
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–property relationships to direct the dynamic properties of acylsemicarbazide-based materials†
Stefan J. D.
Maessen
a,
Siebe
Lekanne Deprez
 b,
Pascal
Vermeeren
b,
Bart W. L.
van den Bersselaar
a,
Martin
Lutz
c,
Johan P. A.
Heuts
a,
Célia
Fonseca Guerra
b and
Anja R. A.
Palmans
*a
b,
Pascal
Vermeeren
b,
Bart W. L.
van den Bersselaar
a,
Martin
Lutz
c,
Johan P. A.
Heuts
a,
Célia
Fonseca Guerra
b and
Anja R. A.
Palmans
*a
aDepartment of Chemical Engineering & Chemistry and Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl
bDepartment of Chemistry and Pharmaceutical Sciences, Amsterdam Institute of Molecular and Life Sciences (AIMMS), Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HZ Amsterdam, The Netherlands
cStructural Biochemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
First published on 4th December 2024
Abstract
Secondary interactions, such as hydrogen bonding or phase separation, can enhance the stability of dynamic covalent materials without compromising on desired dynamic properties. Here, we investigate the combination of multiple secondary interactions in dynamic covalent materials based on acylsemicarbazides (ASCs), with the aim of achieving tunable material properties. The effects of different ASC substituents on the dynamic covalent and hydrogen bonding capabilities were investigated in a small molecule study using a combined experimental and theoretical approach, and revealed the presence of cooperative hydrogen-bonding interactions in 2 directions in one of the derivatives. The different motifs were subsequently incorporated into polymeric materials. Combining ASC motifs capable of strong, multiple hydrogen bonding with a polydimethylsiloxane backbone introduces structure-dependent, ordered nanophase separation in polymeric materials. The thermo-mechanical properties of the materials reveal a strong dependance on the hydrogen-bonding structure and exact nature of the ASC bond. The dynamic behavior in bulk shows that bond exchange depends on the dissociation rate obtained from ASC model compounds, as well as the strength of the secondary interactions in these materials. Differences in hydrogen-bonding structures of the ASC motifs also cause differences in creep resistance of the materials. Interestingly, the materials with strong, ordered and cooperative hydrogen-bonded clusters show the highest creep resistance. Our results demonstrate that tuning both the dissociation rate and the secondary interactions by molecular design in dynamic covalent materials is important for controlling their thermal stability and creep resistance.
Introduction
Dynamic covalent bonds (DCBs) are covalent bonds that are able to break and reshuffle in response to certain stimuli, most commonly by applying heat.1 Numerous dynamic covalent chemistries have been identified that differ in rearrangement mechanisms, which has led to the recent emergence of DCBs as a versatile toolbox for designing reprocessable or healable materials.2,3 Despite the beneficial properties that DCBs bring, the application of dynamic covalent materials is limited by their lower mechanical strength and/or stability compared to conventional polymeric materials.4 Recent research in the field has focused on overcoming these limitations by tuning the material's molecular structure. It was shown that the addition of secondary interactions is a valid strategy towards improving the mechanical properties and stability in dynamic covalent materials, while good dynamic properties are retained.5–8 One way to achieve this is by creating distinct hard and soft phases in materials, either through supramolecular interactions, such as hydrogen bonding, or by phase separation.9–13 Systems utilizing these secondary interactions have the ability to increase the thermal stability of materials, for example by increasing their flow temperature or by reducing creep at elevated temperatures.14–19An interesting DCB that combines dynamic covalent chemistry with supramolecular chemistry is the acylsemicarbazide (ASC), which is formed by the reaction of an acyl hydrazide and isocyanate (Scheme 1A).20,21 The resulting motif resembles an urea and amide moiety fused through an N–N linkage, and can dissociate back into its precursors at elevated temperatures (Scheme 1B).22 Incorporating ASCs in covalently-crosslinked networks gives strong materials with both good (re)processability and healability.22–25 Furthermore, ASCs have received attention in supramolecular networks due to their ability to form multiple hydrogen bonds, leading to densely packed hydrogen-bonded clusters with remarkable mechanical properties.26–29 The strength of the hydrogen bonds in ASCs can be further enhanced by introducing another secondary interaction: phase separation. In systems where ASCs have been combined with hydrophobic polymer backbones such as polydimethylsiloxane (pDMS), the localization of hydrogen-bonding units in phase-separated stacks gave rise to improved tensile strength compared to non-phase-separated systems.25,30–32 The introduction of the relatively soft pDMS phase also prevented crystallization and improved the healability of the materials due to increased chain mobility. Compared to established pDMS-based urethane- or urea-containing elastomers,33,34 ASC groups offer unique hydrogen-bonding patterns in multiple directions. This leads to increased hydrogen-bonding strength and stability, while at the same time introducing a dynamic covalent bond for additional dynamicity.
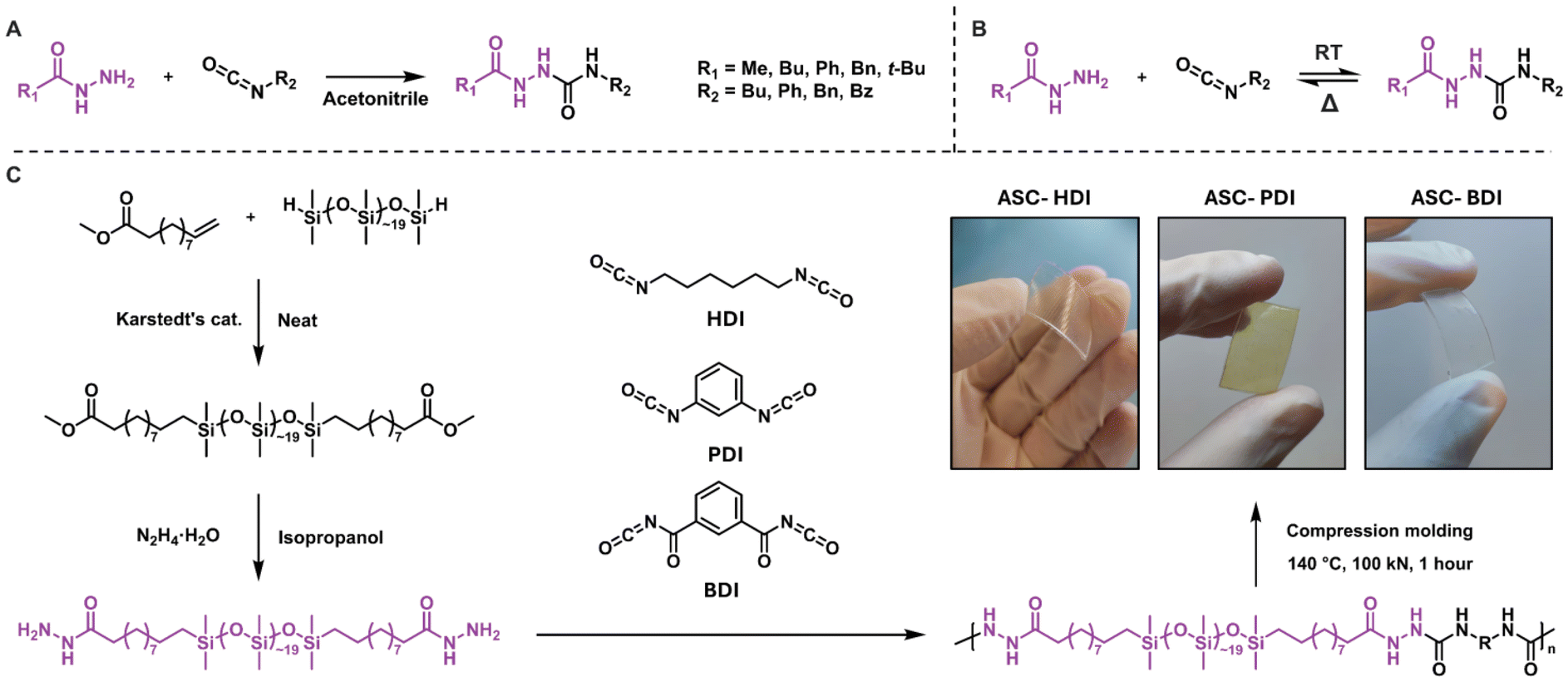 | ||
| Scheme 1 A) Synthetic pathway towards model ASC compounds. (B) Dynamic covalent dissociation of ASC groups. (C) Synthetic pathway towards dihydrazide functionalized pDMS and subsequent polymerization with three isocyanates to yield polymer films after compression molding. | ||
In general, tuning the dynamic covalent character of DCBs has been achieved by introducing (internal) catalysts or by altering the electronic structure around the DCB via various substituents.30,35–41 When using ASCs, the broad availability of different (di)isocyanates and the ease of preparation of acyl hydrazides permits to tune both their electronic structure as well as the nature of the hydrogen bonds. In this work, we present a bottom-up approach to unveil the versatility of ASC groups for dynamic covalent materials. We systematically investigate their structure–property relationships by modifying their molecular structure. We show the quantification of dissociation rates of a library of ASCs comprising a range of substituents using 1H NMR kinetic experiments. In addition, crystal structures from selected molecules were obtained to probe the formed hydrogen-bonding arrays, and evaluating if cooperative hydrogen bonding is present using the framework of Kohn–Sham density functional theory (KS-DFT).42–45 Selected ASC motifs were then incorporated into linear pDMS-based block copolymers, which allowed us to study the thermal, morphological and dynamic covalent properties of the polymer films. Finally, the resistance of the materials towards creep was determined. Our results point towards an interesting role of cooperativity of hydrogen bonds to help reduce creep in materials.
