
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-photon phosphorescence lifetime imaging of cells and tissues using a long-lived cyclometallated Npyridyl^Cphenyl^Npyridyl Pt(II) complex†
Elizabeth
Baggaley
*a,
Igor V.
Sazanovich
ad,
J. A. Gareth
Williams
b,
John W.
Haycock
c,
Stanley W.
Botchway
d and
Julia A.
Weinstein
*a
aDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, U.K.. E-mail: liz.baggaley@sheffield.ac.uk; julia.weinstein@sheffield.ac.uk
bDepartment of Chemistry, Durham University, DH1 3LE, Durham, UK
cDepartment of Engineering Materials, The Kroto Research Institute, University of Sheffield, S3 7HQ, Sheffield, UK
dCentral Laser Facility, Science and Technology Facilities Council, Harwell Science and Innovation Campus, OX11 0QX, UK
First published on 29th July 2014
Abstract
Using a combination of multiphoton excitation, confocal scanning, TCSPC and beam blanking in conjunction with a cyclometallated Npyridyl^Cphenyl^Npyridyl Pt(II) complex (1) with a long luminescence lifetime, we demonstrate lifetime mapping of living cells and histological tissue sections over a time-frame of 50 microseconds, using a laser on/off “beam blanking” approach. This method of performing phosphorescence lifetime imaging microscopy (PLIM) represents an order of magnitude enhancement of the two-photon time-resolved emission imaging microscopy (TP-TREM) method, where in order to achieve a longer imaging window, the excitation laser repetition rate was reduced by cavity dumping [Chem. Sci., 2014, 5, 879]. The method complements and expands other existing imaging methodologies by enabling simultaneous PLIM and FLIM (fluorescence lifetime imaging microscopy – recorded between beam blanking), whilst maintaining essential sub-micron spatial resolution. We demonstrate how the Pt(II) complex can be used to distinguish between cell nuclei and matrix proteins on the basis of emission lifetime, in both structured and homogeneous tissue sections; whilst also revealing how the Pt(II) emission lifetime varies with tissue matrix composition. The proposed imaging approach can be used in conjunction with any biocompatible emissive probe with a long emission lifetime – exemplified here by (1) – and for an array of fluorescent/phosphorescent labels, where discrimination is lifetime-based.
Introduction
The use of transition metal complexes in bio-imaging applications has increased over the past decade.1,2 The synthetic versatility, facile tuning of emission properties through ligand design, photostability, large difference between absorption and emission wavelengths, and long emission lifetimes often associated with emissive transition metal complexes offer a plethora of advantages over conventional organic dyes. Moreover, designing such complexes for targeted labelling can be readily achieved without compromising their photophysical properties, via modification of a peripheral ligand. The majority of such complexes are phosphorescent, not fluorescent, due to the triplet nature of their lowest excited states, and – owing to the formally forbidden nature of the triplet-to-singlet transition – the emission lifetime is comparatively long.3 This is one of the major features differentiating transition metal complexes from organic dyes. Yet the majority of imaging applications of transition metal complexes have been limited to the steady-state regime.1 Lifetime-based imaging, in contrast, offers many advantages with regard to providing information on the local environment through the effect on the kinetics of the excited state processes of the probe that are difficult to measure via steady-state methods, as well as being largely independent of concentration. However, the move towards lifetime imaging of phosphorescent complexes has been relatively slow, largely due to technical limitations associated with the measurements of lifetimes longer than a few nanoseconds in life sciences. Several significant contributions of new methods have been made recently,4,2b demonstrating the importance of the field, and calling for the full potential of imaging with such compounds to develop.Conversely, fluorescence lifetime imaging microscopy, FLIM,5 using traditional fluorescent dyes5b,6,7 and GFP-tagged proteins5a,8 has been known for several decades, and is now a mainstream tool in the bio-sciences (GFP = green fluorescent protein). However, traditional FLIM is limited to pico-to-nanosecond changes in emission lifetimes, which can restrict the detection of analytes at physiological concentrations and is susceptible to interference from autofluorescence – emission form naturally occurring fluorophores,6a such as tryptophan. Autofluorescence is a persistent problem in bio-imaging9 and significantly compromises image contrast, as it occurs in the same (visible) region and within the same (nanosecond) timescale as many of the conventional fluorescent dyes. Accessing the microsecond domain for lifetime imaging potentially offers advantages over FLIM, allowing imaging in the absence of autofluorescence and providing an order-of-magnitude increase in sensitivity for the detection of biological analytes, including oxygen.4f–i,10
Platinum porphyrins were amongst the first metal-based imaging agents to be used in time-resolved, autofluorescence-free imaging.11 In these pioneering experiments,11 a bespoke set-up comprised of mechanical shutters to block excitation and emission beams was used. The minimum delay time between excitation and detection was relatively long (2 μs), and the overall time resolution limited to 0.5 μs.
