
DNA-amphiphilic nanostructures: synthesis, characterization and applications
Nishkarsh
Jain†
 a,
Ankur
Singh†
b and
Dhiraj
Bhatia
*b
a,
Ankur
Singh†
b and
Dhiraj
Bhatia
*b
aDepartment of Biotechnology, Thapar Institute of Engineering and Technology, Prem Nagar, Patiala, Punjab 147004, India
bDepartment of Biological Sciences & Engineering, Indian Institute of Technology Gandhinagar, Palaj, Gujarat 382355, India. E-mail: dhiraj.bhatia@iitgn.ac.in
First published on 4th November 2024
Abstract
DNA's extraordinary potential reaches far beyond its role as a carrier of genetic information. It serves as a remarkably adaptable structural foundation for constructing intricate nanostructures with a diverse range of functionalities. This inherent programmability sets DNA apart from other biomolecules like peptides, proteins, and small molecules. By covalently attaching DNA to synthetic hydrophobic moieties, researchers create DNA amphiphiles capable of interacting with artificial lipid bilayers and cell membranes. These hybrid structures have rapidly gained prominence due to their promising potential in the medical field. This review provides a comprehensive overview of the latest advancements in the synthesis of DNA amphiphiles and their assembly into well-defined nanostructures. It explores the diverse applications of these nanostructures across various medical domains, including targeted drug delivery, innovative immunotherapies, and gene-silencing techniques. Moreover, the review delves into the current challenges and prospects of this rapidly evolving field, highlighting the potential of DNA hybrid materials to revolutionize medical treatments and diagnostics. By addressing the limitations and exploring new avenues of research, scientists aim to unlock the full potential of DNA nanotechnology for the benefit of human health.
Introduction
In the natural world, every organism has a multi-tiered and organized structure. Maintaining this complexity at the nanoscale has given rise to highly functional nanostructures, which have garnered significant interest from chemists due to their complexity and functionality. These supramolecular structures arise from self-assembling nucleic acids, proteins, and other (macro)molecular components through hydrogen bonding, metal coordination, hydrophobic interactions, pi–pi interactions, and other non-covalent forces. A widely used and tested method for constructing diverse nanostructures of size ranging from 5 to 100 nm is the bottom-up strategy (Fig. 1).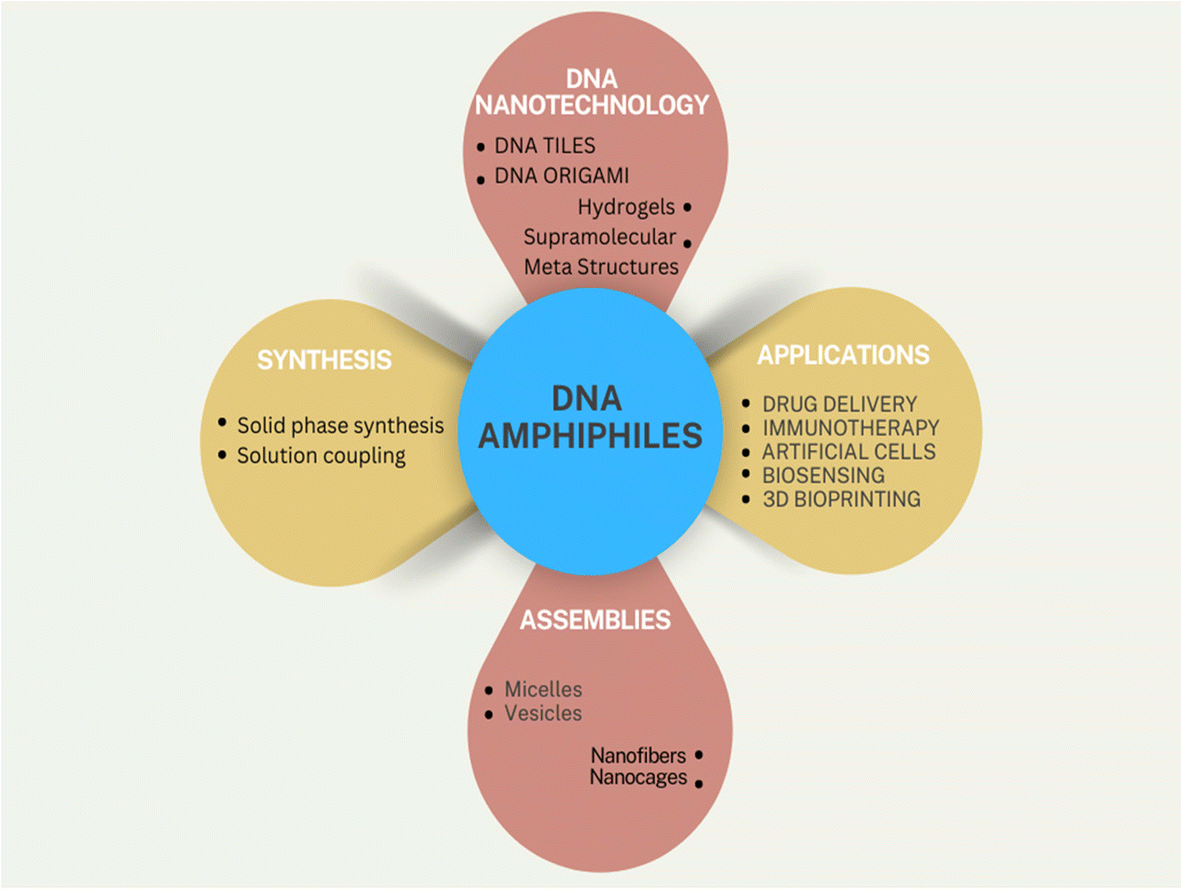 | ||
| Fig. 1 Schematic illustration of DNA amphiphiles, their synthesis methods and applications. | ||
Beyond their numerous biological roles, nucleic acids are among the most promising biomacromolecules for applications in biotechnology,1–3 medicinal chemistry,4 and materials science.5–7 As therapeutic agents, nucleic acids work by inhibiting either DNA or RNA expression, thereby preventing the production of disease-associated proteins.8 The distinctive programmability of DNA gives it an advantage over other molecules, such as peptides, proteins, lipids, and small molecules. DNA exhibits unique properties, such as sequence specificity, molecular recognition, and programmable self-assembly.9–11 These properties allow DNA to be organized into various sizes and shapes, creating addressable and programmable nanostructures with features like high drug loading, enhanced tumor targeting, and improved enhanced permeability and retention (EPR) effects.12–14 Each strand of DNA is formed by several components, including a pentose sugar (deoxyribose), a phosphodiester backbone, and a nitrogenous base (purine and pyrimidines). Taking advantage of these specific functional groups and the underlying chemical interactions, we can synthesize functionalized biopolymeric hybrids with desirable therapeutic and biological effects using DNA strands and another desirable biomaterial candidate. The range of functionality and potential application of the resultant hybrid biomaterial varies with varying counterparts, including but not limited to inorganic nanoparticles,15 organic molecules, and biological macromolecules like peptides or proteins.16 Dr Ned Seeman realized the great potential of DNA as a polymeric material and demonstrated it by building unusual DNA structures in his pioneering work in 1991.17 His contribution created the groundwork that laid the foundations for generating a myriad of intricate DNA architectures, ranging from one-dimensional constructs and complex 2D patterns to highly precise nanoscopic tubes, convex polyhedra, strings, 3D origami,18–23 and other dynamic nanodevices.24–27 These innovative structures, along with the deepened understanding of their assembly mechanisms, created new possibilities for combinations of various molecules that have paved the way for using this material in multidimensional systems like DNA origami28–30 and potential applications in emerging fields such as nanoreactors,31,32 sensors,33 logic operations,34,35 molecular computers,36,37 and diagnostics.38–40 The primary structural feature enabling these designs is the persistence length of double-stranded DNA (dsDNA), which is 50 nm or 150 bp (base pairs). This implies that the DNA strand behaves like a rigid-rod-like polymer at lengths lower than 50 nm, serving as an excellent mechanically stable polymer platform for building higher-order supramolecular assemblies. Additionally, technologies have been developed to engineer DNA molecules that can strongly bind to various target molecules (aptamers) or exhibit catalytic activity (DNAzymes).41–44 Despite their potential, the clinical use of therapeutic nucleic acids (TNAs) is limited by challenges in delivery, reduced bioavailability, and quick renal clearance. For example, negatively charged cell membranes naturally block the entry of foreign polyanionic nucleic acids.45 Even when these nucleic acids enter cells, they are often degraded by DNases or RNases to prevent their integration into the genome.46 Additionally, TNAs must be targeted accurately to specific cells while minimizing side effects on non-target cells.47 These obstacles significantly hinder their efficacy as therapeutic agents and limit their broader in vivo applications.48,49
One possible solution to this is the integration of amphiphilic properties in the DNA nanostructures. Amphiphiles are molecules characterized by the presence of both hydrophilic and hydrophobic groups.50 In aqueous solutions, the hydrophobic groups tend to cluster together, while the hydrophilic groups interact with the water. This behavior leads to the formation of various structures such as vesicles, micelles, tubes, and fibers,51–54 depending upon the specific interaction between the solute and the solvent, solute concentrations, temperature, pH, etc. These amphiphiles, although having numerous applications, have certain limitations. Amphiphiles in medical biology face challenges related to stability and aggregation, biocompatibility and potential toxicity, targeted delivery, premature drug release, and unwanted immune responses, which can be overcome by introducing DNA nanostructures, making them ideal platforms for enhancing the therapeutic potential of amphiphiles in medicine. To enhance the delivery of TNAs and facilitate DNA insertion into cell membranes, increasing the hydrophobicity of nucleic acids is a common strategy. This involves chemically conjugating DNA with hydrophobic groups, creating DNA amphiphiles that can efficiently and stably integrate into live cell membranes.55–58 The self-assembly of polymer–nucleic acid conjugates, driven by hydrophobic, electrostatic, and other intermolecular interactions, results in various morphologies, including spherical micelles, worms, and vesicles.48 These DNA amphiphiles can also be functionalized with groups that enable specific targeting and biocompatibility, offering great potential for biomedical applications.59–62 They possess intrinsic properties such as redox, photophysical, and biological functions, allowing for the rational design of complex assemblies. The scope of this review is to broaden the horizons of DNA amphiphile-based therapeutic systems in biology, where both the DNA and the amphiphilic counterpart synergistically stabilize and enhance the functionality of the hybrid macromolecule. This review also delineates the synthesis and applications of amphiphilic DNA conjugates in biomedical and clinical settings.
Synthesis
DNA amphiphiles typically comprise DNA as a hydrophilic segment, while a small molecule or polymer comprises the hydrophobic segment. Lipids suit the best as an ideal hydrophobic molecule, and the DNA-lipidification process yields such amphiphilic hybrid structures. The insertion of the lipid moiety can take place either within the DNA strands or at either of its ends,53,63–66 allowing the formation of structurally diverse complexes. Solution-phase coupling and solid-phase synthesis are two general methods to prepare modified DNA-conjugated amphiphiles. However, solution-phase coupling is widely utilized for conjugating hydrophilic molecular groups67–69 and shows very low hydrophobic molecular conjugation yields due to solvent incompatibility.70,71 Therefore, solid-phase modification, which often requires organic solvents and solid support, is highly suitable for preparing DNA–organic molecule amphiphiles. With the advancements in the field of de novo chemical synthesis of DNA strands, the robustness and automation of site-specific coupling reactions have experienced a major growth spurt. Implementing specialized solvents, high turn-over catalysts, extreme reaction conditions in controlled environments, and automated robots for high-throughput response has yielded precise control, high yield, and more stable DNA amphiphilic products.66 Here in this section, we will briefly introduce the solid-phase modification, and moreover, the recently developed methods, including polymerase chain reaction (PCR), post-polymerization, and organic-phase reaction, will also be discussed.Solid-phase modification
Four decades ago, Michelson and Todd demonstrated the formation of dinucleotide containing a 3′ to 5′ internucleotide linkage.72 Compared with the success back then, now it is possible to synthesize DNA strands of up to 200 nucleotides in a single run, which has widely broadened the field, thanks to the higher stability of deoxyribonucleoside phosphoramidites. Currently, the commercial-scale synthesis of oligonucleotides is automated and prepared by iterative chemical synthesis on solid supports (controlled pore glasses, [CPG]) using activated phosphoramidite materials.73The de novo synthesis of DNA strands using phosphoramidite chemistry can be broken down into a four-step cycle, adding nucleotides one at a time to a growing oligonucleotide chain coupled to a solid scaffold. The first step incorporates the removal of the dimethoxy trityl (DMT) group from the DMT-protected nucleoside phosphoramidite by treating it with trichloroacetic acid. This step is, therefore, aptly called deblocking or deprotection. The second step consists of base coupling using a fresh DMT–nucleoside phosphoramidite complex onto the freshly exposed 5′-hydroxyl ends of the first nucleotide (bound to a solid scaffold). In the third step, the capping of the unreacted 5′hydrozyl terminal takes place by acetylating and making the oligonucleotide chain insert for further nucleoside addition reactions. The final step involves an oxidation step using iodine, which converts the phosphite (PO33−, trivalent) to a phosphate (PO43−, pentavalent) group, producing a cyanoethyl phosphate group. These four steps are repeated multiple times to yield the DNA strands of the intended length. Once the strands reach the desired length, the completed oligonucleotides are washed off from the solid scaffold, and the base–phosphate backbone protecting groups are removed to yield the final product. This reaction chemistry offers a higher yield at 99% at each cycle.
Using the same phosphoramidite chemistry, scientists were able to synthesize a phosphorothioate backbone based on oligonucleotides. The phosphorothioate DNA (PSDNA) offers exceptional resistance against nucleases, which can easily cleave normal DNA with a phosphodiester (PODNA) backbone. The PS group within the PSDNA backbone offers higher reactivity as compared with the PO group, thereby opening doors to exciting and fruitful chemical reactions with groups including electrophiles like alkyl halides, maleimides, bromoacetic functional groups, etc. Furthermore, the end-terminal PS group is more reactive than PS groups in the middle of the oligonucleotide chain, making it ideal for end-to-end chemical modification. Zhang and team leveraged this concept and demonstrated for the first time PSDNA-Paclitaxel-based spherical nucleic acid (SNA)-like micellar nanoparticles as an antitumor agent.74 They formed a hybrid di-block of normal DNA (PODNA) with phosphorothioate DNA (PSDNA). This di-block copolymer actively reacted with the bromo-benzyl group of a modified paclitaxel (hydrophobic molecule), especially at the PS-rich backbone, forming a hybrid DNA amphiphile that could further form a spherical core. Enriching the SNA with tumor-targeting aptamer oligonucleotides made this an ideal drug delivery nanoparticle against a tumor site. They showed a 53% drug loading efficacy of SNA, active tumor targeting, enhanced tumor inhibition, and reversal of drug resistance in both in vivo and in vitro systems. Their work demonstrated the formation of a DNA amphiphile using a facile synthesis of PSDNA and the PSDNA-based quick conjugation strategy with efficient hydrophobic drug loading capacity, illustrating its significant help in drug delivery applications. Their work paves the way for an easier and more convenient conjugation strategy using phosphorothioate's chemical reactivity, which opens the doors for the next generation of DNA amphiphiles.
In commercial synthesis, the DNA is synthesized from 3′-end to 5′-end due to the standard procedure, but it should be mentioned that it is also possible to synthesize DNA from 5′ to 3′ with special reagents. Solid-phase functionalization is mainly based on the same phosphoramidite chemistry;75 the hydrophobic groups could be easily coupled with the 2-cyanoethyl-N,N-diisopropylphosphamidite group, which is then modified to the DNA on the solid support CPG. Due to the compatibility of different organic solvents in the solid phase method, it is believed to be a universal method for hydrophobic functionalization. It should be noted that this coupling reaction is sensitive to water and oxygen, so it needs to be operated under anhydrous and anaerobic conditions. The solvent used in a commercial synthesizer is anhydrous acetonitrile; therefore, molecules for intended conjugation to DNA should possess decent solubility in acetonitrile. When the hydrophobic groups exhibit low solubility in acetonitrile, the coupling could also be performed manually in any other available solvents. However, the choice of solvent might vary the yield of products formed. Following the DNA synthesis step by the well-studied and well-optimized phosphoramidite chemistry, several other chemical methods facilitate conjugation between the DNA and the amphiphilic molecules. In 2006, Nardin et al. demonstrated the formation of amide bonds between amino-terminated poly(butadiene) [PB, hydrophobic component] and the carboxy-modified nucleotide strand molecules by using a common solid-phase synthesis approach before the cleavage step of the oligonucleotide from the solid support.76 Huisgen cycloaddition (copper-catalyzed alkyneazide cycloaddition) between azido-terminated DNA and alkyne functionalized hydrophobic molecules77 was demonstrated by the Barthélémy group in 2010. Sonogashira coupling78 and the functionalization of 3′-terminus DNA with phosphite amide chemistry79 can also functionalize DNA at the end with hydrophobic molecules, broadening the application of solid-phase synthesis (see Fig. 2A).
 | ||
| Fig. 2 Methods of synthesis of DNA amphiphiles. (A) Solid-phase synthesis methods of DNA-amphiphile conjugates, utilizing click chemistry post-synthesis of DNA strands. (B and C) PCR and HCR-based chain elongation methods to synthesize DNA-amphiphile conjugates of longer length segments. Reproduced with permission. (D) Surfactant-based facile synthesis of amphiphile conjugates at the DNA terminus. Reproduced with permission. Copyright 2014, American Chemical Society. (E) Solution-phase synthesis. Reproduced with permission. Copyright 2014, Wiley-VCH. Frame-guided assembly (FGA) of DNA amphiphilic structures. (F) Inner-frame type FGA-based DNA-amphiphilic molecule consisting of DNA nano-octahedron (N-DNO) backbone encapsulated within the lipid bilayer. Reproduced with permission. Copyright © 2014 American Chemical Society. (G) Inner-frame type assembly based on the FGA approach using cuboidal DNA origami scaffold with precise loading of a polyT-amphiphilic strand (DTDOEG as the LHG) followed by surface coating with G2Cl-18 (principle amphiphile). Reproduced with permission. © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (H) Schematics of an outer-frame type assembled amphiphilic nanostructure using FGA approach. The DNA origami ring projects an ssDNA linker arm (handle), which binds to the anti-handle (DNA-DOPE amphiphile acting as the LHG). Additional lipids and detergents provide the necessary amphiphiles needed for the formation of vesicles assisted by DNA origami rings. The origami rings can be further cleaved to rescue the vesicles. The size of the DNA origami rings modulates the size of the vesicles formed. Reproduced with permission. Copyright © 2016, Springer Nature Limited (I) Schematics of a planar DNA origami platform to form a continuous 2D nanosheet layer of desired shape. Reproduced with permission. © 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In solution-phase couplings, the hydrophobe (hydrophobic molecule) usually undergoes conjugations at the terminal ends of the DNA strands because of the availability of the free terminal ends in the post-processing steps after DNA synthesis. The solution-phase synthesis, therefore, targets the interactions between exposed functional groups on either DNA or the hydrophobe. The chemical groups targeted via this approach include amines,80 thiols,81 or maleimides.82 The opposite nature of polar DNA as compared with the non-polar hydrophobe presents the problem of incompatibility which also can be seen with respective solvents, thereby affecting the overall production yield. To bypass this problem, we need to mask the surface charge of the DNA which makes it polar. We can do this by complexing the DNA strands with positively charged quaternary ammonium salt ions present in the amphiphilic surfactant molecules, making polar DNA solubilize in organic solvents and thus providing a common ground for both the DNA and the hydrophobe to interact and combine (Fig. 2D). By using this strategy, a wide variety of diverse DNA-amphiphilic products could be created. However, further optimization is needed to make the solution-based synthesis process more versatile and equally yield desirable.
Recently developed modification methods
Both the solid-phase and the solution-phase synthesis process often rely on the availability of free DNA strands, which is limited by the efficiency of the phosphoramidite chemical synthesis process. One major caveat pertaining to the chemical synthesis of DNA strands de novo is the limit of degree of polymerization (∼50 nucleotides). Exceeding this limit causes rapid deterioration in the specificity of nucleotide addition reaction steps. Strategies involving polymerase chain reactions (PCR) or the hybridization chain reactions (HCR) are being used to circumvent this problem. Through these approaches, the size-limiting length of the DNA–hydrophobe conjugate can be elongated to our desired length and does not perturb the yield much.PCR-based method for DNA-amphiphile synthesis
By utilizing either solid-phase or solution-based approaches, one can easily synthesize a DNA–amphiphile copolymer block, where the size of the DNA lies within 50 nucleotides. Use of the DNA strand as the template and a pair of forward and reverse primers in the presence of a DNA polymerase enzyme can catalyze the extension of the DNA strands in an enzymatic fashion. The efficiency of the most commonly used Taq DNA polymerase enzymes is very high (∼60 nucleotides per s per enzyme unit) with an error rate of 1 in 4000–5000 nucleotide insertions.83 Note that Taq polymerase does not have proofreading activity. Implementing a PCR-based amplification regime, we can extend the lengths of the DNA strands to a desirable length, further opening up doors for novel nanostructures of therapeutic significance (see Fig. 2B).The PCR-based method is highly suitable; however, it lacks specificity regarding the size of the DNA amplified. There is no control mechanism that monitors size uniformity with DNA strands. The polymerase will keep adding raw dNTPs (deoxynucleotide triphosphates) until it runs out of the dNTP stock. To maintain size uniformity and precise fabrication of hybrid DNA-amphiphilic block copolymers, another technique has been implemented, namely Hybridization Chain Reaction (HCR).
