
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReplacing H+ by Na+ or K+ in phosphopeptide anions and cations prevents electron capture dissociation†
Eva-Maria
Schneeberger
 and
Kathrin
Breuker
*
and
Kathrin
Breuker
*
Institute of Organic Chemistry, Center for Molecular Biosciences Innsbruck (CMBI), University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria. E-mail: kathrin.breuker@uibk.ac.at; Web: http://www.bioms-breuker.at/
First published on 26th July 2018
Abstract
By successively replacing H+ by Na+ or K+ in phosphopeptide anions and cations, we show that the efficiency of fragmentation into c and z˙ or c˙ and z fragments from N–Cα backbone bond cleavage by negative ion electron capture dissociation (niECD) and electron capture dissociation (ECD) substantially decreases with increasing number of alkali ions attached. In proton-deficient phosphopeptide ions with a net charge of 2−, we observed an exponential decrease in electron capture efficiency with increasing number of Na+ or K+ ions attached, suggesting that electrons are preferentially captured at protonated sites. In proton-abundant phosphopeptide ions with a net charge of 3+, the electron capture efficiency was not affected by replacing up to four H+ ions with Na+ or K+ ions, but the yield of c, z˙ and c˙, z fragments from N–Cα backbone bond cleavage generally decreased next to Na+ or K+ binding sites. We interpret the site-specific decrease in fragmentation efficiency as Na+ or K+ binding to backbone amide oxygen in competition with interactions of protonated sites that would otherwise lead to backbone cleavage into c, z˙ or c˙, z fragments. Our findings seriously challenge the hypothesis that the positive charge responsible for ECD into c, z˙ or c˙, z fragments can generally be a sodium or other metal ion instead of a proton.
Introduction
Radical ion chemistry plays an increasingly important role in biomolecular mass spectrometry (MS).1–5 A mechanistic understanding of the various types of unimolecular ion dissociation reactions that are utilized in different MS dissociation techniques is highly critical to evaluating the potential of a technique for any given purpose. For example, insight into the mechanisms of collisionally activated dissociation (CAD) and electron detachment dissociation (EDD) of ribonucleic acids (RNA) provided a rationale for why CAD but not EDD can be used for the site-specific, relative quantitation of RNA nucleobase methylations,6 and why the extent of sequence information from EDD of proteins depends on the number of acidic residues.7 Moreover, for each mechanism, it is important to consider the type of ion that undergoes dissociation, e.g., (M + H)+versus (M + Na)+ or (M − H)−versus (M − 2H + Na)− ions, as their reactivity can be significantly different depending on whether or not and how the charged sites are involved in the dissociation reaction.For the characterization of peptides and proteins and their posttranslational modifications, electron capture dissociation (ECD)8 or electron transfer dissociation (ETD)9 of (M + nH)n+ ions is now routinely used.10,11 Alternatively, (M − nH)n− ions can be studied by EDD,7,12 negative electron transfer dissociation (nETD),13,14 ultraviolet photodissociation (UVPD) at 193 nm,15 or activated electron photodetachment dissociation (EPD) at 260 nm,16 all of which produce complementary a˙ and x fragments from peptide Cα–C backbone bond cleavage (Scheme 1).17 The major fragmentation channel in ECD and ETD of (M + nH)n+ peptide and protein ions is N–Cα backbone bond cleavage into complementary c and z˙ or c˙ and z fragments (Scheme 1) that can provide extensive sequence information.18,19 As the names suggest, electron capture and transfer dissociation involve capture (ECD) or transfer (ETD) of an electron by or to a (M + nH)n+ ion, respectively, and subsequent unimolecular dissociation of the radical (M + nH)(n−1)+˙ ions formed. In 2011, Håkansson and co-workers have discovered that (M − nH)n− peptide anions can also capture electrons to form (M − nH)(n+1)−˙ ions that dissociate into c and z˙ fragments, and termed this previously unknown phenomenon negative ion electron capture dissociation, niECD.20 They have proposed a zwitterionic peptide anion structure as a requirement for niECD, i.e., that a protonated, basic site or a fixed positive charge in the form of a quaternary amine must be present in the (M − nH)n− peptide anions.20,21
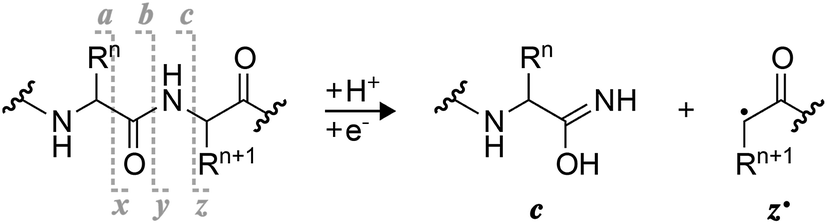 | ||
| Scheme 1 Complementary c and z˙ fragments from N–Cα backbone bond cleavage between residues n and n + 1 in ECD, niECD, or ETD; z˙ fragment structures were confirmed by infrared ion spectroscopy,22c fragments with imidic acid structure can tautomerize to the amide form,23 and H˙ transferring from a c to its complementary z˙ fragment can produce a pair of complementary c˙ and z fragments.24,25 | ||
Two general mechanisms for N–Cα backbone bond cleavage into c and z˙ fragments by ECD have been discussed in the literature, the ‘Cornell mechanism’ and the ‘Utah–Washington model’.1 The major difference between them is that the Cornell mechanism assumes ionic hydrogen bonding between a backbone amide oxygen and a protonated site such as a lysine sidechain, and electron attachment to this positively charged entity,26 whereas the Utah–Washington model proposes electron attachment to a neutral backbone amide in the presence of a remote positive charge.1,27 With the Cornell mechanism, the minor ECD pathway leading to dissociation into a˙ and y fragments can be rationalized by ionic hydrogen bonding between the protonated site and a backbone amide nitrogen.26 Both mechanisms agree in that they assume initial capture of the electron in a Rydberg state,26,28 but the former model is based on direct charge recombination whereas the latter involves formation of an enol-imidate anion radical in the presence of a positive charge that can be up to 10 Å away from the electron capture site, and can be Na+ or another metal ion instead of a proton.28
Mechanistic aspects of both ECD and ETD are still under investigation,1 and most studies to date have used peptide ions that carry a net positive charge. However, experiments with peptide cations, especially when the net charge is high, can be difficult to interpret because salt bridge structures and internal charge solvation can introduce ambiguity in the assignment of possible protonation sites. Our strategy here was to study both peptide anions and cations by niECD and ECD, respectively, and to successively replace protons by alkali metal ions to test the hypothesis that a charge other than H+ can effect N–Cα backbone bond cleavage into c and z˙ fragments.
The question that we address specifically is whether or not the charge carrier that is involved in N–Cα backbone cleavage into c and z˙ fragments in ECD or ETD can be a Na+ or K+ ion instead of a H+ ion. To this end, we have studied both the electron capture efficiency and the efficiency of fragmentation into c, z˙ or c˙, z fragments by N–Cα backbone bond cleavage of peptide cations (2+, 3+) and anions (2−) with up to four Na+ or K+ ions attached. As the model peptide, we used bovine β-casein peptide F48–K63 (FQpSEEQQQTEDELQDK, 16 residues, referred to hereafter by their indices 1–16) that was previously shown to undergo dissociation into c, z˙ or c˙, z fragments by ECD,29–31 ETD,32 and niECD.20 This peptide is phosphorylated (pS3) and has two basic (N terminus and K16) and eight acidic (pS3, E4, E5, E10, D11, E12, D15, and the C terminus) sites. As cations for successive replacement of H+, we chose Na+ and K+ because they can form salt bridges with phosphates and carboxylates, and have high preference for the formation of ionic hydrogen bonds with backbone amide oxygen33 rather than nitrogen (by replacement of the amide proton) as can be observed with divalent and trivalent metal ions.34–36 Moreover, previous studies indicated that Na+ and K+ attachment may interfere with peptide ion dissociation into c, z˙ or c˙, z fragments by both ECD and ETD.37,38
Na+ and K+ bind to carboxylates and backbone amide oxygen of peptides and proteins both in solution and in the gas phase,33,35,39–41 and thus can interfere with the formation of salt bridges between protonated basic and deprotonated acidic residues, and ionic hydrogen bonds between protonated sites and backbone amide oxygen, in gaseous peptide and protein ions.42–48 Deprotonation of acidic sites by Na+ and K+ is, however, thermodynamically unfavorable as proton affinities (PA) of anions are generally far higher than their sodium affinities, which in turn generally exceed their potassium affinities. For example, the calculated affinities of methanide (CH3−) for H+, Na+, and K+ are 1732, 633, and 541 kJ mol−1, respectively.49 Likewise, acetate (CH3COO−) as a model for E4, E5, E10, D11, E12, D15, and the C terminus has a proton affinity of 1453 kJ mol−1, and its calculated affinities for Na+ and K+ are 608 and 534 kJ mol−1, respectively.50 The PA of dihydrogen phosphate (H2PO4−) as a model for the phosphate group of pS3 is lower than that of acetate by 70 kJ mol−1, 1383 kJ mol−1, and its calculated Na+ and K+ affinities, 551 and 454 kJ mol−1, are lower by 82 and 87 kJ mol−1, respectively.51
By successively replacing H+ by Na+ or K+ ions in the phosphopeptide ions, we can not only test the hypothesis that a protonated site in (M − nH)n− or (M + nH)n+ ions is required for niECD or ECD into c, z˙ or c˙, z fragments, respectively, but can also study how breaking up salt bridges between protonated basic and deprotonated acidic residues and ionic hydrogen bonds between protonated sites and backbone amide oxygen affects niECD and ECD. Moreover, the relatively small number of residues of the phosphopeptide studied limits the number of possible intramolecular interactions that could potentially prevent the separation of fragments from niECD or ECD.44,45,52–56
Experimental
Bovine β-casein peptide F48–K63 was purchased from AnaSpec (San Jose, USA) and electrosprayed (1.5 μL min−1) from 1 μM solutions in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O (18 MΩ cm, Millipore, Vienna, Austria) and CH3OH (HPLC-grade, Acros, Vienna, Austria) at pH ∼ 9.0 (adjusted by addition of 5 mM Z-2,6-dimethylpiperidine, 98%, Sigma-Aldrich, Vienna, Austria) for ESI in negative ion mode57 or pH ∼ 4.0 (adjusted by addition of 10 mM CH3COOH, LC/MS grade, Fisher Scientific, Vienna, Austria) for ESI (electrospray ionization) in positive ion mode. NaCl and KCl (Sigma-Aldrich, Vienna, Austria) concentrations (150–600 μM) were adjusted for maximum abundance of the ions of interest in each experiment, e.g. 150 μM for peptide ions with one Na+ or K+ ion attached and 600 μM for peptide ions with four Na+ or three K+ ions attached.
