
Supramolecular method enabling effective through-space charge transfer in thermally activated delayed fluorescence materials with pure orange emission†
Yufeng
Sang
a,
Ruicong
Feng
a,
Yuqian
Wang
a and
Qiao
Song
 *ab
*ab
aShenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: songq@sustech.edu.cn
bGuangming Advanced Research Institute, Southern University of Science and Technology, Shenzhen 518055, China
First published on 22nd November 2024
Abstract
Achieving efficient charge transfer remains a significant challenge for thermally activated delayed fluorescence (TADF) materials that rely on through-space charge transfer (TSCT). In this study, we successfully applied a novel donor–acceptor pair characterized by non-exclusive charge transfer to TSCT-based TADF polymers. We propose a supramolecular strategy to optimize charge transfer by regulating the spatial arrangement of the donor–acceptor pairs. As the order of these pairs increases, localized emission from monomers is gradually quenched, ultimately resulting in pure orange emission. Photophysical studies reveal that improved spatial order accelerates the reverse intersystem crossing process, thereby enhancing the radiative transition rate of TADF emission and enabling efficient TSCT. This research offers a feasible method for designing TSCT-TADF materials. The resulting supramolecular systems with ordered configurations exhibit excellent energy and charge transfer performance, indicating their potential applications in optoelectronic devices, bioimaging, photodynamic therapy, and other fields.
Introduction
Thermally activated delayed fluorescence (TADF) materials based on through-space charge transfer (TSCT) have garnered significant attention in recent years.1–3 Complementing through-bond charge transfer (TBCT), TSCT facilitates an efficient reverse intersystem crossing (RISC) process by the complete spatial separation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), resulting in a minuscule singlet–triplet energy gap (ΔEST).4–6 Additionally, TSCT offers possibilities for flexible and versatile structural design and application.7–9 However, achieving sufficient TSCT requires a strict donor–acceptor distance (0.3–0.5 nm)6,10,11 and an appropriate spatial configuration (face-to-face),12,13 which typically necessitate complex synthesis procedures. This poses an enormous challenge in the development of these materials. Moreover, impure emission caused by incomplete charge transfer often hinders the application of TSCT-based TADF materials.Luminescent polymeric materials exhibit outstanding photophysical properties and processability, providing feasible and flexible platforms for applications such as organic light-emitting diodes (OLEDs),14 sensing,15 bioimaging,16 and theragnostic,17 representing the future direction of low-cost, high-efficiency luminescent materials.18 Recently, impressive progress has been made in TSCT-based TADF polymers.3,19–22 By selecting appropriate monomers with donor or acceptor properties, TSCT-TADF polymers can be constructed conveniently and effectively through polymerization reactions. Nevertheless, existing reports usually emphasize the structure and modification of monomers, while neglecting issues related to loose packing and random donor/acceptor configurations.2 In fact, the influence of the microenvironment and intermolecular interactions on emission performance is just as important as the chemical composition.23 Therefore, investigating the TSCT-TADF backbones and donor/acceptor arrangements is essential for a profound understanding and advancement of these materials.
Supramolecular polymers are defined as polymeric arrays in which monomer units are connected through highly directional and reversible noncovalent interactions.24 Their block-by-block self-assembly and multicompartment structures provide a straightforward approach to regulating the morphology, composition, and function of nanomaterials.25–27 Self-assembling cyclic peptides are an important supramolecular polymer building block, capable of stacking into bottle-brushed nanostructures through multiple hydrogen bonding interactions,28,29 and by attaching polymeric chains onto the cyclic peptides, the assembled supramolecular polymers can be endowed with various functions. Additionally, the unique morphology of bottlebrush polymers has been shown to reduce chain entanglement.30–32 Recently, TSCT-TADF supramolecular polymers have been successfully constructed via cyclic peptide–polymer conjugates.33 Therefore, the distinct architecture of the cyclic peptide-based supramolecular scaffolds may offer a promising platform to explore and optimize the donor–acceptor spatial arrangements, potentially leading to enhanced TADF efficiency and improved material performance.
In this work, 9,9-dimethylacridine (Ac) and 1,8-naphthalimide (NAI) derivatives are chosen to serve as the donor and acceptor, respectively, for constructing TSCT-based TADF materials. Initially, the as-synthesized polymer exhibited dual-band emission: an orange TADF emission and a high-energy peak at around 400 nm, which was identified as localized emission (LE) from the acceptor due to the non-exclusive charge transfer process. To eliminate the LE emission and enhance colour purity, two strategies have been explored. The conventional approach of introducing halogens to strengthen spin–orbit coupling (SOC) and donor–acceptor interaction was found to be unsatisfactory. Instead, a more effective supramolecular strategy involving the construction of bottle-brushed supramolecular polymers with an orderly donor–acceptor arrangement via a cyclic peptide-based scaffold has been proposed (Fig. 1). As anticipated, increasing the degree of order gradually suppressed the LE emission, ultimately achieving a pure orange emission. Photophysical studies revealed that this pure colour emission could be attributed to the ultrafast RISC rate (1.40 × 107 s−1) in the orderly configuration. This highlights the critical role of donor/acceptor arrangements in TSCT-based TADF materials. By optimizing the spatial arrangement of donor and acceptor moieties using the supramolecular method, it is possible to achieve more efficient TADF processes and suppress unwanted emissions, thereby paving the way for the development of high-performance luminescent materials with superior colour purity.