Results and discussion
Design and synthesis of model compounds and polymers
ASC model compounds comprising a range of substituents were synthesized by reacting various hydrazides with different isocyanates to investigate the tunability of the ASC dynamic covalent bond (Scheme 1A and B). It is known that the dynamic covalent character of DCBs can be tuned, also in isocyanate-based systems.46 We therefore include a model compound derived from an isocyanate with the benzoyl group as an electron-withdrawing unit. All model compounds were obtained in high purity (see ESI† for details). In addition, three isocyanates with alkyl, phenyl and benzoyl substituents were incorporated into polymers to study their properties in materials. To achieve this, first pDMS was functionalized with methyl 10-undecenoate, which was converted into a pDMS dihydrazide prepolymer by reacting with hydrazine monohydrate (Scheme 1C). This prepolymer was subsequently reacted in equimolar ratios with three diisocyanates (HDI, PDI, and BDI,47Scheme 1C) in THF. During the polymerization, gelation of the reaction mixture occurred due to the strong organogelating nature of the pDMS-ASC polymers. This led to the formation of oligomers with a number average degree of polymerization of around 10, which were compression molded under 100 kN at 140 °C for 1 h to afford flexible plastic films (ASC-HDI, ASC-PDI and ASC-BDI, Scheme 1C).Kinetic evaluation of bond exchange in ASC model compounds
1H NMR studies were performed to study the effects of various hydrazide and isocyanate substituents on the dissociation rate (kd) of the ASC model compounds. Analogous to the studies done by Xia and coworkers, ASC model compounds were mixed with benzylamine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in DMSO-d6 and were subsequently heated to various temperatures using an oil bath.22 At elevated temperatures, the ASC compounds will dissociate and urea bonds are formed due to the higher nucleophilicity of the amine compared to the hydrazide (Fig. 1A). It was previously determined that this dissociation follows first-order kinetics.22 Thus, by measuring the decrease in ASC concentration (c/c0) over time, the dissociation rate of the ASC model compounds can be determined from the slope of a linear fit in a −ln(c/c0) vs. time plot (eqn (1), Fig. 1B and S6†). By constructing an Arrhenius plot, the activation energy (Eact) of the dissociation can be determined (Fig. 1C).
:1 ratio in DMSO-d6 and were subsequently heated to various temperatures using an oil bath.22 At elevated temperatures, the ASC compounds will dissociate and urea bonds are formed due to the higher nucleophilicity of the amine compared to the hydrazide (Fig. 1A). It was previously determined that this dissociation follows first-order kinetics.22 Thus, by measuring the decrease in ASC concentration (c/c0) over time, the dissociation rate of the ASC model compounds can be determined from the slope of a linear fit in a −ln(c/c0) vs. time plot (eqn (1), Fig. 1B and S6†). By constructing an Arrhenius plot, the activation energy (Eact) of the dissociation can be determined (Fig. 1C).| c = c0e−kdt | (1) |
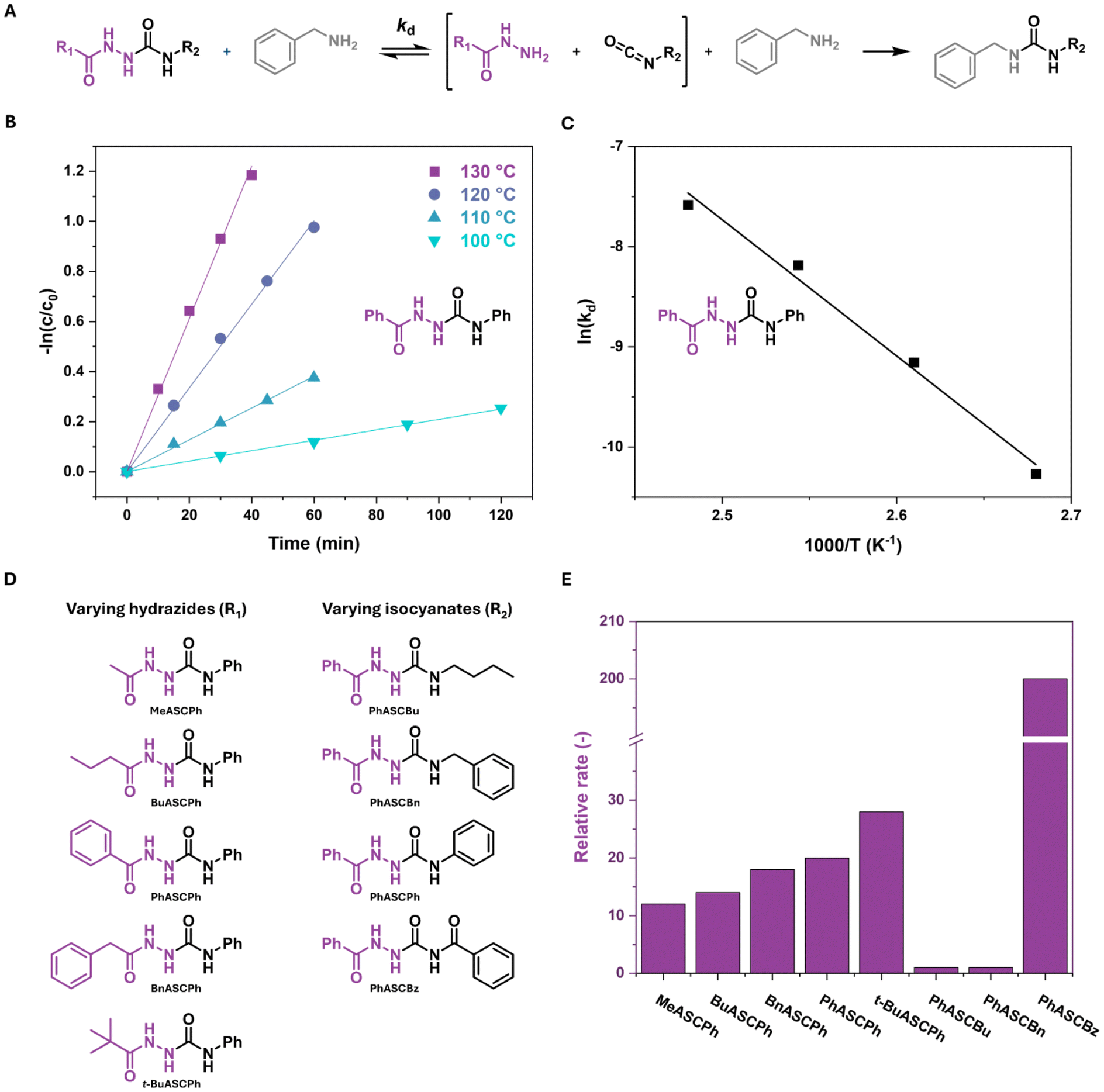 | ||
| Fig. 1 (A) Dissociation of ASC model compounds and trapping of the isocyanate by benzyl amine during the kinetic measurements. (B) Kinetic measurements of PhASCPh at various temperatures. (C) Arrhenius plot of PhASCPh. (D) Library of studied model ASCs where the hydrazide and isocyanate groups have been varied. (E) Relative rates (at 120 °C) of model ASCs. | ||
The kd and Eact were determined for all 8 model compounds, in which the hydrazides were initially varied from methyl to butyl, phenyl and benzyl, and the isocyanates changed from butyl to benzyl, phenyl, and benzoyl (Fig. 1D, Fig. S7, 8 and Table S5†). Fig. 1E shows the comparison of the relative dissociation rate at 120 °C (normalized to that of PhASCBu) of all model compounds.
