Chemical biology toolkit for exploring protein kinase catalyzed phosphorylation reactions
Sanela
Martić
and
Heinz-Bernhard
Kraatz
*
Department of Physical and Environmental Sciences, University of Toronto Scarborough, Toronto, Canada, M1C 1A4; Department of Chemistry, University of Toronto, Toronto, Canada M5S 3H6. E-mail: bernie.kraatz@utoronto.ca; Fax: +1 416 287-7279
First published on 5th October 2012
Abstract
Current interests in biochemical transformations based on protein kinase-catalyzed phosphorylations drive the identification and characterization of biological targets and potential inhibitors of protein kinase activity. A simple transfer of a phosphate group from adenosine triphosphate (ATP) to the Ser/Thr/Tyr residues of target proteins drives cellular processes, including cell expression, growth, and death. Currently, three major experimental approaches towards kinome analysis are available (a) genetic engineering of protein kinases, (b) modifications of target substrates, and (c) derivatization of ATP co-substrate. Each approach offers advantages but also has disadvantages, which are discussed in this perspective, alongside with a rationale for designing and developing biological tools for kinome study.
Introduction
Phosphorylation of proteins drives the cell cycle, cell growth, and death and, alongside dephosphorylation, regulates the metabolic pathways and cell communication.1 The fundamental biochemical reaction depicted in Fig. 1 involves the transfer of the γ-phosphate group from adenosine triphosphate (ATP) to a target protein containing one of the amino acids Thr, Tyr, Ser or His.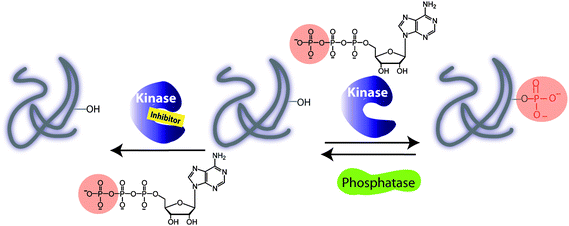 | ||
| Fig. 1 Illustration of a kinase catalyzed phosphorylation of protein substrate in the absence and presence of an inhibitor. A phosphorylation is carried out with adenosine triphosphate (ATP) and a protein kinase. A dephosphorylation by a protein phosphatase leads to removal of a phosphate group and regeneration of a protein. | ||
Among 500 protein kinases, the target specificity is governed by a linear recognition sequence proximal to phosphorylation site of a target, protein docking domains and protein cellular localization. While the consensus sequence may be a useful strategy for identifying the protein kinase substrates, the molecular basis of the substrate recognition by a docking domain of a protein kinase is sometimes required. In addition, the interactions between scaffold proteins and the specific regulatory kinase subunit play a critical role in directing the cellular localization and expression of protein kinase. For example, the cyclins bind cyclin-dependent kinases (CDKs) and induce conformational changes thereby enhancing kinase substrate specificity and subcellular localizations. Other kinases require the prephosphorylation in order to be activated. Dephosphorylation reactions involving phosphatases remove a phosphate group from the substrate and regenerate protein (Fig. 1). Phosphorylation and dephosphorylation reactions are controlled, allowing functions to be turned on and off.2 As a result of protein phosphorylation, changes in the local protein micro-environment take place, giving rise to protein conformation changes, which drive signalling pathways. For example, the cellular progressions during the cell cycle are driven by a number of CDKs and other protein kinases. The cell cycle regulation is important for preserving the genome integrity and serves as a barrier against cancer.3 In general, the interrogation of the cell division is achieved by cellular DNA staining assay, but this method relies heavily on the cellular uptake, and provides only a snapshot of the cellular dynamics, unless the cells are lysed.4 In addition, DNA dyes do not bind the phosphoprotein targets or their respective protein kinases. For protein kinase profiling, the antiphosphoantibodies may be used to target the phospho-Thr/phospho-Tyr/phospho-Ser target proteins (pThr/pTyr/pSer) as a result of the protein kinase activity, or phosphorylated protein kinases downstream the signalling pathway. Hence, the immuno-fluorescence assays with specific antibodies have been explored that target the cell-cycle regulators, overexpressed kinases or phosphorylated protein kinases.5 Several proteins are directly involved in the cellular apoptosis and tumorigenesis.6 For example, the mitogen-activated protein kinases (MAPK)7 pathway, the phosphorylation cascade, is directly linked to human malignancy. A diverse array of growth factors, kinases and phosphatases drives the cell-signalling phosphorylation events making the MAPK cascade a therapeutic target for cancer treatment. Typical strategies employed for the evaluation of signalling pathways use a radio-label, [γ-32P] ATP as a benchmark test, in which the successful transfer of 32P-γ-phosphate to a protein target is monitored.8 Further improvement has been made by using phospho-specific antibodies or target-specific phosphoantibodies.
Since the under/over expression of protein kinase has been linked to diseases, including diabetes, cancer and neurodegenerative disorders,9 probing phosphorylation reactions in vitro and in vivo and their detection are of immediate interest. Importantly, the activity of kinases can be controlled by the interaction with small molecules, some of which are ATP mimics.10 Moreover, the “druggable” characteristic of protein kinases reinforces the need for drug design and inhibitor screening. There are significant challenges associated with the design of selective kinase inhibitors, since kinases possess a nearly identical binding pocket, making selective inhibition by small molecules a formidable task. However, a number of successful strategies have been reported that allow for a selective inhibition and treatment of disorders. Of about eighty small molecule inhibitors that have reached the clinical trials, only a handful, such as Imatinib or Gleevec, has been approved by the U.S. Food and Drug Administration. More recent inhibitor design strategies include the potent organometallic conjugates.11,12 Radiometric or immunoassays are typically used for inhibition studies.
Understanding the kinome is one of the frontiers in the proteomics, but relating all protein targets that are phosphorylated by a specific kinase is for the most part still elusive and has been achieved only in a select few cases. The cellular targets of protein kinases are part of the complex signalling pathways and their identification is challenging due to overlapping substrate specificity. The conventional methods that are used to study the activity of protein kinases and screen potential inhibitors are associated with several setbacks, such as low spatio-temporal resolution, the need to handle and dispose of radioactive material and the cost of antibodies and issues associated with antibodies protocols. In addition, these methods do not allow for detection of the phosphorylation in living cells. There is a clear need for alternative methodologies that provide new insight into signalling pathways under real-to-life conditions, allow to explore protein kinase activity and inhibition and target selectivity. This perspective offers an overview of protein kinase-driven phosphorylations with a focus on understanding protein–co-substrate–target interactions and detection of protein kinase activity and inhibitor screening. Recent reviews focus on high-throughput screening of phosphorylation and dephosphorylation in complex proteomes and living cells, but largely omit the extensive work done on ATP derivatization, substrate modification and their applications.13 As part of this perspective, we focus on new research tools that have been developed to study phosphorylations and kinase–substrate–co-substrate interactions and outline new developments in protein engineering, substrate engineering, and ATP derivative design. The three approaches discussed in this perspective underline the need for kinase, substrate and inhibitor identification which may shed light on the convoluted signalling cascades.
Engineering protein kinases
Protein kinases possess an ATP binding pocket, that is conserved among the hundreds of protein kinases, and make targeting and monitoring the activity of specific protein kinases in vivo a complex task. A number of strategies have been reported that make use of genetic engineering of protein kinases in which the kinase domain can be modified as illustrated in Fig. 2. In this section we will address covalent modifications of different kinase domains for the purpose of studying phosphorylation reactions which will be followed by the kinase-inhibitor complexation in the absence of phosphorylation.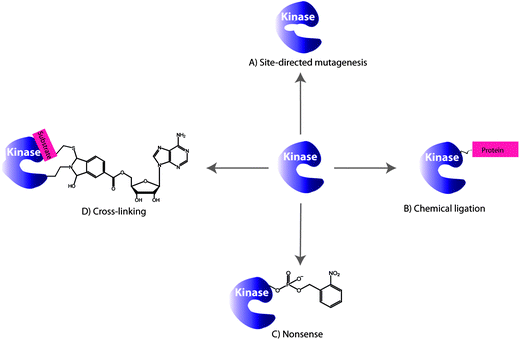 | ||
| Fig. 2 Selected examples of the engineering protein kinase approaches for monitoring phosphorylations: (A) site-directed mutagenesis, (B) chemical ligation, (C) nonsense approach and (D) cross-linking. | ||
Site-directed mutagenesis
Shokat and coworkers explored the site-directed mutagenesis to modify specific amino acid residues within the ATP binding pocket or the catalytic regulation domain of a protein kinase in such a way that the binding pocket has different spatial properties (Fig. 2A). This in turn allows binding of ATP derivatives in the binding pocket with higher affinities compared to ATP and searching for the proteins that are targeted by a specific kinase and selective inhibitors.Typically, ATP is an appropriate substrate for a wild type (WT) protein kinase and fits in the binding pocket of the enzyme. The derivatization of ATP, in some cases, eliminates the binding ability to WT protein kinase and is called “orthogonal” since it is completely silent in normal cells. In turn, the protein kinase of interest may be engineered to accept the modified ATP. An amino acid residue within the ATP binding pocket of the kinase, the first residue of the hinge connecting the C- and N- terminal lobes, is the “gate-keeper” residue and prevents the entry of a chemically N6-modified ATP (vide infra). Originally, the Ile338 was identified as the “gate-keeper” residue in WT Tyr kinases that restricts the utility of N6-ATP. In the “gate-keeper” mode, the bulky substituent effectively closes up the hydrophobic pocket making it inaccessible to the co-substrate. In order to accommodate a modified ATP co-substrate, a modification of the kinase binding pocket, which is remote from the regulatory and catalytic regions, has been achieved by Shokat et al. The “opening the gate” is achieved by amino acid point mutations in kinase. Earliest examples of active site protein kinase modifications include the mutations of the v-Src Tyr kinase because of the existing crystallographic data of Tyr kinases. For example, the Ile338Gly mutation in v-Src affords a mutated protein kinase capable of using N6-ATP derivatives as shown in the crystal structure of v-Src and N6-benzyl-ADP in Fig. 3A.14 The crystal structure also depicts the key interactions between Leu273 and Leu393 and a cosubstrate. The danger in the kinase mutation approach is that the enlarged co-substrate binding site may shut-down the enzymatic activity altogether. To overcome such limitation, a second mutation may be introduced to restore the kinase activity. In some cases, the second mutation enhances the β-sheet stability and promotes the correct folding of mutant kinase. For example, Ile338Gly and Leu393Met mutations increased the selectivity of mutant Src for N6-ATP over unmodified ATP. Interestingly, a protein kinase mutation requires that the N6 modification be relatively bulky, but not too bulky, to achieve favourable kinase/cosubstrate recognition.15 It is important, however, to note that the co-substrate binding site remains unchanged by the point mutations and ATP modifications as demonstrated by a comparative study with the WT kinase–ATP pair. The detailed discussion on the ATP derivatization will be presented in the later section. The utility of this approach lies in the ability to identify cellular substrates of individual protein kinases selectively even in the presence of hundreds of kinases. Shokat et al. used the chemical genetics approach to discover target proteins for a range of Tyr, Thr and Ser protein kinases including v-Src, CDK1 and CDK7 among others.16
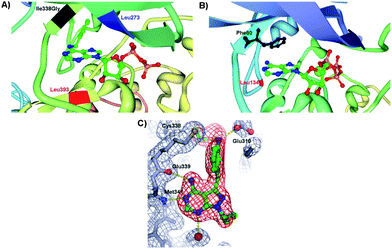 | ||
| Fig. 3 (A) Insight view of mutated v-Src and N6-(benzyl) ADP showing the projection of benzyl ring of the cosubstrate into a kinase pocket and the interactions with the key amino acid residues: Ile338Gly (highlighted in black), Leu273 (highlighted in blue) and Leu393 (highlighted in red) (PDB 1ksw).14,17 (B) Enlargement of the hydrophobic pocket in CDK2 containing the ATP and the key amino acid residues Phe80 (highlighted in black) and Leu134 (highlighted in red) (PDB 1hck).18 (C) Crystal structure of pyrazolopyrimidine inhibitor covalently attached to Thr338Cys c-Src protein kinase. The points of contact of interest are labeled. | ||
In the crystal structure of WT CDK2, Fig. 3B, several “gate-keeper” amino acid residues were identified such as Phe80 and Leu134, which regulate the cosubstrate recognition and binding. A systematic study of the mutant CDK2 with the “gate-keeper” modifications revealed that only Phe80 mutant kinase retained substantial catalytic activity with a number of synthetic N6-modified ATP derivatives.18 The largest N6-(cyclooctyl)ATP was a poorer substrate for a “gate-keeper” kinase than cyclopentyl and cyclohexyl derivatives. ATP derivatives were not good substrates for WT CDK2 since kinase binding site cannot accommodate the N6 modification of the ATP. Importantly, mutated kinases underwent similar enzymatic kinetics as the WT kinases with ATP. Recent investigations into “gate-keeper” biochemistry revealed that more hydrophobic residues in the hinge region are linked to increased levels of catalytic activity for Tyr kinase and have since became targets for inhibitor design.19 The hypothesis was that proteins having the small “gate-keeper” residues (such as Thr) will have larger back cavities and potentially may exhibit greater inhibitor potency and selectivity.20 Since the hydrophobic cavity of the protein kinase may be controlled by the “gate-keeper” residue, the site-directed mutagenesis was next expanded towards the studies of inhibitors of a protein kinase activity. The covalent complementarity approach between an engineered “gate-keeper” and an electrophilic inhibitor allows for the determination of potent inhibitors.21 Instead of incorporating Gly, the Cys “gate-keeper” residue was introduced into c-Src protein kinase (Thr338Cys c-Src) which eliminated the need for enlargement of the ATP binding pocket and has maintained the phosphorylation kinetics that were similar to WT c-Src with ATP. The systematic study of 20 pyrazolopyrimidines with WT and mutated Src demonstrated that these compounds were inefficient inhibitors of the WT kinase, but that their inhibitory activity peaked with engineered Thr338Cys c-Src. The reasons for this dramatic inhibition with the kinase containing the Cys as a “gate-keeper” were ascribed to the interactions of the inhibitor with the kinase pocket. The cocrystal structure of Thr338Cys c-Src protein kinase in Fig. 3C shows the vinylsulfonamide-based pyrazolopyrimidine inhibitor sitting in the protein pocket. The pyrazolopyrimidine interacts with the hinge domain of c-Src (Met341 and Glu339), the oxygen atoms of the sulphonamide interact with the backbone of Asp404, while the sulfonamide group makes a hydrogen bond with Glu310 of the αC helix. In addition, the crystal structure revealed the covalent linkage between Cys338 and the inhibitor. This study demonstrates that the increased inhibition activity may be achieved through the site mutations of “gate-keepers” in vitro and in cells.
The ultimate goal of the genetic modifications is to use mutant kinases specific for ATP derivatives to tag directly kinase substrates in whole cells or cell lysates, and to screen for inhibitors. In this way, the complex signalling pathways may be identified through the substrate labeling and kinase-substrate matching. One of the major challenges associated with the genetic mutations of protein kinases is their use of endogenous ATP in in vivo studies which relies heavily on radio-labeled and immunoassay approaches.
Chemical ligation
Since the modifications at the active site may impede with the kinase activity and target selectivity of some kinases, the chemical ligation (Fig. 2B) at the kinase periphery away from the ATP binding pocket may alleviate these issues. Importantly, the selectivity for the endogenous ATP is maintained. The chemical ligation is a versatile strategy for biochemical and biophysical studies of complex biological processes, such as the protein kinase-driven phosphorylations.22 In this approach, the kinase activity is triggered by the chemical or light stimuli which can be used to monitor phosphorylations as well as the conformational changes associated with the kinase activity. For example, the engineered catalytic domain of the focal adhesion kinase (FAK) was achieved by modifying it with a small protein that inactivates this protein kinase.23 The inactivity was due to the increased flexibility of the catalytic domain, upon ligation, which affected its binding to ATP and the substrate. The FAK activity was simply restored by addition of the rapamycin or non-immunosuppressive rapamycin inhibitors which triggered the FAK autophosphorylation. In another example, the chemical ligation of a peptide to a signal transducer and activator of transcription 6 (STAT6) protein kinase was demonstrated. STAT6 is one of the proteins that reside in the cytoplasm when unphosphorylated, but upon phosphorylation at Tyr641 by a Janus-kinase (JAK), the STAT6 homodimers are formed via their respective SH2 domains.24 The dimerization triggers translocation into nucleus where STAT6 dimer binds DNA and promotes gene expression. To monitor the mobility of the STAT6 dimer in cells, the ligation to a STAT6 protein was achieved through a C-truncated STAT6 with a peptide domain containing the phosphorylated Tyr641 and a fluorescent label (Cy5). The resulting STAT6 construct exists as a dimer and efficiently binds DNA fragments in vitro in a manner similar to the WT STAT6 dimer. The dimerization of STAT6 construct was UV-triggered by nitrophenyl ester deprotection of phosphorylated Tyr641 residue. This process was also evident by monitoring fluorescence of Cy5 and the translocation of STAT6 dimer from cytoplasm into the nucleus of a variety of cell lines, including A431 and COS-7, etc.25The chemical ligation was also explored for determination of the conformational changes associated with kinases in addition to the determination of phosphorylation levels. These assays dramatically differ from the conventional approaches previously described which exclusively monitor kinase activity by sensing the phosphorylation. To illustrate the principle of kinase design for monitoring inhibitor binding, and the subsequent conformational changes of kinase, we will explore a representative example based on the p38α and PKA.26 In order to minimize the interference with respect to normal kinase function, the Gly-rich loop, rather than the activation loop of a kinase, was modified. By placing the fluorophore within the Gly-X-Gly-X-X-Gly (X is any amino acid) motif of the Ser/Thr kinase p38α, the protein conformational changes may be monitored upon inhibitor binding (Fig. 4A and B). For this purpose, several fluorophores were tested, such as acrylodan, fluorescein, pyrene, nitrobenzoxadiazole and 5-((2-[iodoacetyl)amino]ethyl)amino)naphthalene-1-sulfonic acid due to their optical properties. When inhibitor binds p38α, the conformational changes within the Gly-rich and the activation loops results in the reduced fluorescence (Fig. 4C). Unlike traditional activity-based screening assays, this assay uses enzymatically incompetent kinase conformations in order to identify inhibitors, which stabilize the inactive forms of protein kinases and induce fluorescence quenching. While the Gly-rich loop is conserved in 80% of kinases making this method highly general, a careful selection of the kinase-labelling strategies is required. The types of ligands used to label kinases must agree with the conformational environment of a kinase and be tolerated by it for this labeling strategy to be successful. In addition, the decrease in the fluorescence response upon inhibitor binding and conformational change is not ideal and signal-on would be more beneficial.
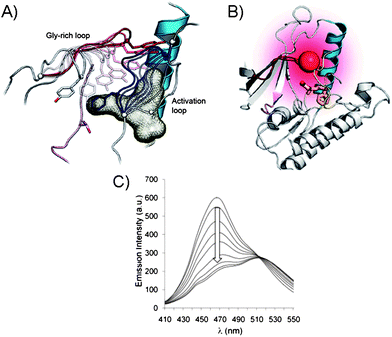 | ||
| Fig. 4 (A) Gly-rich and activation loops in the active p38α protein kinase domain. Conformational changes upon inhibitor binding to the active kinase domain (induced changes are color coded: for activation loop from white to dark blue and for Gly-rich loop from white to dark red). (B) Conformational changes triggered by inhibitor binding in p38α result in protein folding. The Gly-rich loop was labeled with a fluorophore acrylodan (red ball) to generate a direct fluorescence binding assay. (C) Acrylodan emission at 468 nm decreases and is red-shifted upon binding of inhibitor to the fluorescently labeled p38α.26 | ||
Other guest-induced conformational switching in protein kinases has been demonstrated recently in a three-hybrid complex in an attempt to screen inhibitors of protein kinases.27 In this case, a protein kinase PKA was attached to the C-terminal fragment of split-firefly luciferase (Cfluc) which is known for its bright fluorescence. The coiled-coil peptide, acting as a protein support, was attached to the N-terminal fragment of split-firefly luciferase (Nfluc). In order to induce a fluorescence response, the Cfluc and Nfluc domains must be in the close proximity. To achieve this requirement, the Jun-staurosporine peptide conjugate was used. The Jun peptide binds the coiled-coil segment while the staurosporine unit binds the active site of a PKA kinase giving rise to the ternary protein complex. The formation of the ternary complex brings the active luciferase fragments together and restores fluorescence. This assay is extremely useful for monitoring the competitive inhibitor binding and was easily extended to other kinases.31 The obvious drawback of this design is the use of staurosporine, which may have low affinity for MAPKs.28 To address the inhibition of MAPK pathways, alternative inhibitor must be explored.
Nonsense suppression
Genetic protein engineering took a giant leap forward when the nonsense suppression technology was introduced by Schultz and co-workers, which allowed for a site-specific incorporation of phosphoamino acid mimics within a given protein (Fig. 2C).29,30 Today, incorporation of unnatural amino acids can be performed in vivo into both Escherichia coli and eukaryotes. Here, instead of using multiple chemical ligations, the desirable amino acid mutagenesis is achieved by using mischarged tRNAs.31 A detailed discussion on the topic of nonsense codon suppression has been recently published.32 Here, we will focus on the utility of the nonsense methodology for studying protein kinase phosphorylations. To this effect, the replacement of the phosphoamino acid with a mimic in a phosphoprotein creates a phosphoprotein that is resistant to the phosphatase action which may simplify the search for phosphorylated targets in the complex media. The non-hydrolyzable phosphorylated Tyr mimetic, p-carboxymethyl-L-phenylalanine, was introduced in the human signal transducer and activator of transciprtion-1 (STAT1) protein, instead of phosphorylated Tyr701, with site selectivity due to the action of RNA/aminoacyl-tRNA synthetase.33 This approach was useful for monitoring the STAT1 dimerization and DNA binding by using the radiolabeled DNA fragments. It is possible, however, to avoid radiometric detection of STAT family of proteins by introducing the fluorophore as a point mutation in a kinase. For example, in the STAT3 the L-(7-hydrophycoumarin-4-yl)ethylglycine, a fluorescent dye, was positioned at the Trp564 site which is sensitive to the phosphorylation at the neighbouring Tyr705 residue.34 The methodology requires a single point mutation in the correct position and the fluorophore which is sensitive to the STAT phosphorylations which may be monitored by fluorescence. While tRNA has been used for genetically encoding proteins within cells with number of unnatural amino acids with excellent site-specificity,35 the incorporation of the phosphoamino acid remains a challenge. The limitations of the nonsense suppression approach include the low yielding protein product, and the requirement for the extracellular reaction media due to the inability of phosphomimics to penetrate the cell membrane. To extend the utility of the nonsense suppression, the access to the amino acid transporters or the esterase-sensitive protective groups is required. Once the phosphomimics are in the cell, however, the toxicity becomes a challenging issue and the expression of the native proteins may be compromised.Cross-linking
In addition to the point mutation and chemical ligation of protein kinases, the chemical transformation may also be used for kinome analysis. A number of guest-mediated approaches have been exploited for kinase labelling in crude proteome, such as the cross-linking method depicted in Fig. 2D. It is worth mentioning that this protocol requires a substitution of a Ser or Thr by a Cys residue. Prior to cross-linking reactions, the biomolecular recognition between the protein kinase and the guest is achieved by using the ATP derivative or a mimic, such as an inhibitor. The guest molecule carries an additional functionality that is used for signalling, following the covalent binding to kinase.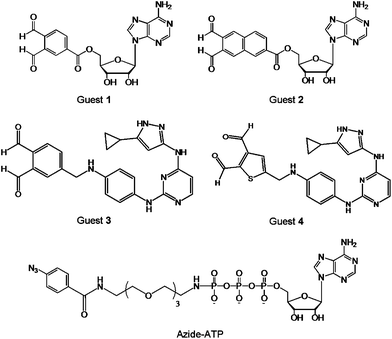 | ||
| Fig. 5 Structures of the cross-linking guests o-phthaldialdehydebenzoyl adenosine (Guest 1), naphthalene-2,3-dicarboxaldehyde adenosine (Guest 2), aminopyrazole o-phthaldialdehydebenzoyl (Guest 3), aminopyrazole thiophene-2,3-dicarboxyaldehyde (Guest 4), and azide-ATP. | ||
Alternatively, the photo-crosslinking moiety can be enzymatically introduced at the phosphate site of a substrate, via protein kinase and γ-labeled ATP analogue.38 To demonstrate this principle, a γ-aryl azide analogue of ATP (azide-ATP), shown in Fig. 5, was selected by Pflum and Suwal as the photo-crosslinker.39 Following the phosphorylation of the rhodamine-labeled substrate by azide-ATP, the UV irradiation induced the cross-linking of the phosphorylated substrate and CK2 protein kinase. The identification of the kinase-substrate product was achieved by mass spectrometry, Coomassie Blue staining, and fluorescence scanning. In addition, the o-phthaldialdehyde may be replaced by a less reactive five-membered heterocyclic dialdehyde. Using this strategy it was possible to cross-link specific Cys-containing substrate to its corresponding kinase in the presence of the complex cellular protein mixture without significant background cross-linking. Since the UV irradiation is necessary to induce the cross-coupling reaction, the utility of this approach with cell lysates or homogenized tissue samples may be limited due to the detrimental effects of UV irradiation on other cellular components.
In all cases, the direct point mutations, chemical ligations, nonsense suppression, chemical derivatizations or guest-induced transformations have been used to probe the kinase activity and identify the phosphorylated targets as well as inhibitors. The careful selection of mutation site or ligation is key to this approach in order to avoid the detrimental effects that the protein manipulation could have on the protein kinase conformation and activity. The following sections on the substrate modification and cosubstrate derivatization bypass the structural kinase requirements for enzymatic activity and offer a different angle for studying the phosphorylation events.
Engineering substrates
An important area of the kinase research is the identification of the target substrates and their phosphorylations, which allows for rapid kinase and substrate profiling. In this context, the labeled substrates may serve as reporters of kinase activities and provide access to the kinase-substrate combinations found in the underlying biochemistry. Commonly, conformational changes in substrates induced by the addition of an anionic phosphate group can be monitored fluorometrically using fluorescently labelled substrates (Fig. 6). In general, two basic principles exist for monitoring phosphorylations and are based on the fluorescence-ON (Types I, III, IV) and fluorescence-OFF signalling (Type II). Peptide and protein-based fluorescent sensors of protein kinase activity have been previously described with respect to design and application.42 Here, we discuss utility of peptide- and protein-based sensors for probing phosphorylations as well as protein kinase conformation.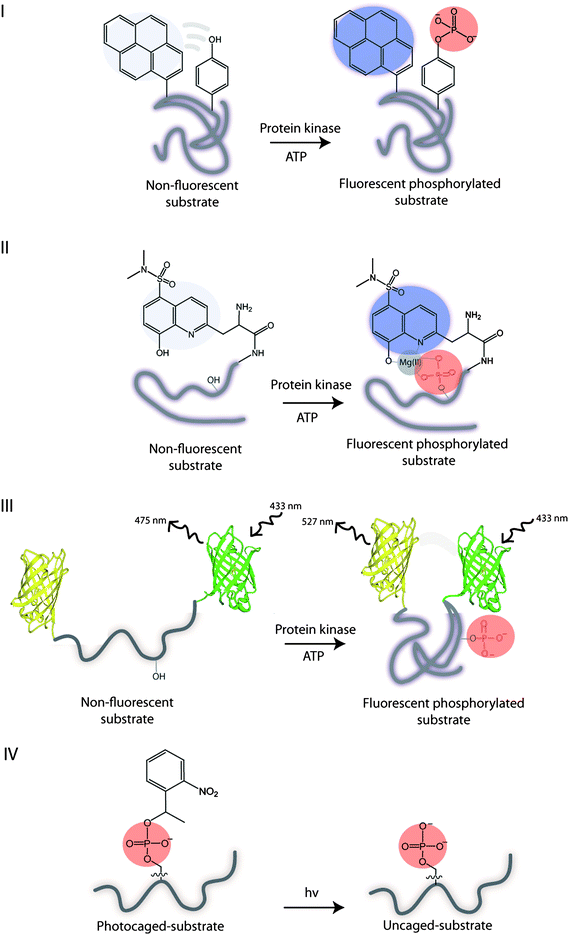 | ||
| Fig. 6 Illustration of the substrate biosensor types: I – phosphorylation results in a self-healing fluorescent substrate. II – guest binding to the fluorescent phosphorylated substrate. III – phosphorylation results in the conformational change of substrate and FRET. IV – UV irradiation of the photocleavable caged phosphorylated substrate yields an uncaged phosphorylated substrate. | ||
Fluorescence spectroscopy is a widely used technique for in vitro and in vivo imaging of biological processes and has been recently applied to the direct and continuous monitoring of phosphorylations. This approach offers a number of advantages, including ease of synthesis, the availability of synthetic fluorophores, and an optical response that is tunable. Contrary to radio-labelling and immunoassays, which detect kinase activity in a static fashion, the fluorescent substrate approach targets kinase selectivity and specificity and allows for measurements in real time as the phosphorylation event is taking place. The main consideration in the fluorescent substrate design is to minimize the interference by a fluorophore and to link the phosphorylation event to the fluorescent change. The number of different approaches have been designed and developed to achieve this requirement and will be discussed next.
Type I sensor
Lawrence and coworkers used a proximal fluorophore, such as pyrene, to monitor the neighbouring Tyr phosphorylation on a single peptide by a range of the non-receptor and receptor Tyr kinases.43 The idea is that the phosphorylation of Tyr disrupts the interactions between pyrene and Tyr and enhances the pyrene fluorescence. Hence, the inherent aromatic amino acid acts as an internal quencher of the fluorescence (Fig. 6, I). More recently, the cascade yellow and oxazine fluorophores were employed, due to their high quantum yields, for monitoring the multiple kinases simultaneously.44 The Lyn and Abl kinase peptide substrates were correspondingly modified with different fluorophores, and when phosphorylated no cross reactivity or interference was observed between these kinases and their substrates. By using the multicomponent fluorescence assay, the dramatic differences in the activity of Lyn and Abl protein kinases were observed in the imatinib-resistant cell line, demonstrating the utility of this approach for monitoring upregulated protein kinases. Despite its versatility, the methodology is limited to the phosphorylation of Tyr residues exclusively.Type II sensor
Alternatively, the fluorescent substrate of a Type II may be used to monitor phosphorylations via intermolecular interactions between the fluorescent phosphorylated peptide substrate and the guest.Hong et al. explored a small bis(Zn(II)-dipicolylamine) complex, containing the dabcyl or phenothiazine quencher unit, for binding to the fluorescent phosphorylated peptides.50 Upon Zn(II) coordination to phosphate group the fluorescent quenching was observed. Unfortunately, these small molecules are signal-off indicators which are less desirable than the ratiometric or signal-on probes.
In a similar fashion, the fluorescent peptide decorated with quantum dots was phosphorylated by Abl and Src protein kinases.51 Upon phosphorylation, the antiphosphotyrosine antibody binds the peptide and results in the FRET between the quantum dot of a phosphopeptide and the FRET-acceptor labeled antiphosphotyrosine antibody. This methodology was however limited to in vitro studies.
Type III sensor
In contrast to the above design which takes advantage of the intermolecular interactions between the phosphate group in a target and a guest, the intramolecular interactions within the phosphorylated substrate, may be used. For example, the phosphorylation of the substrate may be monitored indirectly through the intramolecular protein–protein interactions induced by the phosphorylation event. Hence, the phosphorylation of a substrate induces the conformational changes that alters the distance between the two fluorophores and affects their photophysical properties. The most commonly utilized approach is based on the fluorescence resonance energy transfer (FRET) between a donor and an acceptor to measure the extent of the change, but recently the alternatives to FRET have been explored.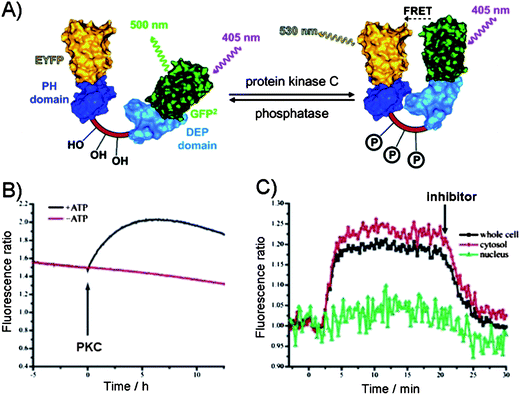 | ||
| Fig. 7 (A) The FRET between the two protein domains (yellow (EYFP) and green (GFP)) on a substrate as the reporter of protein kinase C activity.63 Dishevelled Egl-10 (DEP) domain and p47 homology domains (PH). (B) PKC-catalyzed phosphorylation in the presence of ATP in vitro leading to FRET as evident from the increase in the fluorescence emission ratio. (C) Activation of PKC in N1E-115 neuroblastoma cells. PKC activity was observed in the cytosol but not in the nucleus. Addition of PKC inhibitor down regulated PKC activity.63 | ||
Building on this principle, the imaging of the CREB phosphorylation was achieved in living cells by constructing the protein composed of the phosphorylated kinase-induced domain (KID) and the recognition domain (KIX) sandwiched between the FRET pair. Upon phosphorylation, the KID of CREB binds KIX domain to induce FRET.64 After transfection, the phosphorylation of the construct was comparable to the endogenous CREB phosphorylation and has allowed for measurements in living cells, due to the nontoxicity of the chimeric reporter. In a similar FRET manner, the protein kinase activity was monitored in neuronal compartments in intact tissue, and in living cells, further demonstrating the viability of this approach.65–67
Type IV sensor
While fluorescence is appropriate for sensing the protein modifications, it does not allow for any reaction manipulations. By contrast, the photocaging allows for a direct control over the accessibility of the substrate during the phosphorylation reaction.69 Photocaging introduces a “cage”, Fig. 8, which is a light sensitive protecting group to a side chain of an amino acid of interest by chemical ligation or in vitro transcription and translation methods.70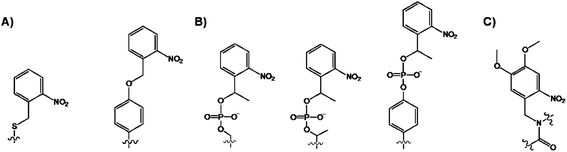 | ||
| Fig. 8 Structures of the caged (A) Ser and Tyr and (B) phospho-Ser/Thr/Tyr via side chain modification, and (C) backbone-caged amino acid. | ||
Initially, the photolabile o-nitrobenzylcysteine or o-nitrobenzyltyrosine groups (Fig. 8A) were used for probing the enzymatic activities that were unrelated to phosphorylation reactions.70 Imperiali and coworkers extended this chemical protocol for the caging of phosphorylated Ser/Thr/Tyr aminoacids with 1-(2-nitrophenyl)ethyl group, and their incorporation into target peptides (Fig. 8B).71 While the most modifications were side chain modifications, the photocaging was also demonstrated through the backbone-caged peptide (Fig. 8C).72 The doubly caged proteins for the phosphorylation studies also exist.73 The obvious advantages associated with the caged substrates are their ability to cross the cell membrane wall and the use of UV light as the trigger for “cage” removal. The sensing design is straightforward. Once in the cell, the caged protein is in its dormant inactive state, but following the photocleavage, the native protein form is liberated. This transformation, in turn, affects the location and the activity of the cellular components which can be easily measured during the cell cycle. For example, Schultz co-workers introduced a 4,5-dimethoxy-2-nitrobenzylserine, using the nonsense methodology, in the Pho4 protein to control its phosphorylation in yeast.74 The cellular location of the caged-Pho4 and uncaged Pho4 was easily controlled by photolysis. In turn, the liberation of Pho4 triggered phosphorylation by CDKs.
The latest advancement in the protein kinase engineering combines the peripheral kinase modification, guest-host recognition and the photo-activatable group based on the coupling of Cys343 group of PKA catalytic subunit to a caging agent shown in Fig. 9A.75 The latest molecular design of caging agent was based on three parts: (1) the combination of two chromophores (carboxytetramethylrhodamine (TAMRA) and QSY7) linked by a photoactivatable linker (PL), (2) the active-site directed inhibitory peptide sequence (PK1) for PKA, and (3) the electrophile for covalent attachment to the Cys343 site of PKA kinase. Firstly, the guest binds the active site of PKA through inhibitory peptide sequence, while the cross-coupling takes place at the remote Cys343 residue of kinase giving rise to the “bound” inactive kinase form (Fig. 9B). The photolabile linker in the PKA construct is designed to generate the fluorescence response upon demand and potentially restore kinase activity.
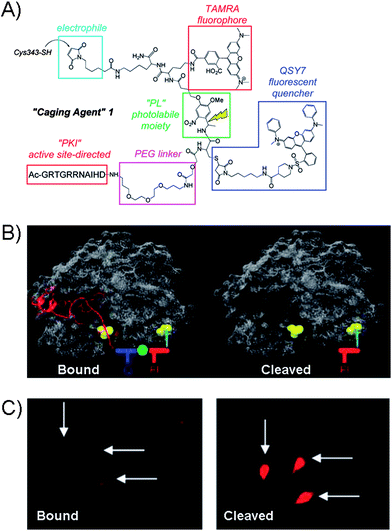 | ||
| Fig. 9 (A) Structure of the Caging Agent 1 which contains protein-directing peptide (PK1), PEG linker, fluorophore (TAMRA) and quencher (QSY7) separated by a photolabile spacer (PL). The electrophilic group serves as the point of attachment to the Cys343 site of the catalytic subunit of PKA. (B) Caging Agent 1 bound to active site of PKA (left) and the cleaved subunit (right). Q = quencher, F = fluorophore. (C) Fluorescence images of microinjected REF52 cells with PKA construct in the “bound” form and after photolysis.76 | ||
Analogues to other caging strategies, the fluorescence restoration was achieved by cleaving off the inhibitory peptide and quencher subunit as shown in Fig. 9B giving rise to the highly fluorescent uncaged peptide. The utility of the PKA was explored with the rat embryonic fibroblast cell line REF52, which were micro-injected with the PKA-construct as shown in the fluorescence image of the “bound” form in Fig. 9C. After photolysis, the fluorescence is restored. At present, the system is limited to micro-injections which may only treat a small number of cells. However, the cell permeable analogue of caging agent may prove useful, but is currently unavailable. While the photolysis in this system restores the overall fluorescence and frees the active site of the kinase, the evidence of the kinase activity has not been demonstrated yet with such a protein.
In order to achieve the control over multiple processes which involve specific sites in a single peptide or protein, different caging groups with distinct photophysical properties are required. Latest attempts to incorporate dual wavelength caging groups, to monitor related signalling pathways, include the caging of 8-Br-cAMP with o-nitrobenzyl moiety and the PKG substitution with the amino-based coumarin derivative.76 PKA kinase catalyses the target phosphorylation while PKG mediates the target phosphorylation. Photolysis with 440 nm light, results in the uncaged 8-Br-cAMP and activation of PKA, while the irradiation with 360 nm light activates PKG. In cells, these probes are selectively photoactive making them stable towards up- or down-regulation by endogenous proteins, and they retain their activity and promote phosphorylation of a specific target. Using similar caging agents, Wip1 phosphatase activity was regulated through selective uncaging of a coumarin-caged phosphorylated target in the absence or presence of the 1-(2-nitrophenyl)ethyl caged phospho-based inhibitor.77 In this system, the 420 nm irradiation produced uncaged phosphosubstrate that was subsequently dephosphorylated by Wip1 phosphatase. In the event that 365 nm irradiation was used, the phosphorylated substrate was not a target of phosphatase due to the presence of the uncaged phospho-containing inhibitor. Continuing on the dual detection of phosphorylation, the photoactive “cage”, carrying a protected but photo-labile Tyr residue, was combined with the cascade yellow fluorophore in a single peptide.48b Here, the fluorophore allows for the visualization of the location and mobility of the substrate, while the caging agent regulates its location and availability. In a non-phosphorylatable (caged) form this peptide can readily cross the cell walls of the human lung cancer A549 cell line, either by microinjection or by the use of cell-permeabilizing agents. Upon photolysis the peptide is transformed into the viable substrate for an intracellular protein kinase. The amount of available peptide may be controlled by varying the light-exposure time. Hence, small quantities of phosphorylatable peptide can be released at multiple time points, thus allowing multiple readouts to be performed in a single experiment. This approach was demonstrated for probing Tyr phosphorylation and has not been applied to non-aromatic amino acid residues.
Photocaging is attractive approach because it offers an on demand release of the target and a control over its cellular localization, however, the photolysis is an irreversible process. In addition, caged compounds should be inert to hydrolysis by cellular media, and the rate of uncaging should be fast for monitoring to take place. While most of the above photocleavable labels are placed as side chains in the given peptide, recently this strategy was extended to include targets with the photocaging of the peptide backbone.78 The caging of certain proteins disrupts the regular cell cycle and leads to the premature mitotic entry and cell arrest.79
Despite the progress made in the fluorescence peptide–protein sensors, this spectroscopic approach often suffer from issues related to the fluorescence interferences and false positives. In addition, the extensive substrate modifications may interfere with the substrate selectivity and binding. Unlike protein-based, the peptide-derived sensors offer ease of large-scale synthesis, handling and storage. Despite their availability, the peptides lack the highly specific kinase recognition domain and may be readily phosphorylated by a range of kinases which clearly complicates the analysis. Moreover, the fluorescent peptides may bind proteins in cell lysates and result in false positive response. Other challenges associated with peptide-sensors are their low cell permeability, which may be resolved by the attachment of Arg-rich sequences. However, Arg-rich peptides may end up stuck in the endosomes and unavailable to interact with the intracellular media. Researchers have since developed other peptide-carriers that efficiently deliver peptides into mammalian cells.80 However, uneven distribution of the peptide-biosensor in cells, which may lead to uneven readouts, can potentially be addressed in the future by using the ratiometric peptide probes, which contain phosphorylation sensitive and insensitive parts. Despite the advancements made in fluorescence monitoring of phosphorylations in vitro and in vivo, the fluorophore attachment site predominantly governs the response and utility of the probe.
Design of adenosine triphosphate derivatives
The use of novel ATP derivatives has provided much insight into protein kinase-catalyzed transformations, their activity, inhibitor screening, and targets in vivo. Useful ATP modifications reported to date include those having modifications at the exocyclic nitrogen N6 of the adenine or substitutions at the γ-phosphate. The substitution at the N6-site of adenine typically requires the mutation of a protein kinase in order to accommodate this analogue. By contrast, the γ-modification introduces the label far away from the binding and catalytic domain of a protein kinase and is largely solvent exposed. The latter is not expected to interfere with the activity and target specificity of the WT protein kinase. Several examples of N6- and γ-modified ATP analogues exist and will be discussed in turn.N 6-modified ATP
Initial work on N6-modifications of the purine ring was reported by Shokat and co-workers, as mentioned in the first section of this review. The substitution at the exocyclic amine of adenine dramatically affects its affinity for a WT protein kinase. Essentially, N6-substituents larger than an isopropyloxy group are detrimental to enzyme activity of WT kinases.81 Early on the inspection of the crystal structures of PKA and CDK2/cyclin A revealed two key residues within a 5 Å sphere of the N6 amino group of bound ATP: Val104/Met120 (PKA) and Val64/Phe80 (CDK2).82 Subsequent sequence alignment of these two kinases with the Src revealed that the Val323 and Ile338 residues of Src must be mutated to Ala in order for this kinase to accept the N6-modified ATP. Original work was based on the use of N6-cyclopentyl-ATP (Fig. 10) which exhibited KM values of ∼50 μM with respect to doubly mutated c-Src and >2000 μM for WT v-Src.83 The phosphorylation levels were detected by using the radiolabeled N6 derivatives. Interestingly, the mutation of Src allowed for the use of N6-modified ATP which was a more efficient substrate than ATP itself since the smaller ATP can no longer establish efficient interactions in the larger mutated binding pocket of the kinase.84 Since then, a number of ATP derivatives, shown in Fig. 10, have been investigated which contain substituents ranging from alkyl, ethers, oximes, cycloalkanes, benzyl and phenyl groups, with WT and Val323Ala, Ile338Ala, Ile338Gly mutant v-Src kinases. Protein kinase engineering was also extended to Fyn, another member of the Tyr protein kinase family. This has been exploited for an interesting approach to kinase monitoring in vivo.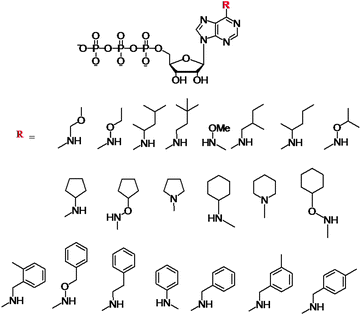 | ||
| Fig. 10 Structures of the N6-modified ATP derivatives showing the variety of R substituents. | ||
Engineering of the ATP binding pocket was also applied to RAF, which is directly involved in the MAPK pathway and which controls proliferation. Visualization of the phosphorylated MEK protein was achieved by immunobloting and anti-phospho MEK antibodies (P-MEK). Phosphorylations were performed in the presence of ATP and a number of N6-modified analogues possessing a range of aliphatic and aromatic substituents. The WT RAF could utilize N6-(ethoxy), N6-(2-methylbutyl), N6-(benzyl) and N6-(2-phenylethyl) ATPs. The double and triple mutations of RAF further revealed that N6-(2-phenylethyl) ATP was the most favoured co-substrate in addition to ATP. Latest demonstration of the N6-benzyl-ATP utility includes the Thr334Ala and Thr361Ala mutated Abl and Arg kinases for direct identification of their substrates.85 The systematic studies revealed several key features with respect to the N6-ATP design: (1) the linker spacer between the N6 and aromatic ring provides flexibility to reach the space at N7 and (2) the 3-methyl substitution at benzyl group leads to lowest steric hindrance.
Gamma-modified ATP
Next, we turn our attention to the use of γ-modified ATP analogues in which the γ-phosphate of ATP is labeled, generally through a phosphoamide or phophoester linkage. The latter covalent modification is less stable under biological conditions. The γ-modification is a synthetically straightforward method that has allowed the synthesis of a series of derivatives in an arsenal of ATP derivatives illustrated in Fig. 11. The presence of a shallow solvent exposed binding pocket for ATP enables kinases to tolerate γ-phosphate derivatization, thereby turning a protein kinase into a catalytic tool for group transfer and labelling of peptides and proteins without the need for laborious genetic engineering of protein kinases.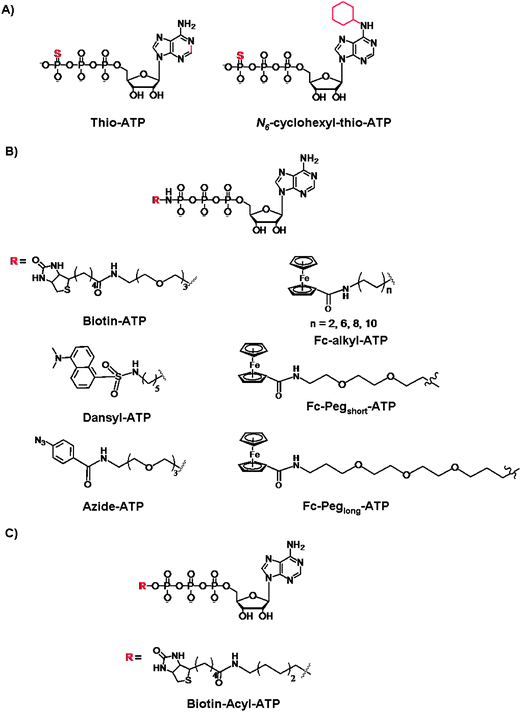 | ||
| Fig. 11 Structures of the γ-modified ATP derivatives: (A) Thio-based ATP, (B) phosphoamide ATP, and (C) phosphoacyl ATP. | ||
The thiophosphorylated substrates may be detected by direct anti-thiophospho antibodies or alkylation and subsequent antigen–antibodies interactions. For example, the thiophosphorylated and p-nitrobenzylmesylate alkylated targets were identified with the polyclonal antibodies raised against these small antigens. Importantly, the immunoassays exhibited desired specificity for the modified targets even in the presence of competing free thiols and nucleophile in the cell lysates. The authors also demonstrated that the antibodies exhibit high affinity for their targets so that the immunoprecipitation may be achieved from complex protein mixture.89 With the thio-alkylation it is possible to label the thiophosphorylated substrates for the purpose of identification and quantification, however all protein targets stand to be thiophosphorylated by various kinases.90 To introduce selectivity, the analog-sensitive kinase (with mutated “gate-keeper” residue) and the WT kinases were compared with the 5′-γ-thiophosphate-N6-(cyclohexyl)ATP, and the subsequent selective transfer of the thiophosphate group was analyzed by mass spectrometry.91 Drawbacks of S-alkylation include a low selectivity in the presence of Cys residues, and the resistance of thiophosphorylated products to dephosphorylation by protein phosphatases. This has been somewhat remedied by an acid-catalyzed hydrolysis which leads to the isolation of the phosphorylated and thio-alkylated proteins. Further efforts were reported by Mann and co-workers who developed a chemoselective thio-alkylation approach that is pH-controlled.92 Under acidic conditions, the Cys and thiophospho residues may be differentiated based on a significant pKa difference (Cys pKa of ∼9 vs. a thiophosphorylated residue pKa < 3). The selective coupling was demonstrated when the thiophosphate-N6-(cyclohexyl)ATP (Fig. 11A) was used for the phosphorylation of p27 and retinoblastoma proteins with CDK2/Cyclin E complex. This work exemplifies the further chemical control over the Thio-ATP methodology, and favourable alkylation of thiophosphate groups over the non-specific and highly undesirable labeling of the Cys residues.
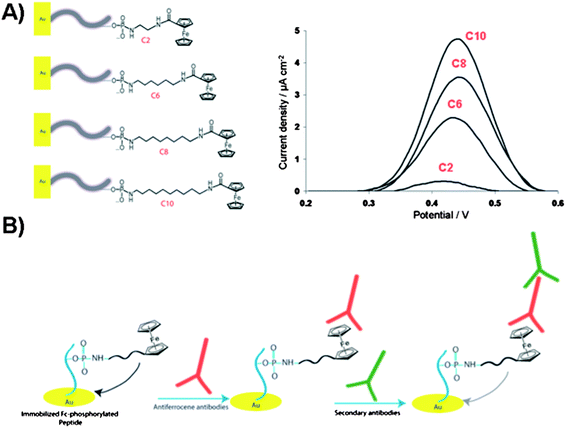 | ||
| Fig. 12 (A) Illustration of the electrochemical biosensor for detection of gold surface-bound peptides using Fc-ATP of different alkyl linker length (C2, C6, C8 and C10). Square-wave voltammograms of the Fc-phosphorylated peptides on gold surfaces showing current density as a function of linker length. (B) Illustration of the ferrocene-phosphorylated peptide on gold surfaces and subsequent binding of antiferrocene and secondary antibodies for monitoring the kinase-catalyzed phosphorylations. | ||
Clear trend was observed in the increase in electrochemical signal with respect to the increase in aliphatic linker length. These findings were related to the Fc-surface coverage rather than to the mobility of the immobilized Fc group. The proposed kinase-co-substrate binding model assumes the steric crowding between the Fc group and kinase binding site for the shorter Fc-ATP analogue. Furthermore, the careful study on the substitution effects of the long alkyl spacer by a long hydrophilic poly(ethylene)glycol (Peg) linker (Fig. 11B) demonstrated that the additional electronic factors play critical role in determining the protein kinase activity.99 The electrochemical signal, i.e. the Fc-phosphorylation, decreased dramatically when hydrophilic Fc-Peg-ATP analogues were used, pointing to the critical nature of the modified-ATP on phosphorylation processes and suggesting a more effective binding of Fc-PEG-ATP derivatives to the binding pocket or a substrate, which reduces the efficiency of group transfer to a peptide target. Despite the fact that the catalytic site of protein kinase, at the γ-phosphate, is shallow and solvent exposed, the inherently polar amino acids are critical for catalysis. The charged residues such as Lys, Glu and Asp appear to play a key role in positioning the co-substrate and one can image that competitive ionic and H-bonding interactions with hydrophilic Fc-ATP may perturb the fine balance required for catalysis. For some kinases, these key charged residues in the catalytic pocket must adopt a specific conformation with respect to the co-substrate and incoming substrate in order for the successful phosphorylation to take place. These Fc-ATP analogues have since been validated with the fluorescence kinase assay as viable co-substrates for a range of kinases in homogenized solutions. In addition, Fc-phosphorylated substrates are sensitive to the phosphatase activity unlike thiophosphorylated substrates. Recently, the anti-ferrocene antibodies were developed and used for detection of Fc-phosphorylated peptide and proteins, such as α-casein, pro-caspase-3 and pro-caspase 8 in a western blot analysis.100 Comparative studies between Fc-ATP and ATP indicate that they have similar kinetics with respect to the substrate and inhibition profiles but exhibit largely different kinetics with respect to the co-substrate (KM values ∼12 and 165 μM for ATP and Fc-ATP, respectively). Importantly, Fc-ATP is used by protein kinases in the whole cell lysate and efficiently Fc-phosphorylates a number of proteins, similarly to ATP. The antiferrocene antibodies strategy with Fc-ATP allows for monitoring protein kinase activity and inhibitor screening in an electrochemical, biochemical and fluorescence assays based on the two step primary antiferrocene and secondary antibodies assay as illustrated in Fig. 12B.
The undesirable feature of using modified ATPs is the use of ATP itself, since numerous highly abundant ATP binding proteins may interfere with protein kinases. In addition, ATP derivatives are always introduced exogenously and are not likely to cross the cell wall.
Conclusions
Chemists have taken a keen interest in exploring protein and peptide phosphorylation reactions, using a synthetic biological toolkit based on protein engineering and chemical derivatization of targets and co-substrates, in order to identify kinase activity and inhibition. This has led to a number of fundamental insights into protein kinase–co-substrate–substrate interactions revealing key events in the regulatory processes governing cellular signalling pathways. Exploration into specific kinase targets remains a challenge. Signalling cascades and new protein targets are being discovered on an ongoing basis, aided by advances in experimental methodologies. Despite our knowledge of basic phosphorylation chemistry, the subtleties of the interplay between protein kinases and phosphatases, and kinases–substrates–cosubstrates are still a focus of research. The role of some kinases as molecular triggers of disfunction and disease has been reported. But given the vast number of protein kinases and their target promiscuity, the interplay between different kinases and their effects and functions remains elusive. This will ensure that this topic will remain at the forefront of research in biological chemistry.Acknowledgements
This work was financially supported by funding from NSERC and the University of Toronto.References
- (a) T. Hunter, Cell, 2000, 100, 113–127 CrossRef CAS; (b) G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS.
- (a) M. R. Domingo-Sananes, O. Kapuy, T. Hunt and B. Novak, Philos. Trans. R. Soc., B, 2011, 366, 3584–3594 CrossRef CAS; (b) G. S. Wu, Cancer Metastasis Rev., 2007, 26, 579–585 CrossRef CAS.
- S. Lapenna and A. Giordano, Nat. Rev. Drug Discovery, 2009, 8, 547–566 CrossRef CAS.
- (a) L. Kurzawa and M. C. Morris, ChemBioChem, 2010, 11, 1037–1047 CrossRef CAS; (b) N. A. St-Denis, D. R. Derksen and D. W. Litchfield, Mol. Cell. Biol., 2009, 29, 2068–2081 CrossRef CAS.
- J. K. Heriche, F. Lebrin, R. Rabilloud, D. Leroy, E. M. Chambaz and Y. Goldberg, Science, 1997, 276, 952–955 CrossRef CAS.
- (a) J. S. Duncan, J. P. Torowec, K. E. Duncan, G. Vilk, C. Wu, B. Luscher, S. S. C. Li, G. B. Gloor and D. W. Litchfield, Sci. Signaling, 2011, 4, 1–13 CrossRef; (b) P. Lahiry, A. Torkamani, N. J. Schork and R. A. Hegele, Nat. Rev. Genet., 2010, 11, 60–74 CrossRef CAS.
- J. S. Sebolt-Leopold and R. Herrera, Nat. Rev. Cancer, 2004, 4, 937–947 CrossRef CAS.
- S. P. Davies, H. Reddy, M. Caivano and P. Cohen, Biochem. J., 2000, 351, 95–105 CrossRef CAS.
- M. Fajolet, G. He, M. Himan, A. Lin, A. C. Narin and P. Greengard, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 4159–4164 CrossRef.
- (a) P. J. Alaimo, M. A. Shogren-Knaak and K. M. Shokat, Curr. Opin. Chem. Biol., 2001, 5, 360–367 CrossRef CAS; (b) A. C. Dar and K. M. Shokat, Annu. Rev. Biochem., 2011, 80, 769–795 CrossRef CAS.
- S. P. Mulcahy and E. Meggers, Top. Organomet. Chem., 2010, 32, 141–153 CrossRef CAS.
- A. Wilbuer, D. H. Vlecken, D. J. Schmitz, K. Kraling, K. Harms, C. P. Bagowski and E. Meggers, Angew. Chem., Int. Ed., 2010, 49, 3839–3842 CrossRef CAS.
- (a) C. H. S. Lu, K. Liu, L. P. Tan and S. Q. Yao, Chem.–Eur. J., 2012, 18, 28–39 CrossRef CAS; (b) M. K. Tarrant and P. A. Cole, Annu. Rev. Biochem., 2009, 78, 797–825 CrossRef CAS; (c) D. R. W. Hodgson and M. Schroder, Chem. Soc. Rev., 2011, 40, 1211–1223 RSC.
- S. M. Ulrich, D. M. Kensi and K. M. Shokat, Biochemistry, 2003, 42, 7915–7921 CrossRef CAS.
- H. Habelhah, K. Shah, L. Huang, A. L. Burlingame, K. M. Shokat and Z. Ronai, J. Biol. Chem., 2001, 276, 18090–18095 CrossRef CAS.
- S. Larochelle, J. Batliner, M. J. Gamble, N. M. Barboza, B. C. Kraybill, J. D. Blethrow, K. M. Shokat and R. P. Fisher, Nat. Struct. Mol. Biol., 2006, 13, 55–62 CAS.
- J. L. Moreland, A. Gramada, O. V. Buzko, Q. Zhang and P. E. Bourne, BMC Bioinf., 2005, 6, 21–26 CrossRef.
- L. M. Elphick, S. E. Lee, E. S. Child, A. Prasad, C. Pignocchi, S. Thibaudeau, A. A. Anderson, L. Bonnac, V. Gouverneur and D. J. Mann, ChemBioChem, 2009, 10, 1519–1526 CrossRef CAS.
- M. Azam, M. A. Seeliger, N. S. Gray, J. Kuriyan and G. Q. Daley, Nat. Struct. Mol. Biol., 2008, 15, 1109–1118 CAS.
- F. Zuccotto, E. Ardini, E. Casale and M. Angiolini, J. Med. Chem., 2010, 53, 2681–2694 CrossRef CAS.
- A. L. Garske, U. Peters, A. T. Cortesi, J. L. Perez and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15046–15052 CrossRef CAS.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS.
- A. V. Karginov, F. Ding, P. Kota, N. V. Dokholyan and K. M. Hahn, Nat. Biotechnol., 2010, 28, 743–748 CrossRef CAS.
- J. E. J. Darnell, Science, 1997, 277, 1630–1635 CrossRef CAS.
- S. Lahiri, R. Seidel, M. Engelhard and C. F. Becker, Mol. BioSyst., 2010, 6, 2423–2429 RSC.
- (a) J. R. Simard, M. Getlik, C. Grutter, V. Pawar, S. Wulfert, M. Rabiller and D. Rauh, J. Am. Chem. Soc., 2009, 131, 13286–13296 CrossRef CAS; (b) J. R. Simard, M. Getlik, C. Grutter, R. Schneider, S. Wulfert and D. Rauh, J. Am. Chem. Soc., 2010, 132, 4152–4160 CrossRef CAS.
- B. W. Jester, K. J. Cox, A. Gaj, C. D. Shomin, J. R. Porter and I. Ghosh, J. Am. Chem. Soc., 2010, 132, 11727–11735 CrossRef CAS.
- R. Krishnamurty and D. J. Maly, Comb. Chem. High Throughput Screening, 2007, 10, 652–666 CrossRef CAS.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34–66 CrossRef CAS.
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS.
- M. K. Tarrant and P. A. Cole, Annu. Rev. Biochem., 2009, 78, 797–825 CrossRef CAS.
- Q. Wang, A. R. Parrish and L. Wang, Chem. Biol., 2009, 16, 323–336 CrossRef CAS.
- J. Xie, L. Superkova and P. G. Schultz, ACS Chem. Biol., 2007, 2, 474–478 CrossRef CAS.
- (a) V. K. Lacey, A. R. Parrish, S. Han, Z. Shen, S. P. Briggs, Y. Ma and L. Wang, Angew. Chem., Int. Ed., 2011, 50, 8692–8696 CrossRef CAS; (b) S. Li, Angew. Chem., Int. Ed., 2010, 12, 2729–2731 Search PubMed.
- Q. Wang, A. R. Parrish and L. Wang, Chem. Biol., 2009, 16, 323–336 CrossRef CAS.
- D. J. Maly, J. A. Allen and K. M. Shokat, J. Am. Chem. Soc., 2004, 126, 9160–9161 CrossRef CAS.
- K. Liu, K. A. Kalesh, L. B. Ong and S. Q. Yao, ChemBioChem, 2008, 9, 1883–1888 CrossRef CAS.
- K. Parang, J. A. Kohn, S. A. Saldanha and P. A. Cole, FEBS Lett., 2002, 520, 156–160 CrossRef CAS.
- S. Suwal and M. K. H. Pflum, Angew. Int. Chem., Ed., 2010, 49, 1627–1630 CrossRef CAS.
- A. V. Statsuk, D. J. Maly, M. A. Seeliger, M. A. Fabian, W. H. Biggs III, D. J. Lockhart, P. P. Zarrinkar, J. Kuriyan and K. M. Shokat, J. Am. Chem. Soc., 2008, 130, 17568–17574 CrossRef CAS.
- K. A. Kalesh, D. S. Sim, J. Wang, K. Liu, Q. Lin and S. Q. Yao, Chem. Commun., 2010, 46, 1118–1120 RSC.
- (a) V. Sharma, Q. Wang and D. S. Lawrence, Biochim. Biophys Acta, 2008, 1784, 94–99 CrossRef CAS; (b) J. A. Gonzalez-Vera, Chem. Soc. Rev., 2012, 41, 1652–1664 RSC.
- (a) Q. Wang, S. M. Cahill, M. Blumenstein and D. S. Lawrence, J. Am. Chem. Soc., 2006, 128, 1808–1809 CrossRef CAS; (b) A. Wakata, S. M. Cahill, M. Blumenstein, R. H. Gunby, S. Jockusch, A. A. Marti, B. Cimbro, C. Gambacorti-Passerini, A. Donella-Deana, L. A. Pinna, N. J. Turro and D. S. Lawrence, Org. Lett., 2008, 10, 301–304 CrossRef CAS.
- Q. Wang, E. I. Zimmerman, A. Toutchkine, T. D. Martin, L. M. Graves and D. S. Lawrence, ACS Chem. Biol., 2010, 5, 887–895 CrossRef CAS.
- E. Lukovic, J. A. Gonzalez-Vera and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 12821–12827 CrossRef CAS.
- E. Lukovic, J. A. Gonzalez-Vera and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 277–283 CrossRef.
- M. D. Schultz and B. Imperiali, J. Am. Chem. Soc., 2003, 125, 14248–14249 CrossRef.
- (a) J. A. Gonzalez-Vera, E. Lukovic and B. Imperiali, J. Org. Chem., 2009, 74, 7309–7314 CrossRef CAS; (b) N. Kumar, K. E. Nielsen, S. Maiti and N. Petersen, J. Am. Chem. Soc., 2006, 128, 14–15 CrossRef CAS.
- C. I. Stains, N. C. Tedford, T. C. Walkup, E. Lukovic, B. N. Gouguen, L. G. Griffith, D. A. Lauffenburger and B. Imperiali, Chem. Biol., 2012, 19, 210–217 CrossRef CAS.
- H. W. Rhee, S. H. Lee, I. S. Shin, S. J. Choi, H. H. Park, K. Han, T. H. Park and J. I. Hong, Angew. Chem., Int. Ed., 2010, 49, 4919–4923 CrossRef CAS.
- J. E. Ghadiali, B. E. Cohen and M. M. Stevens, ACS Nano, 2010, 4, 4915–4919 CrossRef CAS.
- E. Lukovic, E. Vogel Taylor and B. Imperiali, Angew. Chem., Int. Ed., 2009, 48, 6828–6831 CrossRef CAS.
- (a) Q. Ni, D. V. Titov and J. Zhang, Methods, 2006, 40, 279–281 CrossRef CAS; (b) D. M. Rothman, M. D. Schultz and B. Imperiali, Trends Cell Biol., 2005, 15, 502–509 CrossRef CAS.
- F. Sicheri, I. Moarefi and J. Kuriyan, Nature, 1997, 385, 602–609 CrossRef CAS.
- V. Sharma, R. S. Agnes and D. S. Lawrence, J. Am. Chem. Soc., 2007, 129, 2742–2743 CrossRef CAS.
- (a) A. Uri, M. Lust, A. Vaasa, D. Lavogina, K. Viht and E. Enkvist, Biochim. Biophys. Acta, 2010, 1804, 541–546 CrossRef CAS; (b) J. E. Toettcher, D. Gong, W. A. Lim and O. D. Weiner, Nat. Methods, 2012, 8, 837–839 CrossRef.
- (a) Y. Nagai, M. Miyazaki, R. Aoki, T. Zama., S. Inouye, K. Hirose, M. Lino and M. Hagiwara, Nat. Biotechnol., 2000, 18, 313–316 CrossRef CAS; (b) A. Vassa, M. Lust, A. Terrin, A. Uri and M. Zaccolo, Biochem. Biophys. Res. Commun., 2010, 397, 750–755 CrossRef.
- A. Miyawaki and R. Y. Tsien, Methods Enzymol., 2000, 327, 472–500 CAS.
- Y. Wang, E. L. Botvinick, Y. Zhao, M. W. Berns, S. Usami, R. Y. Tsien and S. Chien, Nature, 2005, 434, 1040–1045 CrossRef CAS.
- M. Sato, T. Ozawa, K. Inukai, T. Asano and Y. Umezawa, Nature, 2002, 20, 287–294 CrossRef CAS.
- M. T. Kunkel, A. Toker, R. Y. Tsien and A. C. Newton, J. Biol. Chem., 2007, 282, 6733–6742 CrossRef CAS.
- Q. Wang and D. S. Lawrence, J. Am. Chem. Soc., 2005, 127, 7684–7685 CrossRef CAS.
- J. Brumbaugh, A. Schleifenbaum, A. Gasch, M. Sattler and C. Schultz, J. Am. Chem. Soc., 2006, 128, 24–25 CrossRef CAS.
- M. W. Friedrich, G. Aramuni, M. Mank, J. A. G. Mackinnon and O. Griesbeck, J. Biol. Chem., 2010, 285, 23285–23295 CrossRef CAS.
- C. D. Harvey, A. G. Ehrhardt, C. Cellurale, H. Zhong, R. Yasuda, R. J. Davis and K. Svoboda, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19264–19269 CrossRef CAS.
- T. Ishimoto, H. Mano, T. Ozawa and H. Mori, Bioconjugate Chem., 2012, 23, 923–932 CrossRef CAS.
- K. J. Herbst, M. D. Allen and J. Zhang, J. Am. Chem. Soc., 2011, 133, 5676–5679 CrossRef CAS.
- S. Nyati, R. Ranga, B. D. Ross, A. Rehemtulla and M. S. Bhojani, Anal. Biochem., 2010, 405, 246–254 CrossRef CAS.
- H. M. Lee, D. R. Larson and D. S. Lawrence, ACS Chem. Biol., 2009, 4, 409–427 CrossRef CAS.
- A. Deiters, D. Groff, Y. Ryu, J. Xie and P. G. Schultz, Angew. Chem., Int. Ed., 2006, 45, 2728–2731 CrossRef CAS.
- D. M. Rothman, M. E. Vazquez, E. M. Vogel and B. Imperiali, J. Org. Chem., 2003, 68, 6795–6798 CrossRef CAS.
- S. K. Nandy, R. S. Agnes and D. S. Lawrence, Org. Lett., 2007, 9, 2249–2252 CrossRef CAS.
- M. E. Hahn and T. W. Muir, Angew. Chem., Int. Ed., 2004, 43, 5800–5803 CrossRef CAS.
- E. A. Lemke, D. Summerer, B. H. Geierstanger, S. M. Brittain and P. G. Schultz, Nat. Chem. Biol., 2007, 3, 769–772 CrossRef CAS.
- H. M. Lee, W. Xu and D. S. Lawrence, J. Am. Chem. Soc., 2011, 133, 2331–2333 CrossRef CAS.
- M. A. Priestman, L. Sun and D. S. Lawrence, ACS Chem. Biol., 2011, 6, 377–384 CrossRef CAS.
- B. N. Goguen, A. Aemissegger and B. Imperiali, J. Am. Chem. Soc., 2011, 133, 11038–11041 CrossRef CAS.
- Q. Wang, Z. Dai, S. M. Cahill, M. Blumenstein and D. S. Lawrence, J. Am. Chem. Soc., 2006, 128, 14016–14017 CrossRef CAS.
- Z. Dai, N. G. Dulyaninova, S. Kumar, A. R. Bresnick and D. S. Lawrence, Chem. Biol., 2007, 14, 1254–1260 CrossRef CAS.
- L. Crombez, G. Aldrian-Herrada, K. Konate, Q. N. Nguyen, G. K. McMaster, R. Brasseur, F. Heitz and G. Divita, Mol. Ther., 2009, 17, 95–103 CrossRef CAS.
- K. Shah, Y. Liu, C. Deirmengian and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3565–3570 CrossRef CAS.
- S. S. Taylor and E. Radzio-Andzelm, Structure, 1995, 2, 345–355 CrossRef.
- Y. Liu, K. Shah, F. Yang, L. Witucki and K. M. Shokat, Chem. Biol., 1998, 5, 91–101 CrossRef CAS.
- B. C. Kraybill, L. L. Elkin, J. D. Blethrow, D. O. Morgan and K. M. Shokat, J. Am. Chem. Soc., 2002, 124, 12118–12128 CrossRef CAS.
- S. N. Boyle and A. J. Koleske, Biochemistry, 2007, 46, 11614–11620 CrossRef CAS.
- F. Eckstein, Annu. Rev. Biochem., 1985, 54, 367–402 CrossRef CAS.
- K. C. Facemyer and C. R. Cremo, Bioconjugate Chem., 1992, 3, 408–413 CrossRef CAS.
- J. J. Allen, S. E. Lazerwith and K. M. Shokat, J. Am. Chem. Soc., 2005, 127, 5288–5289 CrossRef CAS.
- J. J. Allen, M. Li, C. S. Brinkworth, J. L. Paulson, D. Wang, A. Hubner, W. H. Chou, R. J. Davis, A. L. Burlingame, R. O. Messing, C. D. Katayama, S. M. Hedrick and K. M. Shokat, Nat. Methods, 2007, 4, 511–516 CrossRef CAS.
- L. L. Parker, A. B. Schilling, S. J. Kron and S. B. Kent, J. Proteome Res., 2005, 4, 1863–1866 CrossRef CAS.
- J. D. Blethrow, J. S. Glavy, D. O. Morgan and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1442–1447 CrossRef CAS.
- S. E. Lee, L. M. Elphick, H. B. Kramer, A. M. E. Jones, E. S. Child, A. A. Anderson, L. Bonnac, N. Suwaki, B. M. Kessler, V. Gouverneur and D. J. Mann, ChemBioChem, 2011, 12, 633–640 CrossRef CAS.
- Z. Wang, R. Levy, D. G. Fernig and M. Brust, J. Am. Chem. Soc., 2006, 128, 2214–2215 CrossRef CAS.
- K. D. Green and M. K. H. Pflum, J. Am. Chem. Soc., 2007, 129, 10–11 CrossRef CAS.
- M. P. Patricelli, A. K. Szardenings, M. Liyanage, T. K. Nomanbhoy, M. Wu, H. Weissig, A. Aban, D. Chun, S. Tanner and J. W. Kozarich, Biochemistry, 2007, 46, 350–358 CrossRef CAS.
- K. D. Green and M. K. H. Pflum, ChemBioChem, 2009, 10, 234–237 CrossRef CAS.
- (a) K. Kerman, H. Song, J. S. Duncan, D. Litchfield and H. B. Kraatz, Anal. Chem., 2008, 80, 9395–9401 CrossRef CAS; (b) S. Martic, M. Labib and H.-B. Kraatz, Electrochim. Acta, 2011, 56, 10676–10682 CrossRef CAS; (c) S. Martic, S. Tackenburg, Y. Bilokin, A. Golub, V. Bdzhola, S. Yarmoluk and H.-B. Kraatz, Anal. Biochem., 2012, 421, 617–621 CrossRef CAS.
- S. Martic, M. Labib, D. Freeman and H. B. Kraatz, Chem.–Eur. J., 2011, 17, 6744–6752 CrossRef CAS.
- S. Martic, M. K. Rains, D. Freeman and H. B. Kraatz, Bioconjugate Chem., 2011, 22, 1663–1672 CrossRef CAS.
- S. Martic, M. Gabriel, J. P. Turowec, D. W. Litchfield and H. B. Kraatz, J. Am. Chem. Soc., 2012 DOI:10.1021/ja302586q.
| This journal is © The Royal Society of Chemistry 2013 |