HCR-based method for DNA-amphiphile copolymer synthesis
The complementary properties of DNA strands are taken advantage of in the case of an HCR-based strategy to synthesize DNA–amphiphile block copolymers. Using this strategy, either the same or different hydrophobe groups can be ligated based on sequence-dependent hybridization of their DNA counterparts. As a result, site-specific addition of hydrophobic moieties could be implemented (as illustrated in Fig. 2C).Post-polymerization
Post-polymerization is another such method to synthesize DNA amphiphiles based on solid phase synthesis. An atom transfer radical polymerization (ATRP) initiator phosphoramidite was functionalized on a 23-mer DNA strand by Matyjaszewski and Das. Activators generated by electron transfer (AGET) ATRP were used to functionalize obtained DNA polymers in solution or alternatively directly on solid support84 (Fig. 2E). Such an approach may provide conjugated polymer segments with well-defined molecular weight and narrow molecular weight distribution, purifying the DNA conjugates rapidly by the solid-support method.Organic-phase reaction
Solution coupling was deemed incompatible with DNA and hydrophobic molecules. To resolve this, Herrmann achieved DNA modification and polymerization through an organic-phase reaction.85 As illustrated in Fig. 2C, the surfactant complexes of DNA formed from the electrostatic interaction of oligonucleotides with cationic surfactants isolated in the aqueous phase are soluble in some organic solvents, such as DMF, DMSO, THF, and CHCl3. These molecular aggregates permit terminal acylation of 3-amine-modified oligonucleotides by different hydrophobic NHS esters, e.g., pyrene, triphenylphosphine (TPP), PPO, PS, etc. Moreover, novel DNA sidechain homopolymers were generated by polymerizing norbornene-functionalized DNA–surfactant complex monomers. The same organic-phase strategy was then utilized by Zhang and his colleagues.86 Nanoscale structures made of poly-DNA were prepared from protected DNAs which had been modified with norbornene units using ring-opening metathesis polymerization. The products included brushes-type DNA graft polymers, diblock copolymers of DNA and polythylene glycol (PEG), and micelles of block copolymers based on DNAs. This shows that organic-phase synthesis can show promise in using DNA in materials science applications.Micelle-templated approach
Sleiman and her colleagues also innovated a micelle-templated method enhancing the reactivity of highly hydrophobic molecules with nucleic acids. This was a part of her work that modified the pristine DNA strand along with the polymer segment (Fig. 2D).87 First, DNA strands were linked to 1,12-dodecanediol phosphoramidite units (hexaethylene, or HE) to obtain DNA amphiphiles. After that, the resulting DNA amphiphiles self-assembled into monodisperse DNA micelles. A complementary non-hydrophobically modified DNA strand may orient its reactive group (amino functional) towards the micelle's centre through DNA hybridization. During this period, however, NHS ester hydrophobic molecules accumulate at the core of DNA micelles to carry out the coupling reaction. In so doing, one can greatly enhance coupling efficiency. The approach of using DNA micelles as templates would lead to an increase in instances where DNA–organic hybrids are used with nanometer pore imitators and nanoparticles for oligonucleotide and drug delivery, along with other applications within DNA nanotech.Frame-guided assembly
The chemistry of synthesizing DNA amphiphiles can form structures at the nanoscale level but also be extrapolated to form microstructures. One such approach achieves this feat by triggering the assembly of amphiphiles by precise positioning of leading hydrophobic groups (LHGs), which are often part of a scaffold or a frame. This process is popularly known as frame-guided assembly, or FGA for short. The inspiration for FGA-based micro and macroscopic molecular assembly has been derived from the structural organization of the cytoskeleton, membrane proteins, and the lipid bilayer.The simplicity of this process makes it highly desirable for scaling up DNA amphiphilic nanostructures to form microscopic structures with desirable biological properties. This method relies heavily on the presence of a frame or a scaffold, where DNA nanotechnology-based molecular platforms can show their prowess. The nanoscale precision of DNA scaffolds coupled with spatially distributed LHGs can trigger a desirable assembly of amphiphilic molecules, which can easily be utilized in areas where membrane fusion is of critical importance. Depending on the positioning of the embedded LHGs, the frames can be classified into the inner frame, outer frame, and planar frame.
First, the inner frame, as the name suggests, is a scaffolding structure that supports the growth of assembly outwards, and the newly formed amphiphilic layer is exposed to the outside (Fig. 2F and G).88–90 This process of assembly is analogous to the bricklaying work, where the freshly added bricks (free amphiphiles) are assembled over a frame made of concrete and steel rods. The second method uses an extrinsic frame where the nucleation happens at the inner side of the frame, and the assembly is constrained by the boundaries defined by the frame used (Fig. 2H).91,92 The assembled amphiphilic molecules acquire the shape of the mold (external frame) just like clay gets the impression of the mold in which it has been cast. The internal assembly of amphiphiles is protected from the external environment and can be leveraged to deliver hydrophobic drugs or small molecules into the cells. The outer frame-based assembled structures retain phenomenal uniform structure while minimizing the batch-to-batch variability of amphiphilic macrostructures. Lastly, the planar frame method provides LHG to seed the assembly of 2D membrane-like sheets of amphiphiles, which are restricted in the frame's shape and follow the frame's geometry in 2D orientations (Fig. 2I).93–95
The underlying mechanism of FGA is based on the electrostatic and van der Waals interactions between LHG and the amphiphiles. By controlling the concentrations, nature, and affinity between LHG and the amphiphiles, we can precisely control the formation and reproducibility of FGA-based amphiphilic microstructures. The success of FGA is often based on the spatial arrangement, density, similarity, and concentrations of the LHG and the amphiphiles. Also, controlling temperature,96 pH,97 and solute concentrations within the reaction system will provide a better grasp of kinetic and thermodynamic interactions between the LHG and amphiphiles.
Factors affecting yield of synthesis
The previously discussed methods can be employed to fabricate a DNA amphiphile with desirable intended applicative need. However, various methods have their own short-falls and technical limitations which further limit the yield of DNA amphiphile synthesis. The yields of DNA amphiphiles with coupling chemistries can vary depending on the specific chemistry used, the reaction conditions, and the efficiency of purification methods. Typically, yields range from 30% to 99% after coupling, though the actual yield depends heavily on the reaction efficiency and the purification strategy (e.g., HPLC, dialysis). Different factors play a role in determining the overall reaction efficiency in the following ways.○ Solvent compatibility – The amphiphilic nature of the molecules involved presents challenges in selecting suitable solvents. DNA is hydrophilic, while the hydrophobic component (e.g., lipids, polymers) typically prefers organic solvents.
○ The solution-phase coupling method has a significantly lesser yield than solid-phase coupling due to the insolubility of the hydrophobic molecule, leading to aggregation or precipitation of hydrophobic molecules before conjugation occurs. Complexation with surfactants can improve yields, but these processes remain lower in comparison with solid-phase techniques. An innovative approach involves using surfactants like cetyltrimethylammonium bromide (CTAB) to enhance solubility in organic solvents, leading to transformation yields exceeding 80%. Using mixed solvent systems or surfactants to solubilize both components can improve reaction yields.98 Traditional aqueous solutions may yield less than 30% for certain hydrophobic modifications due to the poor solubility of the reactants in water.98,99
○ DNA structure and length – The structure of the DNA (single-stranded vs. double-stranded) and its length significantly impact the coupling efficiency. Longer DNA strands may experience steric hindrance or folding, limiting access to reactive groups.
○ Presence of additives – Catalysts or additives, such as copper in click chemistry or salts like NaCl, can enhance the efficiency of certain reactions by stabilizing intermediates or driving the reaction forward. In their absence yields may drop significantly due to incomplete reactions. For example, CuAAC (click chemistry) requires a copper catalyst to proceed efficiently. The incorporation of coupling agents such as HATU has been shown to improve yields significantly. In some cases, yields can reach as high as 80% or more when using optimized conditions and appropriate solvents.98
○ pH of reaction environment – The pH of the reaction environment can affect the ionization state of reactive groups on both DNA and hydrophobic molecules. Many coupling reactions (e.g., amide bond formation) are pH sensitive.
○ Other factors such as the reactivity of functional groups, the purity of starting materials, concentration of reactants, reaction time and temperature also influence yield. Solid-phase synthesis of DNA amphiphiles generally exhibits high reaction efficiencies, often exceeding 95% under optimized conditions. However, specific yields can vary based on structural modifications, coupling strategies, and the complexity of the target molecules. Conventional solid-phase synthesis of oligonucleotides has achieved stepwise coupling efficiencies of approximately 99%.100 In a study focusing on solid-phase organic synthesis on unprotected DNA, coupling and Fmoc-deprotection reactions were reported to proceed with efficiencies greater than 95% within 30 minutes.101 This indicates that solid-phase methods can effectively facilitate rapid and efficient synthesis processes.
The specific efficiencies of some different coupling reactions can be summarised as:
○ Click chemistry (copper-catalyzed azide-alkyne cycloaddition, CuAAC) – Generally, CuAAC reactions can achieve yields of up to 95% under optimized conditions, particularly when using well-defined copper(I) catalysts and appropriate ligands. In practical applications, yields can vary but are often reported around 70–90%.102,103
○ Carbodiimide coupling (EDC/NHS chemistry) – Research indicates that while EDC alone can yield lower efficiencies, the combination with NHS usually improves solubility and reaction outcomes, achieving optimal coupling efficiencies around 85–90% under favorable conditions.104
○ Maleimide–thiol coupling – Studies have reported that maleimide–thiol conjugation can reach up to 84% efficiency when optimized for specific reactant ratios and conditions.105
○ Amide bond formation – Research has demonstrated that using Candida antarctica lipase B can yield over 92% conversion in amide bond formation reactions within a short time frame.106
Other than the above-mentioned coupling chemistry, some non-covalent methods have also evolved for DNA modification. These non-covalent methods include various strategies that leverage molecular interactions without forming permanent bonds. These reaction types involve:
○ Host–guest interactions: This method utilizes the reversible binding between host molecules (like β-cyclodextrin) and guest molecules (such as adamantane) to form supramolecular structures. This approach allows for the formation of DNA-decorated vesicles, which can be useful in drug delivery and diagnostics due to their stimuli-responsive nature.
○ Hydrophobic interactions: Amphiphilic DNA can be synthesized by incorporating hydrophobic groups that promote self-assembly through hydrophobic interactions. For instance, cholesterol-modified siRNA has been constructed using intercalating agents that bind non-covalently to nucleic acids, enabling the formation of stable nanostructures.
○ Electrostatic interactions: Ionic surfactants can interact with the negatively charged phosphate backbone of nucleic acids, facilitating their solubility in organic solvents. This method has been shown to yield high transformation efficiencies while maintaining the structural integrity of the DNA.
○ Self-assembly techniques: DNA amphiphiles can self-assemble into various nanostructures through complementary base pairing and hydrophobic interactions. This includes the use of DNA–dendron hybrids that can guide the assembly of other amphiphilic molecules into hetero-vesicles.
○ π–π interactions: These are a type of non-covalent conjugation method that occurs between the aromatic rings of nucleobases in DNA. These interactions enhance the stacking stability of nucleic acid structures, allowing for effective assembly and functionalization of DNA-based materials. By leveraging π–π interactions, researchers can create more complex and stable DNA architectures without the need for covalent modifications.
The study of non-covalent interactions for DNA modification offers several advantages, including simplicity in synthesis, reversibility, and the potential for dynamic structural changes. These methods can facilitate the creation of stimuli-responsive systems and allow for easier purification processes. It's worth noting that while recent covalent approaches like the micelle-templated and surfactant-aided methods have successfully improved reaction yields, they are currently limited to synthesizing nucleic acid amphiphiles with modifications only at the terminal ends. In theory, non-covalent methods could be used to modify internal regions of nucleic acid amphiphiles more easily. However, research in this area is still in its early stages. With further exploration of new non-covalent interactions, we may soon discover innovative ways to create internally modified nucleic acid amphiphiles.
The scope of scale-up is also possible through choosing the right components to begin and with an appropriate, cost-effective and facile synthesis method. Furthermore, methods such as the FGA are ideal to incorporate directed scale-up to boost yields at commercial scales.
Characterization
DNA amphiphiles come in all shapes and sizes. Depending on the type of component used to represent the hydrophobic part, given that hydrophilic group is represented by DNA, we can start to appreciate the diversity in size, shape, charge, affinity, stiffness, elasticity, etc. As a highly versatile material, various characterization techniques and methods are employed to characterize physical, chemical or biological aspects of it. These aspects may include structure and shape assessment, electrostatic interaction of the nanostructure within itself and with the cells, hydrophobicity/hydrophilicity testing, self-assembly, stability testing, drug loading efficiency and release profiles, biodistribution, pharmokinetics etc.Size assessment
Size estimation of materials at the nanometre scale can easily be performed by the following methods or techniques.Electrophoretic mobility shift assay (EMSA) is an assay to test the relative mobility shift of compounds in various combinations, under the influence of an external electric field while passing through a porous gel base material. Both agarose and polyacrylamide gels could be used to serve as the base within which the material of interest has to be separated. Samples including DNA strands alone, DNA–amphiphile conjugates, or DNA amphiphile + cargo molecules, can be made to run under the effects of electrophoretic movement within polyacrylamide gel to see the increase in the size between all three compounds. The samples with the least weight of size tend to travel faster within the gel, whereas the bulkier or larger-order complexes cannot travel farther. After staining the compounds with specific dyes (Ethidium Bromide for staining DNA, for example) and monitoring their relative band shift coupled with controls and ladders of known marker sizes, it is possible to roughly estimate the average size of the complex, association dynamics, and cargo-loading efficiency. This technique employs a simple-to-use protocol, easy setup and provides a lot of information post-processing of the images taken of the bands. However, the limitation of this technique falls on the extent of hydrophobicity within the sample; as the hydrophobicity increases, the samples cannot properly run through the gel, thereby providing incomprehensible results. The technique can also be employed to work with thin capillaries filled with the base gel instead, giving better control and also being easier to automate.
Dynamic light scattering (DLS) measures the hydrodynamic radius of the samples that are injected into a small chamber (cell) with individual compartments. The injected samples along with the solvent fill up the entire compartment and the movement of samples is purely dictated by Brownian motion. The movement of the samples can be detected by bright laser beam illumination from the bottom and a high-speed camera to record the scattered light from the particles.
The hydrodynamic radius of the injected samples can be calculated using the Stokes–Einstein equation:
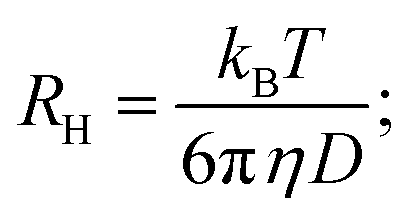
By knowing the diffusion coefficient D, it becomes possible for us to estimate an average size and particle distribution using DLS. Note that the hydrodynamic radius is not the exact size, but an estimated size of the particle encaged by water molecules. The actual size is less than the hydrodynamic size.
High-performance liquid chromatography (HPLC) – is a powerful analytical tool used to identify, separate, and quantify components of a mixture. It incorporates a pressure-driven separation of compounds of a mixture by injecting the samples with the mobile phase (solvent of interest) and making it pass through the stationary phase (granular material packed in a column) where the separation occurs. The nature of the stationary or mobile phase changes with the intent of separating molecules with varying nature. For a hydrophilic material, the mobile phase is organic solvent (non-polar) and the stationary phase is polar (silica gel), while for a hydrophobic material the reverse is true. The injected samples based on their affinity with the stationary phase are separated at different times of elution. Coupled with a UV-Vis detector, a chromatogram of each individual component is achieved. The smaller molecules pass slowly through the pores of the stationary phase, while larger molecules pass through easily and quickly. HPLC is easy to automate and can be coupled with other instruments like Mass-Spectroscopy for detailed analysis of the samples based on their mass to charge ratio.
Shape, morphology and assembly
Techniques like atomic force microscopy, electron microscopy, and confocal laser scanning microscopy are used for studying the morphological features of nanostructures, and verifying their assembly.Atomic force microscopy (AFM) – works by scanning a sharp nanoscale cantilever tip across the surface of a sample, measuring forces between the tip and the sample to generate high-resolution 3D images. It is used to analyse the morphology and verify the assembly of amphiphilic DNA nanostructures by providing detailed topographical images, allowing for the observation of their surface texture, height, overall shape, stiffness and composition.
Scanning electron microscopy (SEM) – uses a focused electron beam that scans the surface of the sample, detecting secondary or backscattered electrons emitted from the sample to produce detailed surface images revealing surface uniformity, structure formation, and potential aggregation.
Confocal laser scanning microscopy (CLSM) – uses a laser to scan the sample point-by-point and collects fluorescence from specific planes within the sample. A fluorescent dye is conjugated with the nanostructure, leading to acquisition of sharp 3D images to confirm appropriate assembly, internal structure, and spatial distribution in real time. It is highly essential to probe the whereabouts of fluorescently tagged molecules within cells or tissue sections.
Electrostatic interaction
Analytical techniques like zeta potential analysis, nuclear magnetic resonance etc., can be used to study the electrostatic interactions among the amphiphiles themselves and with cells along with their hydrophobicity/hydrophilicity.Zeta potential analysis – measures the net surface charge of a particle by measuring the velocity of charged particles under an applied electric field, which gives the zeta potential, an indicator of surface charge. It can reveal how the charged DNA backbone and any modifications contribute to their overall charge and electrostatic interactions in biological environments along with insights into their colloidal stability.
Nuclear magnetic resonance (NMR) operates on the principle of nuclear spin when subjected to a magnetic field, detecting the local magnetic environment of nuclei (usually hydrogen). It can provide molecular-level details of the amphiphilic DNA nanostructures, particularly their dynamic interactions and conformations in response to different environments. It allows the study of electrostatic interactions through shifts in chemical environments and changes in relaxation times, offering a way to observe how these nanostructures interact with cell membranes and other biological molecules.
Conjugation
It is very essential to ensure the correct conjugational patterns of DNA strands with other polymers or molecules forming stable structures. Techniques like Fourier transform infrared spectroscopy (FTIR) and mass spectrometry help evaluate this reaction.Fourier transform infrared (FTIR) spectroscopy operates on the principle of measuring the absorption of infrared light by molecules, causing vibrations in chemical bonds. Each functional group in a molecule absorbs at specific wavelengths, producing a unique spectrum. In studying amphiphilic DNA nanostructures, FTIR can detect changes in characteristic peaks, indicating successful conjugation by identifying new bonds or functional groups introduced during the process.
Mass spectrometry (MS) works by ionizing molecules and measuring the mass-to-charge ratio (m/z) of the resulting ions. It provides precise molecular weight and structural information. MS can confirm successful conjugation by detecting shifts in molecular weight corresponding to the added functional groups or conjugated molecules.
Self assembly, and association
Isothermal Titration Calorimetry (ITC), Circular Dichroism (CD) Spectroscopy, and Small-Angle X-ray Scattering (SAXS) are techniques commonly employed to study the self-assembly behavior of DNA amphiphiles.Isothermal calorimetry (ITC) is used to monitor the heat changes that occur during molecular interactions, such as when amphiphilic DNA nanostructures come together or break apart. By tracking these heat changes, ITC offers detailed insights into the thermodynamics of the process, including the binding enthalpy, entropy, and Gibbs free energy, which help clarify the factors driving the assembly or dissociation.
Circular dichroism (CD) spectroscopy, on the other hand, works by measuring how chiral molecules, like DNA, absorb left and right circularly polarized light differently. This allows scientists to observe changes in the DNA's secondary structure (such as helices or sheets) during assembly or dissociation, providing valuable information about the structural shifts involved.
Small-angle X-ray scattering (SAXS) provides details about the size, shape, and structural arrangement of DNA nanostructures in solution by analyzing how X-rays scatter at small angles. It is particularly useful for observing the formation and structural organization of these nanostructures, as well as any changes in their size or packing during dissociation.
Drug encapsulation capacity and drug release profiles
Various methods, such as UV-visible spectroscopy, HPLC, NMR, and release kinetics studies, can be utilized to evaluate the encapsulation capacity of DNA amphiphiles and their drug release profiles.UV-visible spectroscopy measures how much light a drug molecule absorbs at specific wavelengths. By comparing the absorbance levels before and after encapsulation, researchers can estimate how efficiently the drug is encapsulated within the DNA nanostructures. This technique can also be used to monitor how the drug is released over time by tracking changes in absorbance.