Focusing on the relative rates, varying the hydrazide substituent (R1) causes the dissociation rate to change by less than a factor of 2 going from a methyl to phenyl substituent. This difference is likely related to steric effects, where bulkier substituents result in a higher dissociation rate. This hypothesis was corroborated by an additional model compound, t-BuASCPh, which indeed shows a higher dissociation rate than the other hydrazide substituents. Much larger differences in relative rate are observed when changing the isocyanate (R2), with increases of 20 and 200 times when going from the butyl substituent to the phenyl and benzoyl, respectively. These large differences could be related to differences in electronic stabilization by the substituents. The conjugation of the phenyl and benzoyl substituents most likely activates the dynamic bond, leading to a higher dissociation rate. The variations in Eact between the different model compounds are less pronounced, between 108 and 139 kJ mol−1, and most likely within experimental error (Table S5 and Fig. S8†). The kinetic studies clearly show that the nature of the isocyanate plays a significant role in the dynamic covalent behavior of ASCs. Interestingly, PhASCPh and PhASCBz yielded single crystals suitable for crystal structure analysis and were therefore selected for a more detailed study of their structural properties.
Crystal structure analysis of selected ASC model compounds
The ability to undergo multiple hydrogen bonding is one of the most prominent features of the ASC group.48 To understand and explore the structure and tunability of the hydrogen-bonding patterns in ASCs, crystal structures were obtained of PhASCPh and PhASCBz (see ESI† for details). Crystallization of ASC model compounds with aliphatic substituents was also attempted but unsuccessful and hence will not be discussed. Note that a more detailed theoretical analysis of the different interactions within the crystal structures using the energy decomposition analysis (EDA)42,49 and Vonoroi deformation density (VDD)50,51 can be found in the ESI.†For PhASCPh and PhASCBz, each repeating unit cell of the crystal contains different conformers of the respective monomer (Fig. 2). The urea fragment of PhASCPh is in a trans/trans configuration, while that of PhASCBz is in a cis/trans configuration. This change in preference is caused by the additional carbonyl group in PhASCBz forming an intramolecular hydrogen bond in the PhASCBz crystal. Furthermore, DFT calculations reveal that both PhASCPh monomers in the crystal structure are energetically equivalent while those for PhASCBz are energetically distinct. This is supported by the great similarity of the two conformers in PhASCPh in contrast with those in PhASCBz. From these observations, we infer that the intermolecular hydrogen bonding in a material containing PhASCBz moieties might be weaker due to competition with the intramolecular hydrogen bonds, despite PhASCBz having more functional groups capable of forming hydrogen bonds than PhASCPh.
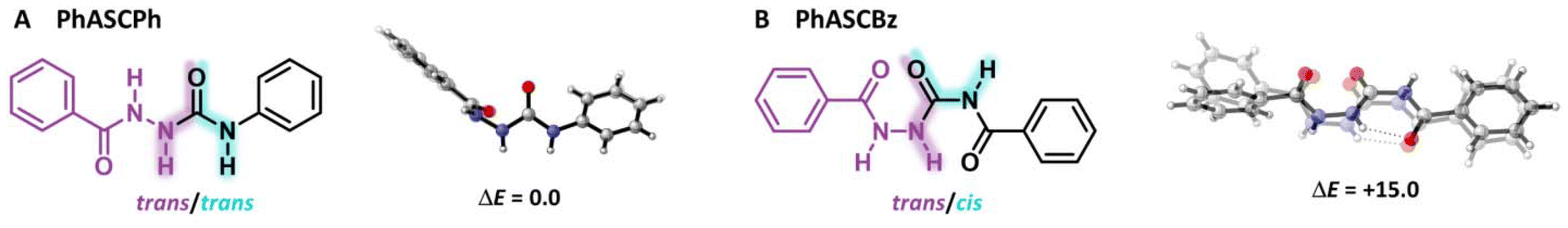 | ||
| Fig. 2 ASC monomers (A) PhASCPh and (B) PhASCBz (trans/trans and trans/cis conformers) shown in the geometries they adopt in the crystal structure. Each unit cell contains two conformers which are superimposed with energy differences in kcal mol−1. | ||
There are two repeating intermolecular hydrogen-bonding patterns observed in the crystal structure of PhASCPh that proceed in perpendicular directions (Fig. 3; see ESI† for analysis of the PhASCBz crystal). The first pattern consists of bifurcated hydrogen bonds formed by the urea fragment (bifurcated HB) while the second pattern consists of a single hydrogen bond formed by the amide fragment (single HB). The combination of these two chains results in a 2D hydrogen-bonded sheet in the crystal structure of PhASCPh (Fig. S9 and S23†). The nature and strength of both patterns was analyzed using DFT, which reveals that the hydrogen bonds account for half of the stabilizing interaction between the interacting monomer in the crystal (see Table S6 in the ESI†).
 | ||
| Fig. 3 Hydrogen-bonding patterns found in the crystal structure of PhASCPh depicting bifurcated (A) and single (B) hydrogen bonds. Hydrogen bond lengths rO–N (in Å), interaction energies ΔEint (in kcal mol−1), and charge transfer values ΔQVDDn (in milli-electrons) are shown, computed at a ZORA-BLYP-D3(BJ)/TZ2P level of theory. The chain elongation direction is from bottom to top (A) and from left to right (B). | ||
The hydrogen bonds, as observed in the crystal structure, exhibit a crucial characteristic for forming chains, namely, cooperativity.52–55 This entails that the interaction between a chain (containing n monomers) and an additional monomer becomes stronger, in a non-additive matter, as the chain becomes longer. In other words, it becomes increasingly more favorable for monomers to aggregate as the chain becomes longer. Fig. 3 illustrates cooperativity in the PhASCPh crystal through considering the interaction energy (ΔEint) in a hydrogen-bonded dimer (1 + 1) to a pentamer (4 + 1). The interaction energy for the bifurcated HB becomes more stabilizing (i.e., more negative) per unit added from −20.3 kcal mol−1 for the 1 + 1 to −23.0 kcal mol−1 for the 4 + 1. Thus, the hydrogen bond becomes more stabilized as the chain elongates, thereby indicating the presence of cooperativity. This is further shown by the increase in charge transfer from the chain to the additional monomer (ΔQVDDn). Note that the cooperative effects approach an asymptote in which the interaction energy and charge transfer converge to values of −23.1 kcal mol−1 and +29 milli-electrons, respectively. Cooperativity is also observed along the single HB chain, although to a lesser extent than the bifurcated HB chain (Fig. 3). This lower degree of cooperativity originates from the hydrogen bonds being weaker along the chain.
