
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt-nanoparticles on ZnO/carbon quantum dots: a trifunctional nanocomposite with superior electrocatalytic activity boosting direct methanol fuel cells and zinc–air batteries†
Anup Kumar
Pradhan
 a,
Sayan
Halder
a and
Chanchal
Chakraborty
*ab
a,
Sayan
Halder
a and
Chanchal
Chakraborty
*ab
aDepartment of Chemistry, Birla Institute of Technology & Science (BITS) Pilani, Hyderabad Campus Jawaharnagar, Samirpet, Hyderabad, Telangana 500078, India. E-mail: chanchal@hyderabad.bits-pilani.ac.in
bMaterials Center for Sustainable Energy & Environment (McSEE), Birla Institute of Technology and Science, Hyderabad Campus, Hyderabad 500078, India
First published on 14th November 2024
Abstract
Architecting efficient, multifunctional, and low-cost nano-electrocatalysts plays a vital role in electrochemical energy conversion and storage systems. Low-Pt hybrid catalysts are in high demand, offering cost-effective solutions for electrode materials in direct methanol fuel cells (DMFCs) and Zn–air batteries (ZABs). Herein, we synthesized a ternary nanocomposite (PtNP-ZnO@CQDs) composed of ultrafine platinum nanoparticles (PtNPs) smaller than 5 nm on photosensitive ZnO and carbon quantum dots (CQDs) via a simple one-pot hydrothermal process for efficient photoinduced electrocatalytic methanol oxidation reaction (MOR), oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) with commendable durability. Comprehensive characterization through Powder X-ray Diffraction (PXRD), Fourier Transform Infrared Spectroscopy (FT-IR), X-ray Photoelectron Spectroscopy (XPS), Brunauer–Emmett–Teller (BET), morphological analysis and electrochemical impedance spectroscopy (EIS) confirms the nanocomposite's structure and properties. The catalyst attains a MOR current density of 9.1 mA cm−2 in photoinduced electrocatalytic methanol oxidation with high CO tolerance and durability. During the OER, the PtNP-ZnO@CQD catalyst reveals a lower overpotential than commercial RuO2 at higher current densities over 30 mA cm−2. In the ORR, the catalyst showed a higher half-wave potential of 0.96 V, higher limiting current density, mass activity, and chronoamperometric stability than commercial Pt/C used as a standard here. PtNP-ZnO@CQDs also exhibited a low peroxide yield, a high number of electron transfers, and photoinduced ORR capability, indicating its superiority over commercial Pt/C catalysts. When used in a rechargeable aqueous ZAB, the PtNP-ZnO@CQD air cathode delivered an open circuit potential of 1.55 V with an impressive energy density of 668 W h kg−1 and a specific capacity of 532 mA h g−1, outperforming ZABs with commercial Pt/C and RuO2. Interestingly, the ZAB composed of the PtNP-ZnO@CQD air cathode shows outstanding long-term cycling stability, maintaining a round trip efficiency of 66.87% after 60 h. ZABs assembled in series successfully powered LED panels, demonstrating the potential of this low-cost, bifunctional Pt-based electrocatalyst for future ZAB commercialization.
1 Introduction
In the modern era, energy and environmental concerns take center stage. The majority of our energy needs are met by burning fossil fuels, resulting in economic imbalances and significant environmental repercussions such as the emission of greenhouse gases and harmful pollutants into the atmosphere. Energy conversion from renewable sources is considered an encouraging solution to overcome dependency on fossil fuels.1 Thanks to continuous efforts, various electrochemical energy storage devices like supercapacitors, primary batteries, rechargeable batteries, and fuel cells have found extensive applications in portable electronics, electric vehicles, and emergency power backup systems, among others.2–4 In the present market scenario, rechargeable lithium-ion batteries dominate the energy storage sector. However, their limited energy densities and the high price of lithium resources owing to the limited abundance of lithium hinder their progression.4 In this regard, fuel cells and metal–air batteries can be suitable alternatives in the energy sector due to their high energy density.4–6 Zinc–air batteries (ZABs) operate through metal oxidation and oxygen reduction reactions, unlike Li-ion batteries, where charge carriers move within lithium. They boast a significantly higher theoretical energy density alongside the inherent low-cost, lightweight, and small-size architecture due to the semi-open architecture with the air cathode.5,7,8 Considering the cost, energy density, voltage output, charging cycle, and environmental aspects, the ZAB is most promising in the air battery category. On the other hand, direct methanol fuel cells (DMFCs) are attracting growing attention due to several specific advantages, such as using methanol as fuel with renewable and abundant resources, high volumetric energy density, portable nature, attractive pricing, and convenient storage and transportation capabilities.9 DMFCs function through the electrocatalytic methanol oxidation reaction (MOR) at the anode, while rechargeable ZABs operate via Zn oxidation/reduction at the metallic zinc anode. However, both systems are common in the ORR, which occurs in the air cathode during the operation of DMFCs and the discharge process of ZABs.10–15 Alongside the ORR during discharging, the OER is the heart of the charging process for rechargeable ZABs.15–18 However, the sluggish kinetics of the ORR and OER at the air electrode predominantly contribute to the limited efficiency in DMFCs, as well as restricted round-trip energy efficiency (55–65%) and low charge/discharge power of ZABs.19 Moreover, corrosion and leaching of the air electrode further diminish the cycling stability of ZABs, typically lasting less than 500 cycles under aqueous alkaline conditions. Although significant progress has been made in developing efficient and durable bifunctional oxygen electrocatalysts in recent years, and ZABs are slowly but steadily improving, they are still in their early stages of development. On the other hand, developing low-cost, highly durable catalysts with high MOR and ORR efficacy is also an everlasting challenge for DMFCs.As state-of-the-art electrocatalysts, Pt-based materials remain the top preference for electrocatalysts as electrode materials for the MOR/ORR in DMFCs and for the ORR in ZABs.17,19,20 Owing to the limited efficacy of OER electrocatalysis, commercial Pt/C is mixed with RuO2 in rechargeable ZABs as it needs a high-performance catalyst for both the OER and the ORR. Nonetheless, Pt is exceptionally costly, largely due to its reliance on approximately 10–20% Pt as the active component supported on conductive carbon materials in commercial Pt/C. Furthermore, Pt nanoparticles (Pt-NPs) tend to agglomerate over prolonged operation, diminishing catalytic efficiency and operational lifespan.20,21 Additionally, high Pt-containing electrodes always suffer from inherent CO-poisoning, which reduces their catalytic efficacy during the MOR.22,23 As a result, researchers face ongoing challenges in developing electrode materials for metal–air batteries that feature uniformly dispersed Pt-NPs with minimal Pt content.
To reduce the Pt-content in the electrocatalyst system, researchers have developed different Pt-based monometallic/bimetallic/trimetallic-based nanostructured electrocatalysts like Pt-Ru, Pt-Mo, and PtCuNi for different energy devices through judicious nanoarchitectonics.24–28 Gong et al. investigated the efficiency of the MOR and ORR using cross-double dumbbell-like Pt-Ni nanostructures.29 Li et al. improved the MOR activity by developing a core–shell Cu–Ni@Pt–Cu nano-octahedral structure, to name a few.30 Although alloying technology holds significant promise, it faces limitations such as synthesis complexity, batch synthesis performance variations, CO-poisoning, and durability issues under harsh electrocatalytic conditions. Moreover, identifying the ideal alloy composition to balance MOR and OER/ORR activities, CO tolerance, and durability is challenging, with different compositions often entailing trade-offs.
Metal-nanoparticle-based composites have been introduced to mitigate the abovementioned issues and enhance the overall efficiency of electrode reactions in DMFCs and ZABs.31,32 Metal oxides contain many oxygen-containing functional groups on their surface, which enhance the catalytic efficacy by increasing –OH adsorption, ameliorating the oxidation of adsorbed species during the MOR, and oxygen electrocatalysis. Pt–metal oxide nanocomposites have been employed as electrode materials to enhance DMFC performance by mitigating CO poisoning and ensuring high catalytic durability. Decorating ZnO composites with metal nanostructures represents a favorable approach to modifying the ZnO interface, indirectly facilitating the interfacial charge transfer mechanism.33–37 Researchers developed Pt-NPs or Pt-based nanoalloy compositing with metal oxides like TiO2, ZnO, SnO2, and Co3O4 on carbon materials to improve the catalytic efficacy and durability by lowering CO poisoning.38–42 Additionally, the inherent semiconducting nature of transition metal oxides, e.g., low-cost and environment-friendly ZnO, can enable photo-induced catalysis alongside the electrocatalytic performance of the nanocomposite.43,44 The heterojunction formed in the ZnO–metal nanoparticle composites can effectively increase the charge transfer and reduce undesired recombinations during photo-induced electrocatalysis.45,46
More efficient heterostructures can be created by incorporating high surface area carbon-based nanomaterials into Pt-composites.47,48 In this regard, carbon quantum dots (CQDs) are innovative zero-dimensional fluorescent nanomaterials with outstanding optical properties, biocompatibility, low toxicity, eco-friendliness, and facile synthesis routes.49–51 Besides bioimaging applications, CQDs are highly promising for energy storage and conversion owing to their strong coordination for adsorption, high surface area, and unique electron-transfer abilities.52,53 Therefore, a ternary nanocomposite of Pt-NPs with ZnO and CQD can be a promising electrocatalyst for the MOR, OER, and ORR with a lower noble metal content in DMFC and ZAB applications, improving the catalyst's efficiency and cost-effectiveness for commercial purposes.
In this work, we explored a PtNP-ZnO@CQD ternary nanocomposite synthesized via a one-pot hydrothermal process, serving as an efficient electrocatalyst for the MOR, OER, and ORR suitable for DMFCs and ZABs. A novel synthesis method was employed, where Pt salt was reduced to Pt-NPs and stabilized with CQDs without additional capping agents. The Pt-ZnO-CQD nanocomposite exhibited lower Pt nanoparticle content than commercially available Pt–C catalysts. Despite this reduction in Pt content, the novel composite demonstrated enhanced trifunctional electrocatalytic performances for the MOR, OER, and ORR, which are very crucial for both DMFCs and ZABs. The reported electrochemical performance suggests the potential for replacing commercially available Pt–C catalysts in commercial applications.
