
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performing and sustainable hard carbons for Na-ion batteries through acid-catalysed hydrothermal carbonisation of vine shoots†
Darío
Alvira
 *ab,
Daniel
Antorán
ab,
Hamideh
Darjazi
cd,
Giuseppe Antonio
Elia
cd,
Claudio
Gerbaldi
*cd,
Victor
Sebastian
befg and
Joan J.
Manyà
ab
*ab,
Daniel
Antorán
ab,
Hamideh
Darjazi
cd,
Giuseppe Antonio
Elia
cd,
Claudio
Gerbaldi
*cd,
Victor
Sebastian
befg and
Joan J.
Manyà
ab
aAragón Institute for Engineering Research (I3A), Thermochemical Processes Group, University of Zaragoza, Escuela Politécnica Superior, Crta. de Cuarte s/n, Huesca, 22071, Spain. E-mail: dalvira@unizar.es
bDepartment of Chemical Engineering and Environmental Technologies, University of Zaragoza, Campus Río Ebro, María de Luna 3, Zaragoza, 50018, Spain
cGAME Lab – Department of Applied Science and Technology (DISAT), Politecnico di Torino, Corso Duca Degli Abruzzi 24, Torino, 10129, Italy. E-mail: claudio.gerbaldi@polito.it
dNational Reference Centre for Electrochemical Energy Storage (GISEL) – INSTM, Via G. Giusti 9, Firenze, 50121, Italy
eInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza, Spain
fNetworking Research Center on Bioengineering Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain
gLaboratorio de Microscopías Avanzadas, Universidad de Zaragoza, Zaragoza, 50018, Spain
First published on 3rd December 2024
Abstract
This study investigates the synthesis of hard carbons via acid-assisted hydrothermal carbonisation (HTC) of vine shoots, followed by thermal annealing, to be used as Na-ion battery anodes. Various carbons with diverse pore structures, ordering degrees, 2D morphologies, and chemical compositions are obtained using 2 mol dm−3 solutions of HNO3, HCl, H2SO4, or H3PO4 as the hydrothermal media. Compared to standard ester-based liquid electrolytes, the use of a diglyme-based electrolyte (NaPF6/DGM) substantially boosts both the initial coulombic efficiency (ICE) and the specific capacity, particularly evident with the highly ordered and porous H3PO4-1000 and HCl-1000 carbons. These materials exhibit outstanding performance, storing 200 mAh g−1 at 2 A g−1 and showing ICE values of 68% and 77%, respectively. The improved stability and capacity are attributed to the formation of a more stable and thinner solid electrolyte interface (SEI), along with sodium storage into graphitic regions through a solvent co-intercalation reaction. While ester-based electrolytes limit hard carbon mesoporosity and require the promotion of turbostratic domains, ether-based electrolytes enable the development of more ordered and porous carbons, enhancing transport kinetics, sodium storage capacity, and electrode stability. Overall, acid-catalysed HTC offers a sustainable pathway for tailoring hard carbon based electrodes, repurposing biomass residues into valuable materials for green and low-cost energy storage applications.
1. Introduction
The increasing demand for energy storage systems has promoted intensive research into advanced materials for post-lithium rechargeable batteries.1–3 Sodium-ion batteries (SIBs) have emerged as promising candidates due to widespread availability and lower cost of sodium resources, with a particular focus on grid-level systems for renewable energy integration, where cost-efficiency and sustainability are critical considerations.4,5 However, the optimisation of SIBs depends on the design and synthesis of advanced electrode materials with superior electrochemical properties. In this regard, hard carbons (HCs) are amongst the most studied anode materials for SIBs, considering their ability to store Na+ ions in their surface functionalities, pores, and pseudographitic domains.6–8 Furthermore, their production from diverse biomass waste resources offers unlimited internal architectural possibilities and enables the valorisation of numerous waste materials to generate high-value-added products in a circular economy approach.9In recent years, hydrothermal carbonisation (HTC) has gained significant attention as a facile and environmentally benign route for the synthesis of carbonaceous materials from lignocellulosic materials.10,11 This method involves the hydrothermal treatment of biomass under mild temperature conditions (180–250 °C) and autogenic pressure (15–30 MPa), leading to the transformation of organic precursors into carbon-rich hydrochars.12,13 The resulting carbonaceous materials—often subjected to further annealing at higher temperatures—exhibit various morphologies, surface chemistries, and structural properties, rendering them suitable for a wide range of applications, including energy storage,14 soil improvement,15 and catalysis.16
In the HTC process, water in subcritical stage acts as both the solvent and catalyst of the reactions. The lower dielectric constant of subcritical water weakens hydrogen bonds and promotes water dissociation into hydronium ions (H3O+) and hydroxide ions (OH−), creating a suitable environment for both acid- and base-catalysed reactions of the organic compounds from biomass.17 Many reactions occur during the HTC of lignocellulosic materials, with hydrolysis governing the reaction since it has a lower activation energy.18
Cellulose undergoes hydrolysis above 200 °C, breaking down cellulose chains into oligomers and glucose. These hydrolysed compounds then undergo dehydration, isomerisation, and fragmentation reactions, yielding soluble products such as furfural-like compounds. The decomposition of monomers also generates organic acids, with the resulting hydronium ions acting as catalysts for further degradation reactions. Subsequent stages involve polymerisation or condensation reactions, forming soluble polymers and spherical shapes, while simultaneous aromatisation of polymers takes place.19 These polymerisation reactions and subsequent nucleation processes are the origin of the typical microspheres observed in hydrochars,20 attractive for the battery field since spherical carbons can shorten the diffusion pathway of Na+ ions compared to conventional 3D porous HCs.21
Hemicelluloses have a lower degree of polymerisation than cellulose and hydrolyse at 180 °C.22 Analogous reactions to those with cellulose, such as dehydration and polymerisation of monosaccharide, also generate hydrochar.23 In contrast, lignin, which consists of crosslinked phenolic polymers, is much more resistant to hydrolysis, and only a small amount is degraded under mild HTC conditions.24 The portion that undergoes hydrolysis and dealkylation can produce phenolic compounds such as guaiacols and catechols, which, through crosslinking reactions, give rise to phenolic hydrochar.25 Meanwhile, water-insoluble macromolecules undergo direct solid–solid transformations to create polyaromatic hydrochar.26 Thus, a high enough lignin content in the raw biomass ensures the persistence of the plant channel-based structure after the HTC pretreatment and final annealing step, making the resulting hard carbon more attractive from a morphological standpoint.
Compared with other thermal conversion technologies, HTC offers several advantages, such as lower ash production and the possibility of using wet biomasses without a prior drying process.27 Moreover, HTC gives rise to carbons rich in surface functionalities, porosity, and spherical shapes.28,29 Another differentiating factor lies in the potential addition of chemical reagents to the aqueous solution, potentially catalysing the inherent reactions of the process and/or promoting the activation or doping of the hydrochar. For instance, in situ catalytic pore formation has been evidenced by adding ZnCl2,30 CaCl2,31 KOH,32 and NaOH,33 while N, P, and S were introduced in the carbon structure by adding melamine, thiourea, (NH4)2SO4, or NaH2PO4.34,35
The inclusion of acids catalyses hydrolysis, dehydration, and decarboxylation reactions in biomass precursors, increasing the carbonisation yield together with chemical and morphological changes.36,37 Different types of acids have been studied, including organic acids (e.g., citric,38 formic,39 and acetic40 acids), Brønsted-Lowry acids (e.g., HNO3,41 H3PO4,42 HCl,43 and H2SO4),44 and Lewis acids (e.g., FeCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 and InCl346). However, as far as we know, only few articles have been published to date studying the acid-assisted HTC of natural lignocellulosic materials to produce HC anodes for SIBs.47,48 In particular, the present study explores the role of four Brønsted-Lowry acids—nitric acid (HNO3), hydrochloric acid (HCl), sulphuric acid (H2SO4), and phosphoric acid (H3PO4)—as catalysts for the hydrothermal carbonisation of vine shoots in order to provide a cost-effective process to obtain high-performance anodes for SIBs.
45 and InCl346). However, as far as we know, only few articles have been published to date studying the acid-assisted HTC of natural lignocellulosic materials to produce HC anodes for SIBs.47,48 In particular, the present study explores the role of four Brønsted-Lowry acids—nitric acid (HNO3), hydrochloric acid (HCl), sulphuric acid (H2SO4), and phosphoric acid (H3PO4)—as catalysts for the hydrothermal carbonisation of vine shoots in order to provide a cost-effective process to obtain high-performance anodes for SIBs.
In addition, we examine the impact of electrolytes on HC anodes by comparing the properties of carbonate-based and ether-based electrolytes, as well as the HC anode interface. Ether-based electrolytes are being studied for SIBs, particularly for their effectiveness in promoting the reversible co-intercalation of solvated sodium ions into graphite anodes. Moreover, when paired with HC electrodes, ether-based electrolytes exhibit notable advantages over their ester-based counterparts. Indeed, they foster the formation of thinner, more compact, ion-conducting, and inherently SEI layers, which is expected to promote higher ICE, stability along prolonged cycling, and enhanced rate capability.49–52 We conducted long-term electrochemical performance tests on HC-based anodes to understand how the choice of electrolyte and the formation of the solid electrolyte interphase (SEI) influence the lifespan of tested materials.
2. Experimental section
2.1 Materials
Vine shoots (Vitis vinifera L.) from the Cabernet Sauvignon variety were collected in the Spanish wine region of Somontano (Huesca province). The raw biomass, generated during winter pruning, underwent sun drying and crushing to achieve a particle size smaller than 0.7 mm. The physicochemical characterisation of the biomass was conducted in a previous study,53 revealing hemicelluloses, cellulose, and lignin compositions of 24.8%, 34.2%, and 20.3%, respectively.2.2 Production of hard carbons
Hard carbons were synthesised using the two-step carbonisation process illustrated in Fig. 1. Initially, 15 g of vine shoots in 50 g of distilled water were hydrothermally treated for 12 hours at 180 °C in a 100 mL Teflon-lined stainless-steel autoclave. The resulting material was then collected via vacuum filtration and dried at 100 °C for 12 hours. Afterwards, the obtained hydrochar underwent carbonisation at 800 or 1000 °C at a heating rate of 5 °C min−1 and a soaking time of 2 hours. This process was conducted under an Ar atmosphere using a tubular reactor (made of mullite) placed inside a furnace (model TF1 16/60/300 from Carbolite, Germany). The resulting hard carbon was ground into powder (ø < 90 μm), washed with a 2 mol dm−3 HCl solution for 2 hours at room temperature, and then rinsed with distilled water until achieving a neutral pH. The final product was labelled as H2O-x, where x indicates the highest carbonisation temperature employed. The same procedure was followed using 2 mol dm−3 Brønsted-Lowry acid solutions (Labbox Labware, Spain) as HTC media, giving rise to HNO3-x, HCl-x, H2SO4-x, and H3PO4-x samples.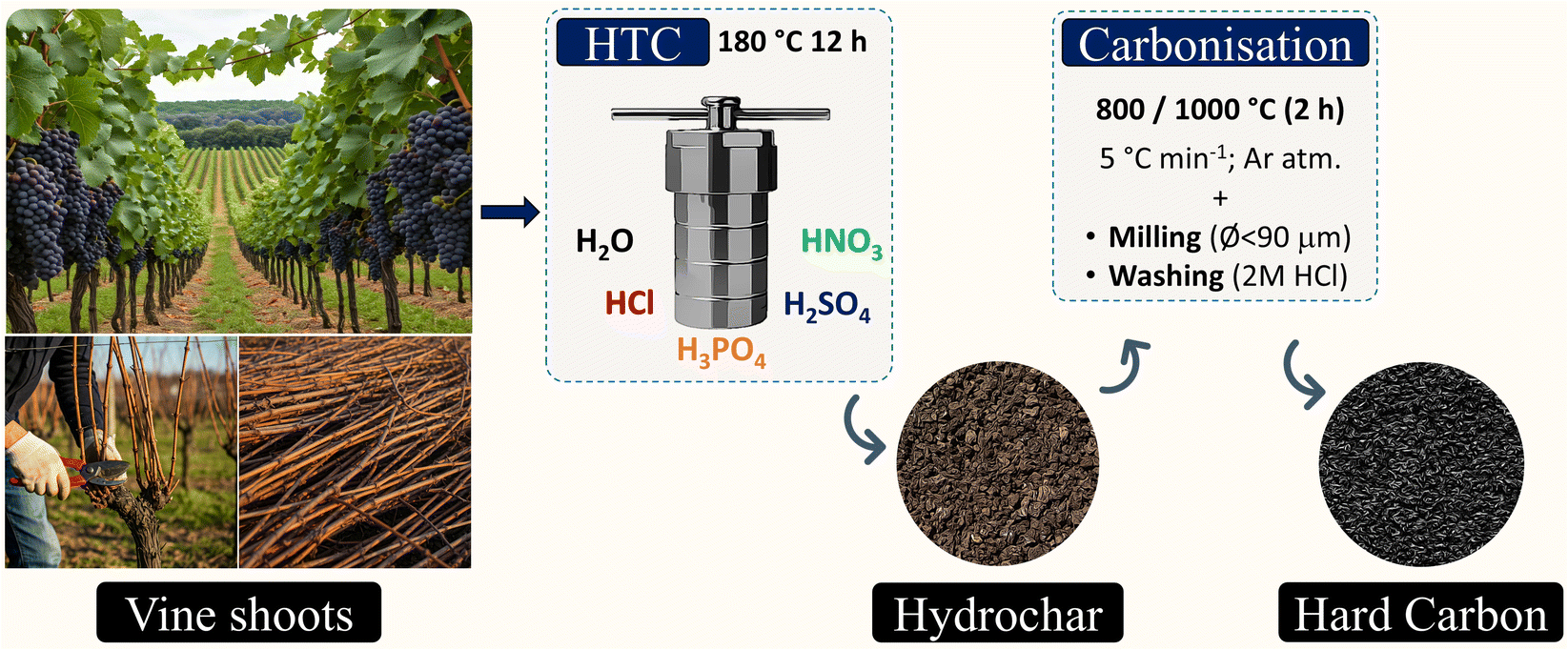 | ||
| Fig. 1 Graphical summary of the hard carbon synthesis via HTC. | ||
2.3 Characterisation of produced hard carbons
The morphologies of hard carbons were observed by an Inspect-F50A scanning electron microscope combined with energy dispersive X-ray spectroscopy (SEM-EDX; FEI, The Netherlands). Additional assessment of the HCs structure was conducted using a Tecnai F30, also from FEI, high-resolution transmission electron microscope (HR-TEM) operated at 300 kV and fitted with a SuperTwin® lens, allowing a point resolution of 0.19 nm. Interplanar distances were determined from HR-TEM images using Digitalmicrograph® software. X-ray photoelectron spectroscopy (XPS; AXIS Supra instrument from Kratos Analytical Ltd, UK) was conducted to investigate the oxygen-, nitrogen-, sulphur-, and phosphorus-containing functional groups on the surface. C 1s, O 1s, N 1s, S 2p, and P 2p regions were deconvoluted using the CasaXPS software package.Structural properties were investigated through X-ray powder diffraction (XRD; Empyrean instrument from Malvern Panalytical, UK, λ = 0.154 nm) and Raman spectroscopy (Alpha 300 model from WITec, Germany, λ = 532 nm). The interlayer spacing between graphene layers (d002), the apparent thickness of crystallites along the c-axis (Lc), and the apparent width of crystallites along the a-axis (La) were calculated from XRD spectra utilising Bragg's law and Scherrer's equation.54 The Raman spectra were subjected to deconvolution into five peaks, as suggested by Sadezky et al.,55 resulting in one Gaussian-shaped band (D3) and four Lorentzian-shaped bands (G, D1, D2, and D4). The peak area ratio (AD1/AG) was calculated as an indicator of the structural disorder degree. The La value was also calculated from the Raman spectra according to the equation proposed by Cançado et al.56
Specific surface areas and pore size distributions were estimated from N2 and CO2 adsorption isotherms at −196 and 0 °C, respectively, after outgassing the samples under vacuum at 150 °C for 8 hours. An Autosorb-iQ analyser from Quantachrome (Germany) was used for the measurement. Specific surface areas (SSA) were calculated using the Brunauer–Emmett–Teller (BET) equation, while pore size distributions were determined using non-local density functional theory (NLDFT) models for slit pore geometries. All the calculations were conducted using the QuadraWin 6.0 software.
2.4 Electrochemical characterisation
The electrochemical performance of hard carbons was measured in three-electrode Swagelok T-cells. The working electrode (WE) comprised the respective hard carbon, carbon black Super P™ (as conductive agent), and sodium carboxymethyl cellulose (Na-CMC; as binder) at mass fractions of 0.8, 0.1, and 0.1, respectively. A uniform slurry was prepared by mixing with deionised (DI) water under magnetic stirring. The slurry was then uniformly coated onto a high-purity aluminium current collector (16 μm thickness) using a baker applicator to create a composite electrode with a thickness of ∼100 μm. Finally, 12 mm diameter electrodes were punched and dried at 120 °C under vacuum for 12 h.The Swagelok cells were assembled in argon-filled gloveboxes from MBraun (Germany) and Jacomex (France), with O2 and H2O contents below 0.5 ppm. Two sodium metal discs with diameters of 12 mm and 5 mm served as counter and reference electrodes (CE and RE), respectively. Additionally, two glass fibre filters with a thickness of 190 μm (Prat Dumas, France) were used as separators. Two different electrolytes (200 μL) were employed in the study: (a) NaTFSI solution (1 mol dm−3) in a mixture (1:1 vol.) of dimethyl carbonate (DMC) and ethylene carbonate (EC), and (b) NaPF6 solution (1 mol dm−3) in the ether-type solvent diglyme (DGM).
Electrochemical measurements were conducted using SP-200 (BioLogic, France) and BT-2000 (Arbin, USA) instruments within a temperature range of 19–22 °C. Galvanostatic charge/discharge (GCD) cycling was performed within a potential window of 0.01–2.5 V vs. Na+/Na, while cyclic voltammetry (CV) profiles were obtained at a scan rate of 0.1 mV s−1 from 0.01 to 2 V vs. Na+/Na. Galvanostatic intermittent titration (GITT) measurements were carried out at a current density of 30 mA g−1, with a pulse time of 20 min and relaxation periods at open circuit of 1 h. Previously, the WE underwent three CV scans to ensure the formation and stabilisation of the solid electrolyte interphase (SEI) layer.
3. Results and discussion
3.1 Physicochemical characterisation of HCs
Fig. 2 and S1 (ESI†) display SEM images of the H2O-x, HNO3-x, and HCl-x samples. Following hydrothermal carbonisation in water (Fig. 2a and b), the original support and transport structures of the grapevine plant (see our previous article53) were preserved, which was facilitated by the relatively high lignin content in the biomass (20.3%) since this biopolymer exhibits limited susceptibility to hydrolysis under mild HTC temperature conditions (180 °C). The preservation of these voids and channels could be advantageous for the electrochemical performance of the resulting HC, enhancing electrolyte penetration and sodium ion diffusion rates. Moreover, compared with carbons produced through direct pyrolysis of vine shoots,53 the HTC pretreatment yielded carbon surfaces with rougher features and smoother contours owing to the hydrolysis of soluble compounds. | ||
| Fig. 2 SEM images of H2O-800 (a), H2O-1000 (b), HNO3-800 (c), HNO3-1000 (d), HCl-800 (e and f), HCl-1000 (g), H2SO4-800 (h and i), H2SO4-1000 (j and k), H3PO4-800 (l and m), and H3PO4-1000 (n and o). | ||
The addition of HNO3 to the aqueous medium induced significant morphological changes in the resulting HCs (Fig. 2c and d). While some plant-derived structures remained (see Fig. S1c and d†), their surfaces became entirely covered by micrometre-sized spherical carbon particles. This transformation is primarily attributed to the high oxidising power of HNO3, which acts as a strong oxidising agent in aqueous solutions.57 The pronounced digestion capacity of this acid was also evidenced by a lower carbon yield (14%) compared to the other studied HTC media (see Table S1†).
In contrast, HCl-mediated HTC gave rise to uncovered carbon surfaces (see Fig. S1e and f†), although clusters of microspheres ranging from 1 to 2.5 μm in diameter and moderate degree of aggregation appeared (Fig. 2e). Upon closer examination of the carbon annealed at 800 °C (Fig. 2f), it was observed that the surface of carbonaceous particles underwent a considerable roughening, which suggests the presence of a mesoporous structure. Both phenomena were attributed to the catalytic role of hydrochloric acid in the hydrolysis and condensation of cellulose and hemicellulose biopolymers, reactions that are less favoured under neutral pH conditions.48 After annealing at 1000 °C (see Fig. 2g and S1f†), no significant morphological changes were observed, with the prevalence of the raw-biomass inherited structures and the small clusters of microspheres originating during the HCl-assisted HTC.
H2SO4- and H3PO4-catalyzed HTC also produced HCs with partially preserved structures from the raw vine shoots, as shown in SEM pictures (at 1000× magnification) given in Fig. S2.† However, at a smaller scale, H2SO4 seems to induce partial hydrolysis of lignin-rich honeycomb complex walls, resulting in cavity expansion and deformation (Fig. 2h). The intrinsic biomass pores surrounded by lignin-rich complexes were maintained, coexisting with clusters of microspheres derived from the more soluble polymers (Fig. 2i). On the other hand, the much rougher texture, also observed upon annealing at 1000 °C (Fig. 2j and k), was attributed to the capacity of sulphuric acid to corrode the surface of lignin-derived hydrochar particles.58 Similar phenomena were observed following the use of H3PO4 (Fig. 2l–o), which also partially degrades the lignin structures,59 resulting in the smoothening of cavities, formation of spherical clusters, and increased surface roughness.
Regarding the textural features of hard carbons, Table S2† summarises the specific surface areas and pore volumes based on N2 and CO2 adsorption isotherms (see also Fig. S3 and S4† for adsorption isotherms and pore size distributions, respectively). As reported in the literature, the hydrothermal pretreatment results in hard carbons with more developed micro- and ultra-microporosities. CO2 BET surface areas of 397 and 445 m2 g−1 were measured for the H2O-800 and H2O-1000 carbons, respectively. Looking at the pore volume, after annealing at 1000 °C, there was also a slight reduction in microporosity and a minor increase in mesoporosity, possibly due to the collapse of small pore structures to create larger cavities. The influence of acid treatments on pore development is illustrated in Fig. 3. The use of HNO3 had a negative effect on the porosity extent, with N2 BET surface areas practically disappearing. In line with the observations from SEM images, the oxidising action of the HNO3 results in a more compact morphology with fewer diffusion channels. In contrast, using HCl, H2SO4, and H3PO4 led to significant development of mesoporosity and microporosity, especially in the case of HCl, while ultra-microporosity remained relatively constant. The higher catalytic activity of HCl in developing porosity can be attributed to its strength as an acid, fully dissociating in a single step and rapidly releasing protons (H+) in aqueous solutions. This contrasts with H2SO4 and H3PO4, which dissociate in two and three steps, respectively, and exhibit lower acid dissociation constants.60
 | ||
| Fig. 3 (a) BET surface areas and (b) pore volumes for all the hard carbons under study. | ||
The acquired HR-TEM images revealed that H2O-800 and H2O-1000 samples exhibited a mostly disordered structure with randomly oriented pseudographitic domains, alongside mesopores and interconnected rounded particles resulting from the partial hydrolysis of more soluble biomass constituents (see Fig. S5†). Upon closer examination (Fig. 4a and b), d002 values exceeding 0.36 nm were observed, therefore granting Na+ intercalation.61 Increasing the temperature from 800 to 1000 °C led to enhanced structural order with curved graphene layers surrounding closed (or quasi-closed) pores and slightly reducing in the d002 interlayer spacing. These long stripes should provide enough electronic conductivity while increasing the closed pore volume, which is supposed to improve the specific capacity of the active material.62
 | ||
| Fig. 4 HR-TEM images of H2O-800 (a), H2O-1000 (b), HNO3-800 (c), HNO3-1000 (d), HCl-800 (e), HCl-1000 (f), H2SO4-800 (g), H2SO4-1000 (h), H3PO4-800 (i), and H3PO4-1000 (j). | ||
Following treatment with HNO3 and thermal processing at 800 °C, rounded surfaces featuring expanded turbostratic domains appeared (d002 ≈ 0.5), but also 2D regions characterised by interlayer distances lower than those of crystalline graphite (Fig. S6†). These ordered zones, with d002 values ranging from 0.23 to 0.30 nm (Fig. 4c), probably originated from polymerisation phenomena that occur subsequent to the nitric acid catalysed digestion of biomass. The formation of graphene quantum dots exhibiting interlayer distances of 0.21 nm, has been previously reported in the literature after lignin treatment with HNO3 and subsequent HTC.63 Moreover, the exfoliation of carbon through oxidation processes may also contribute significantly to the 2D morphology, resulting in the formation of graphite oxides.64 Upon annealing at 1000 °C (Fig. S7†), no substantial differences were observed, the carbon morphology mainly consisted of both spherical domains and interconnected 2D nanosheets (Fig. 4d).
Catalytic hydrolysis with HCl resulted in amorphous carbons composed of turbostratic domains and abundant spherical shapes (see Fig. S8†). Also noticeable is the widening of the interlayer space between graphene layers—which should provide faster intercalation kinetics—with d002 values ranging from 0.41 to 0.49 nm (Fig. 4e). Following annealing at 1000 °C (Fig. S9†), while amorphous regions with turbostratic domains persisted (d002 ≃ 0.40 nm), other local domains underwent significant ordering, giving rise to fully graphitic 2D zones. As shown in Fig. 4f, these domains consist of crystalline carbon rings mostly arranged in a hexagonal lattice, with d002 values of ca. 0.24 nm. This phenomenon aligns with the HCl exfoliant role and ability to remove impurities (ashes and metals), enabling carbon structure formation at higher purity and order.
On the other hand, the catalytic effect of H2SO4 did not appear to promote the formation of large and well-ordered 2D regions (Fig. S10 and S11†). Whether subjected to annealing at 800 or 1000 °C, both carbons exhibited rounded morphologies with rather rough surfaces. Furthermore, turbostratic domains with an extended lattice development were observed in both carbons, giving rise to curved domains enclosing abundant closed porosity (Fig. 4g and h). In contrast, the employment of H3PO4 promoted the development of ordered lamellar regions, although amorphous and porous areas are also present (Fig. S12 and S13†). Even at 800 °C, fully ordered regions emerged (Fig. 4i), confirming the role of phosphorus in promoting graphitisation as suggested in the literature.65,66 Nevertheless, at 1000 °C (Fig. 4j), turbostratic domains with interplanar distances exceeding 0.40 nm also persisted, resulting in a mixture of more amorphous regions alongside fully ordered ones.
The obtained XRD patterns and Raman peaks deconvolution are given in Fig. S14–S17.† As expected, the XRD spectra showed two broad peaks around 23° and 44° corresponding to the (002) and (100 and 101) planes. The Raman spectra presented two peaks at 1350 cm−1 (D band) and 1600 cm−1 (G band), associated with amorphous and crystalline carbon, respectively. From Table 1, it can be deduced that an increase in carbonisation temperature, regardless of the HTC solvent, resulted in enhanced degrees of graphitisation. This was evidenced by increased Lc and La values, as well as decreased d002 values when the carbonisation temperature increased from 800 to 1000 °C. Furthermore, acid-assisted HTC further enhanced the carbon ordering, leading to HCs with narrower interlayer spacings.
| XRD | Raman | |||||
|---|---|---|---|---|---|---|
| d 002 (nm) | L c (nm) | L a (nm) | n | A D1/AG | L a (nm) | |
| H2O-800 | 0.385 | 0.887 | 2.875 | 2.305 | 8.053 ± 0.037 | 2.387 ± 0.011 |
| H2O-1000 | 0.381 | 0.874 | 3.822 | 2.294 | 5.689 ± 0.225 | 3.379 ± 0.135 |
| HNO3-800 | 0.372 | 0.948 | 3.379 | 2.545 | 5.111 ± 0.139 | 3.761 ± 0.101 |
| HNO3-1000 | 0.373 | 0.939 | 3.694 | 2.519 | 4.526 ± 0.066 | 4.248 ± 0.062 |
| HCl-800 | 0.374 | 0.922 | 3.334 | 2.465 | 5.436 ± 0.410 | 3.537 ± 0.256 |
| HCl-1000 | 0.373 | 1.011 | 3.784 | 2.711 | 4.763 ± 0.248 | 4.036 ± 0.215 |
| H2SO4-800 | 0.382 | 0.866 | 3.348 | 2.270 | 5.840 ± 0.159 | 3.292 ± 0.088 |
| H2SO4-1000 | 0.376 | 0.959 | 3.493 | 2.553 | 5.119 ± 0.296 | 3.755 ± 0.217 |
| H3PO4-800 | 0.378 | 0.888 | 3.495 | 2.350 | 4.620 ± 0.140 | 4.161 ± 0.124 |
| H3PO4-1000 | 0.372 | 0.902 | 4.122 | 2.428 | 4.710 ± 0.230 | 4.081 ± 0.209 |
Table 2 reports the elemental composition of the HCs from both EDX and XPS analyses. Only silicon was detected as an impurity, indicating an effective removal of other inorganic elements present in the raw biomass (e.g., Ca, K, and Mg), which may worsen the reversible storage capacity of Na ions.67 Catalytic HTC increased surface oxygen-containing functional groups due to the promotion of acid hydrolysis and the release of oxygenated groups (e.g., COOH and OH). The oxidising nature of some of the acids may also promote this phenomenon. With the increase in annealing temperature, except in the case of the HNO3-x samples, a decrease in the content of oxygenated groups was observed, as expected with the increased ordering of the material and the related loss of surface functional groups. Moreover, using HNO3, H2SO4, and H3PO4 resulted in the introduction of N, S, and P heteroatoms, respectively. Those functional groups could improve the electrochemical properties of the resulting HC due to the formation of additional carbon vacancy defects, improved electronic conductivity, and even the incorporation of redox-active covalent bonds.68
| EDX | XPS | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C | O | P | C 1s | O 1s | Si 2p | N 1s | S 2p | P 2p | |
| H2O-800 | 94.3 | 5.70 | ND | 93.5 | 4.06 | 1.47 | 0.97 | ND | ND |
| H2O-1000 | 95.5 | 4.52 | ND | 94.6 | 3.38 | 1.02 | 0.98 | ND | ND |
| HNO3-800 | 92.4 | 7.65 | ND | 87.0 | 7.39 | 2.89 | 2.72 | ND | ND |
| HNO3-1000 | 89.6 | 10.42 | ND | 87.7 | 7.46 | 3.04 | 1.85 | ND | ND |
| HCl-800 | 90.1 | 9.93 | ND | 89.7 | 7.70 | 2.36 | 0.21 | ND | ND |
| HCl-1000 | 95.7 | 4.31 | ND | 93.1 | 4.90 | 1.95 | 0.10 | ND | ND |
| H2SO4-800 | 95.0 | 4.98 | ND | 90.4 | 6.09 | 2.54 | 0.85 | 0.16 | ND |
| H2SO4-1000 | 95.4 | 4.65 | ND | 91.1 | 5.85 | 2.37 | 0.41 | 0.23 | ND |
| H3PO4-800 | 91.0 | 7.00 | 2.00 | 87.6 | 8.00 | 2.86 | 0.81 | ND | 0.70 |
| H3PO4-1000 | 91.2 | 6.75 | 2.06 | 88.6 | 7.72 | 2.67 | 0.42 | ND | 0.60 |
The high-resolution spectra of C 1s region were deconvoluted into four peaks: aromatic sp2 carbons with C–C bonds (281.6–281.9 eV), sp3 carbons and defective carbon structures with C–C bonds (282.1–282.6 eV), defective carbons and C–O groups (283.3–283.7 eV), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (286.1–287.3 eV). Similarly, the high-resolution spectra of the O 1 s region showed three peaks corresponding to CO (529.3–529.6 eV), C–O–C/COOH (529.7–530.0 eV), and C–O(H) bonds (532.35–535.1 eV). The high-resolution scan of the N 1s region was also deconvoluted into N6 (pyridinic) (394.8–396.1 eV), N5 (pyrrolic) (397.5–398.5 eV), QN (quaternary) (400.6–401.2 eV), and N–O bonds (402.3–405.2 eV). For the H2SO4-x and H3PO4-x carbons, the S 2p and P 2p regions were also examined, revealing peaks corresponding to S–C and S–O bonds, and P–C and P–O bonds, respectively. Results from deconvolution are summarised in Tables 3 and 4 (see also Fig. S18–S27† for deconvoluted XPS spectra).
O groups (286.1–287.3 eV). Similarly, the high-resolution spectra of the O 1 s region showed three peaks corresponding to CO (529.3–529.6 eV), C–O–C/COOH (529.7–530.0 eV), and C–O(H) bonds (532.35–535.1 eV). The high-resolution scan of the N 1s region was also deconvoluted into N6 (pyridinic) (394.8–396.1 eV), N5 (pyrrolic) (397.5–398.5 eV), QN (quaternary) (400.6–401.2 eV), and N–O bonds (402.3–405.2 eV). For the H2SO4-x and H3PO4-x carbons, the S 2p and P 2p regions were also examined, revealing peaks corresponding to S–C and S–O bonds, and P–C and P–O bonds, respectively. Results from deconvolution are summarised in Tables 3 and 4 (see also Fig. S18–S27† for deconvoluted XPS spectra).
| C 1s | O 1s | ||||||
|---|---|---|---|---|---|---|---|
| sp2C | sp3C | C–O | CO |
CO |
C–O–C/COOH | C–O(H) | |
| H2O-800 | 48.6 | 21.4 | 15.6 | 14.4 | 50.3 | 45.7 | 3.97 |
| H2O-1000 | 50.0 | 20.9 | 12.9 | 16.2 | 54.8 | 39.9 | 5.29 |
| HNO3-800 | 56.1 | 23.4 | 9.38 | 11.2 | 48.2 | 50.6 | 1.19 |
| HNO3-1000 | 59.3 | 20.1 | 4.72 | 15.9 | 48.5 | 51.0 | 0.51 |
| HCl-800 | 54.9 | 22.5 | 8.20 | 14.4 | 19.2 | 75.7 | 5.10 |
| HCl-1000 | 55.0 | 18.6 | 15.0 | 11.4 | 23.1 | 73.1 | 3.85 |
| H2SO4-800 | 58.2 | 21.7 | 9.41 | 10.7 | 51.6 | 45.2 | 3.27 |
| H2SO4-1000 | 60.0 | 19.8 | 7.96 | 12.3 | 52.8 | 45.5 | 1.78 |
| H3PO4-800 | 61.1 | 24.8 | 7.27 | 6.90 | 47.3 | 49.4 | 3.31 |
| H3PO4-1000 | 63.8 | 13.0 | 11.4 | 11.7 | 47.4 | 50.6 | 2.05 |
| N 1s | S 2p | P 2p | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N6 | N5 | QN | N–O | S 2p3/2 (S–C) | S 2p1/2 (S–C) | S 2p3/2 oxides | S 2p1/2 oxides | P–C | P oxides | |
| H2O-800 | 15.2 | 60.1 | 10.9 | 13.9 | ND | ND | ND | ND | ND | ND |
| H2O-1000 | 14.2 | 65.7 | 13.4 | 8.61 | ND | ND | ND | ND | ND | ND |
| HNO3-800 | 24.4 | 49.8 | 23.1 | 2.80 | ND | ND | ND | ND | ND | ND |
| HNO3-1000 | 0.00 | 25.6 | 50.9 | 23.5 | ND | ND | ND | ND | ND | ND |
| HCl-800 | 13.2 | 37.4 | 28.9 | 20.5 | ND | ND | ND | ND | ND | ND |
| HCl-1000 | 32.8 | 29.3 | 37.9 | 0.00 | ND | ND | ND | ND | ND | ND |
| H2SO4-800 | 13.1 | 51.5 | 19.0 | 16.5 | 36.9 | 27.8 | 13.5 | 21.8 | ND | ND |
| H2SO4-1000 | 7.15 | 71.8 | 21.1 | 0.00 | 42.1 | 25.3 | 22.3 | 10.3 | ND | ND |
| H3PO4-800 | 5.63 | 15.3 | 65.2 | 13.9 | ND | ND | ND | ND | 90.6 | 9.45 |
| H3PO4-1000 | 13.4 | 41.9 | 38.3 | 6.39 | ND | ND | ND | ND | 91.0 | 8.97 |
Compared with the water-based HTC, all the carbons produced in acid media contain a more significant proportion of sp2-bonded C atoms and fewer sp3-bonded C atoms, indicating a substantial reduction in defects and an increase in structural order. This observation aligns well with the findings obtained from XRD, Raman spectroscopy, and HR-TEM images, which indicated a more significant development of the graphene lattice and the emergence of full-ordered regions facilitated by acid catalysis. The presence of acids in the reaction medium also triggers an increase in C–O–C and COOH oxygenated groups due to enhanced hydrolysis, release of free radicals, and potential oxidation and condensation reactions.
Regarding the doping phenomena induced by acid treatments, the increased presence of S–C bonds, as detected in the S 2p region for H2SO4-x carbons, is expected to enhance their electrochemical performance. This is attributed to the reversible redox reactions between Na+ and the covalent C–Sx–C bonds (x = 1–2), which can provide additional active sites for sodium storage. Furthermore, sulphur doping introduces structural defects and promotes a lower diffusion barrier and better Na+ mobility between graphene layers.69,70 For the phosphorus doping observed in H3PO4-x carbons, the predominance of P–C bonds over phosphorus oxide species is particularly advantageous. Phosphorus atoms integrated into the carbon matrix disrupt the sp2 carbon network, generating additional defects that can facilitate the reversible binding of Na+ ions. In contrast, phosphorus oxide groups are known to promote irreversible reactions, potentially reducing the overall efficiency of sodium-ion intercalation.71 Finally, nitrogen doping via HNO3 treatment appears to favour the formation of quaternary nitrogen and nitrogen oxides, while pyridinic and pyrrolic nitrogen species are present in lower quantities. Pyridinic and pyrrolic nitrogen are particularly desirable for enhancing sodium storage due to their electron-deficient nature, which can improve the reversible adsorption of Na+ ions on the carbon surface. However, the predominance of quaternary nitrogen and oxides suggests a potential trade-off between defect generation and the availability of these electrochemically active nitrogen species.68
3.2 Electrochemical performance
After annealing at 1000 °C, the reversible charge capacities obtained for H2O-1000, HNO3-1000, HCl-1000, H2SO4-1000, and H3PO4-1000 carbons at 0.1 A g−1 were 246 mAh g−1 (ICE: 71%), 219 mAh g−1 (ICE: 64%), 167 mAh g−1 (ICE: 39%), 232 mAh g−1 (ICE: 53%), and 194 mAh g−1 (ICE: 38%), respectively. Galvanostatic profiles showed an increase in the plateau located at potentials below 0.1 V (see Fig. S28 and S29†) when the HCs were annealed at 1000 °C. This increase in capacity could be attributed to greater Na ion intercalation resulting from improved structural order, while maintaining d002 values above 0.37 nm (see Table 1). Increasing the closed pore volume by raising the temperature also boosts the plateau capacity and enhances the ICE and volume energy density. However, for the HCl-1000 sample, the trend of capacity improvement with temperature was not followed, and the ICE remained relatively low. CV curves of HCl-1000 (Fig. S30†) revealed the high irreversibility during the first sodiation in the voltage range of 0.1–1 V, which could be associated with the pronounced degradation of the ester-based electrolyte within the still high mesoporosity of the electrode (0.212 cm3 g−1). The CV curves for the HCl-800 carbon did not exhibit low-voltage peaks. Since the carbon precursor is pyrolyzed at a relatively low temperature, the resulting carbon matrix in some carbonaceous materials generates a highly disordered structure with an interlayer distance larger than 0.40 nm. This phenomenon leads to “pseudo-adsorption” behaviour and a sloping voltage profile.72 Regarding the low ICE values also observed for the H3PO4-1000 carbon, we hypothesise that this could be assigned to irreversible or partially irreversible side reactions, the occurrence of which could explain the broader cathodic peak observed below 0.1 V, similar to that observed in the reaction involving the formation of Na3P.73
Fig. 5 shows the results from the rate capability tests for the HC-based anodes (see also Tables S3 and S4† for detailed results). Among the carbons produced at 800 °C, HCl-800 exhibited the best rate performance, with reversible capacities of 195, 146, 126, and 102 mAh g−1 when cycled at 0.1, 0.5, 1, and 2 A g−1, respectively. On the other hand, H2SO4-1000 showed the best results among carbons annealed at 1000 °C, with reversible capacities of 232, 173, 126, and 92 mAh g−1 at 0.1, 0.5, 1, and 2 A g−1. This HC exhibited abundant turbostratic domains with elongated graphene-like carbon layers, however, unlike the rest of acid-catalysed carbons annealed at 1000 °C, it does not display fully developed graphitic zones (see Fig. 4). Since sodium cannot intercalate into the graphitic interlayer spaces (d002 < 0.37 nm), the lower presence of graphene-like carbon layers in H2SO4-1000 guarantee better performances. Additionally, the presence of C–S–C covalent bonds could enable the reversible absorption of sodium, a fast surface redox process less hindered than intercalation at high current densities. The poorer performance of HNO3-1000, HCl-1000, and H3PO4-1000 carbons at 0.1 A g−1 compared to that of H2O-1000 could also be justified by an excess of graphitization, meanwhile, the improvement of HCl-1000 and H3PO4-1000 carbons at higher current densities can be ascribed to greater mobility of Na+ through the developed hierarchical porosity.
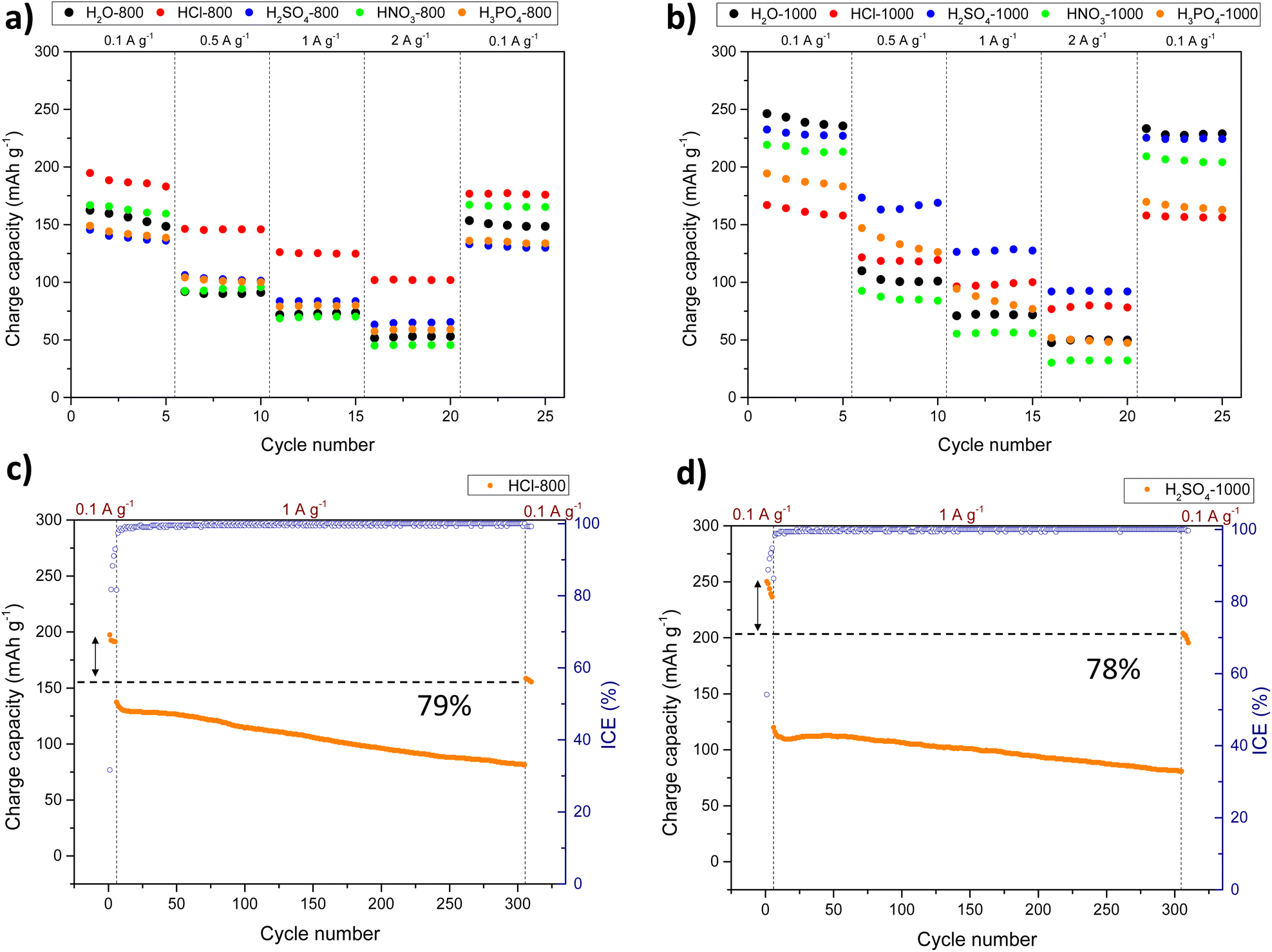 | ||
| Fig. 5 (a and b) Specific charge capacities vs. cycle number at various current densities ranging from 0.1 to 2 A g−1 and (c and d) cycling performance of HCl-800 and H2SO4-1000 for 300 cycles at 1 A g−1 in NaTFSI/EC:DMC. | ||
The best-performing materials, viz. HCl-800 and H2SO4-1000, were subjected to 300 cycles at 1 A g−1 to get insights into the stability upon prolonged cycling. As shown in Fig. 5c and d, an acceptable capacity retention of 79% and 78% was observed for HCl-800 and H2SO4-1000, respectively. The apparent diffusion coefficients (DNa+) for samples annealed at 1000 °C are shown in Fig. S31† to get insight into the different sodium storage mechanisms. As expected, a decrease in ion diffusivity was observed during sodiation when the voltage decreased from the sloping region to the plateau area. Conforming to the three-stage model74 and consistently with our previous studies, the DNa+ profile was divided into three regions: (1) initial stage with high DNa+ values above 0.1 V attributed to the surface adsorption of Na ions, (2) decrease in DNa+ values at voltages below 0.1 V corresponding to intercalation processes, and (3) a last stage linked to the filling of micropores and ultramicropores. During sodiation, the DNa+ for all anodes steadily declined until the voltage reached ∼0.1 V. Upon reaching the cutoff voltage, an increase in the DNa+ value can be observed. During the desodiation process, the diffusion coefficient initially decreases, then increases, and subsequently decreases again before reaching the cutoff voltage. Due to enhanced structural properties, the value for DNa+ is higher and more stable for the H2SO4-1000 electrode upon sodiation and desodiation. For the H3PO4-1000 carbon, a fourth process near 0.01 V with very slow kinetics was observed in both sodiation and desodiation profiles. As mentioned previously, the H3PO4-1000 CV profile likely reflects some kinetically unfavourable phenomenon related to phosphorus presence on the surface.
| NaTFSI/EC:DMC | NaPF6/DGM | |||
|---|---|---|---|---|
| 1st charge capacity (mAh g−1) | ICE (%) | 1st charge capacity (mAh g−1) | ICE (%) | |
| H2O-1000 | 246 | 71 | 215 | 69 |
| HNO3-1000 | 219 | 64 | 229 | 64 |
| HCl-1000 | 167 | 39 | 283 | 77 |
| H2SO4-1000 | 232 | 53 | 228 | 72 |
| H3PO4-1000 | 194 | 38 | 281 | 68 |
Fig. 6 shows the rate capability of HCs when the NaPF6/DGM electrolyte was used as the electrolyte (see Table S5† for capacity results). The outstanding performance of carbons, particularly HCl-1000 and H3PO4-1000, can be attributed to their highly developed hierarchical porosity, facilitating efficient ion diffusion throughout the carbon bulk from the surface to the storage active sites: interlayers of disordered and graphitic domains (via intercalation and co-intercalation, respectively) and both open and closed pores (via pore filling at the low voltage plateaus, can be clearly observed in Fig. S32†). Consequently, these two HCs demonstrated the capability to reversibly store ca. 200 mAh g−1 at a current density of 2 A g−1, which are remarkable results according to the literature,76 especially considering the corresponding high ICE values of 68% and 77%, respectively. On the other hand, HNO3-1000 exhibited a slight improvement in capacity at low current densities, possibly due to a higher extent of co-intercalation. However, starting from 1 A g−1, it yielded poorer results than those obtained through hydrothermal carbonisation in water, which was attributed to lower porous development and hindered ion transport.
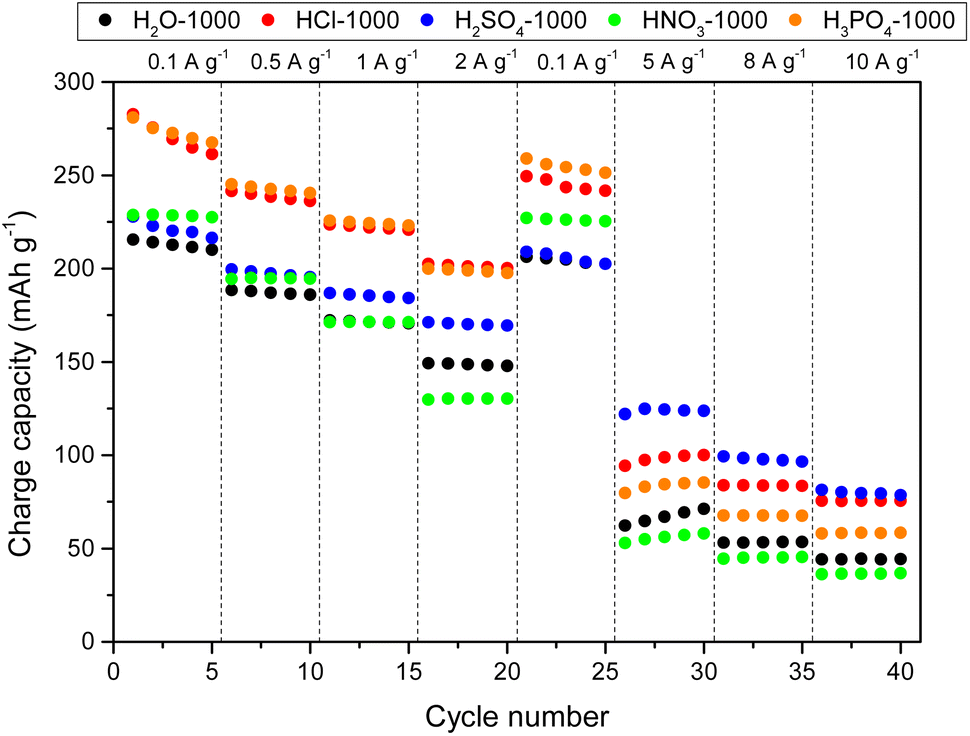 | ||
| Fig. 6 Specific charge capacities vs. cycle number at various current densities ranging from 0.1 to 10 A g−1 in NaPF6/DGM. | ||
Although the H2SO4-1000 shows inferior capacity at low current densities due to the limited availability of graphitic domains, its high porosity, and storage involving surface sulphur groups lead to better rate capability, with noteworthy capacities of 122 mAh g−1 and 81 mAh g−1 at 5 and 10 A g−1, respectively. The long-term cycling of carbons in the ether-type electrolyte was evaluated after 250 cycles at 1 A g−1 (Fig. S34†), revealing good stability compared to that obtained with the ester-type electrolyte. H2O-1000, HNO3-1000, HCl-1000, H2SO4-1000, and H3PO4-1000 carbons exhibited capacity retentions of 90, 93, 86, 87, and 87%, respectively.
4. Conclusions
This study has demonstrated how acid-assisted HTC of vine shoots, followed by an additional annealing step, yielded a wide range of HC materials with attractive electrochemical features as Na-ion battery anodes. Depending on the acid (HCl, H2SO4, HNO3, H3PO4) used in the hydrothermal process, the reactions were favoured to different extents, particularly the hydrolysis. This process led to a series of carbonaceous materials exhibiting different levels of pore development, amorphous/ordered regions, 2D structures, and heteroatom incorporation. The simple, easily scalable, tuneable, and low-cost preparation of carbons may represent an interesting approach to converting biomass waste into high-value-added active materials for electrochemical energy storage applications.In addition, we demonstrated that compared to the ester-type electrolyte, the use of the diglyme-based electrolyte significantly enhanced the electrochemical performance of the laboratory-scale cells. This electrolyte led to a substantial increase in the ICE, addressing a key challenge related to micro- and mesopore-rich carbons, which are required to improve the transport kinetics and rate capability of HC/based anodes. Additionally, thanks to the solvent co-intercalation mechanism, the NaPF6/DGM electrolyte notably improved the anode capacity by facilitating sodium storage within the highly graphitic regions. Thus, the chosen electrolyte accounts for determining the HC properties to be promoted. While the use of ester-based electrolytes should restrict mesoporosity development and maximise turbostratic domain presence, ether-based electrolytes allow the development of more ordered and porous carbons, leading to enhanced transport kinetics, sodium storage capacity, and electrode stability. Within this framework, HCl-1000 and H3PO4-1000 carbons in combination with the diglyme-based electrolyte proved to be the most effective anode materials for SIBs, exhibiting excellent compatibility and outstanding electrochemical properties.
This work highlights the importance of achieving a mixed structure in hard carbons of amorphous and crystalline zones for sodium storage in ether-based electrolytes. Crystalline phases appear to provide higher storage capacity, primarily due to the extensive phenomenon of co-intercalation, where sodium ions intercalate between the graphitic layers. Simultaneously, the coexistence of highly porous amorphous regions plays a critical role by enabling enhanced sodium-ion diffusivity, which significantly improves the high-rate capability of the material. This is particularly advantageous, as overly compact and highly ordered hard carbons often exhibit considerable diffusion resistance. Furthermore, disordered carbon structures offer additional storage sites for reversible Na-ion adsorption, contributing significantly to capacity, particularly at higher potentials. This makes a mixed-phase architecture exceptionally well-suited for optimising sodium-ion battery performance. Overall, we can conclude that the acid-catalysed HTC process offers a promising pathway for customising the production of HC-based electrodes, fostering a sustainable approach that can transform biomass residues into these added-value materials for energy storage/production in a circular economy approach.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Darío Alvira: conceptualization, methodology, investigation, writing – original draft. Daniel Antorán: investigation. Hamideh Darjazi: investigation, methodology, review & editing. Giuseppe Antonio Elia: conceptualization, supervision, review & editing. Claudio Gerbaldi: conceptualization, supervision, review & editing. Víctor Sebastián: conceptualization, investigation, supervision. Joan J. Manyà: conceptualization, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the research project PID2022-137218OB-I00, funded by MCIN/AEI/10.13039/501100011033 and “ERDF A way of making Europe”. VS acknowledge funding from project PID2021-127847OB-I00 MCIN/AEI/10.130 and PDC2022-133866-I00 MCIN/AEI/10.13039/501100011033. The authors also acknowledge the funding from the Aragon Government (Ref. T22_23R). DA also acknowledges the funding from the Regional Government of Aragon (Spain) with a grant for postgraduate research contracts (2019–2023). LMA-ELECMI and NANBIOSIS ICTs are gratefully acknowledged. This study was also carried out under the MOST – Sustainable Mobility Center and received funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA – PNRR eMission 4, Component 2, Investment 1.4 and D.D. 1033 17/06/2022 of the Ministero dell’Università e della Ricerca (MUR), CN00000023). This manuscript reflects only the authors' views and opinions, neither the European Union nor the European Commission can be considered responsible for them. Graphical abstract and Fig. 1 were partly generated by the Google Gemini tool.References
- G. Nazir, A. Rehman, J.-H. Lee, C.-H. Kim, J. Gautam, K. Heo, S. Hussain, M. Ikram, A. A. AlObaid, S.-Y. Lee and S.-J. Park, Nano-Micro Lett., 2024, 16, 138 Search PubMed.
- X. He, N. Iqbal, U. Ghani and T. Li, J. Alloys Compd., 2024, 981, 173680 Search PubMed.
- R. Yokoi, R. Kataoka, T. Masese, V. Bach, M. Finkbeiner, M. Weil, M. Baumann and M. Motoshita, Resour., Conserv. Recycl., 2024, 204, 107526 Search PubMed.
- S. Zhang, R. Cao, X. Pu, A. Zhao, W. Chen, C. Song, Y. Fang and Y. Cao, J. Energy Chem., 2024, 92, 162–175 Search PubMed.
- H. Bai, X. Zhu, H. Ao, G. He, H. Xiao and Y. Chen, J. Energy Chem., 2024, 90, 518–539 Search PubMed.
- J. Wang, L. Xi, C. Peng, X. Song, X. Wan, L. Sun, M. Liu and J. Liu, Adv. Eng. Mater., 2024, 26, 2302063 Search PubMed.
- H. Zheng, J. Zeng, X. Wan, X. Song, C. Peng, J. Wang, L. Sun, H. Wang, M. Zhu and J. Liu, Mater. Futures, 2024, 3, 032102 Search PubMed.
- C. Matei Ghimbeu, A. Beda, B. Réty, H. El Marouazi, A. Vizintin, B. Tratnik, L. Simonin, J. Michel, J. Abou-Rjeily and R. Dominko, Adv. Energy Mater., 2024, 14, 2303833 Search PubMed.
- M. Bartoli, A. Piovano, G. A. Elia, G. Meligrana, R. Pedraza, N. Pianta, C. Tealdi, G. Pagot, E. Negro, C. Triolo, L. V. Gomez, N. Comisso, A. Tagliaferro, S. Santangelo, E. Quartarone, V. Di Noto, P. Mustarelli, R. Ruffo and C. Gerbaldi, Renew. Sustainable Energy Rev., 2024, 194, 114304 Search PubMed.
- J. Petrović, M. Ercegović, M. Simić, M. Koprivica, J. Dimitrijević, A. Jovanović and J. Janković Pantić, Processes, 2024, 12, 207 Search PubMed.
- S. A. Nicolae, H. Au, P. Modugno, H. Luo, A. E. Szego, M. Qiao, L. Li, W. Yin, H. J. Heeres, N. Berge and M. M. Titirici, Green Chem., 2020, 22, 4747–4800 Search PubMed.
- J. A. Libra, K. S. Ro, C. Kammann, A. Funke, N. D. Berge, Y. Neubauer, M. Titirici, C. Fühner, O. Bens, J. Kern and K. Emmerich, Biofuels, 2011, 2, 71–106 Search PubMed.
- H. S. Kambo and A. Dutta, Renew. Sustainable Energy Rev., 2015, 45, 359–378 Search PubMed.
- M. P. Maniscalco, M. Volpe and A. Messineo, Energies, 2020, 13, 4098 Search PubMed.
- M. N. Mahmood Al-Nuaimy, N. Azizi, Y. Nural and E. Yabalak, Environ. Res., 2024, 250, 117923 Search PubMed.
- K. I. John and M. O. Omorogie, Green Chem. Lett. Rev., 2022, 15, 162–186 Search PubMed.
- P. E. Savage, Chem. Rev., 1999, 99, 603–622 Search PubMed.
- M. Heidari, A. Dutta, B. Acharya and S. Mahmud, J. Energy Inst., 2019, 92, 1779–1799 Search PubMed.
- M. Sevilla and A. B. Fuertes, Carbon N. Y., 2009, 47, 2281–2289 Search PubMed.
- A. Sanchez-Sanchez, F. L. Braghiroli, M. T. Izquierdo, J. Parmentier, A. Celzard and V. Fierro, Ind. Crops Prod., 2020, 154, 112564 Search PubMed.
- L. Fan, Z. Shi, Q. Ren, L. Yan, F. Zhang and L. Fan, Green Energy Environ., 2021, 6, 220–228 Search PubMed.
- O. Bobleter, Prog. Polym. Sci., 1994, 19, 797–841 Search PubMed.
- X. Zhang, T. Cao, G. Zhang, Q. Liu, G. Kong, K. Wang, Y. Jiang, X. Zhang and L. Han, J. Mater. Chem. A, 2024, 12, 4996–5039 Search PubMed.
- A. M. Borrero-López, E. Masson, A. Celzard and V. Fierro, Ind. Crops Prod., 2018, 124, 919–930 Search PubMed.
- Z. Fang, T. Sato, R. L. Smith, H. Inomata, K. Arai and J. A. Kozinski, Bioresour. Technol., 2008, 99, 3424–3430 Search PubMed.
- H. Dang, R. Xu, J. Zhang, M. Wang and K. Xu, Chem. Eng. J., 2023, 469, 143980 Search PubMed.