Technological advances in optics, electronic shutters and fast laser sources over the past 15–20 years now allow the delay time between the pulse and the detection to be reduced to virtually zero, significantly widening the range of metal-based complexes potentially suitable for time-resolved imaging. Application of these technological enhancements was demonstrated in 2008, using a family of cell-permeable Npyridyl^Cphenyl^Npyridyl-coordinated platinum(II) complexes. Emission of these photostable probes on the timescale of nanoseconds to microseconds was recorded in vitro using Time-Resolved Emission Imaging Microscopy (TREM).12 The initial technique of TREM was performed under one-photon excitation with a nanosecond pulsed laser as the excitation source, and a gated CCD camera to record images. As a result, the spatial resolution was limited to several microns.
Using two-photon excitation enables one to achieve the essential for life sciences sub-micron spatial resolution and NIR-excitation in the range of relative tissue transparency. However, two-photon excitation also requires a femtosecond laser source, which typically has a time interval of around 10 ns between pulses. Whilst perfectly suitable for FLIM where nanosecond lifetimes are being monitored, such an interval is clearly incompatible with the microsecond lifetimes of phosphorescent transition metal complexes. Several methods have emerged which combine multiphoton excitation with longer-lifetime detection, including frequency multiplex approach for oxygen mapping in mouse brain with >1 μm resolution.4b Reducing the repetition rate of the excitation laser at the sample by employing a Pockels cell, combined with microscope stage scanning in the method termed 2PLM (two-photon phosphorescence lifetime microscopy)4g,h or two-photon laser scanning microscopy10e has achieved submicron resolution and developed 3D imaging.4f,10f
In order to achieve the sub-micron spatial resolution, we recently reported a further development of the TREM method, initially reported by us: two-photon time-resolved emission imaging microscopy, TP-TREM.13 This technique uses a combination of confocal scanning, multiphoton excitation (with the femtosecond Ti-sapphire laser running in a cavity dumped mode to reduce the repetition rate of the laser to allow lifetime imaging on the microsecond domain whilst maintaining the energy-per-pulse) and detection via time correlated single photon counting (TCSPC). This made it possible to achieve a phosphorescence lifetime imaging (PLIM) window of ∼3 μs between laser pulses, yet retaining sufficient power to allow two-photon excitation at 760 nm.
Thus, in a similar fashion to FLIM, TP-TREM involves the determination of the lifetime at all points within the image via TCSPC, where each point corresponds to a volume of ∼1 μm3 (depending on NA of objective and excitation wavelength).14a But, TP-TREM provides a >1000-fold extension to the usual FLIM timescale. Naturally, however, due to the need to maintain the laser stability and power-per-pulse, there is still a technical limit to how far the temporal window can be extended using cavity dump method.
Results and discussion
Extending the lifetime imaging time-scale
In order to extend the two-photon PLIM imaging window further than 3 μs – essential for complexes that emit with lifetimes into the tens of microseconds – whilst maintaining submicron spatial resolution, an alternative approach is required, such as those described in the work on oxygen sensing using Pt-porphyrin based dendritic nanoparticles.10e This paper describes the use of a complementary method for extended the microsecond imaging window, demonstrated with the use of a long-lived luminescent Pt(II) complex, which is compatible with commercial multiphoton microscopes.A standalone lifetime imaging module with the capability to extend the imaging window a further 100 fold (into hundreds of μs) and to perform combined FLIM + PLIM studies has recently become commercially available from Becker & Hickl Gmbh.14b,c Similarly to TP-TREM, this set-up uses a confocal scanning microscope, multiphoton excitation and TCSPC detection to determine the emission lifetime at each point in the image.
The FLIM/PLIM technique is based on a multi-dimensional time-correlated single photon counting (TCSPC) process in combination with either confocal or multiphoton laser scanning.14b To record FLIM and PLIM data simultaneously, a high-frequency pulsed laser is on-off modulated synchronously with the pixels at a period in the microsecond range. For every photon detected, the TCSPC process determines the time in the laser pulse period and the time in the laser modulation period, and builds up photon distributions over these times and the scan coordinates. The results can be interpreted as two pixel arrays that contain fluorescence or phosphorescence decay curves in each pixel.14b–e
Apart from the capability to record FLIM and PLIM simultaneously, an advantage of the technique is that the laser is run at its standard high repetition rate, (∼80–100 MHz). At a given peak power, the large number of laser pulses during the ‘laser on’ periods builds up more triplet population than a single laser pulse would do. Thus, exceedingly high laser peak power, leading to possible higher order excitation or saturation effects, is avoided. Moreover, the length of the microsecond PLIM imaging window can be easily changed by adjusting the laser modulation period or duty cycle without changing parameters of the laser pulse itself.