2 Experimental section
2.1 Materials
Zinc acetate dihydrate [Zn(CH3COO)2·2H2O] (CAS no. 5970-45-6, ≥98%), chloroplatinic acid hexahydrate (H2PtCl6·6H2O) (CAS no. 18497-13-7, ≥37.50% Pt basis), D-glucose (C6H12O6) (CAS no. 50-99-7, ≥99.5%), and sodium hydroxide (NaOH) (CAS no. 1310-73-2, ≥98% pellets) are purchased from Sigma Aldrich. The chemicals were used in the experiments without further purification. Distilled or Milli Q water was used for the experimental process wherever necessary.2.2 Structural characterization
The characterization techniques used in this work included PXRD, performed using a Rigaku Ultima IV X-ray diffractometer with Cu Kα radiation at a scan rate of 1° min−1 and a step size of 0.01. XPS was used for elemental composition analysis. The XPS data were collected using a Thermo Scientific MultiLab 2000 at 15 kV and 10 mA (150 W) with Al Kα radiation (1486.6 eV). For XPS analysis, the powder samples were positioned on conducting carbon tape and placed in a preparation chamber at an ultrahigh vacuum (UHV) of 8.0 × 10−9 torr for 5 hours. The sample was transferred to the analyzer chamber at 5.0 × 10−9 torr. All the spectral data were documented with a step increment of 0.05 eV and a pass energy of 30 eV. The XPS data were plotted, and background correction was performed using Avantage software (Avantage 5.9931), retaining smart background correction. Peak fitting employed Gaussian functions with parameters set at convergence: 0.0001, maximum iterations: 100, fitting algorithm: Powel, and Gauss–Lorentz: product. High-resolution transmission electron microscopy (HRTEM) was performed using an FEI Tecnai G2 20 STEM instrument at 200 kV acceleration voltage. Surface morphology analysis was performed using FEI Apreo Scanning Electron Microscopy (SEM) at an operating voltage of 30 kV. Moreover, thermogravimetric analysis (TGA) of PtNP-ZnO@CQD materials was conducted using a Shimadzu DTG-60 instrument under an N2 atmosphere with a heating rate of 10 °C min−1. The concentrations of Pt and Zn were evaluated using an Atomic Absorption Spectrophotometer (AAS) (Shimadzu AA-7000).2.3 Synthesis of the PtNP-ZnO@CQD nanocomposite
As illustrated in Fig. 1a, a one-pot synthesis method was adopted for synthesizing the PtNP-ZnO@CQD nanocomposite. Initially, 10 mL of 500 mM D-glucose and 10 mL of 500 mM NaOH were homogenized for 10 min at 50 °C. After that, 0.1 g (0.244 mmol) H2PtCl6·6H2O was added as a Pt-metal precursor. The solution was mixed for another 15 minutes. Then, a 20 mL portion of 125 mM Zn(CH3COO)2·2H2O was added to the previous solution and stirred for 30 min at room temperature. Consequently, NaOH solution (5 M) was added drop-wise to adjust the pH value to 10. The solution mixture was transferred to a Teflon-coated autoclave and heated at 85 °C for 3.5 h. The sediment particles were collected by washing several times with distilled water and ethanol and dried in an oven at 200 °C for 12 h. Individual CQDs and ZnO nanoparticles were synthesized using the same procedure, maintaining similar reaction conditions without including the other precursor.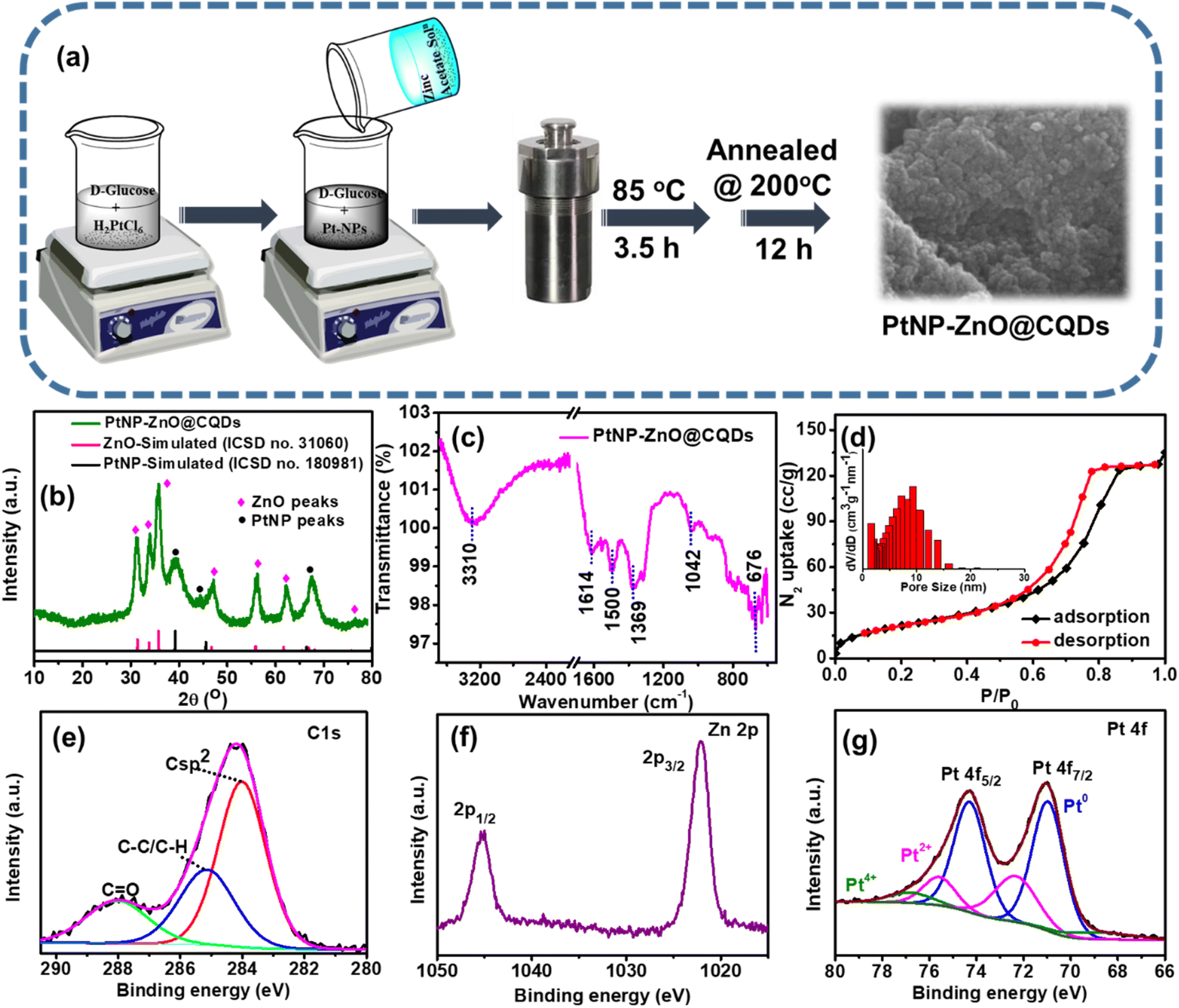 | ||
| Fig. 1 (a) Schematic diagram of the synthesis of PtNP-ZnO@CQDs. (b) X-ray diffraction pattern of PtNP-ZnO@CQDs compared with simulated ZnO and PtNP patterns. (c) FT-IR of PtNP-ZnO@CQDs. (d) N2-isotherm of PtNP-ZnO@CQDs. The pore size distribution plot is given in the inset. Core level XPS spectra of (e) C 1s, (f) Zn 2p, and (g) Pt 4f. | ||
2.4 Preparation of electrodes for electrocatalysis
The catalyst was prepared by adding 3 mg of PtNP-ZnO@CQD nanocomposite to 1 mL water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) solution to study the electrocatalytic behavior. To increase its adhesion with the electrode, 20 μL of Nafion as a binder was used. Then, the whole dispersion was kept under sonication for 1 h to make it homogeneous. After that, 5 μL of catalyst ink from the prepared homogeneous dispersion was coated on the surface of a glassy carbon electrode (GCE) having an area of 0.071 cm2 and then dried in a vacuum oven for 24 h and then used for electrocatalytic applications. For the OER experiment, 20 μL of catalyst was coated on a 0.8 × 0.8 cm2 area of 2 × 1 cm2 nickel foil.
:2) solution to study the electrocatalytic behavior. To increase its adhesion with the electrode, 20 μL of Nafion as a binder was used. Then, the whole dispersion was kept under sonication for 1 h to make it homogeneous. After that, 5 μL of catalyst ink from the prepared homogeneous dispersion was coated on the surface of a glassy carbon electrode (GCE) having an area of 0.071 cm2 and then dried in a vacuum oven for 24 h and then used for electrocatalytic applications. For the OER experiment, 20 μL of catalyst was coated on a 0.8 × 0.8 cm2 area of 2 × 1 cm2 nickel foil.
2.5 MOR study
To evaluate the methanol oxidation performance of the PtNP-ZnO@CQD nanocomposite, we used a modified glassy carbon electrode (GCE) as the working electrode, along with a Pt wire as the counter electrode and an Ag/AgCl electrode as the reference. Prior to each analysis, the GCE surface was meticulously cleaned with different grits of alumina slurry on nylon cloth, followed by thorough rinsing with deionized water. The catalyst ink was prepared in a 1 mL water–ethanol (1:2) solution with 20 μL of Nafion, as previously described. The methanol oxidation reaction (MOR) study was conducted using an Autolab PGSTAT128N potentiostat controlled using Nova 2.1 software. The electrochemical analysis was performed in a methanol solution mixed with 1 M KOH as the electrolyte over a potential range of −0.6 to 0.2 V versus Ag/AgCl, with a scan rate of 20 mV s−1.
2.6 OER and ORR studies
Oxygen evolution reaction (OER) measurements were conducted using an Autolab PGSTAT128N potentiostat controlled using Nova 2.1 software. The electrochemical experiments were performed in a conventional three-electrode system within a single-compartment cell. This setup included a glassy carbon electrode (GCE) with a drop-coated catalyst as the working electrode (WE), a platinum wire as the counter electrode, and an Ag/AgCl/3 M KCl electrode as the reference. To investigate the electrocatalytic oxygen evolution behavior of the PtNP-ZnO@CQD nanocomposite, linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were utilized. The OER was carried out using the PtNP-ZnO@CQD film on nickel foil in a 1 M KOH solution at room temperature. LSV was performed over a potential range of 0 to 1.2 V with a scan rate of 10 mV s−1. All potentials were converted to the reversible hydrogen electrode (RHE) scale using the following equation: ERHE = EAg/AgCl(sat. KCl) + 0.197 + 0.0591 pH.To assess the ORR performance of the PtNP-ZnO@CQD nanocomposite, an electrochemical workstation equipped with a rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) was employed. The ORR was examined using an RDE modified with the nanocomposite electrocatalyst in a 0.1 M KOH solution. The ORR performance of our developed catalyst was compared with that of commercial 10% Pt/C. Hydrodynamic experiments, including RDE and RRDE measurements, were conducted using a modular bi-potentiostat (Metrohm Autolab) with data analysis performed via DropView 8400 software. The experiments were carried out in 0.1 M KOH at various rotation speeds ranging from 400 to 2000 rpm, with a scan rate of 10 mV s−1, using a glassy carbon–platinum (GC-Pt) ring electrode as the WE (with 5 μL of catalyst drop-coated on a GCE disk) in a three-electrode assembly. Measurements were conducted under inert conditions (purged with Ar) and oxygen-saturated conditions by continuously bubbling O2 into the electrolyte for 20 minutes before the start of the experiment and maintaining a constant flow throughout the measurements.