- T. Wang, Y. Zhai, Y. Zhu, C. Li and G. Zeng, Renew. Sustainable Energy Rev., 2018, 90, 223–247 Search PubMed.
- P. Zheng, T. Liu, X. Yuan, L. Zhang, Y. Liu, J. Huang and S. Guo, Sci. Rep., 2016, 6, 26246 Search PubMed.
- F. Veltri, F. Alessandro, A. Scarcello, A. Beneduci, M. Arias Polanco, D. Cid Perez, C. Vacacela Gomez, A. Tavolaro, G. Giordano and L. S. Caputi, Nanomaterials, 2020, 10, 655 Search PubMed.
- F. Li, A. R. Zimmerman, X. Hu, Z. Yu, J. Huang and B. Gao, Chemosphere, 2020, 254, 126866 Search PubMed.
- Y. Li, F. M. Hagos, R. Chen, H. Qian, C. Mo, J. Di, X. Gai, R. Yang, G. Pan and S. Shan, Bioresour. Bioprocess., 2021, 8, 99 Search PubMed.
- H. Fu, Z. Xu, R. Li, W. Guan, K. Yao, J. Huang, J. Yang and X. Shen, ACS Sustainable Chem. Eng., 2018, 6, 14751–14758 Search PubMed.
- S. D. Xu, Y. Zhao, S. Liu, X. Ren, L. Chen, W. Shi, X. Wang and D. Zhang, J. Mater. Sci., 2018, 53, 12334–12351 Search PubMed.
- X. Zou, C. Dong, Y. Jin, D. Wang, L. Li, S. Wu, Z. Xu, Y. Chen, Z. Li and H. Yang, Colloids Surf., A, 2023, 672, 131715 Search PubMed.
- C. Liu, K. Wang, Y. Du, Y. Shan, P. Duan and N. Ramzan, Polymers, 2023, 15, 4478 Search PubMed.
- E. García-Bordejé, E. Pires and J. M. Fraile, Carbon N. Y., 2017, 123, 421–432 Search PubMed.
- A. Funke and F. Ziegler, Biofuels, Bioprod. Biorefin., 2010, 4, 160–177 Search PubMed.
- R. H. Fitri Faradilla, L. Lucia and M. Hakovirta, Environ. Nanotechnol., Monit. Manag., 2021, 16, 100571 Search PubMed.
- Z. Gruber, A. J. Toth, A. Menyhárd, P. Mizsey, M. Owsianiak and D. Fozer, Bioresour. Technol., 2022, 365, 128071 Search PubMed.
- T. Wang, Y. Zhai, Y. Zhu, C. Peng, B. Xu, T. Wang, C. Li and G. Zeng, Energy Fuels, 2017, 31, 12200–12208 Search PubMed.
- D. Liu, Y. Tang, J. Li, Z. Hao, J. Zhu, J. Wei, C. Liu, L. Dong, B. Jia and G. Chen, J. Environ. Chem. Eng., 2021, 9, 105972 Search PubMed.
- B. Ibrahim, M. Schlegel and N. Kanswohl, Chem. Ing. Tech., 2018, 90, 340–347 Search PubMed.
- F. M. Jais, S. Ibrahim, C. Y. Chee and Z. Ismail, Sustainable Chem. Pharm., 2021, 24, 100541 Search PubMed.
- M. Wilk, M. Śliz, K. Czerwińska and M. Śledź, J. Environ. Manage., 2023, 345, 118820 Search PubMed.
- L. T. Tran, H. T. Hoang, M. Q. Nguyen, N. D. Dao and T. H. T. Vu, BioEnergy Res., 2022, 15, 1996–2005 Search PubMed.
- B. Ercan, Y. O. Ajagbe, S. Ucar, K. Tekin and S. Karagoz, Wood Sci. Technol., 2023, 57, 945–964 Search PubMed.
- L. Cao, W. Hui, Z. Xu, J. Huang, P. Zheng, J. Li and Q. Sun, J. Alloys Compd., 2017, 695, 632–637 Search PubMed.
- C. C. Wang and W. L. Su, Surf. Coat. Technol., 2021, 415, 127125 Search PubMed.
- D. Alvira, D. Antorán, H. Darjazi, G. A. Elia, V. Sebastian and J. J. Manyà, J. Power Sources, 2024, 614, 235043 Search PubMed.
- X. Yin, Z. Wang, Y. Liu, Z. Lu, H. Long, T. Liu, J. Zhang and Y. Zhao, Nano Res., 2023, 16, 10922–10930 Search PubMed.
- F. Zeng, L. Xing, W. Zhang, Z. Xie, M. Liu, X. Lin, G. Tang, C. Mo and W. Li, J. Energy Chem., 2023, 79, 459–467 Search PubMed.
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Chem. Soc. Rev., 2022, 51, 4484–4536 Search PubMed.
- D. Alvira, D. Antorán, M. Vidal, V. Sebastian and J. J. Manyà, Batteries Supercaps, 2023, 6, e202300233 Search PubMed.
- D. Alvira, D. Antorán and J. J. Manyà, J. Energy Chem., 2022, 75, 457–477 Search PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon N. Y., 2005, 43, 1731–1742 Search PubMed.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, A. Jorio, L. N. Coelho, R. Magalhães-Paniago and M. A. Pimenta, Appl. Phys. Lett., 2006, 88, 163106 Search PubMed.
- K. Fischer and H.-P. Bipp, Bioresour. Technol., 2005, 96, 831–842 Search PubMed.
- S. Zhang, K. Sheng, W. Yan, J. Liu, E. Shuang, M. Yang and X. Zhang, Chemosphere, 2021, 263, 128093 Search PubMed.
- F. Quesada-Plata, R. Ruiz-Rosas, E. Morallón and D. Cazorla-Amorós, ChemPlusChem, 2016, 81, 1349–1359 Search PubMed.
- D. D. Perrin, Pure Appl. Chem., 1969, 20, 133–236 Search PubMed.
- N. Sun, Z. Guan, Y. Liu, Y. Cao, Q. Zhu, H. Liu, Z. Wang, P. Zhang and B. Xu, Adv. Energy Mater., 2019, 9, 1901351 Search PubMed.
- D. Chen, W. Zhang, K. Luo, Y. Song, Y. Zhong, Y. Liu, G. Wang, B. Zhong, Z. Wu and X. Guo, Energy Environ. Sci., 2021, 14, 2244–2262 Search PubMed.
- Z. Ding, F. Li, J. Wen, X. Wang and R. Sun, Green Chem., 2018, 20, 1383–1390 Search PubMed.
- M. A. Ahmad Farid and Y. Andou, J. Clean. Prod., 2022, 380, 135090 Search PubMed.
- Z. Jinli, W. Jiao, L. Yuanyuan, N. Ning, G. Junjie, Y. Feng and L. Wei, J. Mater. Chem. A, 2015, 3, 2043–2049 Search PubMed.
- L. Yu, D. Tatsumi, S. Zuo and M. Morita, BioResources, 2015, 10, 2406–2417 Search PubMed.
- T. Zhang, J. Mao, X. Liu, M. Xuan, K. Bi, X. L. Zhang, J. Hu, J. Fan, S. Chen and G. Shao, RSC Adv., 2017, 7, 41504–41511 Search PubMed.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Adv. Energy Mater., 2020, 10, 2000927 Search PubMed.
- W. Li, M. Zhou, H. Li, K. Wang, S. Cheng and K. Jiang, Energy Environ. Sci., 2015, 8, 2916–2921 Search PubMed.
- Q. Jin, W. Li, K. Wang, P. Feng, H. Li, T. Gu, M. Zhou, W. Wang, S. Cheng and K. Jiang, J. Mater. Chem. A, 2019, 7, 10239–10245 Search PubMed.
- K. Lu, S. Lu, T. Gu, X. Zheng, K. Ke, X. Li and R. Yang, Electrochem. Commun., 2019, 103, 22–26 Search PubMed.
- X. Chen, C. Liu, Y. Fang, X. Ai, F. Zhong, H. Yang and Y. Cao, Carbon Energy, 2022, 4, 1133–1150 Search PubMed.
- S. Said, Z. Zhang, R. R. C. Shutt, H. J. Lancaster, D. J. L. Brett, C. A. Howard and T. S. Miller, ACS Nano, 2023, 17, 6220–6233 Search PubMed.
- X. Yin, Z. Lu, J. Wang, X. Feng, S. Roy, X. Liu, Y. Yang, Y. Zhao and J. Zhang, Adv. Mater., 2022, 34, 2109282 Search PubMed.
- M. Liu, L. Xing, K. Xu, H. Zhou, J. Lan, C. Wang and W. Li, Energy Storage Mater., 2020, 26, 32–39 Search PubMed.
- D. Alvira, D. Antorán and J. J. Manyà, Chem. Eng. J., 2022, 447, 137468 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07393b |
| This journal is © The Royal Society of Chemistry 2025 |