Here, we show how a long-lived Pt imaging agent can be used in conjunction with the above described “blinking” method in order to image live cells and fixed tissue sections under two-photon excitation over an imaging window of 50 μs. The technique is compatible with two-photon excitation, thus keeping the spatial resolution, large penetration depth, and optical sectioning capability of multiphoton microscopy. With the capability to record FLIM and PLIM in the same imaging process, it complements existing imaging methods.4b,f,g,h,10e
Lifetime mapping with Pt(II) complex 1
Live rat Schwannoma cells (RN22) were incubated with the charge-neutral N^C^N-coordinated Pt(II) complex 1 (Fig. 1), which was successfully taken up within 5 minutes at 37 °C (50 μM, 0.5% DMSO in PBS), with no toxic effect to cells over 24 h. These conditions are very similar to those used in previous studies with human dermal fibroblasts (HFD), human melanoma (C8161) and Chinese hamster ovary (CHO) cells.12 Emission of 1 was observed predominantly from the nucleus, namely the nucleoli and nuclear membrane, with some additional diffuse cytoplasmic staining (Fig. 1, intensity image). This intensity-based staining pattern is identical to that observed in previous studies using CHO, HDF or C1861 cells, and is typical of this family of N^C^N cyclometalating Pt complexes.12,13 The PLIM lifetime map of Pt-labelled cells (Fig. 1, centre) clearly demonstrates how the emission lifetime of 1 varies with intracellular localisation. We note that the cellular emission decay of 1 is not monoexponential, reflecting a distribution of immediate microenvironments of the Pt-complex in cells. The two-exponential approximation is usually sufficient to satisfactorily fit the emission decay data.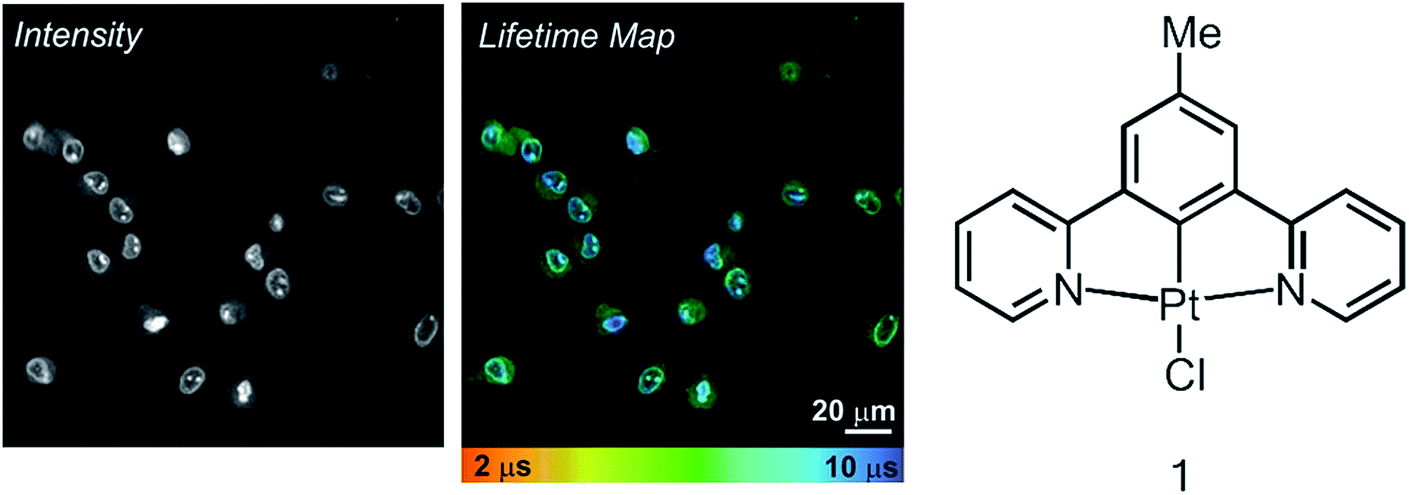 | ||
| Fig. 1 TP-PLIM imaging of live RN22 Schwannoma cells stained with complex 1 (50 μm, 5 min at 37 °C). The chemical structure of 1 is shown on the right. | ||
A significantly longer lifetime is observed in the nucleus and sub-nuclear structures (∼9 μs) compared to the cell cytoplasm. This distribution in lifetime is most likely due to oxygen quenching. DNA binding studies12 have shown that 1 can bind to DNA. The DNA-bound complex is believed to be more protected from collisions with surrounding oxygen, hence the complex that resides in the nucleus exhibits a longer emission lifetime. As is typical for long-lived triplet excited states, e.g. for Pt-porphyrins and assemblies thereof4g,h,10e used for oxygen sensing, the emissive3 π–π* state of 1 and its analogues in solution is strongly quenched by oxygen, in this case with a Stern–Volmer constant of 108–109 M−1 s−1.16 Oxygen-induced changes in the emission lifetime of 1 have also been measured in a gas chamber on the microscope (Fig. S1†) using TP-TREM. The sensitivity of Pt(N^C^N) derivatives to oxygen in cells has been demonstrated by a significant increase of the phosphorescence lifetime in the cytoplasm when equilibrated in an atmosphere of 95%N2/5% CO2.13
The characteristic distribution of emission lifetime extends to fixed sections of skin and liver tissue prepared histologically (Fig. 2 and 3). In both epithelial and extracellular matrix tissue, cell nuclei can be identified with respect to surrounding matrix proteins on the basis of a longer emission lifetime. Detailed structure of the cell nuclei and surrounding matrix can be clearly visualised using 1, and in addition, both intensity images and lifetime maps can be obtained in the absence of autofluorescence. The images are discussed in detail below.