:1 H2O (18 MΩ cm, Millipore, Vienna, Austria) and CH3OH (HPLC-grade, Acros, Vienna, Austria) at pH ∼ 9.0 (adjusted by addition of 5 mM Z-2,6-dimethylpiperidine, 98%, Sigma-Aldrich, Vienna, Austria) for ESI in negative ion mode57 or pH ∼ 4.0 (adjusted by addition of 10 mM CH3COOH, LC/MS grade, Fisher Scientific, Vienna, Austria) for ESI (electrospray ionization) in positive ion mode. NaCl and KCl (Sigma-Aldrich, Vienna, Austria) concentrations (150–600 μM) were adjusted for maximum abundance of the ions of interest in each experiment, e.g. 150 μM for peptide ions with one Na+ or K+ ion attached and 600 μM for peptide ions with four Na+ or three K+ ions attached.
Experiments were performed on a 7 T Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker, Vienna, Austria) equipped with an ESI source, and a hollow dispenser cathode for ECD or niECD, and a CO2 laser (10.6 μm, 35 W at 100% power) for infrared multiple photon dissociation (IRMPD). Phosphopeptide ions from ESI were accumulated in a first hexapole for 0.35–3.0 s, isolated by m/z in a quadrupole, accumulated in a second hexapole for 0.35–6.5 s (see ref. 58 for a scheme of the experimental setup), and transferred into the ICR cell for dissociation by ECD (electron energy 0.8–1.0 eV, irradiation time 70–150 ms), niECD (electron energy 6.2 eV, irradiation time 20 s), or IRMPD (laser power 25%, irradiation time 180 ms), and ion detection. Within each series of experiments in which the number of Na+ or K+ ions was increased from 0 to up to 4 (niECD of 2− ions at 10 V and 80 V skimmer potential, ECD of 2+ ions at 80 V skimmer potential, ECD of 3+ ions at 10 V skimmer potential), all experimental parameters were kept the same except for the ion accumulation time, which was adjusted such that the number of peptide ions varied by less than ∼25%. However, between the series of experiments (2−, 2+, 3+ ions), different ECD/niECD parameters (electron energy, irradiation time, and cathode current) were used and thus electron capture efficiency values are not comparable. For each series of experiments (2−, 2+, 3+ ions), ECD/niECD parameters were adjusted for maximum electron capture of the (M − 2H)2−, (M + 2H)2+, or (M + 3H)3+ ions while minimizing the capture of a second electron. The largest source of error in the determination of electron capture efficiency values was the varying number of ions trapped in the ICR cell, and thus error bars are given as the standard deviations of the relative number of peptide ions within each series of experiments. Between 30 (niECD) and 100 (ECD and IRMPD) scans were summed for each spectrum, and data reduction utilized the SNAP2 algorithm (Bruker, Austria).
Yields of fragments from N–Cα backbone bond cleavage were calculated as percentage values relative to all ECD or niECD products (excluding a˙ and y fragments), considering that N–Cα backbone bond cleavage of a parent ion (with charge ±n) produces a pair of complementary c, z˙ or c˙, z fragments (100% = 0.5 [c and c˙ with charge ≠(±n − 1)] + [c and c˙ with charge (±n − 1)] + 0.5 [z˙ and z with charge ≠(±n − 1)] + [z˙ and z with charge (±n − 1)] + [other products], in which other products are reduced molecular ions and products from loss of small neutral species from the latter).44 Errors for yields of c, z˙ or c˙, z fragments, reduced molecular ions, and products from loss of small neutral species from reduced molecular ions were calculated from standard deviations of residuals from non-linear least square fitting (to quadratic polynomial functions, f(x) = A + Bx2) as described in ref. 55. Because this method relies on the number of data points being significantly larger than the number of coefficients in the fit function, only data for Na+ with x = 0–4 were used. This conservative analysis provided upper error limits of ±5.6% and ±1.5% for yields in niECD of 2− ions and ECD of 3+ ions, respectively. Based on similar yields of c, z˙ or c˙, z fragments, the error for yields in ECD of 2+ ions was assumed to be similar to that in niECD of 2− ions. Electron capture efficiency values were calculated as percentage values of all ECD or niECD products (excluding a˙ and y fragments) relative to all ions (including molecular ions that did not capture an electron), and the fragmentation efficiency into c, z˙ or c˙, z fragments was calculated as percentage values of c, z˙ or c˙, z fragments relative to all ions,59 again considering that N–Cα backbone bond cleavage of a parent ion produces a pair of complementary c, z˙ or c˙, z fragments.
Results and discussion
niECD of phosphopeptide ions with a net charge of 2−
Fig. 1A shows a spectrum from niECD of (M − 2H)2− ions of the β-casein phosphopeptide studied, similar to that reported by Håkansson and co-workers,20 with >98% c, z˙ and <2% c˙, z fragments (Scheme 1) from N–Cα backbone bond cleavage at sites 1–3 and 7–15 along with a pair of a122−˙ and y4− fragments from cleavage at site 12; fragments from cleavage at sites 4–6 were not observed. The fragmentation efficiency was comparable for sites 1 and 7–15, and decreased nearly linearly from sites 1 through 3 (Fig. 1C). This fragmentation pattern can be rationalized by assuming that both the N terminus and K16 are protonated.30 Protonation of the basic N-terminal amino group (Table 1) is evident from the presence of abundant (M − 2H − NH3)3−˙ ions formed by niECD (Fig. 1A),60 and protonation of K16 is indicated by both high pKa and proton affinity values (Tables 1 and 2). Accordingly, four out of the eight acidic sites (pS3, E4, E5, E10, D11, E12, D15, and the C terminus) of the (M − 2H)2− ions should be deprotonated (Fig. 1E). A zwitterion structure with two protonated and four deprotonated sites, featuring salt bridges and ionic hydrogen bonds42,46 between the protonated N terminus and the first three residues, and between protonated K16 and residues 7–15, makes positive charge available for niECD at sites that are up to nine residues apart in sequence from the protonated sites. The wider reach of K16 compared to that of the N terminus is consistent with its long and flexible sidechain, although it is unlikely that a single peptide ion structure can account for the observed fragmentation pattern as the simultaneous binding of nine residues to K16 is geometrically infeasible. Instead, the niECD data indicate multiple phosphopeptide ion structures that could either be separated by high barriers61–65 or rapidly interconvert at room temperature66 depending on the stability of each structure and thus the strength and number of intermolecular hydrogen bond and salt bridge interactions.44,45,67,68 However, the lack of c, z˙ or c˙, z fragments from cleavage at sites 4–6 suggests that the (M − 2H)2− zwitterion structures are similar in that the protonated N terminus interacts only with the first three residues, and protonated K16 interacts only with residues 7–15, thereby forming two clusters of salt bridge and ionic hydrogen bond networks around the positively charged sites (Scheme 2) that are separated by at least residues 4–6. Similar structures have previously been proposed for doubly charged peptide cations.69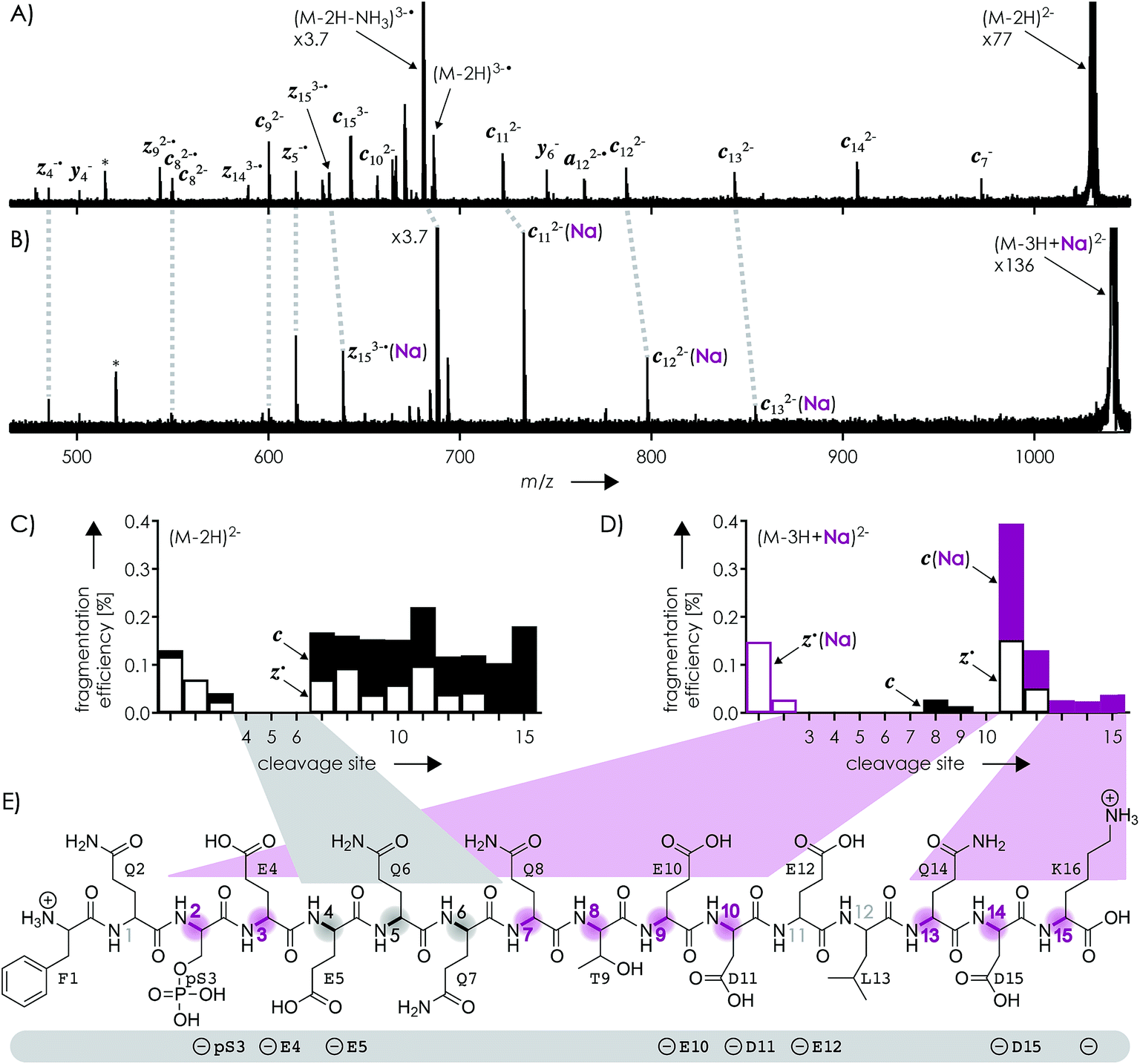 | ||
