
Effects of crosslink density and plasticizer on thermorheological properties of dissociative guanidine-based covalent adaptable networks†
Adelle L.
Koenig
a,
Kelsey M.
Allis
b,
John S.
Lehr
b and
Michael B.
Larsen
 *a
*a
aDepartment of Chemistry, Western Washington University, Bellingham, WA 98225, USA. E-mail: mike.larsen@wwu.edu
bDepartment of Engineering and Design, Western Washington University, Bellingham, WA 98225, USA
First published on 26th November 2024
Abstract
We report the effects of varying crosslink density and plasticizer loading on the thermorheological properties of guanidine-based covalent adaptable networks (CANs). CANs engage in dynamic bond-exchange reactions above Tg, resulting in shifts between thermoset-like materials and states capable of flow that can be greatly impacted by network characteristics beyond the exchange reaction itself. The synthesis of guanidine-based CANs by combination of carbodiimide-containing oligomers and various ratios of amine-containing crosslinker molecules and phthalate plasticizer was used to create a library of CANs with varying crosslink density and equal concentrations of guanidine functionalities. Additionally, Tg was tuned by modifying plasticizer loading. CANs at three degrees of crosslink density and three degrees of plasticizer loading were characterized by rheometry and dynamic mechanical analysis. The resulting data indicated that absolute relaxation times varied directly with crosslink density but were largely unaffected by plasticizer content or temperature relative to Tg; thus, plasticizer served to decouple Tg from relaxation dynamics. Moreover, little difference in activation energies was observed between each system, contrasting studies of associative CANs.
Introduction
Covalent adaptable networks (CANs) are a broad class of crosslinked polymers based on the application of dynamic covalent chemistry (DCC). These materials incorporate reversible covalent bonds that undergo dynamic bond-exchange reactions under specific circumstances.1,2 Intrinsic DCC in CANs results in thermoset-like rigidity when reversible bonds are dormant and thermoplastic-like viscoelastic flow upon substantial activation of the dynamic reaction.3 The effective bridge between thermosets and thermoplastics realized in CANs results in properties such as reprocessability, self-healing, and shape-memory, alongside numerous potential applications.4–6 Two broad categories based on the bond-exchange mechanism encompass the majority of CANs: associative CANs, also known as vitrimers, possess constant crosslink density (νe) throughout the duration of the bimolecular exchange reaction and concomitant relaxation or flow,7,8 while dissociative CANs exhibit changing νe upon activation of the dynamic reaction.9,10 Experimentally, the line between these categories can blur substantially, as dissociative CANs can display little variation in Keq of the associated and dissociated crosslink states with temperature. As a result, determining the relationship of νe to bond-exchange dynamics and the resulting rheological properties can be challenging. Among the currently known dissociative DCCs that display this behavior in CANs are triazolium and anilinium transalkylation,10,11 oxime transcarbamoylation,12 and others,13 while CANs based on Diels–Alder chemistry,5,14 disulfide exchange,15–17 thiol-Michael reactions,18 and hindered urea chemistry show measurable changes in crosslink Keq in the studied temperature windows.19–21In addition to the DCC responsible for crosslink exchange, it has been increasingly recognized that the structural and chemical characteristics of the surrounding polymer matrix have significant effects on CAN properties such as characteristic relaxation time (τ*) and flow activation energy (Ea). Prepolymer molar mass,22,23 branching at the point of crosslinking,24 the concentration of reactive species,25 crosslinker molar mass,26,27 polarity,28 and νe have all been found to exert influence over CAN dynamics. In particular, νe has emerged as an important governor of CAN behavior. Targeted approaches to study its effects include the manipulation of DCC-reactive species concentration in tandem with crosslink concentration29–31 and, conversely, decoupling the two by the incorporation of capping agents or similar strategies.32–34 Additionally, variation in νe due to synthetic modifications such as partial permanent crosslinking,35,36 heterogeneous crosslinking of block prepolymers,37 and independently activated bond-exchange reactions38 has been explored.
As dynamic systems, CANs possess intertwining structural, mechanical, and chemical constituents that present challenges in examining specific effects of the polymer matrix. As a result, studies designed to examine particular characteristics like νe have varying degrees of control of other related properties, which complicates the attribution of a definitive impact to a specific factor. For example, νe has largely been found to relate directly to Tg in both vitrimers and dissociative CANs, as in traditional thermosets.11,21,26,31,32,34 The relationship of νe to rheological properties presents a less cohesive image and may be impacted by the inherent differences in Tg and exchangeable functional group concentration among related systems. For example, νe in both vitrimers and dissociative CANs has been found to relate either directly or inversely to τ*,23,26,31,32,34,39,40 and dependence of Ea on νe can vary substantially.11,23,27,31,34 Thus, the significant effects of the polymer matrix on CAN systems, as well as its potential for synthetic tunability, make it a suitable pathway for altering CAN dynamics without necessitating modification of the bond-exchange reaction itself. However, much of the work performed thus far in this area examines vitrimers, and the effects of non-DCC components on thermorheological properties of dissociative CANs remain understudied. In particular, the effects of decoupling νe from functional group concentration have only been explored in a single system.32 Furthermore, few studies have directly examined the role of added plasticizers on CAN dynamics,41 despite their common use to tune thermorheological properties of conventional polymeric materials. We describe herein the synthesis of a library of dissociative guanidine-based CANs varying in νe and plasticizer loading while maintaining a constant guanidine concentration (Fig. 1). Chemical, thermal, and rheological properties including Tg, τ*, Ea, and νe were measured by dynamic mechanical analysis (DMA), stress relaxation, and small amplitude oscillatory shear (SAOS) frequency sweeps. We show that νe is directly related to τ* while Ea is relatively constant across network compositions. Further, our results indicate that plasticizer can be used to tune Tg while minimally impacting CAN rheology.
