
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe challenges and strategies towards high-performance anode-free post-lithium metal batteries
Jiawei
Wang
a,
Yaosong
Zhou
a,
Yanyi
Zhuo
a,
Kun
Fang
a,
Sicong
Wang
b,
Bin
Zhao
 *a,
Jing
Zhou
*c and
Hua
Wang
*b
*a,
Jing
Zhou
*c and
Hua
Wang
*b
aSchool of Materials and Chemistry, University of Shanghai for Science and Technology, Shanghai 200093, China
bSchool of Chemistry, Key Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, Beihang University, Beijing 100191, China. E-mail: wanghua8651@buaa.edu.cn
cSchool of Chemistry Engineering, Northeast Electric Power University, Jilin 132012, China. E-mail: zhoujing@neepu.edu.cn
First published on 11th December 2024
Abstract
With the merits of high theoretical energy density and ease of manufacture, anode-free post-lithium metal batteries have drawn extensive attention and have been rapidly emerging in recent years. However, the poor reversibility of metal anodes has severely hindered the realization of high-performance anode-free batteries. In this review, the critical challenges and strategies for achieving high-performance anode-free metal batteries are first elucidated. Then, the significant research studies devoted to promoting the reversibility of metal anodes for anode-free post-lithium (including sodium-, potassium- and zinc-) metal batteries are exclusively discussed to extract the principles for their practical implementation. Additionally, remedial solutions of supplying metal to the anode for improving the cyclability of anode-free batteries are also introduced. Finally, we summarize the advancements in anode-free post-lithium metal batteries, and propose some promising directions in this area. This review aims to provide a more comprehensive understanding towards the strategies for achieving highly reversible anode-free post-lithium metal batteries and a timely overview of the latest developments in this emerging field.
1 Introduction
Metal batteries, the negative electrodes of which consist of electrochemically active metals, have been experiencing a renaissance over the last decade.1–4 Compared with traditional ion batteries, such as lithium-ion batteries, using metal anodes greatly increases the areal capacity of the negative electrode, thus enhancing the theoretical energy density of the batteries.1 Moreover, metal anodes can readily match with some highly abundant but metal-ion-free cathodes, enabling the construction of high-energy battery systems like metal–sulfur and metal–air batteries.5,6 With the continuous improvement in the safety and reversibility of metal anodes, which are the greatest concerns for the practical application of metal batteries, metal batteries have shown great development promise.7,8 Lithium metal batteries are most extensively developed due to the first-mover advantage of the closely related lithium-ion batteries industry. However, the growing demand for lithium in commercial lithium-ion batteries is driving up costs, making lithium metal batteries less economically attractive.9 In search of more economical alternatives to lithium metal batteries, researchers have turned to post-lithium metal batteries,10 such as sodium-metal,11 potassium-metal12 and zinc-metal13 batteries, with the related progress being rapidly updated. Since post-lithium metal batteries share some common issues with lithium-metal batteries, drawing on the abundant knowledge developed in lithium-metal batteries, post-lithium metal batteries have become important research fields within just a few years.Although post-lithium metal batteries, including sodium-, potassium- and zinc-metal batteries, are seen as promising candidates for next-generation batteries, the metal anodes in these batteries generally act as both the active material and the current collector. This results in excessive metal usage on the anode side, thus significantly reducing the actual energy density and economic benefits of metal batteries. Moreover, the batteries with metal as the negative electrode increase safety issues and process complexity in manufacture, especially for highly reactive alkali metal anodes. In this context, the so-called “anode-free” battery configuration has emerged as a promising solution for safer and more facile construction of high-energy-density metal batteries.14–16 As shown in Fig. 1, in the anode-free metal battery configuration, the initial composition of the negative electrode is merely a current collector without anode materials. The metal anode is generated by metal plating on the current collector during the battery's first charging process, and the pristine cathode material is necessarily in a fully reduced state and generally serves as the ion source for the metal anode. This anode-free configuration prevents the excessive use of metal on the negative electrode side where the capacity of the plated metal is theoretically equal to the charge capacity of the cathode, thus maximizing the energy density of the negative electrode. The advantage of the anode-free configuration in terms of theoretical energy density has been well elucidated in previous works.14,15 Moreover, avoiding the processing of reactive metal anodes greatly simplifies the manufacturing of anode-free metal batteries. Therefore, anode-free post-lithium metal batteries show great potential for high-density energy storage and have generated broad research interest.
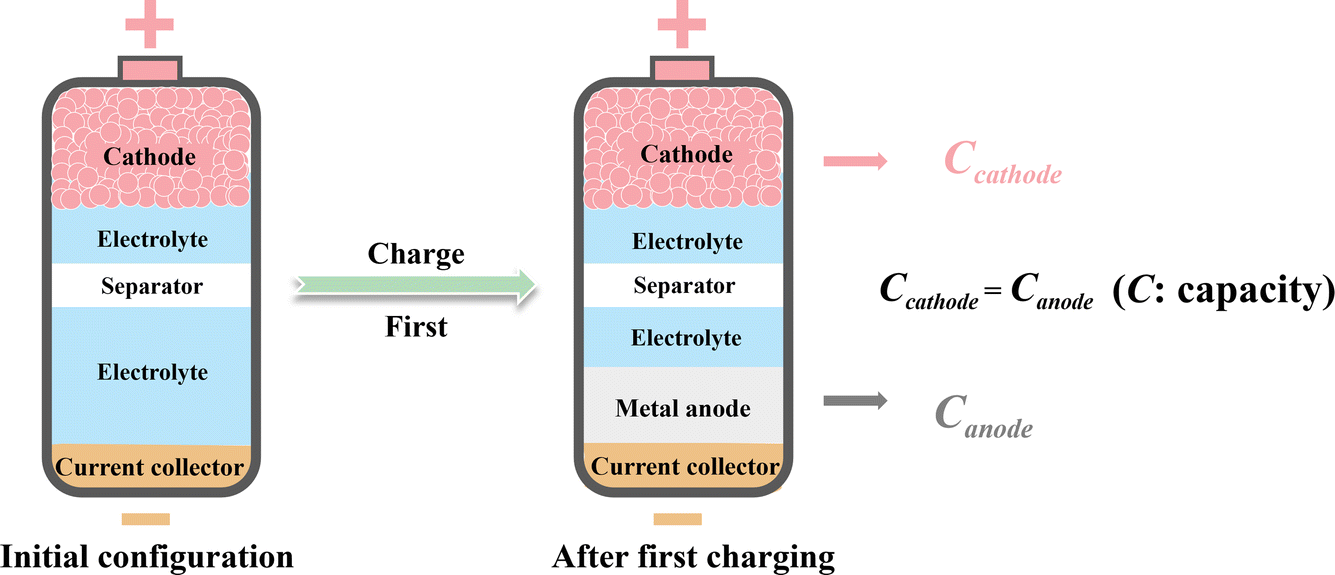 | ||
| Fig. 1 The anode-free metal battery configuration and the relationship between the theoretical capacities of the cathode and anode. | ||
Despite the benefits offered by the appealing structure of anode-free batteries, specific problems with these types of batteries greatly hinder their practical application. For example, due to the lack of excess capacities on both the cathode and anode sides, the battery performance degrades as a result of decay at either electrode, which necessitates high reversibility of both the anode materials and the cathode. Additionally, cathodes in the reduced state should be prepared to satisfy the first charging of anode-free batteries. These requirements pose new challenges to the development of anode-free post-lithium metal batteries. Increasing research studies are being reported in this appealing field, especially in anode-free sodium-,17,18 potassium-19,20 and zinc-metal21 batteries, which need to be timely overviewed to provide a deeper understanding towards their challenges, strategies and recent advances. Compared with the many reviews on anode-free lithium metal batteries,16,22,23 a review that encompasses diverse anode-free post-lithium metal batteries has been rarely reported.24,25 In this review, based on typical research works on anode-free post-lithium (including sodium-, potassium- and zinc-) metal batteries, we aim to provide a comprehensive overview towards the development of anode-free post-lithium metal batteries. First, the general challenges and strategies for constructing anode-free metal batteries are illustrated, and the reversibility issues of the metal anodes are discussed. Then, in response to these challenges, the strategies for constructing high-performance anode-free post-lithium batteries, including sodium-, potassium- and zinc-metal batteries, are classified and introduced. Not only are the strategies towards promoting the reversibility of the metal anodes discussed, but also the remedial strategies for the irreversible metal consumption on the anode are introduced. Finally, prospective views on anode-free post-lithium metal batteries are presented. This work is intended to provide guidance for the researchers in related fields.
2 Challenges for anode-free metal batteries
As the simple configuration of anode-free batteries endows them with easier assembly and higher theoretical energy density compared with traditional metal batteries, numerous challenges arise in the pursuit of high battery performance. Since the theoretical N/P ratio (the capacity ratio of the negative to positive electrode) is equal to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 after the first charging, the degradation of either electrode dictates the battery performance, including capacity and cycling performance (as illustrated in Fig. 2). Therefore, to achieve anode-free batteries with satisfactory capacity and cycling performance, achieving high coulombic efficiency (CE) and capacity retention for both electrodes is essential. In this regard, promoting the reversibility of the metal anode as well as the cathode is the most critical task for high-performance anode-free metal batteries.
:1 after the first charging, the degradation of either electrode dictates the battery performance, including capacity and cycling performance (as illustrated in Fig. 2). Therefore, to achieve anode-free batteries with satisfactory capacity and cycling performance, achieving high coulombic efficiency (CE) and capacity retention for both electrodes is essential. In this regard, promoting the reversibility of the metal anode as well as the cathode is the most critical task for high-performance anode-free metal batteries.
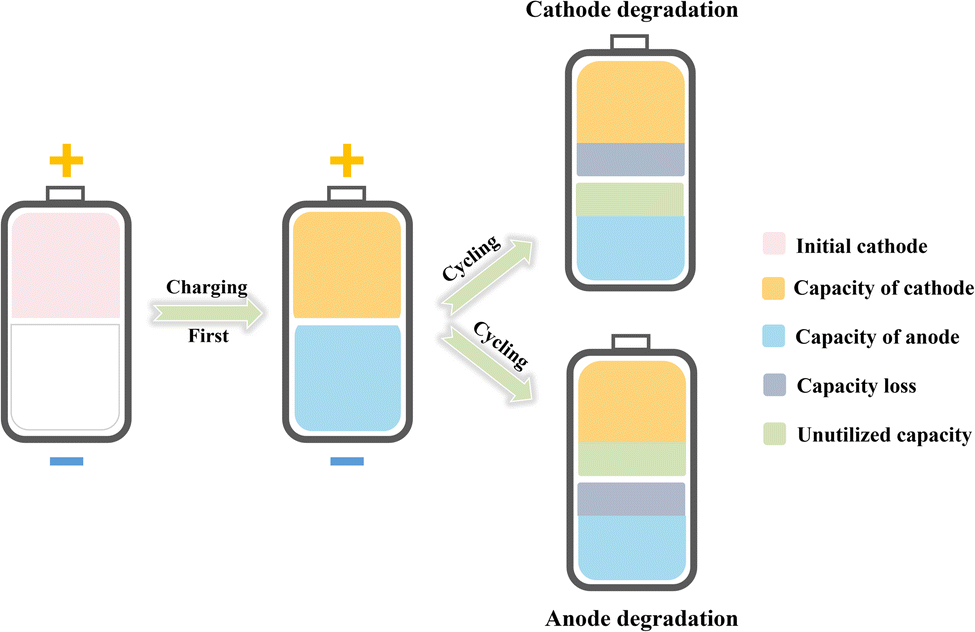 | ||
| Fig. 2 Illustration of the performance of anode-free metal batteries dictated by capacity decay of either electrode. | ||
Generally, the challenges on the anode side are predominant for anode-free metal batteries due to the difficulty in managing reversible metal plating and stripping. Similar to the problems associated with traditional metal anodes,26,27 the non-uniform metal plating and the intractable electrolyte-involved side reactions on the metal anode seriously suppress the reversibility of the electrode (Fig. 3). The persistent loss of active materials on the plated metal anode, such as repeated solid electrolyte interphase (SEI) formation and metal corrosion, results in rapid battery degradation, and the metal dendrites and byproducts can accelerate battery failure. In this regard, strategies that enhance the reversibility of traditional metal anodes also apply to anode-free metal batteries, with the exception that the current collectors have a more critical impact on the anode performance for the latter. Specifically, these strategies involve engineering metal plating substrates and modulating the electrolyte. In addition to improving the reversibility of metal plating/stripping, the remedial strategies that compensate for the active material loss on the anode side are also effective in maintaining the performance of anode-free metal batteries. This kind of strategy is intended to offset the metal loss on anode sides to ensure adequate anode capacity retention after cycling, which might include various means, such as current collector activation, cathode pre-metallization and adopting metal-supply agents. Regarding promoting the reversibility of cathodes, the strategies are similar to those for traditional ion batteries and thus are not exclusively discussed in this work. Therefore, in the following sections, representative studies engaged in promoting the reversibility of metal plating/stripping and compensating for anode loss in anode-free post-lithium (sodium-, potassium- and zinc-) metal batteries are introduced, and the involved strategies are discussed in order to extract the general principles for constructing high-performance anode-free post-lithium metal batteries.
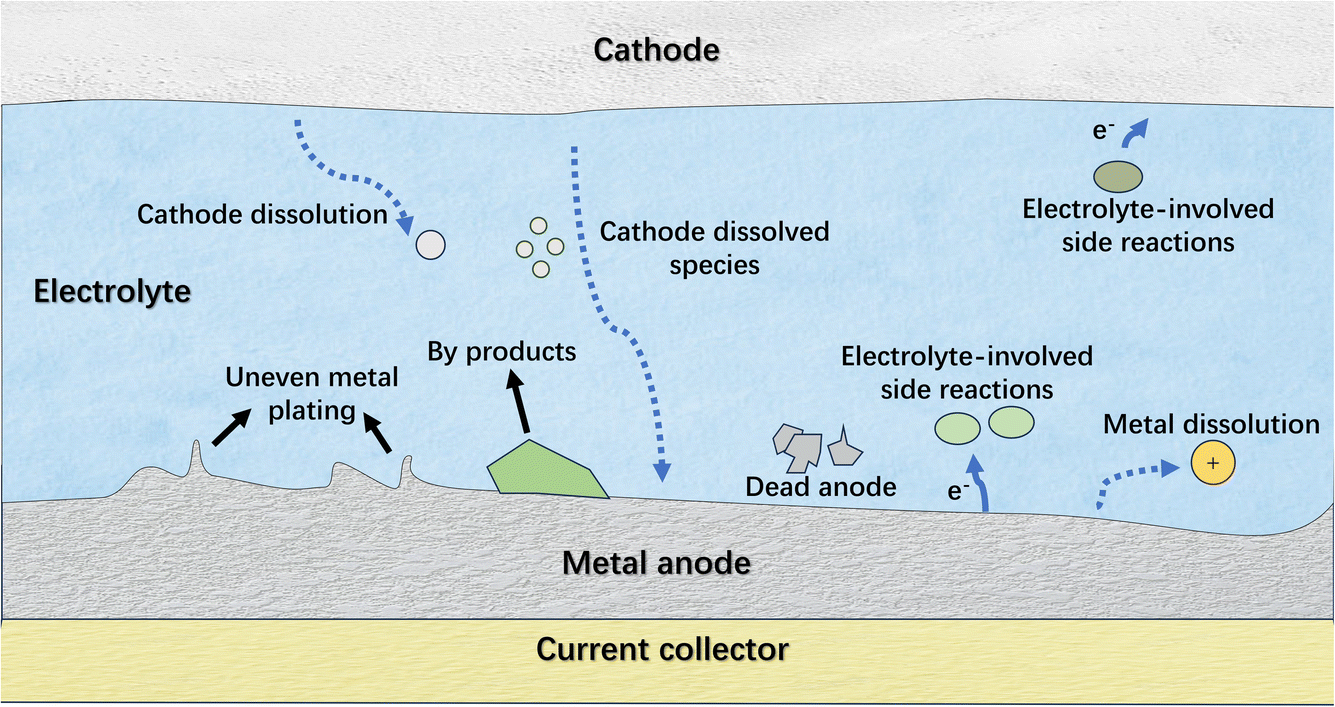 | ||
| Fig. 3 The schematic illustration of the critical issues of anode-free metal batteries. | ||
3 Improving the reversibility of metal plating/stripping
The reversibility of metal anodes is affected by various aspects. In general, efforts to promote the reversibility of metal plating/stripping involve two main strategies, including engineering the current collector and modulating the electrolyte.3.1 Engineering the current collector
As the current collector of the negative electrode acts as the metal plating substrate in anode-free batteries, the physicochemical properties of the current collector directly influence the metal plating behavior and the side reactions on the electrode.28–30 Hence, engineering the current collector has become one of the most widely exploited strategies to promote the reversibility of metal plating/stripping. With regard to metal plating behavior, the properties of the current collector, such as its affinity with the plated metal/metal ion, the degree of crystalline matching with the deposited metal and the nucleation/plating sites provided for metal deposition, have a significant impact on the kinetics of the metal plating process, the crystalline characteristics of the metal deposit and the morphology of the formed metal anode, further affecting the reversibility of the electrode. Besides, the architecture of the current collector, which determines how the deposited metal is accommodated, is closely correlated with the morphology of the metal deposit and the volume change of the anode during charging/discharging.31 With regard to the side reactions on the anode side, the current collector can affect the reactivity of the electrolyte species and even directly participate in the side reactions. Therefore, the current collector can affect the performance of the anode from diverse aspects, giving rise to various strategies of current collector engineering, each emphasizing different aspects.A high affinity between the current collector and the deposited metal can facilitate uniform metal plating. One popular method is to decorate the current collector with materials that have a high binding energy to the deposited metal atoms. Metals, including Au,32 Ag,33,34 Zn35,36 and Sn37 have been demonstrated to have high binding energy with Na atoms (i.e., “sodiophilic”). Mao et al. modified the Cu current collector with Au sputtering. The larger Na binding energy of Au (−1.947 eV) compared to Cu (−1.379 eV) facilitates uniform Na plating, and the in situ formed Au2Na alloy shows even larger sodiophilicity (Na binding energy of −2.431 eV), thus enabling the smooth morphology of the electrode after cycling (Fig. 4a and b).32 Similarly, Zn was found to be a superior current collector for the Na anode compared to conventional Cu and Al foil,35,36 due to the high binding energy of Na atoms with not only Zn but also in situ formed Zn–Na alloy. The Zn current collector exhibited a high Na plating/stripping efficiency of 99.7% (Fig. 4c),35 and the fabricated anode-free Zn//Na3V2(PO4)3 (NVP) cell exhibited 87% capacity retention after 100 cycles (Fig. 4d). For K metal plating, Bi metal was used as a potassiophilic material for current collector modification. Zhang et al. modified Ti3C2/CNT/rGO aerogel with Bi nanoparticles (Bi/CTG), which was adopted as a modification layer on the Cu current collector for K plating.38 The high affinity between K and Bi not only guides the uniform K deposition but also allows for a stable SEI (Fig. 4e and f), thus enabling a smooth and stable electrode interface of the modified current collector after the plating/stripping process compared to the pristine Cu current collector.38 The Bi/CTG@Cu current collector enabled K plating/stripping with a high average CE of 99.4% after 1100 cycles, whereas the fabricated anode-free K full battery with a K1.69Fe[Fe(CN)6]0.90 (PB) cathode exhibited a CE of merely 86.2%, suggesting the necessity of further investigation into its performance. With regard to anode-free zinc metal batteries, metals such as Ag,40 Sb39,41 and Sn,42 have been proven to be zincophilic and have been utilized for current collector modification. For example, a Sb layer was electroplated on Cu foil as a current collector for Zn plating.39 The more delocalized electron distribution on the deposited Zn atoms on the Sb@Cu substrate compared to that on the Zn substrate is demonstrated by density functional theory (DFT) calculations, suggesting enhanced bonding between the Zn and Sb layer (Fig. 4g and h). The formed Sb/Sb2Zn3 interface facilitated Zn nucleation and promoted uniform Zn2+ flux distribution. Benefiting from the above merits, improved morphological uniformity of Zn deposits was observed on the Sb@Cu current collector at a high plating areal capacity of 50 mA h cm−2 (Fig. 4i and j). Notably, with the Sb@Cu current collector, the anode-free Zn–Br2 battery stably operated for 40 cycles at an ultrahigh charging areal capacity of 200 mA h cm−2, showing great potential of this strategy for fabricating current collectors of anode-free zinc–metal batteries.
