
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular tension-driven self-recovering mechanochromism in organic microcrystals†
Weihan Guo cd,
Hua Zhaoa,
Mingda Wangac,
Leilei Siac,
Kaixin Yangac,
Guomin Xia*bc and
Hongming Wang*acd
cd,
Hua Zhaoa,
Mingda Wangac,
Leilei Siac,
Kaixin Yangac,
Guomin Xia*bc and
Hongming Wang*acd
aCollege of Chemistry and Chemical Engineering, Nanchang University, Nanchang 330031, China. E-mail: hongmingwang@ncu.edu.cn
bSchool of Advanced Manufacturing, Nanchang University, Nanchang 330031, China. E-mail: guominxia@ncu.edu.cn
cJiangxi Provincial Key Laboratory of Functional Crystalline Materials Chemistry, Nanchang 330031, China
dInstitute for Advanced Study, Nanchang University, Nanchang 330031, China
First published on 5th June 2025
Abstract
Self-recovering mechanochromic fluorescent (MCF) crystalline materials, requiring no extra treatment for cyclic use, are highly significant for practical applications. However, exploring design principles for these materials remains challenging due to the fracture and disorder of microcrystals. In this study, we present a twisted molecule, Np-H, which exhibits varying rates of self-recovering MCF behavior in its polycrystalline forms, Np-H-1 and Np-H-2, thereby revealing for the first time the crucial effect of intramolecular tension. Single-crystal X-ray diffraction analysis and theoretical calculations reveal that Np-H-1, with smaller torsional angles, exhibits a greater increase in intramolecular tension and enhanced intrinsic vibrations in response to mechanical stimuli, resulting in an accelerated self-recovery rate as compared to Np-H-2. Moreover, the counterparts composed of peripheral methyl or interposed phenyl provide further evidence to the pivotal relationship between intramolecular tension and self-recovering MCF behavior. Notably, Np-H-1 microcrystals exhibit bright emission and can self-recover within 20 minutes after grinding with easy repeatability, enabling the application of chameleon painting inks and edge-ball detection in table tennis.
Introduction
Intelligent responsive materials that generate visual signals in response to complex stimuli have garnered significant attention due to their advanced perception, adaptability, versatility, and wide-ranging applications.1–6 Among these, mechanochromic fluorescent (MCF) substances show particular promise, demonstrating emission changes when subjected to mechanical stimuli (e.g., pressure, scraping, grinding) in solid form.6,7 Unlike their inorganic counterparts, pure organic crystalline MCF materials are environmentally friendly,8 offer diverse molecular design possibilities, and exhibit enhanced sensitivity, making them ideal for applications in force sensors, biomedical sciences, and anti-counterfeiting measures.1,9 Recent developments in the aggregation-induced emission (AIE) concept have rapidly advanced the field of solid-state fluorescent organic materials, often revealing frequent MCF behaviors attributed to their twisted molecular configurations.10 However, many of these materials require additional stimuli, such as heat or solvent exposure, to revert to their original state, and few exhibit intrinsic self-recovery capabilities.11,12Self-recovering MCF materials are crucial for applications that require high durability and repeated use.13 However, achieving self-recovery after mechanical grinding remains a significant challenge due to the fracture and disorganization of microcrystals, in contrast to piezochromic fluorescent (PCF) materials, which respond to quantifiable hydrostatic pressure.2,14–16 A key question arises: How can we design self-recovering MCF materials, and what is the underlying mechanism driving their behavior? From a structural perspective, MCF materials rely on a twisted molecular architecture that can undergo substantial planarization under mechanical stress, leading to changes in electronic configurations and fluorescence characteristics.17–20 It is thus easy conscious of that this planarization process, inevitably, results in significant intramolecular tension accumulation within the microcrystals. By finely self-adjusting intermolecular interactions, this stress accumulation may further facilitate the occurrence and adjustment of the MCF material's self-healing behavior, aligning with the notable phenomena reported in the few existing literature (Fig. 1a and Table S1†).21–27
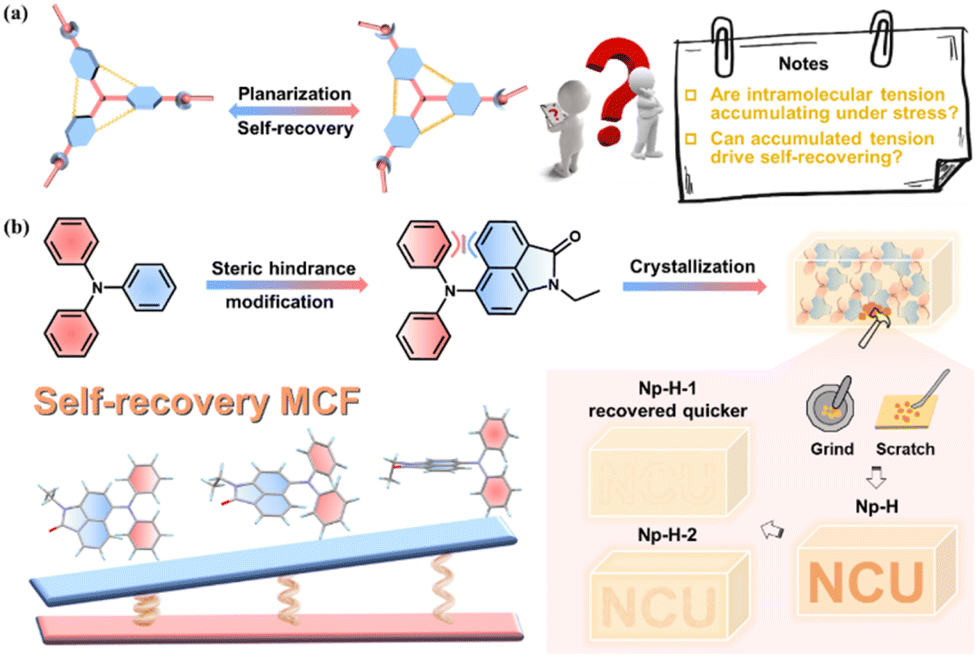 | ||
| Fig. 1 (a) Proposed mechanism of self-recovering MCF materials. (b) Design of Np-H with self-recovering MCF behaviors. | ||
Computational studies indicate that replacing the phenyl group in triphenylamine (TPA), a common MCF building block, with a naphthyl group enhances the response to intramolecular tension due to the increased torsional angles (Fig. S1†). Therefore, we designed and synthesized the counterpart Np-H by further introducing a π-conjugated lactam group, that is 1,8-naphthalenimide (Np). Indeed, Np is well known for its excellent fluorescence, electron-accepting properties, and ability to act as a strong hydrogen-bonding acceptor through its oxygen atom.28 This design enhances efficient emission and sensitivity to conformational changes from intramolecular charge transfer (ICT)29 and potential tension response effects.30,31 By varying self-assembly solvents, Np-H can spontaneously form a range of stable microcrystal structures, each exhibiting notably different self-recovery rates following mechanical grinding (Fig. 1b). These observations are quite rare and provide systematic verification of an intramolecular tension-driven recovery mechanism, supported by both theoretical calculations and single-crystal X-ray diffraction analysis. Furthermore, the optimal Np-H microcrystals were successfully demonstrated in practical applications, including edge-ball detection in table tennis matches and color-changing ink.
Results and discussion
Np-H can be easily synthesized, with the synthesis pathway and detailed structural characterizations provided in the ESI.† To predict its self-recovery MCF behavior, we performed single-molecule calculations (details in the ESI†). The optimized lowest energy ground state structure of Np-H shows a torsion angle of approximately 79.16° between the benzene and Np rings, indicating substantial intramolecular tension as anticipated (Fig. S2a†). The electron density in the HOMO is spread across the entire molecular skeleton, while in the LUMO, it is more localized in the Np ring, indicating an ICT state in the excited state (Fig. S2b†).32 The ICT effect is stimuli-sensitive, as the benzene ring can easily twist under pressure, leading to a red-shift in emission.33,34To verify changes in intramolecular tension at different torsion angles, we calculated the total energy of a single molecule with torsion angles (θ = 0–90°) between the benzene and Np rings (Fig. 2a(i)).35 Generally, as the torsion angle increases, the molecule's total energy decreases significantly, indicating greater stability. However, when the torsion angle reaches 70–80°, the total energy of Np-H levels off, with energy differences within this range being negligible. The torsional angle range corresponding to this energy valley represents the range of the stable crystal conformation. Fortunately, we obtained two high-quality Np-H crystals with torsion angles within this range, named Np-H-1 and Np-H-2 (Fig. 2a(ii)). This is consistent with their stable single-crystal structures. When the Np-H-1 molecule (with torsion angles of 77.13° and 72.79°) is subjected to external force, it tends to planarize, leading to a significant increase in energy. Thus, Np-H-1 is likely to rapidly self-recover to its initial state once the external force is removed. In contrast, Np-H-2 (with torsion angles of 81.32° and 83.36°) shows minimal energy changes and remains stable in the energy valley when planarized under external force. In consequence, Np-H-2 theoretically exhibits slower self-recovery compared to Np-H-1 due to insufficient change of intramolecular tension. Consistently, scatter plot analysis of RDG vs. sign (λ2)ρ confirms the different intramolecular tensions between Np-H-1 and Np-H-2, with higher sign (λ2)ρ values indicating greater tension (Fig. 2a(ii) and S3; † red represents intramolecular tension marked with dotted circles). When the sign of the spike on the right side, denoted as sign(λ2)ρ, is positive, it corresponds to the internal molecular tension (marked with black dotted circles) (Fig. 2a(iii)). Furthermore, the larger the value, the greater the tension (Fig. S4†).36 Therefore, the tension increase in Np-H-1 is higher than in Np-H-2 during planarization under pressure (assuming an extreme planarization value of 60°), resulting in faster self-recovering rate (Fig. 2a(iii)).
 | ||
| Fig. 2 (a) (i) The molecular potential energy surface of Np-H by rotating Np and benzene rings from 0° to 90° (every 10° a point); (ii) the molecular structure and color-filled isosurface for different conformation of Np-H; (iii) the scatter diagram plots of the electron density and its reduced gradient for different conformation of Np-H. (b) Root-mean-square deviation (RMSD) at 10 atm, 30 ns for (i) average 16 molecules and (ii) one of single molecule of Np-H-1 and Np-H-2 in aggregation; (iii and iv) top views of z molecular columns constructed by Np-H-1 and Np-H-2 in aggregation (space-filling models in them are selected single molecule aforementioned); (v and vi) torsion angles (θ1 and θ2) of the selected single molecule in Np-H-1 and Np-H-2 in the pressure equilibration of 1 atm–10 atm–1 atm. (c) (i and ii) 2D fingerprint plot generated by Hirshfeld surface; (iii) relative contributions of various interactions to the Hirshfeld surface area of Np-H-1 and Np-H-2 crystals. | ||
To further understand the aggregated state change of Np-H-1 and Np-H-2 under stress, the molecular dynamics (MD) simulations were performed.37 We choose 16 molecules in one simulation box, and first pressurize it to 10 atm directly. RMSD,38 a commonly employed metric for assessing structural alignment, demonstrates that lower values signify closer structural similarity. Analyzing the dynamic process of entire 16 molecules and one of the molecule, it revealed that both the average Root-mean-square deviation (RMSD) and single-molecule RMSD of Np-H-1 are higher than Np-H-2, which demonstrates that Np-H-1 deviates more from the initial conformation, which becomes more unstable (Fig. 2b(i and ii)). As shown in Fig. 2b(iii and iv), Np-H-1 has more ordered crystal structure than Np-H-2 after pressing at 10 atm for 30 ns, revealing greater potential for self-recovery (space-filling models in them are selected single molecule aforementioned).39 Then we simulated the MCF and self-recovering process of Np-H crystals (1 atm–10 atm–1 atm) to gather the changes in the intramolecular torsion angles throughout the process. As shown in Fig. 2b(v and vi), the torsion angles (θ1 and θ2) of the selected single molecule in Np-H-1 and Np-H-2 both fluctuate between 60° and 90°, which corresponding to the potential energy surface in Fig. 2a(i). The torsion angles of Np-H-1 fluctuates significantly during the pressurization phase (10 atm), indicating strong molecular vibrations and its highly sensitivity to pressure and easy self-recovery. In contrast, the molecular conformation of Np-H-2 shows weaker tendency to vibrate, suggesting that it is less likely to self-recover. In the meantime, the Hirshfeld Surface analysis (Fig. 2c(i and ii) and S5†) was conducted by partitioning electron density within the crystal lattice, exposing fingerprint plots for Np-H-1 and Np-H-2 crystals after pressing at 10 atm for 300 ns.39,40 The sector graphs reflect the relative contributions of interactions, with the total strength of O⋯H, H⋯O, C⋯H and H⋯C interactions in Np-H-1 surpasses that of Np-H-2 under stress, which infers that they are multiple C–H⋯π and C–H⋯O interactions in Np-H-1 after grinding (Fig. 2c(iii)). All these results indicate that the enhanced molecular vibrations and abundant intra/intermolecular interactions can effectively speed up the self-recovery rate of Np-H-1 in aggregation under stress. Detailed crystal structure analyses of pristine Np-H-1 and Np-H-2 crystals are provided in Table S2 and Fig. S6–8.†
With the Np-H on hand, we conducted detailed experiments on its photophysical properties and crystal polymorphs. As shown in Fig. S9,† the absorption spectra of Np-H in different organic solvents indicated a maximum absorption peak around 292 nm, corresponding to the structural peak of diphenylamine and its derivatives, and broad peaks at 444 nm, corresponding to the entire π-system of Np-H. For the photoluminescence (PL) spectra, it showed a gradual redshift and decreased fluorescence intensity with increasing solvent polarity, indicating the excited-state ICT effect in donor–acceptor (D–A) molecular structures (Fig. S10†).41,42 The fluorescence lifetimes of Np-H in different solvents were measured using the single-photon counting method (Fig. S11†). Furthermore, In N,N-dimethylformamide (DMF)/water mixtures, Np-H remains redshifted in wavelength and decreasing in intensity with the increase of water fractions (fw) from 0% to 60%; after that, when the fw exceeds 90%, amorphous nanoparticles' strong emission peaked at 582 nm appeared, suggesting a typical AIE characteristic (Fig. S12†).43 All photophysical properties of Np-H in solution are summarized in Table S3.†
Then Np-H-1 and Np-H-2 microcrystals were prepared using the liquid phase self-assembly method (details in the ESI†). The powder X-ray diffraction (PXRD) patterns and thermogravimetric analysis (TGA) curves coincided with their simulated patterns, suggesting identical molecular packing modes (Fig. S13 and 14†). The microcrystals were loaded on the filter paper after grinding for 10 minutes (Fig. 3a). As shown in Fig. 3b, Np-H-1 microcrystals exhibited a strong yellow emission peaked at 558 nm with a fluorescence quantum yield of 34.5%. Upon slight scratching with a spatula, the fluorescence emission redshifted to approximately 586 nm, and the quantum yield decreased slightly to 27.9%. To our delight, the ground Np-H-1 microcrystals returned to their initial yellow fluorescence emission within 20 minutes at room temperature, demonstrating pronounced self-recovering behavior. Identical tests on Np-H-2 microcrystals revealed a gradual red-shift in fluorescence emission from 575 nm to 590 nm under grinding. This fluorescence behavior almost reverted to the initial orange-yellow emission after 210 minutes (Fig. 3c), demonstrating self-recovery at a slower rate than Np-H-1. These observations align with the calculated results, indicating that incorporating large steric π-conjugated structure in easily twisted molecule (TPA) to increase intramolecular tension is a reliable approach for designing self-recovering MCF materials. Videos of these self-recovery process are available in the ESI.†
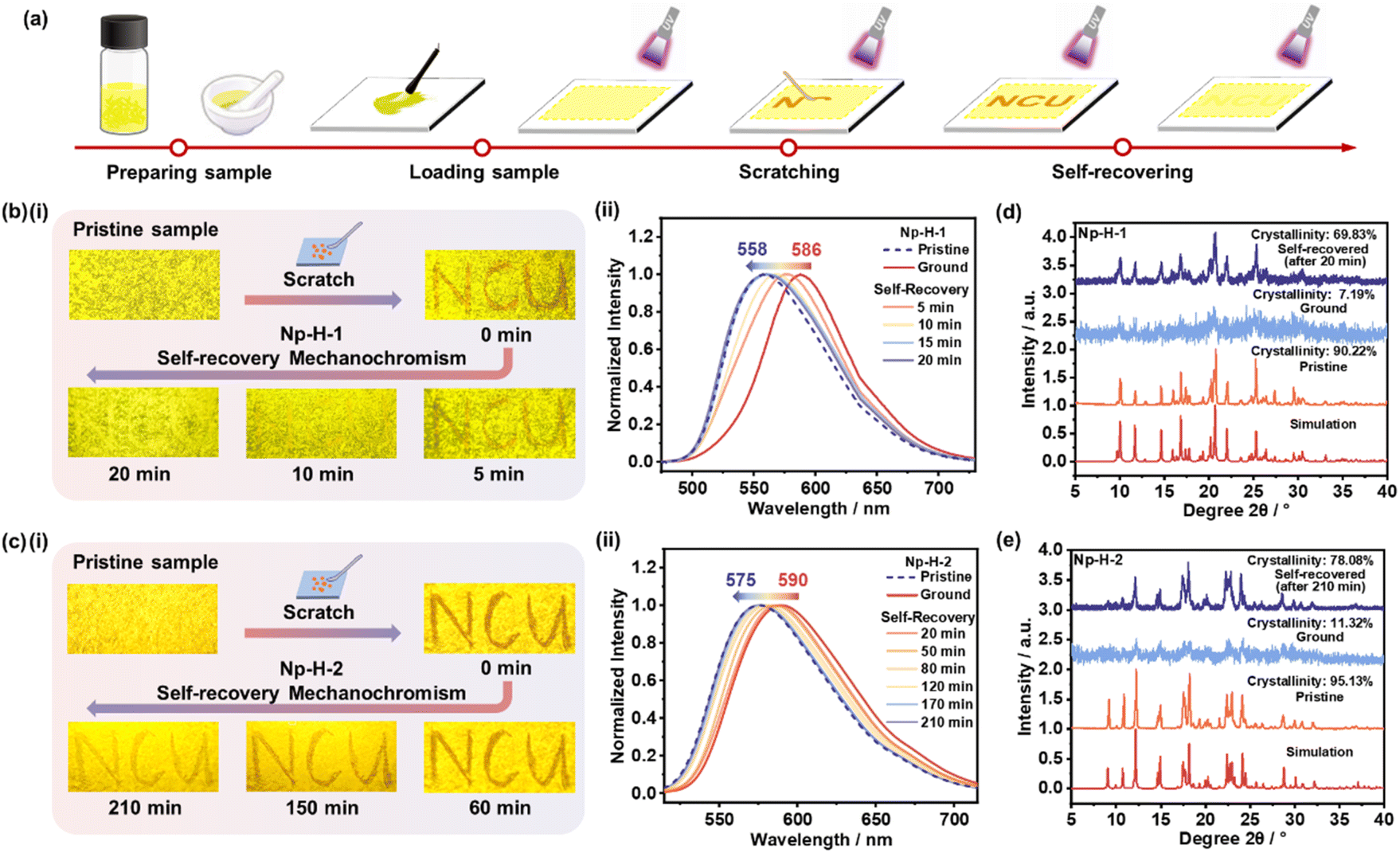 | ||
| Fig. 3 (a) Schematic representation of the self-recovering MCF process based on the representative compound Np-H-1. (b and c) (i) Fluorescence photographs of pristine and treated Np-H microcrystals under UV light illumination (365 nm). (ii) Normalized PL spectra of pristine Np-H microcrystals (dotted line) during the grinding-recovering (solid line) process. PXRD patterns of pristine, ground and self-recovered (d) Np-H-1, (e) Np-H-2 microcrystals. | ||
To reveal their microstructures, PXRD measurements were performed on Np-H-1 and Np-H-2 microcrystals during the grinding-recovery process (Fig. 3d and e). The results showed that both microcrystals transitioned from well-ordered crystalline to amorphous during grinding. Notably, the self-recovered Np-H-1 and Np-H-2 powders returned to crystalline phases with crystallinities of 73.49% and 78.08%, respectively, which would be facilitated by unchanged partial crystalline phases in the vicinity. Moreover, the grinding-recovery test showed that Np-H-1 could cyclically switch between yellow and orange emission at least five times (Fig. S15†), whereas Np-H-2 could only repeat the cycle once (Fig. S16†). In the meantime, the differential scanning calorimetry (DSC) curves were performed on pristine and ground Np-H-1 and Np-H-2 microcrystals from room temperature to 200 °C, demonstrating that there is rarely difference in Np-H-1 but a distinct glass transition peak appeared at 50 °C in ground Np-H-2 powders. As molecules are hard to move below the glass transition temperature, the peak in it to some extent proves the difficult self-recovery process of Np-H-2 that needs 210 minutes together without favorable circulability (Fig. S17†). Single-photon counting of Np-H-1 and Np-H-2 lifetime decay profiles indicated fluorescence from singlet states, with photophysical data summarized in Fig. S18 and Table S4.†
Meanwhile, three structurally similar molecules—Np-OMe, Np-Me, and Np-TPA—were synthesized to further explore the role of intramolecular tension in designing self-recovering MCF materials, and their single-crystal diffraction analyses together with fundamental photophysical characterizations were performed (Fig. S19–S29†). Further tests revealed that Np-OMe exhibit no MCF behavior, as confirmed by their PL spectra and PXRD patterns, which showed no significant change under mechanical pressure (Fig. 4a). To our pleasant surprise, a third crystal polymorph of Np-H, termed Np-H-3, was obtained and also exhibited no MCF behavior. Statistical analyses of intermolecular interactions in Np-H-1, Np-H-2, Np-H-3, and Np-OMe (Fig. 4b) indicated that Np-H-1 and Np-H-2 have relatively soft intermolecular interactions, allowing for not only stable initial crystalline structures but also MCF behaviors. In contrast, strong and abundant Ar–H⋯O/π and π–π interactions in Np-H-3 and Np-OMe greatly hinder their MCF behaviors. Meanwhile, it confirms that the stronger and denser molecular packing of Np-H-1 compared to Np-H-2 ensures structural stability in the initial state. It is demonstrated that the occurrence of MCF behavior is mainly related to the appropriate interactions in molecules.
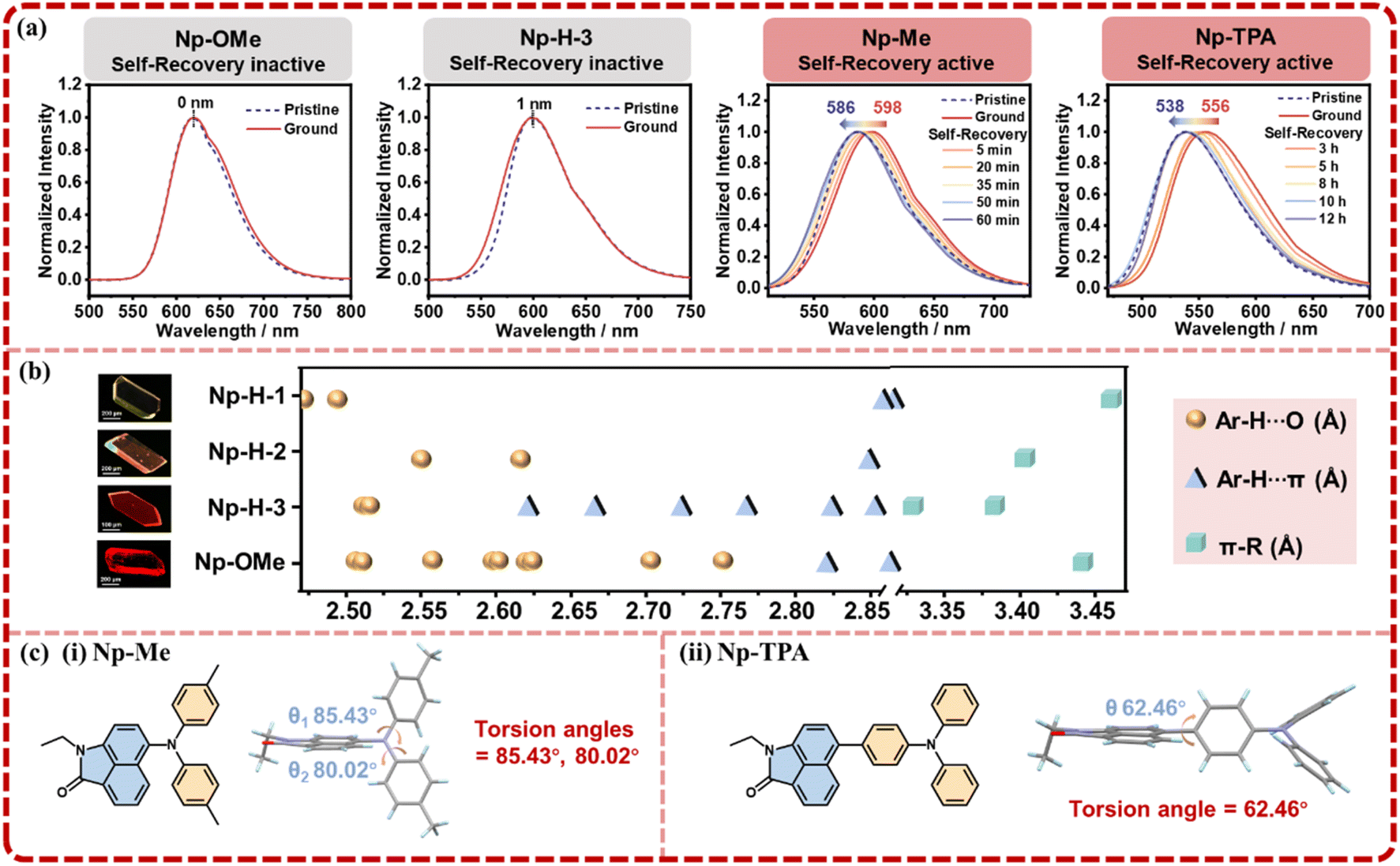 | ||
| Fig. 4 (a) Normalized PL spectra of pristine Np-OMe, Np-H-3, Np-Me, and Np-TPA microcrystals (dotted line) during the grinding-recovering (solid line) process. (b) Statistical chart of intermolecular interactions for locking the phenyl rotations. (c) The molecular formula and conformation of (i) Np-Me, (ii) Np-TPA. | ||
As for Np-Me and Np-TPA, they displayed MCF behaviors naturally, with self-recovery times of 60 minutes and 12 hours, respectively (Fig. S30†). The PXRD changes also align with the crystalline-amorphous-crystalline trend (Fig. S31†). Accordingly, the torsion angles of Np-Me and Np-TPA are approximately 82° and 62°, indicating medium to weak intermolecular tension (Fig. 4c). Notably, the TPA moiety in Np-TPA significantly extends the molecule's spatial range, reducing spatial hindrance and intramolecular tension, which leads to a slower self-recovery rate, consistent with the experimental results. From these observations, the design principles for self-recovering MCF materials are: (1) combining a highly twisted D–A structure with suitable intermolecular interactions ensures a stable crystalline structure and sensitive MCF behavior. (2) Increasing intramolecular tension through configuration planarization under mechanical stimulus facilitates self-recovery in microstructure. The lifetime decay profiles of Np-H-3, Np-OMe, Np-Me and Np-TPA microcrystals in the grinding-recovering process were tested by single photo-counting (Fig. S32). All the photophysical data of Np-H-3, Np-OMe, Np-Me and Np-TPA microcrystals were summarized in Table S5.†
Motivated by this unique self-recovering MCF behavior and bright emission property, we render Np-H-1 as promising writable fluorescent inks for painting. As shown in Fig. 5a, Np-H-1 powders were dispersed in PVA aqueous solution (0.05 g mL−1) and ground for 10 minutes.44 The yellow bottom colour was painted gently on a filter paper with a brush pen, and then a chameleon was painting with the other end of the pen. Owing to the rapid self-recovering performance, a polytropic and colorful painting of chameleon was obtained based on advanced Np-H-1 fluorescent inks. Due to its excellent self-recovering MCF behavior and recyclability, Np-H-1 is also suitable for edge ball detection in table tennis.45 This was demonstrated on a miniature table tennis table (Fig. S33†). As shown in Fig. 5b, when the ball hit the side edge, an orange fluorescent mark appeared, indicating an edge ball. Meanwhile, the trail would disappear in 20 minutes with the self-recovery of Np-H-1 microcrystals. This detection method uses the material's self-recovery property in a solid state, avoiding complex chemical reactions, high temperature heater or solvent evaporation.46 This approach offers a promising application for environmentally friendly self-recovering MCF materials. A video of this application is available in the ESI.†
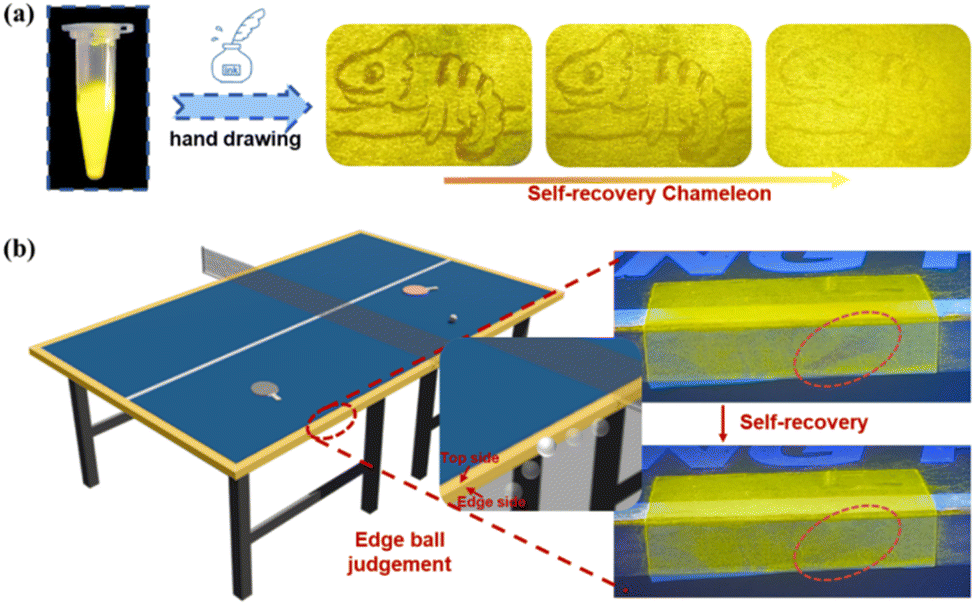 | ||
| Fig. 5 (a) Images of Np-H-1 powders as fluorescent inks under UV lamp and hand drawing on filter paper. (b) Edge ball judgment and self-recovery process. | ||
Conclusions
In summary, we report a twisted organic molecule (Np-H) displaying distinct self-recovering MCF speed in its polymorphic forms (Np-H-1/Np-H-2), highlighting intermolecular tension regulation. Microstructural investigations using single-crystal X-ray diffraction and theoretical calculation reveal that the Np-H-1 phase, characterized by reduced twisted angles and amplified intramolecular tension increment under mechanical loading, achieves faster self-recovery through intensified molecular vibrations. Finely self-adjusting interactions and compact post-pressure molecular aggregation further accelerates this recovering process. Parallel observations in analogous crystalline materials like Np-TPA also confirm the correlation between molecular tension engineering and self-recovering MCF behaviors. Notably, Np-H-1 demonstrates bright fluorescence with self-recovering in 20 minutes and multi-cycle durability, enabling innovative applications in painting inks and table tennis equipment through enhanced edge recognition technology.Data availability
Additional experimental data supporting this article are included in the ESI.†Author contributions
Weihan Guo: conceptualization, data curation, formal analysis, investigation, validation, visualization and writing – original draft. Hua Zhao: data curation and formal analysis. Mingda Wang: data curation and validation. Leilei Si: formal analysis and visualization. Kaixin Yang: formal analysis. Guomin Xia: conceptualization, formal analysis, funding acquisition, investigation, supervision and writing – review & editing. Hongming Wang: funding acquisition, supervision and writing – review & editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22063005, No. 22465023), the Natural Science Foundation of Jiangxi Province (No. 20212ACB203012, 20224BAB214003, 20232BAB203031), the Interdisciplinary Innovation Fund of Natural Science at Nanchang University (No. 9167-27060003-ZD2101, 9167-28220007-YB2113), and the Foundation of Jiangxi Provincial Key Laboratory of Functional Crystalline Materials Chemistry (No. 2024SSY05162).Notes and references
- Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed.
- G. Zhu, Z. Liu, Q. Qi, J. Xing and Q. Li, Angew. Chem., Int. Ed., 2024, 63, e202406417 CrossRef CAS PubMed.
- Z. Li, Y. Wang, G. Baryshnikov, S. Shen, M. Zhang, Q. Zou, H. Agren and L. Zhu, Nat. Commun., 2021, 12, 908 CrossRef CAS PubMed.
- C. Wang, Y. Yu, Y. Yuan, C. Ren, Q. Liao, J. Wang, Z. Chai, Q. Li and Z. Li, Matter, 2020, 2, 181–193 CrossRef.
- S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh and A. Mondal, Science, 2021, 373, 321–327 CrossRef CAS PubMed.
- G. Zhu, Z. Liu, H. K. Bisoyi and Q. Li, Adv. Opt. Mater., 2024, 12, 2301908 CrossRef CAS.
- Y. Yin, Q. Guan, Z. Chen, D. D. Deng, S. Liu, Y. Sun and S. H. Liu, Sci. Adv., 2024, 10, eadk5444 CrossRef CAS PubMed.
- Z. Man, Z. Lv, Z. Xu, Q. Liao, J. Liu, Y. Liu, L. Fu, M. Liu, S. Bai and H. Fu, Adv. Funct. Mater., 2020, 30, 2000105 CrossRef CAS.
- Q. Qi, G. Sekhon, R. Chandradat, N. M. Ofodum, T. Shen, J. Scrimgeour, M. Joy, M. Wriedt, M. Jayathirtha, C. C. Darie, D. A. Shipp, X. Liu and X. Lu, J. Am. Chem. Soc., 2021, 143, 17337–17343 CrossRef CAS.
- Y. Sun, Z. Lei and H. Ma, J. Mater. Chem. C, 2022, 10, 14834–14867 RSC.
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
- Y. Yang, X. Yang, X. Fang, K. Z. Wang and D. Yan, Adv. Sci., 2018, 5, 1801187 CrossRef.
- J. Zou, Y. Fang, Y. Shen, Y. Xia, K. Wang, C. Zhang and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207426 CrossRef CAS.
- Y. Chen, A. Li, X. Li, L. Tu, Y. Xie, S. Xu and Z. Li, Adv. Mater., 2023, 35, 2211917 CrossRef CAS.
- S. Liu, S. Sun, C. K. Gan, A. G. Del Águila, Y. Fang, J. Xiong, T. T. H. Do, T. J. White, H. Li, W. Huang and Q. Xiong, Sci. Adv., 2019, 5, eaav9445 CrossRef CAS PubMed.
- Y. Xu, K. Wang, Y. Zhang, Z. Xie, B. Zou and Y. Ma, J. Mater. Chem. C, 2016, 4, 1257–1262 RSC.
- Y. Dai, S. Zhang, H. Liu, K. Wang, F. Li, B. Han, B. Yang and B. Zou, J. Phys. Chem. C, 2017, 121, 4909–4916 CrossRef CAS.
- A. Li, Z. Ma, J. Wu, P. Li, H. Wang, Y. Geng, S. Xu, B. Yang, H. Zhang, H. Cui and W. Xu, Adv. Opt. Mater., 2018, 6, 1700647 CrossRef.
- S. Zhang, Y. Dai, S. Luo, Y. Gao, N. Gao, K. Wang, B. Zou, B. Yang and Y. Ma, Adv. Funct. Mater., 2017, 27, 1602276 CrossRef.
- H. Liu, Y. Gu, Y. Dai, K. Wang, S. Zhang, G. Chen, B. Zou and B. Yang, J. Am. Chem. Soc., 2020, 142, 1153–1158 CrossRef CAS PubMed.
- P. Xu, Q. Qiu, X. Ye, M. Wei, W. Xi, H. Feng and Z. Qian, Chem. Commun., 2019, 55, 14938–14941 RSC.
- P. S. Hariharan, N. S. Venkataramanan, D. Moon and S. P. Anthony, J. Phys. Chem. C, 2015, 119, 9460–9469 CrossRef CAS.
- T. T. Divya, K. Ramshad, V. C. Saheer and L. Chakkumkumarath, New J. Chem., 2018, 42, 20227–20238 RSC.
- M. Fan, Y. Cheng, B. Fang, L. Lai and M. Yin, Dyes Pigm., 2021, 190, 109311 CrossRef CAS.
- W. Guo, M. Wang, L. Si, Y. Wang, G. Xia and H. Wang, Chem. Sci., 2023, 14, 6348–6354 RSC.
- Z. Lv, Z. Man, Z. Xu, L. Fu, S. Li, Y. Zhang and H. Fu, Adv. Opt. Mater., 2021, 9, 2100598 CrossRef CAS.
- C. Zhu, Q. Luo, Y. Shen, C. Lv, S. Zhao, X. Lv, F. Cao, K. Wang, Q. Song, C. Zhang and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 8510–8514 CrossRef.
- F. Yu, H. Zhao, Y. Li, G. Xia and H. Wang, Mater. Chem. Front., 2022, 6, 155–162 RSC.
- M. Tan, Y. Li, W. Guo, Y. Chen, M. Wang, Y. Wang, B. Chi, H. Wang, G. Xia and H. Wang, Dyes Pigm., 2022, 201, 110243 CrossRef CAS.
- X. Wang, Q. Liu, H. Yan, Z. Liu, M. Yao, Q. Zhang, S. Gong and W. He, Chem. Commun., 2015, 51, 7497–7500 RSC.
- T. Suzuki, H. Okada, T. Nakagawa, K. Komatsu, C. Fujimoto, H. Kagi and Y. Matsuo, Chem. Sci., 2018, 9, 475–482 RSC.
- H. Gao, Y. Gao, C. Wang, D. Hu, Z. Xie, L. Liu, B. Yang and Y. Ma, ACS Appl. Mater. Interfaces, 2018, 10, 14956–14965 CrossRef CAS PubMed.
- Q. Qi, J. Zhang, B. Xu, B. Li, S. X.-A. Zhang and W. Tian, J. Phys. Chem. C, 2013, 117, 24997–25003 CrossRef CAS.
- A. Shimizu, Y. Misaki, C. Kondo, A. Kayama, T. Hirose, H. Sato and R. Shintani, Angew. Chem., Int. Ed., 2025, e202422448 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.02), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- E. A. Coutsias, C. Seok and K. A. Dill, J. Comput. Chem., 2004, 25, 1849–1857 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Sachdeva and M. D. Milton, Dyes Pigm., 2020, 181, 108539 CrossRef CAS.
- J. Long, F. Habib, P. H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed.
- S. Cho, T. S. Shim, J. H. Kim, D. H. Kim and S. H. Kim, Adv. Mater., 2017, 29, 1700256 Search PubMed.
- Y. Huang, Q. Gong, J. Ge, P. Tang, F. Yu, L. Xiao, Z. Wang, H. Sun, J. Yu, D. S. Li, Q. Xiong and Q. Zhang, ACS Nano, 2020, 14, 15962–15972 CrossRef CAS PubMed.
- J. Luo, Z. Wang, L. Xu, A. C. Wang, K. Han, T. Jiang, Q. Lai, Y. Bai, W. Tang, F. R. Fan and Z. L. Wang, Nat. Commun., 2019, 10, 5147 CrossRef PubMed.
- Y. Wang, I. Zhang, B. Yu, X. Fang, X. Su, Y. M. Zhang, T. Zhang, B. Yang, M. Li and S. X. A. Zhang, J. Mater. Chem. C, 2015, 3, 12328–12334 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 2352008–2352012. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02457a |
| This journal is © The Royal Society of Chemistry 2025 |