| Fig. 1 Spectra from niECD of (A) (M − 2H)2− and (B) (M − 3H + Na)2− ions using a skimmer potential of 10 V (asterisks indicate harmonic signals)70 and (C, D) corresponding site-specific fragmentation efficiency (N–Cα backbone bond cleavage, >98% c, z˙ and <2% c˙, z fragments); (E) primary structure of the phosphopeptide with proposed protonation sites (N terminus and K16) and possible locations of the four negative charges (pS3, E4, E5, E10, D11, E12, D15, and the C terminus) indicated below; highlighted in gray are sites from which no fragments were observed in niECD of (M − 2H)2− ions, and in purple sites at which fragment ion formation was substantially reduced or prevented in niECD of (M − 3H + Na)2− ions. | ||
| Group | pKa in protein/pentapeptide |
|---|---|
| Protonated N terminus | 7.7 ± 0.5/8.00 ± 0.03 |
| Protonated K sidechain | 10.5 ± 1.1/10.40 ± 0.08 |
| D sidechain | 3.5 ± 1.2/3.67 ± 0.04 |
| E sidechain | 4.2 ± 0.9/4.25 ± 0.05 |
| C terminus | 3.3 ± 0.8/3.67 ± 0.03 |
| Compound (model for) | PA [kJ mol−1] | Na+ affinity [kJ mol−1] |
|---|---|---|
| N-Methylacetamide, N-acetylated methyl ester of G (backbone amide) | 889 (ref. 73), 905 ± 3 (ref. 74) | 165 (ref. 75) |
| F (N terminus) | 923 (ref. 73) | 198 ± 8 (ref. 75), 206 ± 7 (ref. 76), 198 ± 12 (ref. 77) |
| N-Acetylated methyl ester of T (T residue) | 935 ± 3 (ref. 74) | 197 ± 8 (ref. 75), 203 ± 10 (ref. 78) (T) |
| N-Acetylated methyl ester of Q (Q residue) | 948 ± 12 (ref. 74) | 212 ± 8 (ref. 75), 211 ± 6 (ref. 79) (Q) |
| N-Methyl K (K residue) | ∼1000 (ref. 80) | >213 (K)75 |
| Deprotonated phosphoserine acid amide (deprotonated pS residue) | 1341 (ref. 81) | 551 (dihydrogen phosphate)51 |
| Deprotonated glutamic acid amide (deprotonated E residue) | 1388 (ref. 81) | 611 (acetate)82 |
| Deprotonated aspartic acid amide (deprotonated D residue) | 1393 (ref. 81) | |
| Deprotonated K (C terminus) | 1410 (ref. 81), 1414 ± 10 (ref. 73) |
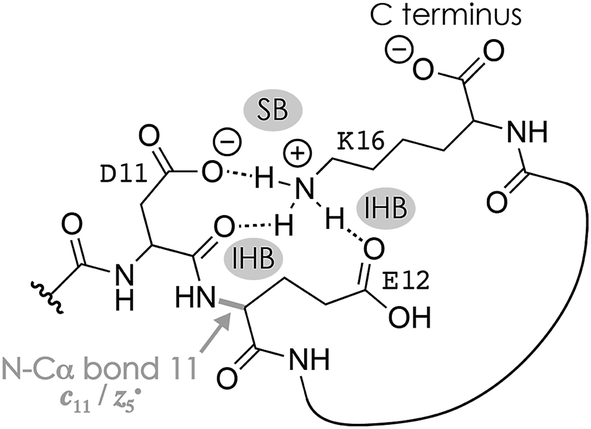 | ||
| Scheme 2 Possible salt bridge (SB) and ionic hydrogen bond (IHB) network illustrated for K16, D11, and E12 that accounts for the formation of c11 and complementary z5˙ fragments from cleavage of the N–Cα backbone bond 11 in niECD of phosphopeptide anions. Similar structures that involve other residues, e.g., the protonated N terminus and the phosphate of pS3, could account for backbone cleavage at other sites. | ||
The spectrum from niECD of (M − 3H + Na)2− ions in Fig. 1B showed far fewer c, z˙ and c˙, z fragments, and none from sites 3–7 and 10 (Fig. 1D). Whereas the fragmentation efficiency in niECD of (M − 2H)2− ions (Fig. 1C) was very similar for cleavage sites 1 and 7–15, that in niECD of (M − 3H + Na)2− ions was highest at site 11 (Fig. 1D). This suggests preferential intramolecular bonding of K16 to the residues adjacent to this cleavage site, D11 and/or E12, in the (M − 3H + Na)2− ions, presumably by both salt bridge and ionic hydrogen bond formation (Scheme 2). The location of Na+ at E10 and/or D11, at least in the majority of the (M − 3H + Na)2− ions, is indicated by the observation of c82− and c92− fragments without sodium, and c112− through c152− with sodium attached. Apparently, sodium binding to E10 and/or D11 interferes with the formation of intramolecular interactions of K16 and residues 7–10, thereby preventing – or at least substantially reducing – niECD backbone cleavage at sites 7–10 (Fig. 1D).
Is it possible that c, z˙ and c˙, z fragments were formed by niECD, but not separated because they were held together by noncovalent bonds?54 To test this hypothesis, we used collisional activation in the source region of the mass spectrometer to disrupt noncovalent bonds within the peptide anions prior to niECD. Previous studies have shown that vibrational activation of gaseous peptide and protein ions by collisions or absorption of infrared photons can result in partial or full unfolding,52,62,83 and that the stability of intramolecular interactions generally follows the order salt bridges > ionic hydrogen bonds > neutral hydrogen bonds > hydrophobic bonds.44,45,53,55,84 Instead of the 10 V skimmer potential in the experiments for Fig. 1, a skimmer potential of 80 V was applied to maximize vibrational activation and ion unfolding while preventing covalent bond dissociation that was observed at skimmer potentials above 80 V. Fig. 2A shows the yields of different products from electron capture, i.e., c, z˙ and c˙, z fragments, reduced molecular ions, and products from loss of small molecules from reduced molecular ions, calculated as percent values relative to all ions that underwent electron capture (see the Experimental section for details). The electron capture efficiency values in Fig. 2B were calculated as percent values of all products from electron capture relative to all ions (including molecular ions that did not undergo electron capture). For example, at 10 V skimmer potential, the yields of c, z˙ and c˙, z fragments, reduced molecular ions, and products from loss of small molecules from niECD of (M − 2H)2− ions were 28%, 4%, and 68%, respectively, and the electron capture efficiency was 4.7%. In other words, only 4.7% of the (M − 2H)2− ions did capture an electron, and out of these 4.7%, only 28% underwent dissociation into c, z˙ and c˙, z fragments.
 | ||
| Fig. 2 (A) Percentage stacked area plots illustrating the yield of c, z˙ and c˙, z fragments (filled circles), products from loss of small molecules (>70% NH3; <30% CO, CONH3, C2H6O, C2H4O2) from reduced molecular ions (open circles), and reduced molecular ions (triangles) of which ∼20% showed loss of H˙ but not Na˙, from niECD of phosphopeptide anions with a net charge of 2− at 10 V (left) and 80 V (right) skimmer potential, and (B) corresponding electron capture efficiency versus the number of Na+ ions attached; solid lines in (A) are quadratic polynomial functions used for error analysis and dashed lines in (B) are exponential fit functions. | ||
The fragmentation patterns (Fig. S1†) and yields of c, z˙ and c˙, z fragments (Fig. 2A) from niECD of (M − 2H)2− ions at 10 and 80 V skimmer potential were virtually the same, and only a few additional fragments of relatively low abundance were found in the spectra from niECD of (M − 3H + Na)2− ions at 80 V (Fig. S1†). However, the electron capture efficiency was somewhat higher at 80 V compared to that at 10 V for both (M − 2H)2− and (M − 3H + Na)2− ions (Fig. 2B), and the fragmentation efficiency (Fig. S1†), i.e., the number of c, z˙ and c˙, z fragments relative to all ions (including molecular ions that did not undergo electron capture), was increased accordingly. We attribute the higher electron capture efficiency at 80 V skimmer potential compared to that at 10 V to an increased spatial separation of the N-terminal residues from the C-terminal residues by breaking of the weaker hydrophobic and neutral hydrogen bonds without significantly disrupting the salt bridge and ionic hydrogen bond networks of K16 (Scheme 2) and the N-terminus. This partial elongation of the peptide structures should decrease the overall negative charge density and thus increase the probability for electron capture.