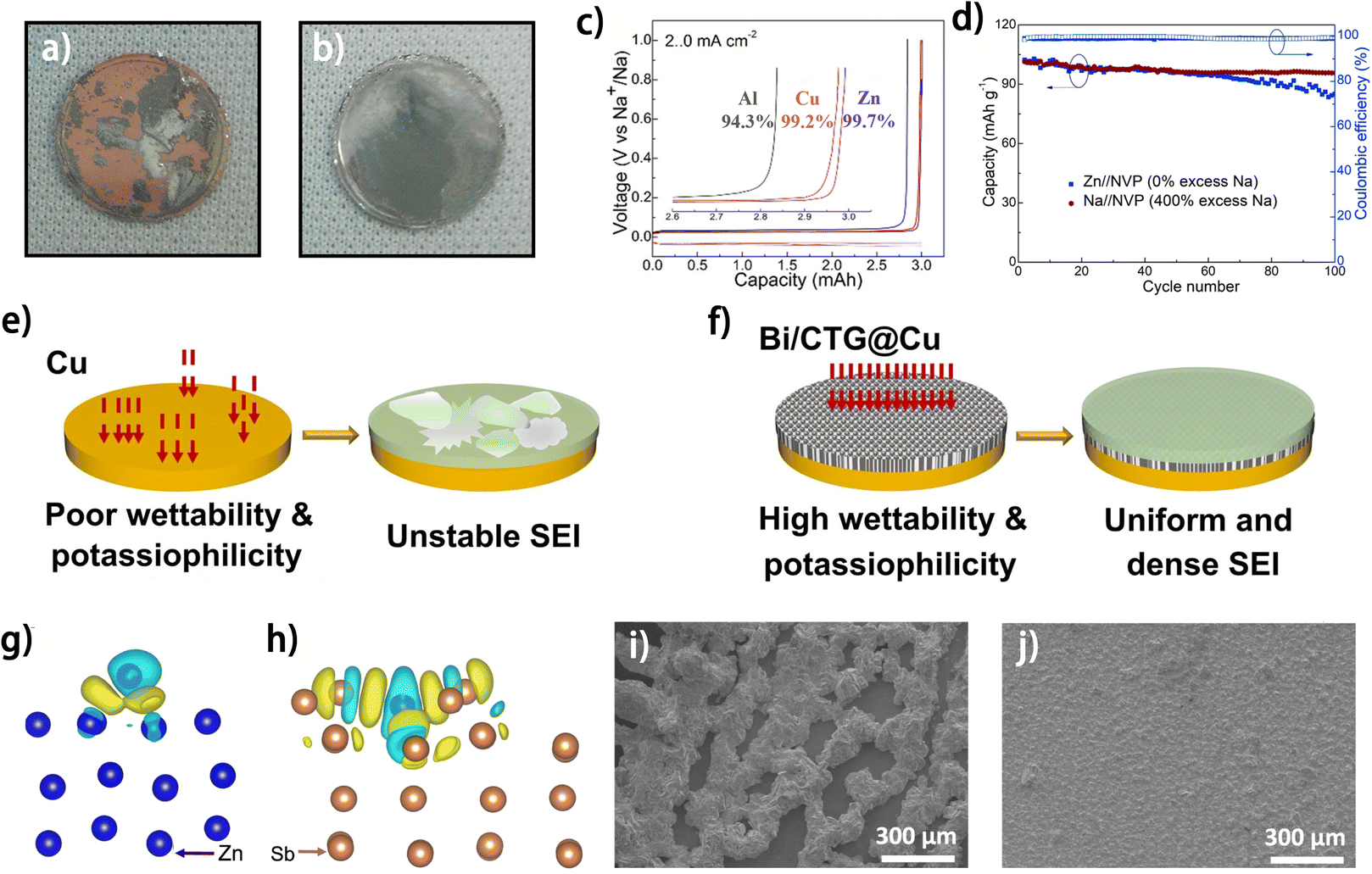 | ||
| Fig. 4 Optical photographs of Na plating with 1.0 mA cm−2 at the 10th cycle on (a) Cu and (b) Cu@Au; reproduced from ref. 32 with permission from Elsevier, copyright 2018. (c) Voltage profiles of half cells showing the plating–stripping efficiencies of Zn, Cu, and Al at 2 mA cm−2 and 3 mA h cm−2. (d) Capacity retention and CE of Zn//NVP and Na//NVP full cells. (c and d) Are reproduced from ref. 35 with permission from The Royal Society of Chemistry, copyright 2023. Schematic diagram of K metal plating/stripping on (e) Cu and (f) Bi/CTG@Cu; reproduced from ref. 38 with permission from Elsevier, copyright 2024. DFT calculation models with differential charge density of (g) Zn (100) and (h) Sb (104).39 Zn electrodeposited on (i) Zn and (j) Sb@Cu substrates with areal capacities of 50 mA h cm−2.39 | ||
Apart from metals and in situ formed alloys, the direct use of alloy matrices or intermetallic compounds has been proven to be feasible for guiding uniform metal plating in anode-free batteries. The alloys with high affinity towards the deposited metal generally function to increase metal nucleation sites and to mitigate the nucleation energy barrier, thus modulating the current distribution on the substrate during metal plating. Chen et al. constructed a potassiophilic Cu6Sn5 layer on commercial Cu foil (Cu6Sn5@Cu) as the current collector for K plating.19 At a plating capacity of 2 mA h cm−2, the K deposit exhibits a considerably more compact and smoother morphology on Cu6Sn5@Cu compared to that on commercial Cu foil (Fig. 5a and b).19 This result is explained by the higher adsorption energy (Ea) of K-atoms on Cu6Sn5 crystal planes compared with that on K metal, as demonstrated by DFT calculations (Fig. 5c). The highest Ea of K-atom adsorption can be found at a single K atom on Cu6Sn5, suggesting a uniform nucleation process on this alloy layer from a thermodynamic perspective. The dimensionless current–time transient profiles of K deposition show that, compared with the 2D progressive K nucleation (2DP) on Cu foil, Cu6Sn5@Cu exhibits 3D instantaneous (3DI) nucleation (Fig. 5d), which is theoretically beneficial for achieving uniform K metal nucleation on the substrate (Fig. 5e). Similarly, Cu3Zn alloy-coated Cu powder (Cu@Cu3Zn) was prepared as a Zn plating substrate for anode-free zinc metal batteries (Fig. 5f).43 The Cu@Cu3Zn modified-carbon-coated Cu foil (CCF) shows decreased Zn nucleation overpotential compared to the pristine CCF, and the “dead” Zn problem is also alleviated. As a result, the Cu@Cu3Zn modified CCF//Zn3V3O8 anode-free zinc metal battery exhibits 80% capacity retention after 200 charge/discharge cycles,43 demonstrating the effectiveness of this alloy layer in improving the reversibility of the Zn anode. Wang et al. prepared a sodium antimony telluride intermetallic (NST)–Na metal composite as the sodium metal anode.45 By further electrochemically stripping the active Na metal from the composite, a thermodynamically stable intermetallic matrix was obtained. The NST skeleton showed preferential formation of individual Na atoms instead of clusters as indicated by the calculated binding energies, thus enabling a complete Na coverage on the substrate during the Na nucleation period and planar deposition subsequently.45 During anodic polarization, the NST skeleton exhibits negligible structural changes, thus ensuring its stable function as the plating substrate. Consequently, the anode-free NST//NVP cell shows an initial CE of 92.3% and a capacity retention of approximately 65% after 100 galvanic cycles at 1C. Besides increasing binding energies towards deposited metal atoms, the affinity with metal ions can also be modulated by intermetallic compounds. Copper phosphide (Cu3P) nanowires were constructed on Cu foil as a Na plating substrate by an in situ growth and transformation method.44 It was found that Cu3P not only decreased the Na nucleation overpotential, but also exhibited lower Na+ adsorption energy and smaller Na+ diffusion energy compared to Cu (Fig. 5g), which together contribute to more uniform Na deposition (Fig. 5h and i). This unique current collector supports Na plating/stripping for 250 cycles with an average CE of 99.12% at 4 mA cm−2 and 4 mA h cm−2. The anode-free NVP//Cu3P@Cu full cell shows an initial capacity decay followed by stable cycling, which may be related to the moderate initial CE of 81.1% (Fig. 5j). The alloys and intermetallic compounds show great potential for guiding uniform metal plating and promoting CE of the metal anodes for anode-free metal batteries. Nevertheless, the inadequate mechanical strength or structural instability of the alloy matrixes may impede their application as current collectors, while adding supporting substrates would severely decrease the energy density of the anode. Considering the abundance of these kinds of materials, deeper and extensive research into these materials as metal plating substrates for anode-free batteries can be expected.
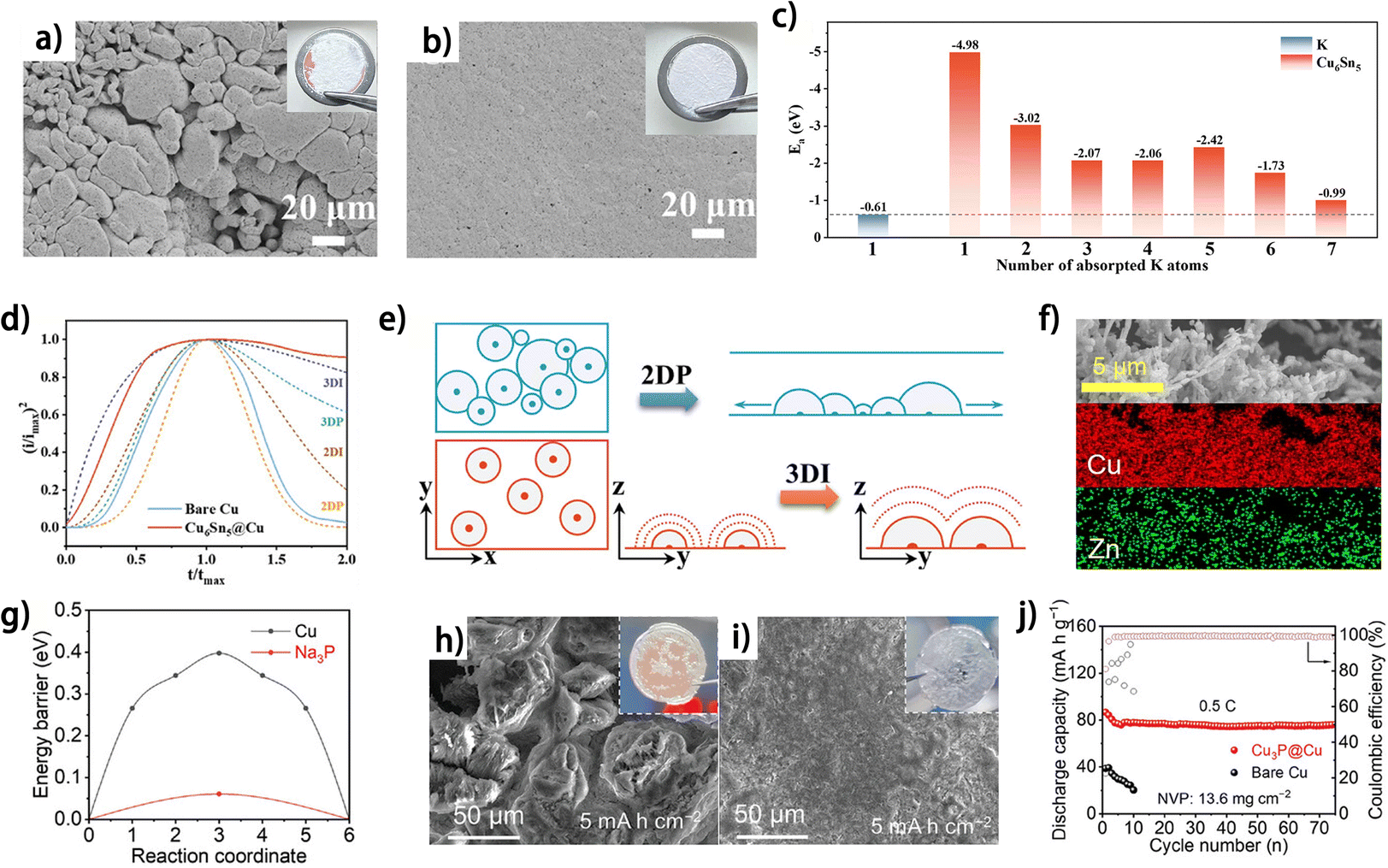 | ||
| Fig. 5 SEM images of K electrodeposition on (a) bare Cu and (b) Cu6Sn5@Cu with a capacity of 2 mA h cm−2 (the inset shows the optical photographs of K deposition morphology on (a) bare Cu and (b) Cu6Sn5@Cu current collectors). (c) Adsorption energy (Ea) of K-atoms on K (110) and Cu6Sn5 (22−1) crystal planes. (d) Dimensionless current–time transient for the K deposition process in comparison with theoretical 2D and 3D models (imax: peak current; tmax: time needed to achieve the peak current). (e) Schematic illustration of 2DP (BFT models) and 3DI (SH models) (x–y is parallel to the substrate; y–z is vertical to the substrate). (a–e) Are reproduced from ref. 19 with permission from Wiley, copyright 2024. (f) EDS mapping images of the Cu@Cu3Zn powder; reproduced from ref. 43 with permission from Science Press and Dalian Institute of Chemical Physics, Chinese Academy of Sciences, copyright 2022. (g) Na+ diffusion energy barrier of Cu and Na3P. SEM and optical images of (h) Cu3P@Cu and (i) bare Cu anodes at 5 mA h cm−2. (j) Cycling performance of Cu3P@Cu//NVP anode-free pouch cells at 0.5 C. (g–j) Are reproduced from ref. 44 with permission from Wiley, copyright 2024. | ||
Carbon materials have been widely adopted as metal nucleation layers on the current collector for metal plating in anode-free sodium-,46–49 potassium-50 and zinc-metal 51,52 batteries. Carbon black was reported as a superior Na plating substrate to Al foil, which was reflected by the increased nucleation sites and improved CE of Na plating/stripping.46 Hu et al. demonstrated the advantages of graphite carbon (GC) as a Na plating substrate compared to active carbon (AC) in diverse dimensions.47 First, the higher porosity of GC is conducive to electrolyte wetting, and the fewer defects of GC endow it with larger electron conductivity, thus facilitating electrode reactions on the GC substrate. Second, instead of ion adsorption-like behavior occurring on AC, the GC exhibits both ion adsorption and ion/solvent co-intercalation behaviors, as evidenced by the existence of both sloping and plateau regions in the initial charging profile of GC in an ether-based electrolyte,53 which accounts for the higher initial CE of GC (82%) compared to that of AC (44%) (Fig. 6a). Moreover, the Na nucleation overpotential on GC (16.4 mV) is also smaller than that on AC (25.7 mV), indicating the more sodiophilic nature of GC. These factors collectively contribute to uniform and reversible Na plating/stripping. The promoting effect of the defective structures in carbons on metal nucleation has been elucidated in anode-free sodium-48,49,54,55 and potassium-metal batteries.50 By treating carbon-coated aluminum (Al@C) foil with high-energy air plasma, our group introduced active sites into the carbon material for Na atom adsorption and nucleation.48,49 It was found that the air plasma treatment produced abundant pyridinic N and pyrrolic N structures (Fig. 6b),48 with the electron-deficient states improving the affinity between these sites and Na atoms. The defects endow the carbon matrix with higher adsorption energy to Na (Fig. 6c and d).48 The improved sodiophilicity of the defect-rich carbon contributes to a much smoother and denser Na deposit (Fig. 6e) compared to the rough and inhomogeneous Na morphology on pristine carbon (Fig. 6f) at a current density of 7 mA cm−2, and the CE of Na plating/stripping was also promoted by the defect-rich carbon, especially at high current densities.49 Sun et al. fabricated a large-scale defect-rich graphene-coated Al foil sheet (Al@G) by roll-to-roll plasma-enhanced chemical vapor deposition on commercial Al foil.50 Compared with pristine Al foil and carbon-coated Al foil (Al@C), the defect-rich graphene layer with high surface energy exhibited superior wettability of liquid potassium and decreased K nucleation energy. Consequently, the Al@G current collector showed a high average CE of 99% for K plating/stripping over 1000 h. Nevertheless, the advantage of this Al@G current collector is not as great as expected in the anode-free K–FeS2//Al@G full cell (Fig. 6g),50 implying large room for improvement in its practical application. Carbon nanodiscs have also been demonstrated as a nucleation layer on Cu foil (C/Cu) for Zn plating in aqueous electrolyte.51 More evenly distributed Zn plating was observed on C/Cu in contrast to Cu foil (Fig. 6h and i) owing to the increased nucleation sites. The smooth Zn deposition results in an increase in the average CE of Zn plating/stripping from 93.4% (on Cu) to 99.2% (on C/Cu). Paired with a pre-zincificated MnO2 cathode, the C/Cu current collector supported the cycling of an anode-free zinc metal battery for 80 cycles with a capacity retention of 68.2% (Fig. 6j). Interestingly, this capacity retention surpassed the theoretical value calculated based on the average CE of the half cell (52.6% capacity retention after 80 cycles with a CE of 99.2%) (Fig. 6k), which requires a deeper investigation into the effect of different battery types on the reversibility of the anode.