2.7 Fabrication and evaluation of ZABs
A lab-made liquid Zn–air battery was assembled using PtNP-ZnO@CQDs on Ni foam as the air cathode and a polished Zn plate as the anode, with an electrolyte solution of 6 M KOH and 0.2 M zinc acetate. The air cathode was prepared by drop-casting the catalytic ink onto one side of the Ni foam. The catalyst ink was prepared as previously described, and 20 μL of the resulting ink was applied to one side of the nickel foam and dried at 60 °C in a hot air oven. For comparison, another Zn–air battery was assembled using a commercially available RuO2 and 10% Pt/C mixture (1:1) as the air cathode, with the same catalyst loading. All Zn–air battery experiments were conducted using an OrigaFlex OGF500 electrochemical workstation. The specific capacity and energy density were calculated by using the following equation:| Specific capacity = (current × time)/weight of zinc consumed |
| Energy density = (current × time × average discharge voltage/weight of zinc consumed) |
3 Results and discussion
3.1 Structural analysis
A one-pot hydrothermal method has been introduced to synthesize the PtNP-ZnO@CQD nanocomposite, as shown in Fig. 1a. After adding the Pt-precursor to the alkaline D-glucose solution, the light yellow color turned dark brown (as shown in Fig. S1 in the ESI†), indicating the formation of PtNPs in the solution. As a reducing sugar, D-glucose reduced the Pt-precursor to PtNPs and served as a stabilizing agent for PtNPs. The previously prepared zinc precursor solution was mixed with PtNP-containing alkaline glucose solution to prepare the PtNP-ZnO@CQD nanocomposite through a green, in situ hydrothermal reaction for 3.5 h, followed by annealing at 200 °C for 12 h. Overall, D-glucose was a precursor for CQDs and acted as a capping agent for stabilizing PtNPs, demonstrating a versatile and cost-effective approach for nanomaterial synthesis with potential applications in various fields.54The XRD pattern of the PtNP-ZnO@CQD nanocomposite is illustrated in Fig. 1b. The exhibited peaks in the ternary nanocomposite exactly match the simulated patterns of ZnO (ICSD no. 31060) and PtNPs (ICSD no. 180981). ZnO in the composite confirms the wurtzite structure as the PXRD of the composites reveals the peaks at 2θ values of 32.09°, 34.85°, 36.77°, 48.02°, 57.22°, 63.05° and 77.57° corresponding to the planes of (100), (002), (101), (102), (110), (103), and (202), respectively. The peak intensity of ZnO is high at the plane (101) due to the more preferable orientation. Additionally, the PXRD pattern of CQDs in Fig. S2a in the ESI† revealed their amorphous nature, attributed to the lack of long-range crystalline order. As a result, the absence of characteristic peaks for CQD in the XRD pattern of the PtNP-ZnO@CQD composite is expected, as CQDs typically exhibit an amorphous structure. Nonetheless, incorporating CQDs in the PtNP-ZnO@CQD composite is strongly supported by complementary techniques such as HRTEM, which provides detailed imaging, and FTIR, which confirms the presence of CQD-specific functional groups. The face-centered cubic (fcc) structures of PtNPs can be confirmed by evaluating the planes (111) and (220) at 2θ values of 40.29° and 68.35°, respectively.55 Additionally, the stability of ZnO in alkaline media is a concern, as ZnO tends to dissolve in highly alkaline environments due to the formation of soluble zincate species [Zn(OH)42−]. The stability of ZnO of the synthesized PtNP-ZnO@CQD composite in alkaline media is further confirmed by dipping the composite in 1 M KOH for 50 hours. The unchanged PXRD pattern (in Fig. S2b in the ESI†) and the appearance of all the XRD peaks responsible for ZnO were observed for the PtNP-ZnO@CQD composite, indicating the stability of the synthesized catalyst in an alkaline medium. The exceptional alkaline stability of the PtNP-ZnO@CQD catalyst arises from the protective roles of CQDs and PtNPs, which enhance ZnO's stability by forming a stabilizing layer, reducing surface defects, and minimizing exposure to alkaline conditions.56 At the same time, PtNPs contribute to improving the stability of ZnO through a strong metal–support interaction (SMSI). The synergistic interactions within the composite ensure its effective stability under alkaline electrolyte conditions.
FT-IR analysis is performed to define the interaction between nanocomposite heterostructures. Fig. 1c shows the characteristic peak for Zn–O vibration at 676 cm−1. PtNP-ZnO@CQDs also revealed the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching band for aromatic hydrocarbons at 1614 cm−1. As the material has oxygen-containing groups and surface-adsorbed water molecules, the stretching vibrations of C–O and –OH are observed at 1042 cm−1 and 3310 cm−1, respectively. Additionally, the peak at 1369 cm−1 is attributed to C–O stretching from surface carboxyl groups (–COOH) on CQDs, while the 1500 cm−1 peak corresponds to CC stretching in aromatic rings, linked to the sp2-hybridized carbon structure. So, the appearance of the characteristic FTIR peaks confirms the presence of CQDs in the composite structure. The specific surface area and porosity were examined using a BET surface analyzer, and the results are tabulated in Table S1 in the ESI.† The PtNP-ZnO@CQD nanocomposite reveals a type IV isotherm in the adsorption and desorption curve, as shown in Fig. 1d. The specific surface area of the nanocomposite is calculated to be 81.56 m2 g−1, with a total pore volume of 0.203 cm3 g−1. The pore distribution plot, as shown in the inset of Fig. 1d, discloses the mesoporous nature of the composite with a mean pore diameter of ∼6.93 nm. The composition of the surface atoms of the PtNP-ZnO@CQD nanocomposite was determined through an XPS study. The XPS survey spectra of the nanocomposite (Fig. S3a in ESI†) demonstrate Zn, C, O, and Pt as the constituent elements. Fig. 1e represents the core level XPS spectra of C 1s, which are fitted into three distinct peaks at 283.8, 285.2, and 288 eV, corresponding to sp2 (C–C), sp3 (C–C, C–H) and CO, respectively. In C 1s spectra, Zn–C does not exist, denoting that the CQDs are not present as a dopant in the PtNP-ZnO@CQD nanocomposite. The core level spectra of O 1s in Fig. S3b in the ESI† reveal three peaks at 529.2, 530, and 531.4 eV corresponding to CO, C–OH or C–O–C, and Zn–O. The peak assigned to Zn–O in the O 1s spectrum is tentatively attributed to the oxygen–zinc bond, influenced by the varying electronic environment of the zinc atom due to its unique electronic properties.57 Additionally, the core level XPS spectra of Zn 2p (Fig. 1f) reveal two peaks observed at 1022.1 and 1045.2 eV related to Zn 2P3/2 and Zn 2p1/2, respectively, with an intensity ratio of approximately 2:1.58 The Pt 4f XPS spectra (in Fig. 1g) can be resolved into two characteristic peaks corresponding to the spin–orbit split states, 4f7/2 and 4f5/2, with an intensity ratio of approximately 4:3. The deconvoluted peaks at binding energies of 71.0 eV (4f7/2) and 74.32 eV (4f5/2) are attributed to Pt in its metallic state (Pt0). In addition, peaks at 72.41 eV (4f7/2) and 75.59 eV (4f5/2) are associated with the Pt2+ oxidation state.59,60 Notably, a minor contribution from Pt4+ is also observed, indicated by a binding energy peak at 76.77 eV.
C stretching band for aromatic hydrocarbons at 1614 cm−1. As the material has oxygen-containing groups and surface-adsorbed water molecules, the stretching vibrations of C–O and –OH are observed at 1042 cm−1 and 3310 cm−1, respectively. Additionally, the peak at 1369 cm−1 is attributed to C–O stretching from surface carboxyl groups (–COOH) on CQDs, while the 1500 cm−1 peak corresponds to CC stretching in aromatic rings, linked to the sp2-hybridized carbon structure. So, the appearance of the characteristic FTIR peaks confirms the presence of CQDs in the composite structure. The specific surface area and porosity were examined using a BET surface analyzer, and the results are tabulated in Table S1 in the ESI.† The PtNP-ZnO@CQD nanocomposite reveals a type IV isotherm in the adsorption and desorption curve, as shown in Fig. 1d. The specific surface area of the nanocomposite is calculated to be 81.56 m2 g−1, with a total pore volume of 0.203 cm3 g−1. The pore distribution plot, as shown in the inset of Fig. 1d, discloses the mesoporous nature of the composite with a mean pore diameter of ∼6.93 nm. The composition of the surface atoms of the PtNP-ZnO@CQD nanocomposite was determined through an XPS study. The XPS survey spectra of the nanocomposite (Fig. S3a in ESI†) demonstrate Zn, C, O, and Pt as the constituent elements. Fig. 1e represents the core level XPS spectra of C 1s, which are fitted into three distinct peaks at 283.8, 285.2, and 288 eV, corresponding to sp2 (C–C), sp3 (C–C, C–H) and CO, respectively. In C 1s spectra, Zn–C does not exist, denoting that the CQDs are not present as a dopant in the PtNP-ZnO@CQD nanocomposite. The core level spectra of O 1s in Fig. S3b in the ESI† reveal three peaks at 529.2, 530, and 531.4 eV corresponding to CO, C–OH or C–O–C, and Zn–O. The peak assigned to Zn–O in the O 1s spectrum is tentatively attributed to the oxygen–zinc bond, influenced by the varying electronic environment of the zinc atom due to its unique electronic properties.57 Additionally, the core level XPS spectra of Zn 2p (Fig. 1f) reveal two peaks observed at 1022.1 and 1045.2 eV related to Zn 2P3/2 and Zn 2p1/2, respectively, with an intensity ratio of approximately 2:1.58 The Pt 4f XPS spectra (in Fig. 1g) can be resolved into two characteristic peaks corresponding to the spin–orbit split states, 4f7/2 and 4f5/2, with an intensity ratio of approximately 4:3. The deconvoluted peaks at binding energies of 71.0 eV (4f7/2) and 74.32 eV (4f5/2) are attributed to Pt in its metallic state (Pt0). In addition, peaks at 72.41 eV (4f7/2) and 75.59 eV (4f5/2) are associated with the Pt2+ oxidation state.59,60 Notably, a minor contribution from Pt4+ is also observed, indicated by a binding energy peak at 76.77 eV.
The SEM image study of the PtNP-ZnO@CQD nanocomposite reveals the formation of spherical structures of nanocomposite materials, as shown in Fig. 2a. Energy dispersive X-ray spectroscopy (EDX) (Fig. 2b) coupled with SEM discloses the presence of C, Zn, Pt, and O as the primary elements in the nanocomposite. Additionally, the elemental mapping from the EDX study demonstrates the homogeneous distribution of all the constitution elements throughout the nanocomposite (Fig. 2c–g). The individual elemental weight percentage and atomic percentages of all the constituents from the EDX study are summarized in Table S2 in the ESI.† Interestingly, the Pt and ZnO contents in the PtNP-ZnO@CQD nanocomposite were determined via EDX analysis to be 3.65 wt% and 17.97 wt%, respectively. These elemental analysis results were compared with the XPS elemental analysis results, as shown in Table S3 in the ESI,† indicating a strong agreement between both measurement techniques. However, these two techniques can provide the weight percentage of the surface elements, not the bulk. Atomic absorption spectroscopy (AAS) was performed to evaluate the bulk Pt and Zn loading. The AAS study reveals (Table S3†) the Pt and Zn loadings of 4.75% and 19.22%, respectively, in the composite. Accordingly, the weight percentage loading of ZnO was determined to be 23.92%. HR-TEM was performed to study the detailed morphology of the PtNP-ZnO@CQD nanocomposite. The TEM images in Fig. 2h and i reveal the spherical PtNPs below 5 nm and ZnO nanoparticles of ∼10 nm. Besides the PtNPs, shaded circles in Fig. 2i are observed with an average particle size ranging from approximately 2 to 3 nanometers. These circles exhibit an amorphous nature and lack distinct lattice fringes, thus confirming the presence of CQDs. Fig. 2j displays the lattice fringes of ZnO nanoparticles with a fringe spacing of approximately 0.32 nm, preferably oriented to the (101) plane to confirm the wurtzite structure of ZnO nanoparticles. The lattice fringes of PtNPs are also observed with a fringe spacing of approximately 0.22 nm, which is in accordance with the (111) atomic plane.