Release kinetics studies examine how quickly and through what mechanisms the drug is released from the nanostructures under various conditions, such as changes in pH or temperature. Techniques like dialysis or membrane diffusion can be applied to mimic physiological environments, allowing scientists to observe the drug's diffusion and release patterns over time.
Pharmacological studies
Characterization of DNA amphiphiles, including their distribution in the body and pharmacokinetics, can be studied using various biological assays, biodistribution studies, bioluminescence imaging, and other techniques.Bioluminescence imaging takes advantage of luciferase enzymes, which emit light when they catalyze the oxidation of luciferin. By tagging DNA nanostructures with luciferase, researchers can visualize and track their distribution in live animals in real time, offering insights into how these nanostructures accumulate in different tissues.
Radiolabeling involves attaching radioactive isotopes, such as iodine-125 or carbon-11, to DNA nanostructures. This allows scientists to track their distribution and metabolism within the body using imaging techniques like SPECT or PET. These methods detect emitted radiation, providing detailed data on the nanostructures’ pharmacokinetics and tissue accumulation over time.
Bioanalytical assays, like enzyme-linked immunosorbent assays (ELISAs), use specific antibodies to detect and measure target molecules linked to DNA nanostructures in biological samples. These assays offer valuable information about how the nanostructures interact with biological systems, helping to assess their uptake, efficacy, and therapeutic potential.
All methods and techniques have their respective pros and cons. Depending on the aspect of study, various modes or techniques can be employed to convey the physical and biochemical properties of DNA amphiphiles. Furthermore, because of two conflicting groups present in the DNA amphiphile, oftentimes a reductionist approach is employed for studying their behaviour separately.
Applications
Due to their inherent hydrophobic properties, nucleic acids can seamlessly integrate into lipid bilayers, thus enhancing their absorption by living cells. The shape, size, charge and the affinity of the hybrid DNA amphiphiles render them amenable for use in various biological situations. For example, a small cage-like morphology or an appropriate surface charge makes select DNA amphiphiles highly desirable for drug delivery applications. On the opposite spectrum a large, highly self-assembled micellar or bi-lamellar morphology is more desirable for use in membrane fusion, cell transfections, artificial cell fabrications, etc. Clearly the type of shape and physico-chemical attributes of the DNA amphiphiles drive the utility of these materials for several applications in diverse fields. These fundamental interactions, be they physical or chemical, including programmability, self assembly, and response to environmental conditions, form the basis for developing amphiphilic DNA structures with various biomedical applications, which we will delve into in this discussion.Drug delivery
Delivery of drugs inside living cells has been extensively conducted through the use of amphiphilic micelles and bilayer vesicles over the past few decades. The hydrophobic and hydrophilic parts of the amphiphilic nanostructures play a vital role in stabilizing the corresponding drugs and their delivery under in vitro and in vivo conditions.57,107–110 In one of the earliest attempts to develop DNA micelles, by Ji and Tae,70 a micelle block co-polymer structure for the delivery of antisense oligonucleotide (ODN) was made by conjugating biodegradable poly(D,L-lactic-co-glycolic acid) (PLGA) with ODN and delivered to murine fibroblast cells. PLGA formed the hydrophobic core, while ODN was the hydrophilic corona, as illustrated in (Fig. 3D). It had a size of less than 100 nm and had a relatively low critical micelle concentration (cms) which averaged to be 7.5, which was much less than previously reported oligo(methyl methacrylate)-poly(acrylic acid) micelles at that time.111 This made it eligible for renal exclusion, helped it escape the reticuloendothelial system, and provided enhanced cell permeability.112 The ODN-PLGA block copolymer showed excellent biodegradability and slowly degraded in the intracellular environment, and did not show an initial burst release but exhibited controlled release rates as opposed to ODN-encapsulated PGLA microspheres.113 It was also observed that the ODN-PLGA block copolymers were localized within the nucleus, and showed much improvement in cellular uptake compared with naked ODN. However, the exact mechanism for cellular entry and endosomal escape was not elucidated, and specific targeting of cells was not possible. Another group tried to elucidate the cellular uptake machinery by targeting Caco2 cells with folic acid-conjugated polypropylene oxide (PPO)-b-DNA block copolymeric micelles (Fig. 3C). They observed that fluorescently labelled PPO-b-DNA micelles revealed receptor-mediated endocytosis uptake, with an average diameter of 10 nm being the most efficient.60 The administration of Doxorubicin (Dox), an anticancer drug, was facilitated by loading the drug in the hydrophilic core, followed by the administration of Dox-loaded PPO-b-DNA micelles to Caco2 cells, resulting in high mortality rates and effective cytotoxicity amongst the cancerous cells.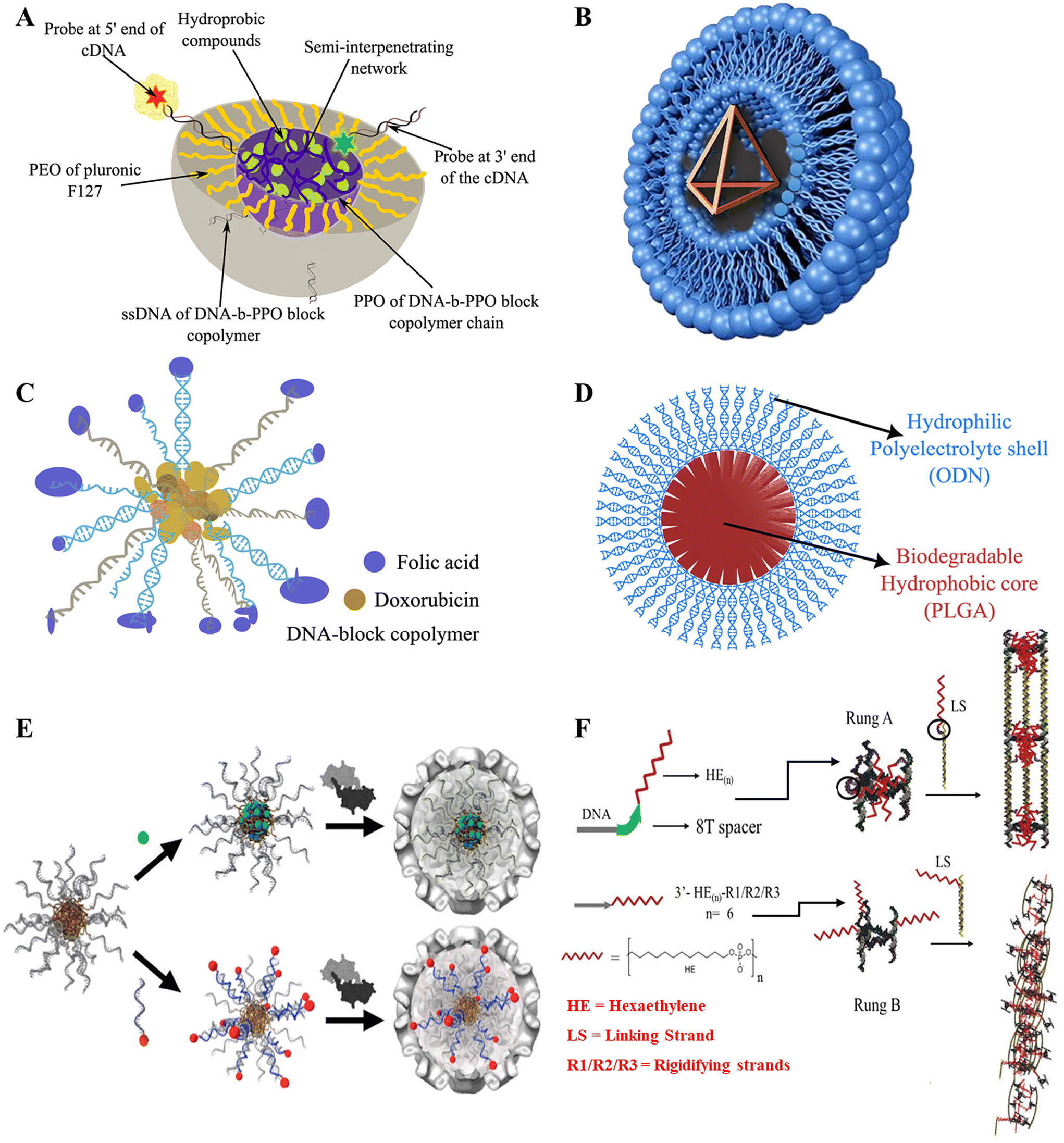 | ||
| Fig. 3 Different DNA-amphiphilic structures for drug delivery. (A) Mixed micelle of DNA-b-PPO and PEO-b-PPO-b-PEO forming an interpenetrating network in the core of the micelle with potential for dox delivery. Reproduced with permission. Copyright 2010, The Royal Society of Chemistry. (B) Lipoplex of DNA tetrahedron encaged within Lipofectin. (C) DNA block copolymer micelle with targeting unit folic acid (purple) and Doxorubicin (yellow ochre). (D) PLGA-ODN block copolymer micelle. (E) DNA Micelle-Templated VC Formations. Top: Loading of the hydrophobic drug in the core. Bottom: Loading hydrophilic compound by DNA-templated hybridization. Reproduced with permission. Copyright 2010, American Chemical Society. (F) Nanotube assembly based on DNA spacer conformations. Top: With spacer–intermolecular hydrophobic interaction at the core, it is more stable. Bottom: Without a spacer the hydrophobic core is unstable and compromises the stability of the DNA nanotube. Reproduced with permission. Copyright 2018, John Wiley and Sons. | ||
Although these are powerful new tools for cancer therapy, there is an inherent limitation to using micellar carriers in vivo: upon dilution below the critical micelle concentration (CMC), they tend to disassemble. One possible solution to this was found by Herrmann and group by creating micelles encapsulated by viral capsid.114 This study successfully demonstrated the encapsulation of DNA-amphiphilic micelles by viral capsid for better functionalization and stability. CCMV viral capsid was used to encapsulate different micelle compositions along with hydrophobic drugs in the core and hydrophilic dyes attached to the DNA surface with the help of cDNA (Fig. 3E). The study reported well-defined structures, with coat protein manipulation potential by chemical modification or mutagenesis, which allowed precise individual cell-targeting capabilities. It was an upgrade from the previous versions that made leakage difficult to contain from the porous capsid surface and only allowed small hydrophobic cargo without severe modifications.115 The capsid coat assembled under normal conditions and pH with the help of the DNA-amphiphilic micelle scaffold, unlike the previous examples.116 However, the size inconsistency of the particles might lead to non-uniform results, and needs to be studied further. In another notable work by the same group, complex micellar structures using Pluronic F-127 and DNA block copolymer were used.117 The Pluronic F-127 was formed by a triblock conjugated framework of poly(ethyleneoxide) (PEO) and poly(propyleneoxide) (PPO) blocks forming (PEOn-PPOm-PEOn). The PPO from the Pluronic and the DNA block copolymer formed the hydrophobic core, while PEO and DNA formed the hydrophilic corona, as described in Fig. 3A. PEO is a biocompatible and thermally responsive copolymer118 often used in various amphiphilic structures,119 but lacks specific cell targeting,120 while DNA block Copolymers (DBC) show instability at low critical micelle concentration (CMC)121 and higher DNA concentrations might trigger immune responses.122 It also can easily be functionalized due to hybridization by cDNA.58 In a mixture these polymers complement each other in overcoming each other's limitations. A UV-induced semi-interpenetrating network ensured the stability of the mix. Despite the advantages, the stability of the micelle for longer duration and in varying environments needs to be accounted for. Also, methods to reduce the complexity in the assembly and cross-linking of the mix should be developed.
Since then, many innovative nanostructures with different functionalities have been devised, including shape-shifting micelles,123 lipid-encapsulated DNA nanooctahedron,124 light-triggered disassembly of nanostructures made of payload molecules,125 dendritic alkyl chain-conjugated DNA nanocages,126 and many more. Similarly, in a notable example, fluorescently labeled DNA nanocages were inserted into cultured embryonic human kidney cells by Walsh et al.127 The efficiency of uptake by the cells was compared with and without the usage of a transfecting reagent (lipofection). The cationic lipid from the lipofection forms ionic bonds with the negatively charged DNA backbone, forming amphiphilic structures called lipoplexes (Fig. 3B). The nanocage was formed from 4 × 63 nucleotide strands. The formation of the DNA tetrahedron amphiphilic structure increased the efficiency of the uptake of cells. It provided more resistance against nuclease degradation than three 63 nucleotide-long ssDNA strands that were single-stranded, partially hybridized, and fully hybridized, each aided with and without lipofection. The size of the DNA tetrahedron formed was about 7 nm, which could not only cross through the cellular membranes but could also lead to increased renal clearance. Nanocages have been previously used to deliver therapeutics, enzymes, etc., as ref. 128–130, and their delivery efficiency could be increased by this method. However, the study showed less significant localization of the tetrahedron inside the nucleus and showed punctate localization within cellular compartments, which may include lysosomes. A lot of work has been done in the past to incorporate different conjugates with DNA nanostructures to enhance their efficiency.126,131 One such piece of work done by Sleiman et al. explored the development of DNA nanotubes with hydrophobic alkyl chains joined together by a spacer region to deliver a hydrophobic dye (Nile red) inside HeLa cells.53 The spacer region could be removed and reinstated by strand displacement, which changed the nanostructure configurations. With the DNA spacer between the alkyl chains and the region hybridized to the nanotube, the chains met on the inside of the structure, forming an internal hydrophobic space that could encapsulate and conditionally release hydrophobic molecules upon the addition of a specific DNA eraser, as illustrated in Fig. 3F. The structure and efficiency of accumulation inside the cells of these nanotubes remained similar to those of empty nanotubes, previously studied by the same group. This indicated there were no negative impacts of adding the hydrophobic moieties.132 Upon removal of the spacers, the hydrophobic molecules assisted in forming a one-dimensional (1D) nanotube network through intermolecular associations, which reduced non-specific cellular entry. This ability to change its configuration in two defined ways aimed to optimize its selection based on the delivery mechanism and the target site. However, drug accumulation was found to be restricted to mitochondria,133 and the structures without the spacer were not uniform and might cause inconsistent results. Furthermore, functionalizing DNA to incorporate more drug types, their combinations, and systematic release would push this field further in the right direction.
Immunotherapy
Immunotherapy is a process of treating diseases by modulating the native immune system of the body. Activation of immune cells is performed by immune cell modification57 or adjuvant delivery134 to selectively target diseased cells like cancer cells135 and pathogens. It may also involve opsonization of the target cell136 assistance to immune cells in direct killing.137 The unique properties of DNA amphiphiles enable them to selectively target a cell type, deliver a cargo of interest (in either burst or sustained release), and easily penetrate the cell membrane followed by intracellular cargo delivery, making it an ideal technology for modulating immune responses.In one such work by Xiong et al., immune cell surfaces were modified with diacyl lipid–DNA aptamer conjugates for specific targeting of cancer cells.57 The aptamer-oligonucleotide sequences selectively bound to a target while the coupled lipid tail anchored these sequences to cell membranes, enabling the modified cells to recognize and engage cancer cells efficiently, as described in Fig. 4B. This method offers a more targeted approach compared with traditional physical delivery methods, such as using medical devices,138 native cell-homing machinery139 and coating cells with carbohydrates,140 peptides,141 or extracellular receptor domains.142 It enhances therapeutic efficacy while minimizing potential side effects, such as off-target impacts and immunogenic reactions. Aptamers make an attractive alternative to antibodies due to their smaller size and non-immunogenic nature. Despite its promising results, the experiment faced limitations such as the potential for incomplete target engagement or unanticipated cellular interactions, which could be mitigated by refining aptamer sequences or exploring different lipid conjugation strategies to increase binding efficiency and specificity. Further research could expand the aptamer library to target a broader range of pathological cells. Looking forward, this aptamer-mediated targeting strategy holds significant potential for advancing cell-based therapies, particularly in oncology, where precise targeting and minimal side effects are crucial.
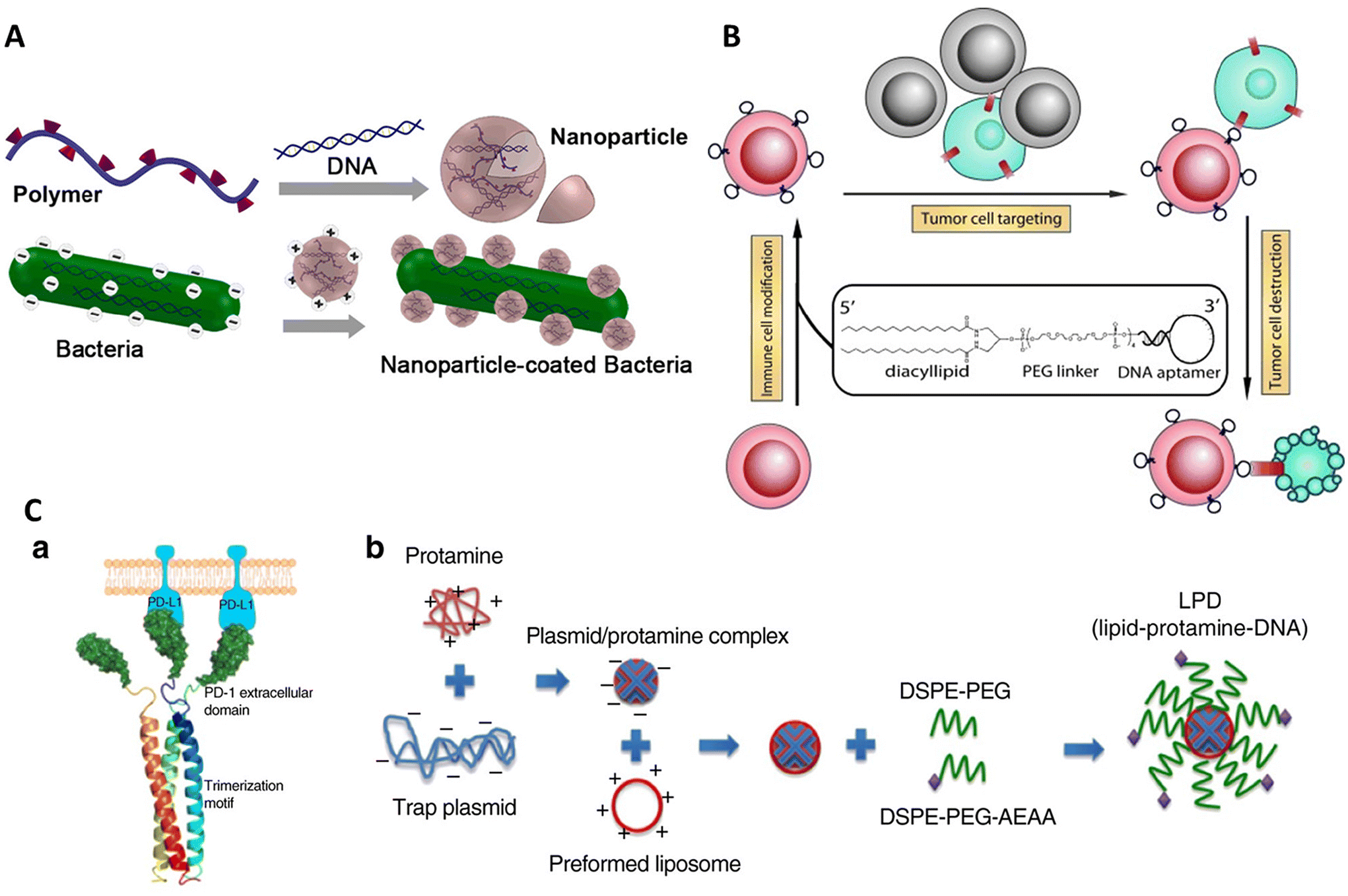 | ||
| Fig. 4 Schematics of DNA amphiphiles used in immunotherapy. (A) Nanoparticle-coated oral DNA vaccine. Reproduced with permission. Copyright 2015, American Chemical Society. (B) Modification of immune cell membrane with lipo-aptamer probes to target cancer cells. Reproduced with permission. Copyright 2012, John Wiley and Sons. (C) Design of locally and transiently expressed PD-L1 trap. (a) Scheme showing the tribody interaction of PD-L1 trap protein. (b) Preparation scheme of PD-L1. Reproduced with permission. Copyright 2018, Springer Nature. | ||
In another notable example, Song et al. investigated the synergistic effects of combining oxaliplatin (OxP)-based chemotherapy with a locally expressed PD-L1 trap fusion protein for enhancing cancer immunotherapy, specifically targeting non-hypermutated microsatellite instability (MSI)-high or mismatch repair (MMR)-deficient solid tumors.143 The complex structure developed comprised a lipid-protamine-DNA (LPD) nanoparticle system encapsulating a plasmid encoding the PD-L1 trap protein. This protein encoded the extracellular domain of PD-1 fused with a trimerization domain, allowing for the transient and local production of the PD-L1 trap protein within the tumor microenvironment (Fig. 4C). This system synergized with the chemotherapeutic agent oxaliplatin (OxP) to enhance anti-tumor efficacy, effectively converting the immunosuppressive tumor environment into an immunogenic one. The findings of this study provided better results than the use of an anti-PD-L1 monoclonal antibody (mAb), which, unlike the PD-L1 trap, induced Th17 cell accumulation in the spleen, suggesting a higher tendency towards immune-related adverse effects (irAEs).144 The novelty of this study lies in its capacity to minimize these adverse effects while enhancing the therapeutic efficacy of PD-L1 checkpoint blockade, thereby improving the safety profile for patients with non-hypermutated, microsatellite-stable (MSS) cancers, particularly colorectal cancer. However, the study acknowledged limitations, such as the need for further clinical trials to determine the optimal timing and dosage of the combined therapy, as well as the dependency on the tumor mutation burden for successful immunotherapy outcomes. Future research directions include exploring the combination of this therapy with other immunogenic agents and evaluating its efficacy in a broader range of cancer types.
