
Optically active helical polymers bearing cinchona alkaloid pendants: an efficient chiral organocatalyst for asymmetric Henry reaction†
Xing-Yu
Zhou
a,
Wen-Gang
Huang
a,
Xue-Cheng
Sun
a,
Hui
Zou
 a,
Li
Zhou
*a and
Zong-Quan
Wu
*b
a,
Li
Zhou
*a and
Zong-Quan
Wu
*b
aDepartment of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, and Anhui Province Key Laboratory of Value-Added Catalytic Conversion and Reaction Engineering, Hefei University of Technology, Hefei 230009, Anhui Province, China. E-mail: lizhou@hfut.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, Jilin 130012, China. E-mail: zqwu@jlu.edu.cn
First published on 19th March 2025
Abstract
Inspired by the highly efficient and enantioselective reactions catalyzed by biomacromolecules, the development of artificial helical polymer-supported catalysts is an attractive and meaningful field. In this work, a series of helical polymers, poly-1ns, with controlled molecular mass (Mns) and narrow molecular mass distribution (Mw/Mns) bearing cinchona alkaloid pendants were obtained by asymmetric polymerization of the corresponding monomer. The poly-1ns exhibited an intense positive Cotton effect at 364 nm, indicating that a preferred right-handed helix was formed in their backbone. Due to the catalytic groups on the pendants and helix in the backbone, the poly-1ns exhibited satisfactory catalytic efficiency in the asymmetric Henry reaction. Compared to small molecule (1) with a similar structure, the enantioselectivity of the Henry reaction was significantly enhanced using poly-1n as catalyst. The enantiomeric excess (ee) value of the Henry reaction could be up to 75%. Furthermore, the helical polyisocyanide catalyst could be recovered and reused facilely for at least five cycles without apparent significant loss of its enantioselectivity.
Introduction
Chiral biomacromolecules such as DNA, enzymes and proteins play extremely important roles in living organisms due to their unique helical structures.1–3 Inspired by their unique structures and functions, many chemists have devoted efforts to the field of design and synthesis of artificial helical polymers.4–8 Therefore, various applications of helical polymers in different fields including chiral separation, chiral recognition, asymmetric catalysis, circularly polarized luminescence (CPL), etc. have been demonstrated in the past few decades.9–14 Introducing organic catalytic groups on the side chains of chiral helical polymers is an important strategy for obtaining a new generation of chiral catalysts.15–17 There are obvious advantages of these polymer catalysts. For example, it is convenient to separate and recover these catalysts due to their high molecular mass.18 Importantly, unique advantages are exhibited when organocatalysts are immobilizing onto helical polymers compared to polymer supports like PS and PMMA. The helical sense of the polymer backbone can not only serve as a scaffold but also provide an additional asymmetric environment, which may amplify the stereoselectivity of asymmetric reactions because of the synergistic effect between the catalytic groups and helix backbone.19–22Because of the chirality transfer and amplification of helical polymers, a wide range of chiral catalysts with diverse structures and functions can be constructed.23–25
Asymmetric catalysis, as one of the most active research areas in chemistry, has attracted ever increasing interest since it provides an inexpensive and convenient approach to yield enantiomerically pure compounds.26–28 Over the past few decades, organocatalysts have emerged as an important branch in the field of asymmetric catalysis due to their mild reaction conditions and efficiency.29–32 Many chemists are dedicated to investigating the applications of organocatalysts such as proline, thiourea, cinchona alkaloids, and organophosphorus compounds, and have achieved a series of excellent works.33–36 However, there are still some inherent shortcomings in these small molecule organocatalysts, such as large usage amount, difficulty in recovery, and possible contamination. Meanwhile, artificial helical polymer catalysts possess excellent catalytic performance, similar to enzymes, and can be recovered and reused conveniently. The works reported by the groups of Suginome, Yashima, Wu, Wan, etc. demonstrated the outstanding catalytic ability of helical polymer catalysts.37–40 Nevertheless, designing and preparing single-handed helical catalysts with specific structures is still a challenge. Polyisocyanides, as representatives of static helical polymers, have attracted the wide interest of chemical researchers.41–45 The main chain of polyisocyanides consists of carbon–carbon single bonds, and each carbon atom carries a substituent. Due to the steric effects of the pendants, the main chain adopts a rod-like structure and twists into a helical conformation.46,47 Therefore, helical polyisocyanide is one of the desirable supporting materials for organocatalysts.
The asymmetric Henry reaction, as an important C–C bond-forming reaction, plays an important role in synthetic organic chemistry and the pharmaceutical industry.48,49 However, there still remain some challenges regarding the catalyst of the Henry reaction, such as tedious synthesis and difficult recovery, which limits its application in industry.50,51 Therefore, it is urgent to develop a new generation of catalysts for the asymmetric Henry reaction. In this work, single-handed helical polyisocyanides bearing cinchona alkaloid pendants were prepared through the living polymerization of the corresponding chiral isocyanide monomers using a Pd(II)-catalyst. During the polymerization, the chirality of the monomer was transferred to the helical backbone and was greatly amplified. All of the isolated poly-1ns showed high optical activity. Interestingly, they could catalyze the asymmetric Henry reaction successfully and afford satisfactory yields and enantiomeric excess (ee) values. The ee of the product could be up to 75% under the optimal experimental conditions, which was much higher than that achieved when catalyzed by monomer 1. Furthermore, the polymer catalysts exhibited good tolerance to a broad substrate scope and could be recycled and reused at least 5 times without apparent decrease of the efficiency and enantioselectivity.