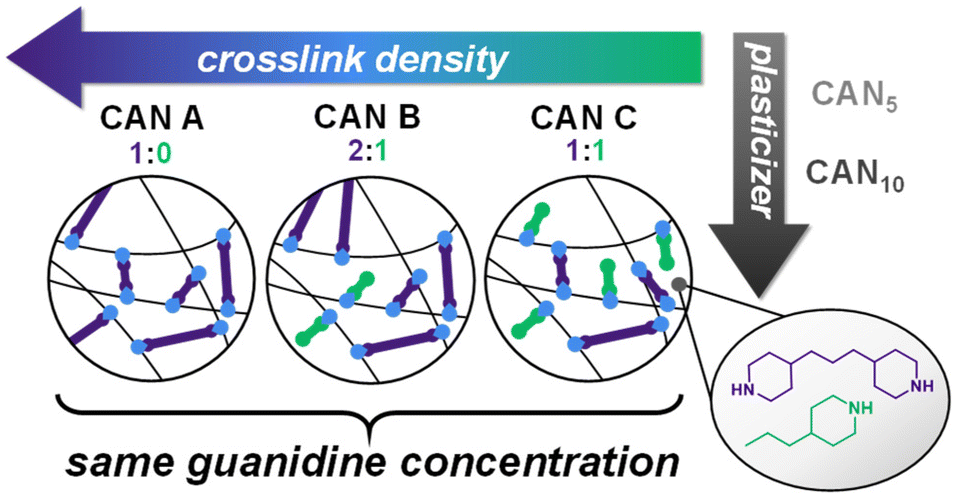 | ||
Fig. 1 Synthetic framework of CANs with varying νe and plasticizer loading. Difunctional amine crosslinker and monofunctional amine capping agent are shown in purple and green respectively; molar ratios of amine functionalities are given as difunctional![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :monofunctional species. Plasticizer mass loading (w/w) is given as a subscript, where CAN0 = 0% w/w plasticizer, CAN5 = 5% w/w plasticizer, and CAN10 = 10% w/w plasticizer. :monofunctional species. Plasticizer mass loading (w/w) is given as a subscript, where CAN0 = 0% w/w plasticizer, CAN5 = 5% w/w plasticizer, and CAN10 = 10% w/w plasticizer. | ||
Experimental section
Materials
Unless otherwise noted, all chemicals were purchased from commercial sources and used as received. Dry tetrahydrofuran (THF) was obtained from an Inert PureSolv solvent purification system.Instrumentation
1H nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker Avance III 500 MHz FT-NMR spectrometer. Chemical shifts are reported in delta (δ) units, expressed in parts per million (ppm) downfield from tetramethylsilane using the residual protio-solvent as an internal standard (CDCl3, 7.26 ppm). Infrared (IR) spectroscopy was performed on a Thermo iS10 FT-IR with single-bounce diamond ATR. Thermogravimetric analysis (TGA) was performed on a TA Instruments Q500 with a platinum pan. Temperature ramp experiments were heated at a rate of 10 °C min−1 from room temperature to 500 °C under nitrogen flow (40 mL min−1). Dynamic mechanical analysis (DMA) temperature ramp experiments were performed on a TA Instruments Q800 in tension mode. Rectangular samples (approximately 21 mm × 5 mm × 1 mm) were deformed by a 0.025% sinusoidal tensile strain at a frequency of 1 Hz while heating from ambient temperature to 190 °C at a rate of 3 °C min−1. Tg was taken as the peak of tanδ. Rheometry was performed on a TA Instruments DHR-2 Discovery Hybrid Rheometer with 25 mm parallel plate geometry. For strain sweep experiments, samples were held at 170 °C for 300 s, then deformed at 0.001%–5.0% strain at 20 points per decade logarithmically at a constant frequency of 1.0 Hz and a constant axial force of 5 N. For stress relaxation experiments, samples were held at temperature for 60 s, then deformed at 1.0% strain for 1200 s with a constant axial force of 5 N throughout a selected temperature range in 5 °C increments. Continuous relaxation spectra were calculated in TRIOS from the raw stress relaxation data at a given temperature. In spectra with multiple peaks in the continuous relaxation spectrum, the principal peak was determined as that with the largest integration value. For frequency sweep experiments, samples were held at temperature for 60 s, then deformed at an oscillation strain of 1.0% at frequencies of 0.001 Hz–100.0 Hz at 5 points per decade logarithmically with a constant axial force of 5 N throughout a selected temperature range in 5 °C increments. Relaxation times were calculated from the modulus crossover point (G′ = G′′) as 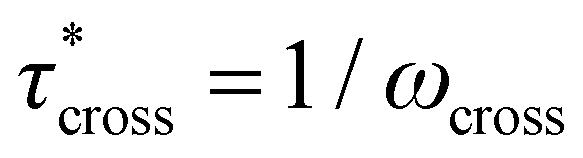 . For frequency sweep experiments, horizontal shift factors (aT) were determined by manually shifting tanδ versus ω curves while vertical shift factors (bT) were determined by manually shifting G* versus ω curves. For stress relaxation experiments, aT was determined by manually shifting relaxation curves and bT was obtained by shifting curves such that G(t) at 1 s was equal.
. For frequency sweep experiments, horizontal shift factors (aT) were determined by manually shifting tanδ versus ω curves while vertical shift factors (bT) were determined by manually shifting G* versus ω curves. For stress relaxation experiments, aT was determined by manually shifting relaxation curves and bT was obtained by shifting curves such that G(t) at 1 s was equal.
Synthesis and sample processing
:1 hexanes:methanol using a separatory funnel. The precipitate was recovered by vacuum filtration and dried under reduced pressure at 40 °C overnight to yield oligomer as a white solid (4.53 g, 46.2%) with an average degree of polymerization of 2.5 ± 0.40 by endgroup analysis. Prolonged reaction times led to persistently insoluble material, potentially due to side reactions that result in crosslinking.421H NMR (500 MHz, CDCl3) δ 7.12–7.04 (m, 29H), 3.93 (s, 5H), 2.33 (s, 6H). ATR-IR: N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN 2117 cm−1.
N 1575 cm−1. The gel fraction was obtained by soaking a pre-weighed sample of CAN in THF for 24 h, then replacing the THF and soaking another 24 h followed by drying in a vacuum oven at 90 °C for 24 h.
CN 2117 cm−1.
N 1575 cm−1. The gel fraction was obtained by soaking a pre-weighed sample of CAN in THF for 24 h, then replacing the THF and soaking another 24 h followed by drying in a vacuum oven at 90 °C for 24 h.
 | (1) |
 | (2) |
Results & discussion
Previous work in our group focused on the synthesis and rheological characterization of guanidine-based CANs.40,43 Upon sufficient heating, N,N′,N′′-trisubstituted guanidines undergo a dissociative, reversible exchange of N-substituents termed thermal guanidine metathesis (TGM).44 To prepare CANs, precursors containing multiple carbodiimide (CDI) functionalities can be combined with alkyl diamine crosslinkers to yield guanidine-crosslinked networks. A library of seven guanidine-based CANs was devised to vary νe and plasticizer loading while maintaining consistency across other aspects of the polymer matrix. Multifunctional CDI oligomers were synthesized by combining monofunctional aryl isocyanates and diisocyanates in the presence of a phospholene oxide catalyst (Scheme 1).43 The resulting oligomers were found to have an average degree of polymerization of 2.51 ± 0.40 by 1H NMR endgroup analysis (Fig. S1†). CANs were synthesized by nucleophilic addition of different ratios of difunctional amine crosslinkers and monofunctional amine capping agents together with specific loadings of dioctyl phthalate plasticizer. CANs A, B, and C were prepared in descending order of νe: CAN A was synthesized with full crosslinking (difunctional amine only), CAN B was synthesized at a 2:1 molar ratio of crosslinker to capping agent, and CAN C was synthesized at a 1:1 molar ratio of crosslinker to capping agent based on amine functionalities. Using this synthetic framework, νe was changed while maintaining total guanidine concentration. Plasticizer loading was varied in three degrees, with CAN0, CAN5, and CAN10 variants possessing 0%, 5%, and 10% w/w dioctyl phthalate, respectively. Previous studies on guanidine-based CANs have shown that the addition of phthalates – a common class of plasticizers used in industry – results in lower Tg with no evidence of potential deleterious effects such as side reactions at high temperatures or microphase separation.40 Accordingly, the chosen plasticizer loadings resulted in an approximately 50 °C range of Tg values for the CAN variants under study (see below). In all cases, ATR FT-IR spectroscopy indicated full conversion of CDI to guanidine, as evidenced by the disappearance of the prominent NCN stretching frequency at 2117 cm−1 and appearance of a CN stretching frequency at 1575 cm−1 (Fig. S2–S9†). Network formation was further confirmed by sol–gel analysis (Table S1†), with gel fractions generally decreasing with higher monofunctional amine and/or plasticizer loading across CAN formulations. Thermal analysis indicated modest stability as the average temperature at 5% mass loss was found to be 196 °C by TGA (Fig. S10–S16†).