In 2015, Tang and group presented a groundbreaking approach in cancer immunotherapy by engineering cationic nanoparticle-coated bacterial vectors for oral DNA vaccine delivery.145 This innovative method employed live attenuated Salmonella bacteria coated with synthetic nanoparticles assembled from cationic polymers such as 25 kDa polyethyleneimine (PEI) and plasmid DNA encoding autologous vascular endothelial growth factor receptor 2 (VEGFR2) (Fig. 4A). The structure formed was a hybrid system where the positively charged polyplex nanoparticles self-assembled on the negatively charged bacterial surface, creating a dense, protective layer around the rod-shaped Salmonella. This nanoparticle coating enhanced bacterial acid tolerance, facilitated phagosomal escape via the “proton sponge” effect,146,147 and promoted dissemination into the bloodstream. This strategy demonstrated significant T-cell activation and cytokine production, effectively inhibiting tumor growth by suppressing angiogenesis and inducing tumor necrosis.148–150 Although more efforts are required in further optimization of the system to enhance survival rates through the digestive tract, increase nanoparticle stability and investigate more bacterial strains for vaccine delivery, it shows promising results. Compared with intravenous delivery of vaccines, oral vaccines are found to be more cost-effective, easily absorbable, less invasive, and less toxic,151–153 and show a strong immunological response after colonizing the gut-associated lymphoid tissue, where they are engulfed and processed by dendritic cells or macrophages.154 The current study's nanoparticle-coated bacterial system substantially improved the structural stability and functional capabilities of the bacterial vectors.
3D bioprinting
3D bioprinting is employed in creating cell-holding scaffolds and functional tissue constructs and uses bio-ink for its function. DNA amphiphiles are an ideal bio-ink due to their biocompatibility and cross-linking capabilities due to DNA hybridization.Li et al. (2015) developed a novel supramolecular polypeptide-DNA hydrogel for 3D bioprinting.155 The hydrogel was prepared by mixing two complementary bio-inks: a polypeptide–DNA conjugate (bio-ink A) and a complementary DNA linker (bio-ink B). Bio-ink A was synthesized by grafting single-stranded DNAs (ssDNAs) onto a polypeptide backbone through a copper(I)-catalyzed click reaction, while bio-ink B comprised double-stranded DNA with sticky ends complementary to the ssDNA motifs156 (Fig. 5A). This hydrogel demonstrated rapid gelation under physiological conditions, high mechanical strength, self-healing properties, and biocompatibility.157–159 Unlike other hydrogels used in bioprinting, this polypeptide-DNA hydrogel overcame limitations such as thermal triggering, shape deformation, and lack of responsiveness.160–165 It also allowed for controlled degradation and potential customization of 3D tissue structures due to its responsiveness to proteases and nucleases.166,167 The study successfully encapsulated living cells within the hydrogel, maintaining their viability and function. However, the study was limited to a specific cell line and did not investigate the long-term stability or functionality of the printed tissues. Future research could focus on optimizing the hydrogel composition for different cell types, assessing the long-term performance of the printed tissues in vivo, and exploring the potential of this technology for drug delivery and regenerative medicine.
 | ||
| Fig. 5 Bioprinting strategies utilizing DNA amphiphiles. (A) 3D printing of polymer–DNA hydrogels using two bio-inks (blue, polymer–DNA; red, double-stranded DNA linker) sensitive to nuclease and protease degradation. Reproduced with permission. Copyright 2015, John Wiley and Sons. (B) Fabrication of bioinspired 3D-printed hydrogels using DNA-induced biomineralization. Reproduced with permission. Copyright 2023, John Wiley and Sons. (C) DNA-functionalized 3D-printed Hydrogel (a) Bioprinter setup with an external syringe pump and hydraulic coupling to a syringe containing a small sample volume. (b) Components of Bio-ink. (c) & (d) Stability of Bioink and the printed structures over 10 months. Reproduced with permission. Copyright 2020, John Wiley and Sons. | ||
Very recently, a work demonstrated by Kim et al. highlighted a unique 3D-printable hydrogel bioink made from salmon sperm DNA doped with silica168 (Fig. 5B). This bioink, composed of functionalized alginate, DNA, and biomineralized silica, was targeted for fabricating wound dressing material. The main selling point of their work revolved around the DNA strand-mediated silica biomineralization process, where the DNA acted as a scaffold for silica formation, resulting in a hydrogel with enhanced mechanical properties and biological activity. The 3D-printed dressings presented promising results in accelerating both acute and diabetic wound healing, attributed to the synergistic effects of DNA and bio silica in promoting angiogenesis and reducing inflammation. These results are far superior to previous attempts using alginate hydrogels, which lacked the enhanced biological properties and mechanical stability.169–171 Future research could focus on optimizing the DNA–silica ratio for enhanced therapeutic efficacy and exploring the applicability of this technology to other tissue engineering applications.
In a more elegant example, the researchers developed a novel low-cost bioprinting technique using a commercially available extrusion printer and a specially formulated DNA-functionalized bioink.172 This bioink comprised gelatine, alginate, and agarose, providing printability, structural integrity, and biocompatibility. The agarose component was modified with DNA using click-chemistry, enabling sequence-specific functionalities within the printed structures. The experiment aimed to integrate dynamic DNA nanotechnology with 3D bioprinting to create complex, programmable hydrogel structures capable of sequence-specific diffusion control, gradient formation, and localization of DNA signals (Fig. 5C). Previous attempts used photolithography, and DNA-bound polystyrene beads used DNA as a glue to create DNA-based hydrogels.173,174 The novelty of this method lies in the ability to control the diffusion coefficients of DNA strands through transient hybridization and strand displacement reactions. As a result, the precision at which DNA strands could be localized within the bioprinted design goes to a whopping millimetre-scale accuracy, highlighting its potential to spatially control the cellular growth pattern at mm or even lower-scale resolutions. However, limitations, such as the challenges in maintaining homogeneous diffusion and the stability of DNA-functionalized structures over time, still exist. Future research directions include improving the resolution and stability of printed structures, exploring enzyme-assisted processes for pattern refinement, and expanding the range of applications to include soft robotics and biocompatible scaffolds for tissue engineering. Furthermore, incorporating a diverse range of DNA-amphiphilic bio-inks might provide good depth to the field of 3D cell and tissue engineering.
Genetic manipulation
Gene manipulation holds promise for treating certain diseases by reducing the expression of disease-causing genes and the production of associated proteins,46 increasing the expression of necessary proteins directly175 and supporting machinery that can edit the genome itself.176 However, the delivery of extrinsic DNA to intracellular targets is often hindered by its inherent physicochemical properties, such as negative charges, high hydrophilicity, and substantial molecular weight.177 To address this challenge, researchers have explored the conjugation of hydrophobic groups to the DNA strands as a method to enhance cellular uptake and increase their stability in a safe and efficient manner.178–180 Initial studies from the 1980s utilized cholesterol-conjugated oligonucleotides to inhibit HIV infections181,182 or to target the intercellular adhesion molecule-1 gene.183 Subsequently, hydrocarbon lipids have also been conjugated to oligonucleotides to improve antisense efficacy.Working on this line of thought, Sinegra et al. (2021) developed and optimized lipid nanoparticle–spherical nucleic acids (LNP-SNAs) for the intracellular delivery of DNA and RNA, leveraging the advantages of both lipid nanoparticles and spherical nucleic acids.184 The researchers employed Design of Experiment (DoE) methodologies, including definitive screening and fractional factorial designs, to systematically explore a wide parameter space of Lipid Nanoparticles (LNP) compositions and surface DNA sequences. Key materials used in the experiment included various ionizable lipids, phospholipids, lipid-PEGs, and cholesterol, with the ethanol dilution method being employed for the synthesis of LNPs (Fig. 6C). The novelty of this experiment lies in its combinatorial approach of optimizing LNP-SNA formulations, which significantly enhanced the delivery efficiency and activity of nucleic acids compared with conventional liposome-based SNAs. Notably, optimized LNP-SNAs demonstrated a two-order magnitude reduction in the siRNA concentration required for effective mRNA silencing and exhibited organ-specific biodistribution profiles in mice. Despite these advancements, the study identified limitations such as the need for further optimization to achieve global maxima in activity and the necessity to investigate the immune cell populations targeted by LNP-SNAs for genome editing applications. Future directions include enhancing the structural diversity of DNA and RNA sequences used in LNP-SNAs, improving their targeting specificity and efficacy, and exploring their potential in delivering a broader range of genetic medicines.
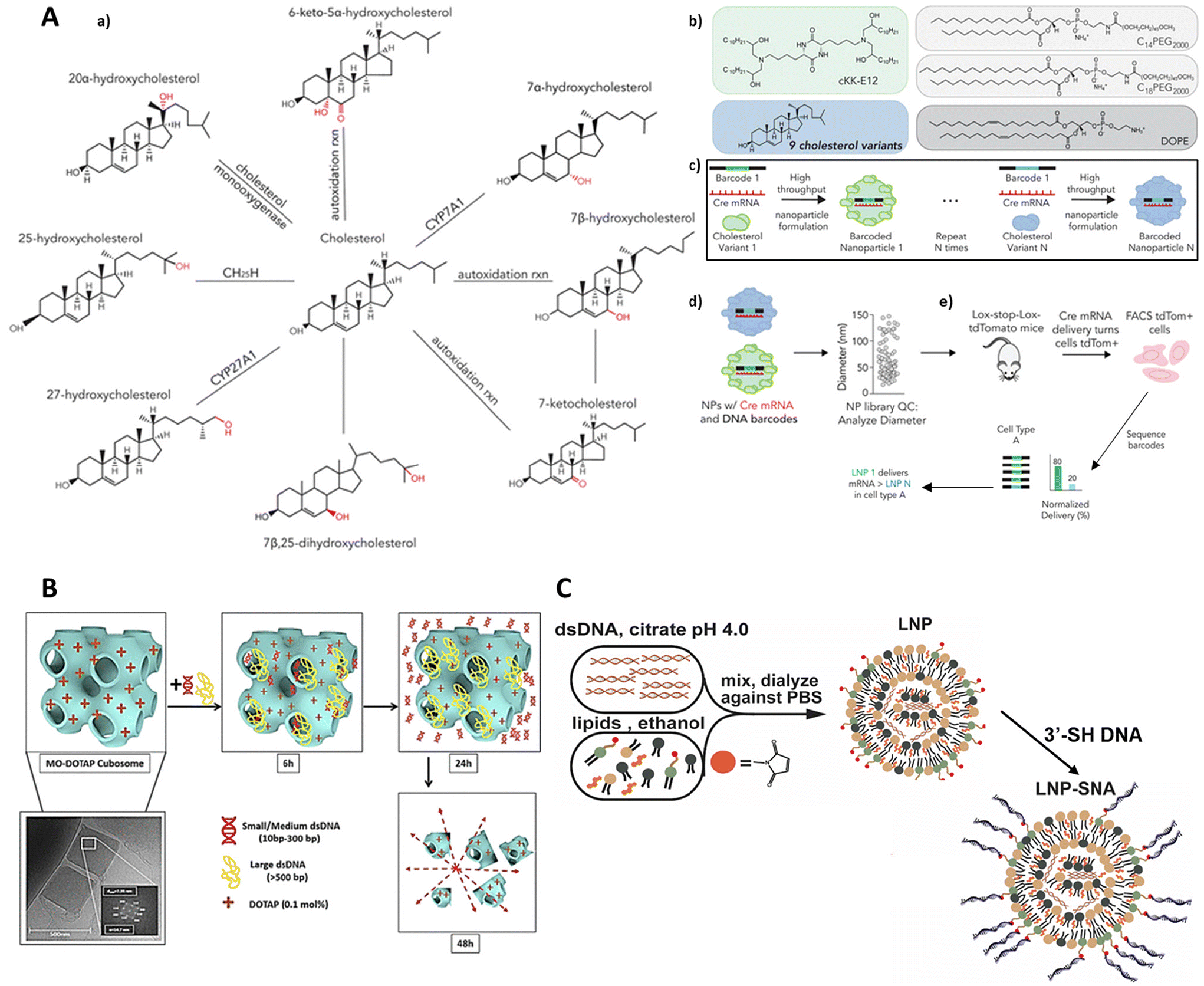 | ||
| Fig. 6 DNA amphiphilic structures for delivering genetic material. (A) Synthesizing a library of nanoparticles containing oxidized cholesterols. (a) Sterol variants made from cholesterol. (b) Formation of 125 nanoparticles by combining 9 cholesterol variants with DNA scaffolds. (c) Barcoding and screening of nanoparticles using FIND. (d) Quality control and pooling (e) intravenous administration of nanoparticles to Ai14 mice. Reproduced with permission. Copyright 2019, John Wiley and Sons. (B) Encapsulation and release of Ds DNA by monoolein-based cationic cubic phases. Reproduced with permission. Copyright 2021, American Chemical Society. (C) Synthesis of Lipid Nanoparticle Spherical Nucleic Acids. Reproduced with permission. Copyright 2020, American Chemical Society. | ||
A team of scientists, led by Paunovska, recently explored how the structure of cholesterol affects the delivery of mRNA to cells within living organisms. They used tiny packages called lipid nanoparticles (LNPs) to deliver the mRNA. The researchers had a hunch that tweaking the cholesterol's structure could change how these LNPs home in on specific cells and deliver their mRNA cargo. To test this idea, they created a collection of LNPs containing nine different versions of cholesterol, some with alterations to the side chain and others with modifications to the ring structure. Each LNP was loaded with a unique DNA barcode (like a molecular address label) and a special type of mRNA called Cre mRNA, which acted as a signal to show if the delivery was successful (Fig. 6A). These LNPs were then injected into mice, and the researchers tracked where the Cre mRNA ended up in different cells throughout the body. This allowed them to see how changes in cholesterol structure affected the LNPs’ ability to deliver mRNA to specific locations. The innovative aspect of this study lies in its use of the FIND (Functional in vivo Delivery) screen, which allowed for the simultaneous evaluation of mRNA delivery by numerous LNPs in a single mouse.185 The researchers found that LNPs containing oxidized cholesterol, particularly 25-hydroxycholesterol and 20α-hydroxycholesterol, exhibited enhanced delivery to Kupffer cells and hepatic endothelial cells compared with hepatocytes. The study's novelty lies in its demonstration that cholesterol modifications could significantly alter LNP targeting in vivo. The ability to target non-parenchymal cells in the liver has significant implications for treating diseases affecting these cell types. The study's strength lies in its direct in vivo assessment of functional mRNA delivery, overcoming the limitations of in vitro assays that may not accurately predict in vivo behaviour due to the absence of a complete immune system,186 variations in blood flow,187 and other factors.188,189 Previous studies have primarily focused on in vitro evaluation of LNPs formulated with unmodified cholesterol.190,191 While other methods like JORDAN have been used to study LNP biodistribution,192 they do not directly measure functional mRNA delivery, which is crucial as less than 4% of exogenous RNA escapes endosomes,193 and this process varies with cell type.194 The current study addressed this gap by directly measuring functional mRNA delivery in vivo using the FIND screen. The study did have some limitations, particularly the need to further investigate the mechanism behind the observed changes in targeting. It's also important to validate the results using a broader selection of ionizable lipids. Moving forward, future research could focus on understanding how modified cholesterol LNPs interact with the immune system, and on exploring the potential of mRNA delivery to Kupffer cells and liver endothelial cells. These findings open new possibilities for developing LNP-based therapies tailored for different liver diseases.
In another suitable example, Sarkar et al. (2020) investigated the potential of cationic cubosomes, formed from monoolein (MO) and various cationic lipids, as nanocarriers for DNA delivery.198 The cubosomes self-assembled into bicontinuous cubic phases, which are fascinating 3D nanostructures characterized by a unique, periodic arrangement of lipid bilayers that create a network of interconnected water channels (Fig. 6B). The hydrophilic nature of these channels makes them ideal for encapsulating water-soluble molecules like DNA. The authors employed a high-throughput approach, utilizing synchrotron small-angle X-ray scattering (SAXS) to characterize the nanostructure of the cubosomes and confirm dsDNA encapsulation. This study stands out for its detailed examination of how various cationic lipids influenced the encapsulation and release of dsDNA, shedding light on the important relationship between structure and function. The results showed that cubosomes were able to encapsulate dsDNA fragments of different sizes effectively, with the cationic lipids improving encapsulation efficiency while keeping toxicity levels low. The use of cationic lipids in cubosomes presents an advantage over traditional cationic liposomes, potentially requiring a lower percentage of cationic lipids for effective cellular interactions, thereby reducing toxicity.195 Furthermore, the cubosomes preserved the structural integrity of the encapsulated dsDNA196 and exhibited controlled release profiles,197 suggesting their potential for protecting and delivering DNA-based therapeutics. The study also highlighted potential limitations, such as the possibility of larger DNA fragments adsorbing onto the cubosome surface rather than being encapsulated within the cubic matrix. The authors suggested that the use of highly swollen cubic phases may be necessary for the encapsulation and effective delivery of functional DNA, which is typically much larger than the dsDNA fragments used in this study.198,199 To improve future research, efforts could be directed towards refining cubosome formulations to better encapsulate larger DNA molecules, and examining their ability to transfect specific cell lines. Sarkar et al. (2020) provided valuable insights into lipid-based nanocarriers for gene therapy, contributing to advancements in the field. Their work built on earlier studies that explored nonlamellar phases, such as hexagonal and cubic phases, as promising avenues for DNA delivery,200–203 offering a more systematic evaluation of cationic lipid influence on cubosome-mediated DNA encapsulation and release.204,205
Biosensors
Tracking cellular functions, metabolic activities, protein homeostasis, and cell–cell signaling in disease conditions and otherwise is crucial for advancing cell biology, developmental biology, and regenerative medicine.206 Traditionally, molecular sensors207,208 or nanoparticles209 attached to the cell membrane have been employed to meet these demands. However, these methods have their own caveats, including restricted targeting capabilities, the necessity for complex chemical modifications, and the inability to provide real-time monitoring in native conditions.206 In contrast, amphiphilic DNA framework-based biosensors offer several advantages over these traditional methods. First, aptamers can be selected and designed via systematic evolution of ligands by exponential enrichment (SELEX) to specifically bind to certain target molecules, such as metal ions, small organic molecules, and proteins with high affinity. Second, the straightforward functionalization of DNA with fluorophores facilitates signal readout employing photoluminescence. Furthermore, hydrophobic tags permit anchoring of the biosensor to the cell membrane. Finally, and importantly, DNA hybridization or the fast response of DNA aptamers to their targets allows monitoring in real time and in situ, with high spatiotemporal resolution feasible.In 2014, Chaoyong Yang's group devised an extracellular pH-sensing probe based on the principle of ratiometric fluorescence analysis, capable of detecting extracellular pH in the range of 6–8.210 The probe was made from a single-stranded DNA ligated with a diacyllipid tail, which helped in surface anchoring to the cellular membrane. The DNA strand was conjugated with a green-emission pH-sensitive dye (FAM), which emits variable frequencies of light depending on the pH of the surrounding medium. A second strand with an orange-emission pH-insensitive dye (TAMRA) was utilized to obtain the fluorescent ratio of both dyes (FAM over TAMRA), which was calibrated to the pH change in the extracellular environment, as described in Fig. 7A. The cells used for this study were live HeLa cells, and later, a 3D culture of HeLa cells was performed in collagen gel to test the efficacy of the DNA-amphiphilic biosensor in a 3-dimensional medium, which resembles in vivo conditions. Previous efforts were generally targeted towards sensing the intracellular environment,211–213 thereby providing a fresh perspective on pH sensing.214 One such similar work focused on expressing pH-sensitive and pH-insensitive fluorescent protein domains on the cellular surface via translation of exogenously introduced DNA, but it was too large and difficult to manipulate. Ratiometric analysis exhibited high sensitivity and live response, but detailed work on the effect on the readings based on in vivo parameters like the impact of environmental variations like temperature changes, light exposure, and the presence of quenching molecules has not been evaluated. Further efforts can be implemented into increasing the range of pH detection, and future perspectives in incorporating detection and measurement of multiple extracellular sensing parameters simultaneously, like temperature, ionic concentration, etc., for advanced cellular targeting and studying can be pondered over.
 | ||
| Fig. 7 Functionalised DNA-based assemblies for Biosensing. (A) Cell-anchored lipid–DNA probes for ratiometric fluorescent sensing of extracellular pH. (a) Structure of the lipid–DNA probes. (b) Working principal of probes. Reproduced with permission. Copyright 2014, American Chemical Society. (B) Structure of tocopherol-labelled fast responsive fluorescent DNA aptamer. Reproduced with permission. Copyright 2012, American Chemical Society. (C) Chemical structure of DNA probe forming molecules for membrane lipid domain studies. | ||
The study by You et al. presented a novel DNA probe designed to monitor dynamic and transient molecular encounters among different lipids and proteins on live cell membranes.215 Two types of ssDNA strands S1 and S2, complementary to each other, were conjugated to different lipids by using suitable anchors made of Tocopherol, Cholesterol, or Diacyllipid (Fig. 7C) with S1 conjugated with a fluorescent dye, like Quasar 670, Carboxytetramethylrhodamine (TMR) or 6-Carboxyfluorescein (FAM), and S2 conjugated with a quencher molecule like BHQ-2 (Black Hole Quencher 2) or BHQ-2 (Black Hole Quencher 2). S1 was hybridized with a walker strand displaced by S2 through a toe hold-mediated strand-displacement reaction. The amount of quenching of fluorescence and the time required for it quantified the interaction between individual lipid types. A blocker strand was attached to the S2, which was removed by an initiator strand for accurate control over the reaction initiation. The experiments gave fascinating results showing the preferred interaction between homologous lipids with each other compared with different lipid types spread across and within lipid domains. The pattern of spreading of the domains also affected the results, as the lipid pair with relatively lower diffusion rate showed more frequent interactions than the one with much faster diffusion rates, indicatively due to relatively closer localization of those lipid domains leading to frequent interaction. The lipid domain theory dictates the presence of multiple domains comprising similar lipid conjugates spread across the cell membrane unevenly instead of a homogeneous distribution, proving special physiological properties in particular sections. Diacyllipids (L) showed a strong preference for interacting with cholesterol (C) over tocopherol (T). The encounter preference was quantified, showing that diacyllipids preferred to interact with cholesterol about three times more than with tocopherol. The experiment revealed a hierarchy in interaction preferences: for diacyllipids, the order of preference was diacyllipid-cholesterol (LC) > diacyllipid-tocopherol (LT). For cholesterol, the preferred interactions were cholesterol-cholesterol (CC) > cholesterol-diacyllipid (CL) > cholesterol-tocopherol (CT).