All in all, the PhASCPh crystal shows cooperativity in a 2D hydrogen-bonded sheet (Fig. S9†), which we hypothesize to result in the formation of strongly hydrogen-bonded clusters if this moiety is utilized in materials. For the PhASCBz crystal structure, extensive hydrogen-bonded networks are also present (Fig. S25†). However, the repeating motifs do no exhibit cooperativity due to hydrogen bonds opposing each other, leading to a net charge transfer of zero (Table S9†). We hypothesize that this may result in weaker overall hydrogen bonding in materials comprising an ASC motif similar to that of PhASCBz.
Thermal and structural properties of pDMS-ASC oligomers
The above results show that both the dissociation dynamics and hydrogen-bonding structure can be tuned by varying the isocyanate substituent in ACS-based model compounds. To understand how these effects translate into materials, the synthesized pDMS-ASC oligomers were investigated using a combination of thermographic analysis (TGA), differential scanning calorimetry (DSC), variable temperature infrared spectroscopy (VTIR), dynamic mechanical thermal analysis (DMTA) and X-ray scattering. TGA showed no considerable degradation below 200 °C, highlighting the thermal stability of the dynamic ASC groups (Fig. S10†).VTIR was used to study changes in hydrogen-bonding structure as a function of temperature on compression-molded samples of the three polymers. At room temperature, ASC-BDI shows a broad N–H stretch peak and two distinct C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches (Fig. 4A). At elevated temperatures, the peaks shifted to higher wavenumbers, indicating weakening of the hydrogen bonds. This weakening was also confirmed by DFT analyses (see ESI† section 5 for more details). Furthermore, a stark decrease in the CO stretch intensity at 1712 cm−1 was observed. Plotting the intensity at 1712 cm−1 as a function of temperature (normalized on the C–H stretch at 2960 cm−1) shows a clear deviation from linearity above 130 °C, indicating a large change in the hydrogen-bonding strength (Fig. 4B). A similar trend is observed by tracking the peak maximum of the N–H stretch region (Fig. S11†). Upon cooling, the N–H stretch region becomes slightly more defined, and a small shoulder in the CO stretch region at 1646 cm−1 appears due to some optimization of the hydrogen-bonding structure after thermal treatment (Fig. S12†). Contrary to other works on ASC-based systems,22,32 we do not observe an NCO-stretching vibration around 2250 cm−1 at elevated temperatures, which could be explained by a poor signal-to-noise ratio in the VTIR spectra around this region. Unlike ASC-BDI, ASC-PDI shows well-defined peaks in the N–H and CO stretch regions, which also shift to higher wavenumbers upon heating (Fig. S13†). The positions and relative intensities of the peaks in ASC-PDI before and after a heating cycle up to 160 °C are equivalent, indicating that there is no pronounced reorganization of the hydrogen bonds before and after heating. When heating above 160 °C however, more pronounced changes were observed (Fig. S14†). Again, a drop in CO stretch intensity is present, now above 160 °C (Fig. 4B). The VTIR of ASC-HDI shows a more complex behavior which is discussed in detail in the ESI (Fig. S15).† In general, all pDMS-ASC materials show a clear transition upon heating at temperatures of 140, 160 and 178 °C for HDI, BDI and PDI respectively, corresponding to weakening of the hydrogen bonds (Fig. 4B).
O stretches (Fig. 4A). At elevated temperatures, the peaks shifted to higher wavenumbers, indicating weakening of the hydrogen bonds. This weakening was also confirmed by DFT analyses (see ESI† section 5 for more details). Furthermore, a stark decrease in the CO stretch intensity at 1712 cm−1 was observed. Plotting the intensity at 1712 cm−1 as a function of temperature (normalized on the C–H stretch at 2960 cm−1) shows a clear deviation from linearity above 130 °C, indicating a large change in the hydrogen-bonding strength (Fig. 4B). A similar trend is observed by tracking the peak maximum of the N–H stretch region (Fig. S11†). Upon cooling, the N–H stretch region becomes slightly more defined, and a small shoulder in the CO stretch region at 1646 cm−1 appears due to some optimization of the hydrogen-bonding structure after thermal treatment (Fig. S12†). Contrary to other works on ASC-based systems,22,32 we do not observe an NCO-stretching vibration around 2250 cm−1 at elevated temperatures, which could be explained by a poor signal-to-noise ratio in the VTIR spectra around this region. Unlike ASC-BDI, ASC-PDI shows well-defined peaks in the N–H and CO stretch regions, which also shift to higher wavenumbers upon heating (Fig. S13†). The positions and relative intensities of the peaks in ASC-PDI before and after a heating cycle up to 160 °C are equivalent, indicating that there is no pronounced reorganization of the hydrogen bonds before and after heating. When heating above 160 °C however, more pronounced changes were observed (Fig. S14†). Again, a drop in CO stretch intensity is present, now above 160 °C (Fig. 4B). The VTIR of ASC-HDI shows a more complex behavior which is discussed in detail in the ESI (Fig. S15).† In general, all pDMS-ASC materials show a clear transition upon heating at temperatures of 140, 160 and 178 °C for HDI, BDI and PDI respectively, corresponding to weakening of the hydrogen bonds (Fig. 4B).
