
A surface modified laser-induced graphene based flexible biosensor for multiplexed sweat analysis†
Sudipta
Choudhury
a,
Saad
Zafar
b,
Deepak
Deepak
a,
Abhishek
Panghal
a,
Bimlesh
Lochab
 b and
Susanta Sinha
Roy
*a
b and
Susanta Sinha
Roy
*a
aDepartment of Physics, School of Natural Sciences, Shiv Nadar Institution of Eminence (SNIoE), Deemed to be University, Delhi-NCR, Greater Noida, 201314, India. E-mail: susanta.roy@snu.edu.in
bMaterials Chemistry Laboratory, Department of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence, Delhi-NCR, Greater Noida, 201314, India
First published on 1st November 2024
Abstract
The growing popularity of electrochemical sensors featuring non-invasive biosensing technologies has generated significant enthusiasm for continuous monitoring of bodily fluid biomarkers, potentially aiding in the early detection of health issues in individuals. However, detection of multiple biomarkers in complex biofluids often necessitates a high-density array which creates a challenge in achieving cost-effective fabrication methods. To overcome this constraint, this work reports the fabrication of an electrochemical sensor utilizing a NiO–Ti3C2Tx MXene-modified flexible laser-induced graphene (LIG) electrode for the separate and concurrent analysis of ascorbic acid (AA), dopamine (DA), and uric acid (UA) in human sweat and also addresses the deficiencies in the existing state of the art by offering a cost-efficient and high-performance sensor that mitigates the degrading constraints of conventional LIG electrodes. Cyclic voltammetry and differential pulse voltammetry measurements reveals that the electrochemical properties of the modified electrode, attain a low detection limit and great sensitivity for the target biomarkers. The NiO–Ti3C2Tx/LIG sensor demonstrated enhanced electrocatalytic activity for the oxidation of ascorbic acid, dopamine, and uric acid, and proved useful for analysing these biomarkers in synthetic sweat samples. Under the optimized conditions, the LOD values were estimated to be 16, 1.97 and 0.78 μM for AA, DA and UA, respectively. The developed high-efficiency sensor holds significant promise for applications in flexible and wearable electronics for health monitoring.
1. Introduction
Health monitoring systems for individuals are anticipated to be crucial in the upcoming fields of personalized health and on-site diagnostic facilities.1,2 Wearable electrochemical sweat sensors provide a non-invasive sweat analysis approach that shows promise as a replacement for traditional blood sampling in the detection of various physical and chemical biomarkers.3–14 This approach enables the assessment of biomolecular states for health monitoring in point-of-care settings.15 Among various biomolecules, three major ones are ascorbic acid (AA), dopamine (DA), and uric acid (UA) found together in biological matrices.16,17 They collectively contribute significantly to numerous physiological processes within the metabolism of the human body.18 UA (2,6,8-trihydroxypurine) serves as the final product of purine metabolism in the liver of humans, which is generally found in body fluids like sweat, blood and urine.19,20 Many illnesses, including gout, hyperuricemia, leukemia, type 2 diabetes, pneumonia, Lesch–Nyhan syndrome, etc., have been linked to abnormally high UA levels.19–23 Due to these reasons, it is vital to monitor UA levels for effective disease management. In animals, the anterior pituitary lobe, leukocytes, and liver exhibit the highest concentrations of AA.24 This compound is vital in the human diet and is commonly employed as an antioxidant in various foods and beverages.25 Additionally, it is commonly used to treat conditions like scurvy, colds, cancer, and hepatic diseases.26–28 DA, a crucial neurotransmitter, plays a significant role in the mammalian central nervous system, and a decrease in its levels is linked to neurological disorders such as Parkinson's disease, Huntington's disease and schizophrenia.29–35 Therefore, it is essential to develop a fast and highly sensitive technique that is capable of simultaneously detecting AA, DA and UA. But precisely measuring each of the three signals AA, DA and UA in the body fluids is difficult due to their overlapping nature.36 Many techniques have been invented such as chemiluminescence, high performance liquid chromatography, capillary electrophoresis, colorimetry and ultraviolet-visible spectroscopy to detect these biomarkers.36,37 Due to the electroactive properties of these substances, electrochemical approaches have garnered significant interest in clinical diagnostics. This is due to their simple operation, cost-effectiveness, fast response, high sensitivity, and good selectivity.38–43 But it is challenging to measure the AA, DA, and UA concentrations individually on any bare electrode without any modifications or functionalization. As a result, several materials for electrode modification have been developed recently to address this issue. Carbon based electrodes exhibit great potential without any added enzyme due to their superior performance, cost-effectiveness and environment friendly nature.8 Among carbon-based samples, graphene-based electrodes are widely recognized for their significant potential in electrochemical sensors due to attributes such as high electron mobility, excellent electrical and thermal conductivity, mechanical flexibility, and, notably, high surface area and porosity.44–48 However, achieving large-scale, chemical-free production of porous graphene presents a considerable challenge. A recent advancement addresses this challenge with the development of a straightforward, scalable, and cost-effective technique for synthesizing three-dimensional porous few-layered graphene through a single-step laser-writing process.49,50 This synthesized graphene, commonly known as laser-induced graphene (LIG), has demonstrated promising outcomes in various applications, including electrocatalysts, sensors, energy storage, and robots.51–60 LIG substrates have emerged as highly effective electrode frameworks for wearable electrochemical sensing applications due to their rapid fabrication process and exceptional electrochemical capabilities.61–66 But the effectiveness of biosensor applications is hindered by the unstable surface conductivity of LIG.36 These disadvantages might be overcome by applying 2D active nanosheets through various interfacial interactions like ionic, covalent, hydrogen and π–π bridging onto the 3D microporous structures of LIG films.40In recent times, there has been a widespread global interest in two-dimensional (2D) MXenes, represented as Mn+1XnTx (where M stands for a transition metal, X for C and/or N, and Tx for surface functional groups such as –OH, –O, or –F).67 Since their discovery, they have attracted increased attention because of their promising features, including a high surface area and metallic conductivity (2400 S cm−1) coupled with a hydrophilic nature due to a hydroxyl or an oxygen-terminated surface.68–70 During the synthesis process, diverse functional groups, typically hydroxyl (–OH), oxygen (–O), and fluorine (–F) and/or Cl−, can be incorporated onto the MXene surface.71,72 Among all the MXene materials, the Ti3C2Tx MXene has emerged as a potential candidate for application in sensors, biosensors, energy storage and diverse fields.73–79 The functional groups of LIG substrates such as carboxyl (–COOH) and hydroxyl groups (–OH) undergo a reaction with Ti3C2Tx MXene nanosheets, resulting in the formation of a covalently crosslinked MXene–LIG composite through C–O–metal bonds.36 This hybrid framework enhances abundant surface functionalities which facilitates the detection of a wide range of biological signals.36 In recent times, several electrochemical biosensors employing Ti3C2Tx MXene nanosheets have been developed. Because of the unavoidable self-stacking behaviour of MXene nanosheets which leads to a decreased active surface area, this phenomenon results in a transition of MXene properties from metallic to semiconducting.74 Therefore, to enhance the catalytic activity and detection capability of a biosensor significantly, the Ti3C2Tx MXene can be incorporated with transition metal oxides.80–82 Numerous types of metal oxide nanoparticles (NPs) have garnered growing attention in electrochemical research. Electrochemical biosensor performance can be improved by combining the Ti3C2Tx MXene with pseudocapacitive metal oxide nanoparticles such as NiO, Co3O4, CuO, ZnO and SnO2.83,84 Among all these metal oxide materials, NiO is considered a promising substitute due to its availability, affordability, high surface area and superior electrochemical performance. Despite the extensive studies on various NPs, there is limited research on the electrochemical behavior of nickel oxide nanoparticles (NiO NPs) and their potential applications in electrochemical biosensors. Electrostatic self-assembly is considered as a straightforward and highly efficient method for making the advanced composites of the aforementioned oxide nanoparticles and MXene nanosheets. Because of its surface functional groups, an MXene is a negatively charged material and NiO nanoparticles are positively charged.85 The opposing surface charges of these two materials initiate a self-assembly process via electrostatic interaction which leads to the formation of a NiO–Ti3C2Tx MXene composite. This self-assembly synthesis that is facilitated by the electrostatic attraction between the nanosheets significantly enhances the conductivity of the composite.74 Notably, to the best of our knowledge, the application of the electrostatic self-assembly composite of NiO–Ti3C2Tx MXene for electrochemical sweat sensing has not been reported previously.
In this work, we present an environmentally friendly method for fabricating a flexible biosensor based on a NiO–Ti3C2Tx MXene modified LIG electrode. The modified sensor exhibited a high electrochemical active surface area and electrocatalytic activity. Electrochemical analyses, specifically cyclic voltammetry (CV) and differential pulse voltammetry (DPV), were utilized to explore the electrochemical responses of AA, DA and UA on the fabricated sensor. This biosensor shows the capability of simultaneously detecting these three molecules in artificial sweat. The as-fabricated flexible biosensor demonstrated superior selectivity, sensitivity, long-term stability, and a wide range of detection with a low detection limit. These properties suggest its suitability as a non-invasive wearable sensing platform for monitoring personal health. To the best of our knowledge, the combination of NiO–Ti3C2Tx MXene/LIG has not been reported for simultaneous monitoring of AA, DA and UA in human sweat. It has been successfully established that the developed sensor could serve as a compact, skin-adherable, and miniaturized device for clinical diagnostic purposes.
2. Experimental section
2.1. Materials
Uric acid (C5H4N4O3), ascorbic acid (C6H8O6), dopamine hydrochloride (C8H12ClNO2), Ti3AlC2 (98%), and NiCl2·6H2O were purchased from Sigma-Aldrich (India) and used as received without any prior treatment. Potassium phosphate monobasic (KH2PO4), potassium phosphate dibasic (K2HPO4), potassium ferricyanide (K3[Fe(CN)6]), potassium ferrocyanide (K4[Fe(CN)6]), hydrochloric acid (HCl), LiF, KOH, KCl, CaCl2, NH4Cl, lactic acid, glucose and urea were obtained from Fisher Scientific. All the aforementioned chemicals were of analytical grade and used without any purification processes. Phosphate buffer solutions (PBS; 0.1 M) with various pH values (from 5 to 8) were prepared with different amounts of KH2PO4 and K2HPO4 and adjusted with 1 M HCl or 1 M KOH solutions. Ultrapure deionized water from a Milli-Q integral water purification system was used in all aqueous solution experiments (resistivity: 18.2 MΩ cm).2.2. Instrumental
The surface morphology and elemental composition of the samples were investigated by using field emission scanning electron microscopy (FE-SEM) (JEOL JSM-7610F Plus) and energy dispersive X-ray spectroscopy (EDS) (EDAX AMETEK). X-ray diffraction (XRD) spectra of the nanocomposites were recorded using a Bruker D8-Discover with Cu-Kα radiation (λ = 1.5406 Å) using a Lynx Eye detector. Raman spectroscopy was conducted by using a micro-Raman spectrometer (STR) equipped with a 532 nm argon-ion laser source with a power of 2.5 mW and a 50× magnification objective lens. Transmission electron microscopy (TEM) images, high-resolution transmission electron microscopy (HRTEM) images, and selected area electron diffraction (SAED) patterns of the samples were captured at 200 kV on a JEOL JEM-F200 TEM. X-ray photoelectron spectroscopy (XPS) spectra were acquired with a Thermo Fisher ESCALAB XI instrument. A monochromated Al (operating at a voltage of 15 kV with a current of 15 mA and a power of 225 W) was used as the excitation source. A flood gun was used for charge compensation purposes. All the electrochemical measurements were carried out using an Autolab potentiostat/galvanostat 302N instrument (Metrohm B.V., Utrecht, The Netherlands) controlled by Nova (version 1.10). For the CV study, a standard three-electrode setup was used, which included a working electrode, a Pt wire as the counter electrode, and Ag/AgCl (with 3 M KCl electrolyte) as the reference electrode. A redox-active electrolyte of 5 mmol L−1 [Fe(CN)6]3−/4− in 0.1 M KCl was used to study ECSA and for electrochemical impedance spectroscopy (EIS). EIS measurements were executed in FRA potential scan mode with the same electrode–electrolyte arrangement. A sinusoidal signal having a root-mean-square value of 10 mV was applied as a perturbation within the frequency range of 0.1 Hz to 100 kHz. The as-obtained Nyquist plots were fitted using the NOVA (version 1.10) software.2.3. Fabrication of the flexible laser-induced graphene electrode
Laser-induced graphene was synthesized using an MV laser system over a 125 μm thick as-received Kapton polyimide (Cole-Parmer, an Antylia Scientific company, India) sheet. The laser treatment was carried out under ambient atmospheric conditions using an MV laser, India (CO2 laser). For the conversion of polyimide into LIG, the laser power was kept constant at 6 W and the lasing speed was controlled at 240 mm s−1. The standard electrode dimension was 0.8 × 0.8 cm2. The electrode, along with the connection lines and pads, was fabricated by laser patterning of the polyimide substrate using the same protocol. After fabrication, we performed the ozone treatment of LIG in a Novascan PSD Pro Series digital UV-Ozone cleaner where the ozone was produced by ultraviolet irradiation (254 nm UV light with an intensity of 20 mW cm−2) under ambient conditions. The ozonization of LIG was carried out for 30 min at room temperature. Ozone molecules were physisorbed on the LIG surface as a result of this treatment. As a result, the hydrophobic surface of LIG became hydrophilic. The hydrophilic surface of ozone treated LIG allows easier penetration of any solvent into the porous LIG film and thus creates a strong attachment.2.4. Synthesis of NiO nanoparticles
All reagents in this work used to prepare the NiO nanoparticles were of analytical grade and used without any further purification. In a typical hydrothermal procedure,86 0.475 g nickel chloride hexahydrate (NiCl2·6H2O) was added into 20 mL mixed solution composed of 10 mL of pure water and 10 mL of ethanol. Then, 0.2 g polyvinylpyrrolidone (PVP) and 10 mL of ethylene glycol (EG) were added into the mixture under stirring. The pH value of the solution was adjusted to about 10 by dropping ammonia solution (NH3·H2O). After that, the mixture was magnetically stirred for 5 min to form a homogeneous solution and then poured into a 50 mL Teflon-lined stainless autoclave which was kept at 140 °C for 10 h. After the autoclave cooled to room temperature, the green sample was harvested by centrifugation and washing with pure water and ethanol several times. After drying at 60 °C for 10 h, the expected product was obtained by calcination at 400 °C for 2 h.2.5. Synthesis of Ti3C2Tx MXene nanosheets
The synthesis of MXene follows a top-down approach in which the parent precursor is selectively etched to form the desired product, MXene.76,77 The parent precursor Ti3AlC2 (MAX phase) was selectively etched by using the minimally intensive layer delamination (MILD) method. Instead of using highly corrosive HF directly, we generated HF in situ using a lithium fluoride salt. In brief, the selective etching of the MAX phase was carried out by adding 0.8 g of LiF salt into 10 mL HCl (12 M). After 5 min of continuous stirring, 0.5 g Ti3AlC2 precursors were slowly added in small portions to the etching solution and kept under stirring for 24 hours at a temperature of 35 °C to obtain the multi-layered Ti3C2Tx. The benefit of using the MILD method is that we do not have to delaminate the multi-layered MXene separately as it will be delaminated in situ by the Li+ ions generated. The synthesized material was centrifuged at 3500 rpm and 25 °C for 10 minutes. The centrifugation process was repeated exhaustively to remove any trapped reagents, and the dark supernatant was collected to prepare the composite membrane. It was also ensured that the pH of the solution (determined from pH paper) was neutralized before collecting supernatants. The leftover precipitate was freeze-dried and lyophilized for further use.2.6. Fabrication of the NiO–Ti3C2Tx MXene loaded LIG
Herein, we developed a facile ultrasonication approach for synthesizing the NiO–Ti3C2Tx composite. The NiO–Ti3C2Tx MXene composite was successfully synthesized by a one-step electrostatic self-assembly method. The composite was prepared by very slowly mixing two dispersion solutions of positive NiO nanoparticles (1 mg mL−1) and negative Ti3C2Tx MXene nanosheets (1 mg mL−1). The composite quickly transforms into a uniform dispersion in DI water due to electrostatic self-assembly. The ultrasonication process was carried out at room temperature for 2 h. After that, 10 μL of the mixed solution was drop-coated onto the ozonized LIG substrate and air dried at 50 °C for 20 minutes. For electrochemical comparison, different weight percentages (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:1 and 2:1) of NiO and Ti3C2Tx were taken. The superior electrochemical performance was observed for the 1:1 composite.
:2, 1:1 and 2:1) of NiO and Ti3C2Tx were taken. The superior electrochemical performance was observed for the 1:1 composite.
2.7. Calculation of the electrochemically active surface area
The electrochemical active surface area (ECSA) was also calculated for all the samples using the Randles–Sevcik equation:87| ip = 2.69 × 105n3/2AD1/2Cv1/2 | (1) |
3. Results and discussion
Fig. 1 shows the schematic illustration of the synthesis of the positive NiO and negative Ti3C2Tx MXene composite which was achieved through an electrostatic self-assembly process. The zeta potentials of NiO nanoparticles and MXene nanosheets were 8.3 mV and −34 mV, respectively, which are shown in Fig. S2 (ESI†). | ||
| Fig. 1 Schematic illustration of the fabrication of the NiO–Ti3C2Tx MXene composite. | ||
When introducing positively charged NiO nanoparticles into a solution containing negatively charged MXene nanosheets, the NiO nanosheets undergo electrostatic self-assembly, adhering to the MXene nanosheets. As a result, a uniform mixture is formed through an ultrasonication process. Fig. 2b shows the zeta potential of individuals in DI water with equal weights (1:1).
 | ||
| Fig. 2 FESEM images of (a) Ti3C2Tx MXene flakes, (b) NiO nanoparticles and (c) and (d) the NiO–Ti3C2Tx@LIG composite at different magnifications. (e) EDS elemental spectra of the NiO–Ti3C2Tx@LIG nanocomposite. | ||
The morphological study of the NiO–Ti3C2Tx/LIG based multiplexed sweat sensing platform was observed using field emission scanning electron microscopy (FESEM) analysis. Fig. 2a shows multilayered Ti3C2Tx MXene nanosheets with flat surfaces and significant lateral dimensions were synthesized through selectively etching out Al layers from the pristine Ti3AlC2. Fig. 2b illustrates the even dispersion and aggregation of NiO nanoparticles in a uniform distribution.
NiO nanoparticles with a very small size have the capability of infiltrating the interlayer regions of multi-layered Ti3C2Tx MXene nanosheets or adhering to their surface through van der Waals interactions.87 The self-assembly process prevents the restacking of MXene nanosheets and facilitates enhanced contact between NiO and the Ti3C2Tx MXene. Therefore, the effective interaction of these two may potentially enhance the conductivity of the composite material. Fig. 2c and d presents the morphology of the NiO/Ti3C2Tx decorated porous LIG films. The elemental composition of the synthesized NiO–Ti3C2Tx/LIG composite is further supported by EDS analysis as illustrated in Fig. 2e.
X-ray powder diffraction was utilized to investigate the crystalline structures and morphologies of the composite sample. Fig. 3a and c shows the XRD patterns of the Ti3AlC2 MAX phase, Ti3C2Tx, NiO and NiO–Ti3C2Tx/LIG composite nanomaterials. The XRD spectrum of NiO nanoparticles (Fig. 3c) displays distinct peaks and these were identified as arising from the face-centered cubic (FCC) structure of the NiO particles.74 The absence of the prior phase (nickel hydroxide) suggests that the samples demonstrate a high degree of crystallinity of NiO nanoparticles. Distinctive sharp peaks were identified at 2θ ≈ 37.2°, 43.3°, 62.9° and 75.4° which showed strong agreement with the crystallographic planes (111), (200), (220) and (311) of NiO nanoparticles.74,88 The diffraction peaks observed in the XRD spectra of the freshly synthesized Ti3C2Tx MXene closely correspond to the distinctive structure observed in the etched Ti3C2Tx MXene by LiF and HCl. Following the MILD treatment, the peak at 2θ = 39° corresponding to the (104) plane of Ti3AlC2 disappeared, which suggests the successful removal of Al layers (Fig. 3a).89,90 Also, the peak corresponding to the Ti3C2Tx MXene (002) plane was observed to move towards a lower 2θ value, shifting from 9.5° to 6.7°, attributed to an increase in the d spacing. Both results indicate successful removal of the Al layer, leading to the exfoliation of the Ti3AlC2 phase into 2D layered Ti3C2Tx MXene nanosheets with no crystallographic stacking.91 In the case of LIG (Fig. 3c), a prominent peak was detected at a 2θ angle of 25.7°, corresponding to the (002) planes of LIG, with an associated interlayer spacing of approximately 0.34 nm (d002).49 Additionally, the peak at 2θ = 43.3° is assigned to the (100) reflections, indicating an in-plane structure. The NiO–Ti3C2Tx/LIG composite sample exhibits nearly all of the distinctive peaks found in NiO, Ti3C2Tx–MXene and LIG. From this analysis, it can be concluded that the composite of NiO–Ti3C2Tx/LIG was successfully synthesized.
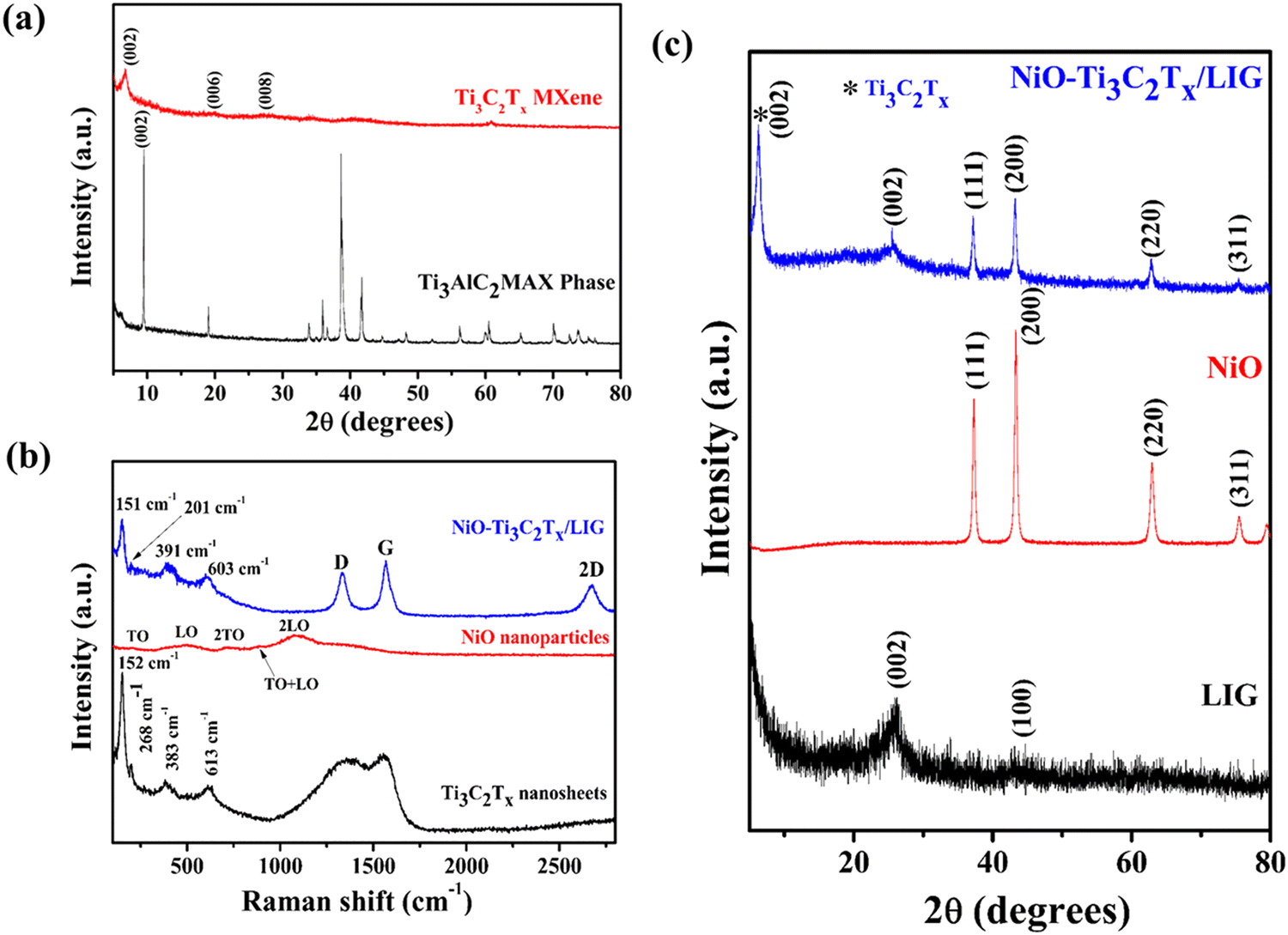 | ||
| Fig. 3 (a) XRD patterns of the precursor Ti3AlC2 MAX phase and delaminated Ti3C2Tx MXene nanosheets. (b) Raman spectra and (c) XRD spectra of LIG, NiO nanoparticle and NiO–Ti3C2Tx/LIG composite electrodes. | ||
Raman measurements were conducted to identify the types of defects and disorders in the 3D porous graphene network resulting from the incorporation of the NiO–MXene composite. As displayed in Fig. 3b, the delaminated Ti3C2Tx MXene nanosheets display bands at approximately 268, 383 and 613 cm−1. The bands at 268 and 383 cm−1 correspond to the in-plane Ti–C vibration (Eg symmetry of Ti3C2) and the in-plane vibration of oxygen atoms (Eg symmetry) in the hydrogen-terminated MXene Ti3C2(OH)2. The peak at 613 cm−1 is primarily due to the Eg vibrations of C atoms in the OH-terminated Ti3C2Tx MXene. The band observed at 152 cm−1 corresponds to the Eg vibrational mode, which originates from the termination with the oxygen functional groups.92–94 From the Raman analysis of NiO nanoparticles, five bands are observed due to phonon scattering. A weak but visible band is observed at 201 cm−1 associated to the zone-boundary phonon mode. The band at 491 cm−1 originates from the longitudinal optical (LO) phonon modes of NiO nanoparticles. Additionally, peaks at 715, 885 and 1080 cm−1 are attributed to the two-phonon (2P) 2TO (transverse optical) modes, TO + LO, and 2LO modes, respectively.95–97 Fig. S5 (ESI†) presents the Raman spectrum of the pristine LIG surface. For pristine LIG, the prominent D, G and 2D peaks are observed at approximately 1343 cm−1, 1581 cm−1, and 2695 cm−1, respectively. The D peak corresponds to the breathing mode of the k-point phonon with A1g symmetry, while the G peak signifies the E2g phonon of sp2 hybridized carbon atoms. The 2D peak, an overtone of the D peak, represents a two-phonon lattice vibration.49 The position and shape of the 2D peak provide insights into the graphene layer thickness and the doping level of graphene. In the composite sample, the bands of LIG, the Ti3C2Tx MXene and NiO are clearly visible, which further confirmed the successful synthesis of NiO–Ti3C2Tx/LIG composites.
FTIR spectra were also acquired to further validate the formation of the NiO–Ti3C2Tx/LIG composite. The typical FTIR spectra of the Ti3C2Tx, NiO and NiO–Ti3C2Tx/LIG samples are displayed in Fig. S3 (ESI†). The FTIR spectrum of MXene nanosheets displays absorption peaks around 3487 cm−1 and 1348 cm−1 which are attributed to surface hydroxyl group terminations.74 This indicates the presence of water adsorption and strong hydrogen-bonded OH groups. Additionally, weak absorption peaks at 1135 cm−1 and 1651 cm−1 are associated with C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively.74 The FTIR spectrum of NiO nanoparticles exhibited two absorption peaks around 3475 and 1374 cm−1 due to the stretching vibrations of O–H bonds.74 Additional stretching vibrations including CO at 1631 cm−1 and C–O at 1013 cm−1 were detected. These were ascribed to moisture adsorbed on the surface and physical absorption of CO2 during sample preparation.98–100 The FTIR spectrum of the NiO–Ti3C2Tx/LIG composite did not change significantly, except the minor variation in peak intensity.
O stretching vibrations, respectively.74 The FTIR spectrum of NiO nanoparticles exhibited two absorption peaks around 3475 and 1374 cm−1 due to the stretching vibrations of O–H bonds.74 Additional stretching vibrations including CO at 1631 cm−1 and C–O at 1013 cm−1 were detected. These were ascribed to moisture adsorbed on the surface and physical absorption of CO2 during sample preparation.98–100 The FTIR spectrum of the NiO–Ti3C2Tx/LIG composite did not change significantly, except the minor variation in peak intensity.
The surface chemical composition, oxidation states and binding energy of the NiO–Ti3C2Tx/LIG composite were analyzed by XPS. The survey spectrum (Fig. 4a) of the synthesized NiO–Ti3C2Tx/LIG composite exhibits five distinct peaks corresponding to Ti 2p, C 1s, Ni 2p, F 1s and O 1s, confirming the presence of these elements in the nanocomposites. It is further clarified by the deconvolution of C 1s, O 1s, and Ti 2p. Also, it directs successful removal of Al layers and the incorporation of oxygen and fluorine groups during MILD treatment.
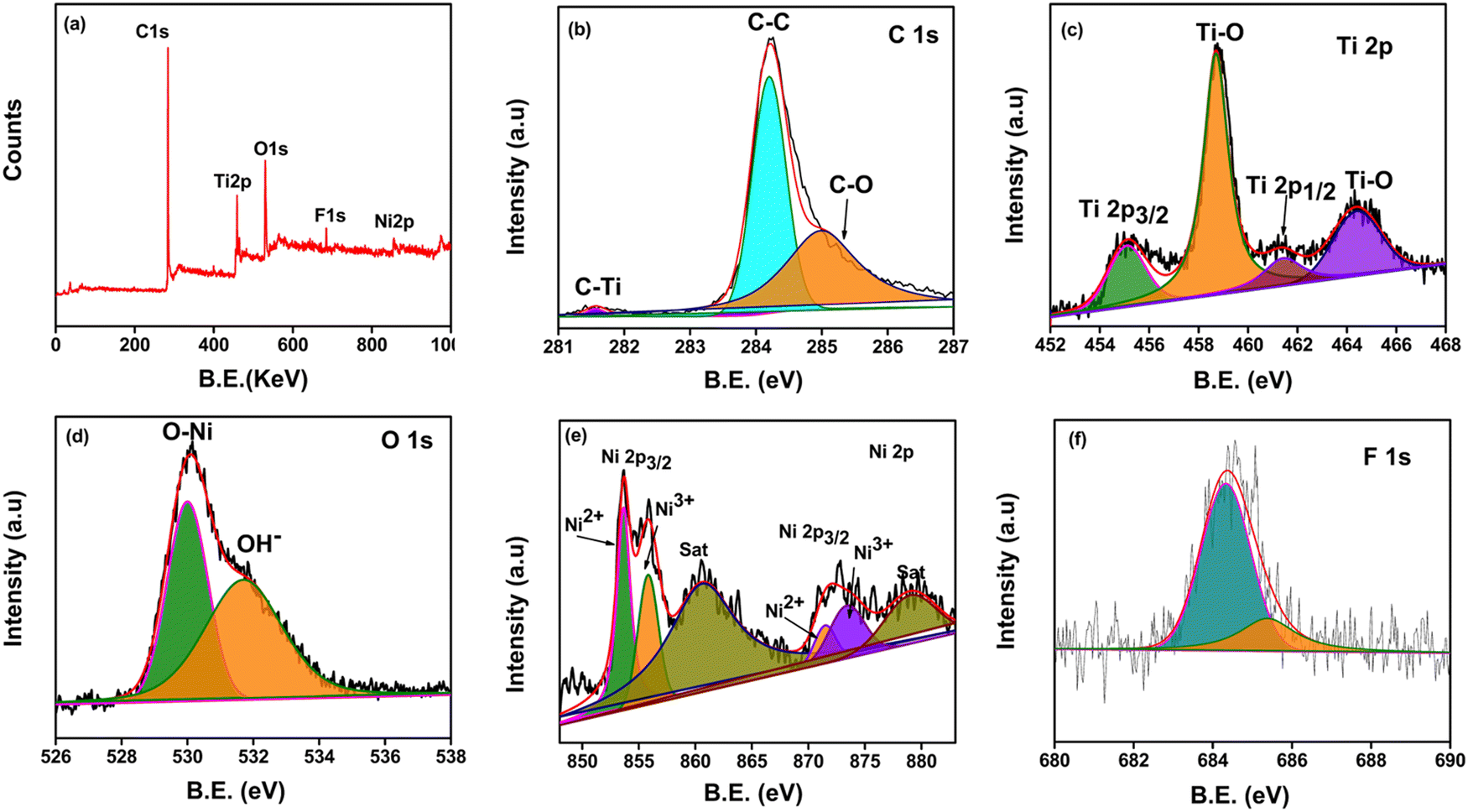 | ||
| Fig. 4 (a) XPS survey spectra of the NiO–Ti3C2Tx/LIG composite and high-resolution deconvoluted XPS spectra of (b) C 1s, (c) Ti 2p, (d) O 1s (e) Ni 2p, and (f) F 1s. | ||
The XPS survey spectrum of NiO–Ti3C2Tx/LIG confirms the presence of Ni in the composite which serves as evidence of NiO intercalation on the Ti3C2Tx MXene/LIG composite (Fig. 4a). The C 1s spectrum (Fig. 4b) displayed three deconvoluted peaks at 281.5, 284.2, and 285 eV corresponding to the characteristic bonds of C–Ti, C–C, and C–O, respectively.74,87 In particular, the sharp peak at 281.5 eV suggests the existence of C–Ti bonds implying the structural composition of the Ti3C2Tx MXene.88Fig. 4c displays the high-resolution core level XPS spectrum of the Ti 2p signal which shows two distinct peaks of Ti 2p3/2 and Ti 2p1/2 at binding energies 455.1 eV and 464.3 eV, respectively. A spin-energy difference of 9.2 eV was estimated between the Ti 2p3/2 and Ti 2p1/2. Additionally, two peaks are observed at 458.6 eV (sp3) and 464.2 eV (sp1), which indicates the presence of Ti–O bonds.74Fig. 4d shows the presence of numerous hydroxyl groups (Ti–OH) which form the Ti3C2(OH)2 coating on the Ti3C2Tx MXene surface. This is validated by the O 1s peak observed at 530.1 eV (O–Ni) and another peak at 531.7 eV (O–H). Fig. 4e shows the core level spectrum of Ni 2p, displaying two spin–orbit doublets labeled as Ni 2p3/2 and Ni 2p1/2 at 853.5 and 872.1 eV, respectively.74 Furthermore, two obvious satellite peaks appeared at 860.7 eV and 879.1 eV, respectively. All the abovementioned peaks are attributed to Ni2+ and Ni3+ oxidation states.74 The fitted peaks for Ti 2p, C 1s, Ni 2p, and O 1s in the spectrum align well with the earlier reports.101 The results above indicate successful synthesis of NiO nanoparticles anchored onto Ti3C2Tx MXene nanosheets using a straightforward ultrasonication technique.
The crystalline structures and morphologies of the NiO–Ti3C2Tx/LIG sample were further studied using TEM, HRTEM, and SAED, as illustrated in Fig. S4 (ESI†). TEM images of the composite sample are shown in Fig. S4a and b (ESI†). The as prepared Ti3C2Tx nanosheets, NiO nanoparticles and LIG powder can be clearly seen in Fig. S4a and b (ESI†). The HRTEM image shown in Fig. S4c (ESI†) reveals that the Ti3C2Tx MXene is composed of only a few layers with a thickness of approximately 0.66 nm. The selected area electron diffraction pattern (Fig. S4d, ESI†) of the NiO–Ti3C2Tx/LIG composite displays distinct diffraction circles corresponding to the crystallographic planes of NiO and Ti3C2Tx which is consistent with the XRD analysis.
Fig. 5a displays the cyclic voltammograms of LIG, Ti3C2Tx/LIG and NiO–Ti3C2Tx/LIG electrodes using the [Fe(CN)6]3−/4− redox couple within the potential range of −0.2 to 0.6 V at a scan rate of 10 mV s−1. In comparison to LIG and Ti3C2Tx/LIG electrodes, the CV response for NiO–Ti3C2Tx/LIG shows a lower ΔEp value with a higher peak current density (Ip) which suggests a mechanism for faster electron transfer. The NiO–Ti3C2Tx modified LIG electrode exhibited quasi-reversible one-electron redox behavior with a peak separation of 0.158 V. For comparison, we also performed the same experiment with different weight ratios (Fig. 5b) of Ti3C2Tx and NiO. The higher current density was obtained for a 1:1 weight ratio of these two. Overall, the NiO–Ti3C2Tx modified LIG electrode exhibited higher catalytic activity than the other electrodes and can be successfully employed in electrochemical sensing applications. Moreover, ECSA was determined for the modified sample using the Randles–Sevcik equation which considers mass transport only through the diffusion process.102 The increased ECSA of NiO–Ti3C2Tx/LIG (0.19 m2) as shown in Fig. 5c offers a superior surface area for the electrolyte, enabling more efficient ion-to-electron transduction.
 | ||
| Fig. 5 Cyclic voltammograms and kinetic analyses of LIG, Ti3C2Tx/LIG and NiO–Ti3C2Tx/LIG. (a) CV curves of different biosensors in 5.0 mM [Fe(CN)6]3−/4− containing 0.1 M KCl at different scan rates. (b) CV curves of the optimized NiO–Ti3C2Tx/LIG electrode at different scan rates. (c) CV curves of the NiO–Ti3C2Tx/LIG electrode at different weight ratios of Ti3C2Tx and NiO. (d) Electrochemical impedance spectroscopy (EIS) of the electrodes in the abovementioned electrolyte. The inset shows the equivalent electrical circuit model. | ||
Electrochemical impedance spectroscopy (EIS) was employed to observe and analyze the interface characteristics of all the electrodes. EIS analysis was performed in the 0.1 Hz to 1 kHz frequency range. Additionally, a model equivalent circuit was utilized to fit the impedance spectrum of each sample. The fitting spectra of the samples are shown in Fig. 5d. The inset of Fig. 5d illustrates the equivalent circuit model. The charge transfer resistance, Rct (Table S1, ESI†), is minimal for the NiO–Ti3C2Tx/LIG electrode which indicates superior electron transfer kinetics and better conductivity. The NiO–Ti3C2Tx-modified LIG electrode demonstrates superior catalytic activity compared to the other electrodes, making it highly suitable for electrochemical sensing applications.
3.1. Optimization of experimental parameters
To achieve optimal sensing performance such as high sensitivity and a low detection limit, we conducted optimization of several experimental parameters. These included adjustments to the pH of the electrolyte, the deposition potential and the deposition time. For this optimization experiment, the concentration of the AA, DA and UA mixture was kept at 2 mM, 1 mM and 1 mM, respectively, in potassium phosphate buffer solution (0.1 M). Fig. S6a (ESI†) shows the variation of peak current for AA, DA and UA molecules as a function of the pH value of the electrolyte. The effect of pH on the current response in the DPV study was obtained for the range of 5 to 8 (Fig. S6a, ESI†). Anodic peak current rises with an increase in pH up to 7.4, but beyond this, a further increase in pH leads to a decrease in the current response. Maximum peak current was observed at pH = 7.4. Therefore, to achieve optimal sensing performance, the analyte solutions were prepared using a 0.1 M phosphate buffer solution with a pH of 7.4. To achieve the best sensing performance in DPV analysis, the deposition potential plays a crucial role that must be optimized. Fig. S6b (ESI†) illustrates how the DPV response of AA, DA and UA analytes is affected by the deposition potential. In order to optimize the deposition potential, several scans were carried out in the potential between −0.2 and −1.0 V. It was found that voltammetric peak current appeared to reach its maximum at −0.2 V, with the peak current decreasing at other deposition voltages. Therefore, optimum deposition potential was chosen at −0.2 V. Optimizing the deposition time in DPV is also essential to achieve both a low detection limit and higher sensitivity. Fig. S6c (ESI†) displays the effect of deposition time on DPV response towards AA, DA and UA molecules. Increasing the deposition time from 100 to 300 seconds resulted in a corresponding increase in the voltammetric peak current. This can be attributed to the increased accumulation of analytes on the electrode surface with longer deposition times. However, extending the deposition time beyond 200 seconds led to a decrease in the current response. This decrease may be attributed to electrode surface saturation caused by the high concentration of these analytes, which may reduce the upper detection limit for AA, DA and UA. Therefore, for the individual and simultaneous detection of these three biomolecules, we have determined that 200 s is the optimal deposition time.3.2. Electrochemical performance of the biosensor
Cyclic voltammograms were recorded to investigate the electrochemical activity as well as sensing ability of the LIG, Ti3C2Tx/LIG and NiO–Ti3C2Tx/LIG electrodes toward the oxidation of AA, DA and UA in phosphate buffer solution (0.1 M) as the supporting electrolyte. From Fig. 6a, it is evident that the three oxidation peaks related to AA, DA and UA exhibit distinct separation on the LIG, Ti3C2Tx/LIG and NiO–Ti3C2Tx/LIG electrodes. In the case of NiO–Ti3C2Tx/LIG, the oxidation current showed an improvement in comparison to that observed with the LIG and Ti3C2Tx/LIG electrodes. In order to precisely determine the oxidation peak differentiation between the three biomarkers, the electrochemical response of the electrode was additionally investigated in PBS solution containing a mixture of the three analytes (2 mM AA, 1 mM DA and 1 mM UA) as depicted in Fig. 6a. As a result, the differences in peak-to-peak separation potentials (ΔE) for AA–DA, DA–UA and AA–UA are recorded at 207 mV, 104 mV and 314 mV, respectively. Also, the individual cyclic voltammetry responses of each analyte are displayed in Fig. 6b–d for all three electrodes. For individual detection of AA at the NiO–Ti3C2Tx/LIG electrode, the oxidation peak potential occurs at −0.02 V. The cathodic and anodic peaks of DA are observed at 0.09 V and 0.23 V, respectively, with a peak potential (ΔE) difference of approximately 0.14 V. In the case of UA, a sharp oxidation peak at 0.36 V and a minor reduction peak at 0.25 V are obtained. The above results indicate that NiO–Ti3C2Tx/LIG possesses an increased electrochemical active surface area and enhanced electron transfer capabilities for the oxidation of AA, DA and UA. | ||
| Fig. 6 Cyclic voltammograms of LIG, Ti3C2Tx/LIG and NiO–Ti3C2Tx/LIG electrodes in 0.1 M PBS (pH 7.4) containing (a) a mixture of 2 mM AA, 1 mM DA and 1 mM UA, (b) 2 mM AA, (c) 1 mM DA and (d) 1 mM UA at a scan rate of 10 mV s−1 compared to the response with only buffer solution. | ||
Since the NiO–Ti3C2Tx modified LIG electrode exhibited a superior response, it was chosen for further DPV analysis. The DPV technique was utilized for the detection of individual analytes and simultaneous monitoring of a mixture of analytes effectively. Individual determination of AA, DA and UA at the NiO–Ti3C2Tx/LIG electrode was carried out under optimal conditions in the potential range of −0.2 to 0.6 V in phosphate buffer solution (pH 7.4). Fig. 7a–c displays the standard DPV reaction of the NiO–Ti3C2Tx/LIG electrode upon the introduction of various concentrations of AA, DA and UA within a phosphate buffer solution. As shown in Fig. 7a–c, three distinct anodic peaks corresponding to the oxidation of AA, DA and UA are observed at −0.05 V, 0.13 V and 0.26 V, respectively. The particular voltage waveform in DPV enables the electroactive substances to undergo oxidation during which they lose hydrogen atoms and electrons on the electrode at a specific potential. The current at the oxidation peak increases in correlation with the concentration of the analyte. The oxidation current increases linearly with increasing concentrations in the ranges of 10 μM–2 mM for AA, 0.1–200 μM for DA, and 0.5–100 μM for UA. This indicates the stable and enhanced catalytic activity of the NiO–Ti3C2Tx/LIG electrode. Calibration plots for analytes are generated by plotting the concentration of analytes against the current density which are shown in Fig. 7d–f. The sensitivity is represented by the slope value of the linear regression plot which is estimated from the calibration plots to be 0.14, 2.02 and 5.94 μA μM−1 for AA, DA and UA, respectively. According to the equation for the limit of detection (LOD = 3σ/m, where ‘σ’ is the standard deviation and m is the slope of the linear plot obtained in the lower concentration region), the estimated LOD values are 16, 1.97 and 0.78 μM for AA, DA and UA, respectively.
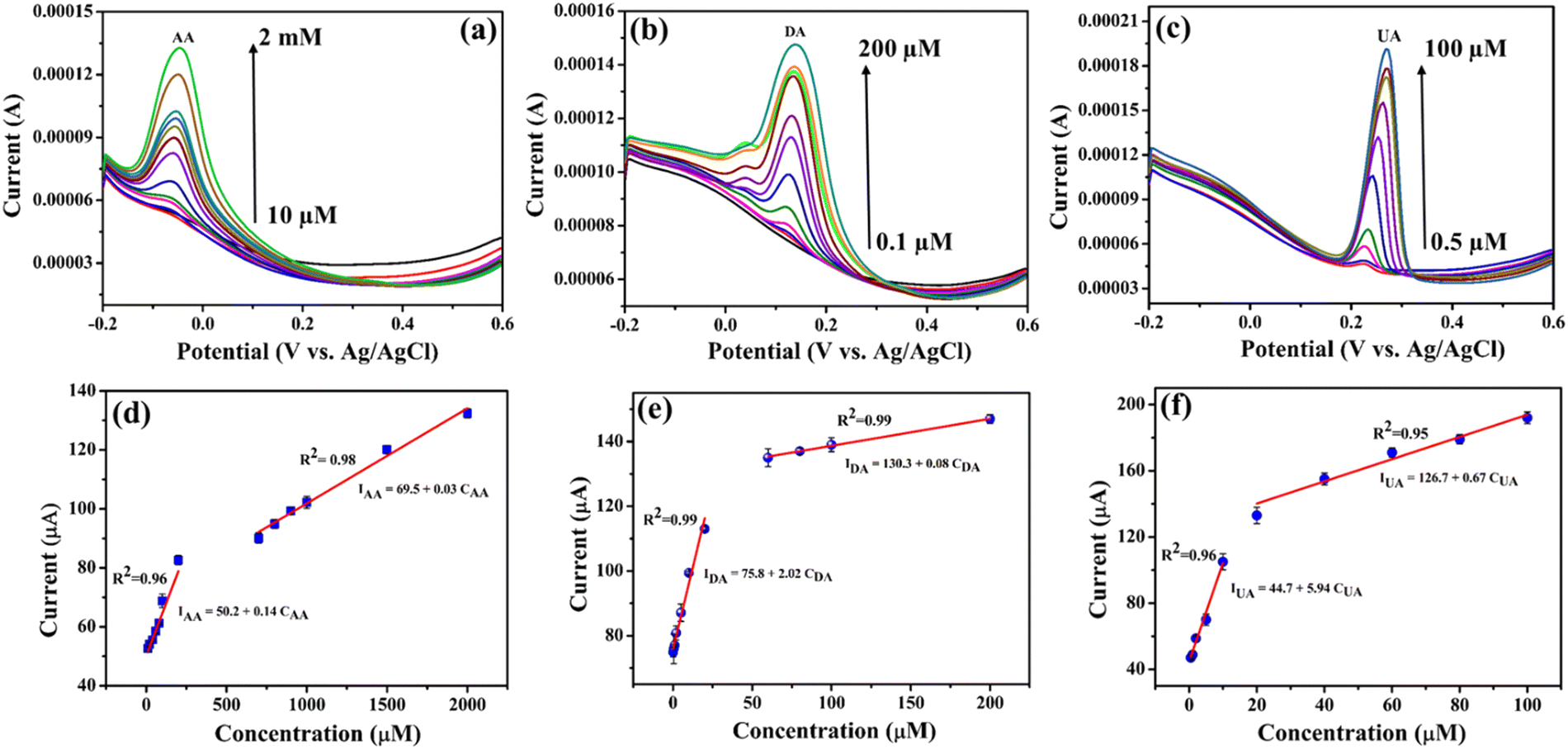 | ||
| Fig. 7 Electrochemical performance of the modified 3D porous laser-induced graphene sensor. Differential pulse voltammetry (DPV) measurements at different concentrations of individual (a) AA, (b) DA and (c) UA in 0.1 M PBS (pH 7.4) as the supporting electrolyte. (d)–(f) Calibration plots of the oxidation peak current versus concentration of each biomarker. Linear fitting is used to determine the sensitivity of the electrodes for each biomarker. | ||
During the simultaneous measurements, only the concentrations of the specific molecule were varied, while the concentrations of the other two analytes were fixed. Fig. 8a–c presents the DPV after repeated addition of varying concentrations of AA, DA and UA while maintaining the other two constant. The DPV results clearly show that the electrochemical oxidation peaks associated with each analyte rise proportionally with an increase in the concentration of the respective species. The peak currents demonstrate a linear rise when the concentration is elevated within the ranges of 20 μM–2 mM for AA, 0.1–100 μM for DA and 0.2–100 μM for UA on the NiO–Ti3C2Tx/LIG electrode. This observation suggests the absence of any influence or mutual interaction of one analyte on the other two species. The limit of detection (LOD) was also determined, and for the NiO–Ti3C2Tx/LIG sensor, the LOD values were 35, 0.4 and 0.08 μM for AA, DA and UA, respectively. The sensitivities were also obtained from the slope of the calibration curve, resulting in values of 0.03, 7.38 and 2.32 μA μM−1 for AA, DA and UA, respectively. Because of the enhanced sensitivity and selectivity, NiO–Ti3C2Tx modified LIG-based electrodes represent a promising choice for biosensing applications.
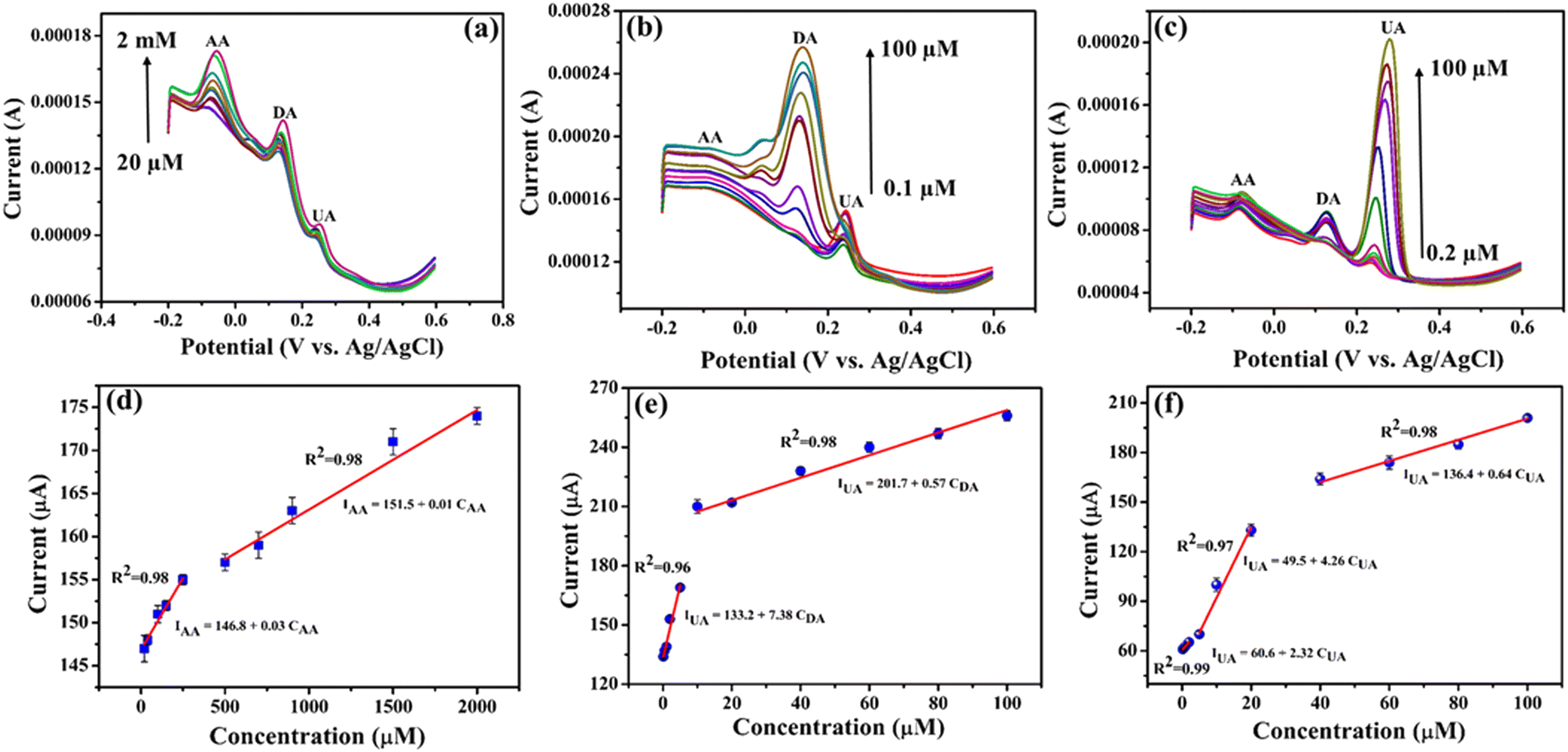 | ||
| Fig. 8 Demonstration of simultaneous detection using the NiO–Ti3C2Tx/LIG sensor in 0.1 M PBS (pH 7.4) (a) containing 4 μM DA and 4 μM UA and different concentrations of AA (20 μM to 2 mM); (b) containing 100 μM AA and 4 μM UA and different concentrations of DA (0.1 μM to 100 μM) and (c) containing 100 μM AA and 4 μM DA and different concentrations of UA (0.2 μM to 100 μM). (d)–(f) Calibration plots of the peak current as a function of analyte concentrations. | ||
3.3. Reproducibility, stability, selectivity and artificial sweat test of the modified electrode
The repeatability and reproducibility of the modified electrodes (n = 8) was demonstrated using the mixture of 2 mM AA, 1 mM DA and 1 mM UA in the 0.1 M PBS (pH = 7.4) solution. The reproducibility of the sensor was tested with eight electrodes that were all fabricated in a similar way. The result indicates that the NiO–Ti3C2Tx/LIG based sensor exhibits excellent reproducibility (Fig. 9a). The anti-interference capability of the fabricated NiO–Ti3C2Tx/LIG sensor was assessed by adding various ions and small molecules into a pH 7.4 phosphate buffer solution containing 1 mM each of AA, DA and UA (as shown in Fig. 9c). No interference was detected in the presence of NaCl, CaCl2, NH4Cl, KCl, lactic acid, urea or glucose. From the figure, the oxidation peaks of AA, DA and UA remain largely unchanged even when exposed to various common interferents. These results indicate that our developed sensors demonstrated good selectivity in accurately detecting each of the three analytes.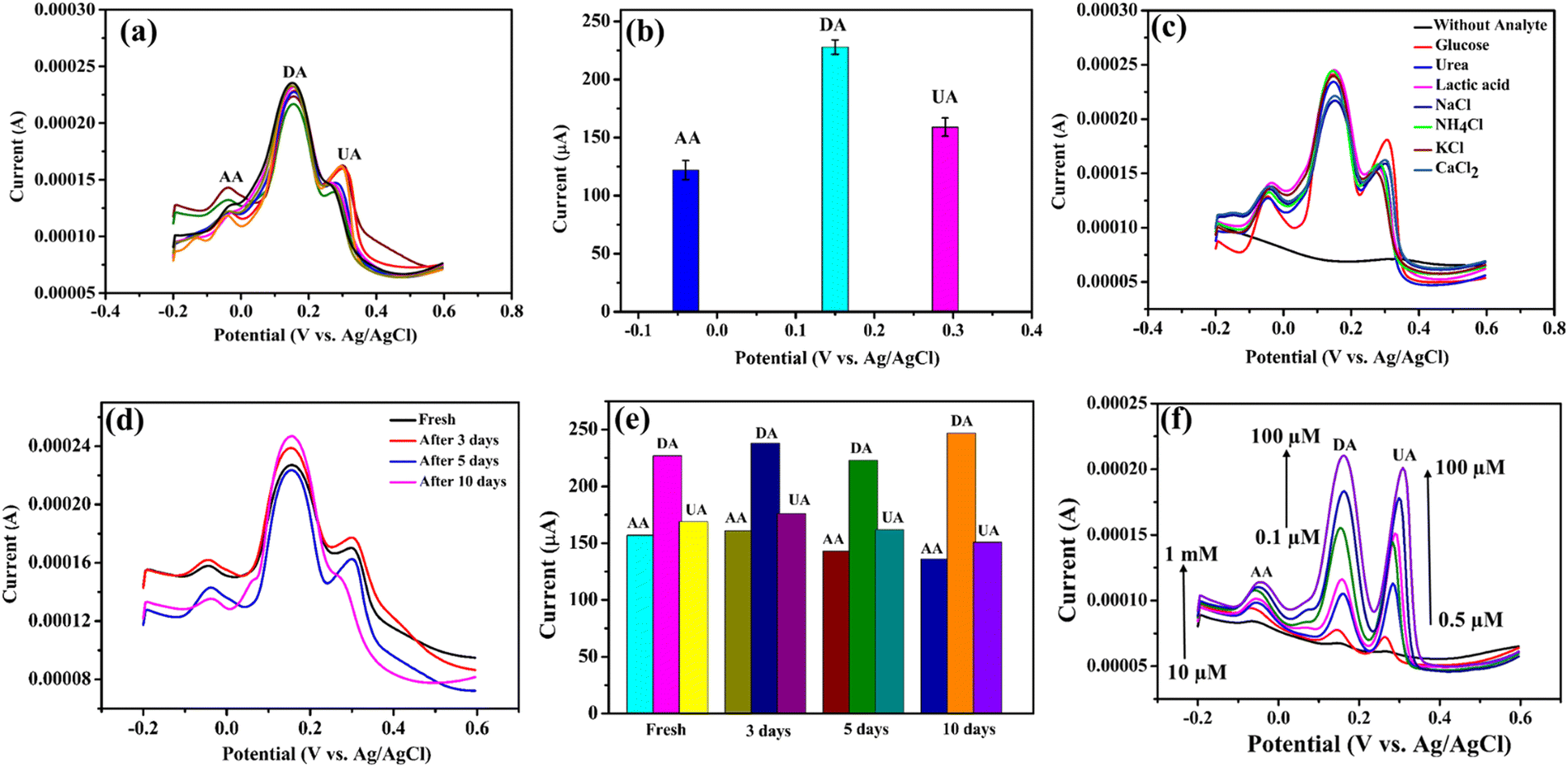 | ||
| Fig. 9 Reproducibility, stability, selectivity and investigation in artificial sweat of the NiO–Ti3C2Tx modified sensor: (a) reproducibility of the 8 sensors, (b) corresponding current vs. potential, (c) response of the fabricated sensor in the presence of other sweat ions and biomolecules, (d) stability of the as-fabricated biosensor, (e) corresponding current vs. number of days, and (f) response of the fabricated sensor in the presence of artificial sweat. | ||
The long-term stability test of the fabricated sensor was also performed, and the results are displayed in Fig. 9d. After storing the fabricated electrodes at room temperature, the current response towards AA, DA, and UA remained consistent with their initial values even after a period of 10 days. This result indicates the excellent stability of the sensors.
We have also explored the feasibility of the biosensor which was accomplished by evaluating its performance in artificial sweat solutions. Fig. 9f illustrates the characterization results of the fabricated multiplexed sensor in artificial sweat solution. The European reference method EN1811:2011103 was followed in the preparation of the artificial sweat solution which involved mixing urea (0.1 wt%) and lactic acid (0.1 wt%) in phosphate buffer solution with a pH of 7.4. After that, different concentrations of AA (10 μM–1 mM), DA (0.1–100 μM) and UA (0.5–100 μM) were mixed with the aforementioned synthetic sweat solution to perform the DPV. According to the above result, the current increases with higher concentrations of the three analytes. Moreover, the accuracy of the detection remained unaffected by any potential interferents that might have been present in the sample during the analysis. Therefore, it can be concluded that the NiO–Ti3C2Tx/LIG electrode demonstrates suitable performance for real-time sweat analysis. According to our study, the electrode's high sensitivity, repeatability, reproducibility, and feasibility make it suitable for noninvasive and wearable systems designed for continuous, real-time monitoring of personal healthcare.
In summary, the present study demonstrates that NiO–Ti3C2Tx/LIG electrodes can act as flexible sensing platforms for AA, DA and UA detection and are also capable of detecting other analytes present in human sweat.
4. Conclusion
Continuous and precise monitoring of AA, DA, and UA in human biofluids is essential for detecting and treating numerous medical diseases. This article presents a flexible and highly sensitive sensor utilising NiO–Ti3C2Tx MXene-modified LIG for the individual and simultaneous detection and quantification of AA, DA, and UA in sweat. The feasibility of the biosensor was accomplished by evaluating its performance in artificial sweat solutions in addition to the repeatability and reproducibility tests. Moreover, electrochemical investigation reveals that the improved sensor demonstrates higher electrical conductivity and an increased electrochemically active surface area. The NiO–Ti3C2Tx/LIG electrode enables accurate assessment of the oxidation potential of individual analyte molecules without interference or overlap from others. The modified electrode exhibits exceptional selectivity and reproducibility in the detection of AA, DA, and UA, characterised by low detection limits and an extensive detection range. Under optimised conditions, the limits of detection (LOD) were determined to be 16, 1.97, and 0.78 μM for ascorbic acid (AA), dopamine (DA), and uric acid (UA), respectively. This study demonstrates the considerable potential of the sensor for the continuous and instantaneous monitoring of many physiological components in human sweat, including ascorbic acid, dopamine, and uric acid. Furthermore, adaptable sensor design can be integrated into a next-generation, compact, dermally compatible device for clinical diagnostics without intrusive procedures.Author contributions
All authors have contributed sufficiently to the work. Sudipta Choudhury: conceptualization, methodology, validation, writing – original draft, writing – review & editing. Saad Zafar: investigation, methodology, data curation. Deepak Deepak: conceptualization, methodology, validation, graphics, data curation. Abhishek Panghal: validation, graphics, data curation. Bimlesh Lochab: methodology, validation, data curation, investigation. Susanta Sinha Roy: conceptualization, methodology, validation, investigation, data curation, writing – review & editing, supervision.Data availability
The results of this research are corroborated by the data which can be obtained from the corresponding author upon request. The data that support these findings can be found in the paper.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
Sudipta Choudhury acknowledges the financial support from Shiv Nadar Institution of Eminence (SNIoE), India through scholarship.References
- S. Choudhury, D. Deepak, G. Bhattacharya, J. McLaughlign and S. S. Roy, Macromol. Mater. Eng., 2023, 308, 2300007 CrossRef CAS.
- J. S. Nah, S. C. Barman, M. A. Zahed, M. Sharifuzzaman, H. Yoon, C. Park, S. Yoon, S. Zhang and J. Y. Park, Sens. Actuators, B, 2021, 329, 129206 CrossRef CAS.
- H. Lee, C. Song, Y. S. Hong, M. Kim, H. R. Cho, T. Kang, K. Shin, S. H. Choi, T. Hyeon and D.-H. Kim, Sci. Adv., 2017, 3, e1601314 CrossRef.
- A. J. Bandodkar, P. Gutruf, J. Choi, K. Lee, Y. Sekine, J. T. Reeder, W. J. Jeang, A. J. Aranyosi, S. P. Lee, J. B. Model, R. Ghaffari, C.-J. Su, J. P. Leshock, T. Ray, A. Verrillo, K. Thomas, V. Krishnamurthi, S. Han, J. Kim, S. Krishnan, T. Hang and J. A. Rogers, Sci. Adv., 2019, 5, eaav3294 CrossRef.
- Y. Qiao, Q. Liu, S. Lu, G. Chen, S. Gao, W. Lu and X. Sun, J. Mater. Chem. B, 2020, 8, 5411 RSC.
- W. Lu, S. Xue, X. Liu, C. Bao and H. Shi, Microchem. J., 2024, 196, 109606 CrossRef CAS.
- A. Koh, D. Kang, Y. Xue, S. Lee, R. M. Pielak, J. Kim, T. Hwang, S. Min, A. Banks, P. Bastien, M. C. Manco, L. Wang, K. R. Ammann, K.-I. Jang, P. Won, S. Han, R. Ghaffari, U. Paik, M. J. Slepian, G. Balooch, Y. Huang and J. A. Rogers, Sci. Transl. Med., 2016, 8, 366ra165 Search PubMed.
- M. Bariya, H. Y. Y. Nyein and A. Javey, Nat. Electron., 2018, 1, 160 CrossRef.
- K. Kwon, J. U. Kim, Y. Deng, S. R. Krishnan, J. Choi, H. Jang, K. Lee, C.-J. Su, I. Yoo, Y. Wu, L. Lipschultz, J.-H. Kim, T. S. Chung, D. Wu, Y. Park, T. Kim, R. Ghaffari, S. Lee, Y. Huang and J. A. Rogers, Nat. Electron., 2021, 4, 302 CrossRef.
- J. Choi, R. Ghaffari, L. B. Baker and J. A. Rogers, Sci. Adv., 2018, 4, eaar3921 CrossRef PubMed.
- W. Gao, S. Emaminejad, H. Y. Y. Nyein, S. Challa, K. Chen, A. Peck, H. M. Fahad, H. Ota, H. Shiraki, D. Kiriya, D.-H. Lien, G. A. Brooks, R. W. Davis and A. Javey, Nature, 2016, 529, 509 CrossRef CAS PubMed.
- L. Wang, J. Lu, Q. Li, L. Li, E. He, Y. Jiao, T. Ye and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2200922 CrossRef CAS.
- M. Wei, Y. Qiao, H. Zhao, J. Liang, T. Li, Y. Luo, S. Lu, X. Shi, W. Lu and X. Sun, Chem. Commun., 2020, 56, 14553 RSC.
- W. Lu, R. Zhang, X. Zhang, Y. Shi, Y. Wang and H. Shi, Analyst, 2023, 148, 5469 RSC.
- L. Yang, H. Wang, A. M. Abdullah, C. Meng, X. Chen, A. Feng and H. Cheng, ACS Appl. Mater. Interfaces, 2023, 15, 34332 CrossRef CAS.
- H. Huang, Y. Yue, Z. Chen, Y. Chen, S. Wu, J. Liao, S. Liu and H. Wen, Microchim. Acta, 2019, 186, 189 CrossRef PubMed.
- Y. Wu, P. Deng, Y. Tian, J. Feng, J. Xiao, J. Li, J. Liu, G. Li and Q. He, J. Nanobiotechnol., 2020, 18, 112 CrossRef CAS PubMed.
- P. K. Aneesh, S. R. Nambiar, T. P. Rao and A. Ajayaghosh, Anal. Methods, 2014, 6, 5322 RSC.
- X. W. Qiangwei Wang and J. Kong, Crit. Rev. Anal. Chem., 2020, 50, 359 CrossRef PubMed.
- D. Lakshmi, M. J. Whitcombe, F. Davis, P. S. Sharma and B. B. Prasad, Electroanalysis, 2011, 23, 305 CrossRef CAS.
- Y. Yang, Y. Song, X. Bo, J. Min, O. S. Pak, L. Zhu, M. Wang, J. Tu, A. Kogan, H. Zhang, T. K. Hsiai, Z. Li and W. Gao, Nat. Biotechnol., 2020, 38, 217 CrossRef CAS.
- V. Bhole, J. W. J. Choi, S. Woo Kim, M. de Vera and H. Choi, Am. J. Med., 2010, 123, 957 CrossRef CAS PubMed.
- N. Murugan, M. B. Chan-Park and A. K. Sundramoorthy, J. Electrochem. Soc., 2019, 166, B3163 CrossRef CAS.
- O. Arrigoni and M. C. De Tullio, Biochim. Biophys. Acta, Gen. Subj., 2002, 1569, 1 CrossRef CAS PubMed.
- S. Liu, X. Jiang and M. Yang, Microchim. Acta, 2019, 186, 445 CrossRef PubMed.
- W. Argoubi, A. Rabti, S. Ben Aoun and N. Raouafi, RSC Adv., 2019, 9, 37384 RSC.
- K. Dhara and R. M. Debiprosad, Anal. Biochem., 2019, 586, 113415 CrossRef CAS.
- P. Kalimuthu and S. A. John, Talanta, 2010, 80, 1686 CrossRef CAS PubMed.
- M. A. Kurian, P. Gissen, M. Smith, S. J. R. Heales and P. T. Clayton, Lancet Neurol., 2011, 10, 721 CrossRef CAS.
- S. Reyes, Y. Fu, K. L. Double, V. Cottam, L. H. Thompson, D. Kirik, G. Paxinos, C. Watson, H. M. Cooper and G. M. Halliday, Neurobiol. Aging, 2013, 34, 873 CrossRef CAS PubMed.
- P. N. Tobler, C. D. Fiorillo and W. Schultz, Science, 2005, 307, 1642 CrossRef CAS.
- S. Hou, M. L. Kasner, S. Su, K. Patel and R. Cuellari, J. Phys. Chem. C, 2010, 114, 14915 CrossRef CAS.
- X. Liu and J. Liu, View, 2021, 2, 20200102 CrossRef.
- D.-S. Kim, E.-S. Kang, S. Baek, S.-S. Choo, Y.-H. Chung, D. Lee, J. Min and T.-H. Kim, Sci. Rep., 2018, 8, 14049 CrossRef.
- J. Jiang and X. Du, Nanoscale, 2014, 6, 11303 RSC.
- Y. Wang, P. Zhao, B. Gao, M. Yuan, J. Yu, Z. Wang and X. Chen, Microchem. J., 2023, 185, 108177 CrossRef CAS.
- H. Chen, R. Li, L. Lin, G. Guo and J.-M. Lin, Talanta, 2010, 81, 1688 CrossRef CAS PubMed.
- A. Nemiroski, D. C. Christodouleas, J. W. Hennek, A. A. Kumar, E. J. Maxwell, M. T. Fernández-Abedul and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11984 CrossRef CAS PubMed.
- Y. Wu, L. Cui, Y. Liu, G. Lv, T. Pu, D. Liu and X. He, Analyst, 2013, 138, 1204 RSC.
- G. Maduraiveeran, M. Sasidharan and V. Ganesan, Biosens. Bioelectron., 2018, 103, 113 CrossRef CAS PubMed.
- Y. Yu, H. Y. Y. Nyein, W. Gao and A. Javey, Adv. Mater., 2020, 32, 1902083 CrossRef CAS PubMed.
- J. Du, R. Yue, F. Ren, Z. Yao, F. Jiang, P. Yang and Y. Du, Biosens. Bioelectron., 2014, 53, 220 CrossRef CAS.
- A. J. Bandodkar and J. Wang, Trends Biotechnol., 2014, 32, 363 CrossRef CAS.
- F. Li, J. Ye, M. Zhou, S. Gan, Q. Zhang, D. Han and L. Niu, Analyst, 2012, 137, 618 RSC.
- S. Alwarappan, R. K. Joshi, M. K. Ram and A. Kumar, Appl. Phys. Lett., 2010, 96, 263702 CrossRef.
- F. Ren, H. Wang, C. Zhai, M. Zhu, R. Yue, Y. Du, P. Yang, J. Xu and W. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 3607 CrossRef CAS.
- X. Liu, H. Zhu and X. Yang, RSC Adv., 2014, 4, 3706 CAS.
- S. Eissa, L. L’Hocine, M. Siaj and M. Zourob, Analyst, 2013, 138, 4378 RSC.
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. G. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed.
- R. Ye, D. K. James and J. M. Tour, Adv. Mater., 2019, 31, 1803621 CrossRef.
- Y. Ling, W. Pang, X. Li, S. Goswami, Z. Xu, D. Stroman, Y. Liu, Q. Fei, Y. Xu, G. Zhao, B. Sun, J. Xie, G. Huang, Y. Zhang and Z. Yan, Adv. Mater., 2020, 32, 1908475 CrossRef CAS.
- E.-C. Cho, C.-W. Chang-Jian, W.-L. Syu, H.-S. Tseng, K.-C. Lee, J.-H. Huang and Y.-S. Hsiao, Appl. Surf. Sci., 2020, 518, 146193 CrossRef CAS.
- S. P. Singh, Y. Li, J. Zhang, J. M. Tour and C. J. Arnusch, ACS Nano, 2018, 12, 289 CrossRef CAS PubMed.
- W. Chen, R. V. Salvatierra, M. Ren, J. Chen, M. G. Stanford and J. M. Tour, Adv. Mater., 2020, 32, 2002850 CrossRef CAS PubMed.
- J. Zhang, M. Ren, L. Wang, Y. Li, B. I. Yakobson and J. M. Tour, Adv. Mater., 2018, 30, 1707319 CrossRef.
- L. Ge, Q. Hong, H. Li, C. Liu and F. Li, Adv. Funct. Mater., 2019, 29, 1904000 CrossRef.
- L. Lan, X. Le, H. Dong, J. Xie, Y. Ying and J. Ping, Biosens. Bioelectron., 2020, 165, 112360 CrossRef CAS PubMed.
- A. Dallinger, K. Keller, H. Fitzek and F. Greco, ACS Appl. Mater. Interfaces, 2020, 12, 19855 CrossRef CAS PubMed.
- L. Li, J. Zhang, Z. Peng, Y. Li, C. Gao, Y. Ji, R. Ye, N. D. Kim, Q. Zhong, Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2016, 28, 838 CrossRef CAS.
- A. Chhetry, S. Sharma, S. C. Barman, H. Yoon, S. Ko, C. Park, S. Yoon, H. Kim and J. Y. Park, Adv. Funct. Mater., 2021, 31, 2007661 CrossRef CAS.
- M. Wang, Y. Yang, J. Min, Y. Song, J. Tu, D. Mukasa, C. Ye, C. Xu, N. Heflin, J. S. McCune, T. K. Hsiai, Z. Li and W. Gao, Nat. Biomed. Eng., 2022, 6, 1225 CrossRef CAS PubMed.
- G. Xu, Z. A. Jarjes, H.-W. Wang, A. R. J. Phillips, P. A. Kilmartin and J. Travas-Sejdic, ACS Appl. Mater. Interfaces, 2018, 10, 42136 CrossRef CAS.
- S. Sharma, S. K. Ganeshan, P. K. Pattnaik, S. Kanungo and K. N. Chappanda, Mater. Lett., 2020, 262, 127150 CrossRef CAS.
- B. A. Getachew, D. S. Bergsman and J. C. Grossman, ACS Appl. Mater. Interfaces, 2020, 12, 48511 CrossRef CAS.
- J. Liu, H. Ji, X. Lv, C. Zeng, H. Li, F. Li, B. Qu, F. Cui and Q. Zhou, Microchim. Acta, 2022, 189, 54 CrossRef CAS PubMed.
- M. Bauer, L. Wunderlich, F. Weinzierl, Y. Lei, A. Duerkop, H. N. Alshareef and A. J. Baeumner, Anal. Bioanal. Chem., 2021, 413, 763 CrossRef CAS PubMed.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- M. Mathew and C. S. Rout, Curr. Opin. Electrochem., 2021, 30, 100782 CrossRef CAS.
- M. Su, H. Lan, L. Tian, M. Jiang, X. Cao, C. Zhu and C. Yu, Sens. Actuators, B, 2022, 367, 132019 CrossRef CAS.
- H. Wang, H. Li, Y. Huang, M. Xiong, F. Wang and C. Li, Biosens. Bioelectron., 2019, 142, 111531 CrossRef CAS.
- R. Zhou, B. Tu, D. Xia, H. He, Z. Cai, N. Gao, G. Chang and Y. He, Anal. Chim. Acta, 2022, 1201, 339653 CrossRef CAS PubMed.
- Y. Lei, W. Zhao, Y. Zhang, Q. Jiang, J.-H. He, A. J. Baeumner, O. S. Wolfbeis, Z. L. Wang, K. N. Salama and H. N. Alshareef, Small, 2019, 15, 1901190 CrossRef.
- C. Lin, W. Zhang, L. Wang, Z. Wang, W. Zhao, W. Duan, Z. Zhao, B. Liu and J. Jin, J. Mater. Chem. A, 2016, 4, 5993 RSC.
- R. A. Chavan, G. P. Kamble, S. B. Dhavale, A. S. Rasal, S. S. Kolekar, J.-Y. Chang and A. V. Ghule, Energy Fuels, 2023, 37, 4658 CrossRef CAS.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., 2015, 127, 3979 CrossRef.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322 CrossRef CAS.
- H. Liu, W. Qin, X. Li, L. Feng, C. Gu, J. Chen, Z. Tian, J. Chen, M. Yang, H. Qiao, X. Guo, Y. Zhang, B. Zhao and S. Yin, Anal. Chem., 2023, 95, 16079 CrossRef CAS.
- U. Amara, I. Hussain, M. Ahmad, K. Mahmood and K. Zhang, Small, 2023, 19, 2205249 CrossRef CAS.
- B. Mendoza-Sánchez and Y. Gogotsi, Adv. Mater., 2016, 28, 6104 CrossRef.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, MXenes, Jenny Stanford Publishing, 2023, pp. 379–399 Search PubMed.
- P. Lian, Y. Dong, Z.-S. Wu, S. Zheng, X. Wang, Sen Wang, C. Sun, J. Qin, X. Shi and X. Bao, Nano Energy, 2017, 40, 1 CrossRef CAS.
- Z. Lin, D. Barbara, P.-L. Taberna, K. L. Van Aken, B. Anasori, Y. Gogotsi and P. Simon, J. Power Sources, 2016, 326, 575 CrossRef CAS.
- J. Kumar, R. R. Neiber, A. Nafady, M. D. Albaqami, R. A. Soomro, M. Baraka, N. Ahmed and S. Karakus, ACS Appl. Nano Mater., 2022, 5, 11352 CrossRef CAS.
- G. Liu, T. Xia, X. Liang, S. Hou and S. Hou, ChemElectroChem, 2022, 9, e202200848 CrossRef CAS.
- Z. Ling, C. E. Ren, M.-Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676 CrossRef CAS.
- G. Qian, Q. Peng, D. Zou, S. Wang and B. Yan, Front. Mater., 2020, 7 Search PubMed.
- S. Chen, Y. Xiang, W. Xu and C. Peng, Inorg. Chem. Front., 2019, 6, 199 RSC.
- X. Li, C. Wen, M. Yuan, Z. Sun, Y. Wei, L. Ma, H. Li and G. Sun, J. Alloys Compd., 2020, 824, 153803 CrossRef CAS.
- L. Chen, F. Chen, G. Liu, H. Lin, Y. Bao, D. Han, W. Wang, Y. Ma, B. Zhang and L. Niu, Anal. Chem., 2022, 94, 7319 CrossRef CAS.
- Y. Zhang, Z. Wang, X. Liu, Y. Liu, Y. Cheng, D. Cui, F. Chen and W. Cao, FlatChem, 2023, 39, 100503 CrossRef CAS.
- C. Zhao, Q. Wang, H. Zhang, S. Passerini and X. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 15661 CrossRef CAS.
- S. Deshmukh, K. Ghosh, M. Pykal, M. Otyepka and M. Pumera, ACS Nano, 2023, 17, 20537 CrossRef CAS PubMed.
- T. Hu, J. Wang, H. Zhang, Z. Li, M. Hu and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 9997 RSC.
- A. Sarycheva, T. Makaryan, K. Maleski, E. Satheeshkumar, A. Melikyan, H. Minassian, M. Yoshimura and Y. Gogotsi, J. Phys. Chem. C, 2017, 121, 19983 CrossRef CAS.
- A. Kotta and H. K. Seo, Appl. Sci., 2020, 10, 5415 CrossRef CAS.
- N. Mironova-Ulmane, A. Kuzmin, I. Sildos, L. Puust and J. Grabis, Latv. J. Phys. Tech. Sci., 2019, 56, 61 CAS.
- M.-L. Lu, T.-Y. Lin, T.-M. Weng and Y.-F. Chen, Opt. Express, 2011, 19, 16266 CrossRef CAS.
- Y. Li, X. Zhou, J. Wang, Q. Deng, M. Li, S. Du, Y.-H. Han, J. Lee and Q. Huang, RSC Adv., 2017, 7, 24698 RSC.
- E. Lee, A. VahidMohammadi, B. C. Prorok, Y. S. Yoon, M. Beidaghi and D.-J. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 37184 CrossRef CAS.
- S. Xu, G. Wei, J. Li, Y. Ji, N. Klyui, V. Izotov and W. Han, Chem. Eng. J., 2017, 317, 1026 CrossRef CAS.
- Q. X. Xia, J. Fu, J. M. Yun, R. S. Mane and K. H. Kim, RSC Adv., 2017, 7, 11000 RSC.
- S. A. Maier, Plasmonics: fundamentals and applications, Springer, 2007, vol. 1 Search PubMed.
- K. Midander, A. Julander, J. Kettelarij and C. Lidén, Regul. Toxicol. Pharmacol., 2016, 81, 381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01936a |
| This journal is © The Royal Society of Chemistry 2025 |