In another approach for monitoring extracellular chemical transmitter dynamics, a tocopherol-labeled fluorescent aptamer sensor was directly anchored onto the cell surface, enabling real-time fluorescence imaging of adenine compounds released from brain astrocytes.216 This experiment utilized a fast-responsive DNA aptamer that binds adenine, specifically designed for high spatiotemporal resolution without the need for structural alterations upon binding, allowing it to respond rapidly to changes in extracellular adenine concentrations (Fig. 7B). The simplicity of the sensor's design and its integration into the cell surface was a notable innovation, maintaining sensor performance and cellular integrity. Despite the advancements demonstrated, the experiment faced limitations such as the reliance on relative quantification of transmitter release, the specificity for adenine compounds, and potential challenges in broader application scopes. Future studies could expand on this by exploring the use of ratiometric sensors for absolute quantification and designing aptamers with varied specificities for different neurotransmitters. Moreover, refining the anchoring mechanism could enhance the adaptability and efficiency of the sensor for real-time imaging in diverse cellular environments, broadening the applicability of this technology in neurochemical and cell signaling research.
Cell adhesion and assembly
When tethered onto the cell membrane, amphiphilic DNA facilitates cell capture and assembly through the specific and precise recognition properties of the nucleic acids. The length of the DNA strands is crucial for successful cell-to-cell contact by hybridization.131 A 20-mer DNA strand on the cell surface cannot hybridize with its complementary sequence due to steric hindrance provided by the dense glycocalyx layer. However, a 60- and 80-mer poly(dT) spacer inserted between the lipid anchor and the DNA recognition element that will hybridize significantly increases the cell adhesion to other surfaces. Eventually, DNA anchored on cell surfaces can be linked to surface-anchored complementary DNA. The accessibility of cell membranes with anchored DNA amphiphiles also facilitates cell assembly and microtissue formation. The study by Chan et al. investigated the dynamics of vesicle fusion mediated by lipid-anchored DNA oligonucleotides, exploring the role played by linker sequences in facilitating membrane proximity and fusion processes217 (Fig. 8C). By modifying the length of non-complementary bases at the membrane-proximal ends of DNA sequences, the research highlighted that increasing linker lengths generally hindered the rate and extent of lipid bilayer fusion and content mixing despite enhancing vesicle docking rates. This finding contrasts with the more efficient docking for sequences requiring less overlap, which suggests that the DNA flexibility facilitated by longer linkers aids initial docking but increases vesicle spacing post-docking, thereby impeding fusion. The novelty of this study lies in its systematic exploration of DNA sequence modifications to understand bilayer distance effects on fusion, using lipid–DNA conjugates as a simplified model system that mirrors the functionality of complex fusion proteins. Future improvements could involve more refined assays to isolate and quantify individual fusion steps and the use of different DNA sequences or chemical modifications to tailor the hybridization energetics and docking mechanics, potentially enhancing the understanding and efficiency of vesicle fusion in synthetic biological systems. | ||
| Fig. 8 Illustration of models for cell adhesion and assembly. (A) Programming the reconstitution of fully ECM-embedded 3D microtissues by DNA-programmed assembly (DPAC). (a) Relationship between DNA spots (colored squares), DNA-programmed connectivity (colored lines), and multistep assembly (b) incubation of cells with lipid-modified oligonucleotides. (c) Patterning of DNA spots on glass slides. Reproduced with permission. Copyright 2015, Springer Nature. (B) Nanomatrix scaffold for cartilage tissue engineering, based on Janus base nanotubes (JBNTs). (a) Chemical structure of JBNTs. (b) The formation process and structural illustration of the layer-by-layer J/T/M NM. (c) Biofunctional exhibition of J/T/M as a cartilage construction. Reproduced with permission. Copyright 2021, American Chemical Society. (C) DNA-mediated fusion of a pair of lipid vesicles. | ||
In a different approach, Chen and co-workers developed a novel injectable nano matrix (NM) scaffold for cartilage tissue engineering, leveraging the unique properties of Janus base nanotubes (JBNTs) combined with matriline-3 and TGF-β1.218 The materials used included JBNTs, matrilin-3, and TGF-β1, which were assembled in a layer-by-layer fashion to create a scaffold that mimicked the extracellular matrix (ECM) of cartilage, providing anchorage and promoting chondrogenesis while preventing hypertrophy (Fig. 8B). This NM scaffold demonstrated significant advancements over previous constructs by ensuring the localized and sustained release of bioactive molecules, thus enhancing stem cell differentiation and maintaining cartilage homeostasis. The experiment's novelty lies in its innovative self-assembly technique, which controlled the nanomatrix structure at the molecular level, ensuring the stability and bioactivity of incorporated growth factors and ECM proteins. However, the study identified several limitations, such as the potential for variable degradation rates of the JBNTs in different physiological conditions, which may affect the long-term stability and bioactivity of the scaffold. Future studies should focus on optimizing the degradation profiles and further evaluating the in vivo efficacy and safety of the NM scaffold. Additionally, expanding the range of bioactive molecules incorporated into the JBNTs could enhance the scaffold's versatility for other tissue engineering applications. Overall, this work provides a promising foundation for developing advanced, injectable scaffolds for cartilage repair and potentially other regenerative medicine fields.
Todhunter et al. aimed at reconstituting multicellular tissue structures with precise control over size, shape, composition, spatial heterogeneity, and embedding extracellular matrix (ECM).219 The DPAC (DNA-programmed assembly of cells) method chemically functionalized dissociated cells with degradable oligonucleotide as a “velcro”, enabling rapid, specific, and reversible cell adhesion. The materials used included aldehyde-silanized glass slides, various reagents like sodium borohydride, and cell lines such as MCF-10A and HUVECs. This method allowed for the patterning of DNA spots on substrates, followed by the layer-by-layer assembly of cells into three-dimensional structures, which were then embedded in ECM gels like Matrigel or collagen, as illustrated in Fig. 8A. The novelty of DPAC lies in its ability to maintain high cell viability and incorporate any combination of cell types without relying on their native adhesive properties. It also enables detailed exploration of tissue structure and cellular behavior, offering substantial advancements over traditional cell-printing techniques by retaining high cell viability and achieving higher throughput. However, limitations included its dependence on cells that can survive dissociation and be labeled by DNA. Future improvements could integrate DPAC with microfluidic technologies to deliver structured signals or with 3D printing to control ECM spatial heterogeneity. Additionally, incorporating stem cells or organoids could enhance the study of organoid development and disease processes in a reproducible 3D setting. Future directions involve further exploring the effects of tissue structure on cell behavior, improving the method's integration with other technologies, and extending its application to a broader range of biological studies and tissue engineering.
Enhancing enzymatic activity
Alemdaroglu et al. (2006) introduced an innovative approach for DNA-templated organic synthesis using self-assembling DNA-polypropylene oxide (PPO) block copolymer micelles.81 These micelles, with a hydrophobic PPO core and a hydrophilic single-stranded DNA shell, utilize hydrophobic interactions, rather than traditional Watson–Crick base pairing, to organize the DNA template in a three-dimensional space84,87 (Fig. 9B). This allows for various organic reactions to occur either on the micelle surface or at the hydrophobic–hydrophilic interface, potentially shielding them from the aqueous environment.29,30 The authors successfully demonstrated the feasibility of this method with several reactions, including isoindole formation, peptide bond formation, and Michael addition, achieving yields comparable to or exceeding those of traditional DNA templates.4,29,30,81 Notably, the isoindole formation was significantly more efficient at the hydrophobic–hydrophilic interface, highlighting the advantage of this three-dimensional approach for reactions involving hydrophobic reactants.220,221 This approach overcomes the limitations of previous methods using PLGA or PS, which have high glass transition temperatures that hinder the investigation and aqueous solubility.70,75,222,223 Future directions include exploring the use of these micelles for drug delivery, DNA detection, and intracellular chemical synthesis, with potential implications for targeted drug therapies. | ||
| Fig. 9 DNA-conjugated amphiphilic reaction control systems; (A) the self-assembly of DNA–Dendrimer amphiphiles and their catalytic process for substrates catalyzed by H2O2. Reproduced with permission. Copyright 2023, Elsevier. (B) DNA-templated synthesis applying DNA-b-PPO polymers. (C) DNA–Dendrimer Amphiphiles. (a) Chemical structure of DNA-decorated discotic monomers (DNA-Disc) and unmodified discotics (Inert-Disc). (b) Self-assembly of the monomers in water (c) Hybridization between the DNA handles on the nanoscaffold and complementary DNA anti-handle. Reproduced with permission. Copyright 2006, John Wiley and Sons. (D) Structure and synthesis of DNA-programmed liposome nanoreactors and carbohydrate mimetics. Reproduced with permission. | ||
Deng et al. (2023) constructed self-assembling DNA-dendrimer nanofibers with peroxidase-mimicking DNAzyme activity.224 G20-Den, containing a G-quadruplex-forming DNA sequence, and the control D20-Den, with single-stranded DNA, were synthesized as illustrated in Fig. 9A. Both self-assembled into nanofibers, with G20-Den forming smaller diameter fibers due to the compact G-quadruplex structure. When combined with hemin, G20-Den nanofibers showed superior catalytic activity compared with D20-Den/hemin in oxidizing substrates (ABTS2-, TMB, pyrogallol) in the presence of H2O2. This study presented a novel approach for enhancing DNAzyme activity through self-assembly of DNA-dendrimer amphiphiles, offering advantages in simplicity and potential for diverse nanostructures with enhanced catalytic properties, compared with previous methods like modifying G-quadruplex/hemin structures or adding exogenous substances.225–227 However, the study was limited by using only two amphiphiles and three substrates. Future research could explore a wider range of structures and substrates and investigate potential medical applications like biosensing or targeted drug delivery.
Estirado et al. (2020) introduced a novel approach for proximity-induced enzyme activation using synthetic supramolecular polymers as dynamic nanoscaffolds.228 The self-assembling monomers, composed of C3-symmetrical bis-pyridine units and single-stranded DNA handles, formed columnar structures displaying multiple DNA handles to recruit caspase-9, an apoptotic signaling enzyme through DNA-duplex formation. This modular system allowed for tuneable enzyme activation by adjusting the DNA handle density on the nano scaffold, as described in Fig. 9C. Increased enzyme activity was observed with increasing DNA-Disc concentration, peaking at equimolar concentrations of caspase-9 and DNA-Disc, followed by a decrease due to combinatorial inhibition at supra-stoichiometric levels. This study's novelty is in demonstrating a fully synthetic and programmable nano scaffold for proximity-induced enzyme activation, mimicking natural processes. This approach is advantageous compared with previous work using DNA origami as it employs a more synthetically accessible and dynamic system with tuneable physical and chemical properties.229 Additionally, unlike other in vitro systems that often struggle to match the activity of apoptosome-activated caspase-9, this approach achieved a substantial 5-fold increase in enzyme activity at optimal conditions.230–233 However, the system's reliance on DNA hybridization and the potential for non-specific interactions could limit its applicability. Future improvements could involve exploring alternative recruitment strategies, such as those based on protein–protein interactions,234–237 and optimizing the nanoscaffold composition to enhance stability and specificity. The ability to dynamically regulate enzyme activity using supramolecular polymers holds promise for developing targeted therapeutic interventions, particularly in apoptosis-related diseases.
Tian et al. (2023) demonstrated the use of DNA-programmed lipid nanoreactors (PLNs) for the synthesis of carbohydrate mimetics.238 These PLNs are essentially liposomes functionalized with lipidated oligonucleotides (LiNAs) that enable programmable membrane fusion through sequence-specific DNA hybridization.239–241 The LiNA sequences on different PLNs can be designed to be complementary, allowing for controlled fusion and mixing of the contents of different liposomes. The authors used click chemistry to synthesize a library of carbohydrate mimetics, demonstrating the potential of PLNs for multistep synthesis within confined spaces (Fig. 9D). The novelty of this work lies in the use of DNA to program the fusion of nanoreactors, enabling precise control over reaction sequences. This is in contrast to previous methods, such as DNA-encoded chemical libraries (DELs),242,243 where the building blocks were covalently attached to DNA, which can interfere with target binding and limit the chemical diversity of the library.244 In this study, the building blocks were encapsulated within the PLNs, allowing for greater flexibility and control over the reaction process. However, the study was limited to a small library of carbohydrate mimetics, and the yields for some reactions were relatively low, particularly for monosaccharides due to their high membrane permeability. Future work could focus on expanding the chemical diversity of the library and optimizing reaction conditions to improve yields. The ability to program reactions within PLNs has potential applications in drug discovery,245,246 particularly for the synthesis and screening of libraries of drug-like molecules.247 Additionally, the controlled fusion of PLNs could be used for targeted drug delivery, where the contents of different liposomes are released only upon reaching a specific target.
DNA amphiphiles can not only boost the significant expression levels of enzyme of interests, but can also be tweaked to detect fine levels of changes in enzyme in the context of disease onset and physiological disorders. There are many housekeeping genes which perturb their signalling pathways by sensing various external and internal cues. One such enzyme is Alkaline phosphatase (ALP), which is ubiquitously present in bones, liver, kidneys, and the digestive system. In many clinical tests for infectious diseases, the level of ALP is monitored in the blood. Cheng Jin and colleagues pondered upon this logic and developed a DNA-amphiphilic system which underwent self-assembly when the precursor molecules were catalysed by the ALP enzyme.248 The design of their DNA amphiphile consisted of chemically conjugating four end-terminal phosphorylated lipids to the single DNA strand using more than two phosphoramidites as a linker in succession. The hydrophobicity of the phosphorylated end-terminal lipids was compromised due to the availability of negatively charged phosphate groups. Upon ALP incubation, the enzyme dephosphorylated the ends of the lipids, thereby increasing their hydrophobicity, which caused the amphiphiles to self-assemble in micelles. The increased hydrophobicity was measured as an increase in the retention times of dephosphorylated amphiphiles in the HPLC columns as compared with the phosphorylated lipid amphiphiles.
Artificial cell
DNA nanotechnology has offered a wide variety of products and macromolecules which have been either inspired by the natural functioning of a cell/living system or have mimicked them to serve a desired function. Bringing together all these products can give birth to a superficial artificial cell with crude functionality. For example, DNA nanotube can serve as a cytoskeletal substitute for actin, microtubules and other cytoskeletal filaments. Likewise, DNA amphiphile can serve as a very basic cell membrane substitute, where the amphiphile can form the lipid bilayer, and the DNA segments can act as the lipid-bound surface proteins. Integrating the enzymatic functionality within this bilayer can then develop a rudimentary system capable of carrying out desirable reactions, giving way to simpler metabolic fates of the artificial cells. The DNA amphiphiles may further form distinct yet smaller vesicular bodies with DNA-hybridization-enabled fusion or fission capabilities. Additionally, fortifying such artificial cells with DNA nanotechnology-enabled products and research output may further bring this technology to fruition and bring us a step closer to mimicking an actual living cell in its full glory. This technology needs some more decades to close the gap between actual and artificial life. However, we have already established some functionalities, which are elucidated below.Sleiman et al. (2023) demonstrated precise structural and mechanical control of the formation of a 2D supramolecular nanosheet made up of triblock DNA amphiphile with chromogenic Cy3 dyes.249 Their triblock amphiphile consisted of: (A) a 9-mer DNA chain (hydrophilic), (B) twelve linear hexethylene chains (hydrophobic), and (C) two cy-3 chromophore-containing block (π–π stacking). The hexaethylene chains of the hydrophobic block were inserted with a phosphate group between two subsequent chains, ensuring folding of the hydrophobic chains and solubility in water due to the phosphate groups exposing to the outside. The design of the packed amphiphile was such that the terminal cy3 dye would interact with the terminal DNA chain, and hence would affect the stability of the individual amphiphile unit, which is very crucial for the formation of supramolecular 2D nanosheet. Their work shed light on a highly configurable nanosheet formation system, where the length of DNA strands, their sequence and the length of the carbon chains dictated the success of a supramolecular 2D nanosheet which resembled the bilayer assembly of lipids of the cell membrane.
Shohda and Sugawara (2006) explored the potential of liposomes as compartments for artificial cell models by investigating DNA polymerization within these structures.250 The researchers synthesized an informational amphiphile composed of a cholesterol tag, a poly(ethylene glycol) (PEG) spacer and a DNA unit. This amphiphile was designed to anchor to the inner surface of a giant liposome, with the DNA unit hybridizing with a template DNA added to the liposome's interior (Fig. 10A). The study confirmed the successful polymerization of DNA on the liposome's inner surface, marking a significant step towards creating an artificial cell model. The novelty of this work lies in its demonstration of DNA polymerization within a cell-sized compartment, a crucial reaction for advancing artificial cell models. This significantly improved over previous studies where the informational substance and catalyst were uniformly dissolved in the aqueous medium inside liposomes.251–254 The study's approach allowed for a more controlled and defined replication process. However, the study's limitations included the use of a DNA polymerase that may not be ideal for the liposome environment, and the potential for the cholesterol-PEG-DNA complex to hinder polymerization. Future research could focus on optimizing the DNA polymerase and refining the amphiphile design to enhance polymerization efficiency. The potential of this study in the medical field is substantial, as it could pave the way for the development of artificial cells capable of delivering therapeutic agents or replicating biological processes within the human body.
 | ||
| Fig. 10 Artificial cell models. (A) Artificial cell model. Ribbon: 100-mer template DNA (gene model); solid circle: DNA polymerase. Reproduced with permission from. Copyright 2006, Royal Society of Chemistry. (B) Integrated artificial cell based on DNA/RNA nanotechnology. Reproduced with permission. Copyright 2012, American Chemical Society (C) peptide–DNA nanotechnology for the construction of synthetic cytoskeletons. Peptide–DNA filaments crosslinked via programmable complementary DNA interactions (middle), mimicking actin (left), and its associated proteins. Reproduced with permission. Copyright 2024, Springer Nature. | ||
In a reviewed study, Zhao et al. delved into the innovative application of DNA nanotechnology to construct artificial cells capable of mimicking the intricate functions of natural cells.255 The researchers focused on creating DNA-based nanostructures that could simulate membrane proteins, monitor the cellular microenvironment, and establish signal feedback networks (Fig. 10B). The study highlights the successful development of DNA nanochannels that mimic both static and dynamic membrane channel proteins. The static DNA nanochannels were constructed using DNA tubes with cholesterol caps, allowing them to anchor onto the lipid membrane and facilitate the transport of molecules.256 The dynamic DNA nanochannels, on the other hand, were designed to be shape-reconfigurable in response to specific physiological conditions, enabling them to regulate molecular transport.257 The researchers also engineered DNA-based biosensors anchored to the cell membrane. These biosensors utilized DNA aptamers and DNAzymes to detect metal ions56,258 and molecular encounters259 in the cellular microenvironment. Additionally, aptamer-based 3D DNA logic gates were developed to recognize specific cell types with high accuracy.260,261 The integration of DNA-based logic gates further allowed for the creation of signal feedback networks, both on the cell membrane and within the artificial cells. The membrane-anchored signal feedback network utilized DNA strand displacement reactions to dynamically sense and respond to environmental stimuli.262 The intracellular signal feedback network employed DNAzyme-enabled AND gates, CRISPR-Cas9 systems,263 and aptamer-encoded logic gates264 to process various external signals and regulate gene expression or drug release. The novelty of this research lies in its utilization of DNA nanotechnology to achieve precise control over the structure and function of artificial cells. The ability to mimic membrane proteins, monitor the cellular microenvironment, and establish signal feedback networks represents a significant advancement in the field of synthetic biology.