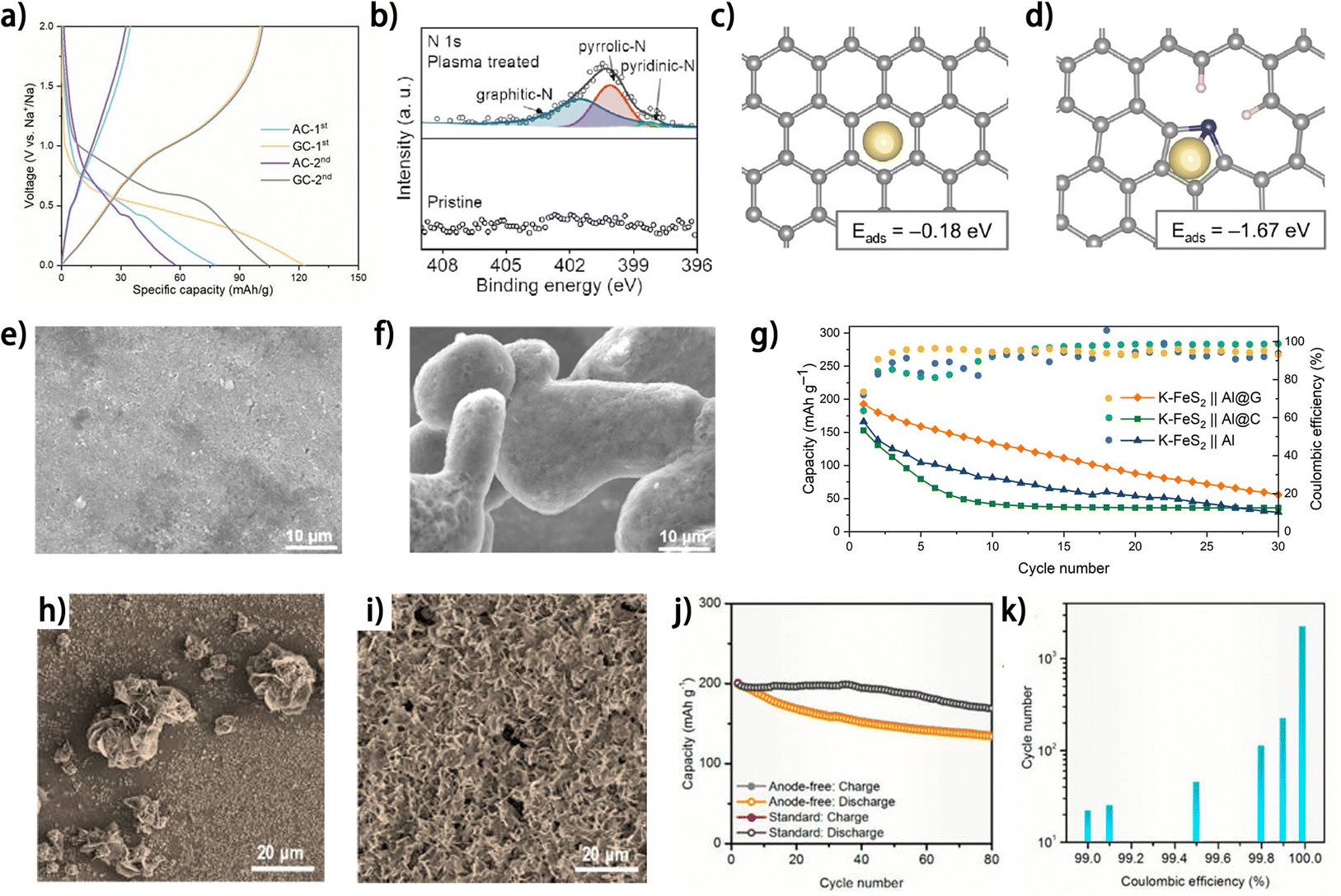 | ||
| Fig. 6 (a) Galvanostatic voltage profiles of AC and GC at 0.6 C (1C = 100 mA g−1) in 1 M NaPF6 in diglyme.47 (b) N 1s XPS spectra of pristine Al@C and plasma-treated Al@C and relaxed configurations of the Na atom absorbed on (c) perfect carbon and (d) N-doped carbon; reproduced from ref. 48 with permission from Elsevier, copyright 2024. SEM images of Na deposition morphology on (e) Al/N–C and (f) Al/C after 5 cycles at 7 mA cm−2; reproduced from ref. 49 with permission from Elsevier, copyright 2024. (g) Cycling performances of different anode-free full-cells at 0.1 A g−1; reproduced from ref. 50 with permission from Wiley, copyright 2022. SEM images of (h) Cu foil after plating for 30 min at 1 mA cm−2; (i) C/Cu after plating for 1 h at 1 mA cm−2. (j) Cycling stability of the pre-zincificated MnO2‖C/Cu anode-free full battery and standard Zn–MnO2 battery at 1 mA cm−2. (k) The predicted cycle life of a battery vs. the CE of its anode. Here, a cathode without performance degradation is assumed, and battery failure happens if its capacity degrades to 80% of the original value. (h–k) Are reproduced from ref. 51 with permission from American Chemical Society, copyright 2021. | ||
The charge state of the carbon substrate has a great impact on the binding energy between the deposited metal atoms and the carbons, which can be regulated by constructing specific heterogeneous structures. The charge transfer between the adjacent components of the heterogenous interface can result in partial charge accumulation/depletion in the carbon component, which has been reported to be conducive to promoting binding energy of deposited metal atoms on it.54,55 Lee et al. prepared porous carbon particles containing carbon-shell-coated Fe nanoparticles (PC-CFe) as a Na plating substrate (Fig. 7a).54 The Fe nanoparticle donates electrons to the surrounding graphene layer and contributes to a stronger bond between Na atoms and the carbon (Fig. 7b). The degree of this charge transfer was found to depend on the type of metal material, as shown by the highest Na adsorption energy on the Fe/C heterogenous interface compared to other metal-based counterpart structures, such as Ag, Sn and Mg, which was attributed to strong hybridization between the C 2p and Fe 3d orbitals. Consequently, the PC-CFe substrate enabled a smooth Na morphology at a high plating capacity of 10 mA h cm−2 (Fig. 7c), and a superior capacity retention of ∼97% for the PC-CFe-based anode-free battery was achieved (Fig. 7d). Besides metals, materials with specific functional groups can also induce charge transfer from/to carbon when they are composited into a heterogenous structure, which was demonstrated with Ti3C2Tx/CNT nano-accordion frameworks (NAFs) (Fig. 7e) by Son et al.55 DFT calculation results indicated that the different functional groups (–OH, –F and –O) endow the surfaces of Ti3C2Tx with divergent work functions, which leads to different charge transfer directions between the CNT and the adjoining Ti3C2Tx as well as distinct partial charges on the CNT (qCNT) (Fig. 7f). Interestingly, both positive and negative divergences in qCNT result in improved sodiophilicity of the material compared to the pristine graphite model, implying the significance of charge density on carbon materials. As a result, the Ti3C2Tx/CNT NAFs exhibited decreased Na metal nucleation overpotential and higher CE for Na plating/stripping in contrast to the single Ti3C2Tx and single CNT counterpart substrate.
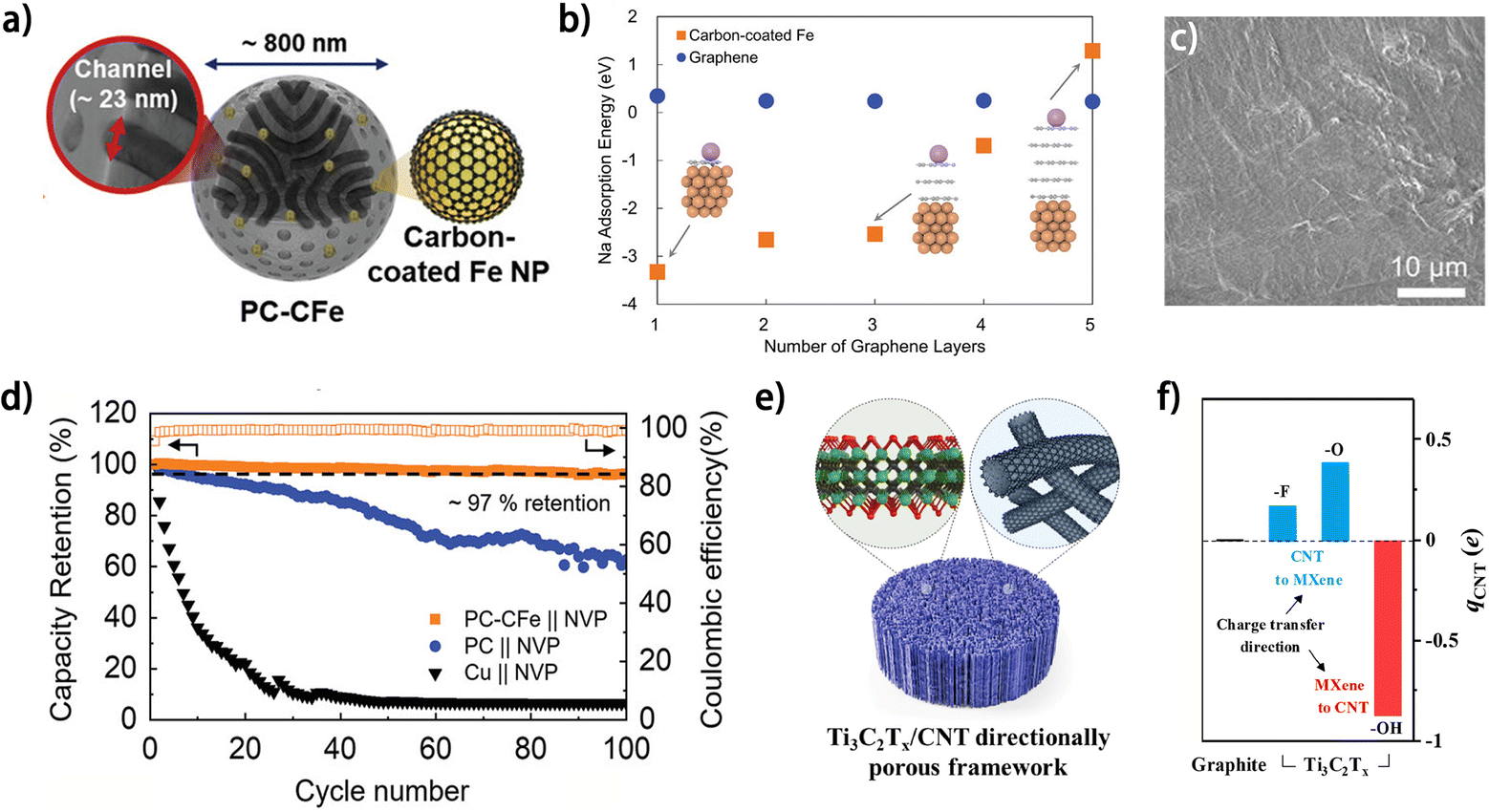 | ||
| Fig. 7 (a) Schematic illustration of the structure of PC-CFe. (b) Na adsorption energy on graphene and carbon-coated Fe as a function of the number of carbon layers. (c) SEM images of plated Na after 40 cycles on PC-CFe at 10 mA cm−2. (d) Cycling stability of the anode-free cells utilizing PC-CFe, PC, and Cu foil with a high-loading NVP cathode (10 mg cm−2) at 1 mA cm−2. (a–d) Are reproduced from ref. 54 with permission from Elsevier, copyright 2023. (e) Schematic illustration of Ti3C2Tx/CNT directionally porous framework.55 (f) The sum of the partial charge of the CNT. (e and f) Are reproduced from ref. 55 with permission from Elsevier, copyright 2023. | ||
Besides the high affinity between metal atoms and the current collector, fast and uniform metal ion diffusion at the electrode/electrolyte interface is also critical for the morphology of the metal deposit and the CE of metal plating/stripping.56 Modifying the current collector with a layer guiding metal ion flux is an effective strategy to promote the reversibility of the metal anode. This design principle for a Na plating substrate was demonstrated by Zhuang et al. using a porous fluorinated covalent triazine framework (FCTF), which possesses both high sodiophilicity and low reactivity with Na+.57 Compared with the nonfluorinated covalent triazine framework (CTF), the FCTF showed a higher affinity with Na atoms, thus enabling smoother Na deposition. More importantly, the depletion of Na+ trapped in the interphase was found to be mitigated on the FCTF (Fig. 8a),57 implying superior reversibility of Na plating/stripping compared to the CTF. This increased Na plating efficiency was attributed to weakened interaction between the FCTF and Na+ due to the integration of fluorine into the framework (Fig. 8b), which enabled a higher proportion of Na0, rather than Na+, to react with the substrate during Na plating (Fig. 8c). However, this deduction has not been theoretically elucidated, and further investigation into the design of substrates with both high affinity for metals and low reactivity with metal ions is still needed. In another work, the fast diffusion of Na+ at the electrode/electrolyte interface was achieved by modifying Cu foil with a HCOONa layer (Fig. 8d).58 The formed homogeneous HCOONa layer not only protected the Na deposit from corrosion caused by the electrolyte but also lowered the Na+ diffusion energy barrier in contrast to some common components in the SEI (Fig. 8e). The fast Na+ diffusion enabled by the HCOONa layer facilitated not only Na plating but also Na stripping, which contributed to more complete Na stripping from Cu foil and greatly alleviated the “dead” Na problem (Fig. 8f and g), indicating the great significance of the ion diffusion kinetics at the electrode/electrolyte interface. For anode-free zinc metal batteries, materials with diverse properties and structures have been adopted as modification layers on current collectors to guide Zn2+ diffusion, including nucleophilic materials,60 hydrophilic zincophilic materials,61 porous ion sieves59 and polymer electrolytes.62 The fast desolvation and diffusion of Zn2+ were reported to be facilitated by an aluminum hydroxide fluoride layer on Cu foil (Cu@AOF), which supported a high average CE of 99.90% for Zn plating/stripping and 80% capacity retention in anode-free Cu@AOF‖Zn0.5VO2 batteries after 300 cycles.61 Wu and Bai et al. manipulated the fluxes of both metal ions and anions at the electrode surface for reversible Zn plating/stripping using a Zn ion-exchanged zeolite layer (ZnA).59 The structure of zeolite was screened based on its pore structure and charge state, with no pores larger than 4.84 Å (the diameter of a SO42− ion) necessary for sieving sulfate ions and thus suppressing anion-involved side reactions (Fig. 8h),59 whereas the negatively charged surface would facilitate Zn2+ diffusion and contribute to smooth Zn deposition (Fig. 8i). The ZnA-coated Cu current collector allowed for 74.6% capacity retention for an anode-free battery (with a pre-zincated α-MnO2 cathode) after 90 cycles. These studies demonstrate the importance of manipulating the diffusion of metal ions for reversible metal anodes and provide inspiration for surface modification of current collectors for anode-free metal batteries.
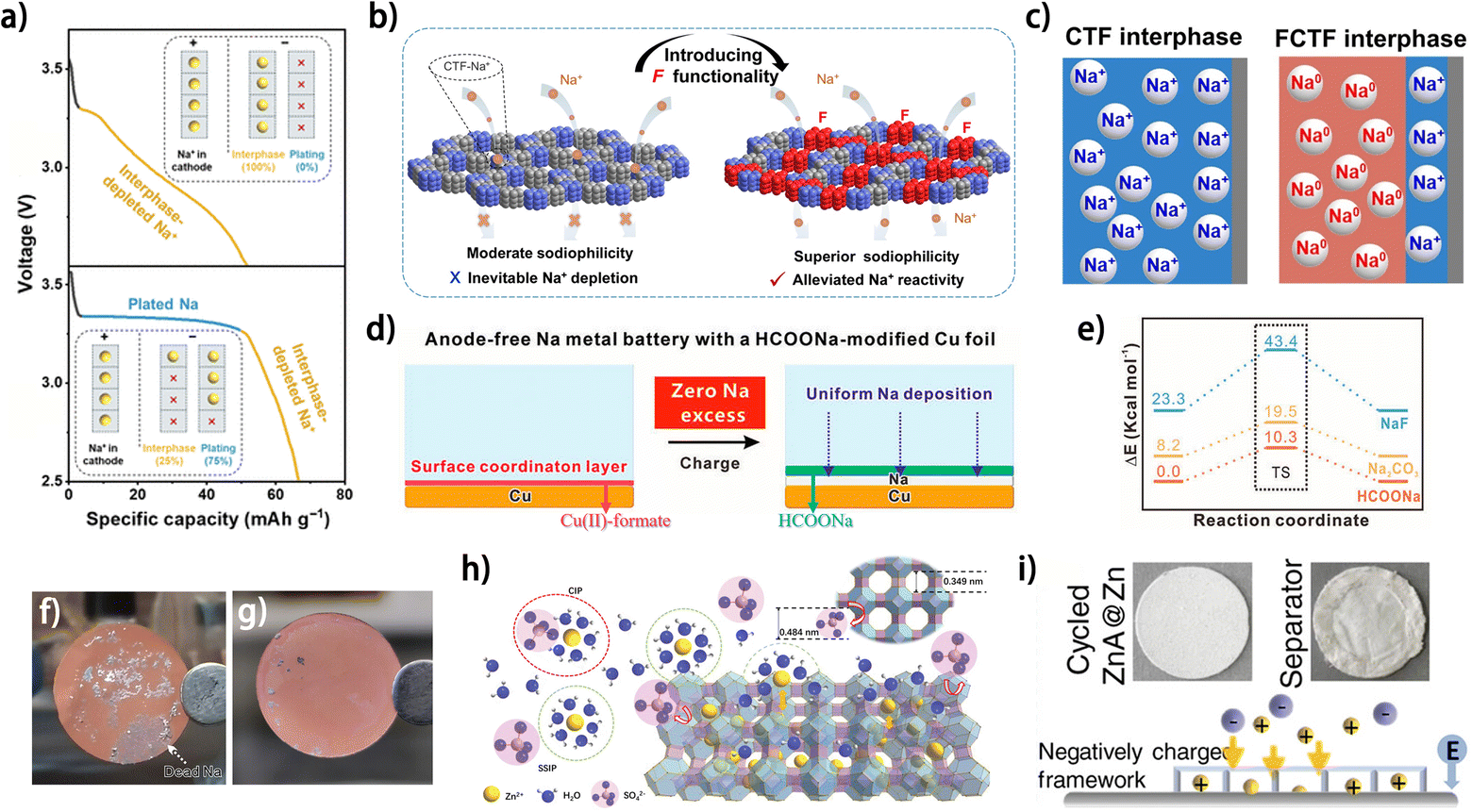 | ||
| Fig. 8 (a) Discharge profiles in anode-free full cells with a covalent triazine framework (CTF) and fluorinated CTF (FCTF) seeding/hosting interphases, and the inset shows the scheme of Na+ distribution for interphase formation and plating. The Na+ utilization for plating is 75% for the FCTF-based full cell and 0% for the CTF based anode-free configurations.57 (b) Schematic illustration of sodiophilicity and Na+ reactivity on the CTF and FCTF before deposition.57 (c) Mechanism of energy storage and reaction process of FCTF/CTF interphases in anode-free configurations.57 (d) Schematic illustration for Na metal batteries with the HCOONa interface. (e) DFT calculations of the Na-ion defect formation energies and the subsequent Na-ion diffusion barriers for three solid electrolytes relative to HCOONa species. (f and g) Optical image of the current collectors for Na anodes after cycling. (f) Cycled Cu foil; (g) cycled SF–Cu foil. (d–g) Are reproduced from ref. 58 with permission from Wiley, copyright 2023. (h) Ion transport illustration in the ZnA protective layer. (i) Photographs of cycled electrodes and illustrations of ionic distributions on the surfaces of ZnA@Zn anodes during the Zn plating process. (h and i) Are reproduced from ref. 59 with permission from Wiley, copyright 2023. | ||
The micromorphology and structure of the current collector have a great impact on the metal plating and stripping behavior.31 In this respect, the most important parameters for the current collector are its pore structure and specific surface area. These parameters do not affect the metal plating/stripping in a monotonic relationship; rather, they have dual effects. With regard to the pore structure of the current collector, high porosity is conducive to accommodating the deposited metal and alleviating volume changes during metal plating/stripping,38,63 but the pores may also trap the metal deposit and result in “dead” metal.64 As for the surface area of the current collector, a high surface area provides more metal nucleation and deposition sites, which generally benefits the formation of a uniform metal plating morphology;65–68 however, the resulting increased contact between the metal anode and the electrolyte may aggravate side reactions and lower the CE of the electrode.63 Therefore, discrepancies exist among different research studies regarding the micromorphology and structural design of the current collector for metal anodes. Nevertheless, systematic research on the effects of the micromorphology and structure of current collectors on the performance of metal plating/stripping has yet to be conducted to decipher the mysteries therein.