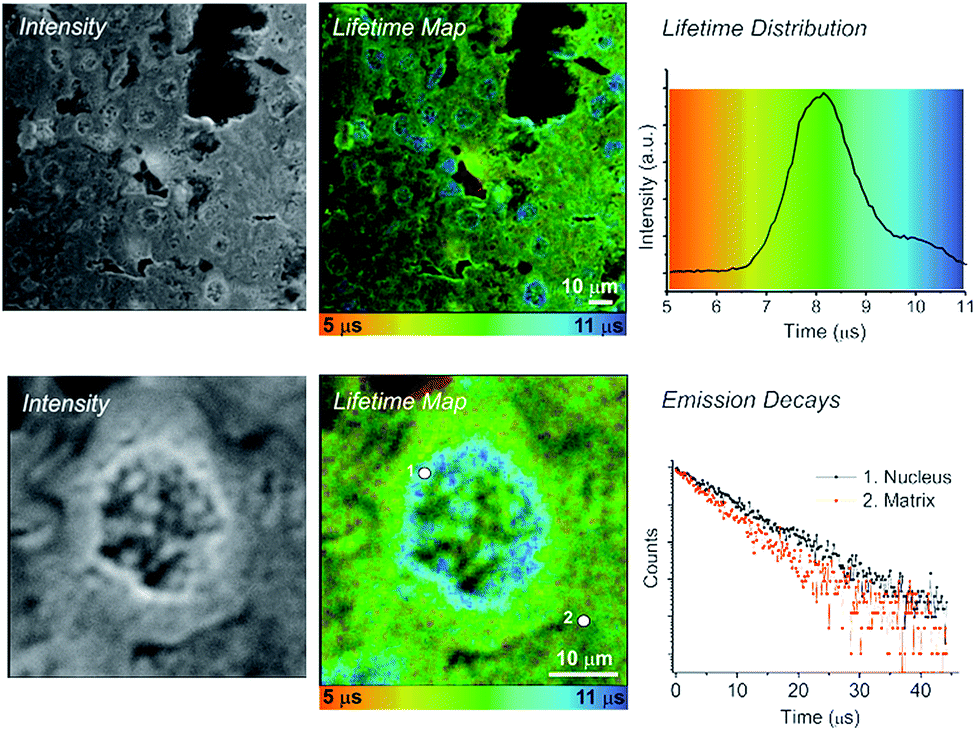 | ||
| Fig. 2 TP-PLIM imaging of fixed rat liver tissue section stained with 1. Bottom: TP-PLIM imaging at higher magnification focusing on just a single nucleus within the tissue. Tissue section thickness 10 μm; scale-bar – 10 μm. | ||
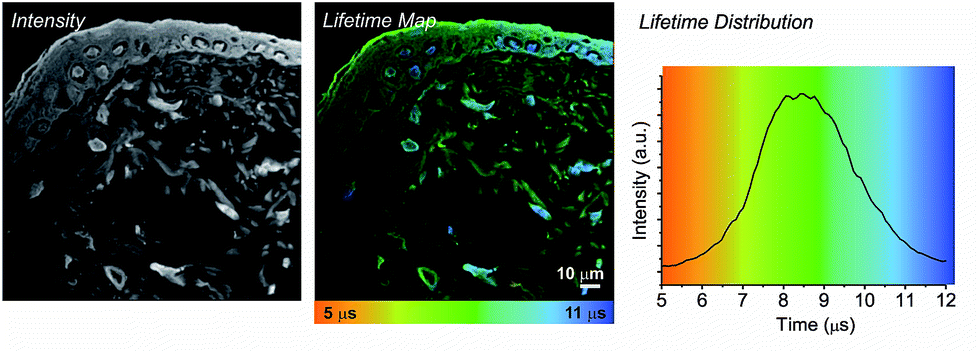 | ||
| Fig. 3 TP-PLIM imaging of histological section (10 μm) of rat skin stained with 1 showing a stratified epithelial upper layer (the epidermis, containing keratinocytes) and dermis (containing fibroblast cells and collagen fibres. | ||
We note that the application of this technology allowed the long lifetime component of the Pt(II) emission to be determined as approximately 10 μs. Intriguingly, this value is longer than that detected in fully de-oxygenated organic solvents at room temperature (7 μs), and close to the natural radiative lifetime τ0 of 12 μs calculated on the basis of the quantum yield Φ and lifetime τ under such conditions (τ0 = kr−1 = τ/Φ assuming that the emissive state is formed with unitary efficiency). This implies a decrease of the non-radiative decay rate compared to solution – probably due to increased rigidity of the complex when intercalated into DNA or bound to proteins, in comparison to fluid solution. The presence of a long lifetime component was suggested by us previously13 but could not be fully resolved using the earlier set-up due to the 3 μs limit of the time-window. In the present study, the imaging window has been extended to 50 μs, which enables the Pt-emission to decay to negligible levels before the arrival of the next laser pulse.