The similarity of the spectra from niECD of (M − 2H)2− ions at 10 and 80 V skimmer potential (Fig. S1†) and the similar yields of c, z˙ and c˙, z fragments (Fig. 2) indicate little noncovalent bonding between complementary fragments that is strong enough to prevent their separation. The strongest interactions within the (M − 2H)2− ions should be the salt bridges and ionic hydrogen bonds between the protonated sites (N terminus and K16) and deprotonated and neutral residues (Scheme 2), respectively, but the strength of these interactions predominantly relies on attractive electrostatic energy85 that is lost upon positive charge neutralization by electron capture. Thus the limiting factor in producing c, z˙ and c˙, z fragments by niECD of (M − 2H)2− ions of the small (16 amino acid residues) phosphopeptide studied here must be the availability of protons next to the cleavage sites rather than separation of c, z˙ and c˙, z fragments. By contrast, in larger peptide and protein ions in which numerous interactions can stabilize a fold after transfer into the gas phase, noncovalent bonding between c and z˙ or c˙ and z fragments can prevent their separation.44,52,54,86,87
The c112− and z5−˙ fragments from cleavage at site 11 were dominant in the spectra from niECD of (M − 3H + Na)2− ions at both 10 and 80 V, but did not stand out in the spectra from niECD of (M − 4H + 2Na)2− ions (Fig. S1†). This relative decrease in fragments from cleavage at site 11 is consistent with binding of the second Na+ to D11 and/or E12 in the majority of (M − 4H + 2Na)2− ions (Fig. S1†), on account of which the SB and IHB network of K16, D11, and E12 that would otherwise facilitate the formation of fragments from cleavage at site 11 (Scheme 2) cannot be formed. Further increasing the number of Na+ ions attached to the peptide ions without changing the net charge of 2− further decreased the fragmentation efficiency (Fig. 2B) until only z143−˙, z142−˙, and z153−˙ fragments were observed in niECD of (M − 6H + 4Na)2− ions (Fig. S1†). In agreement with the order of Na+ affinities (Table 2), this suggests that the H+ on K16 was replaced by Na+ before that on the N terminus. Remarkably, the efficiency of electron capture decreased exponentially with the number of Na+ ions attached to the phosphopeptide anions whereas the yield of c, z˙ and c˙, z fragments was largely unaffected (Fig. 2B). In other words, replacing H+ with Na+ decreased the probability of electron capture, but the fraction of ions that had captured an electron underwent dissociation into c, z˙ and c˙, z fragments to nearly the same extent. The slight (∼10%) decrease in c, z˙ and c˙, z fragment yield with increasing number of Na+ ions attached (Fig. 2A, 80 V) can be attributed to decreased signal-to-noise (S/N) ratios of the fragments at lower electron capture efficiency values. Moreover, dissociation into c, z˙ and c˙, z fragments was generally limited to regions without Na+ attached, i.e., residues 1–2 and 12–16 in the (M − 3H + Na)2− ions, and residues 1–2 in the (M − 6H + 4Na)2− ions (Fig. 1D and S1†). These observations indicate strongly preferred electron capture at or near protonated over sodiated sites, and suggest that dissociation into c, z˙ and c˙, z fragments involves ionic hydrogen bond and salt bridge networks such as that shown in Scheme 2. In support of this hypothesis, Håkansson and co-workers found that neither Na+, Cs+, nor Ca2+ attachment to DYMGWMDF–NH2 peptide anions enabled electron capture in niECD.20 Instead, the introduction of an N-terminal, trimethyl ammonium group, (CH3)3N+–CH2CO–, effected electron capture and N–Cα backbone bond cleavage in niECD of DYMGWMDF–NH2 peptide anions.20 This is consistent with the trimethyl ammonium group forming both salt bridges with deprotonated sites and relatively strong ionic hydrogen bonds with O acceptors such as amide oxygen in the gaseous peptide anions.88 In peptide cations, the introduction of a trimethyl ammonium group, (CH3)3N+–, can in like manner increase the extent of N–Cα backbone bond cleavage,89 whereas a decrease was observed upon introduction of a 2,4,6-trimethylpyridinium group whose positively charged nitrogen does not carry any methyl groups that could act as hydrogen bond donors.90
ESI of phosphopeptide solutions at pH ∼ 9.0 with KCl instead of NaCl produced yields of phosphopeptide anions with K+ attached that were lower by a factor of ∼4 compared to those with Na+ attached, consistent with the higher affinity of aspartate and glutamate in peptides and proteins for Na+ compared to K+.91 Increasing the KCl concentration above 600 μM did not increase the yield of phosphopeptide anions with K+ attached but instead resulted in spectra dominated by cluster ions composed of KCl. The spectra from niECD of phosphopeptide ions with a net charge of 2− and 1–3 K+ attached showed correspondingly smaller S/N ratios that can account for the slightly decreasing yields of c, z˙ and c˙, z fragments with increasing number of K+ ions attached (Fig. S3†). Nevertheless, c, z˙ and c˙, z fragments from backbone cleavage were generally from the same sites as for Na+ (Fig. 1A–D, S1 and S2†), and the electron capture efficiency also decreased exponentially with increasing number of K+ ions attached to the phosphopeptide anions (Fig. S3†).
ECD of phosphopeptide ions with a net charge of 2+
Evidence for salt bridge and ionic hydrogen bond networks that involve protonated residues has also been reported for protein and peptide (M + nH)n+ ions,42,44,46–48,53,92 and we were interested if we could find similar effects of Na+ and K+ attachment in ECD of phosphopeptide cations. ECD of (M + 2H)2+ ions from ESI of solutions at pH ∼ 4.0 using a 10 V skimmer potential produced c, z˙ and c˙, z fragments from backbone cleavage at sites 1–4 (z151+˙, z141+˙, z131+, z121+), 11–12 (z51+˙, z41+˙), and 14–15 (c141+˙, c151+); a similar spectrum was reported by Creese and Cooper.29 This fragmentation pattern (Fig. S4A†) and the presence of abundant (M + 2H − NH3)2+˙ ions60 are consistent with protonation of both the N-terminal amino group and K16 in the (M + 2H)2+ ions, and salt bridge and ionic hydrogen bond networks (Scheme 2) between the protonated N terminus and protonated K16 with residues next to cleavage sites 1–4 (Q2, pS3, E4, and E5), 11–12 (D11, E12, and L13), and 14–15 (Q14 and D15). However, the relatively low abundance of z51+˙ and z41+˙ ions (Fig. S4A†) suggests that interactions with D11, E12, and L13 were present in only a relatively small population of the (M + 2H)2+ ions. ECD of the (M + 2H)2+ ions at 80 V skimmer potential also produced c and z˙ fragments from backbone cleavage at sites 1–2 (z151+˙, z141+˙) and 14–15 (c141+, c151+) but none from sites 3–4 and 11–12 (Fig. S4B–E†), which suggests that the interactions with residues 4–5 and 11–13 were disrupted by collisional activation in the skimmer region. This indicates an overall extension of the (M + 2H)2+ ion structures upon collisional activation while retaining only the interactions between protonated and directly adjacent (Q2, pS3, Q14, D15) residues.Interestingly, Creese and Cooper found that ECD of the (M + 2H)2+ phosphopeptide ions produced c, z˙ and c˙, z fragments (Scheme 1) from backbone cleavage at all except sites 1–2 when an elevated electron energy, ∼10 eV above the <1 eV for regular ECD, was used. They postulated that the deposition of additional energy on electron capture results in cleavage of noncovalent interactions associated with hydrogen rearrangements.29 Although the processes involved in this ‘hot-electron’ capture dissociation (HECD)24,93 at electron energies that are comparable to the ionization energies of (M + 2H)2+ peptide ions94 require further study, we also found evidence for H˙ transfer at elevated energy. Infrared multiphoton dissociation (IRMPD, 25% laser power, 180 ms irradiation time) of (M + 2H)+˙ ions from ECD of phosphopeptide (M + 2H)2+ ions produced spectra similar to those from HECD (Fig. S5†), with fragments from backbone cleavage at sites 3–14. The latter were predominantly c˙, z instead of c, z˙ fragments, suggesting that their formation involved H˙ transfer prior to backbone cleavage.24
The niECD and ECD data indicate that the N terminus and K16 are protonated in both the (M + 2H)2+ and (M − 2H)2− ions. Moreover, from the fragmentation patterns in ECD of (M + 2H)2+ and niECD of (M − 2H)2− phosphopeptide ions and the effect of collisional activation (Fig. S2A, F and S4†), it is evident that the interactions between the protonated (N terminus and K16) and other sites that are more than two residues apart in sequence are either not formed in the (M + 2H)2+ ions, or far less stable than in the (M − 2H)2− ions. Both the (M + 2H)2+ and (M − 2H)2− ions were electrosprayed from denaturing solutions with a high methanol content, but according to pKa values (Table 1), deprotonation of D and E sidechains is much more likely at the higher pH of ∼9.0 used for ESI of (M − nH)n− ions than at the pH of ∼4.0 used for ESI of (M + nH)n+ ions. Changes in pH of the evaporating droplets from ESI, operated in either positive or negative ion mode, are relatively small, on the order of half a pH unit.95–97 Thus in a pH ∼ 9.0 solution, and also in the evaporating droplets from ESI of that solution, the phosphopeptide can form a variety of salt bridges between the protonated sites and even remote (in sequence) deprotonated D and E sidechains (or the C terminus) that should be preserved in the (M − 2H)2− ions because of the high stability of salt bridge interactions in the gas phase.44,53 Moreover, these salt bridges can constitute the basis of networks of electrostatic interactions that include ionic hydrogen bonds between protonated sites and backbone amide oxygen such as that in Scheme 2. Fewer salt bridges can form in a pH ∼ 4.0 solution because fewer carboxylic acid sites are deprotonated, and any ionic hydrogen bonds that may be formed in the gaseous (M + 2H)2+ ions between protonated sites and neutral residues are – in the absence of further stabilization by a salt bridge – not only weaker53 but should also span fewer residues because they generally form in the gas phase instead of in solution.42,43,55 However, the pKa of phosphoserine is 2.19,98 and that calculated for phosphoserine amide is 2.65,81 so the pS3 sidechain should be deprotonated and available for salt bridge formation with the protonated N terminus at either pH. This is consistent with a smaller (by ∼3%) collision cross-section of the (M + 2H)2+ phosphopeptide ions compared to that of the (M + 2H)2+ ions of the unphosphorylated peptide,99 and the observation of fragments from cleavage at sites 1–4 in ECD of (M + 2H)2+ phosphopeptide ions at 10 V skimmer potential (Fig. S4A†) and sites 1–3 in niECD of (M − 2H)2− ions at both 10 and 80 V (Fig. 1C). For the (M + 2H)2+ ions, deprotonation of the pS3 sidechain would require that in addition to the N terminus and K16, another site close to either termini must be protonated, for example, Q2 or Q14. Moreover, unlike the carboxylate sidechains, a singly deprotonated pS3 sidechain still has a proton that could become available for N–Cα backbone bond cleavage in resonance-stabilized networks of electrostatic interactions such as that in Scheme 3.