Results and discussion
First, compound 2 bearing an NH2 group was synthesized from quinine according to the literature with a slight modification.52 Its structure was confirmed by 1H NMR (Fig. S1, ESI†). Then, monomer 1 bearing cinchona alkaloid pendants was generated through the ester exchange reaction between pentafluorophenyl (PFP) ester-functionalized phenyl isocyanide (3) and 2 according to the reported works as outlined in Scheme 1 (see details in the ESI†).53 The structure of monomer 1 was characterized by 1H NMR, 13C NMR, HRMS, FT-IR and elemental analysis (Fig. S2–S5, ESI†). A Pd(II) catalyst was applied to initiate the polymerization of monomer 1 successfully. Because of the living nature of the Pd(II)-initiated polymerization of isocyanide, a series of poly-1ns (the subscript number indicates the initial feed ratio of monomer to catalyst throughout) with controlled molecular masses (Mns) and narrow molecular mass distributions (Mw/Mns) were generated in >92% yields (Table 1). The structure of poly-1150 was confirmed by 1H NMR and FT-IR (Fig. S5 and S6, ESI†). It was demonstrated that symmetric and single model elution peaks of poly-1ns could be distinguished using size exclusion chromatography (SEC) (Fig. 1a). All of the results for the polymerization are summarized in Table 1. It could be concluded that the Mns of the poly-1ns increased monotonically with the degree of polymerization (Fig. 1a and Table 1). Due to the asymmetric induction of chiral monomer 1 during the process of the polymerization, an apparent intense Cotton effect could be observed in the circular dichroism (CD) spectra of poly-1ns in the absorption region of the polymer backbone. The positive Cotton effect corresponding to the n–π transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at 364 nm in Fig. 1c indicated an excess of right-handed helical helix formation.
N at 364 nm in Fig. 1c indicated an excess of right-handed helical helix formation.
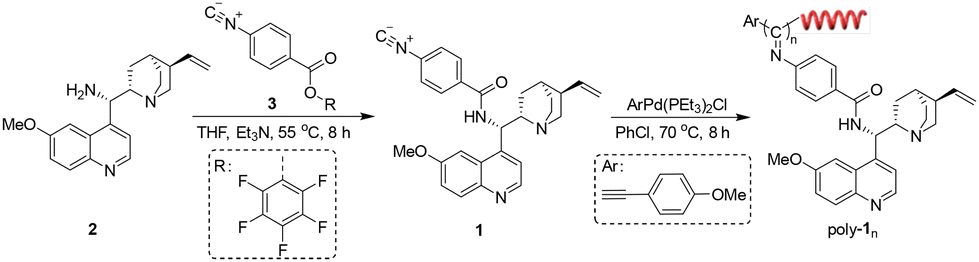 | ||
| Scheme 1 The route for the synthesis of the monomer 1 and poly-1ns. | ||
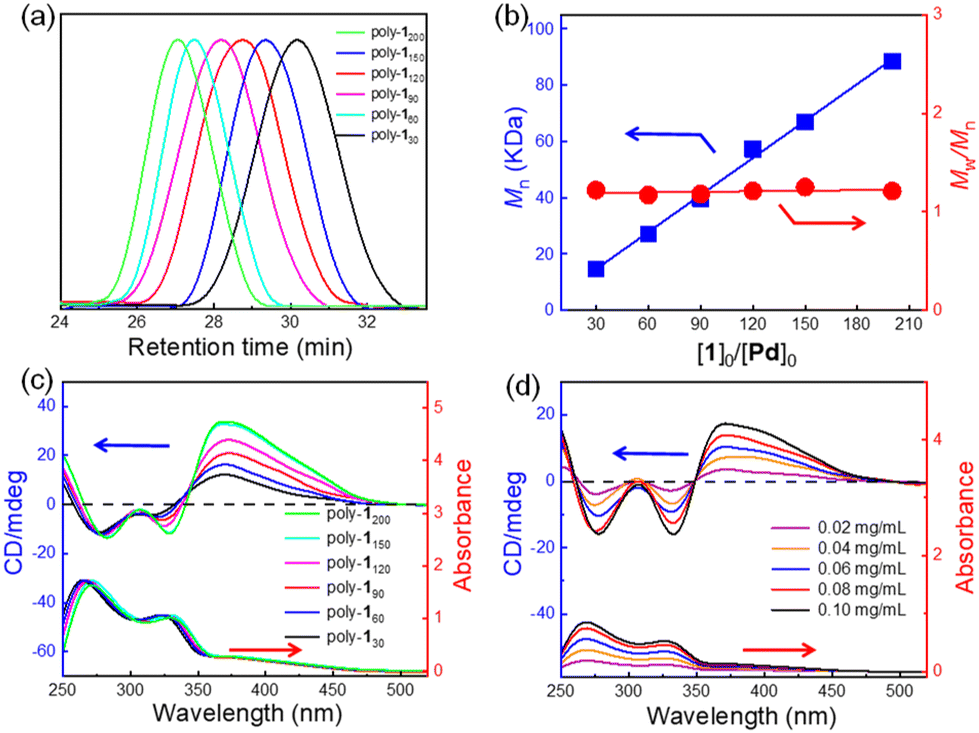 | ||
| Fig. 1 (a) SEC curves of poly-1ns initiated by a Pd(II) catalyst with various initial feed ratios of monomer 1 to Pd(II). (b) Plots of Mn and Mw/Mn values of poly-1ns as a function of the initial 1-to-Pd(II) ratio. The values of Mn and Mw/Mn were recorded by SEC with polystyrene standards using tetrahydrofuran (THF) as an eluent at 40 °C. (c) CD and UV-vis spectra of poly-1ns measured in THF at 25 °C (c = 0.2 mg mL−1). (d) CD and UV-vis spectra of poly-1150 measured in THF at 25 °C at different concentrations. | ||
| Run | Polymer |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (kDa) b (kDa) |
M
w/Mnb |
Yieldc | θ 364 (×103)d |
|---|---|---|---|---|---|
| a The polymers were synthesized according to Scheme 1. b The Mn and Mw/Mn values were determined by SEC with polystyrene standards using THF as an eluent at 40 °C. c Isolated yields. d The CD intensity at 364 nm at 25 °C of poly-1n measured in THF. | |||||
| 1 | Poly-130 | 14.6 | 1.22 | 93 | 9.4 |
| 2 | Poly-160 | 27.1 | 1.17 | 92 | 14.3 |
| 3 | Poly-190 | 39.7 | 1.18 | 94 | 18.2 |
| 4 | Poly-1120 | 57.3 | 1.21 | 92 | 20.9 |
| 5 | Poly-1150 | 66.9 | 1.22 | 95 | 22.4 |
| 6 | Poly-1200 | 88.4 | 1.21 | 92 | 22.6 |