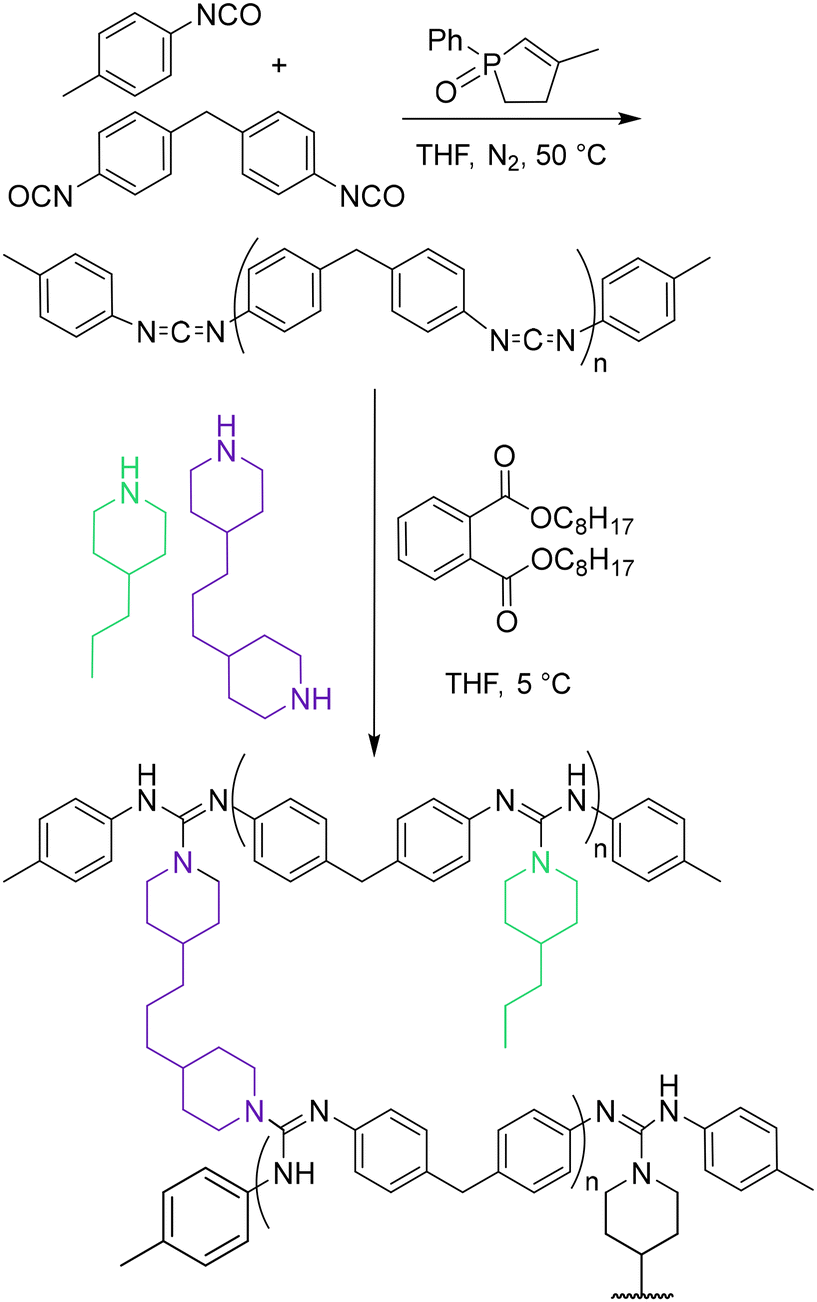 | ||
| Scheme 1 Synthesis of multifunctional carbodiimide oligomer and preparation of guanidine-based covalent adaptable networks. | ||
To determine differences in Tg and νe, DMA temperature ramps were performed on all CANs (Fig. S17–S30†). Tg was found to vary directly with νe, with a larger decrease between CANs A and B than between CANs B and C (Fig. 2a and Table 1). Increased plasticizer was also found to decrease Tg. For example, Tg of CAN A0 was approximately 44 °C higher than that of CAN A5 (176 and 132 °C, respectively). This high Tg precluded further rheological analysis of samples without plasticizer, as the temperature window between Tg and the onset of thermal degradation was very limited. Additionally, trends in νe of plasticized samples (estimated as 3E′/RT)45 in the rubbery plateau at 175 °C were observed to correspond with relative loadings of difunctional:monofunctional amine during synthesis, with little difference between CANs with 5% w/w and 10% w/w plasticizer loadings (Fig. 2b and Table 1).