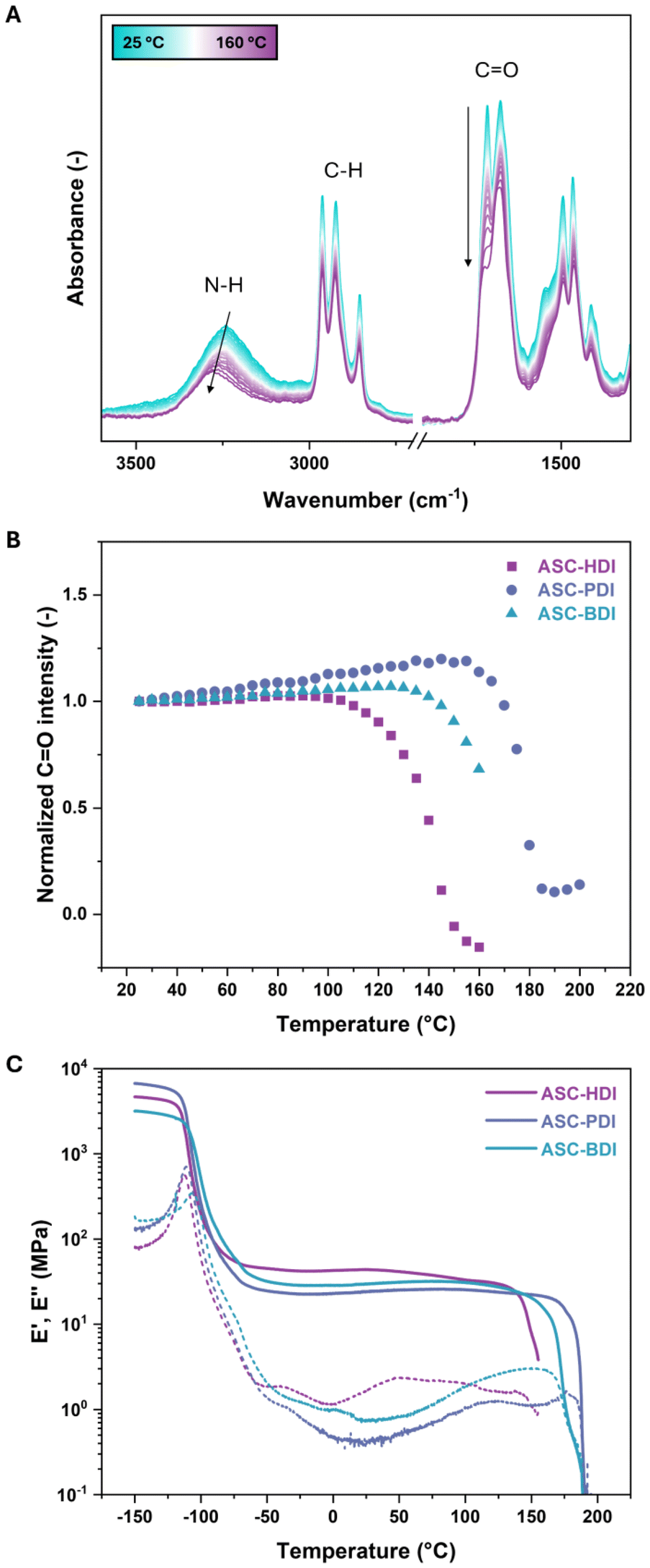 | ||
| Fig. 4 (A) Variable temperature infrared spectroscopy spectra of ASC-BDI (first heating, intervals of 5 °C). (B) Intensity plot of peak maxima of the CO stretches of pDMS-ASC materials over temperature, normalized against the intensity of the C–H stretch at 2960 cm−1. (C) Dynamic mechanical thermal analysis under extension on the ASC oligomers obtained after compression molding representing the storage (solid) and loss (dashed) moduli as a function of temperature. | ||
DMTA gives insight into the thermo-mechanical properties of the ASC-based materials (Fig. 4C). All materials showed a glass transition temperature (Tg) around −110 °C, corresponding to the Tg of the pDMS backbone. This is followed by a rubbery plateau with plateau storage moduli of 43, 29 and 23 MPa for ASC-HDI, ASC-BDI and ASC-PDI, respectively. In these materials, the intermolecular hydrogen bonds act as physical crosslinks keeping the individual chains together. In polymer networks, the plateau modulus is closely connected to the crosslink density of a material.56 The slight differences in plateau modulus between the materials could therefore be related to the differences in hydrogen-bonding patterns. Alternatively, the differences could originate from variations in molecular weights or end-group effects. Remarkably, the rubbery plateaus stretch up to quite high temperatures despite the fact that all pDMS-ASC materials are relatively short-chain oligomers. All three materials showed additional thermal transitions, which were around 145, 170 and 185 °C for ASC-HDI, ASC-BDI and ASC-PDI, respectively. For all materials, the onset of the thermal transition occurs around the same temperature as the pronounced decrease in CO stretch intensity from VTIR (Fig. 4B and C). These results indicate that the thermal transitions of the pDMS-ASC polymers are caused by the weakening or loss of (multiple) hydrogen bonding.
The strength of the hydrogen bonds in these materials can thus be related to the thermal stability of the materials, and consequently the position of the thermal transition. This transition occurs at the highest temperature for ASC-PDI, followed by ASC-BDI and finally ASC-HDI. These observations align well with the results obtained from the crystal structure analysis of PhASCPh, as this additional hydrogen-bond stability could originate from the presence of cooperativity in two directions in ASC-PDI. Curiously, no effect of the dissociation rate of the various ASC motifs on the storage modulus was observed, indicating that the hydrogen-bonding behavior dominates over the dynamic covalent dissociation of the ASCs.
In pDMS-ASC materials, the hydrogen bonding is amplified by the choice of polymeric backbone, where the chemical mismatch between hydrophobic pDMS and hydrophilic ASC groups is expected to induce phase separation. The phase separation in these materials was studied using medium- and wide-angle X-ray scattering (MAXS and WAXS). In the MAXS region (q < 7 nm−1), all pDMS-ASC materials show ordered nanophase separation indicated by the principal scattering peak q and several integer Bragg reflections (2q, 3q) (Fig. 5A). Small variations in domain spacing d (d = 2π/q, d = 5.6 nm, 4.4 nm and 5.1 nm for ASC-HDI, ASC-PDI and ASC-BDI, respectively) were observed between the materials, indicating a difference in the packing of the pDMS chains in the hydrophobic domains. The integer spacing between the scattering peaks indicates lamellar ordering in all materials, which could be caused by the directionality of the hydrogen bonds that are guiding the phase separation. In the WAXS region (q > 7 nm−1) a broad halo between 7 and 10 nm−1 is observed in all materials, which is ascribed to the siloxane backbone.57 Interestingly, ASC-HDI and ASC-BDI show relatively broad features above 10 nm−1, while ASC-PDI shows several well-defined peaks (indicated as “x” in Fig. 5A). The positions of the peaks correspond to distances around 4.2 Å, which are proposed to originate from intermolecular π–π stacking or hydrogen-bond distances. The presence of these peaks is indicative for ordered stacking of the ASC units in the ASC-PDI material, which has been highlighted in Fig. 5B. The crystal structure analysis of PhASCPh also showed high regularity in 2 directions and cooperativity in its hydrogen-bonding structure, which is likely the reason for the ordered stacking in the material. These findings further support that the higher transition temperature observed in DMTA for ASC-PDI is caused by stronger hydrogen bonding compared to ASC-HDI and ASC-BDI.
 | ||
| Fig. 5 (A) MAXS and WAXS 1D transmission scattering profiles of ASC-HDI, ASC-PDI and ASC-BDI. (B) Schematic representation of the ordered phase separation in the pDMS-ASC materials, highlighting the ordering within ASC stacks (purple blocks) for ASC-PDI, with indicated intermolecular spacing x. | ||
Probing exchange dynamics in bulk materials
The above results indicate that combining hydrogen bonds and phase separation leads to rubbery materials even at low polymer molecular weights due to strong secondary interactions. Recent studies indicated that in some cases, strong hydrogen bonding or phase separation can inhibit dynamic bond exchange and therefore bring stability to materials.18,19,58 To probe the exchange behavior of the dynamic covalent bonds in the bulk materials, controlled depolymerization studies were conducted. Hereto, pDMS-ASC oligomers were mixed at 50/50 wt% ratio with corresponding monofunctional pDMS-ASC molecules (mono-ASCs, Fig. 6A). The mono-ASCs were designed to intercalate into the polymer without disrupting the existing hydrogen-bonding and phase-separated structure. Therefore, the hydrogen-bonding units are the same as in the pDMS-ASC oligomers, and the pDMS chain ensures similar phase separation. Upon heating this mixture in bulk, a decrease in the molecular weight of the polymer is expected if bond dissociation and exchange take place. | ||
| Fig. 6 (A) Chemical diagrams of ASC oligomers and mono-ASCs. (B) Workflow for measuring dissociation dynamics in bulk. Normalized mass distributions of ASC-HDI (C, RI detector), ASC-PDI (D, UV detector) and ASC-BDI (E, UV detector) mixtures at 120 °C at various times (in hours). (F) Comparison of the high molecular weight peak (Mp) over time. | ||
The polymer and mono-ASC were dissolved in THF, mixed and dried completely (Fig. 6B). The mixtures were then heated in an oven at 120 °C. SEC traces were measured of the samples on several time intervals. For ASC-HDI, a bimodal distribution at t = 0 h is seen in Fig. 6C, in which the high molecular weight peak (Mp) corresponds to the polymer, and the low molecular weight peak to the mono-ASC. In the first 24 h, the position of the polymer peak shifts to lower molecular weights. In the last 24 h, only a slight further decrease in the intensity of the mono-ASC peak is observed. ASC-PDI also shows a decrease in molecular weight, and the shifts in Mp are similar to that observed for ASC-HDI (Fig. 6D). In the final 24 h, the shape of the high molecular weight peak changes more significantly than of ASC-HDI. Finally, for ASC-BDI the reduction in molecular weight is extremely fast, and in the first two hours the depolymerization is already mostly complete (Fig. 6E).