Further studies indicated that the CD intensities of the poly-1ns were closely related to their Mns. The CD value of poly-1ns at 364 nm increased linearly with the increase of Mns until the Mns reached 66.9 kDa (Fig. 1c). It could be seen that there was a linear relationship between the molar ellipticity at 364 nm (θ364) and Mns of poly-1ns within the Mn range of 14.6 to 66.9 kDa. The θ364 had a maximum value of 22.4 when the Mn reached 66.9 kDa, and then essentially remained on a plateau (Fig. S7a, ESI†). Moreover, the diluting experiment indicated that the CD and absorption intensities of poly-1150 decreased linearly with the diluted concentration, while the θ364 was constant (Fig. S7b, ESI†). It could be concluded that the helix of the main chain was maintained during the dilution. These results further demonstrated that the optical activity of poly-1150 originated from itself rather than intermolecular self-assembly or aggregation. In addition, the helicity of the isolated poly-1150 was also investigated with different temperatures and solvents. The results revealed that the CD values of poly-1150 did not change significantly, indicating that the helical structure of the main chain could remain stable with different temperatures and solvents (Fig. S8, ESI†).
With the polymer catalyst in hand, we attempted to investigate the catalytic efficiency of poly-1ns for the asymmetric Henry reaction using p-nitrobenzaldehyde (4a) and nitromethane (5) as model substrates. As anticipated, the Henry reaction catalyzed by poly-1ns could proceed smoothly and the results are summarized in Table 2. At first, the Henry reaction was conducted tentatively in toluene at room temperature with a 20% (with respect to repeating units) loading of poly-1150. The target product 6a was isolated with 89% yield and 20% enantiomeric excess (ee) (run 1, Table 2). Then, different solvents such as CF3CH2OH, THF, CHCl3, methyl tert-butyl ether (MTBE), etc. were screened to investigate their influences on the stereoselectivity of the reaction. It could be demonstrated that the ee values were not satisfactory (run 2–5, Table 2). We found that using a mixed solution of toluene and CF3CH2OH (3.0 eq.) as the solvent resulted in a slightly higher ee value compared to using toluene or CF3CH2OH separately as the solvent (run 1, 2 and 6, Table 2). Subsequently, various types of additives such as (CF3)2CHOH, CF3SO3H, (CF3)3COH, phenol, etc. were screened carefully and the results are summarized in Table 2 and Table S1 in ESI.† Interestingly, using (CF3)2CHOH as an additive, the ee value and yield of the product were 53% and 84%, respectively, which were higher than those obtained under identical conditions except with the additive CF3CH2OH (run 6 and 7, Table 2). Subsequently, the ee value was further improved to 57% with 87% yield after replacing the additive with (CF3)3COH (run 8, Table 2). The effect of the amount of the additive (CF3)3COH was also carefully studied. Initially, the ee value increased upon increasing the equivalents of (CF3)3COH from 0.2 to 3.0. The ee value increased from 36% to 57%. Unfortunately, upon further increasing the equivalents of (CF3)3COH from 3.0 to 10.0, the ee value of the products decreased to 44% (Table S2, ESI†). The effect of the content of (CF3)3COH on the enantioselectivity may be attributed to the hydrogen bonds formed between the appropriate amount of (CF3)3COH and the intermediates. The hydrogen bonds are beneficial to improving the stability of the intermediates.54,55 Therefore, the optimal choice for the additive amount was fixed at 3.0 eq. The catalyst loading also had an impact on the yield and ee of the product. It could be demonstrated that upon decreasing the catalyst loading from 20% to 15%, the yield and ee of 6a decreased slightly to 84% and 55% (run 9, Table 2). Further reducing the amount of catalyst to 10% led to a further decrease in both yield and ee (run 10, Table 2). However, upon increasing the catalyst loading