 | ||
| Scheme 3 Possible salt bridge (SB) and ionic hydrogen bond (IHB) network involving pS3 and the N terminus that accounts for the formation of z13˙ fragments from cleavage of the N–Cα bond 3 in niECD of phosphopeptide (M − 2H)2− ions. | ||
At 80 V skimmer potential, ECD of phosphopeptide ions with a net charge of 2+ and 1–3 Na+ attached produced spectra very similar to those of the (M + 2H)2+ ions (Fig. S4†). The fragments from ECD of (M + H + Na)2+, (M + 2Na)2+ and (M − H + 3Na)2+ ions were c141+, c151+, z141+˙, and z151+˙ with one, two, and three Na+ ions attached, respectively, consistent with Na+ attachment to the deprotonated sidechains of pS3, E4, E5, E10, D11, and/or E12 but not D15 and the C terminus (Fig. S4†).
ECD of phosphopeptide ions with a net charge of 3+
To further investigate the effect of Na+ and K+ binding, we studied phosphopeptide ions with a net charge of 3+. In these experiments, a skimmer potential of 10 V was used as potentials above 50 V dissociated the 3+ phosphopeptide ions into b and y fragments typical for CAD, and no effect on the ECD fragmentation pattern was found by changing the potential from 10 to 50 V (Fig. S6, S7A and B†). ECD of the (M + 3H)3+ ions produced c, z˙ and c˙, z fragments from backbone cleavage at all sites (Fig. 3A, S7A and B†), which indicates that each backbone amide oxygen interacted with a protonated residue in at least one of the possible (M + 3H)3+ ion structures. The site-specific fragmentation efficiency varied between 0.44% for site 2 and 2.13% for site 11, with an average of 1.02% and a standard deviation of 0.46% (Fig. S8A†). Preferred cleavage at site 11 (Fig. S7A and B†) was also observed in niECD of (M − 3H + Na)2− ions (Fig. 1), suggesting similar electrostatic interactions between K16 and D11 and/or E12 in the (M − 3H + Na)2− and (M + 3H)3+ ions. Out of all products from ECD of the (M + 3H)3+ ions, 61.0% were c, z˙ or c˙, z fragments, 5.9% reduced molecular ions (of which 87% showed H˙ loss, totaling to 5.1% of all ECD products), and 33.1% products from loss of small molecules from reduced molecular ions (Fig. 3B). Thus ECD of (M + 3H)3+ ions at both 10 and 50 V skimmer potential produced only 0.8% (M + 3H)2+˙ ions that could be interpreted as unseparated c and z˙ or c˙ and z fragments54 as discussed in more detail below. Products from loss of small molecules from reduced molecular ions were predominantly (M + 3H − NH3CO)2+˙ and (M + 3H − NH3)2+˙ ions (Fig. S7A and B†), indicating protonation of glutamine100 Q2, Q6, Q7, Q8, or Q14 and the N terminus,60 respectively. Further, the relatively high basicity of lysine (Tables 1 and 2) suggests that K16 is also protonated in the (M + 3H)3+ ions. Assuming that Coulombic repulsion between neighboring residues plays a role in the distribution of charges in the (M + 3H)3+ ions, the most likely protonation site in addition to the N terminus and K16 would be Q6, Q7, or Q8; if pS3 were deprotonated, an additional proton could be located at Q2 or Q14. | ||
| Fig. 3 (A) Site-specific fragmentation efficiency (c, z˙ and c˙, z fragments) from ECD of phosphopeptide ions with a net charge of 3+ and 0–4 Na+ ions attached, with the color scale ranging from red (zero fragmentation) to yellow to green to blue (maximum fragmentation; detailed fragmentation efficiency values can be found in Fig. S7A and B†); (B) percentage stacked area plots illustrating the yield of c, z˙ and c˙, z fragments, products from loss of small molecules (s.m.) from reduced molecular ions, and reduced molecular ions (including loss of H˙; loss of Na˙ was not observed) from ECD of phosphopeptide ions with a net charge of 3+ at 10 V skimmer potential, and (C) corresponding electron capture efficiency versus the number of Na+ ions attached; solid lines in (B) are quadratic polynomial functions used for error analysis and that in (C) is meant to guide the eye. | ||
ECD of 3+ ions with 1–4 Na+ ions attached revealed binding of the first Na+ to pS3 (along with binding to E4 in <10% of the ion population), and binding of the second Na+ to E5 along with binding to Q6, Q7, Q8, and T9 (Fig. S8†). The third and fourth Na+ ions were bound to E10, D11, E12, L13 and Q14, but not D15, K16, and the C terminus (Fig. S8†); similar data were obtained for K+ (Fig. S9†). Strikingly, dissociation into c, z˙ or c˙, z fragments generally decreased next to Na+ or K+ binding sites and was absent or only marginal at sites 3–9 in the (M − H + 4Na)3+ ions (Fig. 3A, S8 and S9†). The only site that showed a substantial increase in fragmentation efficiency by increasing the number of Na+ or K+ ions from zero to one and above was site 15 (Fig. 3A, S8 and S9†). This can be rationalized by Na+ or K+ binding to pS3 in the 3+ ions with 1–4 Na+ ions attached that competes with the formation of networks of electrostatic interactions that involve pS3 and the protonated residues. Such networks could be built around a direct salt bridge, e.g., between pS3 and K16, or around an extended network of salt bridges45–48 with a −/+/−/+ motif, e.g., pS3/Q7/D11/K16. In either case, Na+ or K+ binding to pS3 would result in fewer interactions of K16 with residues further away in sequence, and correspondingly fewer backbone cleavages by ECD. Instead, K16 could form an ionic hydrogen bond with the amide oxygen at site 15 that gives rise to increased backbone cleavage at site 15, as observed by experiment (Fig. 3A, S8 and S9†). Thus a single Na+ or K+ ion attached to pS3 in the 3+ phosphopeptide ions is sufficient to substantially alter the ECD fragmentation pattern. Further Na+ or K+ attachment resulted in an increased c, z˙ or c˙, z fragmentation efficiency at sites 2, 10, 14, and 15 (Fig. S10†), but overall, the fragmentation efficiency decreased, by a factor of ∼2.4 upon increasing the number of Na+ ions attached from 0 to 4 (Fig. 3).
The decrease in c, z˙ or c˙, z fragments with increasing number of Na+ ions attached coincided with a substantial increase in the fraction of reduced molecular ions (Fig. 3B); increasing the number from 0 to 4 Na+ ions increased their fraction from 5.9 to 46.9%. Similar data were obtained in ECD of 3+ ions with 1–3 K+ ions attached (Fig. S9H†). Moreover, the fraction of reduced molecular ions from ECD of (M − H + 4Na)3+ ions that showed H˙ loss was only 7.7% (87% in ECD of (M + 3H)3+ ions, see above), totaling to 3.6% of all ECD products. Thus 43.3% of all products from ECD of (M − H + 4Na)3+ ions were (M − H + 4Na)2+˙ ions that could be interpreted as unseparated fragments, i.e., complementary pairs of c and z˙ or c˙ and z fragments that are still held together by noncovalent bonds.52,54 To test this hypothesis, we isolated the (M − H + 4Na)2+˙ ions (along with the 7.7% (M − 2H + 4Na)2+ ions from H˙ loss) and subjected them to IRMPD, which is a slow heating method101 that effects dissociation via the channels of lowest energy. Thus IRMPD generally cleaves noncovalent before covalent bonds,102 unless the energy required for noncovalent bond cleavage exceeds that for covalent bond cleavage.53,103 Using energies (25% laser power, 180 ms irradiation time) that were sufficient for covalent bond cleavage in 59% of the (M − 2H + 4Na)2+ ions from ESI and 27% dissociation of the (M + 2H)+˙ ions from ECD of (M + 2H)2+ ions resulted in only ∼15% dissociation of the (M − H + 4Na)2+˙/(M − 2H + 4Na)2+ ions. Out of the ∼15% dissociation products, 11.1% were losses of NH3, H2O, and H3PO4 from covalent bond cleavage in (M − H + 4Na)2+˙/(M − 2H + 4Na)2+ ions, and only 3.9% were c, z˙ or c˙, z fragments (b and y fragments from dissociation of the 7.7% nonradical (M − 2H + 4Na)2+ ions were not observed). By contrast, out of the 27% products from IRMPD of the (M + 2H)+˙ ions, 19.3% were c, z˙ or c˙, z (losses of CO2, NH3 and H3PO4 accounted for the other products). Thus by using the exact same laser power (25%) and irradiation time (180 ms), IRMPD of the (M + 2H)+˙ ions produced ∼5 times more c, z˙ or c˙, z fragments (19.3%) than IRMPD of the more highly charged (M − H + 4Na)2+˙ ions (3.9%). The marginal yield of 3.9% at energies that were sufficiently high for covalent bond cleavage (losses of NH3, H2O, and H3PO4) contrasts strongly with an earlier ECD study of ubiquitin in which separation of c, z˙ or c˙, z fragments required only ∼10% of the energy for covalent bond cleavage,52 and with IRMPD experiments in which noncovalently bound c, z˙ and c˙, z fragments of the far larger protein trypsin inhibitor (180 residues) were separated by using the same instrument and laser energy as in the present study (25% power, 180 ms irradiation time).104 We conclude that the (M − H + 4Na)2+˙ ions from ECD of (M − H + 4Na)3+ ions are highly stable radical species of as yet unknown structure rather than complementary pairs of c, z˙ or c˙, z fragments that are still held together by noncovalent bonds.