To visualize the change in molecular weight of the polymers during this experiment, Mp was plotted against time (Fig. 6F). The dramatic loss of molecular weight indicates that ASC-BDI dissociates the fastest, while the more gradual changes in loss of molecular weight are similar for ASC-HDI and ASC-PDI, indicating that they dissociate at similar rates. This is contrary to the results of the model compounds, where PhASCPh dissociated 20 times faster than PhASCBu at 120 °C. The reason for this difference in dissociation rates most likely arises from the hydrogen-bonded, phase-separated regions present in ASC-PDI. The hydrogen bonding of ASC-PDI is more ordered and stronger than that of ASC-HDI and ASC-BDI. It is likely that the dissociation of the ASC bonds in ASC-PDI is inhibited due to restricted motion of the atoms within the hydrogen-bonding stack. This would mean that to have efficient bond exchange in the material, first the hydrogen bonds need to be broken or weakened sufficiently. This “locking” of the dynamic bonds has recently been coined phase locking.58,59
Strong hydrogen bonding aids creep resistance
Phase locking was recently shown to be an effective method towards increasing the thermal stability in dynamic covalent systems, for example by giving creep resistance.18,19,58,59 To determine the stability of the pDMS-ASC materials, rheology measurements were performed. Creep-recovery tests of the compression-molded samples were done between 100 and 140 °C (Fig. S17–19†). Upon imposing a step stress on ASC-HDI, the material first deforms elastically (<100 s), after which permanent plastic deformation occurs (Fig. 7A). The elastic response for the 100 °C measurement is much larger than that measured at higher temperatures, which could be due to passing some sort of phase transition in that temperature range. This also most likely leads to more unrecoverable strain during the recovery phase of this measurement (>1800 s). We determine the slope of the terminal part of the creep compliance (dJ/dt between 1400 and 1800 s) and use it as an indication of the stability towards creep in a material, where a lower value of the slope corresponds to better creep resistance. For all pDMS-ASC materials, the slope increases with increasing temperature, meaning that the materials creep more at higher temperatures, as expected (Fig. 7B).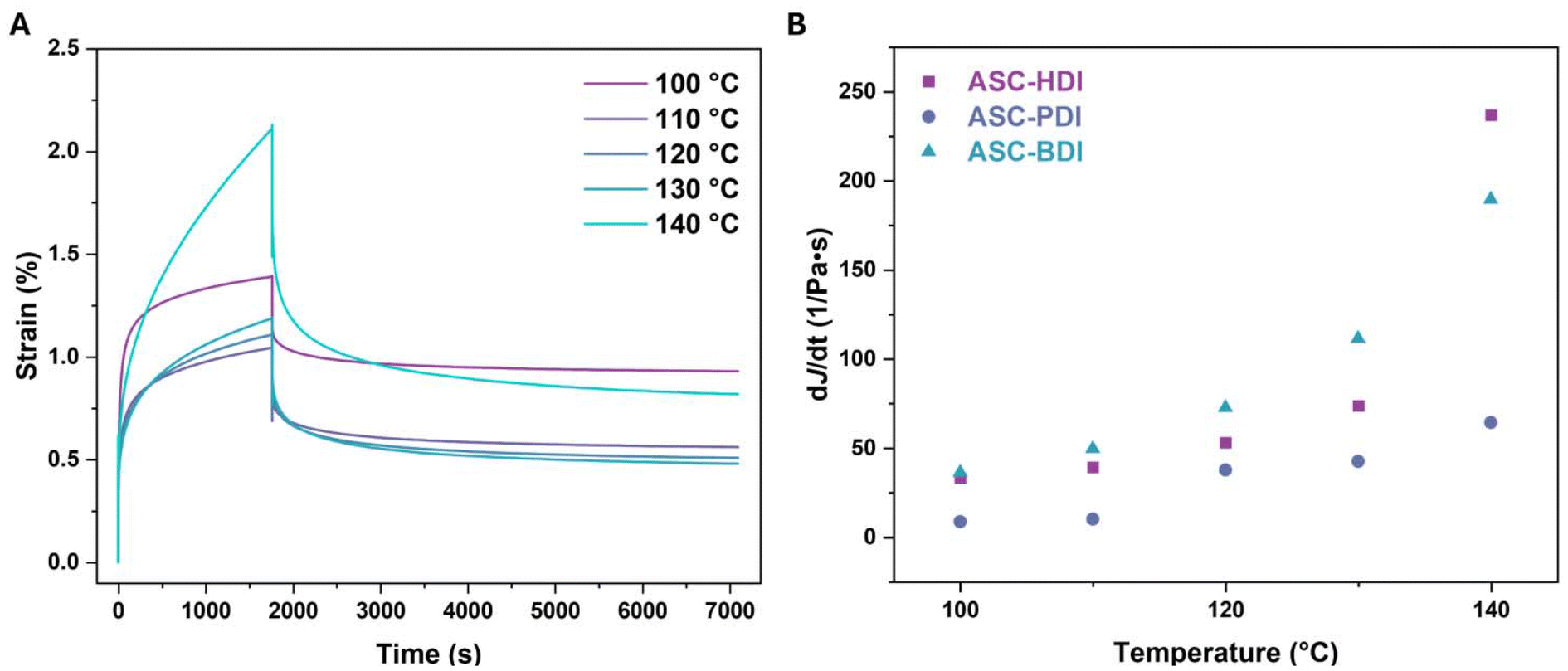 | ||
| Fig. 7 (A) Creep-recovery runs of ASC-HDI at various temperatures at 20 kPa. (B) Temperature dependent slopes of the creep compliance (between 1400 and 1800 s) of ASC oligomers. | ||
The slope of ASC-PDI at 120 °C is similar to that of ASC-HDI and ASC-BDI at 100 °C. From these results, ASC-PDI clearly shows the highest resistance to creep, followed by ASC-HDI, while ASC-BDI has the lowest resistance to creep. The extremely fast dissociation in bulk of ASC-BDI most likely adds to the significant creep observed above 110 °C. The results also show that the creep behavior of the ASC-HDI and ASC-PDI materials is very different, despite having similar dissociation rates in bulk. This means that creep is also determined by other factors such as the presence of secondary interactions. Because of the strong hydrogen bonds in ASC-PDI, the dissociation and also the ability to undergo topology rearrangement is limited, leading to significant creep resistance.
The importance of the design of secondary interactions
All pDMS-ASC materials contain ASC units capable of multiple hydrogen bonding, yet the materials show remarkable differences, especially in their resistance towards creep. ASC-PDI is particularly interesting in this respect: the model ASC shows cooperative hydrogen bonding in two directions and its incorporation into a polymeric material results in the highest ordered as well as the most thermally stable hydrogen bonds, but also the highest creep resistance. This suggests that the exact way how the hydrogen bonds are organized matters. Hydrogen bonds in ASC-HDI and ASC-BDI contribute less to creep stability, most likely because the interaction strength at the temperatures where dynamic covalent exchange takes place is quite low. The choice of polymer matrix also plays and important role; the localization of hydrogen-bonding units in ordered nanophase-separated stacks can effectively enhance the interaction strength and/or cooperativity.We summarize our findings in Fig. 8. Firstly, there is a direct relation between the sharp decrease in storage modulus and loss of hydrogen bonding, highlighting the importance of secondary interactions on the mechanical integrity of pDMS-ASC materials. Next, we clearly see that hydrogen-bond stability at high temperatures can compensate for a higher dissociation rate and result in higher creep stability in ASC-PDI. At some point however, the dissociation rate dominates, leading to a higher susceptibility to creep for ASC-BDI. From this, we learn that creep resistance originates from strong, ordered, cooperative secondary interactions, which can be tuned via structural design of functional groups and polymer matrix.
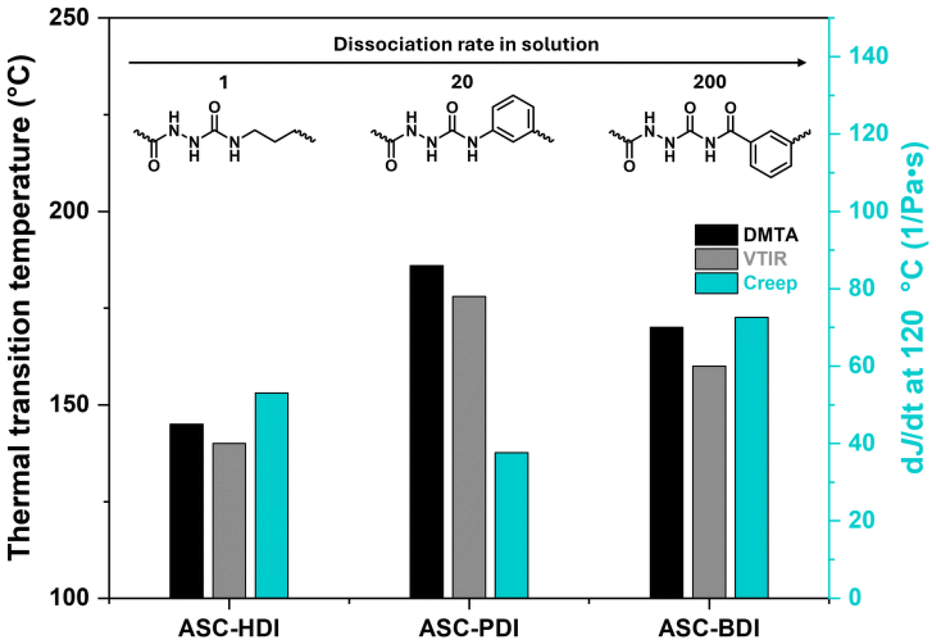 | ||
| Fig. 8 Comparison of transition temperatures from DMTA and VTIR (left axis), the slope of J(t) at 120 °C (right axis) and model compound dissociation rate for ASC-HDI, ASC-PDI and ASC-BDI. | ||
Conclusions
In this work, we explored the dynamic covalent and supramolecular versatility of ASC bonds to create stable dynamic covalent materials. The choice of isocyanate turned out to be crucial for the dissociation rate of the ASC, resulting in dissociation rate differences spanning two orders of magnitude. Structure-dependent hydrogen-bonding patterns were found and analyzed using crystal structure analysis supported by DFT calculations. One of these structures, PhASCPh, contained cooperative hydrogen bonding in two directions, which we assumed to be an important contributor to cluster formation in case of ASC-based materials.Selected ASC motifs were translated to phase-separated polymer films, in which the thermal transitions were related to their hydrogen-bonding strength. Here, ASC-PDI showed the strongest hydrogen bonds, followed by ASC-BDI and ASC-HDI. Due to the hydrophobic pDMS and polar ASC groups, in combination with the directionality of the ASC hydrogen bonding, all pDMS-ASC materials phase separated into lamellae, and ordered hydrogen bonds were observed for ASC-PDI. Depolymerization experiments in bulk allowed to quantify the dynamic covalent dissociation of these materials. Here, ASC-BDI showed extremely fast dissociation, correlating well with the small molecule kinetic experiments. ASC-HDI and ASC-PDI showed similar dissociation behavior, which was attributed to the stronger and more ordered hydrogen bonding in ASC-PDI hindering dissociation of the ASC bonds. This also resulted in higher creep resistance for ASC-PDI compared to the other pDMS-ASC materials. Taken all together, our results show that carefully designing secondary interactions into dynamic covalent systems is of high importance. Additional hydrogen-bonding interactions play a significant role in determining the thermo-mechanical properties and creep resistance while the dynamic character of the materials is retained.
Author contributions
Stefan Maessen: conceptualization, data curation, formal analysis, investigation, validation, visualization, writing – original draft, writing – reviewing and editing. Siebe Lekanne Deprez: data curation, visualization, writing – reviewing and editing. Pascal Vermeeren: supervision, writing – reviewing and editing. Bart van den Bersselaar: data curation, writing – reviewing and editing. Martin Lutz: data curation, writing – reviewing and editing. Johan P.A Heuts: supervision, writing – reviewing and editing. Célia Fonseca Guerra: supervision, writing – reviewing and editing, funding acquisition. Anja R. A. Palmans: supervision, writing – reviewing and editing, funding acquisition.Data availability
Crystallographic data for PhASCPh and PhASCBz have been deposited at the Cambridge Crystallographic Data Centre as CCDC 2377270 and 2377271.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
SM and SLD were supported by funding from the Netherlands Organization for Scientific Research (NWO, grant number OCENW.M.21.035). AP acknowledges support by The Dutch Ministry of Education, Culture, and Science (NWO Gravitation program 024.005.020: Interactive Polymer Materials) and the TU/e. This work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative and VU BAZIS. The authors thank Roy Wink and Rint P. Sijbesma for general discussions about the work and providing feedback.References
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- D. Montarnal, C. Mathieu, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- F. Van Lijsebetten, T. Debsharma, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202210405 ( Angew. Chem. , 2022 , 134 , e202210405 ) CrossRef CAS PubMed.
- H. Zhang, A. van Hertrooij, T. Schnitzer, Y. Chen, S. Majumdar, R. A. T. M. van Benthem, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2023, 56, 6452–6460 CrossRef CAS.
- M. H. P. de Heer Kloots, S. K. Schoustra, J. A. Dijksman and M. M. J. Smulders, Soft Matter, 2023, 19, 2857–2877 RSC.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, K. Huang, Y. Liu and J. Zhu, Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS.
- J. S. A. Ishibashi, I. C. Pierce, A. B. Chang, A. Zografos, B. M. El-Zaatari, Y. Fang, S. J. Weigand, F. S. Bates and J. A. Kalow, Macromolecules, 2021, 54, 3972–3986 CrossRef CAS.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Macromolecules, 2019, 52, 432–443 CAS.
- Y. Oba, T. Kimura, M. Hayashi and K. Yamamoto, Macromolecules, 2022, 55, 1771–1782 CrossRef CAS.
- K. M. Herbert, P. T. Getty, N. D. Dolinski, J. E. Hertzog, D. de Jong, J. H. Lettow, J. Romulus, J. W. Onorato, E. M. Foster and S. J. Rowan, Chem. Sci., 2020, 11, 5028–5036 CAS.
- S. K. Schoustra, V. Asadi, H. Zuilhof and M. M. J. Smulders, Eur. Polym. J., 2023, 195, 112209 Search PubMed.
- L. Wang, Y. Liu, Y. Wei, W. Zeng, Z. Cui and A. Du, Eur. Polym. J., 2023, 193, 112101 CrossRef CAS.
- L. Wang, Y. Liu, Y. Qiao, Y. Wang, Z. Cui, S. Zhu, F. Dong, S. Fang and A. Du, Polym. Chem., 2022, 13, 4144–4153 CAS.
- S. Mondal, A. J. Wong, M. A. Wagh, L. Alperstein, G. J. Sanjayan and B. S. Sumerlin, Polym. Chem., 2024, 15, 1826–1832 CAS.
- S. Engelen, N. D. Dolinski, C. Chen, E. Ghimire, C. A. Lindberg, A. E. Crolais, N. Nitta, J. M. Winne, S. J. Rowan and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 63, e202318412 CrossRef.
- K. Schoustra, M. H. P. de Heer Kloots, J. Posthuma, D. Van Doorn, J. A. Dijksman and M. M. J. Smulders, Macromolecules, 2022, 55, 10341–10355 CrossRef PubMed.
- T. W. Campbell, V. S. Foldi and J. Farago, J. Appl. Polym. Sci., 1959, 2, 155–162 CrossRef CAS.
- A. Palanisamy, V. Ummadisetty and G. Radhakrishan, J. Appl. Polym. Sci., 2002, 83, 86–93 CrossRef CAS.
- D. Fu, W. Pu, J. Escorihuela, X. Wang, Z. Wang, S. Chen, S. Sun, S. Wang, H. Zuilhof and H. Xia, Macromolecules, 2020, 53, 7914–7924 CrossRef CAS.
- S. Wang, D. Fu, X. Wang, W. Pu, A. Martone, X. Lu, M. Lavorgna, Z. Wang, E. Amendola and H. Xia, J. Mater. Chem. A, 2021, 9, 4055–4065 RSC.
- L. Wang, K. Zhang, X. Zhang, Y. Tan, L. Guo, Y. Xia and X. Wang, Adv. Mater., 2024, 36, 2311758 CAS.
- N. Roy, Ž. Tomović, E. Buhler and J. M. Lehn, Chem. – Eur. J., 2016, 22, 13513–13520 CAS.
- Z. Li, Y. L. Zhu, W. Niu, X. Yang, Z. Jiang, Z. Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CAS.
- L. Wang, L. Guo, K. Zhang, Y. Xia, J. Hao and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301762 CAS.
- X. M. An, Y. P. Wang, T. S. Zhu, C. Xing, X. D. Jia and Q. H. Zhang, Chin. J. Polym. Sci., 2024, 42, 1425–1434 CrossRef CAS.
- B. Li, F. Xu, T. Guan, Y. Li and J. Sun, Adv. Mater., 2023, 35, 2211456 CrossRef CAS PubMed.
- R. Sarkar, S. Majumdar, S. Kuil, J. Mallens, J. J. B. van der Tol, R. P. Sijbesma, J. P. A. Heuts and A. R. A. Palmans, J. Polym. Sci., 2023, 61, 1335–1347 CrossRef CAS.
- N. Roy, E. Buhler and J. M. Lehn, Chem. – Eur. J., 2013, 19, 8814–8820 CrossRef CAS PubMed.
- W. Ma, X. Yang, Y. He, J. Lai, Z. Wang, M. Xie, X. Lu and H. Xia, Polymer, 2023, 282, 126164 CrossRef CAS.
- D. Döhler, J. Kang, C. B. Cooper, J. B.-H. Tok, H. Rupp, W. H. Binder and Z. Bao, ACS Appl. Polym. Mater., 2020, 2, 4127–4139 CrossRef.
- W. Zheng, C. Zhang, Y. Han, W. Wang and Z. Li, Small, 2024, 20, 2402124 CrossRef CAS.
- N. D. Dolinski, R. Tao, N. R. Boynton, A. P. Kotula, C. A. Lindberg, K. J. Petersen, A. M. Forster and S. J. Rowan, ACS Macro Lett., 2024, 174–180 CrossRef CAS.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Chem. Sci., 2021, 12, 293–302 RSC.
- V. Zhang, C. Ou, I. Kevlishvili, C. M. Hemmingsen, J. V. Accardo, H. J. Kulik and J. A. Kalow, ACS Macro Lett., 2024, 13, 621–626 CrossRef CAS PubMed.
- H. Zhang, S. Majumdar, R. A. T. M. Van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 9, 272–277 CrossRef CAS.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Polym. Chem., 2020, 11, 5339–5345 RSC.
- Reviews in Computational Chemistry, ed. K. B. Lipkowitz, D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 1–86 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CAS.
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, Wiley, 2013 Search PubMed.
- R. van Meer, O. V. Gritsenko and E. J. Baerends, J. Chem. Theory Comput., 2014, 10, 4432–4441 CAS.
- S. Maes, F. Van Lijsebetten, J. M. Winne and F. E. Du Prez, Macromolecules, 2023, 56, 1934–1944 CAS.
- O. Tsuge, T. Itoh and M. Tashiro, Tetrahedron, 1967, 24, 2583–2590 Search PubMed.
- S. Baddi, S. S. Madugula, D. S. Sarma, Y. Soujanya and A. Palanisamy, Langmuir, 2016, 32, 889–899 CAS.
- T. A. Hamlin, P. Vermeeren, C. Fonseca Guerra and F. M. Bickelhaupt, Energy decomposition analysis in the context of quantitative molecular orbital theory, in Complementary Bonding Analysis, ed. S. Grabowsky, De Gruyter, Berlin. Boston, 2021, pp. 199–212 Search PubMed.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- C. Nieuwland, P. Vermeeren, F. M. Bickelhaupt and C. Fonseca Guerra, J. Comput. Chem., 2023, 44, 2108–2119 CrossRef CAS PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Chem. – Eur. J., 1999, 5, 3581–3594 CrossRef.
- C. Fonseca Guerra, H. Zijlstra, G. Paragi and F. M. Bickelhaupt, Chem. – Eur. J., 2011, 17, 12612–12622 CrossRef CAS.
- L. de Azevedo Santos, D. Cesario, P. Vermeeren, S. C. C. van der Lubbe, F. Nunzi and C. Fonseca Guerra, ChemPlusChem, 2022, 87, e202100541 Search PubMed.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Mater. Today, 2021, 51, 586–625 CAS.
- B. van Genabeek, B. F. M. de Waal, B. Ligt, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2017, 6(7), 674–678 CAS.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 Search PubMed.
- Y. Lai, X. Kuang, W. H. Yang, Y. Wang, P. Zhu, J. P. Li, X. Dong and D. J. Wang, Chin. J. Polym. Sci., 2021, 39, 154–163 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, 1H, 13C NMR spectra, MALDI-ToF-MS spectra, SEC chromatograms, crystal structure determination, details of model compound kinetics experiments, TGA thermograms, VTIR spectra, creep-recovery data, DSC thermograms, tensile tests, frequency sweep measurements and details of DFT analyses. CCDC 2377270 and 2377271. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py01296h |
| This journal is © The Royal Society of Chemistry 2025 |