to 30%, the yield and ee of the product were 88% and 57%, respectively, which were almost the same as those with 20% loading (run 11, Table 2). The results of experiments at various temperature also indicated that lower temperatures were more favourable for improving the stereoselectivity of the reaction. When the temperature was decreased from 25 to 0 °C, the ee value for the model reaction increased from 57% to 62% (run 8 and 12, Table 2). As the temperature continued to decrease, the ee value of the product gradually increased. The ee of 6a reached its maximum value of 75% at −20 °C (run 12–14, Table 2). Further reduction in temperature did not lead to a significant increase in the ee value while the yield also decreased to some extent (run 15, Table 2). In summary, the optimized conditions for the Henry reaction were using 20 wt% catalyst loading in toluene with the additive (CF3)3COH (3.0 eq.) at −20 °C.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Run | Catalyst | Solvent | Additive | X (mol%) | Temp. (°C) | Yieldb (%) | eec (%) |
| a Unless otherwise indicated, all reactions were carried out with 4a (0.20 mmol) and 5 (2.0 mmol) in a specific solvent (0.5 mL). b Isolated yield. c The ee values were determined by HPLC analysis using a chiral stationary phase. | |||||||
| 1 | Poly-1150 | Toluene | — | 20 | 25 | 89 | 20 |
| 2 | Poly-1150 | CF3CH2OH | — | 20 | 25 | 80 | 14 |
| 3 | Poly-1150 | THF | — | 20 | 25 | 84 | 17 |
| 4 | Poly-1150 | CHCl3 | — | 20 | 25 | 87 | 18 |
| 5 | Poly-1150 | MTBE | — | 20 | 25 | 75 | 16 |
| 6 | Poly-1150 | Toluene | CF3CH2OH | 20 | 25 | 87 | 40 |
| 7 | Poly-1150 | Toluene | (CF3)2CHOH | 20 | 25 | 84 | 53 |
| 8 | Poly-1150 | Toluene | (CF3)3COH | 20 | 25 | 87 | 57 |
| 9 | Poly-1150 | Toluene | (CF3)3COH | 15 | 25 | 84 | 55 |
| 10 | Poly-1150 | Toluene | (CF3)3COH | 10 | 25 | 80 | 53 |
| 11 | Poly-1150 | Toluene | (CF3)3COH | 30 | 25 | 88 | 57 |
| 12 | Poly-1150 | Toluene | (CF3)3COH | 20 | 0 | 84 | 62 |
| 13 | Poly-1150 | Toluene | (CF3)3COH | 20 | −10 | 82 | 69 |
| 14 | Poly-1150 | Toluene | (CF3)3COH | 20 | −20 | 80 | 75 |
| 15 | Poly-1150 | Toluene | (CF3)3COH | 20 | −25 | 75 | 76 |
| 16 | Poly-130 | Toluene | (CF3)3COH | 20 | −20 | 81 | 45 |
| 17 | Poly-160 | Toluene | (CF3)3COH | 20 | −20 | 79 | 47 |
| 18 | Poly-190 | Toluene | (CF3)3COH | 20 | −20 | 80 | 60 |
| 19 | Poly-1120 | Toluene | (CF3)3COH | 20 | −20 | 78 | 64 |
| 20 | Poly-1200 | Toluene | (CF3)3COH | 20 | −20 | 81 | 75 |
| 21 | Monomer 1 | Toluene | (CF3)3COH | 20 | −20 | 84 | 40 |

Then, poly-1ns were applied to the catalysis of the model Henry reaction. As summarized in Table 2, it could be found that the higher the Mn of the poly-1ns, the higher the ee of 6a that could be achieved. For example, the ee of 6a was up to 45% using poly-130, while it was increased to 47%, 60%, 64%, and 75% when poly-160, poly-190, poly-1120 and poly-1150 were used as catalysts, respectively (run 14 and 16–19, Table 2). A further increase in the Mn of the catalyst did not result in the ee of the product showing a significant further increase. The ee and yield of the product catalyzed by poly-1200 were almost identical to those of poly-1150 (run 20, Table 2). Meanwhile, the same reaction was also conducted using monomer 1 under identical experimental conditions and the ee of the product was 40%, which was lower than those of poly-1ns (run 21, Table 2). These results were consistent with the Mn-dependent helix of the poly-1ns backbone (Fig. 1c), and it was further confirmed that the improved enantioselectivity came from the synergistic interaction between the catalytic groups and helix of the polymer catalysts.