In addition to their influence on the metal plating/stripping behavior, current collectors may also affect the reactivity of the species in the electrolyte through various means, which would further dictate the side reactions on the metal anode, such as metal corrosion, electrolyte decomposition and SEI formation. In one way, the catalytic nature of the current collector directly influences the electrolyte-involved side reactions on it. Sharon et al. elucidated the selection criteria for Zn plating substrates based on their catalytic effect towards the HER in aqueous electrolytes.52 The amount of HER-induced Zn4SO4(OH)6·4H2O (ZSH) byproducts was found to depend on the current collectors. The XRD results show that the contents of ZSH on Ni, Ti, Cu and graphite foil (GF) are 73.4%, 29.9%, 1.6% and 0.1%, respectively, and the online electrochemical mass spectrometry (OEMS) results revealed that hydrogen evolution on the Cu and GF foil is most insignificant during the first Zn plating (Fig. 9a).52 The distinctions in the catalytic capabilities towards the HER of different substrates could originate from the varying strength of substrate atom-hydrogen bonds, i.e., the stronger bonding between the hydrogen atom and substrate would impede the formation of H2 molecules and thus increase the HER overpotential at a cathodic current, which accounts for the high HER resistance on GF and Cu foil (Fig. 9b). Moreover, the interaction between the substrate and deposited metal affects the metal corrosion, and the GF substrate shows a superior Zn corrosion resistance to Cu foil (Fig. 9c), whereas the mechanism therein needs to be further investigated. The higher CE of the GF//Zn half cell (∼72.3%) (Fig. 9d) in a standing corrosion test than those of Cu foil (∼48.0%), Ti foil (42.9%) and Ni foil-based (failed) counterparts, indicates the great potential of GF as a current collector for anode-free Zn metal batteries. As another way of influencing the side reactions on the metal anode, the current collector can tailor the surface energy of the metal deposit by guiding the metal crystalline orientation, which may influence the tendency or degree of the side reactions occurring on it. Guo et al. prepared a meter-scale graphene-modified copper collector (Cu@G) as the Zn plating substrate to realize the selective growth of the Zn (002) plane (Fig. 9e).69 The DFT results show that the Zn (002) crystalline plane has a higher activation energy for hydrogen evolution (Fig. 9f) and stronger OH− repulsion (Fig. 9g) than (101) and (100) planes, which accounts for the improved corrosion resistance of the Zn deposit and higher average CE (>99.9%) of Zn plating/stripping on Cu@G compared to those on commercial Cu foil. In some cases, the current collector can directly participate in the side reactions and thus change the byproduct components on the metal anode. Knibbe et al. investigated the SEI composition of Na deposits on Cu, α-brass and Zn current collectors.70 It was found that Zn-containing current collectors enable the formation of a ZnF2 component in the SEI by reacting with HF species from the electrolyte (Fig. 9h–j),70 which is beneficial for inhibition of further corrosion of current collectors and ensuring stable metal deposition. Changes in SEI composition after current collector modifications were also reported in a few works,19,48,50 which do not show direct associations between the components of current collectors and the formed SEI, but the related mechanisms remain unrevealed. Overall, the manifold effects of current collectors on metal plating/stripping have manifested their significance in promoting the reversibility of metal plating/stripping for anode-free post-lithium metal batteries.
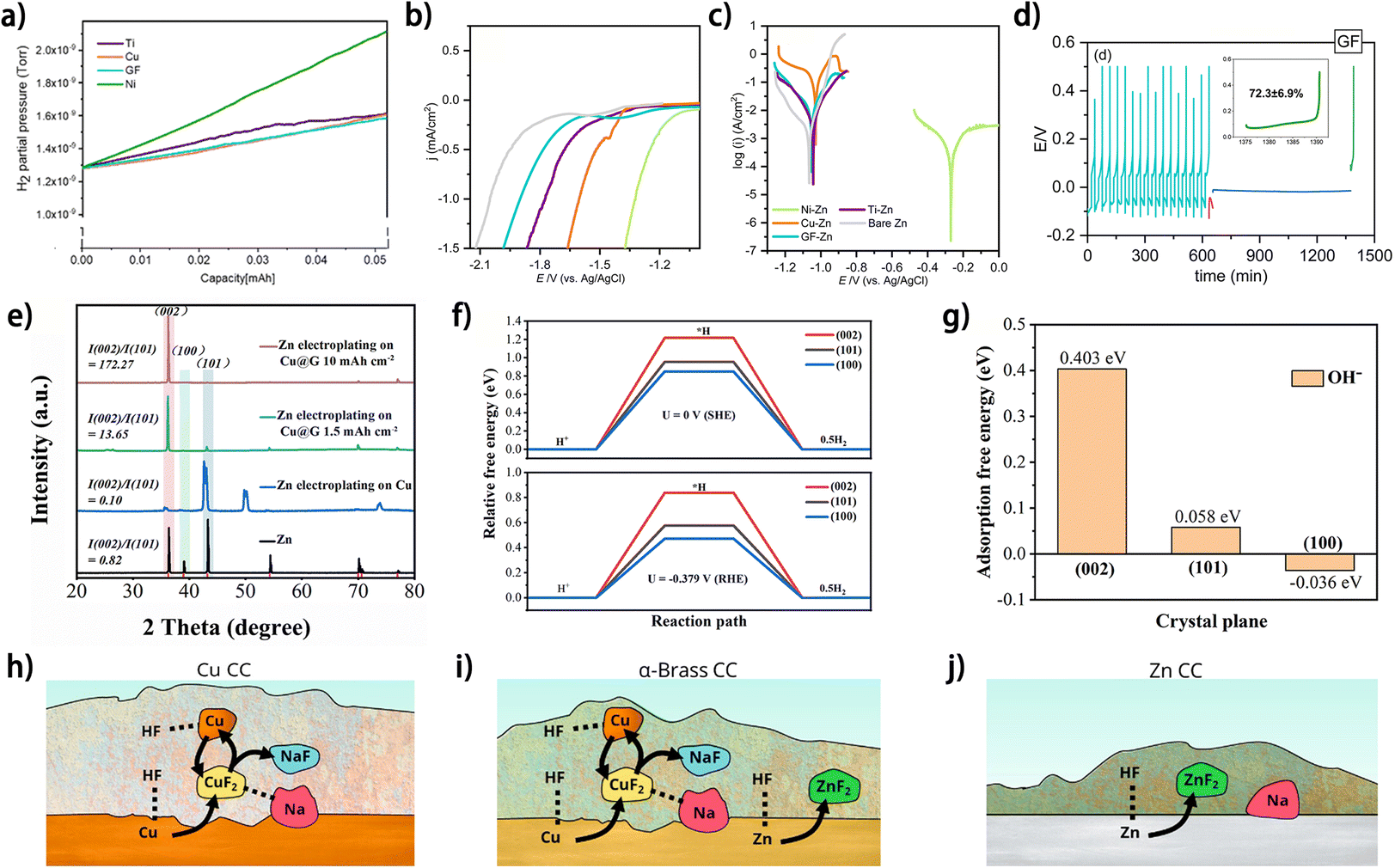 | ||
| Fig. 9 (a) OEMS spectra measured for the examined current collectors. (b) Cathodic polarization analysis measured at 5 mV s−1 and (c) Tafel plots for the examined current collectors. Both experiments were conducted in 1 M Na2SO4 electrolyte. (d) Standing corrosion (self-discharge) experiments for the tested substrates: GF, in 1 M ZnSO4 electrolyte solution. (a–d) Are reproduced from ref. 52 with permission from The Royal Society of Chemistry, copyright 2023. (e) XRD patterns of Zn deposited on Cu and Cu@G. (f) Relative free energies of H+ at Zn (002), (101) and (100) crystal planes at different potentials. (g) Adsorption free energies of OH− on Zn (002), (101) and (100) crystal planes. (e–g) Are reproduced from ref. 69 with permission from The Royal Society of Chemistry, copyright 2024. (h–j) Summary of SEI-related reactions between HF, Na and current collectors (CCs) of Cu, α-brass, and Zn CCs, respectively. (h–j) Are reproduced from ref. 70 with permission from American Chemical Society, copyright 2023. | ||
3.2 Modulating the electrolyte
The reactions involving electrolyte make a critical difference to the reversibility of metal plating/stripping of anode-free metal batteries. The metal corrosion in electrolyte would lead to irreversible metal consumption, and the component and structure of the SEI on the anode, which is generally the result of electrolyte decomposition, would dictate the stability of the metal plating/stripping process. Besides, the desolvation of the metal ions from the electrolyte may affect the electrode kinetics, further influencing the metal plating morphology.71,72 Accordingly, the design of an ideal electrolyte for anode-free metal batteries involves both thermodynamic and kinetic considerations.One rational approach for suppressing electrolyte-involved side reactions is preparing electrolytes that are thermodynamically stable when in contact with the metal anode or at the cathodic potential during metal plating. An ether-based electrolyte (NaPF6-glyme) was experimentally verified by Bai et al. to be thermodynamically stable and free from obviously repeated SEI formation on the Na metal anode (Fig. 10a and b).73 Owing to the reduced Na consumption by SEI formation, the anode-free Cu|NVP full cells showed a high Na inventory retention rate (NIRR) of > 99.8% per cycle at a high cathode loading (6.2 mg cm−2). Although some ether-based electrolytes can minimize side reactions with alkali-metal anodes, the categories of thermodynamically stable electrolytes for alkali-metal anodes are greatly restricted owing to the strong reducing properties of alkali-metals. In contrast, thermodynamically stable electrolytes can be more easily derived for anode-free zinc metal batteries, owing to the much milder reactivity of zinc metal and higher Zn plating potential. The notorious water-induced Zn corrosion and HER in aqueous electrolyte severely suppress the reversibility of Zn anodes.77 Therefore, lowering the water content and the reactivity of H2O molecules in the electrolyte are effective methods for promoting the reversibility of Zn metal plating/stripping of anode-free Zn metal batteries. Owing to the absence of water-induced side reactions, Zn metal anodes exhibited high reversibility in some nonaqueous electrolytes, such as zinc bis(trifluoromethanesulfonyl)imide (Zn(TFSI)2)/acetonitrile,78 Zn(TFSI)2/ethyl methyl carbonate (EMC)79 and ether diluted ionic liquid,80 which supported the stable cycling of the anode-free Zn metal batteries. Nevertheless, compared with aqueous electrolytes, nonaqueous electrolytes suffer from slow electrochemical kinetics, high safety risks and low environmental and economic benefits. Therefore, a popular strategy to achieve high reversibility of Zn plating/stripping while inheriting the merits of aqueous electrolytes is designing co-solvent electrolytes for anode-free Zn batteries. Propylene carbonate (PC),81,82 ethylene glycol (EG)83 and sulfolane (SL)84 were adopted with water to prepare co-solvent electrolytes for anode-free Zn metal batteries. Besides lowering the water content in the electrolytes, the interactions between the co-solvents and water molecules greatly weaken the hydrogen bonds between H2O molecules, thus suppressing water reactivity. Consequently, high CEs (>99.8%) of Zn plating/stripping can be achieved with the co-solvent electrolytes. However, the introduction of organic solvents with higher toxicity, flammability, viscosity and cost would inevitably compromise the advantages of aqueous electrolyte, which may impede their large-scale application in safe and environmental energy storage. Moreover, comprehensive principles for the selection of the co-solvents for the electrolytes of anode-free Zn metal batteries have yet to be established.
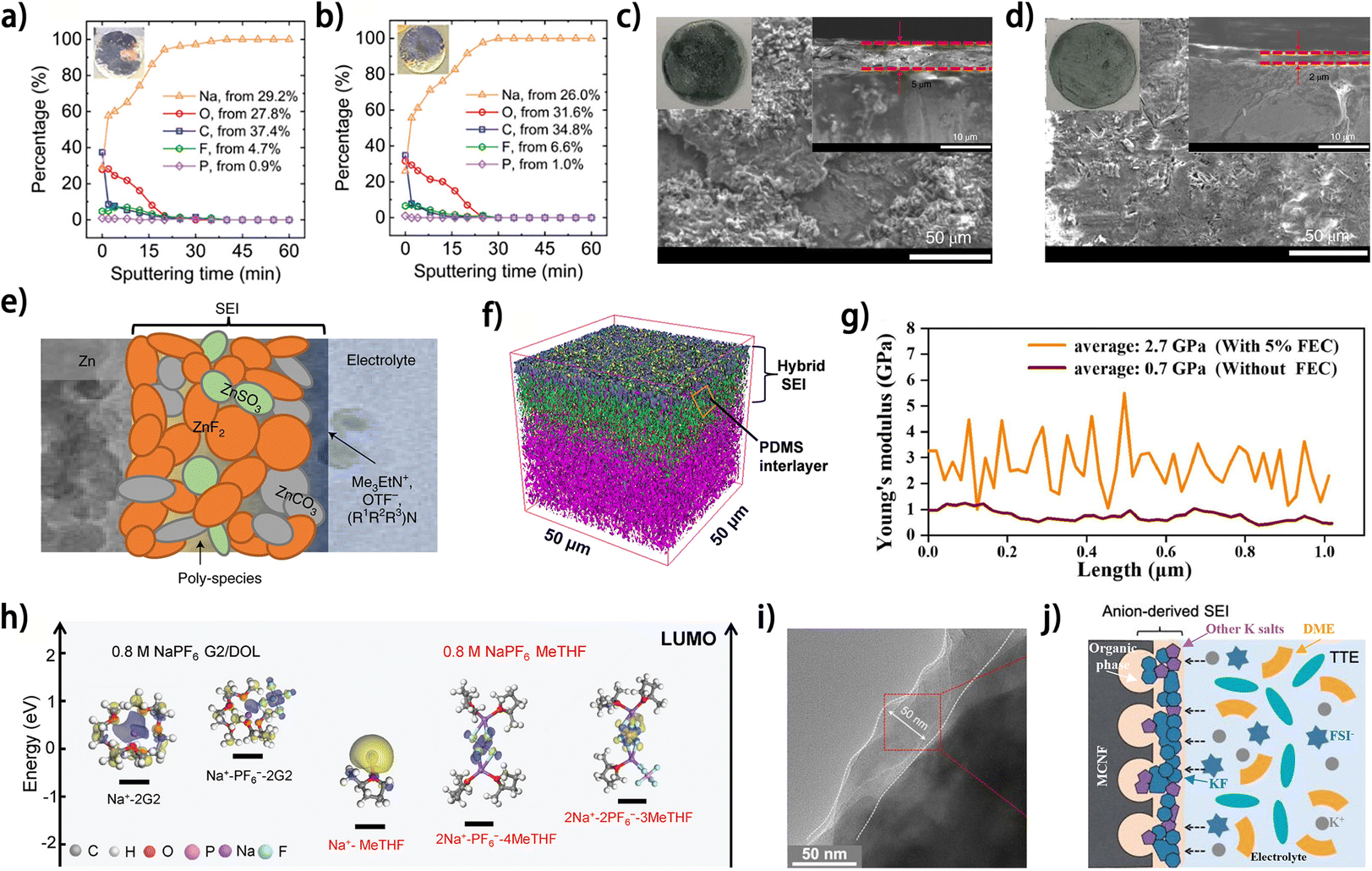 | ||
| Fig. 10 XPS elemental concentration profiles along the thickness of deposited Na on the Cu current collector (a) after completing the first half cycle of plating, (b) after the half cycle of plating following 100 full cycles. (a and b) Are reproduced from ref. 73 with permission from Wiley, copyright 2021. SEM images after 50 plating/stripping cycles in (c) 4 M Zn(OTF)2 and (d) 4 M Zn(OTF)2 + 0.5 M Me3EtNOTF ((right inset): cross-sectional SEM image, (left inset): optical images of Zn foil after cycling). (e) Cartoon of the proposed Zn2+-conducting SEI, characterized by small nodular particles embedded in a polymeric framework. (c–e) Are reproduced from ref. 74 with permission from Springer Nature, copyright 2021. (f) 3D distribution overlay of depth profile curves of TOF-SIMS in negative mode.20 (g) Young's modulus distribution of the SEI covered on nanofiber from the half cell without FEC and with 5% FEC; reproduced from ref. 75 with permission from Elsevier, copyright 2022. (h) LUMO energy levels of solvation complexes of interest in the MeTHF and G2/DOL electrolytes;76 reproduced from ref. 76 with permission from Wiley, copyright 2024. (i) TEM images of MCNF cycled in DHCE. (j) Schematic diagrams of the electrolyte structure and the correspondingly formed SEI on MCNF using DHCE. (i and j) Are reproduced from ref. 63 with permission from American Chemical Society, copyright 2023. | ||
Thin and robust SEI on metal anodes would facilitate the stable metal plating/stripping and can be constructed by introducing appropriate additives into the electrolyte. For aqueous anode-free Zn metal batteries, the SEI construction via electrolyte modulation has been reported in a couple of works.74,82 Wang et al. reported the in situ formation of a ZnF2-rich SEI on the Zn anode by introducing a trimethylethyl ammonium trifluoromethanesulfonate (Me3EtNOTF) additive into 4 M zinc trifluoromethanesulfonate (Zn(OTF)2) aqueous electrolyte.74 A smoother Zn deposit morphology and a thinner SEI layer on the Zn anode were observed after introducing Me3EtNOTF into the electrolyte (Fig. 10c and d), and the CE of Zn plating/stripping also increased from 87.6% to 99.9%. This performance improvement was attributed to the formation of a robust and hydrophobic SEI layer with complex composition (Fig. 10e). The critical effect of the ZnF2 component is elucidated to be shielding water from the Zn anode and enabling uniform lateral Zn deposition. In addition, the Me3EtNOTF additive decreased the wettability of the electrolyte on Zn anodes. Consequently, an anode-free Ti‖ZnxVOPO4 pouch cell with 50 mA h capacity presented 80% capacity retention after 90 cycles at 0.5 mA cm−2.
In contrast to aqueous anode-free Zn metal batteries, constructing the SEI via electrolyte modulation is more widely implemented in anode-free sodium– and potassium–metal batteries. The mechanical strength of the SEI can be promoted by introducing specific components into the in situ formed SEI on metal anodes to improve the stability of the metal/electrolyte interface. NaBF4 has been reported as an effective electrolyte additive to introduce inorganic B–O species into the SEI on Na metal anodes; these species are known for their flexibility and poor crystallinity, thus promising to enhance the toughness of the SEI and facilitate uniform ion flux.47,48 Our group fabricated a hybrid SEI on K metal anodes by adopting polydimethylsiloxane (PDMS) as an electrolyte additive for low-temperature anode-free K batteries.20 The PDMS reacts with the K metal surface and forms a robust organic–inorganic hybrid SEI with the O–Si–O– interlayer (Fig. 10f), which significantly increases the Derjaguin–Müller–Toporov (DMT) modulus of the SEI from ∼1156 MPa (for the PDMS-free electrolyte-derived SEI) to 1489 MPa, thus maintaining a smooth K deposit surface without cracking. Consequently, an average CE of 99.80% for K‖Cu cells was achieved at −40 °C, showing the high reversibility of the SEI-protected K plating/stripping process. Inorganic F− species in the SEI are known as mechanical strength enhancers for Na and K metal anodes.63,75,76 A F-rich SEI could be readily formed on Na metal anodes by adopting fluoroethylene carbonate (FEC) as an electrolyte additive.75 The SEI formed with a 5% FEC additive not only endows the Na metal anode surface with an improved Young's modulus (Fig. 10g) but also provides high corrosion resistance towards the electrolyte.
Besides the SEI modulated by external electrolyte additives, the anion-derived inorganic-rich SEI is generally considered to have high structural stability compared to the solvent-derived organic-rich counterparts. By regulating the solvation structure of cations in the electrolyte, the decomposition preference of electrolyte species can be changed and thus the SEI component on metal anode can be modulated. The contents of different solvation structures (such as solvent-separated ion pairs (SSIPs), contact ion pairs (CIPs) and aggregates (AGGs)) in the electrolyte and their corresponding lowest unoccupied molecular orbital (LUMO) energy are critical considerations for electrolyte modulation,85 which are strongly associated with the composition of the formed SEI on metal anodes. Using a weakly solvating solvent, 2-methyltetrahydrofuran (MeTHF), an AGG-dominated solvation structure was achieved in NaPF6-based electrolyte.76 The predominant AGG (2Na+–1PF6−–4MeTHF) solvate had the lowest LUMO energy (−1.42 eV) and facilitated the formation of an anion-derived inorganic-rich SEI on the Na metal anode, while the small LUMO energy gap between the SSIP solvate and CIP solvate in the diglyme (G2)/1,3-dioxolane (DOL)-based control electrolyte resulted in an organic–inorganic hybrid SEI (Fig. 10h). With a MeTHF-based electrolyte, a high average CE of 99.9% for Na plating/stripping was achieved at −25 °C, and this high reversibility of the Na anode supported the stable operation of an anode-free Al@C//NVP pouch cell for 150 cycles at −25 °C. In another work, Xie et al. reported an anion-derived inorganic-rich SEI on K metal anodes using a diluted high-concentration electrolyte (DHCE) consisting of potassium bis(fluorosulfonyl)imide (KFSI) salt, 1,2-dimethoxyethane (DME) solvent and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoro-propyl ether (TTE) diluent.63 The TTE diluent promoted the participation of the anion into the K+ solvation structure, triggering the formation of an anion-derived inorganic-rich SEI on the K metal anode, which exhibited high toughness and enabled a smooth morphology of the K deposit (Fig. 10i and j). Our group developed an isosorbide dimethyl ether (IDE)/water co-solvent electrolyte for anode-free Zn metal batteries, which increased the anion content in the Zn2+ solvation structure and enabled the formation of a ZnF2-rich SEI on Zn anode.82 Owing to the improved stability of Zn plating/stripping, a high-areal-capacity (∼3 mA h cm−2) anode-free Zn metal battery was fabricated, showing a 60% capacity retention after 300 cycles. The above works demonstrated the significant impact of electrolyte component modulation on SEI properties; however a deeper understanding of the SEI formation mechanism and the specific effects of different components in the SEI on its physicochemical properties still needs to be unveiled.