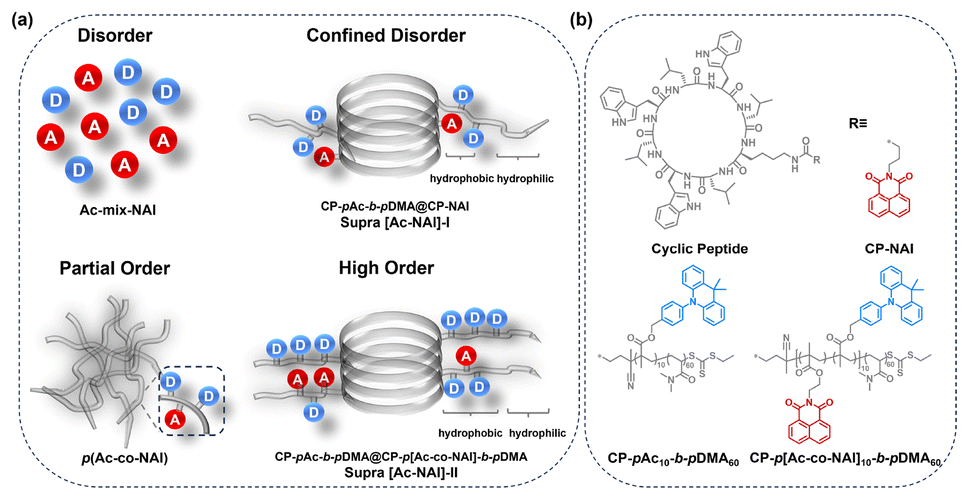 | ||
| Fig. 1 (a) Cartoon illustration of TSCT-TADF materials constructed via covalent and supramolecular polymerization with various degrees of order; (b) chemical structures of cyclic peptide-based TSCT donor/acceptor conjugates. | ||
Results and discussion
Several compounds based on Ac and NAI with different connections were prepared to access the feasibility of constructing TSCT-TADF systems via this novel D–A pair. As shown in Fig. 2a, both the small molecule via TBCT, Ac-NAI, and the TSCT-based polymer, p(Ac-co-NAI), displayed two emission bands. The dominant emission, observed around 600 nm, corresponds to an intense orange colour, similar to previously reported D–A type TADF materials based on Ac and NAI.34,35 The emission of Ac-NAI is red-shifted by approximately 50 nm compared to that of p(Ac-co-NAI), likely due to the better overlap of the HOMO and LUMO electron clouds resulting from covalent connection. Additionally, a high-energy peak at around 400 nm, coinciding with the emission of NAI, was detected. Notably, no long-wavelength emission band was observed in the mixture of small molecule monomers, emphasizing the essential role of the donor–acceptor spatial arrangement in TSCT-TADF systems. The 2D excitation–emission contour map of p(Ac-co-NAI) further confirms that both emission bands originate from the same source (Fig. 2b).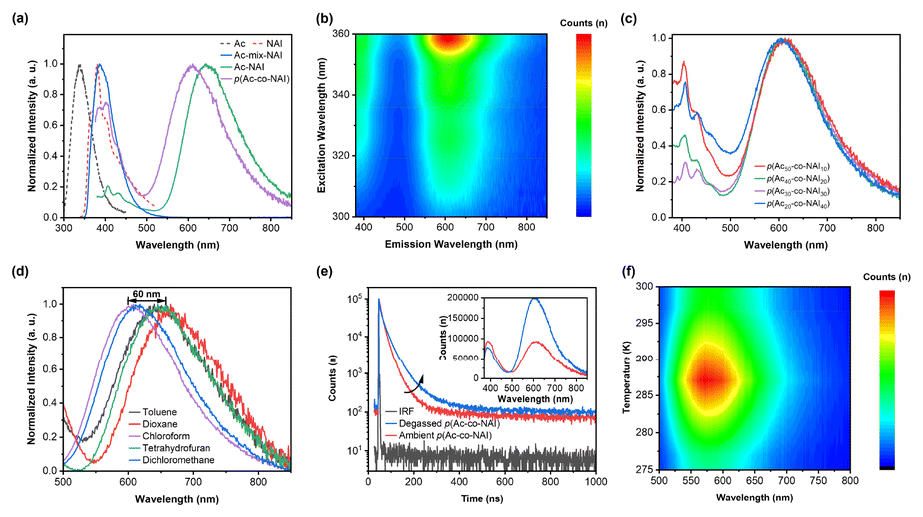 | ||
| Fig. 2 (a) Normalized PL spectra of Ac monomer, NAI monomer, an equimolar mixture of Ac and NAI, covalently connected Ac-NAI, and the TSCT-based polymer p(Ac-co-NAI) in toluene; (b) two-dimensional excitation–emission contour map of p(Ac-co-NAI); (c) normalized PL spectra of p(Ac-co-NAI) copolymers with varying donor/acceptor ratios; (d) normalized PL spectra of p(Ac-co-NAI) in different solvents showing the solvatochromic effect of the TADF peak; (e) PL lifetime decay profiles and steady-state PL spectra (insert) of p(Ac-co-NAI) in ambient and degassed toluene; (f) temperature-dependent emission spectra of p(Ac-co-NAI). | ||
To further explore the origination of dual-band emission, copolymers with different donor/acceptor ratios were synthesized. The orange emission showed no significant shift with the increase of the acceptor ratio, while the high-energy emission decreased continuously until the donor/acceptor ratio reached balance, at which point the relative intensity was at its lowest. Further increase in the acceptor ratio led to a marked enhancement of the high-energy emission (Fig. 2c). Therefore, the high-energy emission could be identified as LE of the acceptor, occurring by non-exclusive charge transfer. Photoluminescence (PL) spectra of p(Ac-co-NAI) were also measured in various organic solvents. The long-wavelength emission band exhibited strong dependence on solvent polarity, with a wavelength shift of up to 60 nm, indicating a distinct CT