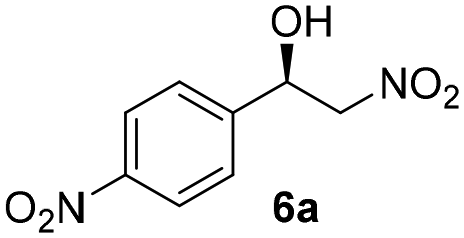
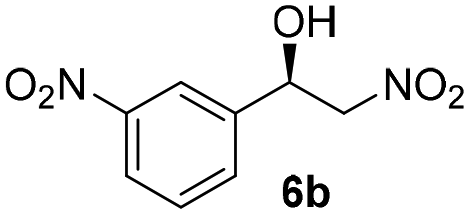
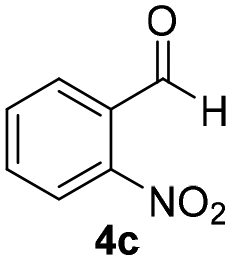
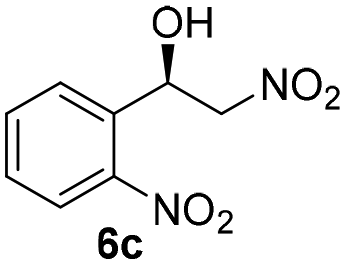
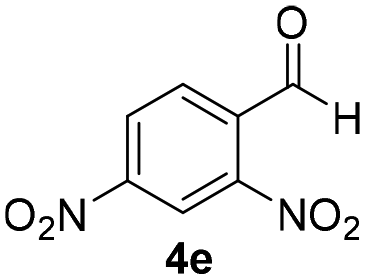
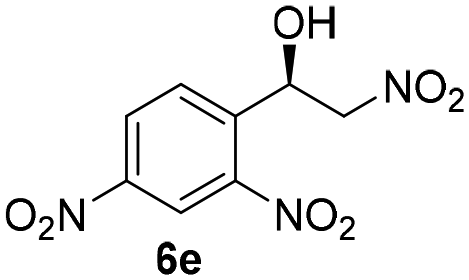
With these results in hand, the substrate scope of the Henry reaction catalyzed by poly-1150 in toluene with (CF3)3COH as an additive at −20 °C was examined in detail. It was demonstrated that the position of the substituents exhibited apparent influence on the ee of the product. A nitro group substituted at the para-position gave acceptable results with 75% ee and 80% yield (run 1, Table 3). Unfortunately, the meta- and ortho-substituted nitrobenzaldehyde gave a relatively low ee of the product (run 2 and 3, Table 3). We also concluded that the high steric hindrance of substituents was not conducive to the improvement of stereoselectivity, and the ee value of the product was slightly reduced (run 1–3, Table 3). The para-substituted benzaldehydes with a cyano group could also afford acceptable ee of the product. The ee and yield of 6d could be up to 65% and 78% (run 4, Table 3). The substrate 4e with two nitro groups at the meta- and para-position afforded 6e in 85% yield and 50% ee (run 5, Table 3). The benzaldehydes bearing para-substituted nitro groups with halogen atoms such as F, Cl and Br substituted in the meta-position could successfully undergo the Henry reaction. The ee values of 6f, 6g and 6h were 69%, 57% and 45%, respectively (run 6–8, Table 3). The Henry reaction could also proceed with para-nitrobenzaldehyde with electron-donating groups. The isolated products generated from benzaldehyde bearing CH3 and OCH3 had 57% and 47% ee, respectively (run 9 and 10, Table 3). It could be concluded that electron-donating groups such as CH3 or OCH3 on substrate 4 were not beneficial to the ee value of the product, and the stronger the electron-donating ability of the substituent, the lower the ee value of the product that was obtained (run 9 and 10). An aromatic aldehyde containing heterocycles, 2-pyridinecarboxaldehyde (4k), was also used as a substrate for the Henry reaction, affording 76% yield and 50% ee (run 11, Table 3).
a
|
|
|||||
|---|---|---|---|---|---|
| Run | 4 | 6 | Time (d) | Yieldb (%) | eec (%) |
| a Unless otherwise indicated, all reactions were carried out with aldehyde (4) (0.20 mmol) and nitromethane (5) (2.0 mmol) in toluene (0.5 mL) using (CF3)3COH (3.0 eq.) as an additive at −20 °C. b Isolated yield. c The ee values were determined by HPLC analysis using a chiral stationary phase. | |||||
| 1 |

|
|
2 | 80 | 75 |
| 2 |

|
|
2 | 82 | 40 |
| 3 |
|
|
2 | 80 | 37 |
| 4 |

|

|
3 | 78 | 65 |
| 5 |
|
|
2 | 85 | 50 |
| 6 |
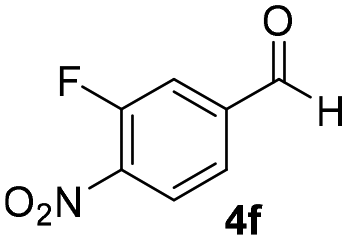
|
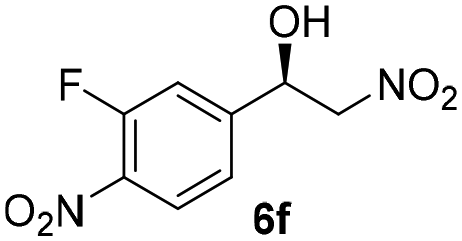
|
3 | 79 | 69 |
| 7 |
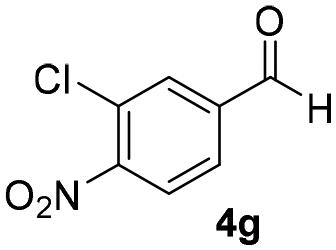
|

|
3 | 83 | 57 |
| 8 |

|

|
3 | 82 | 45 |
| 9 |
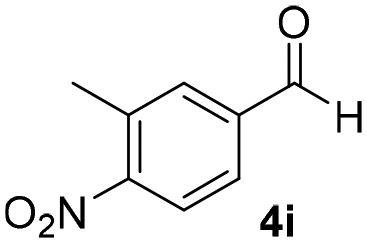
|
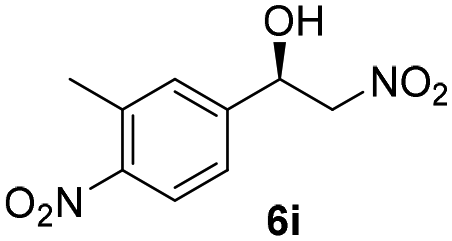
|
3 | 85 | 57 |
| 10 |

|
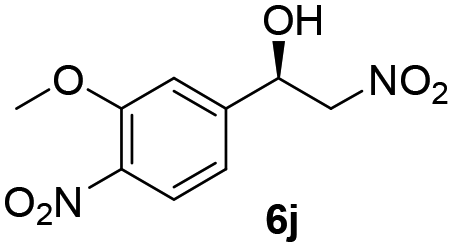
|
3 | 77 | 47 |
| 11 |
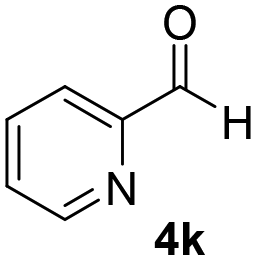
|
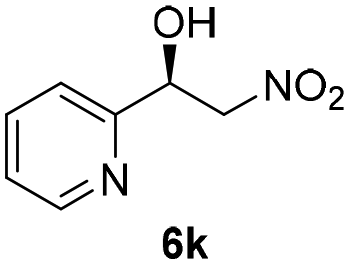
|
3 | 76 | 50 |

Finally, we attempted to investigate the recovery and reuse of the polymer catalyst bearing cinchona alkaloid for the Henry reaction using p-nitrobenzaldehyde (4a) and CH3NO2 (5). After almost completing conversion of the substrates, a poor solvent, Et2O, for poly-1150 was added to the reaction mixture. Then, the poly-1150 could be precipitated and recovered by centrifugation in almost quantitative yield because of its high Mn. The CD spectra showed that the CD pattern of the recovered poly-1150 was the same as that of the fresh one, indicating that the helicity of the recovered poly-1150 backbone remained (Fig. 2a). As expected, the recovered poly-1150 exhibited excellent catalytic activity. The ee and yield of the isolated product using the fourth cycle recovered poly-1150 were 74% and 81%, respectively. After the catalyst was recycled and used four times, there was a slight decrease in the yield of the product. However, through extending the reaction time, a satisfactory yield could be achieved while maintaining the ee of the product with no significant decrease. Thus, it could be concluded that the helical polyisocyanide organocatalyst could be reused at least five times in the Henry reaction without significant loss of its enantioselectivity and efficiency.
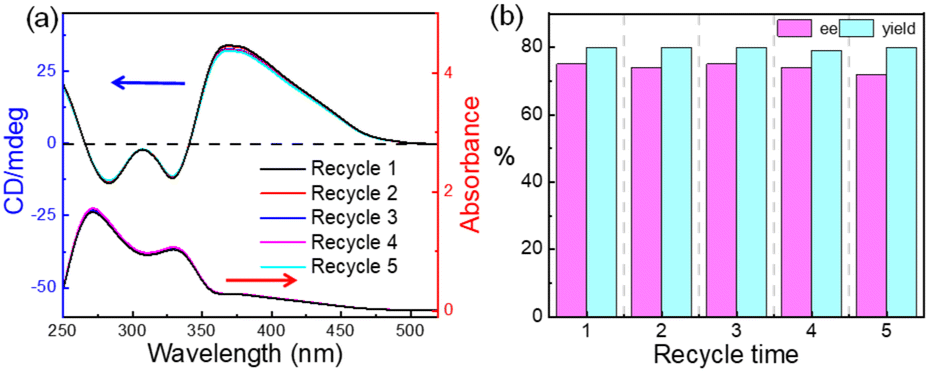 | ||
| Fig. 2 (a) The CD and UV-vis spectra of the recycled poly-1150 measured in THF at 20 °C (c = 0.2 mg mL−1). (b) Yield and ee values of the reaction between 4a (0.20 mmol) and 5 (2.0 mmol) in toluene (0.5 mL) with (CF3)3COH (3.0 eq.) at −20 °C catalyzed by recycled poly-1150 (with 20% loading). | ||
Conclusions
In summary, a family of polyisocyanides bearing cinchona alkaloid pendants was designed and synthesized successfully. The isolated poly-1ns exhibited high optical activity because of the chiral transformation and amplification of the chiral monomer during the polymerization. The asymmetric Henry reaction could be catalyzed successfully by poly-1ns and satisfactory yields and ee values could be achieved. Under the optimal experimental conditions, the ee values of 6a catalyzed by poly-1150 and monomer 1 were 75% and 40%, respectively, indicating that the helix of the main chain of poly-1150 improved the enantioselectivity of the Henry reaction. Meanwhile, the poly-1150 catalyst demonstrated good tolerance to a broad substrate scope and could be reused five times with high efficiency and enantioselectivity. We believe that this study not only develops a novel polymer organocatalyst for the asymmetric Henry reaction, but also provides a direction for constructing novel polymer-supported chiral organocatalysts in the future.Author contributions
X.-Y. Zhou performed the synthesis of polymers, characterized the structures of the polymers and investigated the Henry reaction. W.-G. Huang and X.-C. Sun took part in the reaction development. L. Zhou and Z.-Q. Wu prepared the manuscript, guided and supervised the project. H. Zou reviewed and edited the manuscript. All of the authors discussed the experimental results.Data availability
All experimental procedures, characterisation data, SEC spectra, CD and UV-vis spectra, NMR spectra, and HPLC spectra can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China for financial support (NSFC, No. 52273204, 92256201, 52273006, 22071041, 51903072, 21971052 and 21871073). L. Zhou and H. Zou also thank Anhui Provincial Natural Science Foundation (Grant No. 2008085MB51 and 2308085MB53). L. Zhou thanks the Fundamental Research Funds for the Central Universities of China (Grant No. PA2021GDSK0064).References
- D. P. Glavin, A. S. Burton, J. E. Elsila, J. C. Aponte and J. P. Dworkin, Chem. Rev., 2020, 120, 4660 CAS.
- M. Wang, Y. Zhao, L. Zhang, J. Deng, K. Qi, P. Zhou, X. Ma, D. Wang, Z. Li, J. Wang, J. Yang, J. R. Lu, J. Zhang and H. Xu, ACS Nano, 2014, 15, 10328 Search PubMed.
- X. Zhao, S.-Q. Zang and X. Chen, Chem. Soc. Rev., 2020, 49, 2481 CAS.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CAS.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed.
- H. Dai, R. Hong, Y. Ma, X. Cheng and W. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202314848 Search PubMed.
- S. Cai, J. Chen, S. Wang, J. Zhang and X. Wan, Angew. Chem., Int. Ed., 2021, 60, 9686 CrossRef CAS PubMed.
- X. Cheng and W. Zhang, Angew. Chem., Int. Ed., 2021, 60, 9686 CrossRef PubMed.
- J. Shen and Y. Okamoto, Chem. Rev., 2016, 116, 1094 CrossRef CAS PubMed.
- H. Zou, Q.-L. Wu, L. Zhou, X.-H. Hou, N. Liu and Z.-Q. Wu, Chin. J. Polym. Sci., 2021, 39, 1521 CrossRef CAS.
- X. Wang, X. Gao, H. Zhong, K. Yang, B. Zhao and J. Deng, Adv. Mater., 2025, 37, 2412805 CAS.
- L. Xu, L. Zhou, Y.-X. Li, R.-T. Gao, Z. Chen, N. Liu and Z.-Q. Wu, Nat. Commun., 2023, 14, 7287 CAS.
- T. Ikai, M. Ito, K. Oki, N. Suzuki and E. Yashima, Angew. Chem., Int. Ed., 2023, 62, e202306252 Search PubMed.
- L. Zhou, K. Chen, X.-Y. Zhou and Z.-Q. Wu, Chem. Res. Chin. Univ., 2023, 39, 719–725 CAS.
- T. Yamamoto, R. Murakami, S. Komatsu and M. Suginome, J. Am. Chem. Soc., 2018, 140, 3867 CAS.
- L. Zhou, K. He, N. Liu and Z.-Q. Wu, Polym. Chem., 2022, 13, 3967 CAS.
- Y. Zhang and J. Deng, Polym. Chem., 2020, 11, 5407 CAS.
- R. P. Megens and G. Roelfes, Chem. – Eur. J., 2011, 17, 8514 CAS.
- Y. Zhou, C. Zhang, J. Huang, L. Liu, J. Bai, J. Li, T. Satoh and Y. Okamoto, Anal. Chem., 2024, 96, 2078 CAS.
- L. Liua, Y. Wanga, F. Wanga, C. Zhanga, Y. Zhoua, Z. Zhoua, X. Liua, R. Zhua, H. Donga and T. Satoh, React. Funct. Polym., 2020, 146, 104392 Search PubMed.
- Z. Tang, H. Iida, H.-Y. Hu and E. Yashima, ACS Macro Lett., 2012, 1, 261 CAS.