Fig. 2 shows PLIM lifetime mapping of a histological section of liver tissue, stained with 1. Liver is a homogenous tissue made up of type IV collagen and laminin with randomly interdispersed hepatocyte cells. These cell nuclei become exceptionally clear when stained with 1 and presented as a PLIM lifetime map (Fig. 2). The emission of 1 when bound to nuclear material (DNA, membrane) has a significantly longer lifetime (10.2 ± 0.5 μs) than that bound to surrounding matrix proteins (7.4 ± 0.5 μs); hence cell nuclei are clearly visible, appearing blue on a green-yellow background in the false rainbow colour scheme used to plot the lifetime maps. The distribution histogram shows a bimodal profile, where the intensity of the emission associated with the tissue matrix (yellow-green, ∼8 μs) is greater than that associated with the emission from the nucleus (blue, ∼10 μs) as there is more matrix in the field of view than there are nuclei. The TP-PLIM image recorded at higher magnification (Fig. 2, lower panel) and overlaid decay traces taken from individual pixels further highlight the clear distinction between the emission of 1 when localised in the nucleus compared to the matrix (nucleus = 10.2 μs – position 1; matrix = 7.4 μs – position 2).
The nuclear membrane and organised structure of the tissue matrix are also clearly distinguished when staining with 1. Such structures are not visible when registering the short-lived (nanosecond) emission from liver tissue only. Using compounds which possess >10 ns emission lifetime – such as 1, – as imaging agents provides the capacity to cut out autofluorescence, and therefore to perform imaging of different tissue substructures in the absence of interfering autofluorescence. Apart from removal of autofluorescence, one further advantage of long-lived probes such as 1 is their photostability: two-photon imaging with a laser power of 0.1 mW at the sample (250 KHz, 120 fs)13 has been performed for a period of up to 5 h without any visible difference to the emission intensity.
In contrast to liver, skin is an epithelial tissue made of distinct layers, comprised of a multicellular epidermis (predominantly of keratinocytes) and an underlying dermis comprised predominantly of collagen type I extracellular matrix interspersed with fibroblasts.15 Staining a histological section of rat skin labelled with 1 enables clear structures to be visualised, which is not possible with unlabelled tissue due to strong autofluorescence (Fig. 3). Supra-basal (differentiated) keratinocyte cell nuclei and the stratum corneum (the outermost layer made up of layers of flattened fully differentiated keratinocytes) which make up the epidermis are clearly visible in both the intensity image and the lifetime map. Again, the cell nuclei exhibit a longer emission lifetime (blue, 10.9 ± 0.4 μs), in comparison to the surrounding extracellular matrix (green, 8.9 ± 0.5 μs). In comparison to the epidermis, fibroblast cells contained within the dermis occupy a more dispersed position throughout the extracellular matrix and are difficult to discriminate in the intensity image. Only upon generation of the lifetime map can these cells be observed. The fibroblast cell nuclei (blue) exhibit an emission lifetime virtually identical to that observed for the keratinocyte and hepatocyte cell nuclei, 10.8 ± 0.4 μs. The extra-cellular matrix surrounding the nuclei exhibits a much shorter emission lifetime, 7.8 ± 0.4 μs (yellow-green).
The time-gating function can also be used to remove unwanted autofluorescence and short-lived emission in order to highlight long-lived species only within the field of view. Fig. 4 shows a series of “full” windows (i.e., 0–50 μs) and time-gated images (25–50 μs) for liver (b) and skin (e) tissue sections stained with 1. In both cases, it is the cell nuclei that remain the dominant feature after application of a time gate. In particular, non-differentiated cells in the skin dermis become significantly clearer. As expected, all of the features that remain visible after the long 25 μs time gate correlate to blue (cell nuclei) regions in the lifetime maps (c & f).
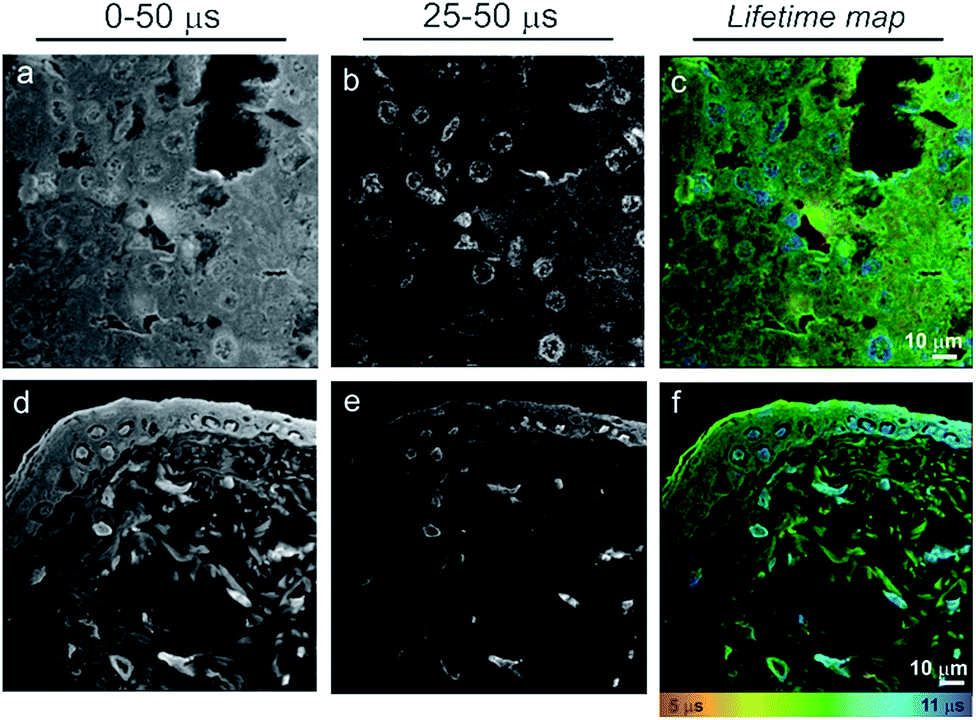 | ||
| Fig. 4 Time gated images of liver (a–c) and skin (d–f) 10 mμ tissue sections, showing how the PLIM method in conjunction with the Pt(II)-based imaging agent 1 can be used to remove autofluorescence, improving image contrast – cell nuclei clearly much more visible in the time-gated image at later times. a, b, d, e – intensity images, c and f – lifetime maps. | ||