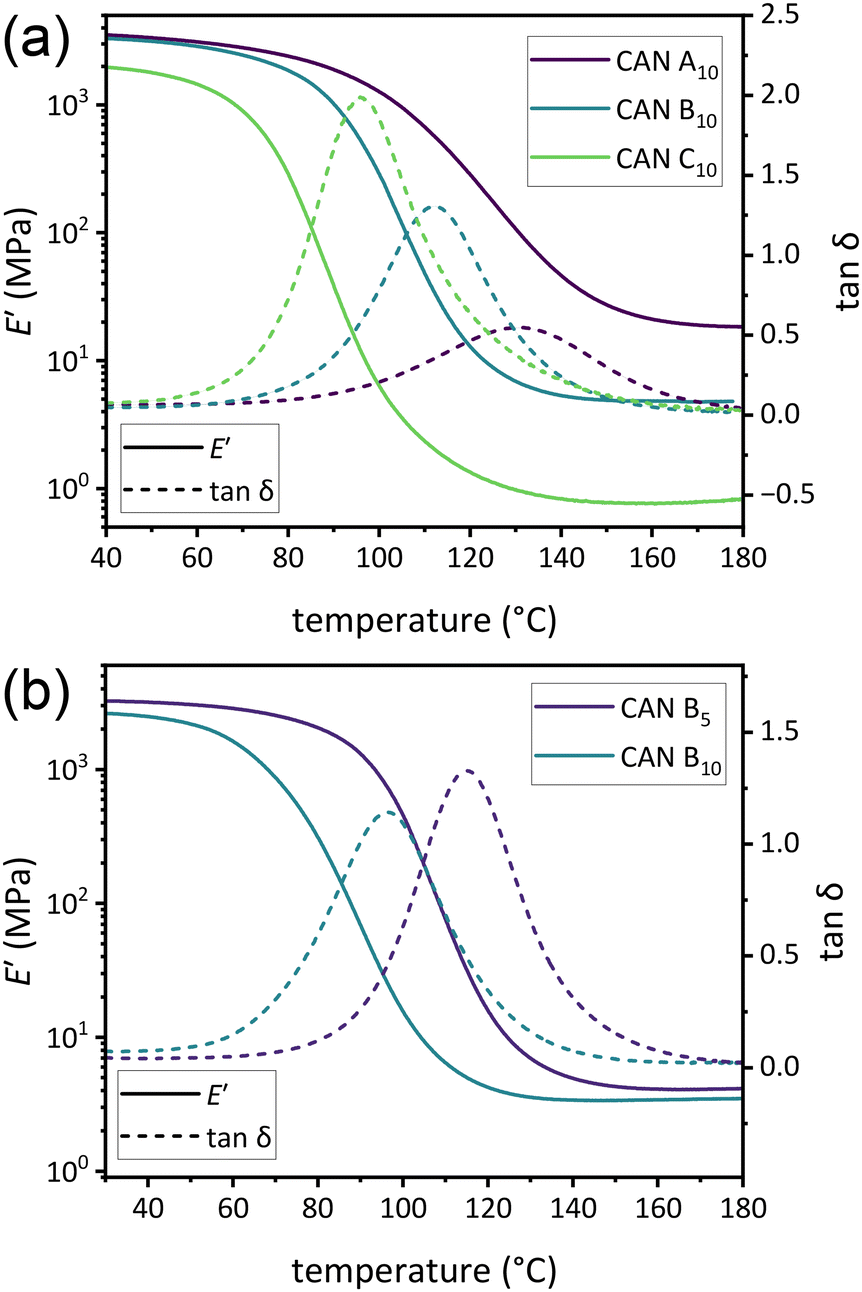 | ||
| Fig. 2 (a) DMA thermograms of E′ versus temperature for CAN A10, CAN B10, and CAN C10. (b) DMA thermograms of E′ and tanδ versus temperature for CAN B5 and CAN B10. | ||
| CAN | T g (°C) | ν e (mmol g−1) |
|
|
|
|---|---|---|---|---|---|
| a Data from three samples. b Data from two samples. c No peak maximum observed in continuous relaxation spectra during experimental window (1200 s). d Limited thermal window for analysis precluded determination. | |||||
| A5 | 132 ± 1.3a | 4.49 ± 0.41a | 129 ± 32a | 126 ± 22a | 405 ± 55a |
| A10 | 126 ± 5.5b | 4.52 ± 0.46b | 122 ± 9b | 159 ± 7a | >1200c |
| B5 | 113 ± 2.8a | 1.23 ± 0.07a | 73 ± 11a | 98 ± 6a | 621 ± 16a |
| B10 | 104 ± 8.0b | 1.11 ± 0.18b | 56 ± 9b | 69 ± 21b | >1200c |
| C5 | 110 ± 4.0b | 0.260 ± 0.06b | 23 ± 9a | 33 ± 2a | 327 ± 32a |
| C10 | 104 ± 7.5b | 0.296 ± 0.08b | 38 ± 9b | 43 ± 14b | >1200c |
| A0 | 176 | —d | —d | —d | —d |
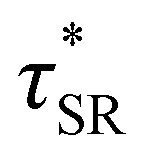
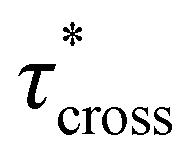
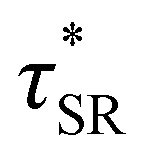
As a means of comparing τ* of each system, a series of small-amplitude oscillatory shear (SAOS) frequency sweeps were performed at 175 °C using parallel plate shear rheometry within the linear viscoelastic region (Fig. S31–S37†). Relaxation times estimated by the modulus crossover 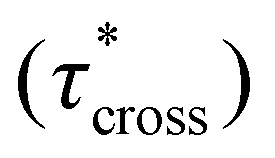 possessed a direct relationship to νe at both 5% w/w and 10% w/w plasticizer (Fig. 3a and Table 1). The relaxation processes at these temperatures thus appear to be dependent upon crosslinking dynamics, as the amount of reactive species remains constant across all CAN compositions. This is similar to findings in other CANs with equal concentrations of reactive groups, which also possess a direct relationship between νe and τ*.32,34 Additionally,
possessed a direct relationship to νe at both 5% w/w and 10% w/w plasticizer (Fig. 3a and Table 1). The relaxation processes at these temperatures thus appear to be dependent upon crosslinking dynamics, as the amount of reactive species remains constant across all CAN compositions. This is similar to findings in other CANs with equal concentrations of reactive groups, which also possess a direct relationship between νe and τ*.32,34 Additionally, 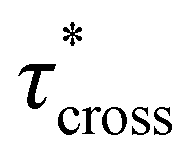 values were not observed to vary consistently as a function of changing plasticizer loading between CAN5 and CAN10 (Fig. 3b and Table 1). As a complementary method of analysis, τ* at 175 °C was also estimated from stress relaxation experiments
values were not observed to vary consistently as a function of changing plasticizer loading between CAN5 and CAN10 (Fig. 3b and Table 1). As a complementary method of analysis, τ* at 175 °C was also estimated from stress relaxation experiments 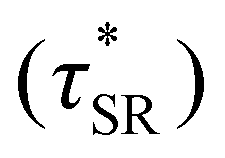 . Some samples exhibited relaxation processes that were not well-captured by fitting the data to a stretched exponential function. As an alternative, we estimated
. Some samples exhibited relaxation processes that were not well-captured by fitting the data to a stretched exponential function. As an alternative, we estimated 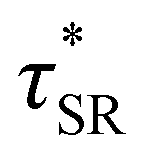 from the principal peak in the continuous relaxation spectra derived from stress relaxation data (Fig. S38–S43†).46–48 These spectra more clearly distinguished the principal relaxation mode from others such as segmental Rouse relaxation, which was observable at short timescales in some compositions. Trends remained similar to those found in frequency sweeps: a direct relationship to νe and no consistent difference due to plasticizer loading was observed (Fig. 3c and d). Additionally, τ* values were generally in relatively good agreement across the two analysis techniques (Table 1). Previous studies have implicated differences in Tg as important governors of CAN relaxation behavior.26,49,50 To examine this possibility, we also compared
from the principal peak in the continuous relaxation spectra derived from stress relaxation data (Fig. S38–S43†).46–48 These spectra more clearly distinguished the principal relaxation mode from others such as segmental Rouse relaxation, which was observable at short timescales in some compositions. Trends remained similar to those found in frequency sweeps: a direct relationship to νe and no consistent difference due to plasticizer loading was observed (Fig. 3c and d). Additionally, τ* values were generally in relatively good agreement across the two analysis techniques (Table 1). Previous studies have implicated differences in Tg as important governors of CAN relaxation behavior.26,49,50 To examine this possibility, we also compared 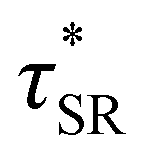 values 35 °C above Tg of each individual network. These values did not exhibit trends similar to those observed at an absolute temperature (Table 1). Thus, temperature relative to Tg does not appear to have predictive value with respect to relaxation behavior, and plasticizer can serve to decouple Tg from CAN dynamics.
values 35 °C above Tg of each individual network. These values did not exhibit trends similar to those observed at an absolute temperature (Table 1). Thus, temperature relative to Tg does not appear to have predictive value with respect to relaxation behavior, and plasticizer can serve to decouple Tg from CAN dynamics.
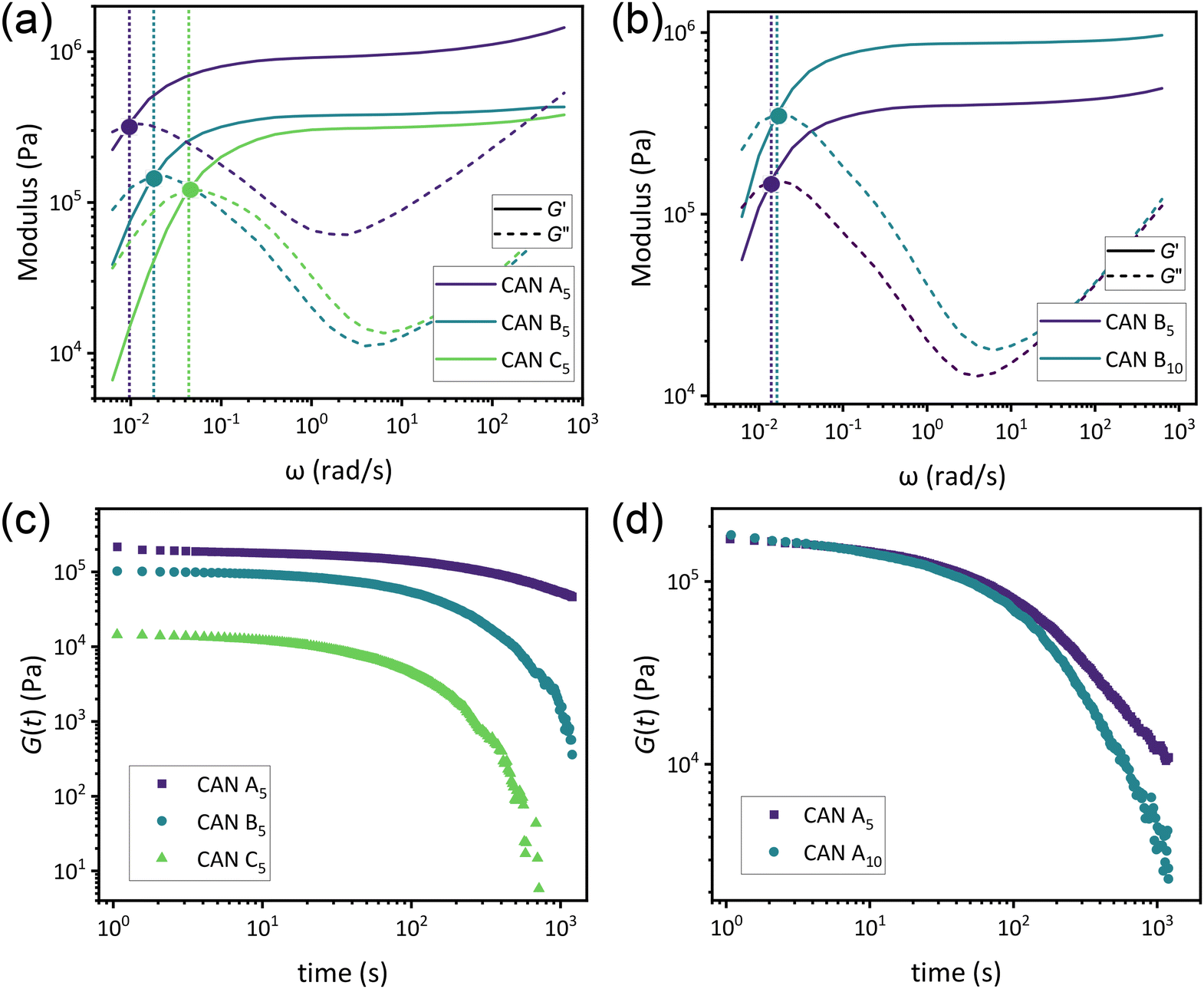 | ||
| Fig. 3 (a) Frequency sweep data for representative samples of CAN A5, CAN B5, and CAN C5 at 175 °C, where vertical lines indicate crossover frequencies. (b) Frequency sweep data for data for representative samples of CAN B5 and CAN B10 at 175 °C, where vertical lines indicate crossover frequencies. (c) Stress relaxation data for representative samples of CAN A5, CAN B5, and CAN C5 at 175 °C. (d) Stress relaxation data for representative samples of CAN A5 and CAN A10 at 175 °C. | ||
Flow activation energies were determined by conducting both frequency sweep and stress relaxation experiments in 5 °C increments above Tg and subsequently constructing master curves by time–temperature superposition (TTS) using both horizontal and vertical shift factors (aT and bT, respectively). While rigorous use of TTS relies on the assumption of thermorheological simplicity, CANs cannot possess this characteristic as they exhibit inherently different temperature dependencies of relaxation processes.46 Nevertheless, previous researchers have found judicious application of TTS to be a valuable means of understanding CAN behavior,10,51,52 and collapse of data in reduced van Gurp–Palmen plots (Fig. 4a and S44a–49a†) supported the validity of TTS in our systems.53 While all CAN B and CAN C variants exhibited superposition of both frequency sweep and stress relaxation data from 145–180 °C, TTS only applied from 165–185 °C in CAN A (Fig. 4b, c and S44b–e to S49b–e†). Across all formulations, aT exhibited an Arrhenius relationship with temperature (Fig. 4d and S44f–S49f†), with similar shifts occurring among SAOS and stress relaxation data. Thus, Ea was calculated using aT compiled from all experiments on each individual CAN formulation (Table 2),10 and little variation was observed based on either νe or plasticizer loading. This contrasts with associative systems, which tend to exhibit changes in Ea with varying νe even with the same concentration of reactive functionalities.33,34 The interpretation of bT in this system is less clear (Fig. S50†). In stress relaxation experiments, G0 and the attendant bT varied as expected for a dissociative system in which νe decreases with increasing temperature. However, bT from SAOS frequency sweeps exhibited more variation. Previous studies of dissociative CANs have found Arrhenius relationships of bT with temperature in both types of measurement corresponding to the enthalpy of the dissociation reaction.10 Such findings were predicted to occur in a limited temperature range and are dependent on the thermodynamics of each specific system. Additionally, bT in associative CANs is highly variable.51,52 As any potential side reaction resulting in permanent crosslinking would occur in both experiments and no significant changes were observed in the IR spectra of CANs after rheological analysis (Fig. S51†), a possible explanation for the lack of observed trends in bT derived from frequency sweeps is a complex interplay between the crosslink dissociation equilibrium, changes in material density, and entropic contributions to elasticity which manifest more substantially on the timescale of the SAOS plateau modulus.
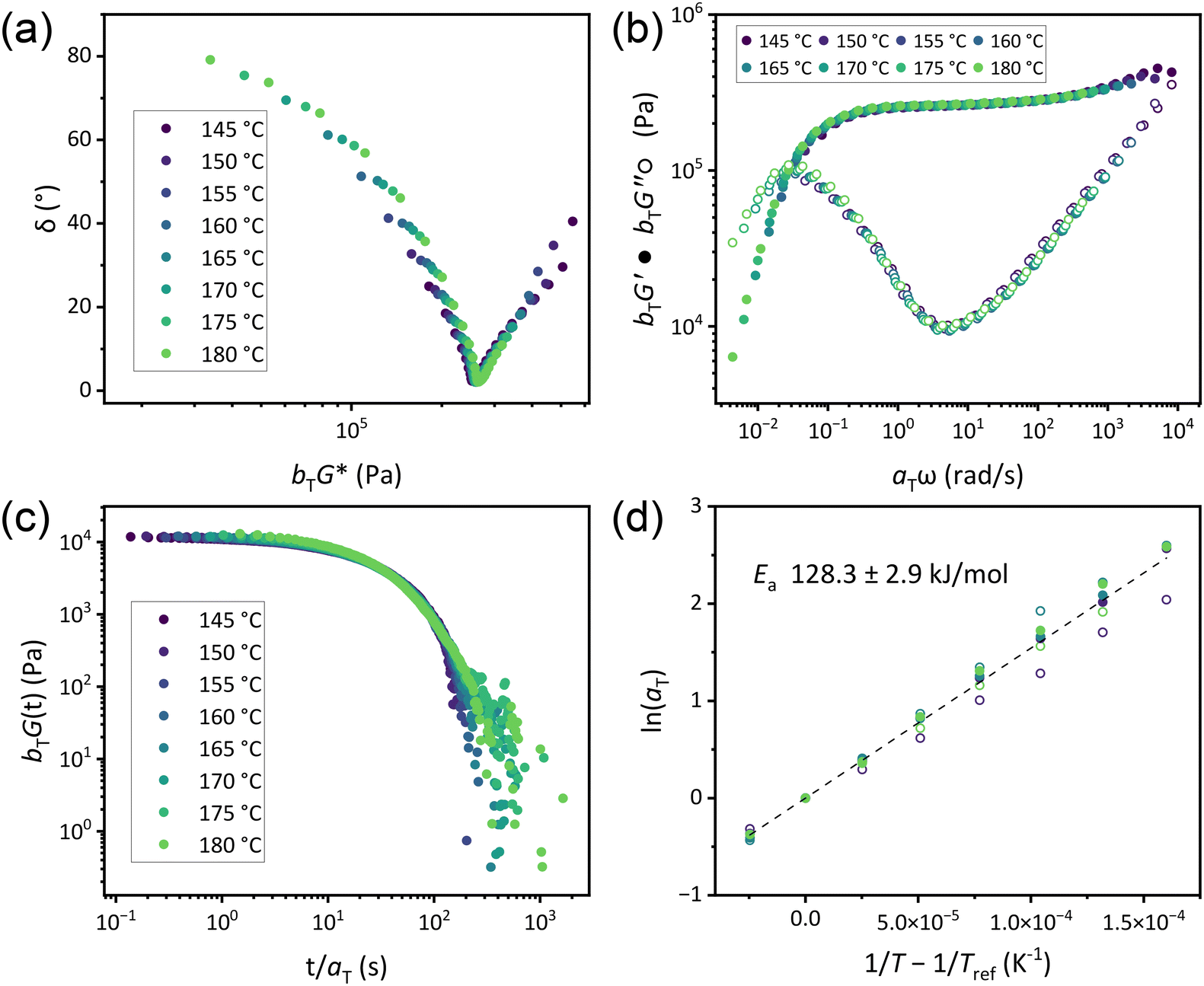 | ||
| Fig. 4 (a) Reduced van Gurp–Palmen plot of a representative sample of CAN C5. (b) Master curve of frequency sweep data constructed via TTS of a sample of CAN C5. (c) Master curve of stress relaxation constructed from TTS of a sample of CAN C5. (d) Arrhenius analysis of horizontal shift factors for all CAN C5 samples. Filled circles are from frequency sweep data and empty circles from stress relaxation; each color is a single sample. Dashed line is line of best fit. Tref = 175 °C for all analyses. | ||
Our findings that τ* varies directly with νe while Ea does not can be rationalized based on the sticky Rouse model developed for unentangled CANs.52,54 A key component of this model is that the longest relaxation times are directly related to both the crosslink lifetime (τxl) and the number of crosslinks per chain, i.e. νe, in accordance with our results (see ESI† for additional discussion).55 In CANs, τxl is determined by contributions of (1) the kinetics of the crosslink exchange reaction; (2) Rouse dynamics, which describe chain diffusion based on repeat unit friction; and (3) the mobility of exchanging functionalities within the polymer matrix. As contributions from (1) and (2) are largely determined by the chemical makeup of the system, their temperature dependence is not expected to vary across different νe. However, the relatively constant Ea we observe implies that the mobility of exchanging functionalities is not substantially impacted by νe in dissociative guanidine-based CANs, similar to another dissociative system with constant functional group concentration32 and contrasting with vitrimers.33,34 We hypothesize this difference originates in the temperature dependence on νe that is not present in associative systems.
Conclusions
In conclusion, we have analyzed the effects of νe and plasticizer on rheological properties of guanidine-based CANs using complementary rheometric techniques. The combination of multifunctional carbodiimide oligomers with specific ratios of difunctional to monofunctional secondary alkyl amine species enabled the synthesis of CANs with varying νe but a constant concentration of reactive groups. By changing νe in three levels and plasticizer loading by weight in three levels, a library of seven CANs was developed. Tg was found to vary directly with νe and plasticizer loading. However, τ* obtained by both stress relaxation and SAOS frequency sweeps varied directly with νe while plasticizer had little effect; thus, plasticizer serves to decouple τ* from Tg. Further, we observed a relatively constant Ea across all network compositions, in contrast with studies of associative CANs. We hope our studies motivate additional investigation of the fundamental properties of vitrimer-like dissociative systems, further illustrate the utility of TTS in rheological analysis of CANs, and add plasticizer content to the growing list of characteristics that can be used to tune CAN dynamics.Data availability
The data supporting this article have been included in the main text and as part of the ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Science Foundation (DMR 2105149) and the Karen and Joseph Morse Summer Research Fellowship for support of this work. The authors thank Dr Ralm Ricarte for helpful discussions of rheological characterization and analysis, Dr Mark Peyron for guidance on statistical analysis, and Kyle Mikkelsen for assistance with characterization techniques.References
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Covalent Adaptable Networks (CANs): A Unique Paradigm in Cross-Linked Polymers, Macromolecules, 2010, 43(6), 2643–2653, DOI:10.1021/ma902596s.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Using the Dynamic Bond to Access Macroscopically Responsive Structurally Dynamic Polymers, Nat. Mater., 2011, 10(1), 14–27, DOI:10.1038/nmat2891.
- C. N. Bowman and C. J. Kloxin, Covalent Adaptable Networks: Reversible Bond Structures Incorporated in Polymer Networks, Angew. Chem., Int. Ed., 2012, 51(18), 4272–4274, DOI:10.1002/anie.201200708.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Enabling Applications of Covalent Adaptable Networks, Annu. Rev. Chem. Biomol. Eng., 2019, 10(1), 175–198, DOI:10.1146/annurev-chembioeng-060718-030217.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, A Thermally Re-Mendable Cross-Linked Polymeric Material, Science, 2002, 295(5560), 1698–1702, DOI:10.1126/science.1065879.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, Inherently Photohealable and Thermal Shape-Memory Polydisulfide Networks, ACS Macro Lett., 2013, 2(8), 694–699, DOI:10.1021/mz400318m.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968, DOI:10.1126/science.1212648.
- W. Denissen, J. M. Winne and F. E. D. Prez, Vitrimers: Permanent Organic Networks with Glass-like Fluidity, Chem. Sci., 2015, 7(1), 30–38, 10.1039/C5SC02223A.
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Toward Stimuli-Responsive Dynamic Thermosets through Continuous Development and Improvements in Covalent Adaptable Networks (CANs), Adv. Mater., 2020, 32(20), 1906876, DOI:10.1002/adma.201906876.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks, Macromolecules, 2020, 53(6), 1884–1900, DOI:10.1021/acs.macromol.9b02204.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Exploring How Vitrimer-like Properties Can Be Achieved from Dissociative Exchange in Anilinium Salts, Macromolecules, 2020, 53(4), 1233–1244, DOI:10.1021/acs.macromol.0c00120.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, Oxime-Based and Catalyst-Free Dynamic Covalent Polyurethanes, J. Am. Chem. Soc., 2017, 139(25), 8678–8684, DOI:10.1021/jacs.7b03967.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets, J. Am. Chem. Soc., 2019, 141(41), 16181–16196, DOI:10.1021/jacs.9b07922.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Rheological and Chemical Analysis of Reverse Gelation in a Covalently Cross-Linked Diels–Alder Polymer Network, Macromolecules, 2008, 41(23), 9112–9117, DOI:10.1021/ma801863d.
- M. Bin Rusayyis and J. M. Torkelson, Recyclable Polymethacrylate Networks Containing Dynamic Dialkylamino Disulfide Linkages and Exhibiting Full Property Recovery, Macromolecules, 2020, 53(19), 8367–8373, DOI:10.1021/acs.macromol.0c01539.
- L. Li, X. Chen and J. M. Torkelson, Covalent Adaptive Networks for Enhanced Adhesion: Exploiting Disulfide Dynamic Chemistry and Annealing during Application, ACS Appl. Polym. Mater., 2020, 2(11), 4658–4665, DOI:10.1021/acsapm.0c00720.
- R. Martin, A. Rekondo, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, The Processability of a Poly(Urea-Urethane) Elastomer Reversibly Crosslinked with Aromatic Disulfide Bridges, J. Mater. Chem. A, 2014, 2(16), 5710–5715, 10.1039/C3TA14927G.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Dynamic Thiol–Michael Chemistry for Thermoresponsive Rehealable and Malleable Networks, Macromolecules, 2016, 49(18), 6871–6878, DOI:10.1021/acs.macromol.6b01061.
- H. Ying, Y. Zhang and J. Cheng, Dynamic Urea Bond for the Design of Reversible and Self-Healing Polymers, Nat. Commun., 2014, 5(1), 3218, DOI:10.1038/ncomms4218.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Malleable and Recyclable Poly(Urea-Urethane) Thermosets Bearing Hindered Urea Bonds, Adv. Mater., 2016, 28(35), 7646–7651, DOI:10.1002/adma.201601242.
- L. Zhang and S. J. Rowan, Effect of Sterics and Degree of Cross-Linking on the Mechanical Properties of Dynamic Poly(Alkylurea–Urethane) Networks, Macromolecules, 2017, 50(13), 5051–5060, DOI:10.1021/acs.macromol.7b01016.
- J. J. Lessard, K. A. Stewart and B. S. Sumerlin, Controlling Dynamics of Associative Networks through Primary Chain Length, Macromolecules, 2022, 55(22), 10052–10061, DOI:10.1021/acs.macromol.2c01909.
- Y. Liu, Z. Tang, J. Chen, J. Xiong, D. Wang, S. Wang, S. Wu and B. Guo, Tuning the Mechanical and Dynamic Properties of Imine Bond Crosslinked Elastomeric Vitrimers by Manipulating the Crosslinking Degree, Polym. Chem., 2020, 11(7), 1348–1355, 10.1039/C9PY01826C.
- T. Isogai and M. Hayashi, Critical Effects of Branch Numbers at the Cross-Link Point on the Relaxation Behaviors of Transesterification Vitrimers, Macromolecules, 2022, 55(15), 6661–6670, DOI:10.1021/acs.macromol.2c00560.
- P. Miao, X. Leng, J. Liu, G. Song, M. He and Y. Li, Regulating the Dynamic Behaviors of Transcarbamoylation-Based Vitrimers via Mono-Variation in Density of Exchangeable Hydroxyl, Macromolecules, 2022, 55(12), 4956–4966, DOI:10.1021/acs.macromol.2c00127.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. D. Prez, Influence of the Polymer Matrix on the Viscoelastic Behaviour of Vitrimers, Polym. Chem., 2020, 11(33), 5377–5385, 10.1039/D0PY00114G.
- B. Soman and C. M. Evans, Effect of Precise Linker Length, Bond Density, and Broad Temperature Window on the Rheological Properties of Ethylene Vitrimers, Soft Matter, 2021, 17(13), 3569–3577, 10.1039/D0SM01544J.
- S. K. Schoustra, T. Groeneveld and M. M. J. Smulders, The Effect of Polarity on the Molecular Exchange Dynamics in Imine-Based Covalent Adaptable Networks, Polym. Chem., 2021, 12(11), 1635–1642, 10.1039/D0PY01555E.
- Y. Feng, H. Qiu, P. Deng, Z. Nie, J. Chen, K. Gong, X. Fan and S. Qi, Tuning the Static and Dynamic Properties of Epoxy Vitrimers through Modulation of Cross-Link Density, Eur. Polym. J., 2023, 196, 112308, DOI:10.1016/j.eurpolymj.2023.112308.
- L. Yang, L. Li, L. Fu, B. Lin, Y. Wang and C. Xu, Design of Ester Crosslinked Rubber with High Dynamic Properties by Increasing Dynamic Covalent Bond Density, Polym. Chem., 2022, 13(48), 6650–6661, 10.1039/D2PY01165D.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Tuning the Viscosity Profile of Ionic Vitrimers Incorporating 1,2,3-Triazolium Cross-Links, Adv. Funct. Mater., 2017, 27(45), 1703258, DOI:10.1002/adfm.201703258.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, Structural Effects on the Reprocessability and Stress Relaxation of Crosslinked Polyhydroxyurethanes, J. Appl. Polym. Sci., 2017, 134(45), 44984, DOI:10.1002/app.44984.