The morphology of metal deposits is dictated by the kinetics of the metal plating process, in which the desolvation of metal ions plays a significant role. A fast metal ion desolvation is beneficial to smooth metal deposition, especially in low-temperature situations where the energy barrier of charge transfer is prominent.86 The desolvation energy of metal ions can be tailored through electrolyte design, including selecting solvents,63,76 anion species,48 electrolyte concentration20 and electrolyte additive species.87 It is commonly believed that the desolvation energy during metal plating can be minimized by adopting weakly solvating agents as electrolyte solvent to decrease the participation of solvents in the solvation shell of the metal ions. Usually, ethers are adopted as electrolyte solvents or diluents owing to their low viscosity, small donor number and low dielectric constant, which endow ethers with weak solvating ability towards metal ions. A high-concentration electrolyte diluted with fluorinated ether TTE (DHCE) was reported to enable easier K+ desolvation compared to the normal HCE, thus facilitating faster K deposition kinetics (Fig. 11a) and contributing to smoother K plating.63 Based on the different coordination abilities of anions to metal ions, the metal ion desolvation energy can also be modulated by selecting anion species in the electrolyte. The higher solvating ability of OTF− and BF4− to Na+ was illustrated by the lower electrostatic potential (ESP) charges of Na+ in the corresponding anion-Na+ complexes, compared to the case of PF6− (Fig. 11b).48 Owing to the decreased solvent-Na+ coordination, the dual-salt (NaOTF and NaBF4)-based electrolyte showed decreased Na nucleation and deposition overpotential in contrast to the NaPF6-based electrolyte at −40 °C. It is worth noting that the increased anion coordination to metal ions does not necessarily reduce the desolvation energy when considering the energy of removing anions from the solvation shell. Our group found that a smaller concentration of KPF6 reduces the coordination of the anion to K+ and thus lowers the desolvation energy (Fig. 11c),20 indicating the necessity of considering metal ion coordination by both solvent and anions to rationally evaluate the kinetics of the desolvation process. As another case, Lee et al. elucidated the significance of the interactions between anions and cations in the electrolyte on the desolvation energy of Zn2+.87 A zwitterion (2-(trimethylazaniumyl)acetate, TAA) containing both soft-acidic and hard-basic moieties was used as an electrolyte additive to weaken the interaction between Zn2+ and OTF− (Fig. 11d). The soft-acidic ([N(CH3)3]+) and hard-basic (COO−) moieties respectively coordinate to soft base OTF− and hard acid Zn2+, and thus the desolvation energy of Zn2+ decreases. The enhanced Zn2+ desolvation kinetics contributed to smooth Zn plating morphology at a low temperature of −40 °C (Fig. 11e and f), while the pristine aqueous electrolyte led to dendritic Zn deposits (Fig. 11g and h). With the modulated anion–cation interaction, the electrolyte enabled 90.4% capacity retention in the anode-free Cu//Znxα-V2O5@Graphene pouch cell after 50 charging/discharging cycles. The electrolyte modulation has been demonstrated as an effective strategy to promote the reversibility of metal plating/stripping in anode-free metal batteries. However, the decreased initial coulombic efficiency (ICE) caused by the inevitable SEI formation on the metal anode remains a vital problem for this strategy. Besides, the commonly used weakly solvating scheme would compromise the electrolyte concentration, which may make ion depletion more likely to occur at the electrode surface, especially at high charging current densities, which is known as a significant factor for metal dendrite formation.2 Undoubtedly, with further exploration and integration with other strategies, electrolyte modulation would play an increasingly significant role in achieving reversible metal plating/stripping for anode-free metal batteries.
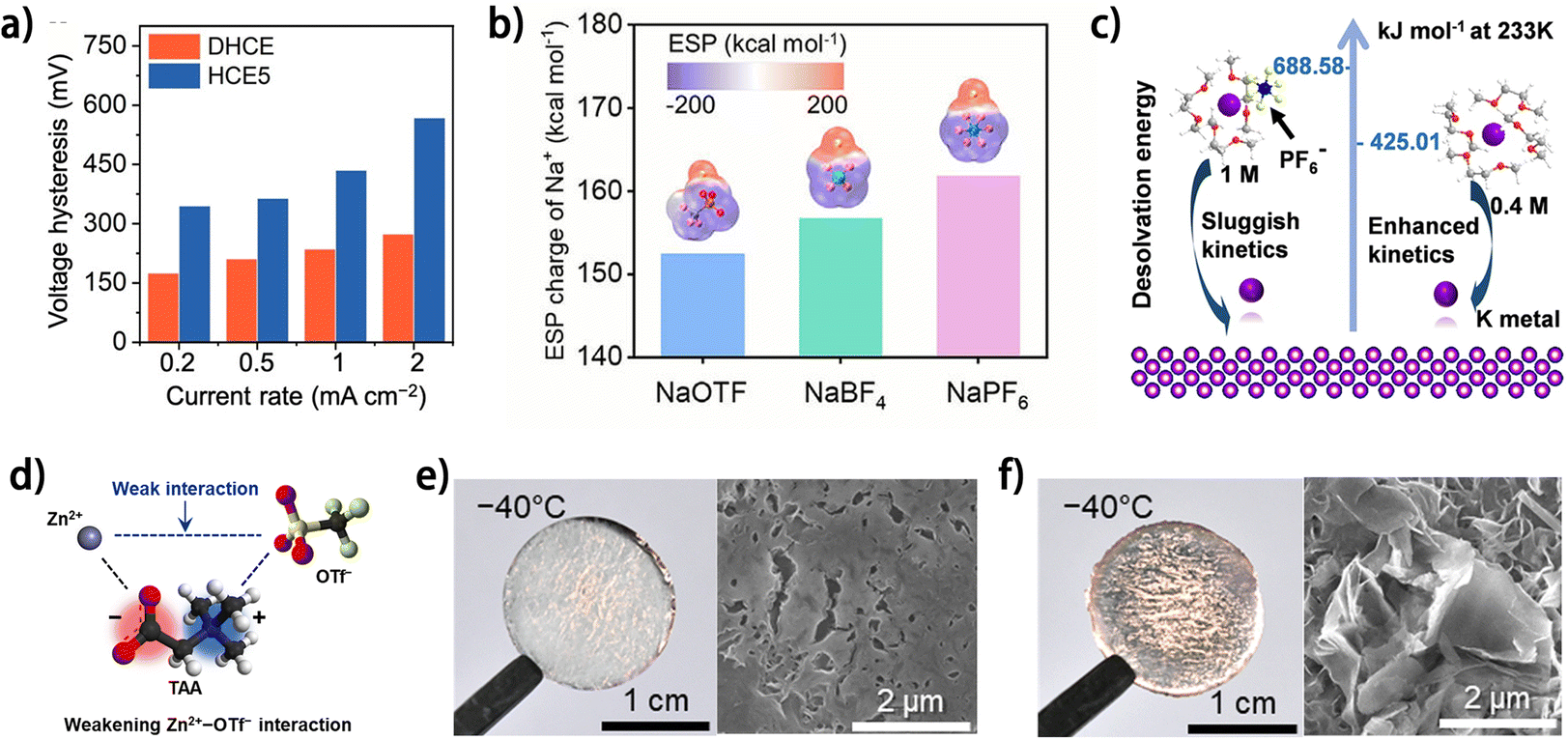 | ||
| Fig. 11 (a) Rate performance of K‖Cu cells in various electrolytes and the corresponding values of voltage hysteresis. DHCE (diluted high-concentration electrolyte):1.6 M KFSI/DME/TTE; HCE5 (high-concentration electrolyte):5.0 M KFSI/DME; reproduced from ref. 63 with permission from American Chemical Society, copyright 2023. (b) ESP mapping of Na+-anion complexes; reproduced from ref. 48 with permission from Elsevier, copyright 2024. (c) Calculated desolvation energies in 0.4 and 1 M KPF6-DME at 233 K. K, C, O, H, F and P are marked with purple, grey, red, white, yellow, and blue, respectively.20 (d) Schematic depicting the roles of soft-acidic/hard-basic TAA in enabling the low-temperature aqueous electrolyte. Photographs (left) and surface SEM images (right) of Zn plated on Cu foil in (e) 4 M Zn(OTf)2 electrolyte and (f) TAA-based electrolyte at −40 °C with 0.2 mA cm−2 and 0.5 mA h cm−2. (d–f) Are reproduced from ref. 87 with permission from The Royal Society of Chemistry, copyright 2024. | ||
4 Compensating for anode loss
In contrast to striving for improving the reversibility of each metal plating/stripping cycle on the anode side, compensating for anode loss emerges as another effective approach to retain the available capacity of the anode. By introducing an additional metal ion source into the battery system during battery preparation, the metal loss on the anode can be either replenished through electrochemical reduction of the supplied ions on the anode, or mitigated by a preformed SEI on the metal plating substrate. The accommodation sites of the extra metal ion source are not restricted to the anode, but can also be within other components of the anode-free metal batteries, such as cathode and electrolyte. Accordingly, the strategies for compensating anode loss are introduced in three parts: electrochemically activating the current collector, pre-metallizing the cathode and introducing metal-supply agents.4.1 Electrochemically activating the current collector
During the first metal plating/stripping process on the current collector of anode-free metal batteries, the metal deposit inevitably reacts with electrolyte species and form an SEI on the anode, which results in metal consumption on the anode and lowered ICE of the battery. To avoid the negative effects of SEI formation on the anode-free metal battery, this metal consumption process can be transferred outside the battery by pre-forming a stable SEI on the current collector before battery assembly. Electrochemically activating the anode current collector (such as through galvanic cycling37,88 and cyclic voltammetry89–91) provides a facile route to this destination, with the feasibility relying entirely on the formation of a stable SEI on the current collector during the activation process. Li et al. investigated the activation process of current collectors for Na metal anodes by in situ electrochemical dilatometry.88 It was found that the initial galvanic charging of the current collector can be divided into two periods: the initial SEI formation (region 1 in Fig. 12a and b) and the subsequent metal deposition (region 2 in Fig. 12a and b) according to the thickness change of the current collector.88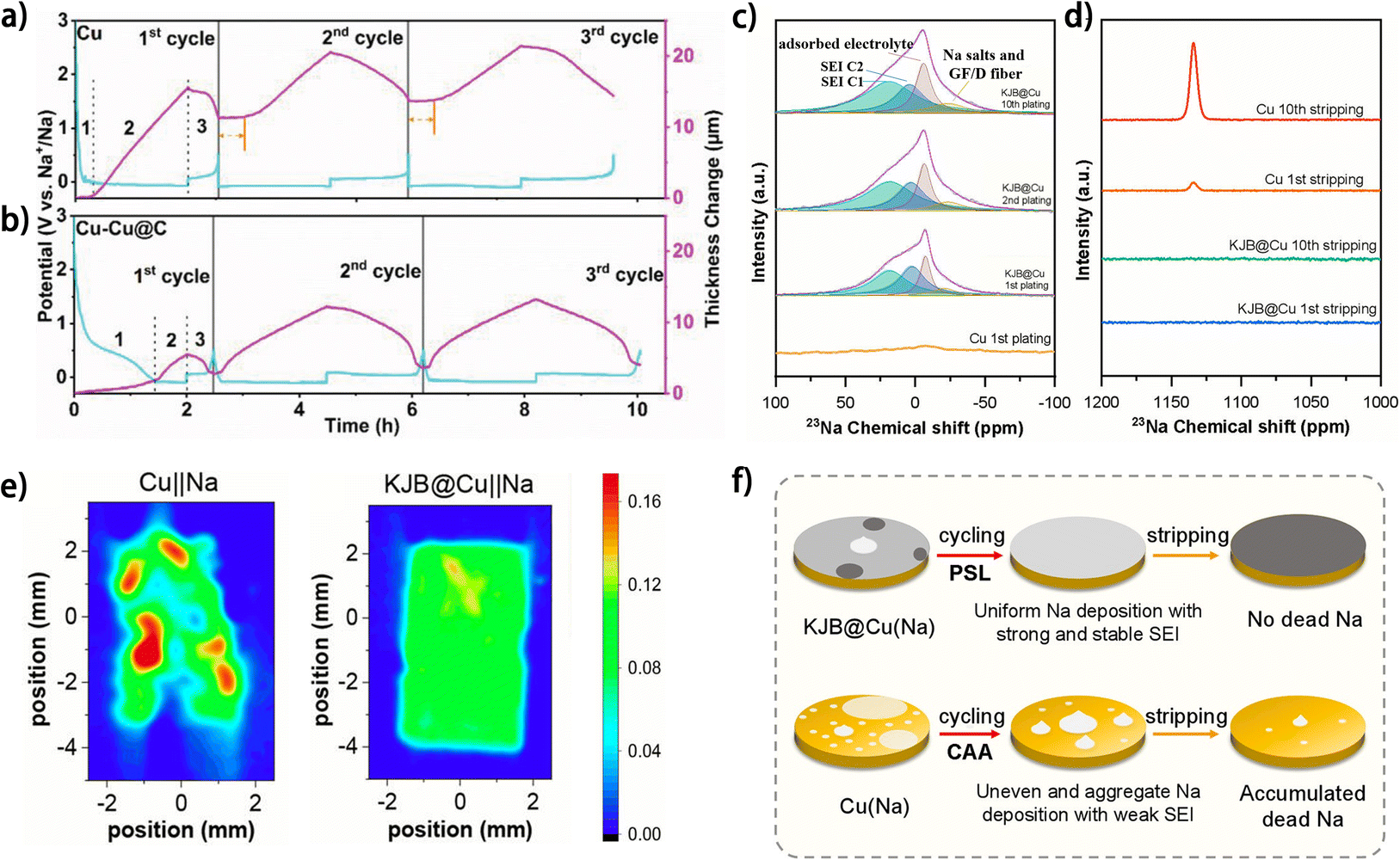 | ||
| Fig. 12 In situ dilatometry investigation of bare Cu and Cu–Cu@C at a current density of 0.5 mA cm−2 and with a Na plating charge fixed at 1 mA h cm−2: voltage profile and corresponding thickness change of (a) bare Cu and (b) Cu–Cu@C during the first three cycles; reproduced from ref. 88 with permission from Wiley, copyright 2022. 23Na solid-state NMR spectra of surface species harvested from Cu and KJB@Cu electrodes in Cu‖Na and KJB@Cu‖Na cells at full (c) plating and (d) stripping states of different cycles. (e) Ex situ EPRI images of bare Cu and KJB@Cu current collectors with 0.25 mA h cm−2 metallic Na deposition disassembled from Cu‖Na and KJB@Cu‖Na half-cells (discharging to 0 V and then plating for 30 min at 0.5 mA cm−2). (f) Corresponding Na deposition mechanism of Cu and KJB@Cu electrodes. (c–f) Are reproduced from ref. 67 with permission from American Chemical Society, copyright 2024. | ||
As an unsuccessful activation process of Cu foil, the negligible region 1 during the first galvanic cycle suggests incomplete SEI formation, which may generate “dead” Na and accounts for the large irreversible expansion in thickness of the current collector. Moreover, the unstable SEI formed in the first cycle resulted in repeated SEI formation during the subsequent cycles, which greatly suppressed the reversibility of the metal anode. In comparison, as a successful activation process, the SEI extensively formed on the carbon-modified Cu current collector (Cu–Cu@C) during the initial galvanic charging, as proved by a large region 1 (Fig. 12b), which ensured reversible metal plating/stripping in the following cycles without significant irreversible thickness expansion of the electrode or repeated SEI formation. Therefore, efficient SEI formation on the current collector during the first activation cycle is vital to the effectiveness of the activation process. In another work, Lou and Hu et al. elucidated the influence of SEI formation during the first metal plating on the subsequent stripping.67 Compared with the weak SEI signals detected on the Cu current collector, the Ketjenblack carbon-coated Cu foil (KJB@Cu) showed evident SEI formation after the first Na plating and the SEI composition remained nearly unchanged in the following metal plating processes (Fig. 12c). The stable SEI on KJB@Cu supported the complete Na stripping, while apparent remnant Na accumulated on Cu foil after galvanic cycles (Fig. 12d). Also, ex situ electron paramagnetic resonance imaging (EPRI) of the current collectors after Na plating suggests that the uniformity of the Na deposit can be determined during the first charging period, as dictated by SEI formation (Fig. 12e). This progressive self-leveling (PSL) behavior of Na plating on KJB@Cu largely promoted the reversibility of the anode, while Na-deposition central accumulative aggregation (CAA) behavior dominated on Cu foil and resulted in poor SEI protection and low reversibility of the anode (Fig. 12f), which was attributed by the authors to the large thermodynamic mismatch or poor compatibility between alkali metals and Cu. Obviously, the activation results are highly dependent on the current collectors, and considering that the SEI properties of metal anodes are also closely associated with the electrolytes,92 electrochemical activation of the current collector is not a fundamental strategy for constructing highly reversible anode-free batteries but rather relies on the knowledge of current collector engineering and electrolyte modulation. Besides, the high device requirements and complex treatment process may impede the large-scale application of electrochemical activation of current collectors.