The noticeable difference in emission lifetime of the dermal and epidermal extra-cellular matrix could be attributed to their different protein composition. The matrix surrounding basal keratinocyte cells, in the epidermis, has a high concentration of keratin – a cysteine rich protein, whereas the dermis predominantly consists of collagen and elastin. Furthermore, the lifetime of the Pt(II) complex 1 detected in the matrix of liver tissue, which is also collagen-rich and keratin-free, is very similar to that of the skin dermis (Fig. S2†). It is possible that groups of proteins and organelles may have a specific lifetime signature when labelled with 1, enabling them to be identified, similar to the way in which cell nuclei can be distinguished from cell cytoplasm and the extracellular matrix. A detailed investigation of binding of Pt(N^C^N) class of compounds to various proteins is currently underway.
Experimental details
The Pt(II) complex 1 was prepared as described previously;16 analytical data match reported values (see ESI†).All imaging experiments have been performed on a Zeiss LSM510 NLO META microscope equipped with a fs Ti:Sapph laser operating at a pulse repetition rate of 80 MHz. Two-photon excitation at a laser wavelength of 780 nm was used to acquire the images. The FLIM/PLIM signals were detected by a Becker & Hickl HPM-100-40 hybrid detector19 connected to the non-descanned port of the LSM510 NLO META. The data were recorded by a Becker & Hickl Simple-Tau 150 TCSPC FLIM system.14b,c The FLIM system contained a DDG-210 pulse generator module for pixel-synchronous on-of modulation of the Ti:Sapp laser. The TP-PLIM data were collected in a 50 μs time window. The pixel dwell time in the LSM510 NLO was set to 60 μs. The data were analysed by Becker & Hickl SPCImage data analysis software14b (see ESI† for further details). Fig. S3† demonstrates, on the example of live cell imaging, that quality of the images obtained in 3 μs window using TP-TREM where laser repetition rate was reduced by cavity dumping,13 and using the FLIM/PLIM set-up described here in 50 μs time-window are very similar.
Details of cell culture, tissue preparation, staining and imaging with complex 1 can be found in the ESI.†
Conclusions
Lifetime imaging microscopy that combines the microsecond domain with sub-micron spatial resolution – two-photon time-resolved emission imaging microscopy TREM (TP-TREM) which allows one to combine FLIM and PLIM – has broadened the scope for transition metal complexes to become key players in biological imaging and diagnostics.4,17 Here, we demonstrate how FLIM + PLIM can be achieved simultaneously under two-photon excitation using an alternative “beam blanking” method, where the frequency of the laser excitation pulse at the sample is reduced. We also show how the use of a probe with large emission lifetime, exemplified by a Pt(II) complex 1, allows cell nuclei to be identified with respect to surrounding matrix proteins on the basis of emission lifetime in both structured and homogenous tissue. Detailed structure of the cell nuclei and surrounding matrix can be clearly visualised using lifetime maps in the microsecond domain. Moreover, both intensity images and lifetime maps can be obtained with high contrast, due to complete cut-off of the short-lived autofluorescence background. Using suitable long-lived probes, visualization of different biologically important entities – DNA, proteins, sub-cellular structures – in live or fixed samples can be achieved, as well as improved contrast and sensitivity in analytical visualization methods, such as histology. The method illustrated here with the use of 1 can potentially be applied to any long-lived, biocompatible labels such as metal complexes, porphyrins, nanoparticle- and dendrimer-based assemblies.1,4,18Acknowledgements
We thank the EPSRC (fellowship to J.A.W. and studentship to E.B.), the BBSRC Tools and Resources Fund, and the Universities of Sheffield and Durham for support. We are most grateful to Dr W. Becker (Becker & Hickl Gmbh) for fruitful collaboration.Notes and references
- (a) M. P. Coogan and V. Fernandez-Moreira, Chem. Commun., 2014, 50, 384 RSC; (b) Y. You, Curr. Opin. Chem. Biol., 2013, 17, 699 CrossRef CAS PubMed; (c) F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12 CrossRef CAS PubMed; (d) D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087 CrossRef CAS PubMed; (e) K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 41, 6021 RSC; (f) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS PubMed; (g) Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; (h) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC ; and references therein; (i) P. A. Waghorn, M. W. Jones, M. B. Theobald, R. L. Arrowsmith, S. I. Pascu, S. W. Botchway, S. Faulkner and J. R. Dilworth, Chem. Sci., 2013, 4, 1430–1441 RSC.
- Complexes of some lanthanide(III) ions, especially those of europium(III) and terbium(III), are also under active investigation, exploiting emission from long-lived metal centred f–f states; e.g.: (a) C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS PubMed; (b) B. Song, C. D. B. Vandevyver, A. S. Chauvin and J. C. G. Bünzli, Org. Biomol. Chem., 2008, 6, 4125 RSC; (c) A. D'Aleo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622 CrossRef PubMed; (d) A. Grichine, A. Haefele, S. Pascal, A. Duperray, R. Michel, C. Andraud and O. Maury, Chem. Sci., 2014, 5, 3475 RSC; (e) A. Beeby, I. M. Clarkson, S. Faulkner, S. Botchway, D. Parker and A. W. Parker, J. Photochem. Photobiol., B, 2000, 57, 83 CrossRef CAS.
- ed. V. Balzani and S. Campagna, Topics in Current Chemistry, 2007, vol. 280, p. 28 Search PubMed.
- (a) T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148 CrossRef CAS PubMed; (b) S. S. Howard, A. Straub, N. G. Norton, D. Kbat and C. Xu, Nat. Photonics, 2013, 7, 33 CrossRef CAS PubMed; (c) N. W. Choi, S. S. Verbridge, R. M. Williams, J. Chen, J.-Y. Kim, R. Schmehl, C. E. Farnum, W. R. Zipfel, C. Fischbach and A. D. Stroock, Biomaterials, 2012, 33, 2710 CrossRef CAS PubMed; (d) L. Murphy, A. Congreve, L. O. Palsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743 RSC; (e) J. Kuil, P. Steunenberg, P. T. K. Chin, J. Oldenburg, K. Jalink, A. H. Velders and F. W. B. van Leeuwen, ChemBioChem, 2011, 12, 1897 CrossRef CAS PubMed; (f) H. Choi, et al. , Opt. Express, 2012, 20, 26219 CrossRef PubMed; (g) L. E. Sinks, E. Roussakis, S. Sakadžić, G. P. Robbins, D. A. Hammer, A. Devor, D. A. Boas and S. A. Vinogradov, Proc. SPIE, 2011, 7903, 79032A CrossRef PubMed; (h) S. Sakadžić, E. Roussakis and M. A. Yaseen, et al. , Nat. Methods, 2010, 7(9), 755–U125 CrossRef PubMed; (i) R. I. Dmitriev, A. V. Kondrashina, K. Koren, I. Klimant, A. V. Zhdanov, J. M. P. Pakan, K. W. McDermott and D. B. Papkovsky, Biomater. Sci., 2014, 2, 853 RSC.
- (a) J. W. Borst and A. J. W. G. Visser, Meas. Sci. Technol., 2012, 21, 102002 CrossRef; (b) M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641 CrossRef CAS PubMed; (c) K. Suhling, P. M. W. French and D. Phillips, Photochem. Photobiol. Sci., 2005, 4, 13 RSC; (d) R. Ebrecht, P. C. Don and F. S. Wouters, Protoplasma, 2014, 251, 293 CrossRef CAS PubMed.
- (a) M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641 CrossRef CAS PubMed; (b) M. Y. Berezin, W. J. Akers, K. Guo, G. M. Fischer, E. Daltrozzo, A. Zumbusch and S. Achilefu, Biophys. J., 2009, 97, L22 CrossRef CAS PubMed; (c) M. J. Ruedas-Rama, J. D. Walters, A. Orte and E. A. H. Hall, Anal. Chim. Acta, 2012, 751, 1 CrossRef CAS PubMed.
- (a) A. Orte, J. M. Alvarez-Pez and M. J. Ruedas-Rama, ACS Nano, 2013, 7, 6387 CrossRef CAS PubMed; (b) J. M. Paredes, M. D. Giron, M. J. Ruedas-Rama, A. Orte, L. Crovetto, E. M. Talavera, R. Salto and J. M. Alvarez-Pez, J. Phys. Chem. B, 2013, 117, 8143 CrossRef CAS PubMed; (c) J. A. Levitt, M. K. Kuimova, G. Yahioglu, P.-H. Chung, K. Suhling and D. Phillips, J. Phys. Chem. C, 2009, 113, 11634 CrossRef CAS; (d) K. Douma, R. T. A. Megens, S. Reitsma, L. Prinzen, D. W. Slaaf and M. A. M. J. Van Zandvoort, Microsc. Res. Tech., 2007, 70, 467 CrossRef CAS PubMed.
- (a) K. Suhling, J. Siegel, D. Phillips, P. M. W. French, S. Lévêque-Fort, S. E. D. Webb and D. M. Davis, Biophys. J., 2002, 83, 3589 CrossRef CAS; (b) I. Gautier, M. Tramier, C. Durieux, J. Coppey, R. B. Pansu, J. C. Nicolas, K. Kemnitz and M. Coppey-Moisan, Biophys. J., 2001, 80, 3000 CrossRef CAS; (c) R. Pepperkok, A. Squire, S. Geley and P. I. H. Bastiaens, Curr. Biol., 1999, 9, 269 CrossRef CAS; (d) H.-J. van Manen, P. Verkuijlen, P. Wittendorp, V. Subramaniam, T. K. van den Berg, D. Roos and C. Otto, Biophys. J., 2008, 94, 67 CrossRef PubMed.
- R. M. Levenson, D. T. Lynch, H. Kobayashi, J. M. Backer and M. V. Backer, ILAR J., 2008, 49, 78 CrossRef CAS.
- (a) W. Zhong, P. Urayama and M. A. Mycek, J. Phys. D: Appl. Phys., 2003, 36, 1689 CrossRef CAS; (b) J. Lecoq, A. Parpaleix, E. Roussakis, M. Ducros, Y. G. Houssen, S. A. Vinogradov and S. Charpak, Nat. Med., 2011, 17, 893 CrossRef CAS PubMed; (c) D. B. Papkovsky and R. I. Dmitriev, Chem. Soc. Rev., 2013, 42, 8700 RSC; (d) R. I. Dmitriev, A. V. Zhdanov, Y. M. Nolan and D. B. Papkovsky, Biomaterials, 2013, 34, 9307 CrossRef CAS PubMed; (e) O. S. Finikova, A. Y. Lebedev, A. Aprelev, T. Troxler, F. Gao, C. Garnacho, S. Muro, R. M. Hochstrasser and S. A. Vinogradov, ChemPhysChem, 2008, 9, 1673 CrossRef CAS PubMed; (f) S. M. S. Kazmi, et al. , Biomed. Opt. Express, 2013, 4, 1061 CrossRef PubMed.
- (a) E. J. Hennink, R. De Haas, N. P. Verwoerd and H. J. Tanke, Cytometry, 1996, 24, 312 CrossRef CAS; (b) R. R. De Haas, R. P. M. van Gijlswijk, E. B. van der Tol, H. J. M. A. A. Zijlmans, T. Bakker-Schut, J. Bonnet, N. P. Verwoerd and H. J. Tanke, J. Histochem. Cytochem., 1997, 45, 1279 CrossRef CAS PubMed; (c) R. R. De Haas, R. P. M. Van Gijlswijk, E. B. Van Der Tol, J. Veuskens, H. E. Van Gijssel, R. B. Tijdens, J. Bonnet, N. P. Verwoerd and H. J. Tanke, J. Histochem. Cytochem., 1999, 47, 183 CrossRef CAS PubMed; (d) A. E. Soini, L. Seveus, N. J. Meltola, D. B. Papkovsky and E. Soini, Microsc. Res. Tech., 2002, 58, 125 CrossRef CAS PubMed.
- S. W. Botchway, M. Charnley, J. W. Haycock, A. W. Parker, D. L. Rochester, J. A. Weinstein and J. A. G. Williams, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16071 CrossRef CAS PubMed.
- E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879 RSC.
- (a) A. Diaspro, G. Chirico and M. Collini, Q. Rev. Biophys., 2005, 38, 97 CrossRef CAS PubMed; (b) W. Becker, The bh TCSPC Handbook, Becker & Hickl GmbH, 5th edn, 2012, on E-mail: http://www.becker-hickl.com. handbookphp.htm Search PubMed; (c) http://www.becker-hickl.com/zeissman1.htm ; (d) http://www.becker-hickl.com/pdf/microsec-flim-08.pdf ; (e) W. Becker, B. Su, A. Bergmann, K. Weisshart and O. Holub, Simultaneous Fluorescence and Phosphorescence Lifetime Imaging, Proc. SPIE, 2011, 7903, 790320 CrossRef PubMed.
- W. Montagna, The Structure and Function of Skin, 3rd edn, 1974, ISBN 0-12-505263-4 Search PubMed.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609 CrossRef CAS PubMed.
- (a) E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock, J. A. Weinstein and J. A. Thomas, Angew. Chem., Int. Ed., 2014, 53, 3367 CrossRef CAS PubMed; (b) R. I. Dmitriev, A. V. Zhdanov, Y. M. Nolan and D. B. Papkovky, Biomaterials, 2013, 34, 9307 CrossRef CAS PubMed.
- (a) S. Banerjee, J. A. Kitchen, S. A. Bright, J. E. O'brien, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Commun., 2013, 49, 8522 RSC; (b) L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocalle and T. E. Keyes, Chem. Commun., 2013, 49, 2658 RSC.
- W. Becker, B. Su, K. Weisshart and O. Holub, Microsc. Res. Tech., 2011, 74, 804 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of complex 1. Details of cell culture, tissue preparation, and staining with 1. Microscope set-up and data analysis details for time-resolved imaging experiments. See DOI: 10.1039/c4ra04489d |
| This journal is © The Royal Society of Chemistry 2014 |