While previous studies have explored the use of diacyllipid–DNA conjugates to construct sensors on cell membranes, the current study's use of DNA nanostructures offers greater versatility and functionality in mimicking membrane proteins and creating intricate signal feedback networks.265 The ability to design and assemble DNA nanostructures with precise control over their size, shape, and functionality allows for the creation of artificial cells that can perform complex tasks and respond to a wider range of environmental cues.
However, the study also acknowledged certain limitations, such as the challenge of simulating more complex membrane protein activities and the need for improved coordination and stability of DNA signal feedback networks. The future directions of this research include the development of more sophisticated DNA-based artificial cells capable of processing multiple biological signals and exhibiting enhanced autonomy and intelligence. The potential applications of this technology in the medical field are vast, ranging from targeted drug delivery and diagnostics to the creation of artificial organs and tissues. The ability to precisely engineer artificial cells with specific functions holds immense promise for revolutionizing medicine and improving human health.
Daly et al. (2024) explored the creation of a synthetic cytoskeleton by combining peptide self-assembly with DNA's programmability.266 The researchers designed peptide–DNA crosslinkers with varying sequence, length, valency, and geometry, inspired by actin-binding proteins.267,268 These crosslinkers could organize peptide nanofilaments into larger, tactoid-shaped bundles, mimicking the organization of a cellular cytoskeleton,269,270 as described in Fig. 10C. The novelty of this work lies in its ability to control the spatial arrangement of these structures within cell-sized droplets and the demonstration of their potential to regulate cargo diffusion and release and even influence droplet shape. While previous studies have bundled DNA nanofilaments in protocells using crowding agents or salt,271,272 these methods are challenging to reverse or fine-tune. Additionally, pH or salt has been used to trigger peptide filament assembly in water-in-oil droplets.273–275 The approach presented by Daly et al. (2024) offers a more controlled and tuneable method for creating synthetic cytoskeletons, potentially opening new avenues for drug delivery, biosensing, and regenerative medicine.266 However, the study was limited to in vitro environments and simple peptide–DNA systems. Future research could focus on more complex peptide sequences, diverse DNA structures, and in vivo applications.
Nanoelectronics
With the advent of nanotechnology and its advancements, the race to discover and invent the next gen of nanomaterials with phenomenal properties is at its all-time high. Various existing materials have been re-explored to test for their potential use in making electronics that could be highly efficient in processing, transmitting, and storing information. One such aspiration is incorporating nanomaterials within electronics to enhance the current state-of-the-art computational power. This vision is embedded in the word Nanoelectronics, which refers to the use of nanotechnology in electronic components, making it possible to scale huge and bulky equipment into small, functional, and efficient instruments for society's needs and consumption. Nanoelectronics is an emerging field that needs growth and further support. However, the scope of this review restricts further discussion about nanoelectronics. The agenda here is to discuss some of the DNA-amphiphilic structures that show promise and potential to be explored for their use in making miniaturized biomimetic sensors and organs.Within the vast range of materials being explored, optoelectronics is focused on finding materials with exceptional light emission and tunability. A wide variety of chromophoric systems have been explored; however, they strongly lack surface addressability, biocompatibility, and surface tunability. The emerging field of DNA nanotechnology and growing interest in DNA amphiphiles have widened this field. Many scientists are now exploring the mechanical assembling properties of DNA with various chromophoric systems. Dr R. Varghese and his lab have successfully demonstrated a general click chemistry-based approach to forming DNA-Oligo(p-phenylene-ethynylene) [DOPE] hybrid amphiphiles into surface-engineered vesicles (Fig. 10A).276 The resultant amphiphile, DOPE, forms higher-order fluorescent nano to micro-vesicles with tuneable emission output. These amphiphiles are very stable and assemble due to hydrophobic and π–π interactions of OPE. The enhancement in the fluorescent signal was achieved by the aggregation-induced enhanced emission (AIEE)-based phenomena. Furthermore, their group demonstrated the thermal reversibility of the fluorescence signal after super quenching the fluorescence activity by a surface decorated with DNA-templated AuNPs. They also showed the formation of a larger light-harvesting micellar fibril by hybridizing DOPE with another DNA modified with ethynyl perylene (DNA-Pe) as a compatible FRET pair. Their results indicate a temperature-controlled association–dissociation dynamics of the light-harvesting complex formed by DOPE-DNA-Pe, which emphasizes the thermal reversibility of the DNA amphiphile complex.
To explore the dynamics of a high aspect ratio surface of DNA in catalytic chemistry, Dr R. Varghese et al. demonstrated the formation of DNA-decorated 2D crystalline nanosheets with a high aspect ratio. They showed a simple and efficient method for forming highly dense DNA-surface coated crystalline nanosheets emerging from π–π interactions of the hydrophobic unit (hexa-peri-benzocoronene as HBC) chemically conjugated to the DNA strand forming an amphiphile (Fig. 10B).277 They showed DNA-driven surface addressability by coating the surface with DNA-hybridization-based AuNP coating as proof of concept. This level of high aspect ratio of accessible DNA chains can be targeted for efficient drug loading and stimuli-responsive drug delivery applications. Furthermore, the densely packed DNA chains offer exciting discoveries in the field of surface catalytic chemistry.
Another fascinating study from the same group focused on the effects of coupling an enantioselective chiral hydrophobic core to the DNA strands.278 They demonstrated the fabrication and synthesis of DNA (hydrophilic)–HPB (hexaphenylbenzene) amphiphiles. These amphiphiles adapted the propeller twist arising from the rotation of C(sp2)–C(sp2) bonds connecting the radial benzene rings to the core benzene rings in the HPB (Fig. 10C). This translated to a helical twist in the DNA amphiphile-based nanoribbons, which has been explored to orient the nanoparticles into 1D assembly using DNA hybridization-based AuNPs (Gold Nanoparticles) and AuNRs (Gold NanoRods) coatings. These 1D-decorated helically twisted DNA amphiphilic Nanoribbons find their applications in the fields of chiral sensing, enantioselective synthesis platforms, photonic and light-harvesting complexes to be used in various biomedical applications, and nanoelectronics fields (Fig. 11).
 | ||
| Fig. 11 DNA amphiphile-based plasmonic nanostructures. (A) Solid-phase “click” chemistry approach for the synthesis of DNA1 amphiphile and their reversible self-assembly into surface-engineered vesicles with enhanced emission. DNA hybridization induced shape conforming a AuNP-loaded micellar sphere and a fluorophore-based design of light-harvesting antennae. Reproduced with permission from ref. 243. © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Representation depicting the amphiphilicity-driven self-assembly of DNA1/2 into DNA-decorated 2D sheets. (a) The 2D crystalline nanosheet is decorated with AuNP through DNA hybridization. Reproduced with permission from ref. 244. Copyright © 2017, American Chemical Society. (C) Schematic for the self-assembly of DNA-based amphiphiles into DNA-decorated, twisted nanoribbons with M-helicity. DNA-directed chiral organization of AuNPs and AuNRs is also shown. (a–c) Shows the SEM images of helically twisted DNA nanoribbons decorated with AuNPs, AuNRs and without coatings, respectively. Reproduced with permission from ref. 245. © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Conclusion and outlook
DNA-based materials exhibit exceptional properties in structural design, offering unique advantages over other building blocks such as peptides, proteins, and synthetic macromolecules. DNA enables the bottom-up construction of intricate architectures and allows for precise tuning of interaction energies between complementary DNA strands. Recent advancements in the design and functionality of DNA amphiphiles capitalize on these properties, promoting their use in diagnostics and biomedicine. The synthesis of amphiphilic DNA molecules is facilitated by well-established protocols, and these materials are commercially available. The topology and interactions of amphiphilic DNA can be precisely controlled, enabling their aggregation into various superstructures like micelles or vesicles, with adjustable sizes and modifiable surfaces through duplex formation. Their hydrophobic units provide an efficient method for modifying the membranes of living cells, which supports further cell surface engineering, cell assembly, and potential sensing applications. However, despite these promising attributes, certain challenges must be addressed before DNA amphiphiles can be fully translated into clinical settings.A significant challenge in this field is ensuring biological stability. While DNA amphiphile micelles have demonstrated improved enzymatic stability,279 minimizing nuclease degradation, particularly in vivo, remains difficult. Additionally, when exposed to biological environments, amphiphilic DNA structures often become coated with a protein corona,280 which may obscure surface recognition elements and reduce targeting efficiency. Maintaining the solubility of amphiphilic DNA in biological media and preserving its membrane activity is also problematic. Serum proteins, such as albumin or lipoproteins, are known to form stable complexes with amphiphilic DNA,281,282 hindering its intended functions. Therefore, strategies to prevent these interactions with serum proteins are crucial for expanding biomedical applications.283 Moreover, amphiphilic DNA molecules in micelle assemblies exist in a dynamic equilibrium with their surroundings. Significant dilution after intravenous administration can reduce concentrations below the critical micelle concentration (CMC), causing micelle disassembly and premature drug release before reaching the target.284,285 To address this, the biological stability of amphiphilic DNA micelles must be optimized for effective delivery. Covalent cross-linking of lipid–DNA molecules has been shown to enhance the stability of these nanostructures,286 indicating that this issue will likely be resolved soon. Incorporating a hydrophobic segment into nucleic acid amphiphiles is crucial for their functionality, but it's also important to consider their biocompatibility and biosafety, particularly for clinical applications. Excessive insertion of lipid–DNA components into membranes can disrupt, damage, or even kill cells.57,287 Since the hydrophobic insertion mechanism does not target specific cell types, additional features for selective targeting are necessary. For instance, labeling DNA amphiphiles with folic acid has been explored to direct them towards cancer cells.288 While most current studies on the biocompatibility of DNA amphiphiles focus on cell cytotoxicity, only a few have assessed their local or systemic toxicity in vivo.61,289 As the development of DNA amphiphiles for biomedical uses continues, it is crucial to expand toxicological evaluations to include aspects such as cell membrane damage, cell signaling disruption, oxidative stress, and genotoxicity to better predict long-term safety. With the progress made in understanding and controlling the synthesis and assembly of DNA amphiphiles, there is now an opportunity to explore their unique properties when combined with hydrophobic molecules. Similar to native protein clusters on cell membranes, DNA nanostructures could function as artificial gates for intracellular/extracellular transport, cellular environment regulation, and signaling control. The interface between DNA amphiphiles and membranes holds promise for advancements in synthetic biology, artificial cell engineering, cell assembly, and tissue formation.290 Additionally, DNA amphiphiles may have potential in cell-based therapies. Beyond immune cells, further research is needed on the use of amphiphilic DNA nanostructures with other circulating cells.291
Overall, DNA amphiphiles are advancing rapidly, with a wide array of nucleic acid materials and structural designs being explored for integration with living systems, particularly in biomedical applications. This field is expected to expand, addressing more complex functions in areas such as oncology, vaccination, and theranostics.
Data availability
This is a review article. No new data were generated for this manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors sincerely thank all of the members of the D. B. group for critically reading the manuscript and for their valuable feedback. The authors recognize IIT Gandhinagar's infrastructure and financial support for this research. NJ and AS acknowledge the financial support from the Ministry of Education, Government of India. AS thanks DST-GoI for PMRF fellowship. DB thanks SERB-ANRF GoI, MoES-GoI, Gujcost, GSBTM for research grants.References
- L. M. Barnes and A. J. Dickson, Curr. Opin. Biotechnol., 2006, 17, 381–386 CrossRef CAS.
- I. Hirao, Curr. Opin. Chem. Biol., 2006, 10, 622–627 CrossRef CAS.
- J. M. Sturino and T. R. Klaenhammer, Nat. Rev. Microbiol., 2006, 4, 395–404 CrossRef CAS.
- R. Corradini, S. Sforza, T. Tedeschi and R. Marchelli, Chirality, 2007, 19, 269–294 CrossRef CAS PubMed.
- P. Alberti, A. Bourdoncle, B. Saccà, L. Lacroix and J.-L. Mergny, Org. Biomol. Chem., 2006, 4, 3383 RSC.
- M. K. Beissenhirtz and I. Willner, Org. Biomol. Chem., 2006, 4, 3392 RSC.
- C. Mao, T. H. LaBean, J. H. Reif and N. C. Seeman, Nature, 2000, 407, 493–496 CrossRef CAS.
- C. J. Wraight and P. J. White, Pharmacol. Ther., 2001, 90, 89–104 CrossRef CAS.
- D. Wang, B. Liu, Y. Ma, C. Wu, Q. Mou, H. Deng, R. Wang, D. Yan, C. Zhang and X. Zhu, J. Am. Chem. Soc., 2017, 139, 14021–14024 CrossRef CAS.
- D. Wang, X. Lu, F. Jia, X. Tan, X. Sun, X. Cao, F. Wai, C. Zhang and K. Zhang, Chem. Mater., 2017, 29, 9882–9886 CrossRef CAS.
- D. Wang, C. Yu, L. Xu, L. Shi, G. Tong, J. Wu, H. Liu, D. Yan and X. Zhu, J. Am. Chem. Soc., 2018, 140, 8797–8806 CrossRef CAS.
- S. Huo, M. Kwak, J. Qin, B. Dittrichn and A. Herrmann, Mater. Today, 2021, 49, 378–390 CrossRef CAS.
- W. Ma, Y. Zhan, Y. Zhang, C. Mao, X. Xie and Y. Lin, Signal Transduction Targeted Ther., 2021, 6, 351 CrossRef CAS.
- L. Xu, C. Yu, D. Wang, J. Pang, L. Shi, Y. Su, L. Gong, D. Yan and X. Zhu, Sci. China: Chem., 2020, 63, 244–253 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- B. Saccà and C. M. Niemeyer, Chem. Soc. Rev., 2011, 40, 5910 RSC.
- D. Bhatia, C. Wunder and L. Johannes, ChemBioChem, 2021, 22, 763–778 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- J. Fu, M. Liu, Y. Liu and H. Yan, Acc. Chem. Res., 2012, 45, 1215–1226 CrossRef CAS.
- P. Wang, T. A. Meyer, V. Pan, P. K. Dutta and Y. Ke, Chem, 2017, 2, 359–382 CAS.
- M. Bathe and P. W. K. Rothemund, MRS Bull., 2017, 42, 882–888 CrossRef CAS.
- S. Nummelin, J. Kommeri, M. A. Kostiainen and V. Linko, Adv. Mater., 2018, 30, 1703721 CrossRef.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- H. Yan, X. Zhang, Z. Shen and N. C. Seeman, Nature, 2002, 415, 62–65 CrossRef CAS PubMed.
- H. Liu and D. Liu, Chem. Commun., 2009, 2625 RSC.
- Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124–3156 CrossRef CAS PubMed.
- G. Tikhomirov, P. Petersen and L. Qian, Nature, 2017, 552, 67–71 CrossRef CAS.
- K. F. Wagenbauer, C. Sigl and H. Dietz, Nature, 2017, 552, 78–83 CrossRef CAS PubMed.
- L. L. Ong, N. Hanikel, O. K. Yaghi, C. Grun, M. T. Strauss, P. Bron, J. Lai-Kee-Him, F. Schueder, B. Wang, P. Wang, J. Y. Kishi, C. Myhrvold, A. Zhu, R. Jungmann, G. Bellot, Y. Ke and P. Yin, Nature, 2017, 552, 72–77 CrossRef CAS.
- M. Liu, J. Fu, C. Hejesen, Y. Yang, N. W. Woodbury, K. Gothelf, Y. Liu and H. Yan, Nat. Commun., 2013, 4, 2127 CrossRef.
- Y. Liu, W. Hou, H. Sun, C. Cui, L. Zhang, Y. Jiang, Y. Wu, Y. Wang, J. Li, B. S. Sumerlin, Q. Liu and W. Tan, Chem. Sci., 2017, 8, 6182–6187 RSC.
- P. Alonso-Cristobal, P. Vilela, A. El-Sagheer, E. Lopez-Cabarcos, T. Brown, O. L. Muskens, J. Rubio-Retama and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2015, 7, 12422–12429 CrossRef CAS.
- D. Han, Z. Zhu, C. Wu, L. Peng, L. Zhou, B. Gulbakan, G. Zhu, K. R. Williams and W. Tan, J. Am. Chem. Soc., 2012, 134, 20797–20804 CrossRef CAS PubMed.
- M. You, G. Zhu, T. Chen, M. J. Donovan and W. Tan, J. Am. Chem. Soc., 2015, 137, 667–674 CrossRef CAS PubMed.
- Y. Song, S. Kim, M. J. Heller and X. Huang, Nat. Commun., 2018, 9, 281 CrossRef.
- J. Y. Kishi, T. E. Schaus, N. Gopalkrishnan, F. Xuan and P. Yin, Nat. Chem., 2018, 10, 155–164 CrossRef CAS PubMed.
- J. Zhang, L. P. Smaga, N. S. R. Satyavolu, J. Chan and Y. Lu, J. Am. Chem. Soc., 2017, 139, 17225–17228 CrossRef CAS PubMed.
- S. Wan, L. Zhang, S. Wang, Y. Liu, C. Wu, C. Cui, H. Sun, M. Shi, Y. Jiang, L. Li, L. Qiu and W. Tan, J. Am. Chem. Soc., 2017, 139, 5289–5292 CrossRef CAS.
- Y. Wang, C. Wu, T. Chen, H. Sun, S. Cansiz, L. Zhang, C. Cui, W. Hou, Y. Wu, S. Wan, R. Cai, Y. Liu, B. S. Sumerlin, X. Zhang and W. Tan, Chem. Sci., 2016, 7, 6041–6049 RSC.
- S. M. Nimjee, R. R. White, R. C. Becker and B. A. Sullenger, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 61–79 CrossRef CAS.
- E. Levy-Nissenbaum, A. F. Radovic-Moreno, A. Z. Wang, R. Langer and O. C. Farokhzad, Trends Biotechnol., 2008, 26, 442–449 CrossRef CAS PubMed.
- P. Röthlisberger, C. Gasse and M. Hollenstein, Int. J. Mol. Sci., 2017, 18, 2430 CrossRef.
- H. Sun and Y. Zu, Small, 2015, 11, 2352–2364 CrossRef CAS PubMed.
- L. Abarca-Cabrera, P. Fraga-García and S. Berensmeier, Biomater. Res., 2021, 25, 12 CrossRef CAS.
- Y. Jiang, S. Huo, J. Hardie, X.-J. Liang and V. M. Rotello, Expert Opin. Drug Delivery, 2016, 13, 547–559 CrossRef CAS.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- A. M. Peterson and J. M. Heemstra, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 282–297 CAS.
- X. Tan, F. Jia, P. Wang and K. Zhang, J. Controlled Release, 2020, 323, 240–252 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- L. Zhao, Y. Lu, L. Zhang and P. Y. K. Chau, Decis. Support Syst., 2012, 52, 645–656 CrossRef.
- J. F. Rahbani, E. Vengut-Climent, P. Chidchob, Y. Gidi, T. Trinh, G. Cosa and H. F. Sleiman, Adv. Healthcare Mater., 2018, 7, 1701049 CrossRef PubMed.
- Y. Wang, X. Zhang, H. Zhang, Y. Lu, H. Huang, X. Dong, J. Chen, J. Dong, X. Yang, H. Hang and T. Jiang, Mol. Biol. Cell, 2012, 23, 3911–3922 CrossRef CAS.