4.2 Pre-metallizing the cathode
Since the metal anodes of anode-free metal batteries are in situ deposited on the anode current collector during the first charging process of the battery, introducing excess capacity during the first charging process (metal plating on the anode and ion extraction from the cathode) relative to the subsequent discharging process (metal stripping on the metal anode and ion insertion into the cathode) would generate a metal reservoir on the metal anode, which can help compensate for metal loss on the anode, thus improving the cyclability of the anode-free battery. Pre-metallizing cathodes with extra metal ions emerges as a feasible implementation for this strategy. The additional ions in the cathodes would be irreversibly extracted from the cathodes during the first charging process and cannot be re-inserted into the cathode thereafter, resulting in a higher initial charging capacity compared to those of the subsequent cycles. At present, this type of cathode has been only reported in a couple of works. Na-rich Na3V2(PO4)3 cathodes93,94 are prepared for anode-free sodium metal batteries. Wu et al. constructed a Na5V2(PO4)3 (Na5VP) cathode by direct-contact presodiation of Na3V2(PO4)3 (Na3VP) (Fig. 13a).93 The Na-rich Na5VP cathode could finally transform into Na1V2(PO4)3 (Na1VP) upon desodiation (Fig. 13b), and the transformation between Na4V2(PO4)3 (Na4VP) and Na1VP (corresponding to a wide voltage window of 1.0–3.8 V) was found to be highly reversible. Therefore, the Na+ ions deintercalated during the transformation from Na5VP to Na4VP could be deposited on the anode side as a Na reservoir, and the anode-free carbon-coated Al foil//Na5VP battery operating between 1.0–3.8 V showed a superior capacity retention of 93% after 130 cycles compared with the lower capacity retention of <70% for the Na3VP-based battery. In another work, a Na-rich Na4VP cathode was prepared by facile chemical solution treatment (Fig. 13c).94 The excess initial charging capacity relative to the subsequent cycling was attributed to the transformation from Na4VP to Na3VP (Fig. 13d), and by setting the cut-off voltage of the subsequent discharging to 2.0 V, the reversible transformation between Na3VP and Na1VP occurs in the following cycles. Benefiting from the excess Na stored on the anode, the Na-rich Na4VP-based anode-free battery exhibited a high capacity retention ratio of 99% after 100 cycles (Fig. 13e), demonstrating the feasibility of cathode pre-metallization in elevating the cyclability of anode-free batteries. Similarly, for anode-free zinc metal batteries, a zinc-rich spinel Zn3V3O8 cathode was synthesized via sol–gel method by Kuang et al.95 During the first charging, Zn3V3O8 undergoes an irreversible phase transformation into ZnyV2O5−x·H2O, and then the ZnyV2O5−x·H2O with decreased capacity acts as the reversible cathode material without changing the voltage window of the battery (Fig. 13f). However, the discharge capacity of the Zn3V3O8//carbon paper anode-free battery is merely 130 mA h g−1 with unsatisfactory cycling stability (26 mA h g−1 after 20 cycles), indicating significant room for improvement in this cathode. Although the above works demonstrated the potential of cathode pre-metallization for improving the cyclability of anode-free metal batteries, some issues are still impeding the practical implementation of this scheme. For example, the lack of theoretical guidance for the selection and synthesis of electrochemically active metal-rich cathodes shows as a critical hurdle. Besides, the irreversible phase transition of metal-rich cathodes during the first charging may lead to huge volume variation93 and undermine the stability of the electrode. More importantly, the low ICE of this type of cathode may decrease the efficiency of cathode loading, which further reduces the energy density of the battery, thus diminishing the performance of the full battery. In view of these issues, introducing a certain amount of metal-rich cathode into the normal cathodes for anode-free batteries may be a more balanced strategy that can simultaneously control metal compensation and mitigate the negative effects introduced by the huge volume variation and low ICE of metal-rich cathodes.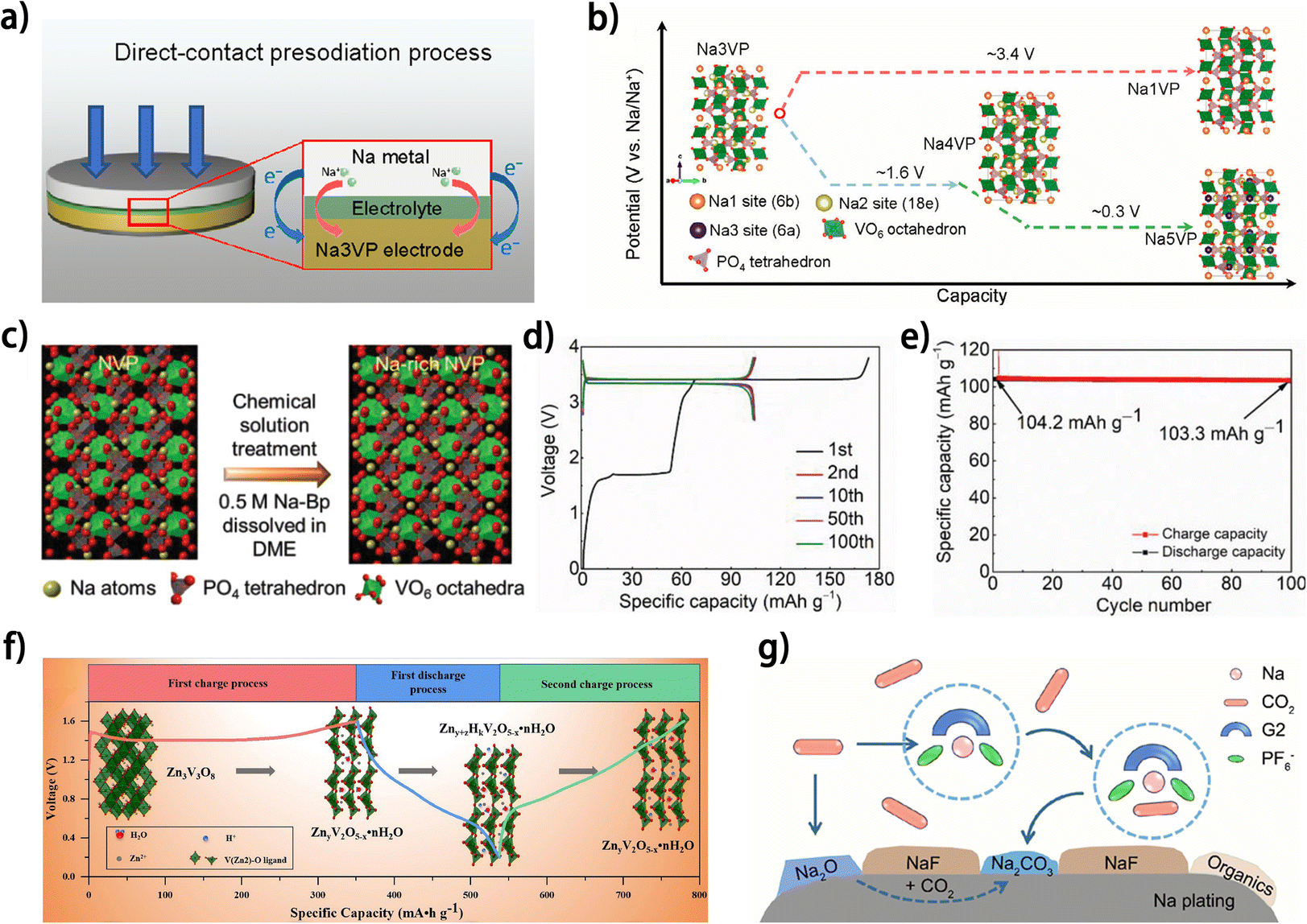 | ||
| Fig. 13 (a) Schematic of the short-circuit presodiation process of the Na3VP cathode.93 (b) Schematic of the phase transitions of Na3VP during sodiation and desodiation.93 (c) The illustration of chemical solution treatment of an NVP electrode. (d) Selected galvanostatic charge and discharge curves and (e) specific capacity versus cycle number of HCH‖Na-rich NVP (30 s) full cells tested at 1C. (c–e) Are reproduced from ref. 94 with permission from Wiley, copyright 2022. (f) Schematic illustration of the significant phase evolution in AZIBs; reproduced from ref. 95 with permission from Elsevier, copyright 2021. (g) Schematic illustration showing possible routes for generating carbonates from dissolved CO2; reproduced from ref. 96 with permission from American Chemical Society, copyright 2023. | ||
4.3 Introducing metal-supply agents
Compared to the strategies that accommodate additional metal ions in pre-metallized cathodes, introducing metal-supply agents into the cathode seems to be a more facile strategy for compensating metal loss on the anode of anode-free batteries. The metal-supply agents function in a way that they are electrochemically oxidized and decomposed during the charging of the battery, simultaneously releasing metal ions into the electrolyte which are then deposited on the anode side. Consequently, additional metal deposition on the metal anode can be derived from the electrolyte and thus the metal loss on the anode can be compensated. Actually, the sacrificial agents for metal ion supplement have been widely adopted in alkali-ion batteries to compensate for metal ion loss during SEI formation on the anode.97,98 In contrast, few research studies on metal-supply agents have been reported for anode-free batteries.96 Elaborate selection of metal-supply agents is necessary considering non-negligible effects of the electrolyte composition on the battery performance. The properties of an ideal metal-supply agent should include: (1) high enough content of metal ions to minimize their dosage, thus maximizing the specific capacity of the cathode; (2) suppressed reactivity towards both electrolytes and the cathode to mitigate side-reactions; (3) ease of decomposition during battery charging to ensure their metal-supply function; (4) the decomposition products should have no negative effects on both the cathode and anode. At present, the metal-supply agents that possess above properties have been rarely reported for anode-free batteries. Zhang et al. introduced sodium oxalate (Na2C2O4) into P2-NaNMT cathodes as the sodium-supply agent for anode-free sodium batteries.96 The gaseous CO2 product of Na2C2O4 decomposition can be eliminated during the battery degassing process, and the dissolved CO2 can react with the Na metal anode to convert NaxO into Na2CO3, which is proposed to be a more robust SEI component (Fig. 13g).96 The anode-free cell with the Na2C2O4 additive achieved 97% capacity retention after 80 cycles. However, it is worth mentioning that the complete decomposition of Na2C2O4 during the first charging process could not be achieved in a 40 mA h pouch cell,96 probably due to the poor reaction kinetics of the metal-supply agent in the thick cathode. This may suggest the limitation of this strategy when applied for large-capacity batteries. Apart from the incomplete decomposition of metal-supply agents, the possible gas emission during their decomposition may be a significantly unstable factor for the cathode especially at a large electrode thickness. Given this, introducing metal-supply agents into the electrolyte may be an alternative option to circumvent these problems, whereas in such cases, the reactivity of the agents with the metal anode would be a critical consideration. Overall, above research works demonstrate the feasibility and great promise of the strategy for compensating anode loss in improving the cyclability of anode-free post-lithium metal batteries, which would also provide inspiration for the practical manufacturing of anode-free batteries. Finally, to provide a brief overview of the development of anode-free post-lithium metal batteries, the advancements in the performance of typical anode-free batteries are presented in Table 1.| Anode current collector | Cathode | Electrolyte | Capacity/mA h g−1 (1st cycle) | Capacity retention | Initial CE | Average CE | Ref. | |
|---|---|---|---|---|---|---|---|---|
| a Data obtained at −25 °C. b Data obtained at −40 °C. | ||||||||
| Sodium batteries | Zn foil | Na3V2(PO4)3@Al | 1 M NaPF6 in diglyme | 109 at 0.5C | 98.5% (40 cycles at 0.5C) | ∼94% | ∼95% | 36 |
| Ag-coated carbon cloth | Prussian white | 1 M NaClO4 in EC/PC with 5 wt% FEC | 116 at 0.5C | 56% (800 cycles at 0.5C) | ∼98% | ∼99% | 33 | |
| Carbon-coated Al foil | P2-NaNMT with 10 wt% Na2C2O4 | 1 M NaPF6 in diglyme | ∼82.5 at 0.1C | 97% (80 cycles at 0.1C) | ∼52% | ∼99.9% | 96 | |
| KJB@Cu | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | ∼112.5 at 0.5C | 94.16% (150 cycles at 0.5C) | ∼94% | ∼99% | 67 | |
| Carbon-coated Al | Na5V2(PO4)3 | 1 M NaPF6 in diglyme | 145 at 2C | 93% (130 cycles at 2C) | ∼99.9% | ∼99.9% | 93 | |
| Zn foil | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | 113 at 0.5C | 87% (100 cycles at 0.5C) | ∼99% | ∼99% | 35 | |
| Graphitic carbon-coated Al foil | Na[Cu1/9Ni2/9Fe1/3Mn1/3]O2 | 0.9 M NaPF6 and 0.1 M NaBF4 in diglyme | 130 at 0.5C | 82% (200 cycles at 0.5C) | 92% | ∼100% | 47 | |
| Al pellets | NaCrO2 | Na0.625Y0.25Zr0.75Cl4.375 catholyte + Na4B10H10B12H12 solid electrolyte | ∼110 at 1 mA cm−2 | 70% (400 cycles at 1 mA cm−2) | 93% | 99.96% | 64 | |
| Al foil | NaCu1/9Ni2/9Fe1/3Mn1/3O2 | 1 M NaPF6 in diglyme | ∼125 at 0.3C | 73.1% (250 cycles at 0.3C) | ∼57% | 99.91% | 99 | |
| Bi modified Cu foil | Na3V2(PO4)3 | NaPF6/DME | 97.6 at 1C | ∼99% (80 cycles at 1C) | 79.5% | 99.2% | 100 | |
| Cu foil | Na3V2(PO4)3 | 1.0 M NaPF6 in diglyme | ∼106 at 0.5C | 70% (100 cycles at 0.5C) | ∼99.9% | Close to 100% | 66 | |
| Cu foils | Na3V2(PO4)3 | 1.0 M NaPF6 in diglyme | 98.1 at 0.5C | 88% (400 cycles at 0.5C) | ∼100% | 99.97% | 58 | |
| Ti3C2Tx/CNT | Na3V2(PO4)3@C | 1.0 M NaPF6 in diglyme | ∼99 at 2C | 53.8% (2500 cycles at 2C) | ∼99% | ∼99.8% | 55 | |
| ∼91 at 5C | ||||||||
| Sn decorated N-doped CNFs | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | ∼102 at 2C (234 mA g−1) | ∼89% (80 cycles at 2C) | ∼89% | ∼97% | 37 | |
| Hard carbon | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | ∼97.5 at 2C | 98.5% (400 cycles at 2C) | ∼98.5% | ∼99% | 94 | |
| Carbon-coated Al foil | Prussian blue-rGO | 1 M NaPF6 in diglyme | 115 at 1C (150 mA g−1) | 79% (250 cycles at 1C) | ∼99% | ∼98% | 46 | |
| Porous carbon particles/carbon-coated Fe nanoparticles | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | 103 at 1 mA cm−2 | 97% (100 cycles at 1 mA cm−2) | ∼95% | ∼99% | 54 | |
| Carbon-coated Al foil | Na3V2(PO4)3 | NaPF6/2-methyltetrahydrofuran | ∼82.5a at 0.5C (after 4 cycles of activation at 0.2C) | 80% (300 cycles at 0.5C)a | ∼89% | ∼99.9% | 76 | |
| ∼60b at 0.5C (after 4 cycles of activation at 0.2C) | 78% (300 cycles at 0.5C)b | ∼89% | ∼99.9% | |||||
| Fluorinated covalent triazine-modified Cu foil | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | 99 at 2C | ∼46% (400 cycles at 2C) | 90.5% | 99.7% | 57 | |
| Sodium–antimony–telluride coated Cu foil | Na3V2(PO4)3 | 1 M NaPF6 in diglyme | ∼90 at 1C | ∼77% (100 cycles at 1C) | 92.3% | 99.7% | 45 | |
| Cu3P nanowire modified Cu foil | Na3V2(PO4)3 | 1 M NaClO4 in EC/PC with 5 wt% FEC | ∼87 at 0.5C | ∼87% (75 cycles at 0.5C) | 81.1% | 99.5% | 44 | |
| Cu foil | Nano-sized Na3V2(PO4)3 | 1 M NaPF6 in diglyme | 103.37 at 3C | 93.4% (100 cycles at 3C) | ∼99% | ∼99.9% | 73 | |
| Plasma-treated-carbon-coated Al foil | Na3V2(PO4)3 | 0.6 M NaOTF and 0.4 M NaBF4 in diglyme | 67b at 0.1C | ∼67% (200 cycles at 0.1C)b | ∼99% | ∼99% | 48 | |
| Carbon nanofibers | Na2S@carbon paper | 1 M NaCF3SO3 in diglyme with 5% FEC | 650 at 0.5C | ∼27% (500 cycles at 0.5C) | ∼70% | ∼97% | 75 | |
| Potassium batteries | Cu foil | K0.51V2O5 | 3 M KTFSI in DME | — | 97% (130 cycles at 0.5 A g−1) | ∼46% | ∼99% | 89 |
| Cu foil | Potassiated PTCDA | 0.4 M KPF6-DME with 2 vol% PDMS additive | ∼73 at 0.2C (26 mA g−1) at −40 °C | 82% (50 cycles at 0.2C)b | ∼89% | ∼97% | 20 | |
| Cu foil | K0.51V2O5 | 3 M KTFSI in DME | ∼70 at 0.5 A g−1 | 95.2% (70 cycles at 0.5 A g−1) | ∼90% | ∼92% | 90 | |
| Graphene modified Al foil | Potassiated FeS2 | 4 M KTFSI in DME | ∼210 at 0.1 A g−1 | ∼24% (30 cycles at 0.1 A g−1) | 73.6% | ∼92% | 50 | |
| Zinc batteries | Cu foil | ZnMnO2 | 3 M Zn(CF3SO3)2 and 0.1 M Mn(CF3SO3)2 aqueous electrolyte | ∼170 at 1 A g−1 | 80% (300 cycles at 1 A g−1) | — | Close to 100% | 62 |
| Ti foil | ZnxVOPO4 | 4 M Zn(OTF)2 + 0.5 M Me3EtNOTF aqueous electrolyte | ∼133 at 0.5 mA cm−2 | 80% (90 cycles at 0.5 mA cm−2) | ∼98% | ∼99.9% | 74 | |
| Zeolite-coated Cu foil | α-MnO2 | 2 M ZnSO4 + 0.2 M MnSO4 aqueous electrolyte | 183.8 at 2 A g−1 | 76.4% (90 cycles at 2 A g−1) | ∼99.9% | ∼99.9% | 59 | |
| Carbon nanodiscs-coated Cu foil | Pre-zincificated MnO2 | Zn(CF3SO3)2 aqueous electrolyte | 200 at 1 mA cm−2 | 68.2% (80 cycles at 1 mA cm−2) | — | — | 51 | |
| Ag-coated Cu foil | Zincificated NaV3O8·1.5H2O | Zn(OTF)2 in isosorbide dimethyl ether-H2O co-solvent | 3 mA h cm−2 at 0.5C | 60% (300 cycles at 0.5C) | ∼99.9% | ∼99.9% | 82 | |
| Aluminum hydroxide fluoride-coated Cu foil | Zn0.5VO2 | 2 M ZnSO4 aqueous electrolyte | 203 at 1 A g−1 | 80% (300 cycles at 1 A g−1) | ∼97% | ∼100% | 61 | |
| 60% (2000 cycles at 1 A g−1) | ||||||||
| Cu foil | Amorphous vanadium oxide grown on graphene | 4 M Zn(OTf)2 + 3 M 2-(trimethylazaniumyl)acetate aqueous electrolyte | ∼320 at 10 A g−1 | 71.4% (100 cycles at 10 A g−1) | ∼99% | 98.7% | 87 | |
| ∼180b at 200 mA g−1 | ∼69% (100 cycles at 200 mA g−1)b | ∼100% | 99.7% | |||||
| Cu nanoclusters@Cu foil | Graphene/PVP@ZnI2 | 5 mM ZnI2 and 10 mM I2 in 2 M ZnSO4 aqueous electrolyte | 125.7 at 1 A g−1 | 63.8% (200 cycles at 1 A g−1) | 96.4% | ∼98% | 101 | |
| Cu foil | Pre-zincated-Zn0.25V2O5·nH2O | 2 mmol Zn(OTf)2 in sulfolane/water eutectic electrolyte | 3.8 mA h cm−2 at 0.15C (1C = 300 mA h gZVO−1) | 85% (100 cycles at 0.15C) | 95% | ∼98% | 84 | |
| Cu foil | Graphite | 0.4 M Zn(TFSI)2 in N-butyl-N-methylpyrrolidinium | 52 at rate of 5C | 90% (100 cycles at 5C) | ∼97% | ∼99.9% | 80 | |
| Ag-coated Cu foil | Graphite | Zn(TFSI)2/ethyl methyl carbonate | 114 at 0.1 A g−1 | 96.2% (50 cycles at 0.1 A g−1) | 94% | ∼95% | 79 | |
| Sb nano-particles modified nano-copper host | Bromine | 0.5 M ZnBr2 and 0.25 M TPABr | 9.8 mA h cm−2 at 10 mA cm−2 | ∼99% (1000 cycles at 10 mA cm−2) | — | 98.95% | 41 | |
| Cu foil | LiFePO4 | 4 M ZnSO4 + 2 M Li2SO4 in 10% DME with SnBr2 | 126 at 0.5C | 35.2% (100 cycles at 0.5C) | ∼88% | 99.1% | 42 | |
| Ag nanowire aerogel | Pre-zincificated MnO2 | 2 M ZnSO4 | 230 at 0.5 A g−1 | 73% (600 cycles at 0.5 A g−1) | ∼100% | ∼100% | 40 | |
| Cu foil | LiFePO4/C | Zn(OTf)2 + 1 M LiOTf in EG/H2O | ∼110 at 1 mA cm−2 | 75.2% (100 cycles at 1 mA cm−2) | ∼90% | 99.32% | 83 | |
5 Conclusion and prospects
In this review, focusing on the critical reversibility issue of metal anodes, the strategies and implementation for realizing high-performance anode-free post-lithium (including sodium–, potassium– and zinc–) metal batteries are discussed, and the state-of-the-art advances in this field are presented. To date, anode current collector engineering and electrolyte modulation are the most popular strategies for constructing anode-free post-lithium metal batteries, generally aiming at achieving uniform metal plating, robust SEI formation, suppressed electrolyte-involved side reactions and enhanced electrode kinetics, which collectively promote the reversibility of metal anodes. Besides, compensating for metal loss on anodes also offers a remedial solution for improving the cyclability of anode-free batteries, which may provide a practical alternative for constructing anode-free batteries before the reversibility issues of the metal anode are completely solved. Overall, considerable progress has been made in anode-free post-lithium metal batteries within a few years; however, this field is still in its infancy and comprehensive investigations are urgently needed to accelerate its development towards practical application. Here, we propose some significant directions for future development in this emerging field.(1) The low reversibility of metal plating/stripping is still a critical hurdle for anode-free metal batteries. Considering the significant influence of current collectors on anode performance in anode-free batteries, some processes involving current collectors, especially the SEI evolution on current collectors, have not been adequately studied. Besides current collector engineering and electrolyte modulation, some effective strategies for metal anodes can be exploited for anode-free metal batteries. For example, considering the suppressed ICE caused by SEI formation on metal anodes of anode-free batteries, fabricating an artificial SEI or preforming a SEI on the anode current collector outside the cell are potential solutions to circumvent related problems. The further development of the above strategies undoubtedly requires deeper comprehension of the SEI formation/evolution mechanism and the relationships between the composition, structure and properties of the formed SEI on the current collector/metal anode. With more efforts devoted to this area, breakthroughs in the reversibility of the metal anodes in anode-free batteries can be expected.