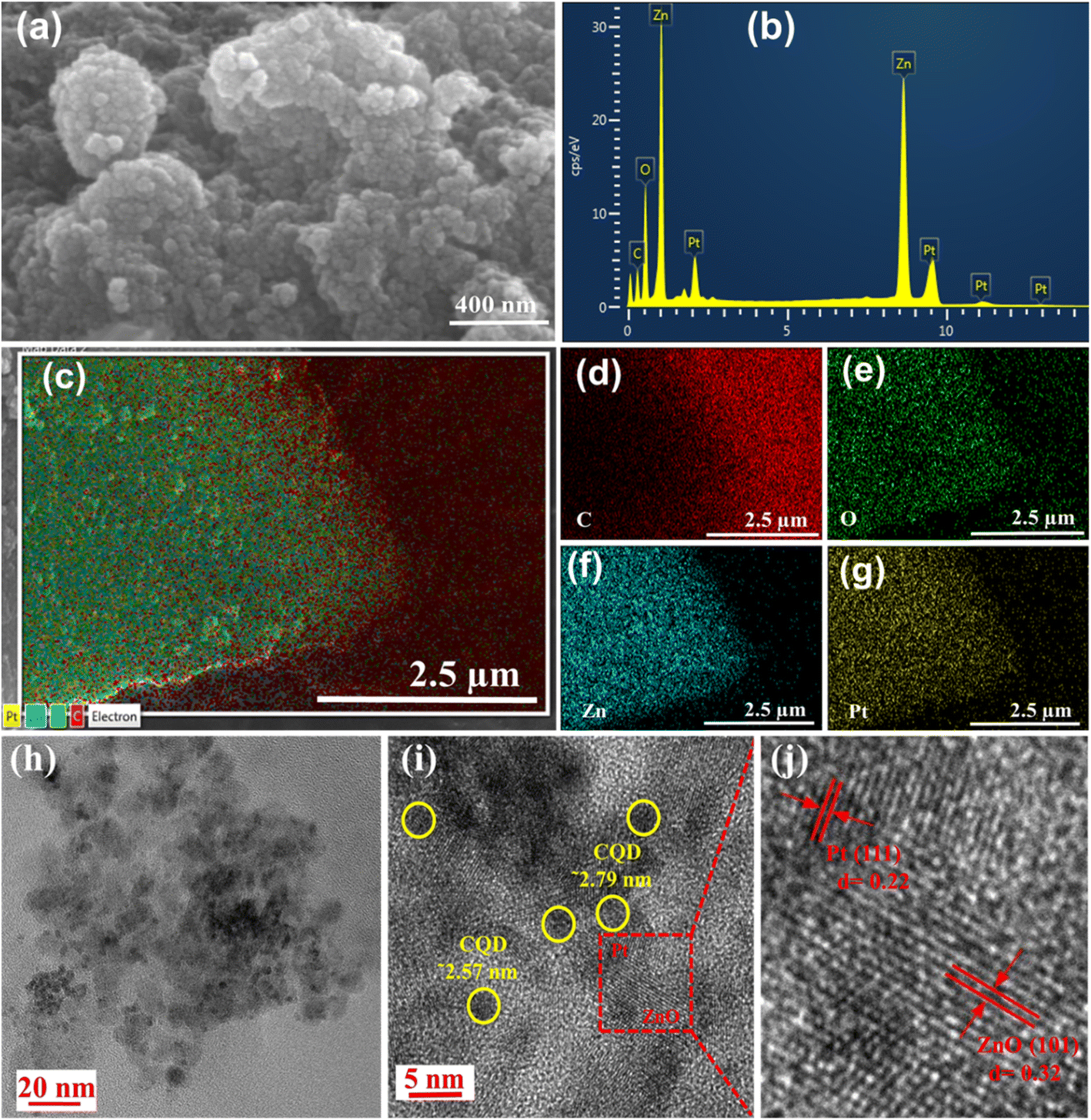 | ||
| Fig. 2 (a) SEM image of PtNP-ZnO@CQDs. (b) EDX spectra of PtNP-ZnO@CQDs. EDX elemental mapping of PtNP-ZnO@CQDs to show (c) all the elements together and individual elements, (d) C, (e) O, (f) Zn, and (g) Pt. TEM of PtNP-ZnO@CQDs in (h) low magnification and (i) high magnification, and (j) HR-TEM image of PtNP-ZnO@CQDs with lattice fringes. | ||
3.2 Electrocatalytic MOR study
As previously mentioned, assessing the performance of the electrocatalytic MOR is crucial for the practical implementation of DMFCs. The MOR entails a challenging six-electron transfer process, representing a key anodic reaction in DMFCs. The DMFC performance of PtNP-ZnO@CQDs was evaluated using a modified glassy carbon electrode (GCE) with a Pt wire counter electrode and Ag/AgCl reference electrode. The catalytic performance of the synthesized catalyst was then compared to that of the Pt/C catalyst. Using an Autolab PGSTAT128N potentiostat, the study utilized methanol and 1 M KOH electrolyte over −0.6 to 0.2 V at 20 mV s−1.As shown in Fig. 3a, the PtNP-ZnO@CQD composite shows no characteristic peak in the CV study without methanol. However, after adding methanol, a significant anodic peak is observed with a current density of 4.93 mA cm−2 at −0.14 V (vs. Ag/AgCl) during the MOR on PtNP-ZnO@CQDs. Additionally, in reverse bias, a small peak is revealed at −0.30 V (vs. Ag/AgCl) with 0.54 mA cm−2 specific current density owing to the oxidation of carbonaceous products, like CO, formed during the incomplete oxidation of methanol. As shown in Fig. 3a, the resulting MOR current density of PtNP-ZnO@CQDs exceeded the MOR current density of commercial Pt/C catalysts. This enhancement is due to the synergistic interaction between PtNPs and the ZnO@CQD support, which provides a high surface area and improves the electron transfer and methanol adsorption kinetics to ameliorate the MOR. In a standard MOR, the peak current density during the forward scan (If) reflects the dehydrogenation of adsorbed methanol to generate Pt-bound carbonaceous species such as CO.61 The ratio of forward scan current density (If) to backward scan current density (Ib) indicates catalytic efficiency and catalyst poisoning. The If/Ib ratio for PtNP-ZnO@CQDs is determined to be 9.12. The high value of the ratio denotes the negligible catalyst poisoning for PtNP-ZnO@CQDs. We performed a cycle study up to the 100th MOR CV cycle to determine the cycle stability. Fig. S4a in the ESI† reveals no such significant decay of the oxidative current density in forward or backward bias even after the 100th CV cycle. To check the stability of the electrode material during methanol oxidation, we performed a chronoamperometry test, as shown in Fig. 3b. The chronoamperometry curve of the PtNP-ZnO@CQD catalyst tested in 1 M KOH containing 1 M methanol at an applied current density of −0.1 V (vs. Ag/AgCl) demonstrates a constant current density throughout the study of 24 h. After 24 hours of chronoamperometric testing, the PtNP-ZnO@CQD-modified electrode maintains a current density of 3.46 mA cm−2, indicating a retention of 77% of its initial current density.
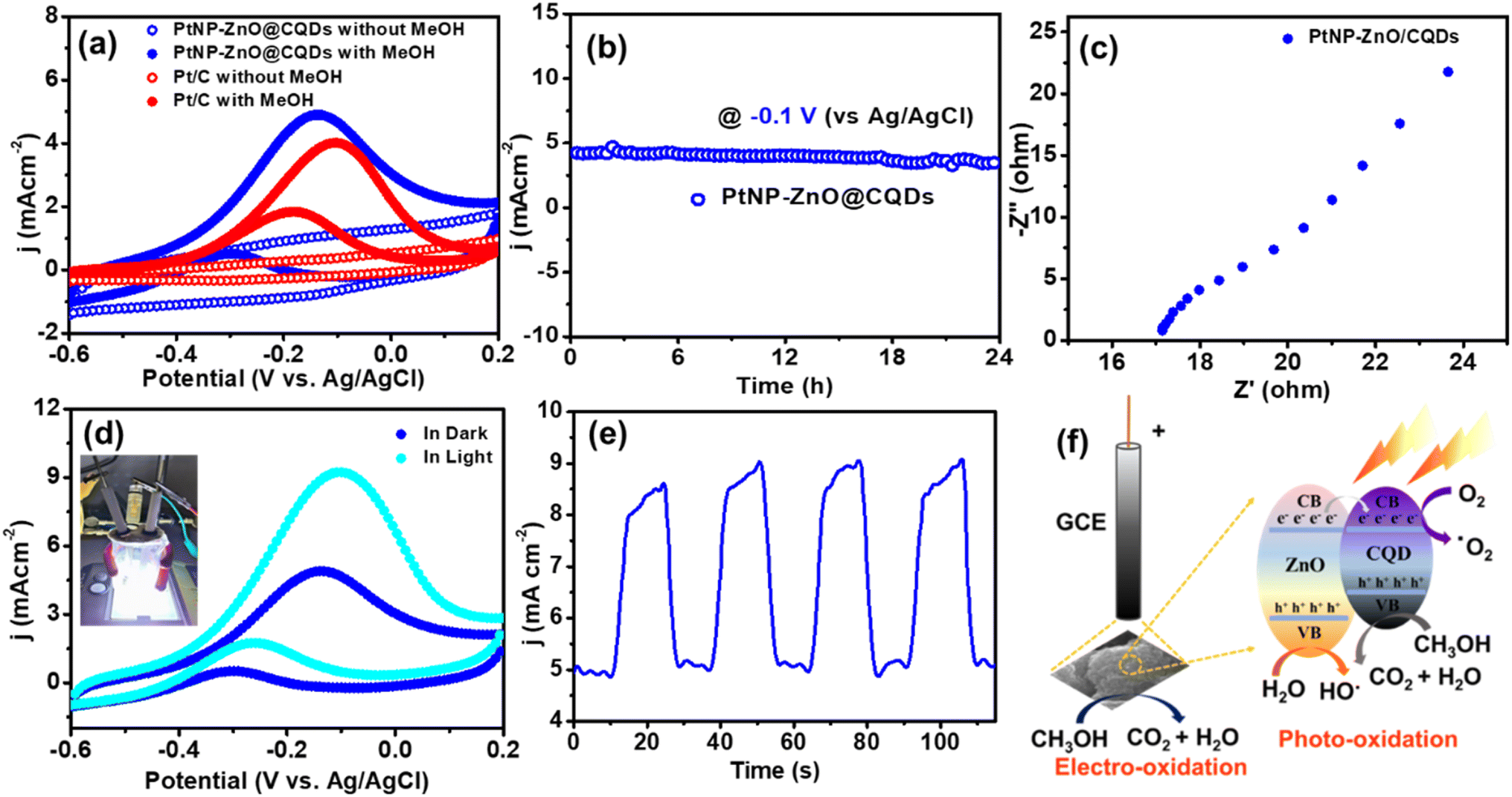 | ||
| Fig. 3 (a) CV plots of PtNP-ZnO@CQDs and commercial Pt/C modified GCE with and without 1 M methanol in 1 M KOH at a scan rate of 20 mV s−1. (b) Chronoamperometry and (c) Nyquist plot of the PtNP-ZnO@CQD modified GCE with 1 M methanol in 1 M KOH. (d) CV plot of the PtNP-ZnO@CQD modified GCE with 1 M methanol in 1 M KOH at a scan rate of 20 mV s−1 in the presence and absence of white LED light. The instrument setup image is given in the inset. (e) Chronoamperometry study of the MOR system under dark and light switching conditions. (f) Band diagram of ZnO and CQDs for the photoinduced electrocatalytic MOR. | ||
The PtNP-ZnO@CQD modified GCE reveals the linear dependency of MOR activity on methanol concentration (Fig. S4b†). Four different molar concentrations of methanol, e.g., 0.5 M, 1 M, 1.5 M, and 2 M, were used to evaluate the MOR efficacy of the PtNP-ZnO@CQD modified electrode on the GCE. The modified nanocomposite loaded GCE was used to evaluate the enhancement of the electrochemical energy conversion reaction. An increase in current densities is observed from 3.79 to 6.41 mA cm−2 by applying the same potential window of −0.6 to 0.2 V (vs. Ag/AgCl) with a scan rate of 20 mV s−1. An EIS study was performed to evaluate the transport properties of the catalyst during the MOR performance. As shown in Fig. 3c, the Nyquist plot reveals a semicircle in the high-frequency region, exhibiting the charge transfer resistance (Rct) to be ∼20 Ω. In the lower frequency region, a Warburg impedance is observed with a straight inclination of ∼45°. The lower Rct exhibited by PtNP-ZnO@CQDs indicates improved kinetics in the context of charge transfer during MOR operation.