- Y. Wu, K. Sefah, H. Liu, R. Wang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5–10 Search PubMed.
- L. Qiu, T. Zhang, J. Jiang, C. Wu, G. Zhu, M. You, X. Chen, L. Zhang, C. Cui, R. Yu and W. Tan, J. Am. Chem. Soc., 2014, 136, 13090–13093 Search PubMed.
- X. Xiong, H. Liu, Z. Zhao, M. B. Altman, D. Lopez-Colon, C. J. Yang, L. Chang, C. Liu and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 1472–1476 Search PubMed.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Macromol. Rapid Commun., 2008, 29, 326–329 Search PubMed.
- A. Biscans, A. Coles, R. Haraszti, D. Echeverria, M. Hassler, M. Osborn and A. Khvorova, Nucleic Acids Res., 2019, 47, 1082–1096 Search PubMed.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899–902 Search PubMed.
- J. Willem De Vries, S. Schnichels, J. Hurst, L. Strudel, A. Gruszka, M. Kwak, K.-U. Bartz-Schmidt, M. S. Spitzer and A. Herrmann, Biomaterials, 2018, 157, 98–106 CAS.
- S. Cansiz, L. Zhang, C. Wu, Y. Wu, I. Teng, W. Hou, Y. Wang, S. Wan, R. Cai, C. Jin, Q. Liu and W. Tan, Chem. – Asian J., 2015, 10, 2084–2094 Search PubMed.
- A. Rodríguez-Pulido, A. I. Kondrachuk, D. K. Prusty, J. Gao, M. A. Loi and A. Herrmann, Angew. Chem., Int. Ed., 2013, 52, 1008–1012 Search PubMed.
- R. J. Banga, N. Chernyak, S. P. Narayan, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 9866–9869 CAS.
- S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan and R. Varghese, Nanoscale, 2017, 9, 5425–5432 Search PubMed.
- M. Kwak and A. Herrmann, Angew. Chem., Int. Ed., 2010, 49, 8574–8587 Search PubMed.
- A. Jäschke, J. P. Fürste, E. Nordhoff, F. Hillenkamp, D. Cech and V. A. Erdmann, Nucleic Acids Res., 1994, 22, 4810–4817 Search PubMed.
- J. H. Jeong, S. W. Kim and T. G. Park, Bioconjugate Chem., 2003, 14, 473–479 Search PubMed.
- R. B. Fong, Z. Ding, C. J. Long, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 720–725 Search PubMed.
- J. H. Jeong and T. G. Park, Bioconjugate Chem., 2001, 12, 917–923 CAS.
- S. A. Bell, M. E. McLean, S.-K. Oh, S. E. Tichy, W. Zhang, R. M. Corn, R. M. Crooks and E. E. Simanek, Bioconjugate Chem., 2003, 14, 488–493 CAS.
- A. M. Michelson and A. R. Todd, J. Chem. Soc., 1955, 2632–2638 CAS.
- C. A. Stein, R. Pal, A. L. DeVico, G. Hoke, S. Mumbauer, O. Kinstler, M. G. Sarngadharan and R. L. Letsinger, Biochemistry, 1991, 30, 2439–2444 CrossRef CAS.
- Y. Guo, J. Zhang, F. Ding, G. Pan, J. Li, J. Feng, X. Zhu and C. Zhang, Adv. Mater., 2019, 31, 1807533 CrossRef.
- Z. Li, Y. Zhang, P. Fullhart and C. A. Mirkin, Nano Lett., 2004, 4, 1055–1058 CrossRef CAS.
- F. Teixeira Jr., P. Rigler and C. Vebert-Nardin, Chem. Commun., 2007, 1130 RSC.
- G. Godeau, H. Arnion, C. Brun, C. Staedel and P. Barthélémy, Med. Chem. Commun., 2010, 1, 76 CAS.
- C. J. Yang, M. Pinto, K. Schanze and W. Tan, Angew. Chem., Int. Ed., 2005, 44, 2572–2576 CrossRef CAS.
- M. W. Reed, E. Lukhtanov, V. Gorn, D. D. Lucas, J. H. Zhou, S. B. Pai, Y. Cheng and R. B. Meyer, J. Med. Chem., 1995, 38, 4587–4596 CrossRef CAS.
- R. J. Banga, B. Meckes, S. P. Narayan, A. J. Sprangers, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 4278–4281 CrossRef CAS.
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed.
- M. Oishi, F. Nagatsugi, S. Sasaki, Y. Nagasaki and K. Kataoka, ChemBioChem, 2005, 6, 718–725 CrossRef CAS.
- M. A. Innis, K. B. Myambo, D. H. Gelfand and M. A. Brow, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9436–9440 CrossRef CAS PubMed.
- S. E. Averick, S. K. Dey, D. Grahacharya, K. Matyjaszewski and S. R. Das, Angew. Chem., Int. Ed., 2014, 53, 2739–2744 CrossRef CAS PubMed.
- K. Liu, L. Zheng, Q. Liu, J. W. De Vries, J. Y. Gerasimov and A. Herrmann, J. Am. Chem. Soc., 2014, 136, 14255–14262 CrossRef CAS.
- X. Tan, H. Lu, Y. Sun, X. Chen, D. Wang, F. Jia and K. Zhang, Chem, 2019, 5, 1584–1596 CAS.
- T. Trinh, P. Chidchob, H. S. Bazzi and H. F. Sleiman, Chem. Commun., 2016, 52, 10914–10917 RSC.
- Y. Dong, Y. Sun, L. Wang, D. Wang, T. Zhou, Z. Yang, Z. Chen, Q. Wang, Q. Fan and D. Liu, Angew. Chem., Int. Ed., 2014, 53, 2607–2610 CrossRef CAS PubMed.
- Y. Dong, Y. R. Yang, Y. Zhang, D. Wang, X. Wei, S. Banerjee, Y. Liu, Z. Yang, H. Yan and D. Liu, Angew. Chem., Int. Ed., 2017, 56, 1586–1589 Search PubMed.
- C. Wang, J. Piao, Y. Li, X. Tian, Y. Dong and D. Liu, Angew. Chem., Int. Ed., 2020, 59, 15176–15180 CrossRef CAS PubMed.
- Y. Dong, S. Chen, S. Zhang, J. Sodroski, Z. Yang, D. Liu and Y. Mao, Angew. Chem., Int. Ed., 2018, 57, 2072–2076 Search PubMed.
- Z. Zhang, Y. Yang, F. Pincet, M. C. Llaguno and C. Lin, Nat. Chem., 2017, 9, 653–659 CrossRef CAS PubMed.
- C. Zhou, Y. Zhang, Y. Dong, F. Wu, D. Wang, L. Xin and D. Liu, Adv. Mater., 2016, 28, 9819–9823 CrossRef CAS PubMed.
- K. Iric, M. Subramanian, J. Oertel, N. P. Agarwal, M. Matthies, X. Periole, T. P. Sakmar, T. Huber, K. Fahmy and T. L. Schmidt, Nanoscale, 2018, 10, 18463–18467 RSC.
- Z. Zhao, M. Zhang, J. M. Hogle, W. M. Shih, G. Wagner and M. L. Nasr, J. Am. Chem. Soc., 2018, 140, 10639–10643 CrossRef CAS PubMed.
- Z. Zhao, C. Chen, Y. Dong, Z. Yang, Q. Fan and D. Liu, Angew. Chem., Int. Ed., 2014, 53, 13468–13470 CrossRef CAS PubMed.
- C. Wang, Y. Zhang, Y. Shao, X. Tian, J. Piao, Y. Dong and D. Liu, Giant, 2020, 1, 100006 CrossRef.
- A. Gubu, X. Zhang, A. Lu, B. Zhang, Y. Ma and G. Zhang, Mol. Ther.–Nucleic Acids, 2023, 33, 144–163 CrossRef CAS PubMed.
- S. Huo, H. Li, A. J. Boersma and A. Herrmann, Adv. Sci., 2019, 6, 1900043 CrossRef.
- C. Agbavwe, C. Kim, D. Hong, K. Heinrich, T. Wang and M. M. Somoza, J. Nanobiotechnol., 2011, 9, 57 CrossRef CAS PubMed.
- D. R. Halpin, J. A. Lee, S. J. Wrenn and P. B. Harbury, PLos Biol., 2004, 2, e175 Search PubMed.
- M. González-Lainez, M. Gallegos, J. Munarriz, R. Azpiroz, V. Passarelli, M. V. Jiménez and J. J. Pérez-Torrente, Organometallics, 2022, 41, 2154–2169 CrossRef.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- S. K. Vashist, Diagnostics, 2012, 2, 23–33 CAS.
- L. Martínez-Jothar, S. Doulkeridou, R. M. Schiffelers, J. Sastre Torano, S. Oliveira, C. F. Van Nostrum and W. E. Hennink, J. Controlled Release, 2018, 282, 101–109 Search PubMed.
- G. Orsy, S. Shahmohammadi and E. Forró, Molecules, 2023, 28, 5706 Search PubMed.
- J. H. Jeong, S. H. Kim, S. W. Kim and T. G. Park, Bioconjugate Chem., 2005, 16, 1034–1037 CrossRef CAS PubMed.
- J. Zhao, J. Liu, T. Wei, X. Ma, Q. Cheng, S. Huo, C. Zhang, Y. Zhang, X. Duan and X.-J. Liang, Nanoscale, 2016, 8, 5126–5138 Search PubMed.
- M. Oishi, Y. Nagasaki, K. Itaka, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2005, 127, 1624–1625 Search PubMed.
- S. F. M. Van Dongen, H.-P. M. De Hoog, R. J. R. W. Peters, M. Nallani, R. J. M. Nolte and J. C. M. Van Hest, Chem. Rev., 2009, 109, 6212–6274 CAS.
- T. Inoue, G. Chen, K. Nakamae and A. S. Hoffman, J. Controlled Release, 1998, 51, 221–229 CAS.
- G. S. Kwon and T. Okano, Adv. Drug Delivery Rev., 1996, 21, 107–116 Search PubMed.
- H. S. Yoo, J. E. Oh, K. H. Lee and T. G. Park, Pharm. Res., 1999, 16, 1114–1118 CrossRef CAS PubMed.
- M. Kwak, I. J. Minten, D.-M. Anaya, A. J. Musser, M. Brasch, R. J. M. Nolte, K. Müllen, J. J. L. M. Cornelissen and A. Herrmann, J. Am. Chem. Soc., 2010, 132, 7834–7835 CAS.
- F. Setaro, M. Brasch, U. Hahn, M. S. T. Koay, J. J. L. M. Cornelissen, A. De La Escosura and T. Torres, Nano Lett., 2015, 15, 1245–1251 CAS.
- E. Strable, J. E. Johnson and M. G. Finn, Nano Lett., 2004, 4, 1385–1389 CrossRef CAS.
- M. Kwak, A. J. Musser, J. Lee and A. Herrmann, Chem. Commun., 2010, 46, 4935 RSC.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CAS.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS.
- Z. Yang, G. Sahay, S. Sriadibhatla and A. V. Kabanov, Bioconjugate Chem., 2008, 19, 1987–1994 CrossRef CAS.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899–902 CrossRef CAS.
- G. Häcker, V. Redecke and H. Häcker, Immunology, 2002, 105, 245–251 CrossRef.
- M. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS PubMed.
- H. Yang, C. K. McLaughlin, F. A. Aldaye, G. D. Hamblin, A. Z. Rys, I. Rouiller and H. F. Sleiman, Nat. Chem., 2009, 1, 390–396 CrossRef CAS.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. A. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- S. H. Kim, K.-R. Kim, D.-R. Ahn, J. E. Lee, E. G. Yang and S. Y. Kim, ACS Nano, 2017, 11, 9352–9359 CrossRef CAS PubMed.
- M. S. Chan, D. Y. Tam, Z. Dai, L. S. Liu, J. W. Ho, M. L. Chan, D. Xu, M. S. Wong, C. Tin and P. K. Lo, Small, 2016, 12, 770–781 CrossRef CAS PubMed.
- S. Raniolo, G. Vindigni, A. Ottaviani, V. Unida, F. Iacovelli, A. Manetto, M. Figini, L. Stella, A. Desideri and S. Biocca, Nanomedicine, 2018, 14, 1181–1190 CrossRef CAS PubMed.
- T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868–875 CrossRef CAS PubMed.
- J. F. Rahbani, A. A. Hariri, G. Cosa and H. F. Sleiman, ACS Nano, 2015, 9, 11898–11908 CrossRef CAS PubMed.
- W. J. Rhee and G. Bao, Nucleic Acids Res., 2010, 38, e109–e109 CrossRef.
- J.-O. Jin, H. Park, W. Zhang, J. W. De Vries, A. Gruszka, M. W. Lee, D.-R. Ahn, A. Herrmann and M. Kwak, Biomaterials, 2017, 115, 81–89 CrossRef CAS PubMed.
- J.-O. Jin, H. Kim, Y. H. Huh, A. Herrmann and M. Kwak, J. Controlled Release, 2019, 315, 76–84 CrossRef CAS PubMed.
- K. N. Katzenmeyer, L. M. Szott and J. D. Bryers, Pathog. Dis., 2017, 75(6), ftx075 Search PubMed.
- M. Walczak, R. A. Brady, L. Mancini, C. Contini, R. Rubio-Sánchez, W. T. Kaufhold, P. Cicuta and L. Di Michele, Nat. Commun., 2021, 12, 4743 CrossRef CAS PubMed.
- A. M. Parr, I. Kulbatski and C. H. Tator, J. Neurotrauma, 2007, 24, 835–845 Search PubMed.
- C. G. Millán, M. L. S. Marinero, A. Z. Castañeda and J. M. Lanao, J. Controlled Release, 2004, 95, 27–49 Search PubMed.
- C. Foxall, S. Watson, D. Dowbenko, C. Fennie, L. Lasky, M. Kiso, A. Hasegawa, D. Asa and B. Brandley, J. Cell Biol., 1992, 117, 895–902 CAS.
- O. Mandelboim, E. Vadai, M. Fridkin, A. Katz-Hillel, M. Feldman, G. Berke and L. Eisenbach, Nat. Med., 1995, 1, 1179–1183 Search PubMed.
- B. Jena, G. Dotti and L. J. N. Cooper, Blood, 2010, 116, 1035–1044 CAS.
- W. Song, L. Shen, Y. Wang, Q. Liu, T. J. Goodwin, J. Li, O. Dorosheva, T. Liu, R. Liu and L. Huang, Nat. Commun., 2018, 9, 2237 CrossRef PubMed.
- M. Tian, Y. Zhang, Z. Liu, G. Sun, G. Mor and A. Liao, Sci. Rep., 2016, 6, 27683 CrossRef CAS PubMed.
- Q. Hu, M. Wu, C. Fang, C. Cheng, M. Zhao, W. Fang, P. K. Chu, Y. Ping and G. Tang, Nano Lett., 2015, 15(4), 2732–2739 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- S. Bao, K. W. Beagley, M. P. France, J. Shen and A. J. Husband, Immunology, 2000, 99, 464–472 CrossRef CAS PubMed.
- D. Li, Y. Li, H. Xing, J. Guo, Y. Ping and G. Tang, Adv. Funct. Mater., 2014, 24, 5482–5492 CrossRef CAS.
- G. L. Beatty and Y. Paterson, Immunol. Res., 2001, 24, 201–210 CrossRef CAS.
- G. Pietersz, C.-K. Tang and V. Apostolopoulos, Mini-Rev. Med. Chem., 2006, 6, 1285–1298 CrossRef CAS.
- J. F. Toso, V. J. Gill, P. Hwu, F. M. Marincola, N. P. Restifo, D. J. Schwartzentruber, R. M. Sherry, S. L. Topalian, J. C. Yang, F. Stock, L. J. Freezer, K. E. Morton, C. Seipp, L. Haworth, S. Mavroukakis, D. White, S. MacDonald, J. Mao and M. Sznol, J. Clin. Oncol., 2002, 20(1), 142–152 CrossRef.
- D. H. Thamm, I. D. Kurzman, I. King, Z. Li, M. Sznol, R. R. Dubielzig, D. M. Vail and E. G. MacEwen, Clin. Cancer Res., 2005, 11, 4827–4834 CrossRef CAS PubMed.
- L. Jia, D. Wei, Q. Sun, Y. Huang, Q. Wu and Z. Hua, Cancer Sci., 2007, 98, 1107–1112 CrossRef CAS PubMed.
- B. D. Jones and S. Falkow, Annu. Rev. Immunol., 1996, 14, 533–561 CrossRef CAS.
- C. Li, A. Faulkner-Jones, A. R. Dun, J. Jin, P. Chen, Y. Xing, Z. Yang, Z. Li, W. Shu, D. Liu and R. R. Duncan, Angew. Chem., Int. Ed., 2015, 54, 3957–3961 Search PubMed.
- C. Li, P. Chen, Y. Shao, X. Zhou, Y. Wu, Z. Yang, Z. Li, T. Weil and D. Liu, Small, 2015, 11, 1224–1224 CrossRef.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- R. Censi, W. Schuurman, J. Malda, G. Di Dato, P. E. Burgisser, W. J. A. Dhert, C. F. Van Nostrum, P. Di Martino, T. Vermonden and W. E. Hennink, Adv. Funct. Mater., 2011, 21, 1833–1842 CrossRef CAS.
- K. Nagahama, T. Ouchi and Y. Ohya, Adv. Funct. Mater., 2008, 18, 1220–1231 CrossRef CAS.
- M. Chaouat, C. Le Visage, W. E. Baille, B. Escoubet, F. Chaubet, M. A. Mateescu and D. Letourneur, Adv. Funct. Mater., 2008, 18, 2855–2861 CrossRef CAS.
- H. Gudapati, J. Yan, Y. Huang and D. B. Chrisey, Biofabrication, 2014, 6, 035022 CrossRef PubMed.
- S. Khalil and W. Sun, J. Biomech. Eng., 2009, 131, 111002 CrossRef.
- J. C. Reichert, A. Heymer, A. Berner, J. Eulert and U. Nöth, Biomed. Mater., 2009, 4, 065001 CrossRef CAS PubMed.
- Y.-C. Chiu, J. C. Larson, V. H. Perez-Luna and E. M. Brey, Chem. Mater., 2009, 21, 1677–1682 CrossRef CAS.
- L. A. Hockaday, K. H. Kang, N. W. Colangelo, P. Y. C. Cheung, B. Duan, E. Malone, J. Wu, L. N. Girardi, L. J. Bonassar, H. Lipson, C. C. Chu and J. T. Butcher, Biofabrication, 2012, 4, 035005 CrossRef CAS PubMed.
- N. Kim, H. Lee, G. Han, M. Kang, S. Park, D. E. Kim, M. Lee, M. Kim, Y. Na, S. Oh, S. Bang, T. Jang, H. Kim, J. Park, S. R. Shin and H. Jung, Adv. Sci., 2023, 10, 2300816 CAS.
- H. Li, Y. J. Tan, K. F. Leong and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 20086–20097 CAS.
- E. B. Heggset, B. L. Strand, K. W. Sundby, S. Simon, G. Chinga-Carrasco and K. Syverud, Cellulose, 2019, 26, 581–595 CrossRef CAS.
- H. Huang, J. Ayariga, H. Ning, E. Nyairo and D. Dean, Addit. Manuf., 2021, 46, 102120 CAS.
- J. Müller, A. C. Jäkel, D. Schwarz, L. Aufinger and F. C. Simmel, Small, 2020, 16, 2001815 CrossRef.
- A. Cangialosi, C. Yoon, J. Liu, Q. Huang, J. Guo, T. D. Nguyen, D. H. Gracias and R. Schulman, Science, 2017, 357, 1126–1130 CrossRef CAS PubMed.
- P. B. Allen, Z. Khaing, C. E. Schmidt and A. D. Ellington, ACS Biomater. Sci. Eng., 2015, 1, 19–26 CrossRef CAS PubMed.
- K. Kusumoto, T. Akao, E. Mizuki and O. Nakamura, Cytotechnology, 2006, 51, 57–66 CrossRef CAS.
- H. Ju, D. Kim and Y.-K. Oh, Asian J. Pharm. Sci., 2022, 17, 641–652 CrossRef CAS PubMed.
- K. Ezzat, Y. Aoki, T. Koo, G. McClorey, L. Benner, A. Coenen-Stass, L. O'Donovan, T. Lehto, A. Garcia-Guerra, J. Nordin, A. F. Saleh, M. Behlke, J. Morris, A. Goyenvalle, B. Dugovic, C. Leumann, S. Gordon, M. J. Gait, S. El-Andaloussi and M. J. Wood, Nano Lett., 2015, 15, 4364–4373 CrossRef CAS PubMed.
- J. M. Tomkins, K. J. Barnes, A. J. Blacker, W. J. Watkins and C. Abell, Tetrahedron Lett., 1997, 38, 691–694 CrossRef CAS.
