
Tailoring the photoluminescence of AIE-type gold nanoclusters via biomineralization-inspired polymorphism†
Sukhendu
Mahata
ab,
Satya Ranjan
Sahoo
ab,
Arun
Mukhopadhyay
ab,
Komal
Kumari
c,
Surajit
Rakshit
 c and
Nirmal
Goswami
*ab
c and
Nirmal
Goswami
*ab
aMaterials Chemistry Department, CSIR-Institute of Minerals and Materials Technology, Bhubaneswar 751013, Odisha, India. E-mail: ngoswami@immt.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, Uttar Pradesh, India
cDepartment of Chemistry, Institute of Science, Banaras Hindu University, Varanasi 221005, Uttar Pradesh, India
First published on 19th November 2024
Abstract
Tailoring the aggregation-induced emission (AIE) characteristics of well-defined metal nanoclusters (MNCs) is highly sought after for numerous practical applications. Studies have primarily focused on assembling AIE-type MNCs using monomorphic molecules. Achieving polymorphic assemblies, with different molecular arrangements could provide valuable insights into the role of external molecular matrices on the photoluminescence (PL) behaviour of these NCs. In this study, by mimicking biomineralization, we successfully embedded AIE-type Au22SG18 NCs within different polymorphic environments of CaCO3. Upon incorporation into CaCO3 matrices such as, calcite, vaterite and a mixture of both, the PL was enhanced in all the inorganic composites accompanied by a significant blue shift. In the metastable vaterite matrix, Au22SG18 NCs exhibited the highest blue shift in the PL spectrum while in the stable crystalline matrix of calcite, the NCs showed the highest PL intensity as well as excited state lifetime. Time-resolved spectroscopic and single-molecule Raman studies revealed that variations in the PL of NCs are linked to the stability of their polymorphic structures, progressing from vaterite to a vaterite/calcite mixture, and finally to calcite. These findings shed light on the crucial role of external molecular arrangement in the AIE behaviour of MNCs.
Introduction
Recently, atomically precise metal nanoclusters (MNCs) have emerged as prime contenders in various application domains, including solid-state lighting and displays, catalysis, biosensing, bioimaging, and therapy, due to their intriguing molecular-like properties such as photoluminescence (PL), HOMO–LUMO transitions, molecular chirality, etc.1–13 Among these features, their well-defined crystal structures and strong PL are particularly noteworthy, enabling researchers to create a range of self-assembled superstructures in three dimensions. This approach has led to significant improvements in properties such as PL, catalytic activity, and biological functions.14–16To construct self-assembled MNCs-based superstructures, various components such as polymers, small organic molecules, metal ions, and biomolecules have been utilized.17–23 These components often act as counterparts, facilitating assembly formation through a range of covalent and non-covalent interactions.24–29 When MNCs are successfully integrated into these assembles, one of the most notable changes in their physicochemical properties is PL, which is highly sensitive to immediate changes in the MNCs environment. This effect is particularly pronounced in MNCs exhibiting aggregation-induced emission (AIE) characteristics, a phenomenon associated with restricted intra and inter-molecular rotational and vibrational motions, that are very responsive to their surroundings.25,30–32 Supramolecular interactions, such as hydrogen-bonding, hydrophobic effects, and electrostatic forces within the assembled superstructures contribute to the rigidity and molecular packing, and thereby restrict the intra- and inter-molecular motions of the ligand shell of the AIE-type MNCs, leading to a significant enhancement in their PL quantum yield.33–35 Many of these supramolecular interactions have been extensively studied in relation to the AIE behavior in MNCs.25 These interactions primarily generate an organic environment or matrix that is uniform and monomorphic in nature.36–38 However, the AIE behavior of MNCs within inorganic solid frameworks or matrices exhibiting polymorphism remains largely unexplored.
Polymorphism arises when a compound adopts at least two different molecular arrangements in the solid state.39–44 These distinct arrangements create varied matrix environments for the assembled MNCs. Integrating AIE-type well-defined MNCs within different polymorphic environments would possibly offer a unique platform to investigate the influence of polymorphism on the AIE behavior of the MNCs. Understanding how polymorphism affects the AIE phenomenon could help establish correlations between specific molecular arrangements and the resulting AIE properties of MNCs. This area of research is crucial for furthering our knowledge of the relationship between molecular structure and AIE behavior in MNCs.
Biomineralization is a natural process where molecules or ions assemble through electrostatic or weak dipole–dipole interactions.45,46 We hypothesize that mimicking this process to create self-assembled inorganic host matrices for AIE-type MNCs could be a promising approach for studying the effects of polymorphism on the AIE behavior of MNCs.47,48 Inorganic solid matrices formed through biomineralization offer tunable cavities and customizable chemistries, which can generate distinct polymorphic environments for AIE-type MNCs.49,50 For example, the mechanism of CaCO3 nucleation via biomineralization is well understood.51 By mimicking this process through different synthetic methods, it is possible to obtain various polymorphs of CaCO3, such as crystalline CaCO3via the coprecipitation method or amorphous CaCO3 (ACC) through vapor deposition.47 These distinct polymorphs could provide unique matrices for the MNCs, offering a valuable opportunity to investigate how different polymorphic environments influence the AIE phenomenon.
In this study, we selected CaCO3 as the inorganic host matrix for assembling pre-synthesized AIE-type AuNCs i.e., Au22SG18 (where –SG stands for reduced glutathione). Using different synthetic routes, we obtained distinct polymorphs of CaCO3: crystalline CaCO3 through a coprecipitation method and vaterite CaCO3 by vapor diffusion method, and a mixture of calcite and vaterite also through an ethanol based coprecipitation method. Upon embedding the Au22SG18 NCs into these polymorphic environments, we investigated their AIE behavior. Remarkably, the PL of the NCs was significantly enhanced in all the CaCO3 matrices, driven by the self-assembling PL enhancement effect. This strong PL enhancement is the characteristic of an AIE-like phenomenon, arising from the rigidification of the ligand shells and conformational orientation of the assemblies. Interestingly, in the vaterite CaCO3 matrix, the NCs exhibited a greater blue shift compared to their behavior in the crystalline matrix, while the PL lifetime of the NCs was longer in the crystalline calcite environment.52 These findings highlight how the distinct polymorphic matrices of CaCO3 affect the AIE behavior of the NCs, offering insight into the relationship between matrix structure and photophysical properties. This study demonstrates that the stability and polymorphic transitions of CaCO3 matrices, from vaterite to calcite, significantly impact the photophysical properties of Au22SG18 NCs, providing new insights into how polymorphism governs AIE behavior.
Results and discussion
To investigate the AIE-behavior of MNCs in an inorganic polymorphic matrix, we selected CaCO3 as an appropriate matrix, due to its straightforward preparation via biomineralization.53 With the right preparation methods and experimental conditions, various crystalline polymorphs of CaCO3 can be produced. Among these, vaterite is the least stable, aragonite is marginally less stable, and calcite is the most thermodynamically stable polymorph at ambient temperature. Additionally, there exists an amorphous polymorph known as amorphous calcium carbonate (ACC).For our model MNCs, we chose atomically precise Au22SG18 NCs (SG = reduced glutathione) due to their well-documented AIE properties, large Stokes shift (>100 nm), excellent photoluminescence quantum yield, biocompatibility, and good photostability.54–56 The successful synthesis of Au22SG18 was confirmed by UV-vis spectroscopy and PL spectroscopy, which showed an absorbance peak at 520 nm along with PL excitation and emission spectra around 520 nm and 660 nm, respectively as illustrated in Fig. S1.† Furthermore, the large Stokes shift (∼140 nm) validated the AIE property of the NCs (Fig. S1†). These NCs underwent rigorous purification before being utilized in subsequent studies.
Au22SG18 NCs were embedded within the polymorphic matrix of CaCO3 through biomineralization (detailed methods are provided in the Experimental section, Fig. S2–S4†). As shown in Field emission scanning electron microscopy (FESEM) and High-resolution transmission electron microscopy (HRTEM) images in Fig. 1A–F, the morphology of the assemblies was distinct and varied. The Au22SG18 NCs were incorporated into different polymorphs of CaCO3: Au22SG18@CaCO3-Cube with crystalline cubic matrix, Au22SG18@CaCO3-Cube/Sphere featuring both crystalline cubic and spherical matrix, and Au22SG18@CaCO3-Sphere with a spherical matrix. Given their varied structures, the surrounding environments of the NCs were expected to differ. Such morphology was consistent with the control structures of respective CaCO3 types without NCs, as shown in Fig. S5–S7.† These findings suggest that different morphologies arose from the inherent synthetic procedure of CaCO3, rather than being induced by the NCs. Additionally, scanning transmission electron microscopy (STEM) images confirmed that the NCs were uniformly embedded within the respective CaCO3 matrices (Fig. 1G–Z–AA).
 | ||
| Fig. 1 FESEM images of (A) Au22SG18@CaCO3-Cube, (B) Au22SG18@CaCO3-Cube/Sphere, and (C) Au22SG18@CaCO3-Sphere. HRTEM images of (D) Au22SG18@CaCO3-Cube, (E) Au22SG18@CaCO3-Cube/Sphere, and (F) Au22SG18@CaCO3-Sphere. STEM images and nanoscale elemental mapping of (G–M) Au22SG18@CaCO3-Cube, (N–T) Au22SG18@CaCO3-Cube/Sphere, and (U–AA) Au22SG18@CaCO3-Sphere. | ||
To reveal the differences in internal crystal structures among the materials with varied morphologies, we conducted powder X-ray diffraction (PXRD) analysis, which allowed us to distinguish the structural differences between the polymorphs of CaCO3 based on these morphological changes. As shown in Fig. 2A, the 2θ peaks for Au22SG18@CaCO3-Cube at 23.01° and 29.41° correspond to the (0 1 2) and (1 0 4) planes, respectively, resembling the calcite structure (JCPDS code 01-072-1937).57 Similar peaks were observed in the control CaCO3-Cube, indicating that the Au22SG18 NCs were embedded within the crystalline matrix. In contrast, the PXRD spectrum for Au22SG18@CaCO3-Sphere was nearly featureless, suggesting an amorphous nature; however, a magnified view revealed small crystalline peaks attributed to the vaterite structure of CaCO3. This indicates that the Au22SG18 NCs were likely embedded in a vaterite matrix, although the presence of some ACC cannot be ruled out.58,59 Alternatively, it is possible that the NCs were initially embedded in an ACC matrix, which gradually converted to vaterite due to the instability of the ACC form under ambient conditions. Interestingly, PXRD results for Au22SG18@CaCO3-Cube/Sphere indicated a mixed state, displaying peaks at 24.91° and 27.05°, corresponding to the (100) and (101) planes of vaterite (JCPDS code – 00-024-0030), alongside a peak at 29.39° associated with the calcite phase (104 plane).60 Similar peaks were also observed in the control CaCO3-Cube/Sphere. These results suggest that in Au22SG18@CaCO3-Cube/Sphere, the NCs were embedded in both calcite and vaterite matrices. The presence of gold in different polymorphs was also probed by inductively coupled plasma-optical emission spectrometer (ICP-OES) measurement (Table S3†). Taken together, we successfully embedded the NCs in distinct polymorphic environments of CaCO3.
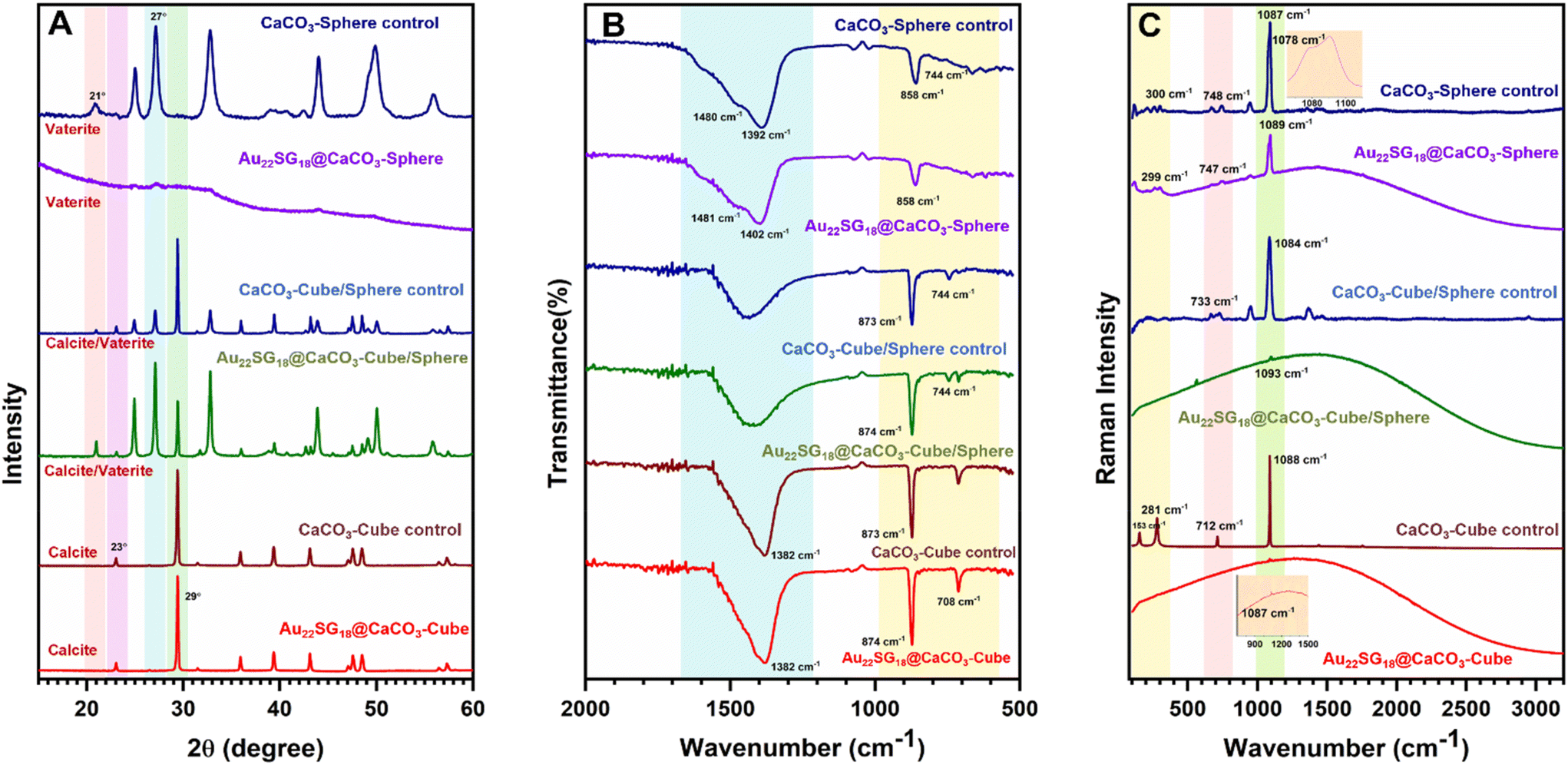 | ||
| Fig. 2 (A) PXRD spectra of Au22SG18@CaCO3-Cube, CaCO3-Cube-control, Au22SG18@CaCO3-Cube/Sphere, CaCO3-Cube/Sphere-control, Au22SG18@CaCO3-Sphere and CaCO3-Sphere-control. (B) FTIR spectra of Au22SG18@CaCO3-Cube, CaCO3-Cube-control, Au22SG18@CaCO3-Cube/Sphere, CaCO3-Cube/Sphere-control, Au22SG18@CaCO3-Sphere and CaCO3-Sphere-control. (C) Raman spectra of Au22SG18@CaCO3-Cube, CaCO3-Cube-control, Au22SG18@CaCO3-Cube/Sphere, CaCO3-Cube/Sphere-control, Au22SG18@CaCO3-Sphere and CaCO3-Sphere-control. | ||
The different polymorphic environments were further characterized by Fourier Transform Infrared Spectroscopy (FTIR) analysis (Fig. 2B). The Au22SG18@CaCO3-Sphere showed the absorption band at 858 cm−1,744 cm−1 and splitting bands at 1402 cm−1 and 1481 cm−1 that reflected its vaterite nature.61 The Au22SG18@CaCO3-Cube showed the absorption bands at 874 cm−1 and 708 cm−1 representing a calcite phase that reflected its crystalline nature.62 The Au22SG18@CaCO3-Cube/Sphere showed the absorption band at 744 cm−1 representing the vaterite phase and bending bands at 873 cm−1 and 710 cm−1 signals its crystalline polymorph calcite.61,62 Raman spectroscopy also reflected similar inorganic assemblies (Fig. 2C). Raman Peak was observed at 1087 cm−1 for Au22SG18@CaCO3-Cube which was aligned with CaCO3-Cube-control. Raman peaks at 281 cm−1, 712 cm−1 and 1088 cm−1 were observed in the CaCO3-Cube-control, resembling those of calcite.63 The spectroscopic signal at 1087 cm−1 cemented out that Au22SG18@CaCO3-cube had a crystalline environment with calcite polymorphism. Again, in the Au22SG18@CaCO3-Sphere, peaks were observed at 300 cm−1, 748 cm−1 and 1089 cm−1 with a shoulder peak at 1078 cm−1, which was similar to CaCO3-Sphere-control. The shoulder peak originated at 1078 cm−1 strongly matched with the spherical vaterite.63–65 Similarly, peaks in Au22SG18@CaCO3-Cube/Sphere resembled those of CaCO3-Cube/Sphere which was previously confirmed that the synthesized material was a mixture of both calcite and vaterite polymorphs embedded with AuNCs.63–65
Following the successful integration of NCs into various polymorphic structures of CaCO3, all materials were subjected to PL analysis. In the solid-state PL spectra, the NCs within the CaCO3 matrix exhibited a blue shift, which was attributed to their structural compactness. Notably, this blue shift was more pronounced as the structure transitioned from cubic (calcite) to spherical (vaterite/ACC) via intermediate forms (calcite/vaterite/ACC). This indicates a clear relationship between the PL properties and the crystallinity or stability of the inorganic matrix. As the crystallinity and stability decreased from cubic to spherical structures, the PL maxima shifted towards lower wavelengths, resulting in a blue shift (Fig. 3A). NCs embedded in spherical solids can be considered isotropic, as they exhibit identical physical properties in all directions due to the random arrangement of particles. However, their crystallinity is expected to be lower than that of the calcite matrix. The PL spectra revealed a maximum blue shift for solid-state Au22SG18 (around 46 nm), likely due to the increased compactness and rigidity in this form. In the solid state, the free molecular movement observed in the liquid state was restricted, as the molecules came closer together, reducing the available free space. The largest blue shift was observed for Au22SG18@CaCO3-Sphere (∼33 nm), while smaller shifts were seen for Au22SG18@CaCO3-Cube (∼18 nm) and Au22SG18@CaCO3-Cube/Sphere intermediate structures (∼28 nm). This difference in blue shift may also be related to the synthesis conditions. Au22SG18@CaCO3-Sphere was synthesized in an ethanolic medium, leading to enhanced aggregation of the AuNCs from the start. Ethanol, being an antisolvent, promoted aggregation by increasing the hydrophobicity of the medium, resulting in a larger blue shift. For Au22SG18@CaCO3-Cube/Sphere, ethanol was introduced after 15 seconds to suspend the reaction, which similarly contributed to the blue shift in the PL emission maxima. In contrast, Au22SG18@CaCO3-Cube was prepared in an ethylene glycol-water medium, which resulted in a smaller blue shift. Notably, using core–shell type glutathione protected AuNCs instead of Au22SG18, resulted in a similar blue shift across all three polymorph structures, suggesting the broad applicability of this method (Experimental section and Fig. S8†).
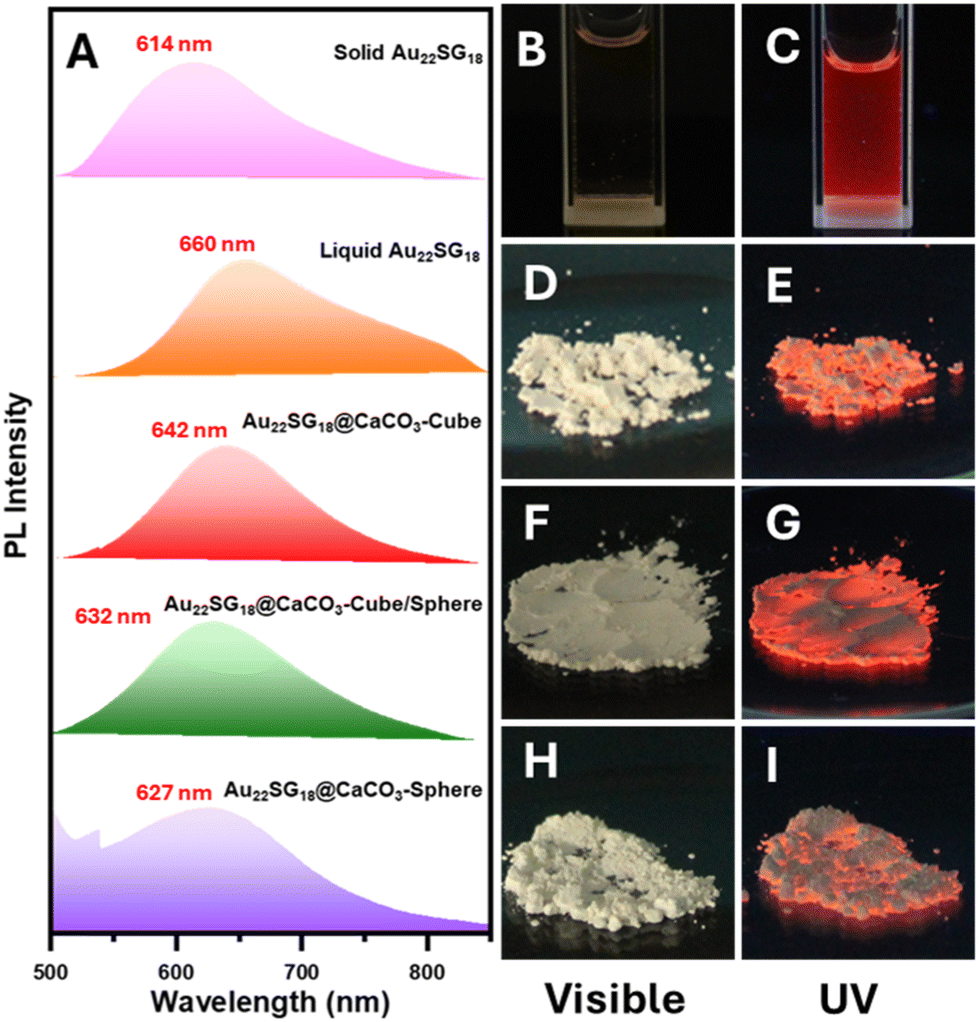 | ||
| Fig. 3 (A) PL intensity of solid Au22SG18, liquid Au22SG18, Au22SG18@CaCO3-Cube, Au22SG18@CaCO3-Cube/Sphere, Au22SG18@CaCO3-Sphere and. Digital images of (B and C) liquid Au22SG18, (D and E) Au22SG18@CaCO3-Cube, (F and G) Au22SG18@CaCO3-Cube/Sphere, (H and I) Au22SG18@CaCO3-Sphere taken under visible and UV (λex = 520 nm). | ||
Images of all cluster-loaded polymorphs were taken under visible and UV light. In the solid state, all powdered samples appeared slightly yellowish (Fig. 3D, F and H), similar to the solution color of Au22SG18 NCs (Fig. 3B), indicating successful encapsulation within the white CaCO3 matrices. Under UV light, the images show that the characteristic reddish emission of the Au22SG18 NCs (Fig. 3C) was retained across all cluster-encapsulated polymorphs (Fig. 3E, G, & I). To confirm the successful incorporation of Au22SG18 NCs into individual cubic or spherical structures, single-particle confocal microscopy was performed. The cubic and spherical morphologies of the Au22SG18@CaCO3-Cube and Au22SG18@CaCO3-Sphere were clearly visible in Fig. S9 and S10.† Further analysis using single-particle Raman spectroscopy provided insight into the surrounding environment of the Au22SG18 NCs (Fig. 4A–D). These data confirmed the polymorphic nature of the CaCO3 matrices around the Au22SG18 NCs in the Au22SG18@CaCO3-Cube, and Au22SG18@CaCO3-Sphere. In Au22SG18@CaCO3-Cube, the Raman spectra showed characteristic calcite peaks at 156 cm−1, 275 cm−1, 710 cm−1 and 1084 cm−1. For Au22SG18@CaCO3-Sphere, Raman peaks at 102 cm−1 and 301 cm−1 were indicative of vaterite.64,66 These results suggest that the Au22SG18 NCs exist within distinct polymorphic environments.
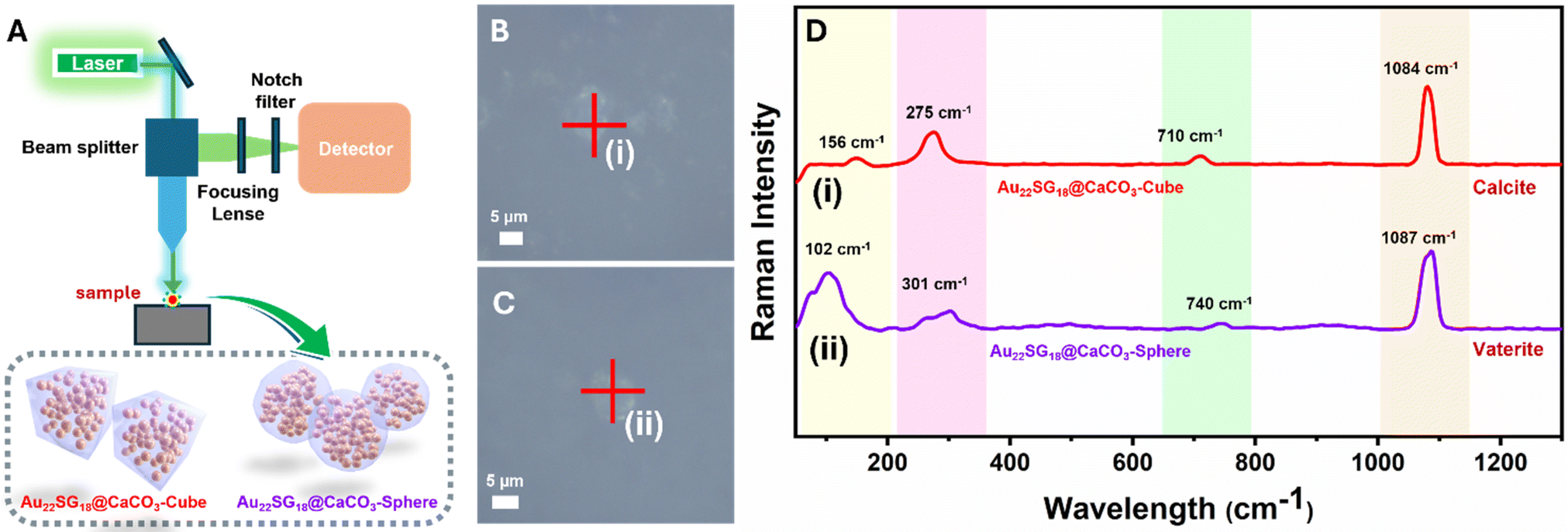 | ||
| Fig. 4 (A) Pictorial diagram of single particle Raman instrumentation. Confocal images of (B) Au22SG18@CaCO3-Cube, (C) Au22SG18@CaCO3-Sphere, (D) single particle Raman spectrum of as-synthesized samples (i) Au22SG18@CaCO3-Cube and (ii) Au22SG18@CaCO3-Sphere. | ||
After analyzing the steady-state PL performance, we performed Time-Correlated-Single-Photon-Counting (TCSPC) experiment in the solid state as shown in Fig. 5. The average lifetimes of the samples were first measured in microsecond windows (Fig. 5A). The decay profiles as well as the average lifetime values followed the trend as Au22SG18@CaCO3-Cube > Au22SG18@CaCO3-Cube/Sphere > Au22SG18@CaCO3-Sphere (Table S1†). Interestingly, Au22SG18 exhibited the shortest average microsecond lifetime among all variants, suggesting a beneficial role of polymorph structures in reducing non-radiative channels. The trend observed in the nanosecond decay mirrored that of the microsecond results. In the nanosecond decay analysis, Au22SG18@CaCO3-Cube exhibited the longest lifetime of 76.71 ns, Au22SG18@CaCO3-Cube/Sphere had a lifetime of 45.75 ns, and Au22SG18@CaCO3-Sphere showed the shortest lifetime of 10.12 ns (Fig. 5B). We hypothesize that as the crystallinity of the matrices increases, the energy of the excited state decreases, leading to longer lifetimes and slower decay rates. As the structure shifts from cubic to spherical, the shorter decay component increases, resulting in faster decay and shorter lifetimes for Au22SG18@CaCO3-Sphere. Conversely, as crystallinity increases in the cubic phase, decay slows down, yielding longer lifetimes (Fig. 5C, Table S2†). This enhancement is likely due to reduced non-radiative decay in more crystalline structures, explaining why the cubic polymorph exhibits the highest excited-state lifetime.
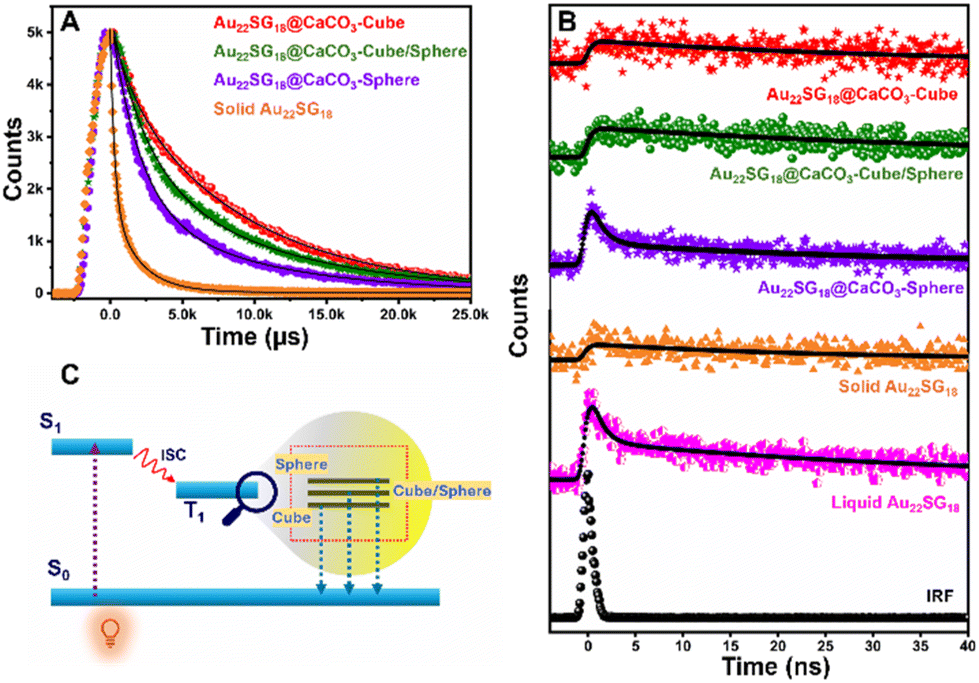 | ||
| Fig. 5 (A) TCSPC lifetime (microsecond) of solid Au22SG18, liquid Au22SG18, Au22SG18@CaCO3-Cube, Au22SG18@CaCO3-Cube/Sphere, Au22SG18@CaCO3-Sphere. (B) TCSPC lifetime (nanosecond) of solid Au22SG18, liquid Au22SG18, Au22SG18@CaCO3-Cube, Au22SG18@CaCO3-Cube/Sphere, Au22SG18@CaCO3-Sphere. (C) Jablonski diagram depicting the different energy levels of Au22SG18@CaCO3-Cube, Au22SG18@CaCO3-Cube/Sphere, Au22SG18@CaCO3-Sphere (λex = 375 nm and λem = 660 nm). | ||
Maintaining the structure and composition of the NCs after integration with various CaCO3 polymorphs is crucial. To confirm this, we conducted a pH-dependent study. Since CaCO3 is known to degrade in acidic environments, lowering the pH of the solution should release the NCs from their respective matrices, allowing us to verify their structural integrity through optical characterization.67 Initially, at pH 9, the dispersion of the composites in ultrapure water (1 mg mL−1) resulted in a cloudy solution, which cleared upon reducing the pH to 5 by adding 1 M HCl. This occurs because the calcium carbonate anions react with protons to form weak carbonic acid. When the pH was raised back to 9 using 1 M NaOH, the solution became cloudy again (Fig. S10†). At pH 5, the PL emission peak of Au22SG18@CaCO3-Cube diminished entirely, but when the pH was increased again, the emission peak reappeared but the intensity of the emission was not recovered completely which was near about half of the intensity peak at the initial stage with ∼5 nm red shift which was approximately 9-fold of PL emission peak at pH 5 (Fig. 6A). Similar results were observed for Au22SG18@CaCO3-Cube/Sphere. However, in this case the PL intensity was recovered up to 90% of the initial intensity at the same position i.e., 629 nm. PL intensity peak was observed at 629 nm for both cases and PL intensity recovered when the pH was raised back to 9 was approximately 6-fold of PL emission peak at pH (Fig. 6B). The trend was similar in the case of Au22SG18@CaCO3-Sphere, however, the metastable nature of these samples makes it difficult to analyse the PL data (Fig. 6C).
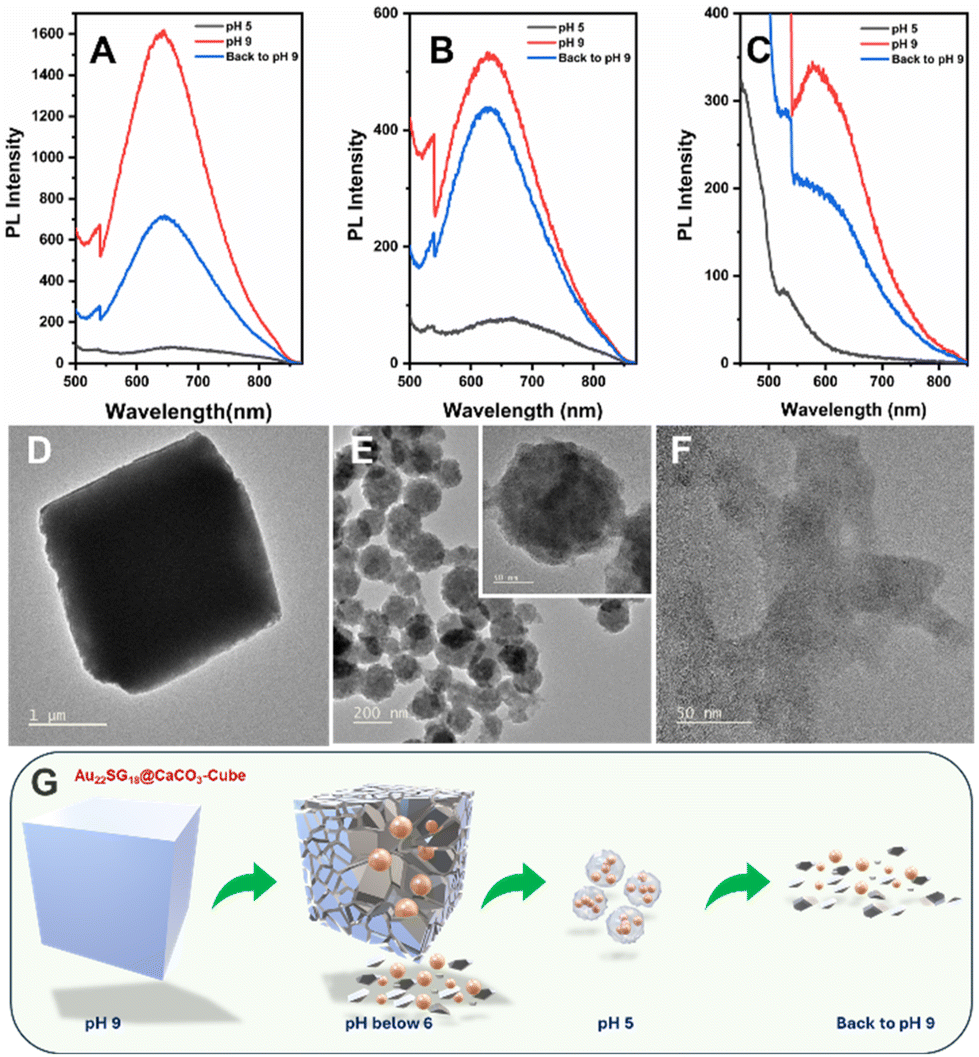 | ||
| Fig. 6 PL intensity of as-synthesized materials in liquid state at different pH environment (A) Au22SG18@CaCO3-Cube, (B) Au22SG18@CaCO3-Cube/Sphere, (C) Au22SG18@CaCO3-Sphere. HRTEM images of Au22SG18@CaCO3-Cube at (D) pH 9, (E) pH 5 and (F) back to pH 9. (G) Schematic showing the morphological change of Au22SG18@CaCO3-Cube at different pH. | ||
Since the concentration of the AuNCs remains constant before and after pH changes, comparing their PL intensity before and after dissociation allowed us to assess the impact of polymorph structure on the PL properties of the NCs. Specifically, embedding within a calcite matrix enhanced PL by up to 20-fold, while in the case of vaterite, PL enhancement was 7-fold. These findings suggest that the greater stability and crystallinity of the matrix provide a more rigid environment for the NCs, thereby improving their PL through AIE mechanism.
Since Au22SG18@CaCO3-Cube was the most stable polymorph among the samples studied, we analyzed its morphology using TEM. At pH 5, the cube's morphology was observed to change into a spherical shape (Fig. 6D and E), with the sphere consisting of smaller-sized NCs (inset of Fig. 6E). This transformation is attributed to the coordination between the carboxylate groups of the GSH ligands and the free Ca2+ ions in solution. Similar spherical aggregates, coordinated by metal ions, have been reported previously for other MNCs.68 Interestingly, when the solution pH was reverted to 9, the cubic morphology did not reappear; instead, aggregated NCs were observed (Fig. 6F), indicating the irreversible nature of the morphological change. A proposed mechanism for this pH-dependent morphology transformation is shown in Fig. 6G.
Experimental
Materials and methods
All the chemicals were purchased commercially and used as received. Hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O), reduced L-glutathione (GSH), ethylene glycol (C2H6O2), calcium chloride (CaCl2), sodium carbonate (Na2CO3), sodium borohydride (NaBH4), ethanol (EtOH) and hydrochloric acid were purchased from Sigma-Aldrich. Sodium hydroxide (NaOH) was purchased from Qualigens. Ammonium bicarbonate (NH4HCO3) was purchased from Thermo Fisher Scientific. All the glassware used was washed with aqua-regia and rinsed with deionized water. Ultra-pure Milli-Q water was used as a solvent throughout the experiments unless mentioned.Characterisation
UV–vis absorption spectra were recorded by JASCO V-750 spectrophotometer. PL spectra of both liquid and solid samples were measured with a JASCO FP-8350 spectrofluorometer. Time-correlated single photon counting (TCSPC) lifetime was measured in an Edinburgh FLS 980 Instrument. Transmission electron microscopy (TEM) images were captured with a JEOL JEM-200 at 200 kV whereas FESEM (Field emission scanning electron microscopy) images were captured in a ZEISS SUPRA 55 Instrument for studying the surface morphology and microstructures of the synthesized samples. For SEM sample preparation, synthesized materials were well dispersed in Milli-Q water and drop-casted on glass slides. Similarly, TEM samples were prepared by drop casting the well-dispersed sample (in Milli-Q water) on copper grids (200 mesh) followed by drying in vacuum. The infrared spectra (FT-IR) of solid samples were recorded by a Thermo Scientific Nicolet iS20 Smart iTX-Diamond ATR-IR spectrometer. Raman spectrum measurements were recorded using a Renishaw microscopic confocal Raman spectrometer at room temperature using a 532 nm diode laser to explore the structural information and chemical environment of the synthesized samples. The confocal images were obtained using an inverted confocal microscope, Leica (Leica TCS SP8). The samples were excited using a diode laser (490 nm) through a water objective (63 × 1.4 NA). After cleansing the glass slide with double-distilled water to remove dirt particles, a 10–15 μL sample solution aliquot was dropped onto it and covered with a clean coverslip. Coverslips were repeatedly washed in double-distilled water to ensure cleanliness. For single particle Raman spectra measurements, the samples were excited with a 532 nm diode laser through a 50× objective, using a WItec GmbH (alpha 300) instrument. Rigaku RINT 2500X diffractometer with monochromate Cu Kα radiation (40 kV, 40 mA) at a scan rate of 0.1° min−1 has been utilized to analyse the wide-angle powder X-ray diffraction (PXRD) pattern for the as-synthesized materials for structural information. The pH of the solutions was measured with an Oakton pH 700 pH meter. Inductively coupled plasma-optical emission spectrometer (ICP-OES) experiments were performed by PerkinElmer Optima 2100 DV.Synthesis of Au22SG18 NCs
The was carried out following a reported procedure with slight modifications.54–56 Briefly, a freshly prepared aqueous solution of HAuCl4 (1.5 mL, 20 mM) was added to 27.6 mL of ultra-pure water under stirring, followed by the addition of GSH (0.9 mL, 50 mM). The solution turned from yellow to a turbid white upon the addition of GSH. After 3–4 minutes, the pH was adjusted to 11, causing the turbidity to disappear, and the mixture was stirred at 600 rpm for 20 minutes. NaBH4 (75 μL, 10 mM) was then added dropwise, and the solution was stirred gently at 200 rpm for 30 minutes. Subsequently, 1 M HCl was added to lower the pH to 2.5, and the mixture was stirred for 24 hours at 200 rpm. After synthesis, the Au22SG18 NCs were purified using a Pur-A-Lyzer (Mega 3500 dialysis kit).Synthesis of Au22SG18@CaCO3-Cube & CaCO3-Cube-control
We had followed a reported protocol with some required alteration.69 Au22SG18@CaCO3-Cube & CaCO3-Cube-control 4 mL of CaCl2 (0.33M, in EG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 5:1) was mixed with 28 mL Milli-Q water in a conical flask under stirring at 600 rpm. After 5 minutes, 4 mL of freshly prepared Au22SG18 NCs solution was added to the flask. Following 20 minutes of stirring, 4 mL of Na2CO3 (0.33M, in EG:H2O = 5:1) was introduced into the mixture causing the solution to turn from clear to cloudy. The mixture was then stirred at 600 rpm for 1 hour. The synthesized product was collected by centrifugation at 10000 rpm for 5 minutes, followed by washing the precipitate twice with Milli-Q water. The washed precipitate was freeze-dried to obtain a solid powder, referred to as Au22SG18@CaCO3-Cube. CaCO3-Cube-control without NCs was also obtained following a similar procedure.
:H2O = 5:1) was mixed with 28 mL Milli-Q water in a conical flask under stirring at 600 rpm. After 5 minutes, 4 mL of freshly prepared Au22SG18 NCs solution was added to the flask. Following 20 minutes of stirring, 4 mL of Na2CO3 (0.33M, in EG:H2O = 5:1) was introduced into the mixture causing the solution to turn from clear to cloudy. The mixture was then stirred at 600 rpm for 1 hour. The synthesized product was collected by centrifugation at 10000 rpm for 5 minutes, followed by washing the precipitate twice with Milli-Q water. The washed precipitate was freeze-dried to obtain a solid powder, referred to as Au22SG18@CaCO3-Cube. CaCO3-Cube-control without NCs was also obtained following a similar procedure.
Synthesis of Au22SG18@CaCO3-Cube/Sphere & CaCO3-Cube/Sphere-control
With some minimal adaptation, Au22SG18@CaCO3-Cube/Sphere & CaCO3-Cube/Sphere-control was synthesized.70 30 mL of CaCl2 (0.1 M in H2O) was placed in a conical flask and stirred at 600 rpm. Freshly prepared 6 mL of Au22SG18 cluster solution was then added. After 5 minutes of stirring, 30 mL of Na2CO3 (0.1 M in H2O) was poured into the reaction mixture under vigorous stirring for 15 seconds. The resulting cloudy suspension was mixed with 60 mL (1:1) of ethanol and centrifuged at 5000 rpm. The precipitate was washed twice with ethanol, and centrifuged again at 5000 rpm, and then dried in a vacuum oven at room temperature overnight to obtain a solid powder, termed as Au22SG18@CaCO3-Cube/Sphere. A control CaCO3-Cube/Sphere was synthesised using a similar synthetic procedure without the addition of NCs.
Synthesis of Au22SG18@CaCO3-Sphere & CaCO3-Sphere-control
Synthesis of spherical CaCO3 assembly was based on a reported procedure for ACC with minor modifications.71 Briefly, 200 mg of CaCl2 was dissolved in 100 mL of absolute ethanol in a beaker with 500 μL Au22SG18 (10 mL of as-synthesized Au22SG18 NCs was concentrated to 1 mL). The beaker was covered with parafilm punctured with several holes and placed in a desiccator. A Petri dish containing crushed NH4HCO3 was placed in the desiccator. After 48 hours, the product was collected by centrifugation at 8000 rpm for 5 minutes. The precipitate was washed three times with ethanol and dried in a vacuum oven at room temperature overnight to yield a solid powder, termed as Au22SG18@CaCO3-sphere. CaCO3-spheres without NCs were synthesized with a similar synthetic procedure.pH-Dependent stability test
All the composites, Au22SG18@CaCO3-Cube, Au22SG18@CaCO3-Cube/Sphere, and Au22SG18@CaCO3-Sphere were dispersed in Milli-Q water (1 mg mL−1) for pH-dependent stability test. Initially, the pH of all the dispersions was around 9 and the solution was cloudy. After reducing the pH to 5 (by adding 1 M HCl) the solution became clear. Furthermore, when the pH was reversed to 9 the cloudiness reappeared. PL intensity of the NCs in solution was monitored at different pH values.Synthesis of core–shell type gold nanoclusters (AuNCs) and AuNCs@CaCO3-composites
We followed a well-established protocol to synthesize core–shell type AuNCs.72 To prepare a 50 mL stock solution containing 2 mM AuNCs, we began by adding 43.5 mL of Milli-Q water to a round bottom flask. Then, 5 mL of freshly prepared 20 mM HAuCl4 solution was added under stirring at 75 °C. After 3 minutes, 1.5 mL of 100 mM GSH was introduced, which turned the initial yellow solution colourless. The mixture was incubated under stirring for 24 h, during which the yellow colour gradually reappeared. The resulting orange emitting AuNCs were stored at 4 °C for future use. We used similar synthetic protocol for AuNCs@CaCO3-Cube, AuNCs@CaCO3-Cube/Sphere and AuNCs@CaCO3-Sphere, where we used AuNCs instead of Au22SG18.Conclusions
In summary, by mimicking biomineralization, we have successfully integrated atomically precise atomically precise AIE-type Au22SG18 NCs into various polymorphic forms of CaCO3. The red-emitting Au22SG18 NCs exhibited distinct PL characteristics: the cubic crystalline calcite matrix displayed the highest PL intensity and excited-state lifetime, while the metastable vaterite matrix induced a pronounced blue shift due to self-aggregation within its spherical structure. Detailed spectroscopic and microscopic analyses confirmed the successful encapsulation and preservation of the PL properties. Single-molecule Raman spectroscopy validated the presence of the NCs within individual nanocubes or nanospheres, and confocal imaging combined with STEM studies revealed the uniform distribution of luminescent NCs within the calcite nanocubes, vaterite nanospheres and in their mixed form. Furthermore, the clusters could be released from the CaCO3 matrices by adjusting the solution pH. With pH-dependent release properties, coupled with the biocompatibility and biodegradability of calcium carbonate-based nanomaterials, our developed cluster-embedded polymorphs could have strong potential as safe and effective drug carriers for biomedical applications. Overall, this work introduces a novel inorganic matrix for studying the behavior of AIE-type atomically precise NCs, potentially paving the way for further exploration of luminescent properties in other MNCs and beyond.Author contributions
S. M. conducted most of the experiments, with assistance from S. R. S. and A. M. in data analysis. K. K. and S. R. performed the Confocal microscopy and single particle Raman experiments. N. G. supervised the entire project. S. M., S. R. S. and N. G. wrote the manuscript, with input from all the authors. S. M. prepared the schemes and figures. All authors have reviewed and approved the final version of the manuscript.Data availability
All relevant data are presented in the main text and ESI† (UV-vis and PL spectra; FESEM images; HRTEM images; lifetime values; confocal microscopic images).All relevant data are within the manuscript and its additional files.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. M., S. R. S., and A. M., and would like to thank the University Grants Commission of India (UGC) for providing a fellowship. K. K. thanks ICMR for offering the Senior Research Fellowship (Lr. No. BMI/11(98)/2020). S. R. wishes to acknowledge BHU Varanasi for providing the seed grant and bridge grant (Dev. Scheme No. 6031) under the IoE scheme. The authors also acknowledge the Department of Chemistry, Central Discovery Centre, BHU, for their in-house support. N. G. acknowledges CSIR, New Delhi for financial support (Grant Number: HCP-30) and thanks CSIR-Institute of Minerals and Materials Technology (IMMT), Bhubaneswar for in-house financial support (Grant Number: OLP-110). We thank Dr Y. S. Chaudhary for access to the TCSPC facility and acknowledge the central instrumental facilities led by the central characterization department at CSIR-IMMT.References
- X. Du and R. Jin, ACS Nano, 2019, 13, 7383–7387 Search PubMed.
- Y. Xiao, Z. Wu, Q. Yao and J. Xie, Aggregate, 2021, 2, 114–132 CAS.
- J. Lin, Q. Zhang, L. Wang, X. Liu, W. Yan, T. Wu, X. Bu and P. Feng, J. Am. Chem. Soc., 2014, 136, 4769–4779 CrossRef CAS.
- T. T. Jia, G. Yang, S. J. Mo, Z. Y. Wang, B. J. Li, W. Ma, Y. X. Guo, X. Chen, X. Zhao, J. Q. Liu and S. Q. Zang, ACS Nano, 2019, 13, 8320–8328 CrossRef CAS PubMed.
- M. Zhou, X. Du, H. Wang and R. Jin, ACS Nano, 2021, 15, 13980–13992 CrossRef CAS PubMed.
- Y. Zhou, L. Liao, S. Zhuang, Y. Zhao, Z. Gan, W. Gu, J. Li, H. Deng, N. Xia and Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 8668–8672 CrossRef CAS PubMed.
- G. Deng, S. Malola, J. Yan, Y. Han, P. Yuan, C. Zhao, X. Yuan, S. Lin, Z. Tang, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2018, 57, 3421–3425 Search PubMed.
- Z. Wang, B. Chen, A. S. Susha, W. Wang, C. J. Reckmeier, R. Chen, H. Zhong and A. L. Rogach, Adv. Sci., 2016, 3, 1600182 Search PubMed.
- S. Gratious, E. N. Nahan, R. Jin and S. Mandal, Acc. Mater. Res., 2024, 10, 1291–1302 CrossRef.
- N. Alam, A. K. Das, P. Chandrashekar, P. Baidya and S. Mandal, Nanoscale, 2024, 16, 10087–10107 Search PubMed.
- K. Sahoo, T. R. Gazi, S. Roy and I. Chakraborty, Commun. Chem., 2023, 6, 157 Search PubMed.
- A. Mukhopadhyay, S. R. Sahoo, S. Mahata and N. Goswami, Anal. Bioanal. Chem., 2024, 416, 3963–3974 Search PubMed.
- G. Yang, Z. Wang, F. Du, F. Jiang, X. Yuan and J. Y. Ying, J. Am. Chem. Soc., 2023, 145, 11879–11898 Search PubMed.
- T. D. Fernández, J. R. Pearson, M. P. Leal, M. J. Torres, M. Blanca, C. Mayorga and X. Le Guével, Biomaterials, 2015, 43, 1–12 Search PubMed.
- E. Porret, X. Le Guével and J. L. Coll, J. Mater. Chem. B, 2020, 8, 2216–2232 Search PubMed.
- X. Pan, Y. Yao, M. Zhang, X. Yuan, Q. Yao and W. Hu, Nanoscale, 2024, 16, 8196–8215 Search PubMed.
- A. Yahia-Ammar, D. Sierra, F. Mérola, N. Hildebrandt and X. Le Guével, ACS Nano, 2016, 10, 2591–2599 Search PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- Q. Zhang, Y. X. Deng, H. X. Luo, C. Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D. H. Qu, J. Am. Chem. Soc., 2019, 141, 12804–12814 CrossRef CAS PubMed.
- K. Tanaka, G. H. Clever, Y. Takezawa, Y. Yamada, C. Kaul, M. Shionoya and T. Carell, Nat. Nanotechnol., 2006, 1, 190–194 CrossRef CAS.
- D. Bera, A. Mukhopadhyay, Nonappa and N. Goswami, J. Phys. Chem. Lett., 2023, 14, 7299–7305 CrossRef CAS PubMed.
- D. Bera, M. Baruah, A. K. Dehury, A. Samanta, Y. S. Chaudhary and N. Goswami, J. Phys. Chem. Lett., 2022, 13, 9411–9421 CrossRef CAS PubMed.
- X. Dou, S. Saalah, C. K. Chiam, J. Xie and C. S. Sipaut, Nanoscale, 2024, 16, 20089–20099 RSC.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- S. Pan, W. Liu, J. Tang, Y. Yang, H. Feng, Z. Qian and J. Zhou, J. Mater. Chem. B, 2018, 6, 3927–3933 RSC.
- H. H. Deng, X. Q. Shi, F. F. Wang, H. P. Peng, A. L. Liu, X. H. Xia and W. Chen, Chem. Mater., 2017, 29, 1362–1369 CrossRef CAS.
- T. Chen, S. Yang, J. Chai, Y. Song, J. Fan, B. Rao, H. Sheng, H. Yu and M. Zhu, Sci. Adv., 2017, 3, e1700956 CrossRef PubMed.
- H. Ao, H. Feng, M. Zhao, M. Zhao, J. Chen and Z. Qian, ACS Sens., 2017, 2, 1692–1699 CrossRef CAS PubMed.
- D. Bera and N. Goswami, J. Phys. Chem. Lett., 2021, 12, 9033–9046 CrossRef CAS PubMed.
- X. Su and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 3902–3910 CAS.
- Z. Wu, J. Liu, Y. Gao, H. Liu, T. Li, H. Zou, Z. Wang, K. Zhang, Y. Wang, H. Zhang and B. Yang, J. Am. Chem. Soc., 2015, 137, 12906–12913 CAS.
- N. Goswami, F. Lin, Y. Liu, D. T. Leong and J. Xie, Chem. Mater., 2016, 28, 4009–4016 CAS.
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- A. Mukhopadhyay, S. Mahata and N. Goswami, J. Phys. Chem. Lett., 2024, 15, 8510–8519 CAS.
- Q. Li, X. Zhou, L. L. Tan and L. Shang, Sens. Actuators, B, 2023, 385, 133695 CAS.
- D. Wei, M. Li, Y. Wang, N. Zhu, X. Hu, B. Zhao, Z. Zhang and D. Yin, J. Hazard. Mater., 2023, 441, 129890 CrossRef CAS PubMed.
- Y. Cai, H. Zhu, W. Zhou, Z. Qiu, C. Chen, A. Qileng, K. Li and Y. Liu, Anal. Chem., 2021, 93, 7275–7282 Search PubMed.
- R. Chang, D. Choi, M. H. Kim and Y. Park, ACS Sustain. Chem. Eng., 2017, 5, 1659–1667 CrossRef CAS.
- M. L. P. Vidallon, F. Yu and B. M. Teo, Cryst. Growth Des., 2020, 20, 645–652 CrossRef CAS.
- G. Falini, S. Fermani, M. Gazzano and A. Ripamonti, Dalton Trans., 2000, 3983–3987 RSC.
- A. Katsman, I. Polishchuk and B. Pokroy, Faraday Discuss., 2022, 235, 433–445 RSC.
- Y. Sonobe, H. Watamura and I. Hirasawa, Chem. Eng. Technol., 2015, 38, 1053–1058 CrossRef CAS.
- F. A. Davila-Hernandez, B. Jin, H. Pyles, S. Zhang, Z. Wang, T. F. Huddy, A. K. Bera, A. Kang, C. L. Chen, J. J. De Yoreo and D. Baker, Nat. Commun., 2023, 14, 8191 CrossRef.
- G. Falini, S. Albeck, S. Weiner and L. Addadi, Science, 1996, 271, 67–69 CrossRef.
- S. Mann, Nature, 1993, 365, 499–505 CrossRef CAS.
- G. X. Gu, C. T. Chen, D. J. Richmond and M. J. Buehler, Mater. Horiz., 2018, 5, 939–945 RSC.
- Y. Jiang, X. Chen, J. Yang, L. Y. Chang, T. S. Chan, H. Liu, X. Zhu, J. Su, H. Zhang, Y. Fan and L. Liu, Phys. Chem. Chem. Phys., 2022, 24, 29034–29042 RSC.
- Q. Yao, Z. Luo, X. Yuan, Y. Yu, C. Zhang, J. Xie and J. Y. Lee, Sci. Rep., 2014, 4, 3848 CrossRef PubMed.
- F. Nudelman and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582–6596 CAS.
- H. Lu, Y. C. Huang, J. Hunger, D. Gebauer, H. Cölfen and M. Bonn, J. Am. Chem. Soc., 2021, 143, 1758–1762 CrossRef CAS PubMed.
- Q. Wu, T. Zhang, Q. Peng, D. Wang and Z. Shuai, Phys. Chem. Chem. Phys., 2014, 16, 5545–5552 CAS.
- J. W. Morse, R. S. Arvidson and A. Lüttge, Chem. Rev., 2007, 107, 342–381 CrossRef CAS.
- Y. G. Srinivasulu, N. Goswami, Q. Yao and J. Xie, J. Phys. Chem. C, 2021, 125, 4066–4076 CrossRef CAS.
- H. Deng, K. Huang, L. Xiu, W. Sun, Q. Yao, X. Fang, X. Huang, H. A. A. Noreldeen, H. Peng, J. Xie and W. Chen, Nat. Commun., 2022, 13, 3381 CrossRef CAS.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- R. Lakshminarayanan and S. Valiyaveettil, Cryst. Growth Des., 2003, 3, 611–614 CrossRef CAS.
- A. F. Suzana, S. S. Lee, I. Calvo-Almazán, W. Cha, R. Harder and P. Fenter, Adv. Mater., 2024, 36, 2310672 CAS.
- W. Li and C. Gao, Langmuir, 2007, 23, 4575–4582 CAS.
- L. Liu, X. Zhang, X. Liu, J. Liu, G. Lu, D. L. Kaplan, H. Zhu and Q. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 1735–1745 CAS.
- M. Sato and S. Matsuda, Z. Kristallogr. – Cryst. Mater., 1969, 129, 405–410 CAS.
- P. K. Ajikumar, L. G. Wong, G. Subramanyam, R. Lakshminarayanan and S. Valiyaveettil, Cryst. Growth Des., 2005, 5, 1129–1134 CrossRef CAS.
- F. C. Donnelly, F. Purcell-Milton, V. Framont, O. Cleary, P. W. Dunne and Y. K. Gun’ko, Chem. Commun., 2017, 53, 6657–6660 RSC.
- M. De La Pierre, R. Demichelis, U. Wehrmeister, D. E. Jacob, P. Raiteri, J. D. Gale and R. Orlando, J. Phys. Chem. C, 2014, 118, 27493–27501 CrossRef CAS.
- R. Liu, F. Liu, Y. Su, D. Wang and Q. Shen, Langmuir, 2015, 31, 2502–2510 CrossRef.
- F. C. Donnelly, F. Purcell-Milton, V. Framont, O. Cleary, P. W. Dunne and Y. K. Gun'ko, Chem. Commun., 2017, 53, 6657–6660 RSC.
- Y. B. Hu, M. Wolthers, D. A. Wolf-Gladrow and G. Nehrke, Cryst. Growth Des., 2015, 15, 1596–1601 CrossRef CAS.
- Z. Liu, X. Jing, S. Zhang and Y. Tian, Anal. Chem., 2019, 91, 2488–2497 CrossRef CAS.
- H. Bahrom, A. A. Goncharenko, L. I. Fatkhutdinova, O. O. Peltek, A. R. Muslimov, O. Y. Koval, I. E. Eliseev, A. Manchev, D. Gorin, I. I. Shishkin, R. E. Noskov, A. S. Timin, P. Ginzburg and M. V. Zyuzin, ACS Sustain. Chem. Eng., 2019, 7, 19142–19156 CrossRef CAS.
- L. Dong, Y. J. Xu, C. Sui, Y. Zhao, L. B. Mao, D. Gebauer, R. Rosenberg, J. Avaro, Y. D. Wu, H. L. Gao, Z. Pan, H. Q. Wen, X. Yan, F. Li, Y. Lu, H. Cölfen and S. H. Yu, Nat. Commun., 2022, 13, 5088 CrossRef CAS PubMed.
- S. F. Chen, H. Cölfen, M. Antonietti and S. H. Yu, Chem. Commun., 2013, 49, 9564–9566 RSC.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04022h |
| This journal is © The Royal Society of Chemistry 2025 |