transition between the Ac and NAI units (Fig. 2d). Conversely, the LE emission displayed only a 10 nm shift, typical of fluorescence (Fig. S1†). Both steady-state PL spectra and lifetime decay profiles of the orange emission revealed high sensitivity to oxygen, with stronger emission intensity and longer lifetimes observed under nitrogen, suggesting the involvement of triplet states (Fig. 2e). The lifetime decay profile of the orange emission band in air was fitted with two distinct lifetimes: a delayed lifetime (τd) of 1312.2 ns and a prompt lifetime (τp) of 24.0 ns. In contrast, the LE emission was nearly unaffected by oxygen (Fig. 2e and Fig. S2†), consistent with typical fluorescence behaviour. Most importantly, the TADF process was directly observed through temperature-dependent fluorescence spectra. As the temperature increased from 275 K to 288 K, the orange emission intensified continuously. Further temperature increases led to a decrease in emission intensity, attributable to enhanced non-radiative transitions caused by molecular thermal motion (Fig. 2f). In summary, Ac/NAI were successfully utilized to activate TSCT-TADF. However, the incomplete charge transfer process resulted in intense LE emission, which substantially reduced both the colour purity and TADF emission efficiency.
To eliminate the LE emission, intramolecular modification is the most classical approach. Halogen atoms (–Cl and –Br) were introduced due to their ability to strengthen D–A interaction and SOC. The absorption spectra of D–A copolymers with halogen-substituted NAI exhibited broader profiles compared to the pristine p(Ac-co-NAI), suggesting a stronger intramolecular interaction in the ground state (Fig. 3a). Additional evidence for the enhanced D–A interaction was provided by 1H NMR spectroscopy. Prior to polymerization, the chemical shifts of the β-position protons on the donor acridine remained consistently at 7.32/7.34 ppm, irrespective of the variations in acceptors (Fig. S3†). However, in the as-synthesized polymers, as the electronegativity of the NAI substituent increased (–Br > –Cl > –H), the signals of the same protons gradually shifted upfield. This shift could be attributed to the deshielding effect caused by the enhanced D–A interaction (Fig. S4†). As excepted, introducing halogens suppressed the proportion of LE emission. However, a significant red-shift in the TADF emission was observed, from 610 nm of p(Ac-co-NAI) to 656 nm of p(Ac-co-NAIBr), accompanied by a notable decrease in photoluminescence quantum yields (PLQY) from 2.17% of p(Ac-co-NAI) to 0.94% of p(Ac-co-NAIBr) (Fig. 3b and Table S1†). Transient fluorescence spectroscopy revealed that as the mass of the substituent atom increased, the proportion of the delayed lifetime (τd) significantly increased, while both decay lifetimes shortened. This is consistent with the characteristics of heavy-atom-accelerated ISC (Fig. 3c).