- S.-M. Kang, X. Song, T.-T. Zhang, L. Xu, Y.-Y. Zhu and Z.-Q. Wu, Inorg. Chem. Front., 2023, 10, 3345 Search PubMed.
- Y. Nagata, R. Takeda and M. Suginome, ACS Cent. Sci., 2019, 5, 1235 CAS.
- A. Desmarchelier, X. Caumes, M. Raynal, A. Vidal-Ferran, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2016, 138, 4908 CAS.
- C. He, G. Yang, Y. Kuai, S. Shan, L. Yang, J. Hu, D. Zhang, Q. Zhang and G. Zou, Nat. Commun., 2018, 9, 5117 Search PubMed.
- T. Imamoto, Chem. Rev., 2024, 124, 8657 Search PubMed.
- Y. Huang and T. Hayashi, Chem. Rev., 2022, 122, 14346 Search PubMed.
- R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373 Search PubMed.
- W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853 CAS.
- F. Scharinger, Á. M. Pálvölgyi, M. Weisz, M. Weil, C. Stanetty, M. Schnürch and K. Bica-Schröder, Angew. Chem., Int. Ed., 2022, 61, e202202189 CAS.
- A. A. Wani, S. S. Chourasiya, D. Kathuria and P. V. Bharatam, Chem. Commun., 2021, 57, 11717 Search PubMed.
- S. Li, C. Zhang, G. Pan, L. Yang, Z. Su, X. Feng and X. Liu, ACS Catal., 2023, 13, 4656 CrossRef CAS.
- P. Kumar and N. Dwivedi, Acc. Chem. Res., 2013, 46, 289 CrossRef CAS PubMed.
- Y. Lin, W. J. Hirschi, A. Kunadia, A. Paul, I. Ghiviriga, K. A. Abboud, R. W. Karugu, M. J. Vetticatt, J. S. Hirschi and D. Seidel, J. Am. Chem. Soc., 2020, 142, 5627 CrossRef CAS PubMed.
- R. Torán, E. Portillo, A. Sanz-Marco, C. Vila and G. Blay, Org. Chem. Front., 2023, 10, 6081 RSC.
- P. Mei, Z. Ma, Y. Chen, Y. Wu, W. Hao, Q.-H. Fan and W.-X. Zhang, Chem. Soc. Rev., 2024, 53, 6735 RSC.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2020, 142, 18317 CAS.
- T. Ikai, M. Ando, M. Ito, R. Ishidate, N. Suzuki, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2021, 143, 12725 CAS.
- Z.-Q. Wu, X. Song, Y.-X. Li, L. Zhou, Y.-Y. Zhu, Z. Chen and N. Liu, Nat. Commun., 2023, 14, 566 CAS.
- G. Shi, X. Dai, Q. Xu, J. Shen and X. Wan, Polym. Chem., 2021, 12, 242 RSC.
- N. Liu, L. Zhou and Z.-Q. Wu, Acc. Chem. Res., 2021, 54, 3953 CAS.
- N. Liu, R.-T. Gao and Z.-Q. Wu, Acc. Chem. Res., 2023, 56, 2954 CAS.
- T. Yonehara, S. Nimori, R. Kumai and H. Goto, Macromolecules, 2023, 56, 2965 CAS.
- C. Du, D. Gao, M. Gao, H. Yuan, X. Liu, B. Wang and C. Xing, ACS Appl. Mater. Interfaces, 2021, 13, 27955 CAS.
- Y. Zhai, Y. Wang, X. Zhu, Z. Xing, S. Qi, S. Wang, Y. Han and Z. Chen, Macromolecules, 2021, 54, 5249 CAS.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226 CAS.
- H. Yuan, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Angew. Chem., Int. Ed., 2020, 59, 2720 CAS.
- S. Fan, T. Jiang, T. Lv, J. Liu and X. Wang, Org. Lett., 2023, 25, 4355 CAS.
- J.-R. Zhuo, J.-Q. Zhao, L. Yang, Y.-L. Wu, Y.-P. Zhang, Y. You, Z.-H. Wang, M.-Q. Zhou and W.-C. Yuan, Org. Lett., 2024, 26, 2623 CAS.
- J. Otevrel, D. Svestka and P. Bobal, Org. Biomol. Chem., 2019, 17, 5244 RSC.
- M. Kawada, R. Tsuyusaki, K. Nakashima, M. Yamada, A. Kozakai, Y. Matsushima, S.-I. Hirashima and T. Miura, Chem. – Asian J., 2022, 17, e202101299 CrossRef CAS PubMed.
- S. Medina, M. J. Harper, E. I. Balmond, S. Miranda, G. E. M. Crisenza, D. M. Coe, E. M. McGarrigle and M. C. Galan, Org. Lett., 2016, 18, 4222 CrossRef CAS PubMed.
- J. Yin, L. Xu, X. Han, L. Zhou, C. Li and Z.-Q. Wu, Polym. Chem., 2017, 8, 545 RSC.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé, Chem. Rev., 2022, 122, 12544 CrossRef CAS PubMed.
- A. Dixit, P. Kumar, G. D. Yadav and S. Singh, Inorg. Chim. Acta, 2018, 479, 240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01284d |
| This journal is © The Royal Society of Chemistry 2025 |