The better CO tolerance, as evidenced by the high If/Ib values in the MOR CV plot, was again assessed by introducing CO over the catalyst-modified GCE for 15 minutes in a 1 M KOH solution, as depicted in Fig. S4c.† CV was conducted at a scan rate of 20 mV s−1 within a potential range of −0.6 to 0.2 V (vs. Ag/AgCl). Fig. S4d in the ESI† shows negligible oxidation at −0.23 V (vs. Ag/AgCl) exclusively attributed to the oxidation of adsorbed CO on the catalyst surface after exposing the PtNP-ZnO@CQD modified GCE to a CO environment for 15 min. The result indicates the effectiveness of the PtNP-ZnO@CQD catalyst in terms of CO tolerance.
The superior MOR performance is credited to the active site of the PtNPs in the ZnO/CQD matrix. The high specific surface area of the composite and low charge transfer resistance coupled with better ion diffusion in the PtNP-ZnO@CQD catalyst facilitate the electrocatalytic MOR performance. Commonly, the MOR electrocatalytic performance starts with the adsorption of methanol on the electrode surface, as shown in eqn (1). In this regard, the high surface area and the presence of heteroatoms in CQDs provide better adsorption of methanol. Afterward, according to eqn (2), the anodic oxidation of methanol happens to release six electrons and carbon dioxide.
| CH3OH → (CH3OH)ad | (1) |
| Anode: (CH3OH)ad + 6OH− → CO2 + 6e− + 5H2O | (2) |
As stated earlier, incomplete oxidation produces CO on the catalyst surface, leading to rapid deactivation of the Pt catalyst to weaken its technological practicality. The oxidation of the adsorbed CO on the PtNP surface by the generated –OH species on the Pt surface at a higher potential undesirably slows down the MOR efficacy.62 However, the presence of ZnO and the CQDs minimizes CO poisoning as they absorbed –OH and consequently converted the catalyst-adsorbed CO to CO2 to restore the active sites of PtNPs for continuous MOR performances.34,39,40
Under visible light illumination, the PtNP-ZnO@CQD modified GCE also exhibits a significant enhancement in electrocatalytic MOR performance, amplifying the current density in forward scan (If) to 9.1 mA cm−2, which is almost double the If measured under dark conditions (Fig. 3d). Notably, this photoinduced MOR enhancement can be attributed to a higher weight percentage of photosensitive ZnO. Further investigation through chronoamperometry measurements shed light on the effects of periodic light irradiation on the catalyst-modified GCE. Initially, chronoamperometry curves, as shown in Fig. 3e, indicate a lower current density due to the MOR without light conditions. However, when white LED light is applied for 10 s to the PtNP-ZnO@CQD-modified electrode, the current density sharply increases while the light is on. This observation underscores the positive impact of light treatment on the electrocatalytic MOR process in PtNP-ZnO@CQDs. The photoinduced electrocatalytic MOR performance was completely reversible, as shown in Fig. 3e.
The enhancement of the photoelectrochemical methanol oxidation activity in PtNP-ZnO@CQDs under light irradiation stems from the strong metal–support interaction (SMSI) between the PtNPs and ZnO/CQD matrix, along with the light-induced enhancement of charge transport properties. Initially, the SMSI between the Pt NPs and the ZnO/CQD matrix was characterized through XPS analysis, as presented in Fig. S5 in the ESI.† The core-level Pt 4f spectra show a negative shift in the PtNP-ZnO@CQD composite compared to the commercial Pt/C reference, indicating charge transfer at the interfaces between the PtNPs and the ZnO/CQDs.63 This negative shift confirms the presence of the SMSI, which enhances the catalytic activity of the material. Similar investigations have previously analyzed the electronic structure to compare SMSI strength across different metals and their respective supports, supporting our findings.64 This synergy facilitates the movement of the Fermi level within wurtzite ZnO and CQDs, assisting charge separation owing to the SMSI interaction of Pt NPs. Positive bias and light irradiation generate electron–hole pairs that participate in surface redox reactions, bolstering photocurrent. Moreover, photogenerated holes migrate to the catalyst surface, transforming OH−/H2O species into ˙OH radicals, potent oxidant species that further oxidize adsorbed species like CH3OH (Fig. 3f).65,66 Additionally, the reactive ˙OH radicals can also oxidize the absorbed CO, if any, to play a key role in suppressing catalyst poisoning.67
3.3 OER activity of PtNP-ZnO@CQDs
OER measurements were conducted using an Autolab PGSTAT128N potentiostat in a three-electrode system, with a Ni-foil coated with PtNP-ZnO@CQDs as the working electrode. Linear sweep voltammetry (LSV) and CV were used to investigate the electrocatalytic behavior in a 1 M KOH solution, with LSV over 0 to 1.2 V at a scan rate of 10 mV s−1. Fig. 4a represents the LSV curve for significant OER activity of the PtNP-ZnO@CQD nanocomposite, as it reveals a low overpotential of 355 mV at 10 mA cm−2, which is only 71 mV higher than the overpotential of the benchmark catalyst RuO2 (284 mV at 10 mA cm−2) under similar conditions. However, PtNP-ZnO@CQDs show less OER overpotential than the constituent materials of ZnO and pristine CQDs. The OER kinetics under alkaline conditions can be observed from the Tafel slope, as shown in Fig. 4b. The Tafel slope of PtNP-ZnO@CQDs is determined to be 61.7 mV dec−1, which is less than that of the commercial benchmark RuO2 (73.5 mV dec−1). From Fig. 4c, the bar diagram of overpotential at various current densities reveals that the overpotential of PtNP-ZnO@CQDs is relatively higher than that of RuO2 at lower current densities (till 30 mA cm−2). However, it becomes lower compared to RuO2 at higher current densities, making it superior for OER operation at higher current densities. A higher current density at low overpotential is always desirable for industrial standards. Additionally, the OER performance of PtNP-ZnO@CQDs is compared with those of recently reported catalysts, as shown in Table S4 in the ESI,† to highlight the comparable electrocatalytic OER activity of the synthesized catalyst. From the table, it is evidenced that our catalysts, PtNP-ZnO@CQDs, exhibited comparable or better OER performance than those in recently published highly reputed articles. The chronoamperometric study, as shown in Fig. 4d, unveils its tremendous durability after 25 h of a continuous OER process, as it retains 98% of its initial current density. The polarization curve after a 25 h chronoamperometry study is slightly lower than the initial LSV of the PtNP-ZnO@CQD modified electrode (Fig. 4e), demonstrating the robustness and resilience of the nanocomposite for the OER in the long run. This decent durability can be achieved due to the unique heterostructure of the nanocomposite material. To correlate the energy efficiency of electrocatalytic oxygen evolution, the Faradaic Efficiency (FE) was measured using a water displacement method adopted from our earlier reports.18,68 In this process, a constant potential of 1.6 v (vs. RHE) was applied for 30 minutes (Fig. S6a†), and the oxygen produced by the PtNP-ZnO@CQD modified anode was measured (as shown in Fig. S6b†). The detailed procedure for determining FE and the corresponding set-up are given in Fig. S6 in the ESI.† The OER process reveals a high FE of 93%, unveiling the significant OER efficacy of PtNP-ZnO@CQDs. An EIS study during the OER was performed to understand the transport properties. The Nyquist plot in Fig. 4f reveals low polarization resistance for the PtNP-ZnO@CQD modified electrode compared to the constituent materials ZnO and CQDs. The significant electrocatalytic behavior of the PtNP-ZnO@CQD nanocomposite was further evaluated by calculating the double-layer capacitance (Cdl), ECSA, and roughness factor (Rf) electrochemically. The Cdl value of PtNP-ZnO@CQDs was obtained from the CV plots in the non-faradic region by applying a potential range of 0 to 0.1 V (vs. Ag/AgCl) at different scan rates of 10–50 mV s−1 in 1 M KOH, as shown in Fig. S7a in the ESI.† The Cdl value is calculated to be 4.1 mF cm−2 (Fig. S7b†), and accordingly, the ECSA and Rf of PtNP-ZnO@CQDs are calculated to be 68.3 cm2 and 106.7, respectively. Furthermore, the ECSA of the intermediate ZnO and CQDs was evaluated using CV studies, following the procedure outlined in Fig. S7c–f in the ESI.† A summary of the electrochemical parameters for the synthesized catalyst and the constituent materials is provided in Table S5 in the ESI.† Notably, the final PtNP-ZnO@CQD composite exhibited the highest ECSA and the lowest Rct value, indicating superior electrochemical performance. The high ECSA value, active sites and mesoporosity in PtNP-ZnO@CQDs for swift ion diffusion and low charge transfer resistance significantly ameliorate the electrocatalytic OER efficacy of PtNP-ZnO@CQDs.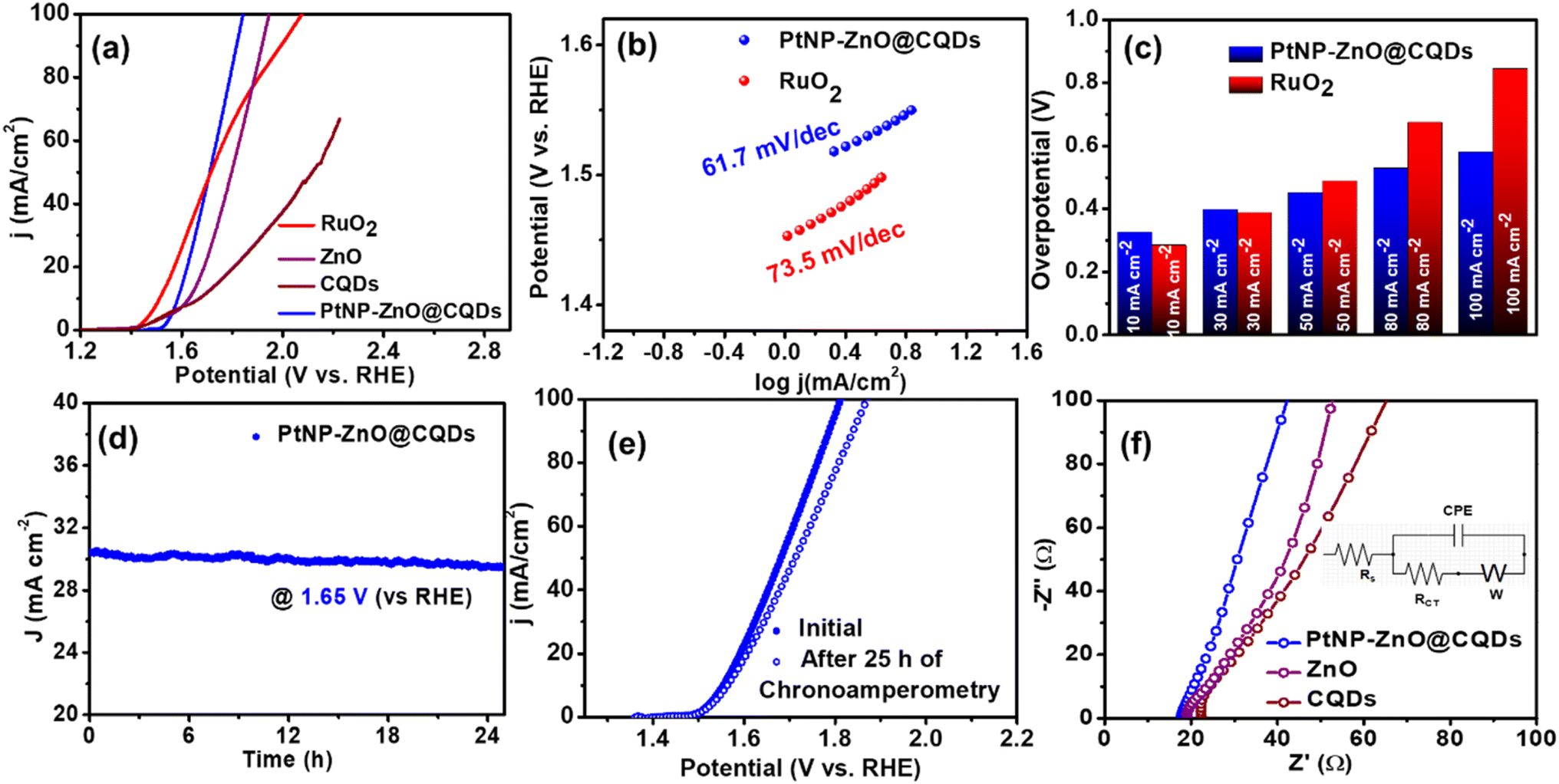 | ||
| Fig. 4 (a) LSV of PtNP-ZnO@CQDs, ZnO, CQDs, and commercially available RuO2 catalyst during the OER. (b) Tafel plots of PtNP-ZnO@CQDs and RuO2. (c) Bar diagram of the OER overpotentials of PtNP-ZnO@CQDs and RuO2 at different current densities. (d) Chronoamperometry study of the PtNP-ZnO@CQD modified electrode. (e) LSV plots of PtNP-ZnO@CQDs at the initial stage and after 25 h of chronoamperometric study. (f) Nyquist plot of PtNP-ZnO@CQDs, ZnO, and CQDs. The equivalent circuit is given in the inset. | ||
3.4 Electrocatalytic ORR study
Initially, a cathodic linear sweep voltammetry (CLSV) plot was obtained in O2-saturated 0.1 M KOH at a 10 mV s−1 scan rate with 1600 rpm. As shown in Fig. 5a, the PtNP-ZnO@CQD modified electrode exhibits a higher limiting current density of 5.0 mA cm−2 than commercial Pt–C (4.2 mA cm−2) and other constituents like ZnO (1.58 mA cm−2) and CQDs (1.53 mA cm−2). Tafel plots were plotted to evaluate the mechanism and kinetic rate of the catalysts relative to ORR efficiency. As depicted in Fig. 5b, PtNP-ZnO@CQD containing electrodes exhibit a comparable Tafel slope of 135.85 mV dec−1 compared to Pt/C (130.28 mV dec−1). PtNP-ZnO@CQDs reveals the highest onset potential of 1.04 V (vs. RHE) and an E1/2 value of 0.96 V (vs. RHE), surpassing ZnO (onset potential = 0.62 V and E1/2 = 0.56 V vs. RHE), CQDs (onset potential = 0.71 V and E1/2 = 0.48 V vs. RHE) and commercial Pt/C (onset potential = 1.02 V and E1/2 = 0.86 V vs. RHE) as represented in Fig. 5c. So, the CLSV plot indicates the superior ORR performance of PtNP-ZnO@CQDs over the constituent materials and the commercial Pt/C. The noticeable reduction peak was also detected in CV profiles, as shown in Fig. S8a in the ESI,† under an oxygen-saturated environment compared to the inert argon environment, indicating the excellent oxygen reduction activity of PtNP-ZnO@CQDs. The mass activities of the Pt-based electrocatalysts are provided in Fig. 5d, revealing a 2.2 times increase in the mass activity of PtNP-ZnO@CQDs (0.426 A mgPt−1) compared to Pt/C (0.196 A mgPt−1). Taking the BET surface area into account, the specific activity of PtNP-ZnO@CQDs was determined to be 0.028 mA cm−2. For better understanding, the ORR performance (limiting current density, mass activity, E1/2, etc.) of PtNP-ZnO@CQDs is compared with that of Pt-containing ORR electrocatalysts in recent reports in Table S6 in the ESI,† demonstrating comparable or better ORR performance in PtNP-ZnO@CQDs despite its low Pt-content. Similar to the MOR, the PtNP-ZnO@CQD modified electrode also demonstrates a significant enhancement in ORR performance under light irradiation as it exhibits a higher limiting current density of 6.1 mA cm−2 (Fig. S8b†). The photogenerated electrons in ZnO or CQDs contribute to oxygen reduction along with the electrocatalytic ORR in the presence of light to enhance the ORR performance of the PtNP-ZnO@CQD composite.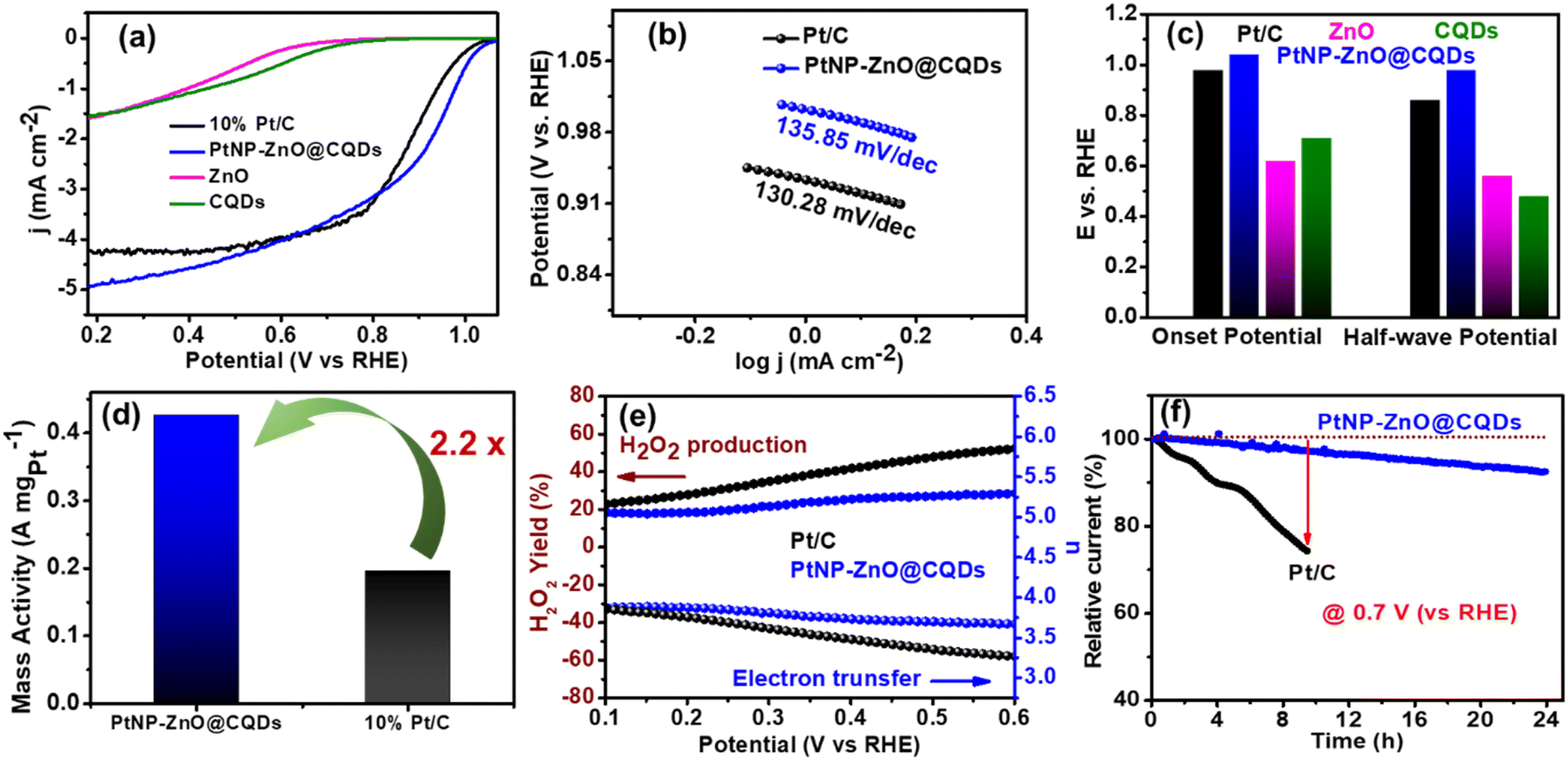 | ||
| Fig. 5 (a) CLSV curve of PtNP-ZnO@CQD, ZnO, CQD, commercial Pt/C catalysts. (b) Tafel slope of the catalysts. (c) Bar diagram to compare different catalysts' onset and half-wave potentials. (d) Bar diagram to compare the mass activity of different catalysts. (e) Number of electrons transferred and H2O2 yield of PtNP-ZnO@CQD and Pt-carbon catalysts. (f) Chronoamperometric plot of PtNP-ZnO@CQD and Pt/C catalysts during the ORR. | ||
The number of electrons transferred and the percentage of peroxide yield are very important to obtain the effective pathway of ORR activity of the nanocomposite. A number of electron transfers close to 4 is much desired as the electrochemical pathway exhibits a lower yield of undesired hydrogen peroxide in the process. The details of the calculation using the disk current and ring current obtained from RRDE measurements are provided in the ESI,† and the data are shown in Fig. 5e. In a wide potential range 0.6 to 0.1 V, the number of electron transfers varies from 3.70 to 3.90 for PtNP-ZnO@CQDs and 3.25 to 3.85 for commercial Pt/C catalysts, as shown in Fig. 5e. Accordingly, the peroxide yield also varies from 28 to 19% in PtNP-ZnO@CQDs and 53% to 24% in commercially available Pt/C. So, a high number of electron transfers and a lower yield of peroxide for PtNP-ZnO@CQDs further advocate the superiority of our nanocomposite catalyst over commercial high Pt-containing Pt/C. The chronoamperometric stability of the prepared PtNP-ZnO@CQD catalysts at an applied potential of 0.7 V (vs. RHE) was examined over 24 hours in an O2-saturated 0.1 M KOH solution at 400 rpm, and the relative percentage of retained current density was compared with that of the commercially available Pt/C catalyst. Remarkably, among the two materials, PtNP-ZnO@CQDs retains ∼90% of initial current density after 24 h (Fig. 5f). However, the commercial Pt/C shows only ∼75% retention of initial current density after 8 h, indicating the robust performance of PtNP-ZnO@CQDs towards the ORR. Additionally, we varied the amounts of Pt and Zn to evaluate the electrocatalytic ORR activity of the ternary PtNP-ZnO@CQD composite, as shown in Fig. S9 in the ESI.† The results clearly demonstrate a direct correlation between Pt content and ORR activity, with an increase in Pt concentration leading to enhanced ORR performance.
The individual OER and ORR activities of ZnO and CQDs, as depicted in Fig. 4a and 5a, show significantly higher overpotentials and lower current densities than the PtNP-ZnO@CQD composite. This highlights the distinct catalytic roles of Pt, ZnO, and CQDs in the composite. PtNPs, with their high surface energy in NPs and superior electron transfer capabilities, serve as the primary catalytic sites, effectively lowering the activation energy for reactions like the ORR, thereby enhancing the overall activity. ZnO, functioning as a semiconductor, not only facilitates electron mobility but also provides a stable matrix for PtNPs, improving charge separation and electron transfer to accelerate reaction kinetics. Its bandgap allows visible light absorption in photocatalytic applications, aiding in electron–hole pair generation. Meanwhile, CQDs act as electron reservoirs, promoting charge transfer within the composite. The high surface area of quantum dots enhances the adsorption to elevate the catalytic activity. Furthermore, CQDs' large surface area increases catalytic site availability and improves PtNP dispersion, preventing aggregation and maintaining high efficiency. In the PtNP-ZnO@CQD composite, a synergistic effect emerges, where PtNPs and CQDs collaboratively enhance catalytic performance. ZnO and CQDs stabilize the PtNPs, preventing degradation, while ZnO and PtNPs together facilitate efficient charge transfer, collectively boosting the overall catalytic efficiency.
3.5 Zinc–air batteries
Considering the superior bi-functional ORR and OER activity of PtNP-ZnO@CQDs, we further investigated a laboratory-made ZAB utilizing the PtNP-ZnO@CQD nanocomposite as electrode materials.69 The rechargeable ZAB was assembled using PtNP-ZnO@CQDs or Pt/C-RuO2 modified Ni-foam as the cathode and a metallic Zn plate as the anode with an aqueous electrolyte containing 6.0 M KOH and 0.2 M Zn(CH3COO)2, as shown in Fig. 6a. Initially, the PtNP-ZnO@CQD containing ZAB exhibits an open circuit potential (OCP) of 1.55 V, as shown in Fig. 6b. The OCP changes very little with operation as the PtNP-ZnO@CQD-based ZAB exhibits an OCP of 1.49 V after 6 h of continuous operation. On the other hand, the Pt/C-RuO2-based ZAB reveals an OCP of 1.42 V, which is reduced to 1.39 V after 6 h of operation. The PtNP-ZnO@CQD-containing ZAB demonstrates the highest areal power density of 148 mW cm−2 at a current density of 225 mA cm−2, exceeding the commercially available Pt/C-RuO2 catalyst-based ZAB (highest power density of 118.6 mW cm−2 at a current density of 120 mA cm−2), as shown in Fig. 6c. Additionally, the mass of the consumed Zn anode was normalized to determine the high specific capacity of the PtNP-ZnO@CQD catalyst, which was 532 mA h g−1 at 2 mA cm−2 with a gravimetric energy density of 668 W h kg−1 (Fig. 6d). These values are much higher than the specific capacity and gravimetric energy density of the Pt/C-RuO2 containing ZAB, which were reported as 510 mA h cm−2 and 586 W h kg−1, respectively. The charge–discharge polarization curves of the two ZABs in Fig. 6e reveal that a smaller charge–discharge interval is required for the PtNP-ZnO@CQD-based ZAB (0.69 V at 50 mA cm−2, 1.47 V at 150 mA cm−2, and 1.87 V at 250 mA cm−2) than the Pt/C-RuO2 catalyst-based ZAB (1.02 V at 50 mA cm−2, 1.76 V at 150 mA cm−2, and 2.18 V at 250 mA cm−2), signifying much better rechargeability in the PtNP-ZnO@CQDs-based ZAB. Furthermore, the PtNP-ZnO@CQDs-based ZAB was examined by impedance measurement, as shown in Fig. 6f. The Nyquist plot indicates the charge-transfer resistance (Rct) of the air electrode to be 6.5 Ω, confirming the better conductivity of the PtNP-ZnO@CQD catalyst as the air cathode. Maintaining long-term cycle stability is a crucial feature of rechargeable ZABs. The long-term rechargeability of the PtNP-ZnO@CQD-based ZAB was determined by a continuous galvanostatic discharge–charge cycling performance study for more than 60 cycles at a current density of 5 mA cm−2, and the plot is presented in Fig. 6f. The PtNP-ZnO@CQD catalyst-based ZAB reveals an initial round trip efficiency of 77.16% and sustains a round trip efficiency of 66.87% even after 60 h. The slight degradation of the battery life can be attributed to the degradation of the zinc anode by dendrite growth on the zinc metal surface (as shown in Fig. S10 in the ESI†) rather than the PtNP-ZnO@CQD-based air cathode, implying that the PtNP-ZnO@CQD air electrode has a much higher cycle life than the zinc anode. However, under the same conditions, the Pt/C-RuO2 catalyst-based ZAB demonstrated a raid degradation of cyclic performance only after 12 h with a wide potential gap (Fig. 6f). To evaluate the chemical stability of the catalyst after use, the post-catalyst characterization after ZAB operation was performed by using XPS and SEM studies. The practical application of the fabricated ZAB with the PtNP-ZnO@CQD air cathode was tested by using two ZABs in a series connection, providing an open circuit potential of 2.7 V, as shown in Fig. 7a. The assembled ZABs in series combinations can light a 1.8 V red LED light panel (18 red LEDs in series for the photo-frame panel), a 3.4 V blue LED light and a 6 V white LED light, indicating the potential commercial application of the PtNP-ZnO@CQD catalyst in the replacement of the commercially available Pt/C-RuO2 mixture as an air electrode for ZAB.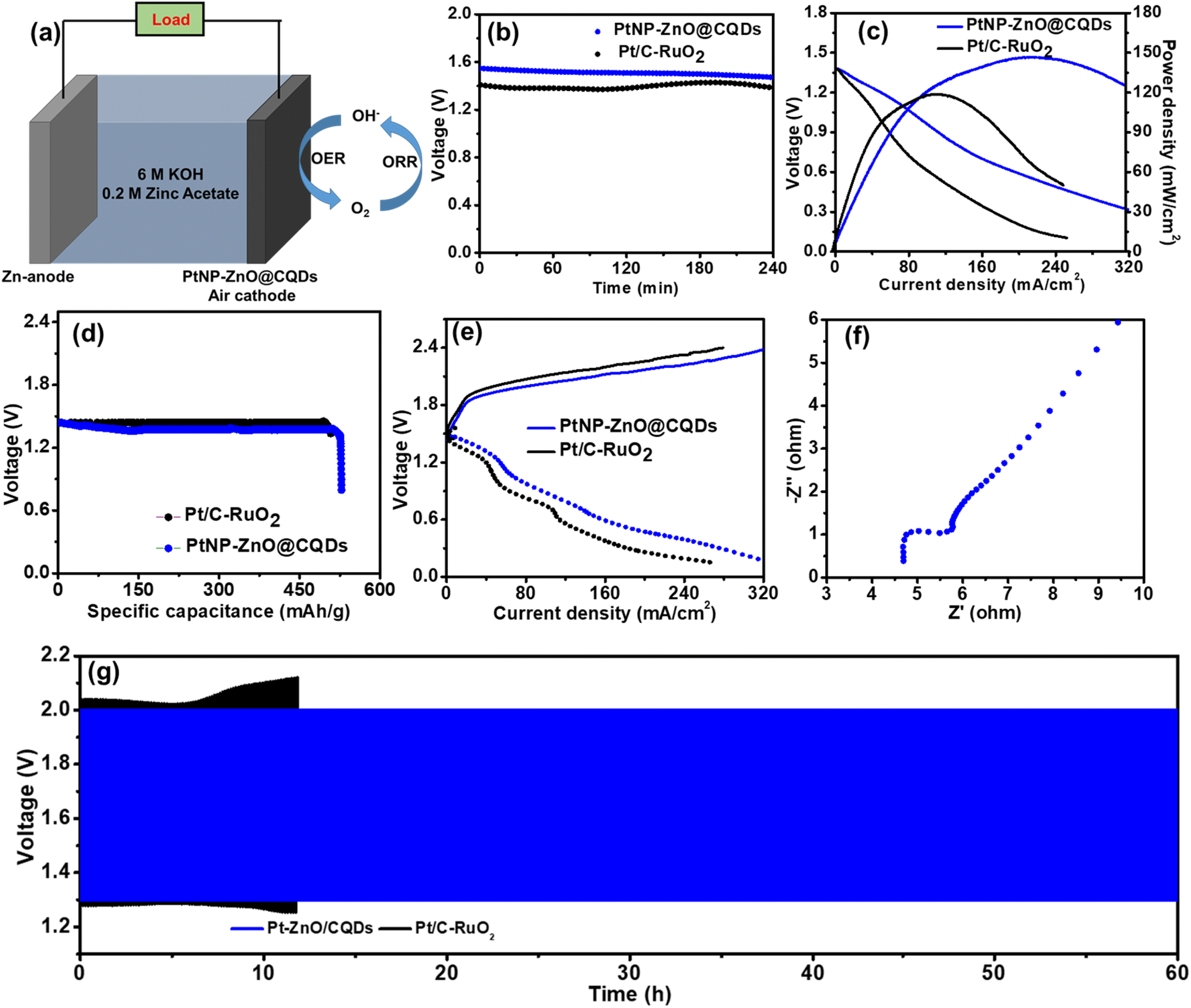 | ||
| Fig. 6 (a) Schematic diagram of the PtNP-ZnO@CQD-based rechargeable ZAB. (b) OCP study and (c) polarization curves with the corresponding areal power density plot of PtNP-ZnO@CQD (blue line) and Pt/C-RuO2-based (black line) ZABs. (d) The specific capacitance plots of the two ZABs at 2 mA cm−2 current density. (e) The charging–discharging polarization curves for the two ZABs. (f) The EIS study of the assembled PtNP-ZnO@CQD-based rechargeable ZAB. (g) Long-term cycling stability of PtNP-ZnO@CQD (blue line) and Pt/C-RuO2 (black line) containing ZABs at a current density of 5 mA cm−2. | ||
 | ||
| Fig. 7 (a) The assembled ZABs are made up of PtNP-ZnO@CQD catalysts in a series combination. The assembled ZAB lights of (b) a 1.8 V red LED light panel, (c) a 3.4 V blue LED light and (d) a 6 V white LED light. | ||
4 Conclusion
In summary, a ternary nanocomposite containing ultrafine PtNPs decorated on photosensitive ZnO and CQDs is synthesized using a simple one-pot hydrothermal synthesis as a trifunctional electrocatalyst for high-performance MOR, OER, and ORR with decent durability. The low Pt-content nanocomposite PtNP-ZnO@CQDs is thoroughly characterized by XRD, FT-IR, XPS, BET analysis, SEM, EDX, HRTEM, etc. During the MOR, the PtNP-ZnO@CQD catalyst exhibited a significant anodic peak current density of 4.93 mA cm−2 at −0.14 V (vs. Ag/AgCl), which was further increased to 9.1 mA cm−2 upon white LED irradiation. The PtNP-ZnO@CQD nanocomposite reveals a low overpotential of 355 mV at 10 mA cm−2 and during the OER. The OER overpotential of PtNP-ZnO@CQDs was comparatively lower than that of commercial RuO2 at higher current densities. Additionally, the electrocatalyst also demonstrates a high half-wave potential of 0.96 V, high limiting current density, and a mass activity of 0.426 A mgPt−1 during the ORR, surpassing the performance of commercial 10% Pt/C in every aspect. The low peroxide yield, high number of electron transfers, and photoinduced ORR capability also highlight the superiority of PtNP-ZnO@CQDs over commercial Pt/C catalysts. The excellent performance of PtNP-ZnO@CQDs is precisely due to the existence of the synergistic effect of the porous structure, high surface area for adsorption, high ECSA, low charge transfer resistance, SMSI between PtNPs and support CQDs, etc., improving the intrinsic trifunctional activity despite the low Pt-content. Furthermore, a rechargeable aqueous ZAB assembled with the PtNP-ZnO@CQD air cathode exhibits an outstanding energy density of 668 W h kg−1 and a high specific capacity of 532 mA h g−1 with superior charge–discharge stability up to 60 h, outperforming the ZAB composed of a commercial 10% Pt/C and RuO2 mixture. Finally, the capability of the assembled ZABs in series to light up different LED panels provides insight into developing bifunctional low-cost Pt-based electrocatalysts to boost the future commercialization of ZABs.Data availability
Data will be available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the PURSE Project No. SR/PURSE/2020/20 (G) funded by the Department of Science and Technology (DST), Govt. of India. The authors are thankful to the central analytical laboratory of the BITS Pilani Hyderabad campus for instrument facilities.References
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS
.
- J. Huang, Y. Xie, Y. You, J. Yuan, Q. Xu, H. Xie and Y. Chen, Adv. Funct. Mater., 2023, 33, 2213095 CrossRef CAS
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu, Y. Chen, L. Qie, Z. Zhu, G. Xu, J. Kong and J. Li, Nat. Energy, 2019, 4, 374–382 CrossRef CAS
- L. Li, X. Tang, B. Wu, B. Huang, K. Yuan and Y. Chen, Adv. Mater., 2024, 36, 2308326 CrossRef CAS PubMed
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC
- Z.-F. Huang, J. Song, K. Li, M. Tahir, Y.-T. Wang, L. Pan, L. Wang, X. Zhang and J.-J. Zou, J. Am. Chem. Soc., 2016, 138, 1359–1365 CrossRef CAS PubMed
- S. Zhao, T. Liu, J. Wang, I. Temitope Bello, Y. Zuo, M. Wei, K. Wang, K. K. S. Lau and M. Ni, Chem.–Eng. J., 2022, 450, 138207 CrossRef CAS
- X. Chen, Z. Yan, M. Yu, H. Sun, F. Liu, Q. Zhang, F. Cheng and J. Chen, J. Mater. Chem. A, 2019, 7, 24868–24876 RSC
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed
- P. C. Meenu, S. Roy, C. Chakraborty and S. Roy, Adv. Powder Technol., 2021, 32, 2663–2689 CrossRef CAS
- A. K. Pradhan, S. Halder and C. Chakraborty, Surf. Interfaces, 2024, 44, 103816 CrossRef CAS
- X. Zhao, X. Maimaitiyiming, M. Tursun and H. Lin, Fuel, 2024, 364, 131089 CrossRef CAS
- X. Cao, R. Song, X. Zhou, X. Wang, X. Dong, N. Yuan and J. Ding, Langmuir, 2022, 38, 4948–4957 CrossRef CAS
- X. Cao, J. Li, X. Dong, R. Song, X. Zhou, X. Wang, N. Yuan and J. Ding, J. Alloys Compd., 2022, 928, 166932 CrossRef CAS
- S. Halder, A. K. Pradhan, P. Sivasakthi, P. K. Samanta and C. Chakraborty, Mater. Today Chem., 2023, 32, 101649 CrossRef CAS
- G. Nazir, A. Rehman, J.-H. Lee, C.-H. Kim, J. Gautam, K. Heo, S. Hussain, M. Ikram, A. A. AlObaid, S.-Y. Lee and S.-J. Park, Nano-Micro Lett., 2024, 16, 138 CrossRef
- N. H. H. Phuc, T. Anh Tu, L. Cam Loc, C. Xuan Viet, P. Thi Thuy Phuong, N. Tri and L. Van Thang, Nanoenergy Adv., 2023, 3, 13–47 CrossRef
- N. Gupta, S. Halder, R. P. Behere, P. Singh, S. Kanungo, M. Dixit, C. Chakraborty and B. K. Kuila, ACS Appl. Mater. Interfaces, 2023, 15, 29042–29051 CrossRef CAS PubMed
- T. Zhou, N. Zhang, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC
- C. Zhang, X. Shen, Y. Pan and Z. Peng, Front. Energy, 2017, 11, 268–285 CrossRef
- M. N. Islam, A. B. Mansoor Basha, V. O. Kollath, A. P. Soleymani, J. Jankovic and K. Karan, Nat. Commun., 2022, 13, 6157 CrossRef CAS
- Y. Zhong, Y. Lu, Z. Pan, J. Yang, G. Du, J. Chen, Q. Zhang, H. Zhou, J. Wang, C. Wang and W. Li, Adv. Funct. Mater., 2021, 31, 2009853 CrossRef CAS
- W. Chen, J. Cao, W. Fu, J. Zhang, G. Qian, J. Yang, D. Chen, X. Zhou, W. Yuan and X. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200190 CrossRef CAS PubMed
- P. Ochal, J. L. Gomez De La Fuente, M. Tsypkin, F. Seland, S. Sunde, N. Muthuswamy, M. Rønning, D. Chen, S. Garcia, S. Alayoglu and B. Eichhorn, J. Electroanal. Chem., 2011, 655, 140–146 CrossRef CAS
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed
- A. Li, J. X. Zhao and D. T. Pierce, J. Colloid Interface Sci., 2010, 351, 365–373 CrossRef CAS
- A. Sharma, S. K. Mehta, S. Singh and S. Gupta, J. Appl. Electrochem., 2016, 46, 27–38 CrossRef CAS
- D. Gao, S. Yang, L. Xi, M. Risch, L. Song, Y. Lv, C. Li, C. Li and G. Chen, Chem. Mater., 2020, 32, 1581–1594 CrossRef CAS
- W. Gong, Z. Jiang, R. Wu, Y. Liu, L. Huang, N. Hu, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2019, 246, 277–283 CrossRef CAS
- C. Li, X. Chen, L. Zhang, S. Yan, A. Sharma, B. Zhao, A. Kumbhar, G. Zhou and J. Fang, Angew. Chem., Int. Ed., 2021, 60, 7675–7680 CrossRef CAS PubMed
- X. Sun, S. Wang, Y. Hou, X. F. Lu, J. Zhang and X. Wang, J. Mater. Chem. A, 2023, 11, 13089–13106 RSC
- X. Wang, J. Zhang, P. Wang, L. Li, H. Wang, D. Sun, Y. Li, Y. Tang, X. F. Lu, Y. Wang and G. Fu, Energy Environ. Sci., 2023, 16, 5500–5512 RSC
- B. Y. Xia, H. B. Wu, J. S. Chen, Z. Wang, X. Wang and X. W. Lou, Phys. Chem. Chem. Phys., 2012, 14, 473–476 RSC
- S. Roy, S. Payra, S. Challagulla, R. Arora, S. Roy and C. Chakraborty, ACS Omega, 2018, 3, 17778–17788 CrossRef CAS PubMed
- W. Shi, H.-U. Park, A.-H. Park, L. Xue, S.-K. Kim, G.-G. Park and Y.-U. Kwon, Appl. Catal., B, 2023, 331, 122692 CrossRef CAS
- S. Kang, H. Kim and Y.-H. Chung, Nano Convergence, 2018, 5, 13 CrossRef PubMed
- Z. Zhang, J. Liu, J. Gu, L. Su and L. Cheng, Energy Environ. Sci., 2014, 7, 2535–2558 RSC
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 RSC
- X.-L. Sui, Z.-B. Wang, M. Yang, L. Huo, D.-M. Gu and G.-P. Yin, J. Power Sources, 2014, 255, 43–51 CrossRef CAS
- M. Huang, W. Wu, C. Wu and L. Guan, J. Mater. Chem. A, 2015, 3, 4777–4781 RSC
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, H. D. Abruña, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317–9332 CrossRef CAS
- X. Zhang, H. Li, J. Yang, Y. Lei, C. Wang, J. Wang, Y. Tang and Z. Mao, RSC Adv., 2021, 11, 13316–13328 RSC
- A. Di Mauro, M. Zimbone, M. Scuderi, G. Nicotra, M. E. Fragalà and G. Impellizzeri, Nanoscale Res. Lett., 2015, 10, 484 CrossRef PubMed
- C.-Y. Su, Y.-C. Hsueh, C.-C. Kei, C.-T. Lin and T.-P. Perng, J. Phys. Chem. C, 2013, 117, 11610–11618 CrossRef CAS
- M. A. Hanif, J. Akter, I. Lee, M. A. Islam, K. P. Sapkota, H. G. Abbas and J. R. Hahn, J. Photochem. Photobiol., A, 2021, 413, 113260 CrossRef CAS
- C. Chen, W. Yu, T. Liu, S. Cao and Y. Tsang, Sol. Energy Mater. Sol. Cells, 2017, 160, 43–53 CrossRef CAS
- J.-J. Chen, J.-B. Tan, C.-F. Li, L.-F. Gu, X.-F. Lu and G.-R. Li, J. Phys. Chem. C, 2020, 124, 13036–13044 CrossRef CAS
- B. Zhao, H. Luo, J. Liu, S. Chen, H. Xu, Y. Liao, X. F. Lu, Y. Qing and Y. Wu, Chin. Chem. Lett., 2024, 109919 CrossRef
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed
- V. C. Hoang, K. Dave and V. G. Gomes, Nano Energy, 2019, 66, 104093 CrossRef CAS
- F. Sher, I. Ziani, M. Smith, G. Chugreeva, S. Z. Hashimzada, L. D. T. Prola, J. Sulejmanović and E. K. Sher, Coord. Chem. Rev., 2024, 500, 215499 CrossRef CAS
- J. Liu, G. Qin, P. Raveendran and Y. Ikushima, Chem.–Eur. J., 2006, 12, 2131–2138 CrossRef CAS PubMed
- X. Kong, H. Cao, C. Li and X. Chen, J. Colloid Interface Sci., 2017, 487, 60–67 CrossRef CAS PubMed
- H. Huang, J. Cui, G. Liu, R. Bi and L. Zhang, ACS Nano, 2019, 13, 3448–3456 CrossRef CAS PubMed
- A. Lewera, L. Timperman, A. Roguska and N. Alonso-Vante, J. Phys. Chem. C, 2011, 115, 20153–20159 CrossRef CAS
- M. Gantumur, M. I. Hossain, Md. Shahiduzzaman, A. Tamang, J. H. Rafij, Md. Shahinuzzaman, H. Thi Cam Tu, M. Nakano, M. Karakawa, K. Ohdaira, H. AlMohamadi, M. A. Ibrahim, K. Sopian, Md. Akhtaruzzaman, J. M. Nunzi and T. Taima, ACS Appl. Mater. Interfaces, 2024, 16, 36255–36271 CrossRef CAS PubMed
- W. Yan, X. Wang, M. Liu, K. Ma, L. Wang, Q. Liu, C. Wang, X. Jiang, H. Li, Y. Tang and G. Fu, Adv. Funct. Mater., 2024, 34, 2310487 CrossRef CAS
- Z. Li, X. Jiang, X. Wang, J. Hu, Y. Liu, G. Fu and Y. Tang, Appl. Catal., B, 2020, 277, 119135 CrossRef CAS
- J.-J. Fan, Y.-J. Fan, R.-X. Wang, S. Xiang, H.-G. Tang and S.-G. Sun, J. Mater. Chem. A, 2017, 5, 19467–19475 RSC
- D. J. Suh, C. Kwak, J.-H. Kim, S. M. Kwon and T.-J. Park, J. Power Sources, 2005, 142, 70–74 CrossRef CAS
- R. Li, Z. Liu, Q. T. Trinh, Z. Miao, S. Chen, K. Qian, R. J. Wong, S. Xi, Y. Yan, A. Borgna, S. Liang, T. Wei, Y. Dai, P. Wang, Y. Tang, X. Yan, T. S. Choksi and W. Liu, Adv. Mater., 2021, 33, 2101536 CrossRef CAS PubMed
- J. Xu, H. Xu, A. Dong, H. Zhang, Y. Zhou, H. Dong, B. Tang, Y. Liu, L. Zhang, X. Liu, J. Luo, L. Bie, S. Dai, Y. Wang, X. Sun and Y. Li, Adv. Mater., 2022, 34, 2206991 CrossRef CAS PubMed
- E. Antolini, Appl. Catal., B, 2018, 237, 491–503 CrossRef CAS
- Y. Nosaka and A. Nosaka, ACS Energy Lett., 2016, 1, 356–359 CrossRef CAS
- I. Kisacik, A. Stefanova, S. Ernsta and H. Baltruschat, Phys. Chem. Chem. Phys., 2013, 15, 4616–4624 RSC
- S. Halder, A. K. Pradhan, S. Khan and C. Chakraborty, Energy Adv., 2023, 2, 1713–1723 RSC
- A. K. Pradhan, S. Halder and C. Chakraborty, J. Energy Storage., 2024, 98, 113008 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental calculation and characterization of materials such as XPS, SEM-EDX data, CV study for the MOR and ORR, instrumentation details for the CO tolerance study, FE study, CLSV for the photoinduced ORR study, comparison table, etc. See DOI: https://doi.org/10.1039/d4ta05630b |
| This journal is © The Royal Society of Chemistry 2025 |