- M. Hayashi and R. Yano, Fair Investigation of Cross-Link Density Effects on the Bond-Exchange Properties for Trans-Esterification-Based Vitrimers with Identical Concentrations of Reactive Groups, Macromolecules, 2020, 53(1), 182–189, DOI:10.1021/acs.macromol.9b01896.
- R. Hajj, A. Duval, S. Dhers and L. Avérous, Network Design to Control Polyimine Vitrimer Properties: Physical Versus Chemical Approach, Macromolecules, 2020, 53(10), 3796–3805, DOI:10.1021/acs.macromol.0c00453.
- L. Chen, L. Zhang, P. J. Griffin and S. J. Rowan, Impact of Dynamic Bond Concentration on the Viscoelastic and Mechanical Properties of Dynamic Poly(Alkylurea-Co-Urethane) Networks, Macromol. Chem. Phys., 2020, 221(1), 1900440, DOI:10.1002/macp.201900440.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Vitrimers Designed Both To Strongly Suppress Creep and To Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments, Macromolecules, 2018, 51(15), 5537–5546, DOI:10.1021/acs.macromol.8b00922.
- M.-J. Xie, C.-C. Wang, R. Zhang, J. Cao, M.-Z. Tang and Y.-X. Xu, Length Effect of Crosslinkers on the Mechanical Properties and Dimensional Stability of Vitrimer Elastomers with Inhomogeneous Networks, Polymer, 2024, 290, 126550, DOI:10.1016/j.polymer.2023.126550.
- O. Konuray, S. Moradi, A. Roig, X. Fernández-Francos and X. Ramis, Thiol–Ene Networks with Tunable Dynamicity for Covalent Adaptation, ACS Appl. Polym. Mater., 2023, 5(3), 1651–1656, DOI:10.1021/acsapm.2c02136.
- A. Safaei, S. Terryn, B. Vanderborght, G. Van Assche and J. Brancart, Toughening and Stiffening in Thermoreversible Diels–Alder Polymer Network Blends, Macromolecules, 2023, 56(11), 4325–4335, DOI:10.1021/acs.macromol.2c02558.
- H. E. Houck, K. A. McConnell, C. J. Klingler, A. L. Koenig, G. K. Himka and M. B. Larsen, Postpolymerization Modification by Nucleophilic Addition to Styrenic Carbodiimides, ACS Macro Lett., 2023, 12(8), 1112–1117, DOI:10.1021/acsmacrolett.3c00382.
- J. Chen, L. Li, J. Luo, L. Meng, X. Zhao, S. Song, Z. Demchuk, P. Li, Y. He, A. P. Sokolov and P.-F. Cao, Covalent adaptable polymer networks with CO2-facilitated recyclability, Nat. Commun., 2024, 15, 6605, DOI:10.1038/s41467-024-50738-7.
- A. P. Gies, W. H. Heath, R. J. Keaton, J. J. Jimenez and J. J. Zupancic, MALDI-TOF/TOF CID Study of Polycarbodiimide Branching Reactions, Macromolecules, 2013, 46, 7616–7637, DOI:10.1021/ma401481g.
- A. J. Melchor Bañales and M. B. Larsen, Thermal Guanidine Metathesis for Covalent Adaptable Networks, ACS Macro Lett., 2020, 9(7), 937–943, DOI:10.1021/acsmacrolett.0c00352.
- V. Ramirez, E. B. V. Pelt, R. K. Pooni, A. J. M. Bañales and M. B. Larsen, Thermodynamic, Kinetic, and Mechanistic Studies of the Thermal Guanidine Metathesis Reaction, Org. Biomol. Chem., 2022, 20(29), 5861–5868, 10.1039/D2OB01036D.
- P. C. Hiemenz and T. Lodge, Polymer Chemistry, CRC Press, Boca Raton, 2nd edn, 2007, pp. 446 Search PubMed.
- R. G. Ricarte and S. Shanbhag, A Tutorial Review of Linear Rheology for Polymer Chemists: Basics and Best Practices for Covalent Adaptable Networks, Polym. Chem., 2024, 15(9), 815–846, 10.1039/D3PY01367G.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Control of hierarchical polymer mechanics with bioinspired metal-coordination dynamics, Nat. Mater., 2015, 14, 1210–1216, DOI:10.1038/nmat4401.
- J. S. A. Ishibashi, I. C. Pierce, A. B. Chang, A. Zografos, B. M. El-Zaatari, Y. Fang, S. J. Weigand, F. S. Bates and J. A. Kalow, Mechanical and Structural Consequences of Associative Dynamic Cross-Linking in Acrylic Diblock Copolymers, Macromolecules, 2021, 54, 3972–3986 CrossRef.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Fire-Safe, Monomer-Recovery, Highly Malleable Thermosets from Renewable Resources, Macromolecules, 2018, 51, 8001–8012 CrossRef CAS.
- G. Lee, H. Y. Song, S. Choi, C. B. Kim, K. Hyun and S. Ahn, Harnessing β-Hydroxyl Groups in Poly(β-Amino Esters) toward Robust and Fast Reprocessing Covalent Adaptable Networks, Macromolecules, 2022, 55, 10366–10376 CrossRef CAS.
- M. L. Martins, X. Zhao, Z. Demchuk, J. Luo, G. P. Carden, G. Toleutay and A. P. Sokolov, Viscoelasticity of Polymers with Dynamic Covalent Bonds: Concepts and Misconceptions, Macromolecules, 2023, 56, 8688–8696, DOI:10.1021/acs.macromol.3c01545.
- R. G. Ricarte, S. Shanbhag, D. Ezzeddine, D. Barzycki and K. Fay, Time-Temperature Superposition of Polybutadiene Vitrimers, Macromolecules, 2023, 56, 6806–6817, DOI:10.1021/acs.macromol.3c00883.
- H. Y. Song, G. Lee, S. Ahn and K. Hyun, Linear Viscoelasticity of Covalent Adaptable Network (CAN) Polymers Comprising β-Amino Esters, Korea-Aust. Rheol. J., 2023, 35(2), 69–79, DOI:10.1007/s13367-023-00054-1.
- R. G. Ricarte and S. Shanbhag, Unentangled Vitrimer Melts: Interplay between Chain Relaxation and Cross-link Exchange Controls Linear Rheology, Macromolecules, 2021, 54, 3304–3320, DOI:10.1021/acs.macromol.0c02530.
- R. H. Colby, X. Zheng, M. H. Rafailovich, J. Sokolov, D. G. Peiffer, S. A. Schwarz, Y. Strzhemechny and D. Nguyen, Dynamics of Lightly Sulfonated Polystyrene Ionomers, Phys. Rev. Lett., 1998, 81, 3786–3789, DOI:10.1103/PhysRevLett.81.3876.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization of oligomers and CANs; data from DMA, stress relaxation, and SAOS analyses; discussion of the sticky Rouse model for unentangled CANs. See DOI: https://doi.org/10.1039/d4py01124d |
| This journal is © The Royal Society of Chemistry 2025 |