(2) The synthesis for metal-rich cathodes beyond electrochemical methods urgently needs to be developed, especially for zinc-rich cathodes of rocking-chair type anode-free zinc batteries. Other battery configurations such as hybrid-ion batteries42,101 and dual-ion batteries49,102 may provide an optional avenue for anode-free batteries, where the charge carriers for the cathodes are not the same ions as in the metal anodes.103 Besides, considering the relatively low cost and mild reactivity of Zn metal as well as the large abundance of Zn-free cathodes for zinc batteries, such as manganese oxides, vanadium oxides and Prussian blue analogues, the necessity and advantages of anode-free Zn metal batteries are greatly undermined compared to those of their alkali-metal counterparts. As a promising alternative for constructing energy-dense Zn batteries, anode-less Zn metal batteries with high Zn utilization (which contain a little excess of metal on the anode) may provide a more viable solution;21,104 these batteries are advantageous in simplified cathode preparation and higher stability of the battery owing to lowered reversibility requirements on the anode, whereas the fundamental issues for them would be similar to those for the anode-free batteries.
(3) The pursuit of high mass loading of cathodes and high-areal-capacity metal plating/stripping on anodes are undoubtedly critical tasks for achieving high-energy-density anode-free metal batteries for practical applications. On the other side, the prominent volume change problem of metal anodes during metal plating/stripping is also noteworthy. The solid-state batteries have manifested great potential for addressing this problem due to a high-pressure environment and high mechanical strength of the solid electrolyte.105–107 The distinct effects of pressure on metal plating in batteries have also been widely demonstrated.108,109 However, considering the relatively low inner pressure of cells for liquid-electrolyte-based batteries, developing porous current collectors to accommodate the metal deposit is necessary for the stable operation of anode-free batteries in practical application scenarios.
(4) Some particular anode-free battery systems have drawn notable attention in recent years. For example, anode-free batteries with high-voltage cathodes are pursued to reach the extreme energy density of metal batteries. One great challenge is the difficulty in endowing electrolytes with high compatibility with both the metal anode and the high-voltage cathodes, especially for alkali-metal batteries. In this area, electrolyte engineering has demonstrated its effectiveness.47,99 Also, the construction of a robust SEI that prevents the electrolyte-induced side reactions is a promising strategy for realizing high-energy-density anode-free batteries. As another emerging field, low-temperature anode-free metal batteries aim to extend the application of high-energy batteries to harsh temperature environments.20,48,76,82,87 Although the electrode kinetics will be largely limited at low temperatures, the suppression of side reactions on metal anodes is beneficial to the reversibility of the battery. At present, a specialized research system with temperature-specific material characterization techniques and battery performance evaluation methods, for example, in situ low-temperature characterizations and adequate reference electrodes for low-temperature application, has yet to be established for low-temperature metal batteries, and efforts in this area would greatly push forward the development of this field.
(5) The discrepancies between the CE of metal plating/stripping derived from half cells and the cycling performance of full batteries have been reported,44,51 which may stem from the differing operation conditions of the anodes in half cells for CE tests from those in full cells, suggesting the necessity of advancing evaluation methods for the reversibility of metal anodes. For example, the CE of metal plating/stripping on a current collector is commonly evaluated through galvanic cycling of metal foil//current collector half cells, with arbitrarily set current densities and high cut-off overpotentials for the stripping process ranging from 0.2–1 V.110 These protocols may differ greatly from the actual application conditions of metal anodes in full cells, and thus the derived CE loses its reliability in predicting the full cells' performance. Therefore, approaches that can more effectively evaluate the reversibility of metal anodes in anode-free batteries demand further exploration.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Jiawei Wang: conceptualization, writing – original draft preparation, writing – reviewing & editing. Yaosong Zhou: writing – reviewing & editing, visualization. Yanyi Zhuo: visualization. Kun Fang: writing – reviewing & editing. Sicong Wang: writing – reviewing & editing. Bin Zhao: writing – reviewing & editing. Jing Zhou: writing – reviewing & editing. Hua Wang: conceptualization, writing – reviewing & editing, supervision.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the International Cooperation Project of the National Key Research and Development Program of China (2022YFE0126300), National Natural Science Foundation of China (21972007, 52172178 and 52302287), Natural Science Foundation of Beijing (2222059) and Natural Science Foundation of Shanghai (21ZR1445700).References
- H. Kim, G. Jeong, Y. U. Kim, J. H. Kim, C. M. Park and H. J. Sohn, Metallic Anodes for Next Generation Secondary Batteries, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC.
- Y. P. Guo, H. Q. Li and T. Y. Zhai, Reviving Lithium-Metal Anodes for Next-Generation High-Energy Batteries, Adv. Mater., 2017, 29, 1700007 CrossRef.
- J. Xiang, L. Yang, L. Yuan, K. Yuan, Y. Zhang, Y. Huang, J. Lin, F. Pan and Y. Huang, Alkali-Metal Anodes: from Lab to Market, Joule, 2019, 3, 2334–2363 CrossRef.
- C. Nie, G. Wang, D. Wang, M. Wang, X. Gao, Z. Bai, N. Wang, J. Yang, Z. Xing and S. Dou, Recent Progress on Zn Anodes for Advanced Aqueous Zinc-Ion Batteries, Adv. Energy Mater., 2023, 13, 2300606 CrossRef.
- S. H. Chung and A. Manthiram, Current Status and Future Prospects of Metal-Sulfur Batteries, Adv. Mater., 2019, 31, e1901125 CrossRef PubMed.
- Y. Chen, J. Xu, P. He, Y. Qiao, S. Guo, H. Yang and H. Zhou, Metal-Air Batteries: Progress and Perspective, Sci. Bull., 2022, 67, 2449–2486 CrossRef PubMed.
- Q. Wang, L. Ren, T. Lu, S. Wang, Z. Jiang, I. A. Chandio, L. Guan, L. Hou, H. Du, H. Wei, X. Liu, C. Yang, Y. Wei, W. Liu and H. Zhou, Air-Stable Lithium Metal Anodes: A Perspective of Surface Engineering from Different Dimensions, ACS Energy Lett, 2023, 8, 4441–4464 CrossRef.
- C. Li, X. Xie, S. Liang and J. Zhou, Issues and Future Perspective on Zinc Metal Anode for Rechargeable Aqueous Zinc-Ion Batteries, Energy Environ. Mater., 2020, 3, 146–159 CrossRef.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Post-Lithium-Ion Battery Cell Production and Its Compatibility with Lithium-Ion Cell Production Infrastructure, Nat Energy, 2021, 6, 123–134 CrossRef.
- C. Ye, H. Li, Y. Chen, J. Hao, J. Liu, J. Shan and S. Z. Qiao, The Role of Electrocatalytic Materials for Developing Post-Lithium Metal‖Sulfur Batteries, Nat. Commun., 2024, 15, 4797 CrossRef PubMed.
- Y. Sun, J. C. Li, H. S. Zhou and S. H. Guo, Wide-Temperature-Range Sodium-Metal Batteries: from Fundamentals and Obstacles to Optimization, Energy Environ. Sci., 2023, 16, 4759–4811 RSC.
- L. K. Zhao, X. W. Gao, T. Z. Ren, D. Wang, D. W. Wang, Z. M. Liu, H. Chen and W. B. Luo, Regulating Ion Transport Behaviors Toward Dendrite-Free Potassium Metal Batteries: Recent Advances and Perspectives, Rare Met., 2024, 43, 1435–1460 CrossRef.
- H. Y. Li, S. J. Li, R. L. Hou, Y. Rao, S. H. Guo, Z. Chang and H. S. Zhou, Recent Advances in Zinc-Ion Dehydration Strategies for Optimized Zn-Metal Batteries, Chem. Soc. Rev., 2024, 53, 7742–7783 RSC.
- W. Yao, P. Zou, M. Wang, H. Zhan, F. Kang and C. Yang, Design Principle, Optimization Strategies, and Future Perspectives of Anode-Free Configurations for High-Energy Rechargeable Metal Batteries, Electrochem. Energy Rev., 2021, 4, 601–631 CrossRef.
- S. Nanda, A. Gupta and A. Manthiram, Anode-Free Full Cells: A Pathway to High-Energy Density Lithium-Metal Batteries, Adv. Energy Mater., 2021, 11, 2000804 CrossRef CAS.
- L. Dong, S. Zhong, S. Zhang, B. Yuan, J. Liu, H. Xie, C. Zhang, Y. Liu, C. Yang, J. Han and W. He, Toward Practical Anode-Free Lithium Pouch Batteries, Energy Environ. Sci., 2023, 16, 5605–5632 RSC.
- Z. Hu, L. Liu, X. Wang, Q. Zheng, C. Han and W. Li, Current Progress of Anode-Free Rechargeable Sodium Metal Batteries: Origin, Challenges, Strategies, and Perspectives, Adv. Funct. Mater., 2024, 34, 2313823 CrossRef CAS.
- T. Yang, D. Luo, Y. Liu, A. Yu and Z. Chen, Review Anode-Free Sodium Metal Batteries as Rising Stars for Lithium-Ion Alternatives, iScience, 2023, 26, 105982 CrossRef CAS.
- N. Ren, L. Wang, X. Li, K. Cao, Z. He, Y. Shao, J. Xiao, Y. Zhu, B. Pan, S. Jiao and C. Chen, Design Principles of Mediation Layer for Current Collectors toward High-Performance Anode-Free Potassium-Metal Batteries: A Case Study of Cu6Sn5 on Copper, Adv. Funct. Mater., 2024, 34, 2313538 CrossRef CAS.
- M. Tang, S. Dong, J. Wang, L. Cheng, Q. Zhu, Y. Li, X. Yang, L. Guo and H. Wang, Low-Temperature Anode-Free Potassium Metal Batteries, Nat. Commun., 2023, 14, 6006 CrossRef CAS.
- X. Zhang, L. Zhang, X. Jia, W. Song and Y. Liu, Design Strategies for Aqueous Zinc Metal Batteries with High Zinc Utilization: from Metal Anodes to Anode-Free Structures, Nano–Micro Lett., 2024, 16, 75 CrossRef CAS.
- W. Z. Huang, C. Z. Zhao, P. Wu, H. Yuan, W. E. Feng, Z. Y. Liu, Y. Lu, S. Sun, Z. H. Fu, J. K. Hu, S. J. Yang, J. Q. Huang and Q. Zhang, Anode-Free Solid-State Lithium Batteries: A Review, Adv. Energy Mater., 2022, 12, 2201044 CrossRef.
- P. Molaiyan, M. Abdollahifar, B. Boz, A. Beutl, M. Krammer, N. Zhang, A. Tron, M. Romio, M. Ricci, R. Adelung, A. Kwade, U. Lassi and A. Paolella, Optimizing Current Collector Interfaces for Efficient “Anode-Free” Lithium Metal Batteries, Adv. Funct. Mater., 2024, 34, 2311301 CrossRef.
- Y. Tian, Y. An, C. Wei, H. Jiang, S. Xiong, J. Feng and Y. Qian, Recently Advances and Perspectives of Anode-Free Rechargeable Batteries, Nano Energy, 2020, 78, 105344 CrossRef.
- Z. Tong, B. Bazri, S. Hu and R. Liu, Interfacial Chemistry in Anode-Free Batteries: Challenges and Strategies, J. Mater. Chem. A, 2021, 9, 7396–7406 RSC.
- C. G. Tang, C. Y. Cai, J. D. Zhang, F. Gao, T. Hu, Z. Pu, J. Z. Weng and M. Q. Zhu, Recent Progress of Regulation Factors on the Deposition of Sodium Anodes, J. Electrochem. Soc., 2024, 171, 070534 CrossRef.
- J. Wang, Y. Yang, Y. Zhang, Y. Li, R. Sun, Z. Wang and H. Wang, Strategies towards the Challenges of Zinc Metal Anode in Rechargeable Aqueous Zinc Ion Batteries, Energy Storage Mater., 2021, 35, 19–46 CrossRef.
- G. Weng, Z. X. Dong, P. Xiang, Y. L. Zhu, C. Wu, X. Z. Yang, H. K. Liu and S. X. Dou, Critical Criteria Depicting the Rational Design of Zn Anode Current Collector, Adv. Funct. Mater., 2024, 34, 2400839 CrossRef.
- Y. X. Gong, B. Wang, H. Z. Ren, D. Y. Li, D. L. Wang, H. K. Liu and S. X. Dou, Recent Advances in Structural Optimization and Surface Modification on Current Collectors for High-Performance Zinc Anode: Principles, Strategies, and Challenges, Nano–Micro Lett., 2023, 15, 208 CrossRef PubMed.
- G. Wang, C. Song, J. Q. Huang and H. S. Park, Recent Advances in Carbon-Based Current Collectors/Hosts for Alkali Metal Anodes, Energy Environ. Mater., 2023, 6, e12460 CrossRef.
- J. Y. Chen, Y. Z. Wang, S. J. Li, H. R. Chen, X. Qiao, J. Zhao, Y. W. Ma and H. N. Alshareef, Porous Metal Current Collectors for Alkali Metal Batteries, Adv. Sci., 2023, 10, 2205695 CrossRef.
- S. Tang, Z. Qiu, X. Wang, Y. Gu, X. Zhang, W. Wang, J. Yan, M. Zheng, Q. Dong and B. Mao, A Room-Temperature Sodium Metal Anode Enabled by A Sodiophilic Layer, Nano Energy, 2018, 48, 101–106 CrossRef.
- H. Wang, Y. Wu, S. Liu, Y. Jiang, D. Shen, T. Kang, Z. Tong, D. Wu, X. Li and C. S. Lee, 3D Ag@C Cloth for Stable Anode Free Sodium Metal Batteries, Small Methods, 2021, 5, 2001050 CrossRef CAS.
- X. Chen, X. Zhou, Z. Yang, Z. Hao, J. Chen, W. Kuang, X. Shi, X. Wu, L. Li and S. Chou, A Conductive and Sodiophilic Ag Coating Layer Regulating Na Deposition Behaviors for Highly Reversible Sodium Metal Batteries, Chem. Sci., 2024, 15, 4833–4838 RSC.
- O. J. Dahunsi, S. Gao, J. Kaelin, B. Li, R. I. Abdul, B. An and Y. Cheng, Anode-Free Na Metal Batteries Developed by Nearly Fully Reversible Na Plating on the Zn Surface, Nanoscale, 2023, 15, 3255–3262 RSC.
- C. Liu, Y. Xie, H. Li, J. Xu and Z. Zhang, In Situ Construction of Sodiophilic Alloy Interface Enabled Homogenous Na Nucleation and Deposition for Sodium Metal Anode, J. Electrochem. Soc., 2022, 169, 080521 CrossRef CAS.
- S. Li, H. Zhu, Y. Liu, Q. Wu, S. Cheng and J. Xie, Space-Confined Guest Synthesis to Fabricate Sn-Monodispersed N-Doped Mesoporous Host toward Anode-Free Na Batteries, Adv. Mater., 2023, 35, 2301967 CrossRef CAS PubMed.
- D. Zhang, M. Liu, M. Li, Z. Yuan, Y. Hu, H. Chen, C. Li, L. Kong, K. Zhao, J. Ren and B. Liu, One-Stone, Two Birds: Three-Dimensional Structure and Rich-Inorganic Solid Electrolyte Interface Derived from Bi Nanoparticles Modified Ti3C2/CNT/rGO Aerogel for Anode-Free Potassium Metal Batteries, Chem. Eng. J., 2024, 482, 148896 CrossRef.
- X. Zheng, Z. Liu, J. Sun, R. Luo, K. Xu, M. Si, J. Kang, Y. Yuan, S. Liu, T. Ahmad, T. Jiang, N. Chen, M. Wang, Y. Xu, M. Chuai, Z. Zhu, Q. Peng, Y. Meng, K. Zhang, W. Wang and W. Chen, Constructing Robust Heterostructured Interface for Anode-Free Zinc Batteries with Ultrahigh Capacities, Nat. Commun., 2023, 14, 76 CrossRef PubMed.
- W. Ling, Q. Yang, F. Mo, H. Lei, J. Wang, Y. Jiao, Y. Qiu, T. Chen and Y. Huang, An Ultrahigh Rate Dendrite-Free Zn Metal Deposition/Striping Enabled by Silver Nanowire Aerogel with Optimal Atomic Affinity with Zn, Energy Storage Mater., 2022, 51, 453–464 CrossRef.
- K. Xu, X. Zheng, R. Luo, J. Sun, Y. Ma, N. Chen, M. Wang, L. Song, Q. Zhao and W. Chen, A Three-Dimensional Zincophilic Nano-Copper Host Enables Dendrite-Free and Anode-Free Zn Batteries, Mater. Today Energy, 2023, 34, 101284 CrossRef.
- T. A. Nigatu, H. K. Bezabh, S. Jiang, B. W. Taklu, Y. Nikodimos, S. Yang, S. Wu, W. Su, C. Yang and B. J. Hwang, An Anode-Free Aqueous Hybrid Batteries Enabled by In-Situ Cu/Sn/Zn Alloy Formation on Pure Cu Substrate, Electrochim. Acta, 2023, 443, 141883 CrossRef.
- S. Xie, Y. Li and L. Dong, Stable Anode-Free Zinc-Ion Batteries Enabled by Alloy Network-Modulated Zinc Deposition Interface, J. Energy Chem., 2023, 76, 32–40 CrossRef.
- W. Zhang, J. Zheng, Z. Ren, J. Wang, J. Luo, Y. Wang, X. Tao and T. Liu, Anode-Free Sodium Metal Pouch Cell Using Cu3P Nanowires in situ Grown on Current Collector, Adv. Mater., 2024, 36(15), 2310347 CrossRef.
- Y. Wang, H. Dong, N. Katyal, H. Hao, P. Liu, H. Celio, G. Henkelman, J. Watt and D. Mitlin, A Sodium–Antimony–Telluride Intermetallic Allows Sodium-Metal Cycling at 100% Depth of Discharge and as An Anode-Free Metal Battery, Adv. Mater., 2022, 34, 2106005 CrossRef.
- F. Mazzali, M. W. Orzech, A. Adomkevicius, A. Pisanu, L. Malavasi, D. Deganello and S. Margadonna, Designing a High-Power Sodium-Ion Battery by in situ Metal Plating, ACS Appl. Energy Mater., 2019, 2, 344–353 CrossRef.
- Y. Li, Q. Zhou, S. Weng, F. Ding, X. Qi, J. Lu, Y. Li, X. Zhang, X. Rong, Y. Lu, X. Wang, R. Xiao, H. Li, X. Huang, L. Chen and Y. Hu, Interfacial Engineering to Achieve An Energy Density of over 200 W h kg−1 in Sodium Batteries, Nat. Energy, 2022, 7, 511–519 CrossRef.