- A. S. Boutorin, L. V. Gus'kova, E. M. Ivanova, N. D. Kobetz, V. F. Zarytova, A. S. Ryte, L. V. Yurchenko and V. V. Vlassov, FEBS Lett., 1989, 254, 129–132 CrossRef CAS PubMed.
- J. Liu, Z. Wang, S. Zhao and B. Ding, Nano Res., 2018, 11, 5017–5027 CAS.
- R. L. Letsinger, G. R. Zhang, D. K. Sun, T. Ikeuchi and P. S. Sarin, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6553–6556 CrossRef CAS PubMed.
- C. A. Stein, R. Pal, A. L. DeVico, G. Hoke, S. Mumbauer, O. Kinstler, M. G. Sarngadharan and R. L. Letsinger, Biochemistry, 1991, 30, 2439–2444 CAS.
- M. Manoharan, K. L. Tivel, T. P. Condon, L. K. Andrade, I. Barber-Peoch, G. Inamati, S. Shah, V. Mohan, M. J. Graham, C. F. Bennett, S. T. Crooke and P. D. Cook, Nucleosides Nucleotides, 1997, 16, 1129–1138 CAS.
- A. J. Sinegra, M. Evangelopoulos, J. Park, Z. Huang and C. A. Mirkin, Nano Lett., 2021, 21, 6584–6591 CAS.
- C. D. Sago, M. P. Lokugamage, K. Paunovska, D. A. Vanover, C. M. Monaco, N. N. Shah, M. Gamboa Castro, S. E. Anderson, T. G. Rudoltz, G. N. Lando, P. Munnilal Tiwari, J. L. Kirschman, N. Willett, Y. C. Jang, P. J. Santangelo, A. V. Bryksin and J. E. Dahlman, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(42), E9944–E9952 CAS.
- S. W. Jones, R. A. Roberts, G. R. Robbins, J. L. Perry, M. P. Kai, K. Chen, T. Bo, M. E. Napier, J. P. Y. Ting, J. M. DeSimone and J. E. Bear, J. Clin. Invest., 2013, 123, 3061–3073 CAS.
- K. M. Tsoi, S. A. MacParland, X.-Z. Ma, V. N. Spetzler, J. Echeverri, B. Ouyang, S. M. Fadel, E. A. Sykes, N. Goldaracena, J. M. Kaths, J. B. Conneely, B. A. Alman, M. Selzner, M. A. Ostrowski, O. A. Adeyi, A. Zilman, I. D. McGilvray and W. C. W. Chan, Nat. Mater., 2016, 15, 1212–1221 CAS.
- K. Paunovska, C. D. Sago, C. M. Monaco, W. H. Hudson, M. G. Castro, T. G. Rudoltz, S. Kalathoor, D. A. Vanover, P. J. Santangelo, R. Ahmed, A. V. Bryksin and J. E. Dahlman, Nano Lett., 2018, 18, 2148–2157 CAS.
- C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, Nat. Rev. Drug Discovery, 2015, 14, 239–247 CAS.
- Y. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. Zhang, D. Chen, R. L. Bogorad, H. Yin, Y. Chen, A. J. Vegas, C. A. Alabi, G. Sahay, K. T. Olejnik, W. Wang, A. Schroeder, A. K. R. Lytton-Jean, D. J. Siegwart, A. Akinc, C. Barnes, S. A. Barros, M. Carioto, K. Fitzgerald, J. Hettinger, V. Kumar, T. I. Novobrantseva, J. Qin, W. Querbes, V. Koteliansky, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3955–3960 CAS.
- J. Hao, P. Kos, K. Zhou, J. B. Miller, L. Xue, Y. Yan, H. Xiong, S. Elkassih and D. J. Siegwart, J. Am. Chem. Soc., 2015, 137, 9206–9209 CAS.
- K. Paunovska, C. J. Gil, M. P. Lokugamage, C. D. Sago, M. Sato, G. N. Lando, M. Gamboa Castro, A. V. Bryksin and J. E. Dahlman, ACS Nano, 2018, 12, 8341–8349 CrossRef CAS.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stöter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS.
- W. Palm and C. B. Thompson, Nature, 2017, 546, 234–242 CrossRef CAS.
- A. Lechanteur, V. Sanna, A. Duchemin, B. Evrard, D. Mottet and G. Piel, Nanomaterials, 2018, 8, 270 CrossRef.
- N. Tran, M. Hocquet, B. Eon, P. Sangwan, J. Ratcliffe, T. M. Hinton, J. White, B. Ozcelik, N. P. Reynolds and B. W. Muir, J. Colloid Interface Sci., 2018, 519, 107–118 CrossRef CAS PubMed.
- J. Clogston and M. Caffrey, J. Controlled Release, 2005, 107, 97–111 CAS.
- S. Sarkar, N. Tran, M. H. Rashid, T. C. Le, I. Yarovsky, C. E. Conn and C. J. Drummond, ACS Appl. Bio Mater., 2019, 2, 182–195 CrossRef CAS PubMed.
- A. I. I. Tyler, H. M. G. Barriga, E. S. Parsons, N. L. C. McCarthy, O. Ces, R. V. Law, J. M. Seddon and N. J. Brooks, Soft Matter, 2015, 11, 3279–3286 RSC.
- C. R. Safinya, K. K. Ewert and C. Leal, Liq. Cryst., 2011, 38, 1715–1723 CrossRef CAS PubMed.
- R. Koynova, B. Tenchov and R. C. MacDonald, ACS Biomater. Sci. Eng., 2015, 1, 130–138 CrossRef CAS PubMed.
- C. Leal, N. F. Bouxsein, K. K. Ewert and C. R. Safinya, J. Am. Chem. Soc., 2010, 132, 16841–16847 CrossRef CAS.
- M. Martínez-Negro, K. Kumar, A. L. Barrán-Berdón, S. Datta, P. Kondaiah, E. Junquera, S. Bhattacharya and E. Aicart, ACS Appl. Mater. Interfaces, 2016, 8, 22113–22126 CrossRef.
- C. Leal, K. K. Ewert, N. F. Bouxsein, R. S. Shirazi, Y. Li and C. R. Safinya, Soft Matter, 2013, 9, 795–804 RSC.
- I. Amar-Yuli, J. Adamcik, S. Blau, A. Aserin, N. Garti and R. Mezzenga, Soft Matter, 2011, 7, 8162 RSC.
- M. M. Ali, D. Kang, K. Tsang, M. Fu, J. M. Karp and W. Zhao, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 547–561 CAS.
- Y. Teramura and H. Iwata, Soft Matter, 2010, 6, 1081 RSC.
- N. A. A. Rossi, I. Constantinescu, R. K. Kainthan, D. E. Brooks, M. D. Scott and J. N. Kizhakkedathu, Biomaterials, 2010, 31, 4167–4178 CrossRef CAS PubMed.
- M. T. Stephan, J. J. Moon, S. H. Um, A. Bershteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed.
- G. Ke, Z. Zhu, W. Wang, Y. Zou, Z. Guan, S. Jia, H. Zhang, X. Wu and C. J. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 15329–15334 CrossRef CAS.
- S. Chen, Y. Hong, Y. Liu, J. Liu, C. W. T. Leung, M. Li, R. T. K. Kwok, E. Zhao, J. W. Y. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926–4929 CrossRef CAS PubMed.
- J. Llopis, J. M. McCaffery, A. Miyawaki, M. G. Farquhar and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6803–6808 CrossRef CAS.
- Y.-H. Chan, C. Wu, F. Ye, Y. Jin, P. B. Smith and D. T. Chiu, Anal. Chem., 2011, 83, 1448–1455 CrossRef CAS.
- J. Urra, M. Sandoval, I. Cornejo, L. F. Barros, F. V. Sepúlveda and L. P. Cid, Pflug. Arch. Eur. J. Physiol., 2008, 457, 233–242 CrossRef CAS.
- M. You, Y. Lyu, D. Han, L. Qiu, Q. Liu, T. Chen, C. Sam Wu, L. Peng, L. Zhang, G. Bao and W. Tan, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- T. Tokunaga, S. Namiki, K. Yamada, T. Imaishi, H. Nonaka, K. Hirose and S. Sando, J. Am. Chem. Soc., 2012, 134, 9561–9564 CrossRef CAS.
- Y.-H. M. Chan, B. Van Lengerich and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979–984 CrossRef CAS.
- L. Zhou, W. Zhang, J. Lee, L. Kuhn and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 51321–51332 CrossRef CAS.
- M. E. Todhunter, N. Y. Jee, A. J. Hughes, M. C. Coyle, A. Cerchiari, J. Farlow, J. C. Garbe, M. A. LaBarge, T. A. Desai and Z. J. Gartner, Nat. Methods, 2015, 12, 975–981 CrossRef CAS.
- M. Roth, Anal. Chem., 1971, 43, 880–882 CrossRef CAS.
- P. De Montigny, J. F. Stobaugh, R. S. Givens, R. G. Carlson, K. Srinivasachar, L. A. Sternson and T. Higuchi, Anal. Chem., 1987, 59, 1096–1101 CrossRef CAS.
- J. Selb and Y. Gallot, Makromol. Chem., 1980, 181, 809–822 CrossRef CAS.
- D. W. Miller, E. V. Batrakova, T. O. Waltner, V. Yu. Alakhov and A. V. Kabanov, Bioconjugate Chem., 1997, 8, 649–657 CrossRef CAS PubMed.
- C. Deng, H. Yang, S. Liu and Z. Zhao, Polymer, 2023, 266, 125621 CrossRef CAS.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS.
- Y. Guo, J. Chen, M. Cheng, D. Monchaud, J. Zhou and H. Ju, Angew. Chem., Int. Ed., 2017, 56, 16636–16640 CrossRef CAS.
- R. Zhong, M. Xiao, C. Zhu, X. Shen, Q. Tang, W. Zhang, L. Wang, S. Song, X. Qu, H. Pei, C. Wang and L. Li, ACS Appl. Mater. Interfaces, 2018, 10, 4512–4518 CrossRef CAS.
- E. Magdalena Estirado, B. J. H. M. Rosier, T. F. A. De Greef and L. Brunsveld, Chem. Commun., 2020, 56, 5747–5750 RSC.
- B. J. H. M. Rosier, A. J. Markvoort, B. Gumí Audenis, J. A. L. Roodhuizen, A. Den Hamer, L. Brunsveld and T. F. A. De Greef, Nat. Catal., 2020, 3, 295–306 CrossRef CAS.
- M. Renatus, H. R. Stennicke, F. L. Scott, R. C. Liddington and G. S. Salvesen, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14250–14255 CrossRef CAS.
- Y. Chao, E. N. Shiozaki, S. M. Srinivasula, D. J. Rigotti, R. Fairman and Y. Shi, PLos Biol., 2005, 3, e183 CrossRef PubMed.
- Y. Li, M. Zhou, Q. Hu, X. Bai, W. Huang, S. H. W. Scheres and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1542–1547 CrossRef CAS.
- D. T. Dang, H. D. Nguyen, M. Merkx and L. Brunsveld, Angew. Chem., Int. Ed., 2013, 52, 2915–2919 CrossRef CAS.
- Q. Luo, Z. Dong, C. Hou and J. Liu, Chem. Commun., 2014, 50, 9997 RSC.
- M. K. Müller, K. Petkau and L. Brunsveld, Chem. Commun., 2011, 47, 310–312 RSC.
- K. Petkau-Milroy, D. A. Uhlenheuer, A. J. H. Spiering, J. A. J. M. Vekemans and L. Brunsveld, Chem. Sci., 2013, 4, 2886 RSC.
- S. P. W. Wijnands, W. Engelen, R. P. M. Lafleur, E. W. Meijer and M. Merkx, Nat. Commun., 2018, 9, 65 CrossRef.
- X. Tian, N. A. Risgaard, P. M. G. Löffler and S. Vogel, J. Am. Chem. Soc., 2023, 145, 19633–19641 CrossRef CAS PubMed.
- S. M. Christensen, P.-Y. Bolinger, N. S. Hatzakis, M. W. Mortensen and D. Stamou, Nat. Nanotechnol., 2012, 7, 51–55 CrossRef CAS.
- P. M. G. Löffler, O. Ries, A. Rabe, A. H. Okholm, R. P. Thomsen, J. Kjems and S. Vogel, Angew. Chem., Int. Ed., 2017, 56, 13228–13231 CrossRef.
- G. Stengel, R. Zahn and F. Höök, J. Am. Chem. Soc., 2007, 129, 9584–9585 CAS.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CAS.
- Y. Li, P. Zhao, M. Zhang, X. Zhao and X. Li, J. Am. Chem. Soc., 2013, 135, 17727–17730 CAS.
- B. Thomas, X. Lu, W. R. Birmingham, K. Huang, P. Both, J. E. Reyes Martinez, R. J. Young, C. P. Davie and S. L. Flitsch, ChemBioChem, 2017, 18, 858–863 CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CAS.
- H. Yanai, S. Obara and T. Taguchi, Org. Biomol. Chem., 2008, 6, 2679 CAS.
- R. A. Goodnow, C. E. Dumelin and A. D. Keefe, Nat. Rev. Drug Discovery, 2017, 16, 131–147 CrossRef CAS.
- J. Zou, Q. Gao, J. Nie, Y. Zhang and C. Jin, RSC Adv., 2021, 11, 18322–18325 RSC.
- M. Ghufran Rafique, J. M. Remington, F. Clark, H. Bai, V. Toader, D. F. Perepichka, J. Li and H. F. Sleiman, Angew. Chem., Int. Ed., 2023, 62, e202217814 CrossRef CAS PubMed.
- K. Shohda and T. Sugawara, Soft Matter, 2006, 2, 402 RSC.
- I. A. Chen and J. W. Szostak, Biophys. J., 2004, 87, 988–998 CrossRef CAS.
- W. Yu, K. Sato, M. Wakabayashi, T. Nakaishi, E. P. Ko-Mitamura, Y. Shima, I. Urabe and T. Yomo, J. Biosci. Bioeng., 2001, 92, 590–593 CrossRef CAS.
- S. M. Nomura, K. Tsumoto, T. Hamada, K. Akiyoshi, Y. Nakatani and K. Yoshikawa, ChemBioChem, 2003, 4, 1172–1175 CrossRef CAS PubMed.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669–17674 CrossRef CAS PubMed.
- N. Zhao, Y. Chen, G. Chen and Z. Xiao, ACS Appl. Bio Mater., 2020, 3, 3928–3934 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- P. Liu, Y. Zhao, X. Liu, J. Sun, D. Xu, Y. Li, Q. Li, L. Wang, S. Yang, C. Fan and J. Lin, Angew. Chem., Int. Ed., 2018, 57, 5418–5422 CrossRef CAS PubMed.
- M. Xiong, H. Zhu, Q. Rong, C. Yang, L. Qiu, X.-B. Zhang and W. Tan, Chem. Commun., 2016, 52, 4679–4682 RSC.
- Y. Zhang, V. Pan, X. Li, X. Yang, H. Li, P. Wang and Y. Ke, Small, 2019, 15, 1900228 CrossRef PubMed.
- X. Chang, C. Zhang, C. Lv, Y. Sun, M. Zhang, Y. Zhao, L. Yang, D. Han and W. Tan, J. Am. Chem. Soc., 2019, 141, 12738–12743 CrossRef CAS.
- R. Peng, X. Zheng, Y. Lyu, L. Xu, X. Zhang, G. Ke, Q. Liu, C. You, S. Huan and W. Tan, J. Am. Chem. Soc., 2018, 140, 9793–9796 CrossRef CAS.
- H. Liu, Q. Yang, R. Peng, H. Kuai, Y. Lyu, X. Pan, Q. Liu and W. Tan, J. Am. Chem. Soc., 2019, 141, 6458–6461 CrossRef CAS.
- Y. Liu, Y. Zeng, L. Liu, C. Zhuang, X. Fu, W. Huang and Z. Cai, Nat. Commun., 2014, 5, 5393 CrossRef CAS.
- T. Fu, Y. Lyu, H. Liu, R. Peng, X. Zhang, M. Ye and W. Tan, Trends Biochem. Sci., 2018, 43, 547–560 CrossRef CAS PubMed.
- N. Wu, F. Chen, Y. Zhao, X. Yu, J. Wei and Y. Zhao, Langmuir, 2018, 34, 14721–14730 CrossRef CAS.
- M. L. Daly, K. Nishi, S. J. Klawa, K. Y. Hinton, Y. Gao and R. Freeman, Nat. Chem., 2024, 16, 1229–1239 CrossRef CAS PubMed.
- M. E. Janson, R. Loughlin, I. Loïodice, C. Fu, D. Brunner, F. J. Nédélec and P. T. Tran, Cell, 2007, 128, 357–368 Search PubMed.
- J. R. Bartles, Curr. Opin. Cell Biol., 2000, 12, 72–78 CrossRef CAS PubMed.
- P. Prinsen and P. Van Der Schoot, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 68, 021701 Search PubMed.
- K. L. Weirich, S. Banerjee, K. Dasbiswas, T. A. Witten, S. Vaikuntanathan and M. L. Gardel, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2131–2136 CAS.
- K. Jahnke, V. Huth, U. Mersdorf, N. Liu and K. Göpfrich, ACS Nano, 2022, 16, 7233–7241 CAS.
- N. Arulkumaran, M. Singer, S. Howorka and J. R. Burns, Nat. Commun., 2023, 14, 1314 CAS.
- R. Booth, I. Insua, S. Ahmed, A. Rioboo and J. Montenegro, Nat. Commun., 2021, 12, 6421 CAS.
- A. Méndez-Ardoy, J. R. Granja and J. Montenegro, Nanoscale Horiz., 2018, 3, 391–396 Search PubMed.
- A. Méndez-Ardoy, A. Bayón-Fernández, Z. Yu, C. Abell, J. R. Granja and J. Montenegro, Angew. Chem., Int. Ed., 2020, 59, 6902–6908 CrossRef PubMed.
- S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan, S. Chaudhary and R. Varghese, Angew. Chem., Int. Ed., 2014, 53, 8352–8357 CrossRef CAS PubMed.
- S. K. Albert, I. Sivakumar, M. Golla, H. V. P. Thelu, N. Krishnan, J. L. K. L. Ashish and R. Varghese, J. Am. Chem. Soc., 2017, 139, 17799–17802 CrossRef CAS PubMed.
- M. Golla, S. K. Albert, S. Atchimnaidu, D. Perumal, N. Krishnan and R. Varghese, Angew. Chem., Int. Ed., 2019, 58, 3865–3869 CrossRef CAS PubMed.
- T. Chen, C. S. Wu, E. Jimenez, Z. Zhu, J. G. Dajac, M. You, D. Han, X. Zhang and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 2012–2016 CrossRef CAS.
- S. C. Owen, D. P. Y. Chan and M. S. Shoichet, Nano Today, 2012, 7, 53–65 CrossRef CAS.
- M. F. Osborn, A. H. Coles, A. Biscans, R. A. Haraszti, L. Roux, S. Davis, S. Ly, D. Echeverria, M. R. Hassler, B. M. D. C. Godinho, M. Nikan and A. Khvorova, Nucleic Acids Res., 2019, 47, 1070–1081 CrossRef CAS PubMed.
- A. Lacroix, T. G. W. Edwardson, M. A. Hancock, M. D. Dore and H. F. Sleiman, J. Am. Chem. Soc., 2017, 139, 7355–7362 CrossRef CAS PubMed.
- A. E. Felber, N. Bayó-Puxan, G. F. Deleavey, B. Castagner, M. J. Damha and J.-C. Leroux, Biomaterials, 2012, 33, 5955–5965 CrossRef CAS PubMed.
- T. Miller, R. Rachel, A. Besheer, S. Uezguen, M. Weigandt and A. Goepferich, Pharm. Res., 2012, 29, 448–459 CrossRef CAS PubMed.
- P. P. Karmali and D. Simberg, Expert Opin. Drug Delivery, 2011, 8, 343–357 CrossRef CAS.
- X. Li, C. A. Figg, R. Wang, Y. Jiang, Y. Lyu, H. Sun, Y. Liu, Y. Wang, I. Teng, W. Hou, R. Cai, C. Cui, L. Li, X. Pan, B. S. Sumerlin and W. Tan, Angew. Chem., Int. Ed., 2018, 57, 11589–11593 CrossRef CAS PubMed.
- M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 6218–6223 CrossRef CAS PubMed.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899–902 CrossRef CAS.
- H. Liu, B. Kwong and D. J. Irvine, Angew. Chem., Int. Ed., 2011, 50, 7052–7055 CrossRef CAS PubMed.
- J. Yang, Z. Meng, Q. Liu, Y. Shimada, R. C. L. Olsthoorn, H. P. Spaink, A. Herrmann and A. Kros, Chem. Sci., 2018, 9, 7271–7276 RSC.
- A. C. Anselmo and S. Mitragotri, J. Controlled Release, 2014, 190, 531–541 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |