphere to control the conditions of the several phases formed by milling treatment.
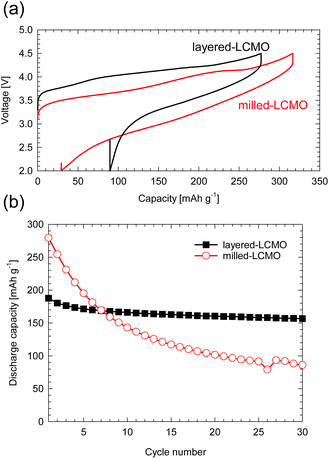 |
| | Fig. 1 (a) Initial and second charge–discharge profiles and (b) cyclability of milled-LCMO and layered-LCMO at a rate of 16 mA g−1 between 2.0 and 4.5 V. | |
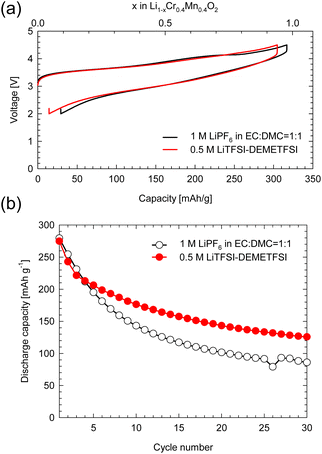 |
| | Fig. 2 (a) The initial charge–discharge profiles and (b) cyclability of milled-LCMO using 1 M LiPF6 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 1:1 v/v electrolyte and 0.5 M LiTFSI in DEMETFSI electrolyte. :DMC = 1:1 v/v electrolyte and 0.5 M LiTFSI in DEMETFSI electrolyte. | |
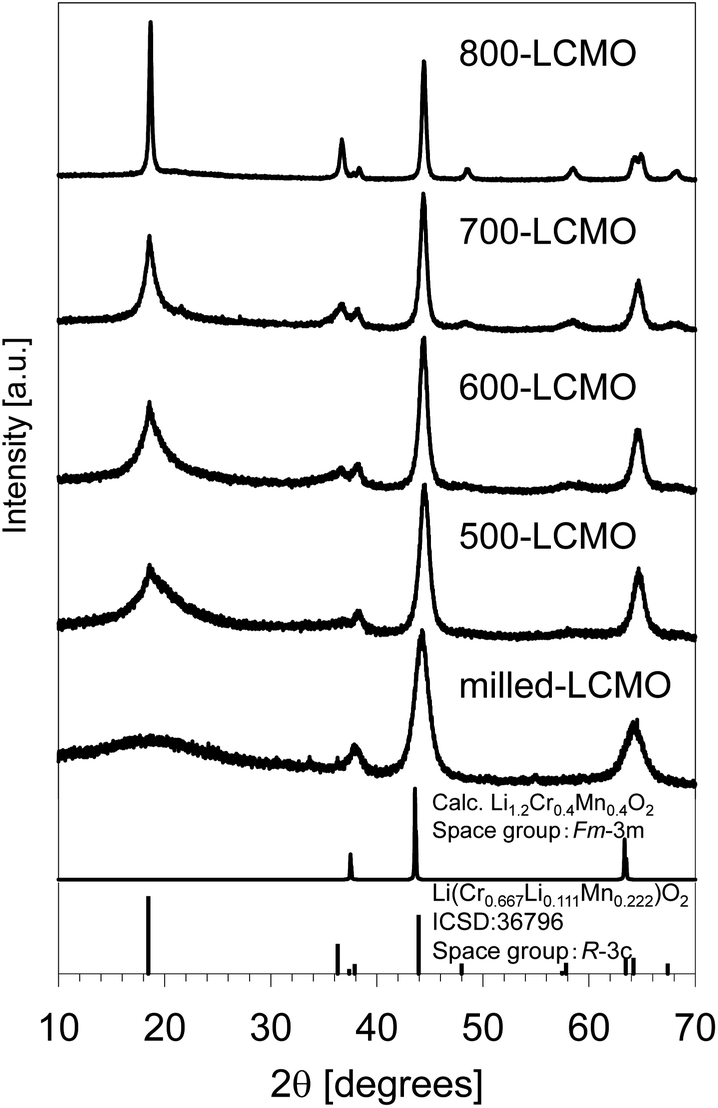
Fig. 3 summarizes the XRD profiles of the obtained samples at various resintering temperatures. The diffraction peak intensity of (0 0 3) increased, and the diffraction peak width became narrower as the thermal-treatment temperature increased. Therefore, this showed that the crystallinity of LCMO was restored and the domain size increased. The crystallinity and domain size before and after the thermal treatments were evaluated based on Fourier analysis of high-resolution TEM images. Fig. S4(a–c)† show the TEM images, their FFT power spectra, and inverse Fourier transform images of masked FFT powder spectra for milled-LCMO, 600-LCMO, and 800-LCMO, respectively. The domain size of milled-LCMO ranges from 1 to several nanometers, whereas the domain size increases with the increase in the post-annealing temperature, compared with that of milled-LCMO. The crystallinity is also improved at higher annealing temperatures, judging from the intensity broadness of the power spectra. It should be noted that the 800-LCMO sample clearly shows a layered structure, albeit with localized lattice distortions. These results are in good agreement with the XRD profiles. Fig. 4a and b show ADF-STEM images and the corresponding elemental distributions for nano-particles of 600-LCMO and 800-LCMO, respectively. The inhomogeneous distributions of Cr and Mn remain unchanged. Note that the valence state of Mn mostly exhibits tetravalent Mn4+, although divalent Mn (Mn2+) is found near the particle surface of 600-LCMO due to a lower oxygen concentration there, resulting in the reduction of Mn. For 800-LCMO, on the other hand, Cr-rich and Mn-rich phases are clearly separated, where Mn and Cr are always tetravalent (Mn4+) and trivalent (Cr3+), respectively. In addition, the pre-peak of the O K edge is split into two by 2.3 eV and their relative intensities differ. The Cr-rich area exhibits a higher peak intensity at the higher energy peak, while the Mn-rich phases exhibit a lower energy peak, as shown in Fig. 4c. To clarify the origin of the spectral difference, O K-edge EELS spectra (together with Cr- and Mn-L2,3 edges) were measured from LiCrO2 and Li2MnO3 as the reference spectra, as shown in Fig. 4d. The pre-peak is located at 530 and 532 eV for Li2MnO3 and LiCrO2, respectively, with both phases exhibiting almost identical main peaks. These results suggested that the local structure of the Cr-rich phase closely resembled that of LiCrO2, while the Mn-rich phase exhibited similarities to Li2MnO3. It is also possible that the two stoichiometric phases overlap in the viewing direction, given the varying relative peak intensity observed in different areas. This consideration is consistent with the fact that the Li concentration tends to be slightly deficient in the areas dominated by the Cr-rich phase. We also measured the Mn K-edge XANES spectra. The Mn oxidation state was returned to Mn4+ from Mn3+ by thermal treatment, as shown in Fig. S5,† consistent with the results of EELS. To clarify the detailed local structures of the obtained samples, the X-ray total scattering measurements and neutron total scattering measurements for the obtained powders (layered-LCMO, milled-LCMO, and 600-LCMO) were carried out at room temperature. Fig. S6† shows the partial pair distribution functions gij(r) (gij(r) is the partial pair distribution function of atoms i and j, respectively) for Li–Li, Li–Cr, Li–Mn, Li–O, Cr–Cr, Cr–Mn, Cr–O, Mn–Mn, Mn–O, and O–O correlations, which were obtained by modelling X-ray and neutron total scattering profiles. The peak positions of most correlations for each sample were not shifted. On the other hand, the peak intensity of the Cr–Cr correlation was the weakest for layered-LCMO and highest for milled-LCMO among all samples. Although this change in peak intensity is small, Mn–Mn and Li–Li correlations exhibited a similar tendency. It is considered that in milled-LCMO, Cr ions are preferentially surrounded by other Cr ions rather than by Li and Mn ions. In other words, Cr and Mn ions are considered not uniformly distributed within the particles and are separated. On the other hand, the peak intensity of the Cr–Mn correlation in milled-LCMO and 600-LCMO was lower than that in layered-LCMO. These results exhibit a trend similar to that obtained with STEM-EELS. It is interesting to observe how the cathode properties of the obtained samples change when the compositional discontinuity within the particles, caused by ball milling treatment, is partially restored through heat treatment.
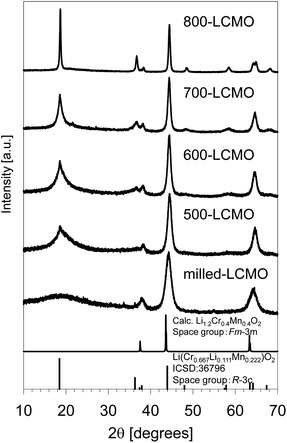 |
| | Fig. 3 XRD profiles of the obtained samples at various resintering temperatures. | |
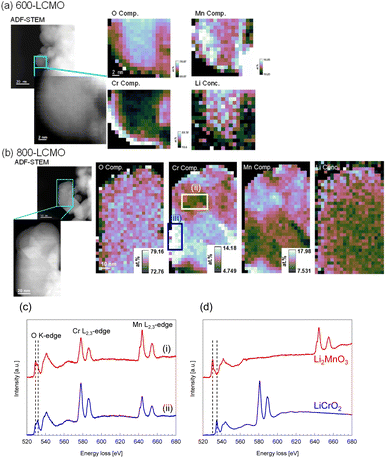 |
| | Fig. 4 Image diagrams based on STEM-EELS elemental mapping results for (a) 600-LCMO (EELS spectra for region (i) are shown) and (b) 800-LCMO. (c) EELS spectra for regions (ii), (iii) and (d) and LiCrO2 and Li2MnO3 as standard samples. | |
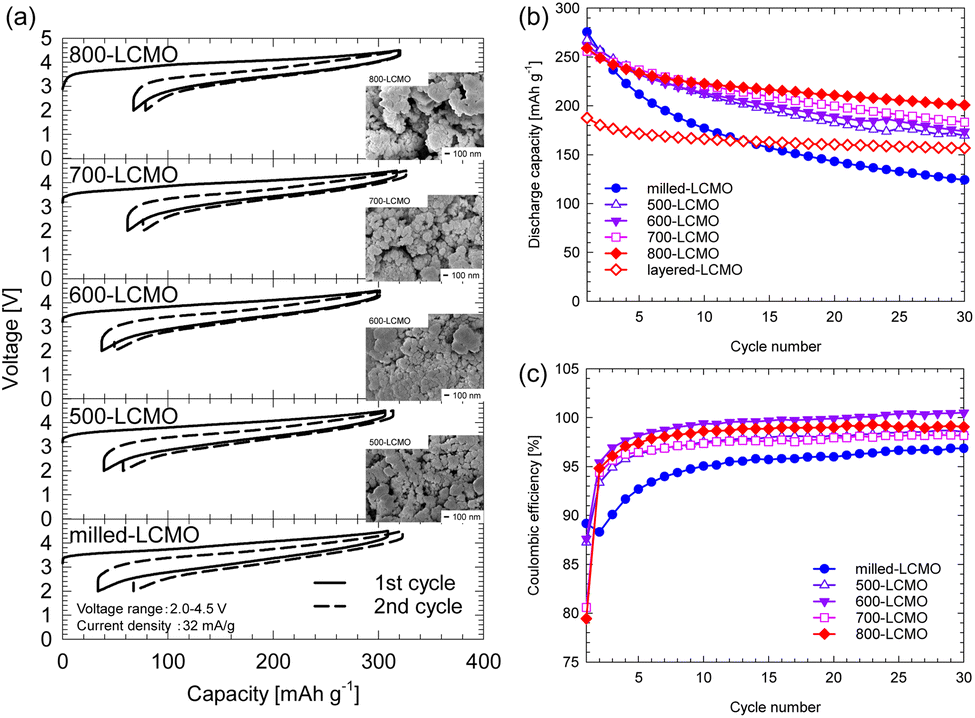
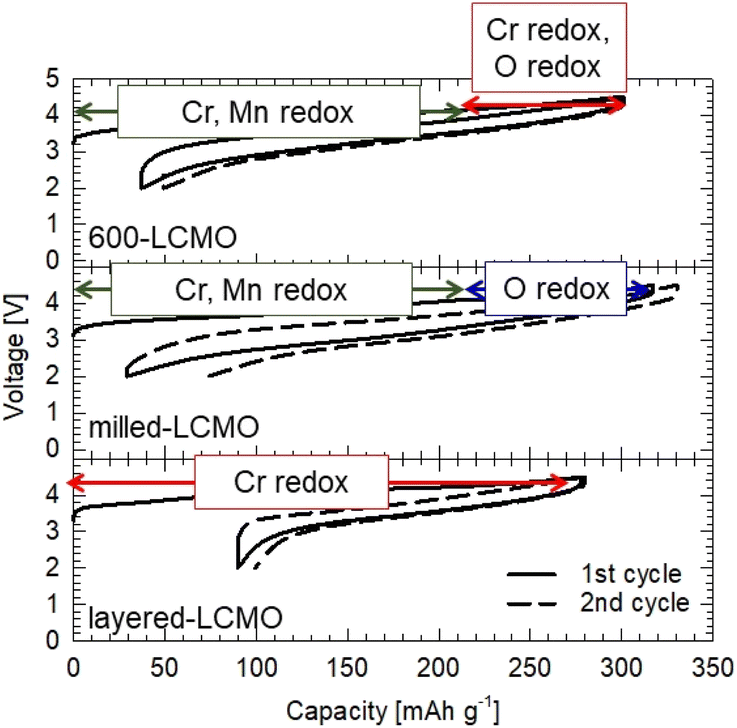
Fig. 5 shows (a) the initial and second charge–discharge curves and (b) the cyclability of the obtained samples at a rate of 32 mA g−1 in the voltage range of 2.0–4.5 V. The test temperature was 25 °C. The initial charge–discharge capacities for the obtained samples were 309/275 mA h g−1 (milled-LCMO), 306/267 mA h g−1 (500-LCMO), 299/262 mA h g−1 (600-LCMO), 317/256 mA h g−1 (700-LCMO), and 319/252 mA h g−1 (800-LCMO), respectively. The irreversible capacities were 34 mA h g−1 (milled-LCMO), 39 mA h g−1 (500-LCMO), 37 mA h g−1 (600-LCMO), 62 mA h g−1 (700-LCMO), and 67 mA h g−1 (800-LCMO), respectively. The initial discharge capacity decreased as the layered rocksalt structure recovered, while the irreversible capacity increased. Here, although the crystal structure of 800-LCMO has returned to the layered rocksalt type, its discharge capacity was larger than that of the obtained layered-LCMO by the solid-state method. It can be seen from Fig. 5 that the particle sizes of the resintered samples did not change much compared to those of milled-LCMO. The reduction in particle size is believed to have significantly increased the discharge capacity of 800-LCMO compared to that of layered-LCMO. The cyclability of the obtained samples is summarized in Fig. 4f. The capacity retentions after 30 cycles were 45% (milled-LCMO), 64% (500-LCMO), 67% (600-LCMO), 71% (700-LCMO), and 78% (800-LCMO). The cyclability improved because of the growth of the layered rocksalt phase through thermal treatment. However, milled-LCMO shows poor cyclability, and layered- or 800-LCMO has a large irreversible capacity. Therefore, the coulombic efficiencies of each sample are compared in Fig. 5c. Milled-LCMO had lower coulombic efficiency among them. This is because, as mentioned above, Mn and Cr ions were eluted from the electrode during charge–discharge cycles. On the other hand, other samples after thermal treatment had improved coulombic efficiency. In particular, it was clarified that the coulombic efficiency of 800-LCMO exceeded 99%. To clarify these details, we investigated the charge compensation mechanism of each sample using Cr K-, L-edge, Mn K-, L-edge, and O K-edge XANES spectra.
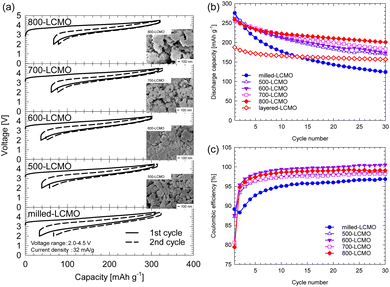 |
| | Fig. 5 (a) SEM image and initial and second charge–discharge profiles for thermally treated LCMO, obtained at a rate of 32 mA g−1 between 2.0 and 4.5 V. (b) Cyclability and (c) coulombic efficiency of the obtained samples. | |
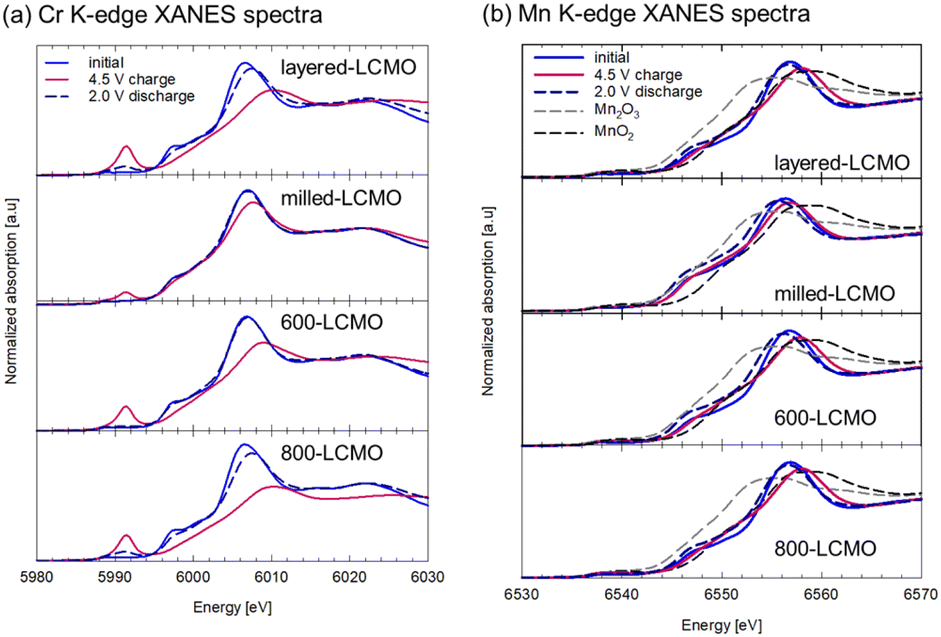
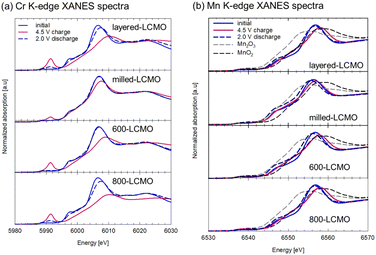
Fig. 6 shows XANES spectra of the (a) Cr K-edge and (b) Mn K-edge for each electrode (layered-LCMO, milled-LCMO, 600-LCMO, and 800-LCMO) after the charge–discharge reaction. The sampling points of the electrodes were the initial state, the charged state up to 4.5 V, and the discharge state down to 2.0 V after the charging process up to 4.5 V, respectively. For Cr K-edge XANES spectra (Fig. 6a), the main peak positions for 1s → 4p transitions shifted to higher energies in the charging process from initial to 4.5 V, but the energy shift for layered-LCMO or 600-LCMO was larger than that for milled-LCMO. In addition, the pre-edge peak intensity corresponding to 1s → 3d transitions increased as the charging reaction progressed for each sample. The peak intensity of layered-LCMO, 600-LCMO and 800-LCMO was also higher than that for milled-LCMO. In other words, milled-LCMO exhibits the smallest contribution from the Cr redox during the charging reaction among them. Conversely, the oxidation reaction of Cr is considered to be the main reaction in the charging process in layered-LCMO and 600-LCMO. On the other hand, the features and intensity of the pre-edge peak were returned to their initial state for milled-LiCrO2 and 600-LCMO, but for layered-LCMO and 800-LCMO, they did not return to the initial state. It is considered that the irreversibility of the Cr redox reaction is responsible for the large irreversible capacity of layered-LCMO. For Mn K-edge XANES spectra (Fig. 6b), the edge positions for each sample did not shift during the initial charging process, but the main peak positions for 1s → 4p transitions shifted to higher energy. However, the Mn oxidation state of layered-LCMO and 800-LCMO is +4, indicating that it is unlikely to change to a higher oxidation state during the charging reaction. Therefore, this was caused by local structural changes around the Mn ions. After the discharge process, the main peak position of the Mn K-edge was returned to the initial state. Therefore, it was considered that the Mn redox reaction did not contribute in the charge–discharge reaction for layered-LCMO and 800-LCMO. These results suggested that the oxidation reaction of Cr was the main reaction in the charging process in layered-LCMO and 800-LCMO. Therefore, we measured Cr, Mn L-edge, and O K-edge XANES spectra for milled-LCMO and 600-LCMO, which are sensitive to changes in oxidation number, and investigated the electronic structures of Cr, Mn, and O. Fig. 7 summarizes the Cr, Mn L-edge XANES spectra and O K-edge XANES spectra of the electrodes for milled-LCMO and 600-LCMO after the initial charge–discharge reaction. The features and peak positions of the Cr L-edge spectra in the initial state for both samples agreed with the data in a previous report.3 Therefore, the oxidation number of chromium in the initial state was Cr3+ in both samples. On the other hand, the features and peak positions of the Mn L-edge spectra in the initial state for both samples were complex. Peaks were observed at 639 eV and 642 eV in the case of milled-LCMO and at 639 eV, 642 eV, and 644 eV in the case of 600-LCMO. It was reported that the peak positions were 639–640 eV for Mn2+, 642–643 eV for Mn3+, and 643–644 eV for Mn4+.7 These results suggested that manganese exhibited mixed oxidation states of Mn2+ and Mn3+ in milled-LCMO, as well as mixed oxidation states of Mn2+, Mn3+, and Mn4+ in 600-LCMO. For the milled-LCMO cathode, the Cr L-edge peak at 577 eV shifted to a higher energy level, and new peaks appeared at 580 eV in the charging process up to 3.95 V. The oxidation state of Cr changed from Cr3+ to Cr4+ or higher. B. Ammundsen et al. reported that there is no evidence of the Cr L-edges of Cr4+ and Cr5+ and that the peak at 580 eV appeared during charging in the Li1.2Cr0.4Mn0.4O2 cathode.3 In addition, they mentioned that the peak at 580 eV indicates Cr6+. These results suggested that the oxidation state of Cr changed from Cr3+ to Cr4+ and then to Cr6+ during charging. Furthermore, the change in the peak position of the Cr L-edge was small even when charged up to 4.5 V. On the other hand, the peak position of the Mn L-edge shifted to higher energy in the charging process up to 3.95 V. Even after charging up to 4.5 V, the peak position at 643 eV was not changed, but a peak at 642 eV appeared. This result suggested that the charging reaction up to 3.95 V proceeded through Cr3+/Cr6+ redox and Mn2+,3+/Mn4+ redox. There results suggested that the redox of Cr and Mn did not contribute to the charging reaction from 3.95 V to 4.5 V, and in contrast the Mn ion was partially reduced from Mn4+ to Mn3+. To clarify the charging reaction from 3.95 V to 4.5 V, we examined changes in the O K-edge XANES spectrum. In the charging process up to 4.5 V, a new peak was confirmed at approximately 530 eV. In the case of the Li1.2Mn0.4Ti0.4O2 cathode,8 this peak has been explained by hole stabilization in oxygen through a π-type interaction between O 2p and Mn e.g. orbitals coupled with re-hybridization of orbitals. These results suggested that the charging reaction from 3.95 V to 4.5 V was driven by oxygen redox and oxygen release. For 600-LCMO, the peak at 577 eV shifted to a higher energy level, and new peaks appeared at 580 eV. The oxidation state of Cr changed from Cr3+ to Cr4+ and then to Cr6+. After the charging reaction up to 4.5 V, the peak intensity at 580 eV was higher than that of the charged state up to 3.95 V. This result suggested that the charging reaction up to 4.5 V included the Cr3+/Cr6+ redox. On the other hand, the Mn L-edge spectra showed changes similar to those in milled-LCMO. However, the peak intensity at 642 eV was lower than that of milled-LCMO in the charged state at 4.5 V. In addition, the peak intensity at 530 eV (O K-edge) was also lower than that of milled-LCMO in the charged state at 4.5 V. These results suggested that the contribution of oxygen redox in the charging reaction of 600-LCMO decreased. In the discharge process, the peak of chromium returned to the original position in both samples. On the other hand, the peak position of manganese differed between milled-LCMO and 600-LCMO. This result suggested that the oxidation state of manganese was a mixture of Mn2+ and Mn3+ in milled-LCMO and Mn3+ in 600-LCMO. The charge compensation mechanism during the charge–discharge reaction of each cathode material is summarized as shown in Fig. 8. This result suggested that milled-LCMO exhibits a large reversible capacity because of not only Cr redox but also Mn redox and oxygen redox. In the case of 600-LCMO, the charge–discharge reaction was driven by Cr redox, Mn redox, and slight oxygen redox. This result suggested that the contribution ratio of oxygen redox during the charging reaction influences the magnitude of the reversible capacity and the low cyclability. However, it was revealed that it is possible to control the contribution rate of oxygen redox, thereby increasing the reversible capacity and improving the cycle stability by using heat treatment to form a nanocomposite state of the Cr-rich phase, Mn-rich phase, and layered rock salt phase within a single particle.
 |
| | Fig. 6 (a) Cr K-edge and (b) Mn K-edge XANES spectra after charge–discharge reactions for layered-LCMO, milled-LCMO, 600-LCMO, and 800-LCMO electrodes. | |
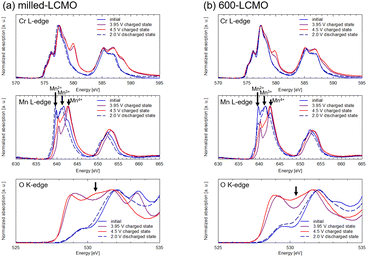 |
| | Fig. 7 Cr L-edge, Mn L-edge, and O K-edge XANES spectra after charge–discharge reactions for (a) milled-LCMO and (b) 600-LCMO electrodes. | |
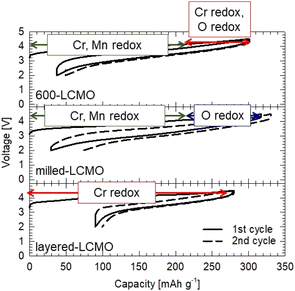 |
| | Fig. 8 Relationship between the redox reaction of each element and the charge–discharge reaction for each cathode. | |
Conclusions
In this manuscript, we proposed milled-LCMO and thermally treated-LCMO as cathode materials that have a large capacity for next-generation LIBs. Milled-LCMO underwent ball-milling treatment resulting in its separation into a Cr-rich phase, a Mn-rich phase, and Li2O, and the oxidation state of the Mn ion was changed to Mn3+ from Mn4+. The thermally treated-LCMO in an Ar atmosphere returned to the layered rocksalt structure on increasing the temperature, but 800-LCMO was partially separated into Li2MnO3 and LiCrO2. Milled-LCMO and thermally treated-LCMO exhibited a large reversible capacity because each phase formed nanointerfaces in the particles. In particular, 800-LCMO had a larger reversible capacity than layered-LCMO and exhibited better cyclability, because the charge and discharge reactions proceeded only through the Cr redox. However, it had the largest irreversible capacity among them because the Cr redox did not completely reverse. Although milled-LCMO and 600-LCMO had a larger reversible capacity than 800-LCMO, milled-LCMO exhibited the lowest cyclability among them. In the charging process, milled-LCMO proceeded by Mn3+/Mn4+, Cr3+/Cr6+, and oxygen redox, while layered- and 800-LCMO proceeded by only Cr3+/Cr6+ redox. It is considered that the difference in redox species, in particular the progression of oxygen redox during the charging process, affects the cyclability of milled-LCMO. However, the amount of oxygen redox reduced on increasing the re-sintered temperature; therefore, 600-LCMO and 800-LCMO showed improved cyclability. From these results, it was clarified that the thermally treated LCMO prepared from milled-LCMO combined the favourable properties of milled-LCMO and layered-LCMO.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP19H05814, JP19H05815, and JP19H05816 (Grant-in-Aid for Scientific Research on Innovative Areas “Interface Ionics”). A part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” and “Microscopic Imaging Solution Platform in Program for Advanced Research Equipment Platforms” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. Neutron total scattering experiments were approved by the Neutron Scattering Program Advisory Committee (Proposal No. 2019S06). Synchrotron radiation experiments were performed with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2023A1001).
References
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783 CrossRef CAS; J. R. Dahn, U. Sacken and C. A. Michal, Solid State Ionics, 1990, 44, 87 CrossRef; Y. Lv, T. Mei, H. Gong, D. Wi, Z. Xig, Y. Zhu and Y. Qian, Micro Nano Lett., 2012, 7, 439 CrossRef; L. Zhang, K. Takada, N. Ohta, M. Osada and T. Sasaki, J. Power Sources, 2007, 174, 1007 CrossRef; S. Komaba, C. Takei, T. Nakayama, A. Ogata and N. Yabuuchi, Electrochem. Commun., 2010, 12, 355 CrossRef; Z. Guo, Z. Cui and A. Manthiram, ACS Energy Lett., 2024, 9, 3316 CrossRef.
- M. Thackeray, S. Kang, C. Johnson, J. Vaughey, R. Benedek and S. Hackney, J. Mater. Chem., 2007, 17, 3112 RSC; F. Ding, J. Li, F. Deng, G. Xu, Y. Liu, K. Yang and F. Kang, ACS Appl. Mater. Interfaces, 2017, 9, 27936 CrossRef CAS PubMed; F. Wu, H. Lu, Y. Su, N. Li, L. Bao and S. Chen, J. Appl. Electrochem., 2010, 40, 783 CrossRef; Y. Wang, S. Liu, X. Guo, B. Wang, Q. Zhang, Y. Li, Y. Wang, G. Wang, L. Gu and H. Yu, J. Mater. Sci. Technol., 2025, 207, 266 CrossRef.
- B. Ammundsen, J. Paulsen, I. Davidson, R.-S. Liu, C. H. Shen, J.-M. Chen, L.-Y. Jang and J.-F. Lee, J. Electrochem. Soc., 2002, 149(4), A431 CrossRef.
- G. X. Feng, L. F. Li, J. Y. Liu, N. Liu, H. Li, X. Q. Yang, X. J. Huang, L. Q. Chen, K. W. Nam and W. S. Yoon, J. Mater. Chem., 2009, 19, 2993 RSC.
- H. Notohara, K. Urita and I. Morinaga, ACS Appl. Mater. Interfaces, 2023, 15, 30600 CrossRef; J. Kikkawa, T. Shiotsuki, Y. Shimo, S. Koshiya, T. Nagai, T. Nito and K. Kimoto, Jpn. J. Appl. Phys., 2018, 57, 035802 CrossRef.
- R. Qi, B. D. L. Campéon, I. Konuma, Y. Sato, Y. Kaneda, M. Kondo and N. Yabuuchi, Electrochemistry, 2022, 90(3), 037005 CrossRef.
- Z. Ji, M. Hu and L. Xin, Sci. Rep., 2023, 13, 14132 CrossRef.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Mater., 2016, 7, 13814 Search PubMed; Y. Kobayashi, M. Sawamura, S. Kondo, M. Harada, Y. Noda, M. Nakayama, S. Kobayakawa, W. Zhao, A. Nakao, A. Yasui, H. B. Rajendra, Y. Yamanaka, T. Ohta and N. Yabuuchi, Mater. Today, 2020, 37, 43 CrossRef CAS; A. Kitajou, T. Tanka, H. Miki, H. Koga, T. Okajima and S. Okada, Electrochemistry, 2016, 84(8), 597 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shohei
Matsuda
a,
Koji
Ohara
b,
Kazutaka
Ikeda
c and
Shunsuke
Muto
*a,
Shohei
Matsuda
a,
Koji
Ohara
b,
Kazutaka
Ikeda
c and
Shunsuke
Muto