- Q. Zhu, D. Yu, J. Chen, L. Cheng, M. Tang, Y. Wang, Y. Li, J. Yang and H. Wang, A 110 W h kg−1 Ah-Level Anode-Free Sodium Battery at −40°C, Joule, 2024, 8, 482–495 CrossRef.
- J. Zhao, M. Tang, H. Lan, Q. Zhu, G. Wang, G. Yang, J. Yang, W. Zhou and H. Wang, An Anode-Free Sodium Dual-Ion Battery, Energy Storage Mater., 2024, 70, 103480 CrossRef.
- Y. Zhao, B. Liu, Y. Yi, X. Lian, M. Wang, S. Li, X. Yang and J. Sun, An Anode-Free Potassium-Metal Battery Enabled by A Directly Grown Graphene-Modulated Aluminum Current Collector, Adv. Mater., 2022, 34, 2202902 CrossRef CAS.
- Y. Zhu, Y. Cui and H. N. Alshareef, An Anode-Free Zn–MnO2 Battery, Nano Lett., 2021, 21, 1446–1453 CrossRef CAS PubMed.
- O. Blumen, G. Bergman, K. Schwatrzman, S. Harpaz, S. H. Akella, M. S. Chae, N. Bruchiel-Spanier, N. Shpigel and D. Sharon, Selection Criteria for Current Collectors for Highly Efficient Anode-Free Zn Batteries, J. Mater. Chem. A, 2023, 11, 1997–1998 RSC.
- A. P. Vijaya Kumar Saroja, Z. Wang, H. R. Tinker, F. R. Wang, P. R. Shearing and Y. Xu, Enabling Intercalation-Type TiNb24O62 Anode for Sodium- and Potassium-Ion Batteries via A Synergetic Strategy of Oxygen Vacancy and Carbon Incorporation, SusMat, 2023, 3, 222–234 CrossRef CAS.
- K. Lee, Y. J. Lee, M. J. Lee, J. Han, J. Lim, K. Ryu, H. Yoon, B. H. Kim, B. J. Kim and S. W. Lee, A 3D Hierarchical Host with Enhanced Sodiophilicity Enabling Anode-Free Sodium-Metal Batteries, Adv. Mater., 2022, 34, 2109767 CrossRef CAS.
- S. Kandula, E. Kim, C. W. Ahn, J. Lee, B. Yeom, S. W. Lee, J. Cho, H. Lim, Y. Lee and J. G. Son, A Resilient MXene/CNT Nano-Accordion Framework for Anode-Free Sodium-Metal Batteries with Exceptional Cyclic Stability, Energy Storage Mater., 2023, 63, 103024 CrossRef.
- F. Huang, P. Xu, G. Z. Fang and S. Q. Liang, In-Depth Understanding of Interfacial Na+ Behaviors in Sodium Metal Anode: Migration, Desolvation, and Deposition, Adv. Mater., 2024, 36, 2405310 CrossRef CAS PubMed.
- R. Zhuang, X. Zhang, C. Qu, X. Xu, J. Yang, Q. Ye, Z. Liu, S. Kaskel, F. Xu and H. Wang, Fluorinated Porous Frameworks Enable Robust Anode-Less Sodium Metal Batteries, Sci. Adv., 2023, 9, eadh8060 CrossRef.
- C. Wang, Y. Zheng, Z. N. Chen, R. Zhang, W. He, K. Li, S. Yan, J. Cui, X. Fang, J. Yan, G. Xu, D. Peng, B. Ren and N. Zheng, Robust Anode-Free Sodium Metal Batteries Enabled by Artificial Sodium Formate Interface, Adv. Energy Mater., 2023, 13, 2204125 CrossRef.
- R. Zhao, J. Yang, X. Han, Y. Wang, Q. Ni, Z. Hu, C. Wu and Y. Bai, Stabilizing Zn Metal Anodes via Cation/Anion Regulation toward High Energy Density Zn-Ion Batteries, Adv. Energy Mater., 2023, 13, 2203542 CrossRef.
- Y. Xu, X. Zheng, J. Sun, W. Wang, M. Wang, Y. Yuan, M. Chuai, N. Chen, H. Hu and W. Chen, Nucleophilic Interfacial Layer Enables Stable Zn Anodes for Aqueous Zn Batteries, Nano Lett., 2022, 22, 3298–3306 CrossRef PubMed.
- C. Wang, D. Wang, D. Lv, H. Peng, X. Song, J. Yang and Y. Qian, Interface Engineering by Hydrophilic and Zincophilic Aluminum Hydroxide Fluoride for Anode-Free Zinc Metal Batteries at Low Temperature, Adv. Energy Mater., 2023, 13, 2204388 CrossRef.
- D. Y. Jeong, W. J. Chang, S. Jang, M. Kim, Y. Kim, B. Kim and W. I. Park, Controlling Dendrite Growth and Side Reactions in Anode-Free Zn-Ion Aqueous Batteries with PMMA:Zn Coated Electrode, J. Energy Storage, 2024, 76, 109791 CrossRef.
- S. Li, H. Zhu, C. Gu, F. Ma, W. Zhong, M. Liu, H. Zhang, Z. Zeng, S. Cheng and J. Xie, Customized Electrolyte and Host Structures Enabling High-Energy-Density Anode-Free Potassium–Metal Batteries, ACS Energy Lett., 2023, 8, 3467–3475 CrossRef.
- G. Deysher, J. A. S. Oh, Y. Chen, B. Sayahpour, S. Ham, D. Cheng, P. Ridley, A. Cronk, S. W. Lin, K. Qian, L. H. B. Nguyen, J. Jang and Y. S. Meng, Design Principles for Enabling An Anode-Free Sodium All-Solid-State Battery, Nat. Energy, 2024, 9, 1161–1172 Search PubMed.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share and C. L. Pint, Anode-Free Sodium Battery Through in situ Plating of Sodium Metal, Nano Lett., 2017, 17, 1296–1301 CrossRef PubMed.
- I. B. Abdul Razak, B. An, B. Li, O. J. Dahunsi, F. Xia, P. Murugan, D. Brown and Y. Cheng, Metallic Cu Surface Enables Reversible Na Metal Anodes and Stabilizes Anode-Free Sodium Metal Batteries, J. Electrochem. Soc., 2023, 170, 110534 CrossRef.
- S. Kang, F. Geng, Z. Li, Y. Jiang, M. Shen, Q. Chen, X. Lou and B. Hu, Progressive Self-Leveling Deposition Improves the Cyclability of Anode-Less Sodium Metal Batteries Revealed by in situ EPR Imaging, ACS Energy Lett, 2024, 1633–1638 CrossRef.
- Y. Zhang, L. Wang, Q. Li, B. Hu, J. Kang, Y. Meng, Z. Zhao and H. Lu, Iodine Promoted Ultralow Zn Nucleation Overpotential and Zn-rich Cathode for Low-Cost, Fast-Production and High-Energy Density Anode-Free Zn-Iodine Batteries, Nano–Micro Lett., 2022, 14, 208 CrossRef PubMed.
- M. Xi, Z. Liu, W. Wang, Z. Qi, R. Sheng, J. Ding, Y. Huang and Z. Guo, Shear-Flow Induced Alignment of Graphene Enables the Closest Packing Crystallography of the (002) Textured Zinc Metal Anode with High Reversibility, Energy Environ. Sci., 2024, 17, 3168–3178 RSC.
- E. R. Cooper, M. Li, Q. Xia, I. Gentle and R. Knibbe, Facilitating Sodium Nucleation in Anode-Free Sodium Batteries, ACS Appl. Energy Mater., 2023, 6, 11550–11559 CrossRef.
- A. J. Hu, F. Li, W. Chen, T. Y. Lei, Y. Y. Li, Y. X. Fan, M. He, F. Wang, M. J. Zhou, Y. Hu, Y. C. Yan, B. Chen, J. Zhu, J. P. Long, X. F. Wang and J. Xiong, Ion Transport Kinetics in Low-Temperature Lithium Metal Batteries, Adv. Energy Mater., 2022, 12, 2202432 CrossRef.
- W. H. Ma, S. Wang, X. W. Wu, W. W. Liu, F. Yang, S. D. Liu, S. C. Jun, L. Dai, Z. X. He and Q. B. Zhang, Tailoring Desolvation Strategies for Aqueous Zinc-Ion Batteries, Energy Environ. Sci., 2024, 17, 4819–4846 RSC.
- B. Ma, Y. Lee and P. Bai, Dynamic Interfacial Stability Confirmed by Microscopic Optical Operando Experiments Enables High-Retention-Rate Anode-Free Na Metal Full Cells, Adv. Sci., 2021, 8(12), 2005006 CrossRef PubMed.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X. Yang, K. Xu, O. Borodin and C. Wang, Fluorinated Interphase Enables Reversible Aqueous Zinc Battery Chemistries, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef PubMed.
- M. Geng, D. Han, Z. Huang, S. Wang, M. Xiao, S. Zhang, L. Sun, S. Huang and Y. Meng, A Stable Anode-Free Na-S Full Cell at Room Temperature, Energy Storage Mater., 2022, 52, 230–237 CrossRef.
- B. Ge, J. Deng, Z. Wang, Q. Liang, L. Hu, X. Ren, R. Li, Y. Lin, Y. Li, Q. Wang, B. Han, Y. Deng, X. Fan, B. Li, G. Chen and X. Yu, Aggregate-Dominated Dilute Electrolytes with Low-Temperature-Resistant Ion-Conducting Channels for Highly Reversible Na Plating/Stripping, Adv. Mater., 2024, 36, 2408161 CrossRef.
- Z. Cai, J. Wang and Y. Sun, Anode Corrosion in Aqueous Zn Metal Batteries, eScience, 2023, 3, 100093 CrossRef.
- W. Kim, H. Kim, K. M. Lee, E. Shin, X. Liu, H. Moon, H. Adenusi, S. Passerini, S. K. Kwak and S. Lee, Demixing the Miscible Liquids: toward Biphasic Battery Electrolytes Based on the Kosmotropic Effect, Energy Environ. Sci., 2022, 15, 5217–5228 RSC.
- G. Wang, M. Zhu, G. Chen, Z. Qu, B. Kohn, U. Scheler, X. Chu, Y. Fu, O. G. Schmidt and X. Feng, An Anode-Free Zn–Graphite Battery, Adv. Mater., 2022, 34, 2201957 CrossRef PubMed.
- N. Li, J. Wang, Q. Zhang, X. Zhou, H. Wang, G. Lu, J. Zhao, Z. Chen and G. Cui, Enhanced Zn2+ Transport in Ionic Liquid Electrolyte by Hydrofluoroether Dilution for High-Power and Long-Life Zn/Graphite Cells, Batteries Supercaps, 2023, 6, e202300058 CrossRef.
- F. Ming, Y. Zhu, G. Huang, A. Emwas, H. Liang, Y. Cui and H. N. Alshareef, Co-Solvent Electrolyte Engineering for Stable Anode-Free Zinc Metal Batteries, J. Am. Chem. Soc., 2022, 144, 7160–7170, DOI:10.1021/jacs.1c12764.
- Y. Li, J. Wang, S. Wang, Y. Wang, Y. Xu, L. Cheng, M. Tang, G. Wang, W. Tian, W. Huang and H. Wang, Realizing High-Areal-Capacity Anode-Free Zn Metal Batteries, Energy Storage Mater., 2024, 66, 103245 CrossRef.
- J. Duan, L. Min, M. Wu, T. Yang, M. Chen and C. Wang, “Anode-Free” Zn/LiFePO4 Aqueous Batteries Boosted by Hybrid Electrolyte, J. Ind. Eng. Chem., 2022, 114, 317–322 CrossRef.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Enabling Selective Zinc-Ion Intercalation by A Eutectic Electrolyte for Practical Anodeless Zinc Batteries, Nat. Commun., 2023, 14, 3067 CrossRef PubMed.
- X. J. Pu, S. H. Zhang, D. Zhao, Z. L. Xu, Z. X. Chen and Y. L. Cao, Building the Robust Fluorinated Electrode-Electrolyte Interface in Rechargeable Batteries: from Fundamentals to Applications, Electrochem. Energy Rev., 2024, 7, 21 CrossRef.
- J. Wang, D. Yu, X. Sun, H. Wang and J. Li, Anodes for Low-Temperature Rechargeable Batteries, eScience, 2024, 4, 100252 CrossRef.
- H. Kim, K. M. Lee, W. Kim, S. H. Kweon, X. Wang, S. Zheng, S. Kim, J. H. Ha, S. J. Kang, Z. Wu, S. K. Kwak and S. Lee, Restructuring of Aqueous Electrolytes Using A Soft-Acidic/Hard-Basic Zwitterion for Low-Temperature Anode-Free Zn Batteries, Energy Environ. Sci., 2024, 17, 1961–1974 RSC.
- H. Li, H. Zhang, F. Wu, M. Zarrabeitia, D. Geiger, U. Kaiser, A. Varzi and S. Passerini, Sodiophilic Current Collectors Based on MOF-Derived Nanocomposites for Anode-Less Na-Metal Batteries, Adv. Energy Mater., 2022, 12, 2202293 CrossRef.
- J. Wang, Y. Zuo, M. Chen, K. Chen, Z. Chen, Z. Lu and L. Si, Bifunctional Separator with A Light-Weight Coating for Stable Anode-Free Potassium Metal Batteries, Electrochim. Acta, 2022, 433, 141211 CrossRef CAS.
- L. Si, J. Wang, M. Chen, K. Chen, Z. Chen, Z. Lu, Y. Zhang, Y. Zhang and H. Liu, Stable Solid Electrolyte Interface Achieved by Separator Surface Modification for High-Performance Anode-Free Potassium Metal Batteries, ACS Appl. Energy Mater., 2023, 6, 326–333 CrossRef CAS.
- M. Tanwar, H. K. Bezabh, S. Basu, W. Su and B. Hwang, Investigation of Sodium Plating and Stripping on A Bare Current Collector with Different Electrolytes and Cycling Protocols, ACS Appl. Mater. Interfaces, 2019, 11, 39746–39756 CrossRef CAS PubMed.
- B. Sayahpour, W. Li, S. Bai, B. Lu, B. Han, Y. Chen, G. Deysher, S. Parab, P. Ridley, G. Raghavendran, L. H. B. Nguyen, M. Zhang and Y. S. Meng, Quantitative Analysis of Sodium Metal Deposition and Interphase in Na Metal Batteries, Energy Environ. Sci., 2024, 17, 1216–1228 RSC.
- J. Wu, C. Lin, Q. Liang, G. Zhou, J. Liu, G. Liang, M. Wang, B. Li, L. Hu, F. Ciucci, Q. Liu, G. Chen and X. Yu, Sodium-Rich NASICON-Structured Cathodes for Boosting the Energy Density and Lifespan of Sodium-Free-Anode Sodium Metal Batteries, InfoMat, 2022, 4, e12288 CrossRef.
- Y. Liu, X. Wu, A. Moeez, Z. Peng, Y. Xia, D. Zhao, J. Liu and W. Li, Na-Rich Na3V2(PO4)3 Cathodes for Long Cycling Rechargeable Sodium Full Cells, Adv. Energy Mater., 2023, 13, 2203283 CrossRef.
- J. Wu, Q. Kuang, K. Zhang, J. Feng, C. Huang, J. Li, Q. Fan, Y. Dong and Y. Zhao, Spinel Zn3V3O8: A High-Capacity Zinc Supplied Cathode for Aqueous Zn-Ion Batteries, Energy Storage Mater., 2021, 41, 297–309 CrossRef.
- Y. Zhang, C. Zhang, Y. Guo, M. Fan, Y. Zhao, H. Guo, W. Wang, S. Tan, Y. Yin, F. Wang, S. Xin, Y. Guo and L. Wan, Refined Electrolyte and Interfacial Chemistry toward Realization of High-Energy Anode-Free Rechargeable Sodium Batteries, J. Am. Chem. Soc., 2023, 145, 25643–25652 CrossRef.
- K. M. Su, Y. Wang, B. Yang, X. Zhang, W. Wu, J. W. Lang and X. B. Yan, A Review: Pre-Lithiation Strategies Based on Cathode Sacrificial Lithium Salts for Lithium-Ion Capacitors, Energy Environ. Mater., 2023, 6, e12506 CrossRef.
- P. Kulkarni, H. Jung, D. Ghosh, M. Jalalah, M. Alsaiari, F. A. Harraz and R. G. Balakrishna, A Comprehensive Review of Pre-Lithiation/Sodiation Additives for Li-Ion and Na-Ion Batteries, J. Energy Chem., 2023, 76, 479–494 CrossRef.
- Z. Lu, H. Yang, Q. H. Yang, P. He and H. Zhou, Building A Beyond Concentrated Electrolyte for High-Voltage Anode-Free Rechargeable Sodium Batteries, Angew. Chem., Int. Ed., 2022, 61, e202200410 CrossRef PubMed.
- X. Cheng, D. Li, S. Peng, P. Shi, H. Yu, Y. Jiang and S. Li, In-Situ Alloy-Modified Sodiophilic Current Collectors for Anode-Less Sodium Metal Batteries, Batteries, 2023, 9, 408 CrossRef.
- H. Wu, S. Luo, W. Zheng, L. Li, Y. Fang and W. Yuan, Metal- and Binder-Free Dual-Ion Battery Based on Green Synthetic Nano-Embroidered Spherical Organic Anode and Pure Ionic Liquid Electrolyte, Energy Mater., 2024, 4, 400015 Search PubMed.
- Y. An, B. Xu, Y. Tian, H. Shen, Q. Man, X. Liu, Y. Yang and M. Li, Reversible Zn Electrodeposition Enabled by Interfacial Chemistry Manipulation for High-Energy Anode-Free Zn Batteries, Mater. Today, 2023, 70, 93–103 CrossRef.
- Y. Zhang, L. Wang, Q. Li, B. Hu, J. Kang, Y. Meng, Z. Zhao and H. Lu, Iodine Promoted Ultralow Zn Nucleation Overpotential and Zn-Rich Cathode for Low-Cost, Fast-Production and High-Energy Density Anode-Free Zn-Iodine Batteries, Nano–Micro Lett., 2022, 14, 208 CrossRef.
- L. Zhang, J. Xiao, X. Xiao, W. Xin, Y. Geng, Z. Yan and Z. Zhu, Molecular Engineering of Self-Assembled Monolayers for Highly Utilized Zn Anodes, eScience, 2024, 4, 100205 CrossRef.
- Z. J. Wu, S. N. He, C. Zheng, J. T. Gan, L. N. She, M. C. Zhang, Y. Gao, Y. X. Yang and H. G. Pan, Fabrication Pressures and Stack Pressures in Solid-State Battery, eScience, 2024, 4, 100247 CrossRef.
- T. Ortmann, T. Fuchs, J. K. Eckhardt, Z. Ding, Q. Ma, F. Tietz, C. Kübel, M. Rohnke and J. Janek, Deposition of Sodium Metal at the Copper-NASICON Interface for Reservoir-Free Solid-State Sodium Batteries, Adv. Energy Mater., 2023, 14, 2302729 CrossRef.
- Z. Li, J. Fu and X. Guo, How to Commercialize Solid-State Batteries: A Perspective from Solid Electrolytes, Nat. Sci. Open, 2023, 2, 20220036 CrossRef.
- X. Yin, W. Tang, I. D. Jung, K. C. Phua, S. Adams, S. W. Lee and G. W. Zheng, Insights into Morphological Evolution and Cycling Behavior of Lithium Metal Anode under Mechanical Pressure, Nano Energy, 2018, 50, 659–664 CrossRef.
- X. Zhang, Q. J. Wang, K. L. Harrison, K. Jungjohann, B. L. Boyce, S. A. Roberts, P. M. Attia and S. J. Harris, Rethinking How External Pressure Can Suppress Dendrites in Lithium Metal Batteries, J. Electrochem. Soc., 2019, 166, A3639–A3652 CrossRef.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Realizing High Zinc Reversibility in Rechargeable Batteries, Nat. Energy, 2020, 5, 743–749 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |