Biocompatible reduction-responsive polypeptide micelles as nanocarriers for enhanced chemotherapy efficacy in vitro
Received
3rd September 2012
, Accepted 1st October 2012
First published on 18th October 2012
Abstract
To develop biocompatible reduction-responsive micellar systems for efficient intracellular drug delivery, disulfide-linked block copolymers of methoxyl poly(ethylene glycol) (mPEG) and poly(ε-benzyloxycarbonyl-L-lysine) (PZLL) were synthesized through ring-opening polymerization of ε-benzyloxycarbonyl-L-lysine N-carboxyanhydride with amino group terminated disulfide functionalized mPEG as macroinitiator. The copolymers self-assembled into micelles in phosphate buffered saline (PBS) at pH 7.4 through direct dissolution and dialysis methods. The micelles were revealed to have excellent hemocompatibilities, and cell and tissue compatibilities, which rendered them with potential for drug delivery applications. Doxorubicin (DOX), an anthracycline antitumor drug, was loaded into the micelles through nanoprecipitation with a drug loading efficiency of about 30 wt%. The in vitro DOX release from all DOX-loaded micelles was accelerated in PBS with 10.0 mM GSH, mimicking intracellular reductive conditions. DOX-loaded micelles showed higher cellular proliferation inhibition towards glutathione monoester pretreated HeLa (a human cervical cell line) and HepG2 cells (a human hepatoma cell line) as compared to unpretreated or buthionine sulfoximine pretreated cells. The above results indicated that the biocompatible reduction-responsive micelles have vast potential in targeted intracellular delivery of antitumor drugs to achieve enhanced efficacy in malignancy therapy.
1. Introduction
Presently, malignancy (i.e. cancer) is one of the most serious diseases around the world, which exhibits high morbidity and mortality.1,2 In spite of the considerable development of various antitumor drugs, such as doxorubicin (DOX), paclitaxel, and chlorambucil, clinical outcomes have been disappointing due to their life-threatening side effects, including leukaemia, cardiotoxicity, etc.3–5 To eliminate or reduce the severe side effects, many kinds of nanocarriers, such as micelles,6,7 vesicles,8,9 liposomes10 and nanogels,2,11,12 have been developed as vehicles for delivery of antitumor drugs. Among all of the aforementioned nanovehicles, polymeric micelles self-assembled from amphiphilic block copolymers have emerged as one of the most promising nanocarriers for various antitumor drugs. Micelles are usually associated with several merits as polymeric drug carriers, such as improved drug solubility in water, prolonged circulation time, enhanced accumulation in tumor sites, decreased side effects, and elevated drug bioavailability and efficacy.5,13
In recent years, stimuli-responsive micelles, which show sharp intelligent response to intracellular microenvironmental stimuli, such as pH,14,15 redox,16,17 temperature18 and enzymes,19 have attracted considerable attention. Just to reach the targeted tumor tissues, the smart nanocarriers can rapidly enter cells through endocytosis and quickly release the payload triggered by intracellular stimuli, resulting in aggressive activity toward tumor cells and maximal chemotherapeutic efficacy with fewer side effects.20 In particular, reduction-responsive micelles containing disulfide bonds have been considered to be the most promising nanovehicles due to the difference in redox potential between the mildly oxidizing extracellular milieu and the reducing intracellular microenvironment. The disulfide bonds can be reductively degraded in the presence of glutathione (GSH), a thiol-containing tripeptide. The intracellular concentration of GSH is ∼0.5 to 10.0 mM and is significantly higher than that outside cells (∼2.0 to 20.0 μM).1 The dramatic variation of GSH concentration provides an opportunity for designing intracellular specific drug delivery systems. Therefore, such smart micelles containing GSH-cleavable disulfide bonds may hold great potential for efficient intracellular delivery of antitumor drugs.
Very recently, several reports have appeared concerning reduction-responsive micelles formed from amphiphilic disulfide-linked block copolymers. Zhong's group prepared reduction-responsive micelles based on disulfide-linked poly(ethylene glycol) (PEG) or dextran and poly(ε-caprolactone) (PCL) block copolymers and applied them for rapid intracellular release of DOX.13,21 Wang et al. developed a micellar system from disulfide-linked block copolymers of PCL and poly(ethyl ethylene phosphate) aimed at intracellular DOX release triggered by GSH in tumor cells.22 Li and co-workers exploited two kinds of reduction-responsive micelles originating from disulfide-linked PEG-polypeptides (i.e. poly(ε-benzyloxycarbonyl-L-lysine) (PZLL) and poly(rac-leucine)) to achieve intracellular delivery of DOX.23,24
In this work, a type of biocompatible reduction-responsive micelle based on disulfide-linked methoxyl poly(ethylene glycol) (mPEG) and PZLL was developed for efficient intracellular delivery of antitumor drugs. The block copolymers were synthesized through ring-opening polymerization (ROP) of ε-benzyloxycarbonyl-L-lysine N-carboxyanhydride (ZLL NCA) with amino group terminated disulfide functionalized mPEG (mPEG-S2-NH2) as macroinitiator. As compared to previous work, the synthesis pathway for the block copolymers was more convenient.23,24 The micelles of copolymers in phosphate buffered saline (PBS) at pH 7.4 were prepared through direct dissolution and dialysis methods. The micelles were revealed to have excellent hemocompatibilities, and cell and tissue compatibilities. DOX was loaded into the micelles through nanoprecipitation, and the in vitro DOX release was accelerated in intracellular reductive conditions. The DOX-loaded micelles showed higher cellular proliferation inhibition towards glutathione monoester (GSH-OEt) pretreated HeLa (a human cervical cell line) and HepG2 (a human hepatoma cell line) cells as compared to unpretreated or buthionine sulfoximine (BSO) pretreated cells. These reducible micelles are highly promising for the targeted intracellular delivery of antitumor drugs.
2. Experimental section
2.1. Materials
ZLL was purchased from GL Biochem Co., Ltd. ZLL NCA was synthesized as described in our previous work with slight modification.25,26 mPEG (Mn = 5000) was purchased from Sigma-Aldrich without further purification. GSH, GSH-OEt, buthionine sulfoximine (BSO) and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. Doxorubicin hydrochloride (DOX·HCl) was purchased from Zhejiang Hisun Pharmaceutical Co., Ltd. N,N-Dimethylformamide (DMF) was dried over CaH2 at 25 °C for 48 h before vacuum distillation. All other reagents and solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. and used as obtained.
2.2. Sample syntheses
The three-step procedure employed for the synthesis of methoxy poly(ethylene glycol)-S-S-poly(ε-benzyloxycarbonyl-L-lysine) (mPEG-S2-PZLL) was shown in Scheme 1.
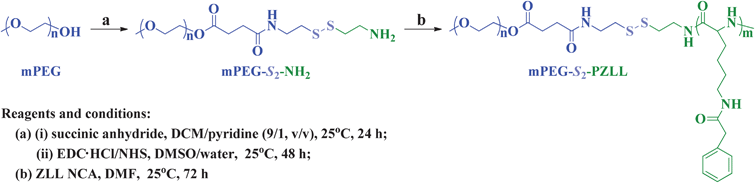 |
| | Scheme 1 Synthesis pathway for mPEG-S2-PZLL. | |
2.2.1. Synthesis of mPEG-S2-NH2 macroinitiator.
mPEG-S2-NH2 was synthesized through a ring-opening reaction of succinic anhydride (SA) and a subsequent condensation reaction. Briefly, mPEG (10.0 g, 2.0 mmol) and SA (0.24 g, 2.4 mmol) were dissolved in 100.0 mL of dichloromethane (DCM)–pyridine (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixed solvent, and the reaction proceeded at 25 °C for 24 h. Then, the solution was precipitated into excess diethyl ether. The obtained product was further washed twice with diethyl ether and dried under vacuum at 25 °C for 24 h, yielding carboxyl group terminated mPEG (mPEG–COOH, yield: 95.3%). Subsequently, mPEG–COOH (5.0 g, 0.98 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 0.94 g, 4.9 mmol) and N-hydroxysuccinimide (NHS, 0.23 g, 1.96 mmol) were dissolved into 50.0 mL of dimethyl sulfoxide (DMSO) and were kept stirring at 25 °C for 12 h. Then, the above reaction mixture was added into a solution of cysteamine hydrochloride (4.42 g, 19.61 mmol) in 10.0 mL of water, dropwise. After the completion of the addition, the reaction proceeded at 25 °C for an additional 48 h. The solution was extracted with 90.0 mL of DCM 3 times (30.0 mL × 3). The DCM phase was dried, concentrated and then precipitated into excess diethyl ether. The final mPEG-S2-NH2 was further washed twice with diethyl ether and dried under vacuum at 25 °C for 24 h (yield: 92.7%).
:1, v/v) mixed solvent, and the reaction proceeded at 25 °C for 24 h. Then, the solution was precipitated into excess diethyl ether. The obtained product was further washed twice with diethyl ether and dried under vacuum at 25 °C for 24 h, yielding carboxyl group terminated mPEG (mPEG–COOH, yield: 95.3%). Subsequently, mPEG–COOH (5.0 g, 0.98 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 0.94 g, 4.9 mmol) and N-hydroxysuccinimide (NHS, 0.23 g, 1.96 mmol) were dissolved into 50.0 mL of dimethyl sulfoxide (DMSO) and were kept stirring at 25 °C for 12 h. Then, the above reaction mixture was added into a solution of cysteamine hydrochloride (4.42 g, 19.61 mmol) in 10.0 mL of water, dropwise. After the completion of the addition, the reaction proceeded at 25 °C for an additional 48 h. The solution was extracted with 90.0 mL of DCM 3 times (30.0 mL × 3). The DCM phase was dried, concentrated and then precipitated into excess diethyl ether. The final mPEG-S2-NH2 was further washed twice with diethyl ether and dried under vacuum at 25 °C for 24 h (yield: 92.7%).
2.2.2. Syntheses of disulfide-linked mPEG and PZLL (mPEG-S2-PZLL) block copolymers.
As shown in Scheme 1, mPEG-S2-PZLL block copolymers were synthesized through ROP of ZLL NCA, initiated by mPEG-S2-NH2 in DMF. The different reactant feeding molar ratios were listed in Table 1. The reaction was conducted at 25 °C for 72 h. Then, the reaction solution was precipitated into an excess diethyl ether–ethanol (2:1, v/v) mixture. The obtained product was further washed twice with diethyl ether and dried under vacuum at 25 °C for 24 h (yield: 88.7%).
| Entry |
Feeding molar ratio of ZLL NCA/mPEG-S2-NH2 |
DP of PZLLa |
M
n
(g mol−1) |
M
n
(g mol−1) |
PDIb |
|
Determined by 1H NMR.
Determined by GPC.
|
| mPEG113-S2-PZLL18 |
20 |
18 |
10000 |
11900 |
1.09 |
| mPEG113-S2-PZLL35 |
40 |
35 |
14400 |
16300 |
1.14 |
2.3. Preparation of micelles
Micelles were prepared by direct dissolution and dialysis methods.
2.3.1. Direct dissolution method.
Typically, the micelles were prepared by directly dissolving 5.0 mg of mPEG-S2-PZLL in 5.0 mL of PBS, marked as micelle1.
2.3.2. Dialysis method.
Briefly, 5.0 mg of mPEG-S2-PZLL was dissolved in 1.0 mL of DMF, and the solution was allowed to stir at 25 °C for 2 h. Then, to this solution, 2.0 mL of PBS was added at a rate of 1.0 mL min−1 under gentle stirring. After standing for 3 h at 25 °C, DMF was removed by dialysis using a dialysis bag (MWCO 3500 Da) against PBS for 24 h and the volume was set to the mark in a 5.0 mL volumetric flask to obtain the micelle solution, marked as micelle2.
2.4. Characterization
1H NMR spectra were recorded on a Bruker AV 400 NMR spectrometer in chloroform-d (CDCl3) or trifluoroacetic acid-d (TFA-d). FT IR spectra were recorded on a Bio-Rad Win-IR instrument using the potassium bromide method. Number-, weight-average molecular weights (Mn, Mw) and molecular weight distributions (polydispersity index, PDI = Mw/Mn) were determined by gel permeation chromatography (GPC) using a series of linear TSKgel Super columns (AW3000 and AW5000) and a Waters 515 HPLC pump, with an OPTILAB DSP Interferometric Refractometer (Wyatt Technology) as the detector. The eluent was DMF containing 0.01 M lithium bromide at a flow rate of 1.0 mL min−1 at 40 °C. Monodispersed polystyrene standards purchased from Waters Co., Ltd. with a molecular weight range of 1790–200000 were used to generate the calibration curve. Transmission electron microscopy (TEM) measurements were performed on a JEOL JEM-1011 transmission electron microscope with an accelerating voltage of 100 kV. A drop of the micelle solution (0.05 g L−1) was deposited onto a 230 mesh copper grid coated with carbon and allowed to dry in air at 25 °C before measurements. Dynamic laser scattering (DLS) measurements were performed on a WyattQELS instrument with a vertically polarized He–Ne laser (DAWN EOS, Wyatt Technology). The scattering angle was fixed at 90°.
2.5. Critical micellization concentration (CMC) measurements
CMC was measured by fluorescence spectroscopy using pyrene as probe on a PTI Fluorescence Master System with the software Felix 4.1.0. Typically, a predetermined amount of pyrene in acetone was added into a series of ampoules, and the acetone was removed first by gently flowing N2 and then by vacuum. A predetermined volume of mPEG-S2-PZLL solution and PBS were added into the ampoules, consecutively, to get solutions with different mPEG-S2-PZLL concentrations ranging from 6.10 × 10−5 to 1.0 g L−1, whereas the concentration of pyrene in each flask was fixed at 6.0 × 10−7 mol L−1, slightly lower than the saturation solubility of pyrene in water. These solutions were shaken vigorously and then allowed to equilibrate at 25 °C for at least 24 h. The excitation spectra of pyrene with various mPEG-S2-PZLL concentrations were measured at the detection emission wavelength (λem = 390 nm). The CMC value was obtained from the intersection of the tangent to the horizontal line of I340/I335 with relative constant value and the diagonal line with rapidly increased I340/I335 ratio.
Bovine serum albumin (BSA) was used as a model protein to determine the protein adsorption of mPEG-S2-PZLL micelles. The micelles were incubated with a solution of BSA in PBS at pH 7.4, with the final concentrations of micelles at 0.125, 0.25 or 0.5 g L−1, and BSA at 0.5 g L−1, respectively. After incubation at 37 °C for a determined time, 1.0 mL of each sample was withdrawn after vortexing to ensure homogeneity, and then centrifuged at 16000 g for 15 min to precipitate the protein-adsorbed micelles. The BSA concentration of the supernatant was determined using UV-Vis spectroscopy (UV-2401PC, Shimadzu) by measuring the maximal absorbance at 280 nm. Then, the amount of BSA adsorbed on the micelle was calculated against a standard calibration curve of BSA using a previously reported method.11 mPEG113 and PEI25k were used as negative and positive controls, respectively.
2.7. Hemolysis activity examination
The hemolysis properties of the mPEG-S2-PZLL micelles were examined by spectrophotometry. Rabbit blood stabilized with dipotassium ethylene diamine tetraacetate was obtained from the Experimental Animal Center of Jilin University. To obtain red blood cells (RBCs), 5.0 mL of a rabbit blood sample was added to 10.0 mL of physiological saline (PS), and then the RBCs were isolated from serum by centrifugation at 1200 rpm for 8 min. The RBCs were further washed 6 times with PS (40.0 mL × 6). Following the last wash, the remaining RBCs were dispersed in 30.0 mL of PS. 0.4 mL of micelles at different concentrations were separately mixed with 0.4 mL of RBCs suspended in PS. The mixtures were then incubated at 37 °C in a thermostatted water bath for 2 h. Then, the RBCs were centrifuged at 3000 rpm for 10 min and 100.0 μL of the supernatant of each sample was transferred into a 96-well plate. Free hemoglobin in the supernatant was measured with a Bio-Rad 680 microplate reader at 540 nm. PS and triton X-100 (10.0 g L−1) were used as negative and positive controls, respectively. All hemolysis experiments were carried out in triplicate. The hemolysis ratio (HR) of the RBCs was calculated using eqn (1):| |  | (1) |
In eqn (1), Asample, Anegitive control, and Apositive control were denoted as the absorbances of the sample, negative and positive controls, respectively.
2.8. Tissue response evaluations
Five-week-old female BALB/c nude mice were obtained and housed from the Animal Holding Unit of Northeast Normal University. The animal procedures were approved by the School of Life Sciences Animal Care and Use Committee of Northeast Normal University. Micelles were dissolved in PBS (1.0 g L−1, 10.0 μL) and injected into the tibialis anterior muscles in the mice. The muscles that received the injections were isolated one week later, fixed in 4% (w/v) paraformaldehyde, washed and embedded in paraffin. Tissue sections were cut into 8.0 μm thick slices, which were placed onto gelatin-coated slides and stained with hematoxylin and eosin (H&E) for histological examinations. The muscles without injection and receiving an intramuscular injection of 10.0 μL of PBS and PEI25k in PBS (1.0 g L−1, 10.0 μL) were used as controls.
2.9. Drug loading and release
DOX was used as a model drug for drug loading. DOX-loaded mPEG-S2-PZLL micelles were prepared by a nanoprecipitation technique (Scheme 2). Typically, mPEG-S2-PZLL (20.0 mg), DOX·HCl (4.0 mg) and triethylamine (0.72 mg) were mixed in 4.0 mL of DMF. The solution was allowed to stand at 25 °C for 2 h. Then, 2.0 mL of deionized water was added dropwise to the solution under stirring. The mixture was stirred at 25 °C for 6 h and the organic solvent was removed by dialysis against deionized water for 24 h. The dialysis medium was refreshed 5 times and the whole procedure was performed in the dark. Then, the solution was filtered and lyophilized. For determination of drug loading content (DLC) and drug loading efficiency (DLE), the drug loaded micelle was dissolved in DMF and analyzed with fluorescence measurements using a standard curve method (λex = 480 nm). The DLC and DLE of the drug loaded micelles were calculated according to eqn (2) and (3), respectively:| | 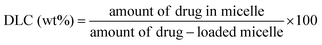 | (2) |
| |  | (3) |
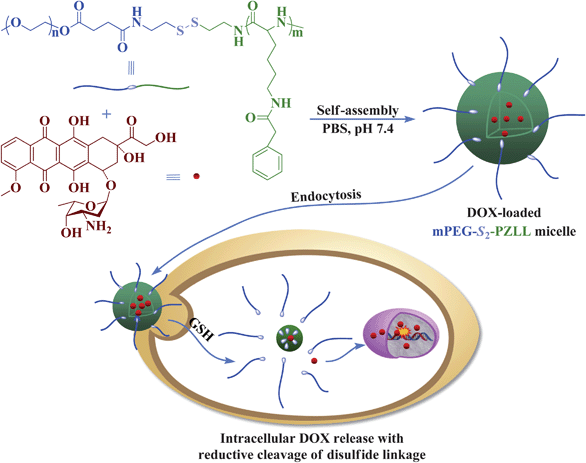 |
| | Scheme 2 Schematic illustration of DOX loading and targeted intracellular release. | |
The release profiles of the DOX-loaded micelles were investigated in PBS (pH 7.4) without or with 10.0 mM GSH. The weighed, freeze-dried DOX-loaded micelle was suspended in 3.0 mL of release medium and transferred into a dialysis bag (MWCO 3500 Da). The release experiment was initiated by placing the end-sealed dialysis bag into 50.0 mL of release medium at 37 °C with continuous shaking at 70 rpm. At predetermined intervals, 2.0 mL of external release medium was taken out and an equal volume of fresh release medium was replenished. The amount of released DOX was determined using fluorescence measurements (λex = 480 nm). The release experiments were conducted in triplicate.
2.10. Intracellular drug release
The cellular uptake and intracellular release behaviors of the DOX-loaded micelles were observed by confocal laser scanning microscopy (CLSM) and flow cytometric analyses toward HeLa or HepG2 cells.
2.10.1. CLSM.
The cells were seeded in 6-well plates at 2.0 × 105 cells per well in 2.0 mL of complete Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, supplemented with 50.0 IU mL−1 penicillin and 50.0 IU mL−1 streptomycin, and cultured for 24 h, and then treated with 0.5 mM BSO for 12 h or treated with 10.0 mM GSH-OEt for 2 h. Cells were washed by PBS and incubated at 37 °C for an additional 2 h with DOX-loaded micelles at a final DOX concentration of 10.0 mg L−1 in complete DMEM. Cells without BSO and GSH-OEt pretreatment were used as control. Then, the culture medium was removed and cells were washed with PBS thrice. Thereafter, the cells were fixed with 4% (w/v) paraformaldehyde for 30 min at 25 °C, and the cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, blue) for the cell nuclei and Alexa Fluor® 488 phalloidin (Alexa 488, green) for the cytoskeleton, according to the standard protocols provided by the suppliers. CLSM images of the cells were obtained through confocal microscopy (Olympus FluoView 1000).
2.10.2. Flow cytometric analyses.
The cells were seeded in 6-well plates at 2.0 × 105 cells per well in 2.0 mL of complete DMEM, and cultured for 24 h, and then treated with 0.5 mM BSO for 12 h or treated with 10.0 mM GSH-OEt for 2 h. The cells were washed by PBS and incubated at 37 °C for an additional 2 h with the DOX-loaded micelles at a final DOX concentration of 10.0 mg L−1 in complete DMEM. Cells without BSO and GSH-OEt pretreatment were used as control. Thereafter, the culture medium was removed and the cells were washed with PBS thrice and treated with trypsin. Then, 1.0 mL of PBS was added to each culture well, and the solutions were centrifuged for 4 min at 3000 rpm. After removal of supernatants, the cells were resuspended in 0.3 mL of PBS. Data for 1.0 × 104 gated events were collected, and analysis was performed by flow cytometry (Beckman, California, USA).
2.11. Cell viability assays
The relative cytotoxicities of the micelles against HeLa and HepG2 cells were evaluated in vitro by a standard MTT assay. The cells were seeded in 96-well plates at 7000 cells per well in 100.0 μL of complete DMEM and incubated at 37 °C in a 5% CO2 atmosphere for 24 h, followed by removing the culture medium and adding micelle solutions at different concentrations (0–0.5 g L−1). The cells were subjected to MTT assay after being incubated for another 72 h. The absorbance of the solutions were measured on a Bio-Rad 680 microplate reader at 490 nm. Cell viability (%) was calculated based on eqn (4):| | 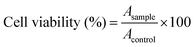 | (4) |
In eqn (4), Asample and Acontrol represented the absorbances of the sample and control wells, respectively.
The cytotoxicities of the DOX-loaded micelles against HeLa and HepG2 cells were also evaluated in vitro by MTT assay. Similarly, cells were seeded into 96-well plates at 10000 cells per well in 200.0 μL of complete DMEM and further incubated for 24 h. Then, the cells were treated with 0.5 mM BSO for 12 h or 10.0 mM GSH-OEt for 2 h. Cells without pretreatment were used as control. After washing the cells with PBS, the DOX-loaded micelles were added at different DOX concentrations (0–10.0 mg L−1 DOX). The cells were subjected to MTT assay after being incubated for another 24, 48 or 72 h. The absorbance of the solutions was measured on a Bio-Rad 680 microplate reader at 490 nm. Cell viability (%) was calculated based on eqn (4).
3. Results and discussion
3.1. Synthesis and characterization of disulfide-linked block copolymers
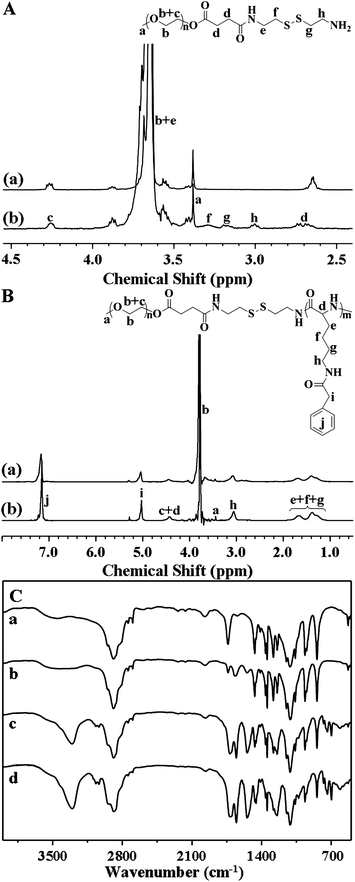
Synthesis and polymerization of α-amino acid N-carboxyanhydrides were first reported by Hermann Leuchs in 1906.27 Since then, these cyclic and highly reactive amino acid derivatives have been used for the formation of polypeptides by ROP with a primary amine initiator in a controlled manner.28,29 In this work, the disulfide-linked mPEG-S2-PZLL block copolymers were synthesized through ROP of ZLL NCA, initiated by the terminated primary amine group in the mPEG-S2-NH2 chain (Scheme 1). First, mPEG-S2-NH2 was synthesized through the ring-opening reaction of SA and a subsequent condensation reaction. The characteristic resonances at 3.29 and 3.18 ppm are attributed to the methylene protons neighboring the disulfide bond (–C(O)NHCH2CH2–S2–CH2CH2NH2, 2H and 2H) in the 1H NMR spectrum (Fig. 1Ab), and the typical amide I and II bands at 1662 (υC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1546 cm−1 (υC(O)–NH) observed from FT IR spectroscopy (Fig. 1Cb) confirmed the successful synthesis of mPEG-S2-NH2. The ZLL NCA reacted with mPEG-S2-NH2 to obtain the mPEG-S2-PZLL block copolymer, whose chemical structure was also characterized by both 1H NMR and FT IR. The 1H NMR spectra verified the successful synthesis of mPEG-S2-PZLL, and all signals were well assigned, as shown in Fig. 1B. The degree of polymerization (DP, Table 1) of PZLL in the resultant mPEG-S2-PZLL was calculated by comparing the integrated area of the peak at 5.06 or 3.08 ppm, attributed to the methylene proton of the side group of the ZLL unit, with the signal at 3.65 ppm of the methylene proton assigned to mPEG. The results of FT IR (Fig. 1Cc and Cd) also demonstrated the generation of a PZLL block, based on the increased strength of absorption at 1654 (υCO) and 1539 cm−1 (υC(O)–NH), attributed to the amide bond in the backbone of PZLL. The successful synthesis of mPEG-S2-PZLL was further confirmed by GPC. The PDI values of the copolymers were around 1.09–1.14. The relatively low molecular weight distribution may be attributed to the living character of the ROP of the NCA monomer, initiated by primary amine. It should be noted that the Mn measured by GPC was relatively higher than that measured by 1H NMR, due to the structural differences between the resultant copolymer and the monodispersed polystyrene standards that were used to generate the calibration curve in GPC analyses.
O) and 1546 cm−1 (υC(O)–NH) observed from FT IR spectroscopy (Fig. 1Cb) confirmed the successful synthesis of mPEG-S2-NH2. The ZLL NCA reacted with mPEG-S2-NH2 to obtain the mPEG-S2-PZLL block copolymer, whose chemical structure was also characterized by both 1H NMR and FT IR. The 1H NMR spectra verified the successful synthesis of mPEG-S2-PZLL, and all signals were well assigned, as shown in Fig. 1B. The degree of polymerization (DP, Table 1) of PZLL in the resultant mPEG-S2-PZLL was calculated by comparing the integrated area of the peak at 5.06 or 3.08 ppm, attributed to the methylene proton of the side group of the ZLL unit, with the signal at 3.65 ppm of the methylene proton assigned to mPEG. The results of FT IR (Fig. 1Cc and Cd) also demonstrated the generation of a PZLL block, based on the increased strength of absorption at 1654 (υCO) and 1539 cm−1 (υC(O)–NH), attributed to the amide bond in the backbone of PZLL. The successful synthesis of mPEG-S2-PZLL was further confirmed by GPC. The PDI values of the copolymers were around 1.09–1.14. The relatively low molecular weight distribution may be attributed to the living character of the ROP of the NCA monomer, initiated by primary amine. It should be noted that the Mn measured by GPC was relatively higher than that measured by 1H NMR, due to the structural differences between the resultant copolymer and the monodispersed polystyrene standards that were used to generate the calibration curve in GPC analyses.
 |
| | Fig. 1
1H NMR spectra of mPEG113-COOH (a) and mPEG113-S2-NH2 (b) (A, in CDCl3), and mPEG113-S2-PZLL18 (a) and mPEG113-S2-PZLL35 (b) (B, in TFA-d); FT IR spectra of mPEG113-COOH (a), mPEG113-S2-NH2 (b), mPEG113-S2-PZLL18 (c) and mPEG113-S2-PZLL35 (d) (C). | |
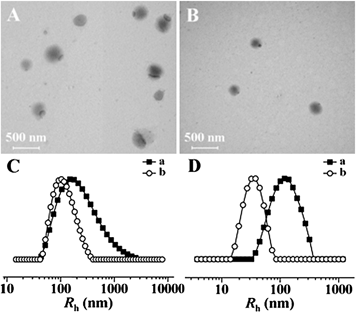 |
| | Fig. 2 Typical TEM micrographs of mPEG113-S2-PZLL18 micelle1 (A) and micelle2 (B), and hydrodynamic radii (Rh) of mPEG113-S2-PZLL18 micelle1 (a) and micelle2 (b) (C), and mPEG113-S2-PZLL35 micelle1 (a) and micelle2 (b) (D). | |
| Entry |
CMCa (mg L−1) |
CMCb (mg L−1) |
R
h
(nm) |
R
h
(nm) |
DLC (wt%) |
DLE (wt%) |
|
Micelle1.
Micelle2.
Micelle1.
Micelle2.
|
| mPEG113-S2-PZLL18 |
18.74 ± 1.28 |
9.63 ± 1.13 |
181.0 ± 15.7 |
125.0 ± 5.9 |
5.35 ± 0.93 |
28.24 ± 1.25 |
| mPEG113-S2-PZLL35 |
13.91 ± 1.07 |
3.68 ± 0.68 |
116.0 ± 10.2 |
60.8 ± 5.0 |
6.51 ± 0.86 |
34.83 ± 1.13 |
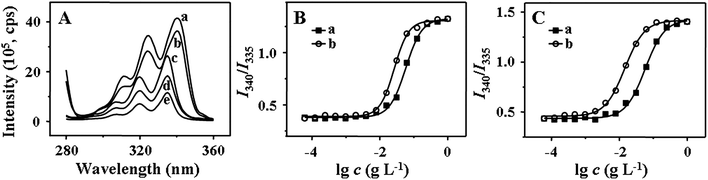 |
| | Fig. 3 Excitation spectra of pyrene in directly dissolved aqueous solutions of mPEG113-S2-PZLL18 at different concentrations (a: 1.0, b: 1.25 × 10−1, c: 1.56 × 10−2, d: 1.95 × 10−3, e: 2.44 × 10−4 g L−1, λem = 390 nm) (A), and the intensity ratio (I340/I335) as a function of the log of the concentration of mPEG113-S2-PZLL18 (B) and mPEG113-S2-PZLL35 (C) (a: micelle1, b: micelle2). | |
CMC is one of the key parameters to describe the physical properties of micelles. At the CMC, the hydrophobic portion starts to associate to reduce the interaction with water molecules, leading to micelle formation. From the plots of the pyrene fluorescence intensity ratio of I340/I335 (Fig. 3B and C) versus the log of copolymer concentration, the CMC values were obtained, taken as the intersection of the tangents to the horizontal line of intensity ratio with relatively constant value and the diagonal line with rapidly increased intensity ratio (Table 2). Both the micelle preparation approach and the copolymer composition affected the CMC value. Micelle1 has a higher apparent CMC value than micelle2, attributed to the non-micellar aggregation and incomplete diffusion of copolymeric chains. The CMC value of mPEG113-S2-PZLL35 was lower than that of mPEG113-S2-PZLL18, induced by the longer PZLL segment, which played an important role in the stability of the micelle. With the lower dispersity and CMC value of micelle2, the dialysis method was more appropriate to prepare the micelles. In addition, the low CMC values below 10 mg L−1 of the copolymers provided the possibility of retaining the stability of the micelles in very dilute aqueous milieu (e.g. blood stream), which is one of the important features for drug delivery.
3.3. Hemocompatabilities of micelles
Excellent hemocompatibilities of nanocarriers are one of the requisites because nanocarriers are designed to be administrated via intravenous injection for most drug delivery applications. Therefore, the interaction of the micelles and blood components should be effectively minimized and the abilities for resistance to nonspecific protein adsorption and elimination of hemolysis were requested.
In this work, the mPEG shell of the micelles can prevent recognition by the reticuloendothelial system and resist nonspecific protein adsorption (i.e. nonfouling properties) in complicated in vivo circumstances, which results in prolonged periods of circulation.34,35 Herein, the interaction of the mPEG-S2-PZLL micelle2s with the model protein (i.e. BSA) was addressed to examine the potential of the micelle2s for in vivo applications. As shown in Fig. 4, for all concentrations (0.125, 0.25 and 0.5 g L−1) and incubation times (2, 4 and 6 h), the micelle2s and mPEG113 showed slight BSA adsorption, while PEI25k interacted strongly with BSA. It is believed that such a characteristic is the result of the formation of a strong hydration layer around the surface of the micelle because of the existence of the hydrophilic mPEG shell. In addition, the HR of the micelle was assessed by a hemolysis assay. An enhanced HR results in a higher level of broken RBCs. The RBCs were exposed to the micelles at different concentrations for 2 h. As shown in Fig. 5, the mPEG-S2-PZLL micelle2s showed negligible hemolysis destruction to RBCs, even at a very high concentration of 0.5 g L−1. These results indicated that the mPEG-S2-PZLL micelle2s were hemocompatible, allowing potential application as drug delivery vehicles.
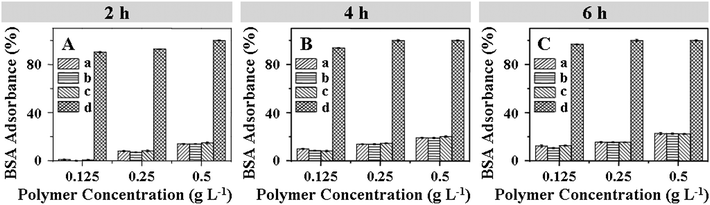 |
| | Fig. 4 BSA adsorption on the mPEG113-S2-PZLL18 micelle2 (a), mPEG113-S2-PZLL35 micelle2 (b), mPEG113 (c) and PEI25k (d) after incubation at 37 °C with different concentrations and periods of time: 2 (A), 4 (B) and 6 h (C). | |
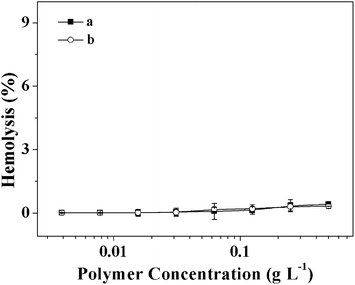 |
| | Fig. 5 Percentage of RBC hemolysis incubated with mPEG113-S2-PZLL18 micelle2 (a) and mPEG113-S2-PZLL35 micelle2 (b). PS (−) and triton X-100 (10.0 g L−1) (+) were used as negative and positive controls, respectively. Data were represent as mean ± standard deviation (n = 3). | |
3.4. Cell and tissue compatibilities of micelles
Evaluation of the cell and tissue compatibilities of micelles is necessary for drug delivery applications. The in vitro cytotoxicities of the micelles to HeLa and HepG2 cells were evaluated by a MTT assay. Fig. 6 shows the viability of cells treated with micelles at different concentrations for 72 h, which was compared with those treated with PEI25k. More than 90% of the cells cultured with the micelles remained viable, even at a concentration of 0.5 g L−1, whereas there were few live cells left after treatment with PEI25k at the same concentration. This suggested that the micelles had low cytotoxicities and could be safely used as biocompatible carriers for delivering bioactive substances.
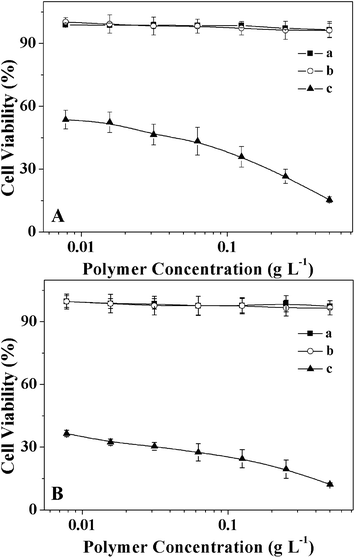 |
| | Fig. 6 Cell viabilities of HeLa (A) and HepG2 (B) cells incubated with mPEG113-S2-PZLL18 micelle2 (a) and mPEG113-S2-PZLL35 micelle2 (b) with PEI25k (c) as control for 72 h. Data were presented as mean ± standard deviation (n = 6). | |
To further demonstrate the biocompatibilities of the micelles in vivo, the acute inflammatory response toward micelles was evaluated in the muscle in female BALB/c nude mouse. As shown in Fig. 7, severe necrosis was observed in the muscle sample after 7 days following PEI25k injection. However, histological analysis revealed no obvious inflammatory reactions at the injection sites with the same dose of the micelles, which were comparable to muscle samples without injection and after receiving PBS injection, indicating the good biocompatibilities of the micelles in vivo.
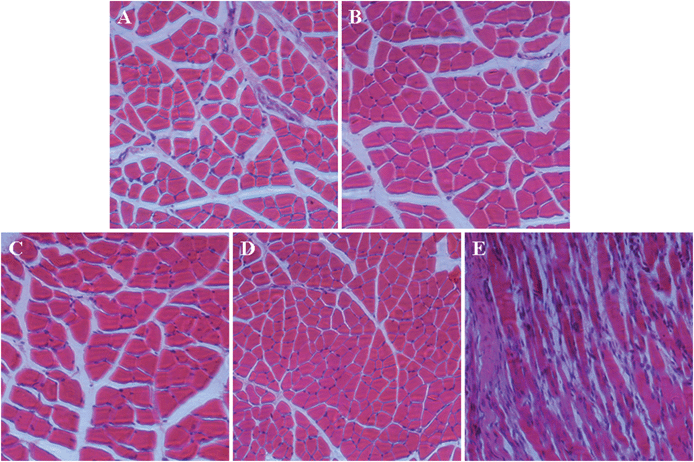 |
| | Fig. 7 Tissue responses after intramuscular injections of mPEG113-S2-PZLL18 micelle2 (A) and mPEG113-S2-PZLL35 micelle2 (B) as compared with that without (C), and with PBS (D) and PEI25k (E) injections. Tissue sections were analyzed by H&E staining after 7 days following injection. | |
3.5. DOX loading and reduction-responsive release
DOX is one of the most effective anthracycline antitumor drugs against a wide range of malignancies, which interacts with DNA through insertion and then inhibits the biosyntheses of bioactive macromolecules.1,13 In the current study, to better understand the potential applications of the obtained micelles in drug delivery, DOX was selected as a model drug to determine the loading and release properties.
DOX was loaded into the micelles by a nanoprecipitation technique (Scheme 2). Comparison of the drug-loading abilities of the micelles indicated that the higher DLC and DLE were attributed to longer hydrophobic PZLL blocks (Table 2). The DLC values for mPEG113-S2-PZLL18 and mPEG113-S2-PZLL35 were 5.35 ± 0.93 and 6.51 ± 0.86 wt%, depending on the compositions of the block copolymers, while the DLE values were 28.24 ± 1.25 and 34.83 ± 1.13 wt%, respectively.
The drug release behaviors of the DOX-loaded micelles with different DP of PZLL were investigated in PBS without and with 10.0 mM GSH at pH 7.4. The cumulative release percentages of DOX loaded in micelles versus time were plotted in Fig. 8. In the absence of GSH, less than 50 wt% loaded DOX was released from the micelles over the test duration (60 h). However, the DOX release rates were accelerated by the presence of GSH, and over 90% of loaded DOX was released with 10.0 mM GSH, analogous to the intracellular reductive microenvironment, in 60 h. The fast DOX release from the micelles under the reductive conditions was most likely due to the degradation of the micelles during cleavage of the disulfide bonds (Scheme 2). The length of PZLL was found to be relevant to the drug release kinetics, and the release rates of DOX from micelles were of the order of mPEG113-S2-PZLL18 micelle (with 10.0 mM GSH) > mPEG113-S2-PZLL35 (with 10.0 mM GSH) > mPEG113-S2-PZLL18 micelle (without GSH) > mPEG113-S2-PZLL35 (without GSH). The increase in the DP of PZLL led to the decrease of the DOX release rate. Therefore, it was possible to modulate the release rate of DOX from the micelles by adjusting the reductive environment and polypeptide content. These release profiles are not only beneficial for minimizing drug loss in circulation, but also for selective accumulation in tumor tissue by the enhanced permeability and retention effect, which may improve the overall therapeutic efficacy in vivo.
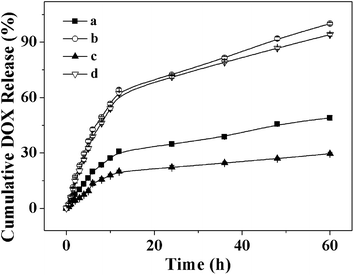 |
| | Fig. 8 DOX release from DOX-loaded mPEG113-S2-PZLL18 micelles (a, b) and mPEG113-S2-PZLL35 micelles (c, d) at pH 7.4 without GSH (a, c) and with 10.0 mM GSH (b, d) in PBS at 37 °C. Data were presented as mean ± standard deviation (n = 3). | |
3.6. Intracellular DOX release and cellular proliferation inhibition
To demonstrate the feasibility of the micelles for intracellular drug delivery in malignancy therapy, the cellular uptake and intracellular release behaviors of the DOX-loaded micelles were evaluated by CLSM and flow cytometry toward HeLa or HepG2 cells. HeLa or HepG2 cells were first pretreated with 0.5 mM BSO for 12 h, or with 10.0 mM GSH-OEt for 2 h, and then incubated with typical DOX-loaded mPEG113-S2-PZLL18 or mPEG113-S2-PZLL35 micelles for 2 h (10.0 mg L−1 DOX). HeLa or HepG2 cells without pretreatment were used as controls. It is documented that BSO is an inhibitor for the intracellular synthesis of GSH, and GSH-OEt can obviously improve the intracellular GSH concentration.1 As expected, the strongest intracellular DOX fluorescence was observed in the GSH-OEt pretreated cells after incubation with the DOX-loaded micelles (Fig. 9). In contrast, the weakest DOX fluorescence was shown in the cells preincubated with BSO. It was noted that stronger intracellular fluorescence was observed in the cells incubated with DOX-loaded mPEG113-S2-PZLL35 micelles due to the larger total amount of intracellular released DOX, which was associated with the difference in the amount of released DOX between those without and with GSH (Fig. 8). The triggered intracellular drug release was further confirmed by flow cytometric analyses. As shown in Fig. 10, the flow cytometric histogram of the GSH-OEt pretreated cells incubated with DOX-loaded micelles shifts clearly in the direction of high fluorescence intensity as compared with that of the control, and the weakest fluorescence intensity was observed in the cells incubated with BSO. The free DOX was reported to have stronger fluorescence as compared with the DOX loaded in nanoparticles at the same absolute DOX concentration due to the self-quenching effects of DOX.21,36 Thus, the enhanced fluorescence intensity in the GSH-OEt pretreated HeLa or HepG2 cells may be the result of the high endocytosis efficiency of the DOX-loaded micelles and the improved intracellular release of DOX, caused by the reduction-responsive degradation of the micelles.
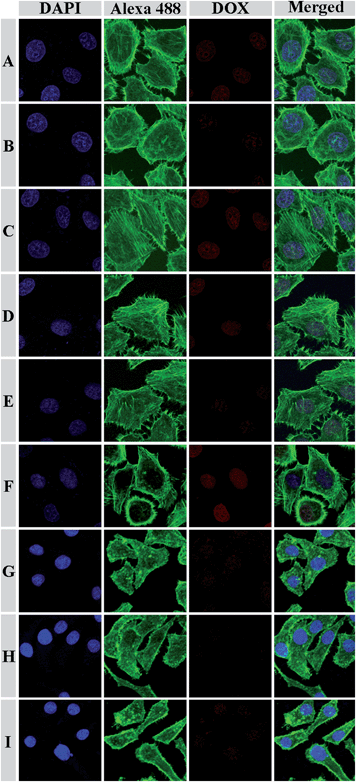 |
| | Fig. 9 Representative CLSM images of HeLa cells incubated with DOX-loaded mPEG113-S2-PZLL18 micelles (A, B and C) and mPEG113-S2-PZLL35 micelles (D, E and F), and HepG2 cells incubated with DOX-loaded mPEG113-S2-PZLL18 micelles (G, H and I) for 2 h: (A, D and G) cells without pretreatment; (B, E and H) cells pretreated with 0.5 mM BSO; (C, F and I) cells pretreated with 10.0 mM GSH-OEt. The cell nuclei and cytoskeleton of the cells were stained with DAPI (blue) and Alexa 488 (green), respectively. | |
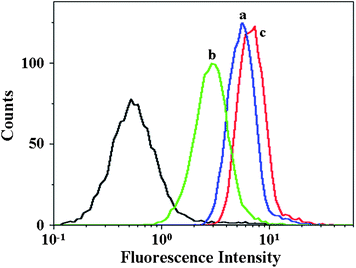 |
| | Fig. 10 Flow cytometric profiles of HeLa cells incubated with DOX-loaded mPEG113-S2-PZLL18 micelles for 2 h: (a) cells without pretreatment; (b) cells pretreated with 0.5 mM BSO; (c) cells pretreated with 10.0 mM GSH-OEt. | |
The in vitro cellular proliferation inhibition of DOX-loaded mPEG-S2-PZLL micelles against HeLa and HepG2 cells was also estimated by a MTT assay. Cells were pretreated with 0.5 mM BSO or 10.0 mM GSH-OEt for 12 h and 2 h, separately, and then incubated with the DOX-loaded micelles for 24, 48 and 72 h. The cells without pretreatment were used as controls. According to the previous literature, 0.5 mM BSO and 10.0 mM GSH-OEt did not show any cytotoxicities toward HeLa and HepG2 cells.1,37 As shown in Fig. 11, in contrast to the control cells, the HeLa and HepG2 cells pretreated with BSO and GSH-OEt exhibited the lowest and highest inhibition efficiency, respectively. The results revealed that the faster DOX release from the DOX-loaded micelles was triggered by higher intracellular GSH concentration, which enhanced the inhibition of the cellular proliferation. In addition, the DOX-loaded mPEG113-S2-PZLL35 micelles exhibited higher cellular proliferation inhibiting ability than the DOX-loaded mPEG113-S2-PZLL18 micelles in all the test conditions and durations. This was due to the slower extracellular DOX release and almost identical intracellular DOX release from the DOX-loaded mPEG113-S2-PZLL35 micelles (Fig. 8). The half maximal inhibitory concentration (IC50) values of the DOX-loaded micelles were listed in Table 3. In contrast to the control cells, the cells pretreated with BSO and GSH-OEt exhibited the highest and lowest IC50 values, respectively. With the same test conditions and duration, all the cells cultured with DOX-loaded mPEG113-S2-PZLL35 micelles showed lower IC50 values in contrast with the DOX-loaded mPEG113-S2-PZLL18 micelles. It quantificationally confirmed the differences in cellular proliferation inhibitions of unpretreated and pretreated cells with various DOX-loaded mPEG-S2-PZLL micelles and revealed the adjustable reduction-responsive ability of the micelles in cells.
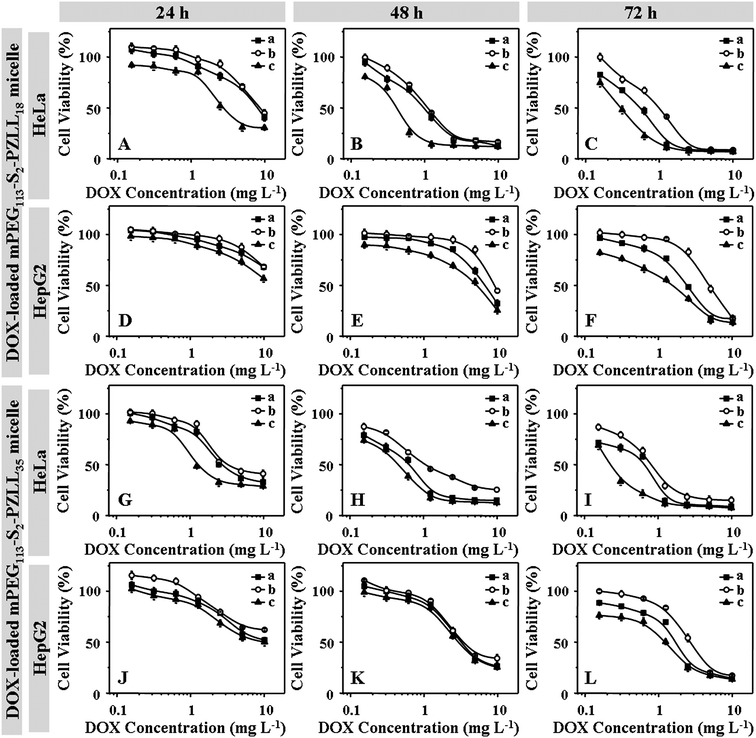 |
| | Fig. 11 Proliferation inhibition towards HeLa (A, B, C, G, H and I) and HepG2 cells (D, E, F, J, K and L) incubated with DOX-loaded mPEG113-S2-PZLL18 micelles (A, B, C, D, E and F) and mPEG113-S2-PZLL35 micelles (G, H, I, J, K and L) with various DOX concentrations for 24 (A, D, J and G), 48 (B, E, H and K) and 72 h (C, F, I and L). The cells were unpretreated (a), pretreated with 0.5 mM BSO (b) and 10.0 mM GSH-OEt (c), respectively. Data were presented as mean ± standard deviation (n = 6). | |
Table 3 IC50 values of DOX-loaded micelles after incubation with unpretreated and pretreated HeLa and HepG2 cells for 24, 48 and 72 h
| Entry |
Pretreatment |
IC50a (mg L−1) |
IC50b (mg L−1) |
IC50c (mg L−1) |
| HeLa |
HepG2d |
HeLa |
HepG2 |
HeLa |
HepG2 |
|
24 h.
48 h.
72 h.
Beyond the test range.
|
| mPEG113-S2-PZLL18 |
Unpretreated |
8.05 |
— |
1.01 |
6.62 |
0.55 |
2.34 |
| 0.5 mM BSO for 12 h |
8.65 |
— |
1.15 |
8.98 |
0.99 |
4.72 |
| 10.0 mM GSH-OEt for 2 h |
2.75 |
— |
0.46 |
5.19 |
0.30 |
1.48 |
| mPEG113-S2-PZLL35 |
Unpretreated |
2.75 |
— |
0.67 |
3.26 |
0.86 |
1.70 |
| 0.5 mM BSO for 12 h |
3.32 |
— |
1.08 |
3.49 |
0.94 |
2.69 |
| 10.0 mM GSH-OEt for 2 h |
1.31 |
— |
0.46 |
3.04 |
0.24 |
1.25 |
4. Conclusions
Two disulfide-linked mPEG-S2-PZLL block copolymers were synthesized through ROP of ZLL NCA, initiated by mPEG-S2-NH2. The block copolymers formed into micelles in PBS at pH 7.4 by direct dissolution and dialysis methods. The micelles prepare by the dialysis method have a lower dispersity and CMC value than those prepared by the direct dissolution approach. The hemocompatibilities, and cell and tissue compatibilities of the micelles were revealed to be excellent, which rendered them with potential for delivering bioactive substances. DOX was loaded into the micelles, and the DOX release was accelerated in the presence of 10.0 mM GSH or from micelles with lower PZLL content. The DOX-loaded micelles exhibited faster DOX release in HeLa and HepG2 cells, which were pretreated with 10.0 mM GSH-OEt or incubated with DOX-loaded mPEG113-S2-PZLL35 micelles. Moreover, a higher cellular proliferation inhibition efficacy was achieved toward GSH-OEt pretreated or DOX-loaded mPEG113-S2-PZLL35 micelle cultured HeLa and HepG2 cells. These above features, including biocompatibilities, stability in storage conditions and intracellular stimuli triggered drug release, bestowed the mPEG-S2-PZLL micelles with unique advantages as vehicles for intelligent intracellular drug delivery in clinical chemotherapy.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (Projects 51233004, 51273196, 51273037, 51203153, 51273080, 51003103, 50973108, 20904053 and 21174142).
Notes and references
- F. Shi, J. Ding, C. Xiao, X. Zhuang, C. He, L. Chen and X. Chen, J. Mater. Chem., 2012, 22, 14168–14179 RSC
 .
.
- J. Ding, F. Shi, C. Xiao, L. Lin, L. Chen, C. He, X. Zhuang and X. Chen, Polym. Chem., 2011, 2, 2857–2864 RSC .
- A. Bhaw-Luximon, L. M. Meeram, Y. Jugdawa, W. Helbert and D. Jhurry, Polym. Chem., 2011, 2, 77–79 RSC .
- J. Liu, W. Huang, Y. Pang, X. Zhu, Y. Zhou and D. Yan, Biomaterials, 2010, 31, 5643–5651 CrossRef CAS .
- J. Cheng, J. X. Ding, Y. C. Wang and J. Wang, Polymer, 2008, 49, 4784–4790 CrossRef CAS .
- W. Wang, J. X. Ding, C. S. Xiao, Z. H. Tang, D. Li, J. Chen, X. L. Zhuang and X. S. Chen, Biomacromolecules, 2011, 12, 2466–2474 CrossRef CAS .
- X. Zhao, Z. Poon, A. C. Engler, D. K. Bonner and P. T. Hammond, Biomacromolecules, 2012, 13, 1315–1322 CrossRef CAS .
- J. Ding, C. Xiao, C. He, M. Li, D. Li, X. Zhuang and X. Chen, Nanotechnology, 2011, 22, 494012 CrossRef .
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS .
- S. H. Jung, S. H. Jung, H. Seong, S. H. Cho, K.-S. Jeong and B. C. Shin, Int. J. Pharm., 2009, 382, 254–261 CrossRef CAS .
- Y. Y. Yuan, J. Z. Du, W. J. Song, F. Wang, X. Z. Yang, M.-H. Xiong and J. Wang, J. Mater. Chem., 2012, 22, 9322 RSC .
- J. Ding, C. Xiao, L. Yan, Z. Tang, X. Zhuang, X. Chen and X. Jing, J. Controlled Release, 2011, 152, E11–E13 CrossRef CAS .
- H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef CAS .
- C. Y. Zhang, Y. Q. Yang, T. X. Huang, B. Zhao, X. D. Guo, J. F. Wang and L. J. Zhang, Biomaterials, 2012, 33, 6273–6283 CrossRef CAS .
- V. N. Rao, S. R. Mane, A. Kishore, J. Das Sarma and R. Shunmugam, Biomacromolecules, 2012, 13, 221–230 CrossRef .
- N. Ma, H. Xu, L. An, J. Li, Z. Sun and X. Zhang, Langmuir, 2011, 27, 5874–5878 CrossRef CAS .
- X. J. Cai, H. Q. Dong, W. J. Xia, H. Y. Wen, X. Q. Li, J. H. Yu, Y. Y. Li and D. L. Shi, J. Mater. Chem., 2011, 21, 14639 RSC .
- X. Jiang, L. Li, J. Liu, W. E. Hennink and R. Zhuo, Macromol. Biosci., 2012, 12, 703–711 CrossRef CAS .
- K. Y. Choi, H. Y. Yoon, J.-H. Kim, S. M. Bae, R.-W. Park, Y. M. Kang, I. S. Kim, I. C. Kwon, K. Choi, S. Y. Jeong, K. Kim and J. H. Park, ACS Nano, 2011, 5, 8591–8599 CrossRef CAS .
- S. R. MacEwan, D. J. Callahan and A. Chilkoti, Nanomedicine, 2010, 5, 793–806 CrossRef CAS .
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS .
- L. Y. Tang, Y. C. Wang, Y. Li, J. Z. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095–1099 CrossRef CAS .
- T. B. Ren, W. J. Xia, H. Q. Dong and Y. Y. Li, Polymer, 2011, 52, 3580–3586 CrossRef CAS .
- H. Y. Wen, H. Q. Dong, W. J. Xie, Y. Y. Li, K. Wang, G. M. Pauletti and D. L. Shi, Chem. Commun., 2011, 47, 3550–3552 RSC .
- J. Ding, C. Xiao, Z. Tang, X. Zhuang and X. Chen, Macromol. Biosci., 2011, 11, 192–198 CrossRef CAS .
- J. Ding, C. Xiao, L. Zhao, Y. Cheng, L. Ma, Z. Tang, X. Zhuang and X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2665–2676 CrossRef CAS .
- H. Leuchs, Ber. Dtsch. Chem. Ges, 1906, 39, 857–861 CrossRef CAS .
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS .
- T. Thambi, H. Y. Yoon, K. Kim, I. C. Kwon, C. K. Yoo and J. H. Park, Bioconjugate Chem., 2011, 22, 1924–1931 CrossRef CAS .
- A. Lalatsa, A. G. Schaetzlein, M. Mazza, L. Thi Bich Hang and I. F. Uchegbu, J. Controlled Release, 2012, 161, 523–536 CrossRef CAS .
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS .
- J. Rodriguezhernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS .
- J. Ding, X. Zhuang, C. Xiao, Y. Cheng, L. Zhao, C. He, Z. Tang and X. Chen, J. Mater. Chem., 2011, 21, 11383–11391 RSC .
- K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto, Y. Anraku, A. Kishimura, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2012, 134, 13172–13175 CrossRef CAS .
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472 CrossRef CAS .
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS .
- J. Y. Liu, Y. Pang, W. Huang, X. H. Huang, L. L. Meng, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.