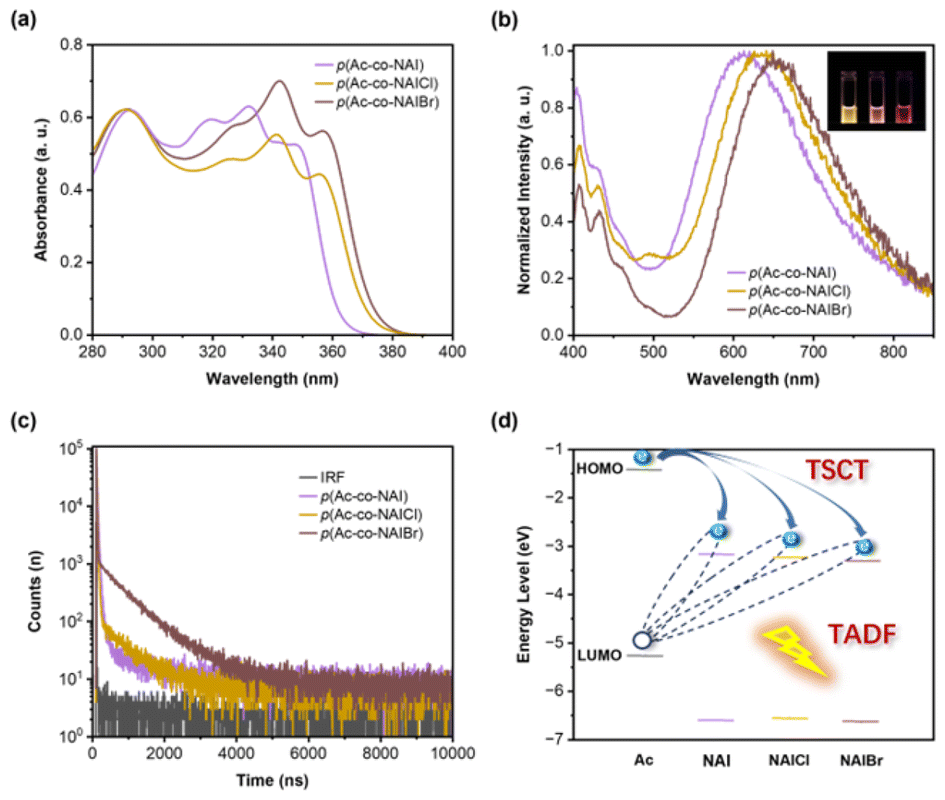 | ||
| Fig. 3 (a) UV-Vis absorption spectra of TSCT-TADF polymers with different acceptor substitutions; (b) normalized PL spectra of TSCT-TADF polymers with different acceptor substitutions (insert: photograph under UV light showing emission shift with different acceptor substitution); (c) PL lifetime decay profiles of TSCT-TADF polymers with different acceptor substitutions; (d) illustration of energy transfer process involved in TSCT. | ||
Cyclic voltammetry provided insight into the energy levels of electrons in these TSCT-TADF systems (Fig. S5–S7†). As shown in Fig. 3d and Table S2,† the LUMO level decreases from −3.17 eV to −3.30 eV as the electron-accepting ability enhances in the order of NAI, NAICl, and NAIBr, which accounts for the observed red-shift in the TADF emission. While the substitution of the acceptor promotes the TSCT process to some extent, the LE emission remains unignorable. Additionally, the disturbance to the energy levels caused by these modifications leads to unavoidable emission shifts and quenching, which hinder the practical applications of TSCT-TADF materials. Thus, it can be concluded that intramolecular modification is not the most ideal approach for optimizing the colour purity of TSCT-TADF materials.
As an alternative approach to enhancing donor–acceptor (D–A) interactions, the photophysical properties of p(Ac-co-NAI) were investigated in its solid-state form. This approach promotes closer packing of the donor and acceptor units without perturbing the energy levels. In comparison to its behaviour in solution, the as-prepared film exhibited a pure blue-shifted TADF emission at 578 nm (Fig. 4a), along with an improved PLQY of 4.39%. In the film state, the prompt lifetime of the orange emission, representing the fluorescence constituent, significantly increased (62.4 ns vs. 24.0 ns), in line with the suppression of non-radiative transitions in a rigid environment. Interestingly, the delayed lifetime slightly decreased (1291.3 ns vs. 1312.2 ns), suggesting that molecular aggregation might play a role in regulating the TSCT-TADF properties (Fig. S8†). Further exploration using concentration-dependent PL spectra confirmed the existence of aggregation-induced emission (AIE) and suggested that this AIE effect likely originates from inter-chain interactions (Fig. S9†). These insights highlight the possibility of using supramolecular chemistry, which focuses on the fabrication of ordered structures via noncovalent interactions, to regulate the emission properties of TSCT TADF materials. By leveraging the unique characteristics of supramolecular assemblies, it might be possible to fine-tune the spatial arrangement of donor and acceptor units, thereby optimizing the photophysical properties without the drawbacks associated with intramolecular modifications.
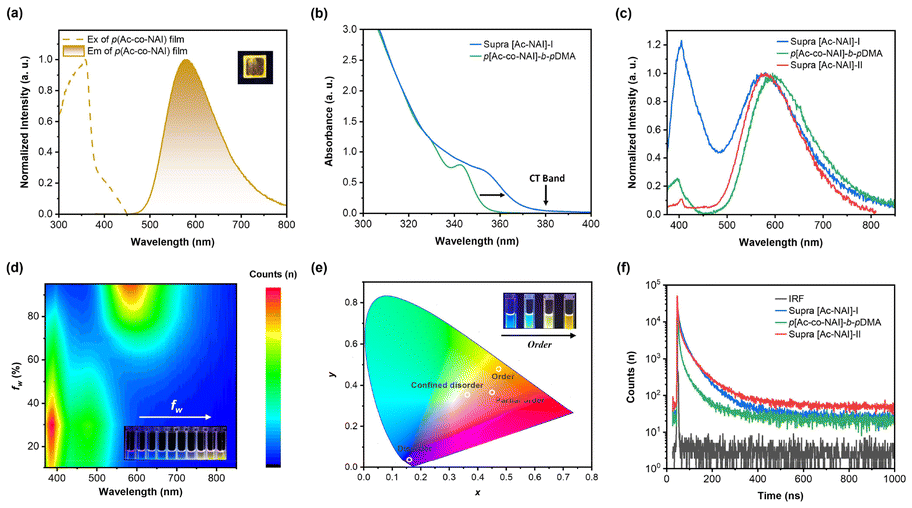 | ||
| Fig. 4 (a) PL excitation and emission spectra of p(Ac-co-NAI) film (insert: photograph of p(Ac-co-NAI) film under a UV lamp); (b) UV-Vis absorption spectra of TSCT-TADF polymers with/without cyclic peptide-based supramolecular scaffold; (c) normalized PL spectra of TSCT-TADF systems with different D/A arrangement; (d) water fraction-dependent PL spectra of supra [Ac-NAI]-II in THF/water mixed solvents (insert: photograph of supra [Ac-NAI]-II in THF/water mixed solvents with increasing fw under a UV lamp); (e) CIE 1931 diagram of TSCT-TADF species with different D/A arrangement (insert: photograph of different TSCT-TADF systems under a UV lamp); (f) PL lifetime decay profiles of TSCT-TADF systems with different D/A arrangement. | ||
To test our hypothesis, two types of assemblies were initially prepared in aqueous solution. The first one was constructed by covalently attaching a hydrophilic polymer chain (poly(N,N-dimethylacrylamide), pDMA) to the D–A copolymer to form an amphiphilic copolymer, p[Ac5-co-NAI5]-b-pDMA60, which subsequently self-assembled into a micellar structure. The second one utilized cyclic peptide-based supramolecular scaffold to co-assemble TSCT donor/acceptor pairs (CP-pAc10-b-pDMA60@CP-NAI, marked as supra [Ac-NAI]-I) (Fig. S10 and Table S3†). When comparing the two, it was found that the cut-off edge of the UV-Vis absorption spectrum for the latter one exhibited a significant red shift at the same concentration. A distinct charge transfer absorption band was observed beyond 380 nm, which did not belong to the absorption of any individual donor or acceptor component (Fig. 4b). This demonstrates that efficient donor–acceptor interactions can be induced within the supramolecular scaffold. However, contrary to expectations, this configuration did not enhance colour purity. In supra [Ac-NAI]-I, the LE emission was even stronger than the TADF emission (Fig. 4c), indicating that the donor–acceptor interactions alone might not be sufficient to optimize emission performance.
To further investigate the role of spatial arrangements in TSCT-based TADF materials, a second supramolecular system, termed supra [Ac-NAI]-II, was designed. This system featured a more ordered donor–acceptor configuration, achieved by co-assembling a cyclic peptide-based donor conjugate (CP-pAc10-b-pDMA60) with a donor–acceptor conjugate (CP-p[Ac5-co-NAI5]-b-pDMA60) (Fig. S10 and Table S3†). As expected, the newly synthesized supramolecular polymer exhibited nearly quenched LE emission and pure TADF emission compared to the previous two, likely due to more efficient intermolecular CT resulting from its more ordered structure (Fig. 4c). Photophysical analysis of supra [Ac-NAI]-II in THF/water mixed solvent systems further confirmed the presence of interchain CT. At lower water fraction (fw), interchain CT is ineffective, resulting in only LE emission. As fw increases, LE emission slightly intensifies due to the growing concentration of the acceptor in the hydrophobic microenvironment. Once fw reaches 40%, interchain CT is activated, enabling TADF emission. With further increases in fw, TADF emission continues to enhance. At very high water fractions (>90%), strong interchain CT interactions completely suppress LE emission, resulting in pure orange emission (Fig. 4d). Additionally, the normalized emission spectra of supra [Ac-NAI]-II in water remained consistent across a wide concentration range, owing to the strong binding affinity of the cyclic peptides (Fig. S11†), further highlighting the advantage of the supramolecular strategy.
By comparing the as-prepared different TSCT systems, it can be concluded that the spatial ordering of the donor/acceptor plays a crucial role in modulating the emission properties. As illustrated in Fig. 1, in a completely disordered state, when donor and acceptor monomers are mixed in an organic solvent (Ac-mix-NAI), TSCT could not be observed. In supra [Ac-NAI]-I, donors and acceptors are randomly distributed but confined within a hydrophobic microenvironment framed by the cyclic peptide-based supramolecular scaffold. While this confinement allows some TSCT to occur, the process is incomplete, resulting in a pronounced LE band. In TSCT copolymers formed through covalent bonding, the donors and acceptors are partially ordered within individual polymer chains. However, the random entanglement between chains leads to a disorder state in the condensed phase, which still results in significant LE. The supra [Ac-NAI]-II system, with its bottle-brushed structure, reduces chain entanglement, and facilitates a more ordered arrangement of donor–acceptor pairs. This more organized structure enhances interchain CT, effectively suppressing LE emission and therefore leading to pure colour emission (Fig. 4e). The results highlight the critical role that donor–acceptor spatial arrangement plays in optimizing TSCT-TADF emission properties.
Time-resolved fluorescence spectroscopy provided insight into the kinetics underlying these observations (Fig. 4f). As the degree of D/A ordering increased, the radiative transition rate (kr) in the TSCT-TADF assemblies showed significant acceleration. Specifically, kr increased from 1.62 × 105 s−1 in a confined disordered configuration, to 1.97 × 105 s−1 in a partially ordered configuration, and finally to 2.70 × 105 s−1 in the fully ordered configuration (Table S4†). A more pronounced effect was observed for the reverse intersystem crossing rate (krisc). In the highly ordered supra [Ac-NAI]-II, krisc reached 1.40 × 107 s−1, which is 11 times faster than that of supra [Ac-NAI]-I (confined disordered configuration, 1.28 × 106 s−1). This marked increase in krisc confirms the enhancement of CT interactions with greater D/A ordering. In contrast, the krisc values in other TSCT-TADF systems are much lower than the kr of NAI (3.79 × 107 s−1, Fig. S12†), resulting in partial exciton recombination through localized exited states and an incomplete TADF process (Table S4†). These findings highlight that achieving a more ordered D/A arrangement is critical for enhancing the efficiency and completeness of the TADF process, effectively eliminating LE and enabling pure colour emission. By precisely manipulating molecular aggregation and packing through this supramolecular strategy, significant improvements in emission properties can be achieved via enhanced interchain CT interactions.
Given the involvement of triplet states in the TADF emission process, we hypothesized that the unsatisfactory PLQY of the AC-NAI-based TSCT-TADF system (below 5%) could be attributed to competing energy transfer pathways, such as the generation of singlet oxygen (1O2). To test this proposition, we evaluated the 1O2 generation capabilities of supra [Ac-NAI]-II and its constituent building units using 9,10-anthracenediyl-bis(methylene) dimalonic acid (ABDA) as a probe. As expected, under continuous irradiation with a 350 nm light, the absorption of ABDA continuously decreased, indicating the generation of 1O2 (Fig. 5a). In contrast, the control group showed negligible decrease upon photoirradiation (Fig. S13†), confirming that 1O2 generation was specific to the tested system. Notably, the 1O2 generation rate of supra [Ac-NAI]-II was significantly accelerated, being 6.4 times faster than that of the donor polymer segment (pAc10-b-pDMA60), and 13.3 times faster than the DA-copolymer segment (p[Ac5-co-NAI5]-b-pDMA60) (Fig. 5b). These results indicate that the self-assembly process in supra [Ac-NAI]-II significantly increases photosensitization efficiency. Given it compatibility with aqueous environment, large Stokes shift, and capability to generate 1O2, supra [Ac-NAI]-II emerges as a promising multifunctional platform for photodynamic therapy (PDT), especially for targeted cellular treatments.36,37
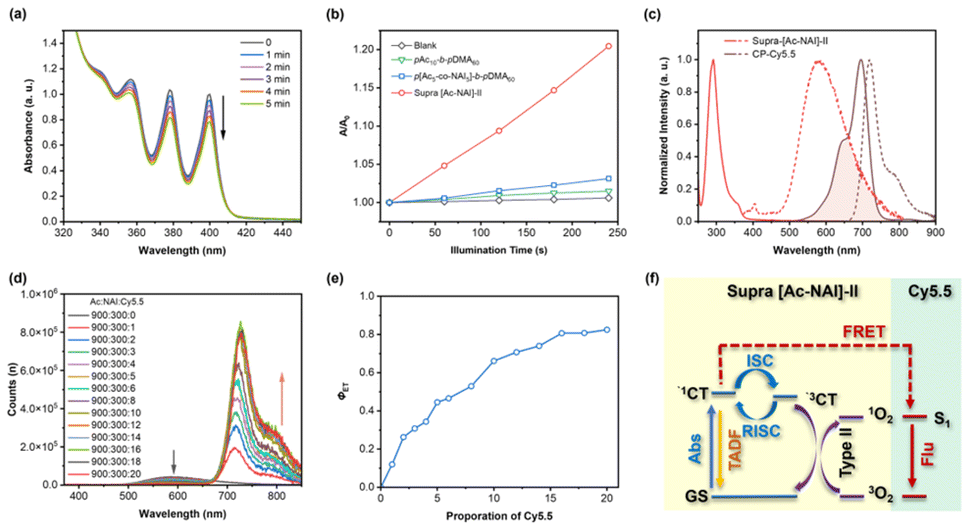 | ||
| Fig. 5 (a) UV-vis absorption spectra of supra [Ac-NAI]-II/ABDA mixture under continuous irradiation (350 nm); (b) 1O2 generation efficiencies of supra [Ac-NAI]-II and its building units monitored by recording the absorbance of ABDA at 400 nm; (c) normalized UV-vis absorption spectra (solid curves) and fluorescence spectra (dash curves) of supra [Ac-NAI]-II and CP-Cy5.5; (d) fluorescence spectra of supra [Ac-NAI]-II co-assembled with different molar ratios of CP-Cy5.5; (e) energy transfer efficiency (ΦET) at different CP-Cy5.5 molar ratios; (f) scheme of multi-channel photo-functional system based on supra [Ac-NAI]-II. | ||
Supramolecular systems based on cyclic peptide-based scaffolds have demonstrated significant efficiency in energy transfer processes.38–40 Inspired by our previous studies, we selected the fluorescent dye Cy5.5, whose absorption spectrum closely matches the emission spectrum of supra [Ac-NAI]-II, as the energy acceptor to construct a thermally assisted fluorescence (TAF) system (Fig. 5c). As excepted, an effective energy transfer process was observed when co-assembling CP-Cy5.5 with supra [Ac-NAI]-II. As the molar ratio of CP-Cy5.5 increased, the TADF emission at 582 nm progressively attenuated, while a concomitant enhancement of a near-infrared (NIR) emission of Cy5.5 at 720 nm was observed, under an excitation of 350 nm (Fig. 5d). Time-resolved fluorescence spectroscopy provided further compelling evidence of Förster resonance energy transfer (FRET). In the as-prepared TAF system, the lifetime of TADF emission was shortened, accompanied by a significant increase in the NIR emission lifetime compared to directly excited Cy5.5 (Fig. S14†). To quantitatively assess the performance of the TAF system, we calculated the energy transfer efficiency (ΦET) at different molar ratios of supra [Ac-NAI]-II and CP-Cy5.5. The ΦET increased with the increase of CP-Cy5.5 molar ratio, reaching a maximum of 82.5% at an Ac/NAI/Cy5.5 molar ratio of 900/300/20 (Fig. 5e). Compared to supra [Ac-NAI]-II, the PLQY of the TAF system at maximum ΦET increased by approximately 4.6 times (5.66% vs. 1.24%), and the full width at half maximum (FWHM) of the emission spectrum narrowed by 91 nm (47 nm vs. 138 nm). The supramolecular TAF system based on TSCT-TADF provides a feasible approach to integrate multiple photo-functions within a single system via electron transition or energy transfer mechanisms, thereby expanding its potential applications (Fig. 5f).
Conclusions
A novel D–A pair was developed to construct TSCT-TADF materials. Due to non-exclusive charge transfer, an extra LE emission was observed alongside the TADF band. To address this, self-assembling cyclic peptide-based supramolecular scaffolds were introduced to promote an ordered D/A arrangement. As the degree of order increased, the LE emission diminished, ultimately yielding a pure orange emission. Photophysical studies revealed that this pure colour emission resulted from efficient inter-chain CT, evidenced by an ultrafast RISC rate (1.40 × 107 s−1) in the highly ordered configuration. Supramolecular polymers with pure orange emission were demonstrated to exhibit multi-channel energy transfer, offering a versatile platform for integrating various photo-functions within a single system. Crucially, this approach provides a facile and universal solution to avoid incomplete charge transfer-cased localized emission in TSCT-based TADF materials, facilitating the realization of pure colour emission. This advancement is expected to accelerate the development and application of TSCT-TADF materials in areas such as display, sensing, bioimaging, and beyond.Author contributions
Methodology and original draft writing were performed by Yufeng Sang; investigation was conducted by Yufeng Sang and Ruicong Feng; formal analysis was done by Yuqian Wang; review & editing, funding acquisition and supervision were performed by Qiao Song. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (22471118), Shenzhen Science and Technology Innovation Program (20220815163454004), and Guangdong Basis and Applied Basic Research Foundation (2022A1515011394). We also thank the China Spallation Neutron Source for allocating beam time (P0124052700001).References
- X.-F. Song, C. Jiang, N. Li, J. Miao, K. Li and C. Yang, Chem. Sci., 2023, 14, 12246–12254 RSC.
- W.-C. Chen, M.-H. Zheng, Y.-L. Wu, R.-J. Wang, J.-M. Jin, S.-W. Chen, B. Liu, J.-X. Chen, Y. Huo and S. Ji, Chem. Eng. J., 2024, 480, 148314 CrossRef CAS.
- J. Hu, Q. Li, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2019, 58, 8405–8409 CrossRef CAS.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CAS.
- S. Shao and L. Wang, Aggregate, 2020, 1, 45–56 Search PubMed.
- G. Zhang, F. Chen, Y. Di, S. Yuan, Y. Zhang, X. Quan, Y. Chen, H. Chen and M. Lin, Adv. Funct. Mater., 2024, 34, 2404123 CAS.
- Y. Tan, A. Ying, Y. Liu, X. Cai, L. Zhan, Z. Bin, J. You, C. Li and S. Gong, Chem. Eng. J., 2024, 487, 150618 CAS.
- B. Zhang, J. Xu, C.-T. Li, H.-L. Huang, M.-X. Chen, M.-H. Yu, Z. Chang and X.-H. Bu, Angew. Chem., Int. Ed., 2023, 62, e202303262 CAS.
- Q. Xue and G. Xie, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- J. Li, P. Shen, Z. Zhao and B. Tang, CCS Chem., 2019, 1, 181–196 CrossRef CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CAS.
- Q. Li, J. Hu, J. Lv, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 20174–20182 CAS.
- W. Luo, T. Wang, Z. Huang, H. Huang, N. Li and C. Yang, Adv. Funct. Mater., 2024, 34, 2310042 CAS.
- Y. Bao, T. Wang, Q. Li, F. Du, R. Bai, M. Smet and W. Dehaen, Polym. Chem., 2014, 5, 792–798 CAS.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CAS.
- S. Matoori, Y. Bao, A. Schmidt, E. J. Fischer, R. Ochoa-Sanchez, M. Tremblay, M. M. Oliveira, C. F. Rose and J. C. Leroux, Small, 2019, 15, 1902347 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- M. Yu, Y. Gao, A. Ying, L. Li, G. Xie, S. Gong, X. Gao, T. Wang and C. Yang, Macromolecules, 2023, 56, 5381–5389 CrossRef CAS.
- R. Hojo, B. T. Luppi, K. Bergmann and Z. M. Hudson, Polym. Chem., 2023, 14, 2742–2749 RSC.
- Y. Xin, Y. Zhu, R. Chi, C. Duan, P. Yan, C. Han and H. Xu, Adv. Mater., 2023, 35, 2304103 CrossRef CAS PubMed.
- S. Shao, J. Hu, X. Wang, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2017, 139, 17739–17742 CrossRef CAS PubMed.
- S. Ye and Y. Bao, Chem. Mater., 2024, 36, 5878–5896 CrossRef CAS.
- B. Qin, Z. Yin, X. Tang, S. Zhang, Y. Wu, J.-F. Xu and X. Zhang, Prog. Polym. Sci., 2020, 100, 101167 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CAS.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed.
- H.-Q. Peng, W. Zhu, W.-J. Guo, Q. Li, S. Ma, C. Bucher, B. Liu, X. Ji, F. Huang and J. L. Sessler, Prog. Polym. Sci., 2023, 137, 101635 CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. Mcree and N. Khazanovich, Nature, 1994, 372, 709–709 CrossRef CAS.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS.
- B. R. Sveinbjörnsson, R. A. Weitekamp, G. M. Miyake, Y. Xia, H. A. Atwater and R. H. Grubbs, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14332–14336 CrossRef.
- Y. Lei, Y. Wang, S. K. Hill, Z. Cheng, Q. Song and S. Perrier, Adv. Mater., 2024, 36, 2401346 CrossRef CAS.
- H. Lu, Y. Wang, S. K. Hill, H. Jiang, Y. Ke, S. Huang, D. Zheng, S. Perrier and Q. Song, Angew. Chem., Int. Ed., 2023, 62, e202311224 CrossRef CAS PubMed.
- Y. Wang, R. Feng, Y. Yu and Q. Song, CCS Chem., 2024, 6, 1165–1173 Search PubMed.
- W. Zeng, H. Lai, W. Lee, M. Jiao, Y. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K.-T. Wong, C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 Search PubMed.
- W. Zeng, T. Zhou, W. Ning, C. Zhong, J. He, S. Gong, G. Xie and C. Yang, Adv. Mater., 2019, 31, 1901404 CrossRef PubMed.
- R. Lv, P. Yang, F. He, S. Gai, C. Li, Y. Dai, G. Yang and J. Lin, ACS Nano, 2015, 9, 1630–1647 CAS.
- R. Lv, C. Zhong, R. Li, P. Yang, F. He, S. Gai, Z. Hou, G. Yang and J. Lin, Chem. Mater., 2015, 27, 1751–1763 CAS.
- Y. Wu, Y. Wang, X. Yu and Q. Song, Adv. Sci., 2024, 11, 2404269 CAS.
- Q. Song, J. Zhang, X. Yu, Z. Cheng, J. Yang, S. C. L. Hall and S. Perrier, Polym. Chem., 2022, 13, 4366–4371 CAS.
- Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2021, 143, 382–389 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01181c |
| This journal is © The Royal Society of Chemistry 2025 |