What limits dissociation next to Na+ and K+ binding sites?
So what is the reason that dissociation into c, z˙ or c˙, z fragments generally decreases next to the sites of Na+ or K+ binding? To address this question, two different types of Na+ or K+ binding must be considered, as discussed in the following for Na+. In the first case (Scheme 4A), Na+ is attached to one or more uncharged sidechains in the binding region (residues 3–14) of high Na+ affinity, e.g., glutamines Q6, Q7, and Q8 (Table 2), and could at the same time interact with backbone amide oxygens.35,61,105–107 Although the interactions of Na+ in Scheme 4A are with the same sites as those expected for protonated residues, replacing the H+ that is bound to Q6, Q7, and/or Q8 in the (M + 3H)3+ ions (see above) by Na+ substantially decreased the number of c, z˙ or c˙, z fragments from backbone cleavage next to these sites (Fig. 3A and S8†).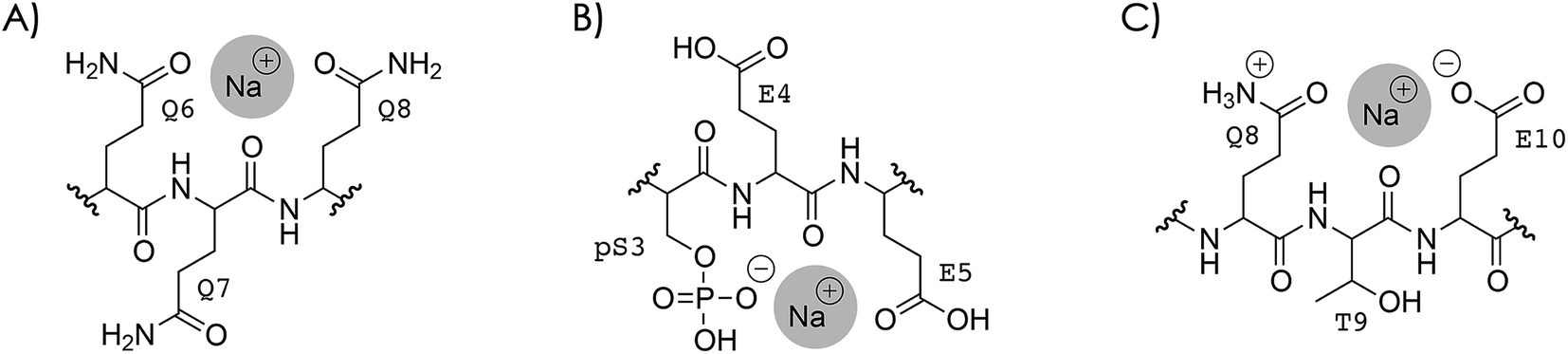 | ||
| Scheme 4 Na+ binding to (A) uncharged Q6, Q7, and Q8, and (B) deprotonated pS3 and uncharged E4 and E5 in phosphopeptide ions with a net charge of 2+ or 3+; similar structures are possible for carboxylates instead of the phosphate and for protonated instead of sodiated residues, (C) Na+ binding that prevents interactions of protonated sites with backbone amide oxygen illustrated for Q8, T9, and E10. | ||
In the second case, Na+ is attached to a deprotonated sidechain in the binding region (pS3, E4, E5, E10, D11, or E12) in a salt bridge motif with a net charge of zero (Scheme 4B). If this were the only binding motif in the 3+ ions with 1–4 Na+ ions attached, three residues were still protonated, and these should still be the most basic sites (Tables 1 and 2): the N terminus, Q6, Q7, or Q8, and K16 (with additional protonation of Q2 or Q14 if pS3 is deprotonated). In other words, the second scenario would neither affect the number of protonated sites nor the sites of protonation within the phosphopeptide ions.
The Na+ locations indicated by the ECD data (Fig. S4 and S8†) and the higher Na+ affinity of deprotonated versus uncharged residues (Table 2) suggest that the second scenario (Scheme 4B) dominates in the 2+ and 3+ ions. Accordingly, the protonated sites are the N terminus and K16 in the 2+ ions, and the N terminus, Q6, Q7 or Q8, and K16 (with additional protonation of Q2 or Q14 if pS3 is deprotonated), with or without up to four Na+ or K+ ions attached. Assuming that electrons are captured preferentially at or near protonated sites as indicated by the niECD data (Fig. 2B), this scenario provides a rationale for the unchanging electron capture efficiency with increasing number of Na+ or K+ ions attached (Fig. S4† and 3C), in agreement with observations by Beauchamp, Loo, and coworkers.30 The decreased efficiency of fragmentation into c, z˙ or c˙, z fragments next to Na+ or K+ binding sites can then be attributed to each alkali metal ion binding to both a deprotonated sidechain and an adjacent backbone amide oxygen,33 thus preventing a direct interaction of the protonated sites with the backbone amide oxygen (Scheme 4C).
Structures such as that shown in Scheme 4C are highly plausible because Na+ binds to both deprotonated sidechains and backbone amide oxygen already in solution108 whereas protonated sites form interactions with backbone amide oxygen after transfer into the gas phase by ESI.42,43 Owing to the positive charge of Na+, both electron capture into the π* orbital of the backbone amides next to the Na+ binding site and subsequent proton transfer from an adjacent Cα or a protonated sidechain as proposed within the framework of the Utah–Washington mechanism1,28 should be highly favored in all structures in Scheme 4, yet dissociation into c, z˙ or c˙, z fragments next to Na+ or K+ binding sites was highly disfavored in all experiments in this study (Fig. 1, 3A, S8 and S9†).
Finally, we noted that with increasing number of Na+ or K+ ions attached to the 2+ and 3+ phosphopeptide ions, the fraction of c and z˙ fragments generally decreased in favor of c˙ and z fragments (Fig. S11†). Transfer of H˙ between c and z˙ fragments is common in both ECD and ETD of peptides without Na+ or K+ attached,25,109,110 and has been attributed to noncovalent bonding between c and z˙ fragments that extends the timescale for fragment separation beyond that for backbone cleavage.24,111 In ECD of the (M + 3H)3+ ions, we observed H˙ transfer between c and z˙ fragments from cleavage at sites 2, 14, and 15 (Fig. S11†) at both 10 and 50 V skimmer potential, indicating noncovalent interactions of the protonated N terminus and protonated K16 with adjacent residues in at least a fraction of the (M + 3H)3+ ions. Likewise, noncovalent interactions of Na+ or K+ (Scheme 4) could delay fragment separation and thus enable H˙ transfer between c and z˙ fragments, as previously suggested by Wada and co-workers for ETD of peptides with K+ and Ca2+ attached.38 However, virtually all (∼99%) c, z˙ and c˙, z fragments were separated in ECD of the (M + 3H)3+ ions, consistent with positive charge neutralization by electron capture at the protonated sites and a substantial weakening of their electrostatic interactions, whereas ECD of the 3+ ions with 1–4 Na+ or 1–3 K+ ions attached showed a decrease in c, z˙ and c˙, z fragment yield and a corresponding increase in the fraction of reduced molecular ions (Fig. 3B and S9H†). Thus Na+ or K+ attachment either interferes with N–Cα backbone bond cleavage, or it increases the strength of noncovalent interactions between c, z˙ and c˙, z fragments to the extent that they are not separated in the ECD experiments.
Two experimental observations argue strongly against the possibility that c, z˙ or c˙, z fragment separation is limited by noncovalent interactions of Na+ or K+. First, there is little correlation between the extent of H˙ transfer and the decrease in fragmentation efficiency (Fig. S11†). For example, H˙ transfer between c and z˙ fragments from cleavage at site 10 in ECD of the 3+ ions increased steadily with increasing number of Na+ ions attached (0%, ∼3%, ∼46%, ∼75%, ∼78% for 0, 1, 2, 3, and 4 Na+ ions attached, respectively), but the fragmentation efficiency showed a significant decrease only for the (M − H + 4Na)3+ ions (Fig. S11†). Further, H˙ transfer between c and z˙ fragments from cleavage at site 15 even decreased with increasing number of Na+ ions attached, and site 12 showed only small changes in fragmentation efficiency even though H˙ transfer between c and z˙ fragments increased from ∼0% at 0 Na+ to ∼80% at 4 Na+ ions (Fig. S11†). Thus the noncovalent bonding indicated by H˙ transfer between c and z˙ fragments is not a limiting factor for fragmentation efficiency. Second, the IRMPD energy (25% laser power, 180 ms irradiation time) required to dissociate ∼15% of the reduced molecular ions from ECD of (M − H + 4Na)3+ ions into a mere 3.9% c, z˙ or c˙, z fragments (Fig. S12†) was sufficient to cause Na+ scrambling (i.e., migration of Na+ away from the original binding site), as indicated by the loss of H3PO4 (but not NaH2PO4) discussed above. Extensive loss of H3PO4 (but not NaH2PO4) was also observed in IRMPD (25%, 180 ms) of (M − 2H + 4Na)2+ (∼70%) and (M − H + 3Na)2+ (∼74%) ions from ESI. As further evidence for Na+ scrambling, IRMPD (25%, 180 ms) of the (M − H + 3Na)2+ ions produced y5, y6, y8 and y14 ions, of which 70, 51, 0 and 97% carried Na+, respectively; ECD of the (M + 3Na)3+ produced z5˙/z5, z6˙/z6, z8˙/z8 and z14˙/z14 ions of which 3, 24, 100 and 100% carried Na+, respectively. Although Na+ or K+ attachment affects both H˙ transfer and fragmentation efficiency, it does so in different ways: in the first case by delaying fragment ion separation sufficiently long for H˙ transfer to occur, and in the second case by blocking amide oxygen that could otherwise bind to protonated sites.
The scrambling of Na+ in both the reduced molecular ions from ECD of (M − H + 4Na)3+ ions and the (M − 2H + 4Na)2+ and (M − H + 3Na)2+ ions from ESI shows that the laser energy used for IRMPD was sufficiently high to disrupt noncovalent interactions of Na+, including its strong salt bridge (Table 2) with the phosphate group of pS3. Thus if the noncovalent interactions of Na+ were to prevent separation of c, z˙ and c˙, z fragments, IRMPD at 25% laser power and 180 ms irradiation time should efficiently dissociate the reduced molecular ions from ECD of (M − H + 4Na)3+ ions, yet only 3.9% c, z˙ or c˙, z fragments were observed. We conclude that instead of preventing their separation, Na+ binding interferes with the formation of c, z˙ or c˙, z fragments. In agreement with electron capture efficiency values (Fig. 3C), the reduced molecular ions observed in ECD of 3+ ions with up to four Na+ ions attached (Fig. 3B) could then result from electron capture at protonated sites that are not hydrogen bonded to backbone amide oxygen. The high energy requirements and the very low yield of c, z˙ or c˙, z fragments in IRMPD of the reduced molecular ions from ECD of (M − H + 4Na)3+ ions (Fig. S12†) further suggest that they are formed by intramolecular H˙ transfer to backbone amide oxygen, similar to the attachment of low-energy (∼0.15 eV) hydrogen atoms to backbone amide oxygen of even-electron peptide ions that was previously observed to initiate dissociation into c, z˙ and c˙, z fragments.112 Moreover, intramolecular H˙ transfer to backbone amide oxygen is consistent with backbone cleavage at sites 5–10 (Fig. S12†), around the postulated protonation site at Q6, Q7, or Q8 in the (M − H + 4Na)3+ ions. Strong evidence for intramolecular H˙ transfer within reduced molecular ions of bovine β-casein peptide F48–K63 derivatives was also reported by Loo, Beauchamp, and co-workers.31
H+versus metal cations: electron affinity limits electron capture efficiency
The findings reported here seriously challenge the hypothesis that the positive charge responsible for ECD into c, z˙ and c˙, z fragments can generally be a sodium or other metal ion instead of a proton.28 As a result of their different electronic structures, protons and alkali cations differ in their electron affinity (Table 3), which is equal to both the charge recombination energy gained by electron attachment and the ionization energy of the corresponding atoms, and in their ability to form interactions with functional groups of the phosphopeptide as discussed above. The electron affinities of Na+ and K+ are a mere 38% and 32% of the electron affinity of H+, respectively (Table 3), which suggests strongly favored electron capture at protonated over sodiated or potassiated sites.69 The similar decrease in electron capture efficiency of the phosphopeptide ions with a net charge of 2− by successive replacement of H+ by Na+ or K+ (Fig. 2B and S3B†), including that on K16 and lastly that on the N-terminal F1 (Fig. S1†), confirms this hypothesis. For the phosphopeptide cations with net charges of 2+ or 3+, the electron capture efficiency was largely unaffected because the up to four protons replaced by Na+ or K+ were bound to the phosphate and carboxylate groups instead of the basic residues (N terminus, Q6, Q7, or Q8, and K16). Our findings are corroborated by an ECD study of peptide ions in the Williams group that showed preferred neutralization of H+ over Li+, and of Li+ over Cs+, during ECD.69| Ion | Electron affinity [eV] |
|---|---|
| H+ | 13.59844 |
| Li+ | 5.39172 |
| Na+ | 5.13908 |
| K+ | 4.34066 |
| Rb+ | 4.17713 |
| Cs+ | 3.89390 |
Divalent (e.g., Ni2+, Cu2+, and Zn2+) and trivalent (e.g., La3+, Yb3+, and Tb3+) metal ions can potentially increase the electron capture efficiency of peptides when bound to either phosphate, carboxylate, or uncharged sites. However, for model peptides derived from bradykinin, Chan and co-workers observed a decrease in electron capture efficiency for La3+, In3+, Al3+, and Ga3+ attached to the peptide ions with a net charge of 3+ compared to that of the (M + 3H)3+ ions, and an increase only for Rh3+.114 Again, the effect on electron capture efficiency is consistent with the order of recombination energies of the metal ions attached to the peptides, i.e., ∼8.0 eV for La3+, ∼9.2 eV for In3+, ∼9.6 eV for Al3+, ∼10.2 eV for Ga3+, and a far higher value of ∼13.0 eV for Rh3+ (Table 4). Strikingly, ECD of the model peptides with Rh3+ attached produced only a, b, and y fragments whereas ECD of the model peptides with La3+, In3+, Al3+, or Ga3+ attached produced almost exclusively c, z˙ or c˙, z fragments.114 In a similar fashion, ECD of substance P ions with Ba2+, Sr2+, Ca2+, Mg2+, Mn2+, Fe2+, or Zn2+ attached (with charge recombination energies ranging from ∼4.8 to ∼8.6 eV, Table 4) and a net charge of 3+ produced c, z˙ or c˙, z fragments similar to those from ECD of (M + 3H)3+ ions whereas attachment of Co2+ (∼9.2 eV) and Ni2+ (∼10.5 eV) led to the formation of fragments from cleavage within and next to the C-terminal methionine residue, and Cu2+ (∼12.6 eV) produced only b and y ions.115 Further, both ECD and ETD of 2+ ions of histidine peptides that lacked both amino groups and acidic sites (acetylation of the N-terminal amine and amidation of the C-terminal carboxylate), with Zn2+ (∼8.6 eV), Ni2+ (∼10.5 eV), or Cu2+ (∼12.6 eV) attached, produced only a and b but no c, z˙ or c˙, z fragments.116 In an ETD study of 15 different peptides, Vachet and Dong observed c and z˙ fragments with Cu2+ instead of Cu+ attached, consistent with electron transfer to protonated sites instead of Cu2+. Moreover, they found a positive correlation between the number of residues that strongly bind Cu2+ (i.e., histidine, methionine, aspartic acid, and glutamic acid) and the yield of c and z˙ versus a, b, and y fragments, which they rationalized by a lowering of the Cu ion recombination energy relative to the recombination energy of the protonated sites such that electron transfer to protonated sites and dissociation into c and z˙ fragments become competitive.117 Likewise, an ECD study of various (∼400 to ∼1800 Da) peptide ions with a net charge of 3+ and trivalent metal ions attached to acidic sites in salt bridge motifs revealed electron capture at the protonated site and dissociation into predominantly c, z˙ or c˙, z fragments for all of the nine metals studied except Eu3+, which instead gave rise to dissociation into b and y fragments.118 Thus trivalent metal ion attachment to highly acidic peptides that resist multiple protonation in electrospray ionization can be exploited to make them amenable to ECD or ETD,119 but the formation of c, z˙ or c˙, z fragments still requires electron capture at protonated sites whereas electron capture at metals leads to dissociation into a, b, and y fragments.
| Metal | 3. IE [eV] | 2. IE [eV] | 1. IE [eV] | ΔE [eV] |
|---|---|---|---|---|
| Ba | 10.00383 | 5.21166 | 4.79216 | |
| Sr | 11.03028 | 5.69487 | 5.33541 | |
| Ca | 11.87172 | 6.11316 | 5.75856 | |
| Mg | 15.03527 | 7.64624 | 7.38904 | |
| Mn | 15.63999 | 7.43404 | 8.20595 | |
| Fe | 16.19920 | 7.90247 | 8.29673 | |
| Zn | 17.96439 | 9.39420 | 8.57019 | |
| Co | 17.08440 | 7.88101 | 9.20339 | |
| Ni | 18.16884 | 7.63988 | 10.52896 | |
| Cu | 20.29239 | 7.72638 | 12.56601 | |
| Lu | 20.95940 | 14.13000 | 6.82940 | |
| La | 19.17730 | 11.18496 | 7.99234 | |
| In | 28.04415 | 18.87041 | 9.17374 | |
| Al | 28.44764 | 18.82855 | 9.61909 | |
| Ga | 30.72576 | 20.51514 | 10.21062 | |
| Tb | 21.82000 | 11.51300 | 10.30700 | |
| Ho | 22.79000 | 11.78100 | 11.00900 | |
| Pm | 22.44000 | 10.93800 | 11.50200 | |
| Tm | 23.66000 | 12.06500 | 11.59500 | |
| Sm | 23.55000 | 11.07800 | 12.47200 | |
| Yb | 25.05300 | 12.17919 | 12.87382 | |
| Rh | 31.06000 | 18.08000 | 12.98000 | |
| Eu | 24.84000 | 11.24000 | 13.60000 |
In ECD of the phosphopeptide ions with a net charge of 3+, the electron capture efficiency did not decrease with increasing number of Na+ or K+ ions attached (Fig. 3C, S4G and S9I†), consistent with electron capture at the unchanging number of protonated sites, but the yield of c, z˙ or c˙, z fragments nevertheless decreased with increasing number of Na+ or K+ ions attached (Fig. 3B and S9†). In peptides and proteins in solution, the alkali metal ions Na+ and K+ preferentially bind to negatively charged sites,121i.e., the carboxylates and phosphates, but at the same time have an appreciable affinity and selectivity for backbone amide oxygen.33 Thus Na+ or K+ can simultaneously bind to acidic residues and adjacent backbone amide oxygen (Schemes 4B and C), thereby competing with both the formation of salt bridges between protonated and deprotonated residues and interactions between protonated residues and backbone amide oxygen, which explains the strong (from 61% to 26%) decrease in yield of c, z˙ or c˙, z fragments with increasing number of Na+ or K+ ions attached in ECD of 3+ ions (Fig. 3B). Depending on whether the peptide ions are proton-deficient (2− ions) or proton-abundant (3+ ions), one of either effects of replacing H+ with Na+ or K+ dominates, that is, to substantially reduce the electron capture efficiency and to interfere with interactions of protonated sites that would otherwise lead to backbone ECD cleavage into c, z˙ or c˙, z fragments. Taken together, the niECD and ECD data strongly suggest that the formation of c, z˙ or c˙, z fragments not only requires electron capture at a protonated site, but also that the protonated site must be bound to a backbone amide oxygen.
Conclusions
Our comprehensive niECD, ECD, IRMPD, and ECD/IRMPD study shows that dissociation into c, z˙ or c˙, z fragments requires electron capture at a protonated site, and that this protonated site must interact with backbone amide oxygen. Replacing H+ by Na+ or K+ in proton-deficient peptide ions decreases the electron capture efficiency whereas replacing H+ by Na+ or K+ at phosphate and carboxylate groups in proton-abundant peptide ions prevents interactions of the protonated sites with backbone amide oxygen because Na+ or K+ ions at the same time bind to adjacent backbone amide oxygen. Vibrational ion activation can scramble Na+ or K+ ions and mobilize hydrogen atoms, formed by electron capture at protonated sites that did not interact with backbone amide oxygen, such that the latter can attach to backbone amide oxygen and effect dissociation into c, z˙ or c˙, z fragments. High sequence coverage in ECD of peptides can be afforded by a high number of protonated residues that bind to backbone amide oxygen, or by multiple interactions of fewer protonated sites, which can be stabilized by hydrogen bonding networks built around salt bridge structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by the Austrian Science Fund (FWF): P27347 and P30087 to K. B., and by the Austrian Research Promotion Agency FFG (West-Austrian BioNMR).References
- F. Turecek and R. R. Julian, Peptide Radicals and Cation Radicals in the Gas Phase, Chem. Rev., 2013, 113(8), 6691–6733 CrossRef PubMed.
- J. S. Brodbelt, Photodissociation mass spectrometry: new tools for characterization of biological molecules, Chem. Soc. Rev., 2014, 43(8), 2757–2783 RSC.
- H. B. Oh and B. Moon, Radical-Driven Peptide Backbone Dissociation Tandem Mass Spectrometry, Mass Spectrom. Rev., 2015, 34(2), 116–132 CrossRef PubMed.
- R. Antoine, J. Lemoine and P. Dugourd, Electron Photodetachment Dissociation for Structural Characterization of Synthetic and Bio-Polymer Anions, Mass Spectrom. Rev., 2014, 33(6), 501–522 CrossRef PubMed.
- S. Schürch, Characterization of nucleic acids by tandem mass spectrometry – the second decade (2004–2013): from DNA to RNA and modified sequences, Mass Spectrom. Rev., 2016, 35(4), 483–523 CrossRef PubMed.
- H. Glasner, C. Riml, R. Micura and K. Breuker, Label-free, direct localization and relative quantitation of the RNA nucleobase methylations m(6)A, m(5)C, m(3)U, and m(5)U by top-down mass spectrometry, Nucleic Acids Res., 2017, 45(13), 8014–8025 CrossRef PubMed.
- B. Ganisl, T. Valovka, M. Hartl, M. Taucher, K. Bister and K. Breuker, Electron detachment dissociation for top-down mass spectrometry of acidic proteins, Chem.–Eur. J., 2011, 17(16), 4460–4469 CrossRef PubMed.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, Electron capture dissociation of multiply charged protein cations. A nonergodic process, J. Am. Chem. Soc., 1998, 120(13), 3265–3266 CrossRef.
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(26), 9528–9533 CrossRef PubMed.
- Y. Y. Zhang, B. R. Fonslow, B. Shan, M. C. Baek and J. R. Yates, Protein Analysis by Shotgun/Bottom-Up Proteomics, Chem. Rev., 2013, 113(4), 2343–2394 CrossRef PubMed.
- A. D. Catherman, O. S. Skinner and N. L. Kelleher, Top Down Proteomics: Facts and Perspectives, Biochem. Biophys. Res. Commun., 2014, 445(4), 683–693 CrossRef PubMed.
- B. A. Budnik, K. F. Haselmann and R. A. Zubarev, Electron detachment dissociation of peptide di-anions: an electron–hole recombination phenomenon, Chem. Phys. Lett., 2001, 342(3–4), 299–302 CrossRef.
- M. Huzarska, I. Ugalde, D. A. Kaplan, R. Hartmer, M. L. Easterling and N. C. Polfer, Negative Electron Transfer Dissociation of Deprotonated Phosphopeptide Anions: Choice of Radical Cation Reagent and Competition between Electron and Proton Transfer, Anal. Chem., 2010, 82(7), 2873–2878 CrossRef PubMed.
- N. M. Riley, M. J. P. Rush, C. M. Rose, A. L. Richards, N. W. Kwiecien, D. J. Bailey, A. S. Hebert, M. S. Westphall and J. J. Coon, The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation (AI-NETD), Mol. Cell. Proteomics, 2015, 14(10), 2644–2660 CrossRef PubMed.
- J. A. Madsen, T. S. Kaoud, K. N. Dalby and J. S. Brodbelt, 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization, Proteomics, 2011, 11(7), 1329–1334 CrossRef PubMed.
- V. Larraillet, R. Antoine, P. Dugourd and J. Lemoine, Activated-Electron Photodetachment Dissociation for the Structural Characterization of Protein Polyanions, Anal. Chem., 2009, 81(20), 8410–8416 CrossRef PubMed.
- I. Anusiewicz, M. Jasionowski, P. Skurski and J. Simons, Backbone and side-chain cleavages in electron detachment dissociation (EDD), J. Phys. Chem. A, 2005, 109(49), 11332–11337 CrossRef PubMed.
- R. A. Zubarev, D. M. Horn, E. K. Fridriksson, N. L. Kelleher, N. A. Kruger, M. A. Lewis, B. K. Carpenter and F. W. McLafferty, Electron capture dissociation for structural characterization of multiply charged protein cations, Anal. Chem., 2000, 72, 563–573 CrossRef PubMed.
- D. M. Good, M. Wirtala, G. C. McAlister and J. J. Coon, Performance characteristics of electron transfer dissociation mass spectrometry, Mol. Cell. Proteomics, 2007, 6(11), 1942–1951 CrossRef PubMed.
- H. J. Yoo, N. Wang, S. Y. Zhuang, H. T. Song and K. Håkansson, Negative-Ion Electron Capture Dissociation: Radical-Driven Fragmentation of Charge-Increased Gaseous Peptide Anions, J. Am. Chem. Soc., 2011, 133(42), 16790–16793 CrossRef PubMed.
- K. E. Hersberger and K. Håkansson, Characterization of O-Sulfopeptides by Negative Ion Mode Tandem Mass Spectrometry: Superior Performance of Negative Ion Electron Capture Dissociation, Anal. Chem., 2012, 84(15), 6370–6377 CrossRef PubMed.
- J. Martens, J. Grzetic, G. Berden and J. Oomens, Structural identification of electron transfer dissociation products in mass spectrometry using infrared ion spectroscopy, Nat. Commun., 2016, 7, 11754–11760 CrossRef PubMed.
- G. Frison, G. van der Rest, F. Turecek, T. Besson, J. Lemaire, P. Maitre and J. Chamot-Rooke, Structure of Electron-Capture Dissociation Fragments from Charge-Tagged Peptides Probed by Tunable Infrared Multiple Photon Dissociation, J. Am. Chem. Soc., 2008, 130(45), 14916–14917 CrossRef PubMed.
- P. B. O'Connor, C. Lin, J. J. Cournoyer, J. L. Pittman, M. Belyayev and B. A. Budnik, Long-lived electron capture dissociation product ions experience radical migration via hydrogen abstraction, J. Am. Soc. Mass Spectrom., 2006, 17(4), 576–585 CrossRef PubMed.
- M. M. Savitski, F. Kjeldsen, M. L. Nielsen and R. A. Zubarev, Hydrogen rearrangement to and from radical z fragments in electron capture dissociation of peptides, J. Am. Soc. Mass Spectrom., 2007, 18(1), 113–120 CrossRef PubMed.
- K. Breuker, H. Oh, C. Lin, B. K. Carpenter and F. W. McLafferty, Nonergodic and conformational control of the electron capture dissociation of protein cations, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(39), 14011–14016 CrossRef PubMed.
- X. Chen and F. Turecek, The arginine anomaly: arginine radicals are poor hydrogen atom donors in electron transfer induced dissociations, J. Am. Chem. Soc., 2006, 128(38), 12520–12530 CrossRef PubMed.
- E. A. Syrstad and F. Turecek, Toward a general mechanism of electron capture dissociation, J. Am. Soc. Mass Spectrom., 2005, 16(2), 208–224 CrossRef PubMed.
- A. J. Creese and H. J. Cooper, The effect of phosphorylation on the electron capture dissociation of peptide ions, J. Am. Soc. Mass Spectrom., 2008, 19(9), 1263–1274 CrossRef PubMed.
- C. H. Sohn, C. K. Chung, S. Yin, P. Ramachandran, J. A. Loo and J. L. Beauchamp, Probing the Mechanism of Electron Capture and Electron Transfer Dissociation Using Tags with Variable Electron Affinity, J. Am. Chem. Soc., 2009, 131(15), 5444–5459 CrossRef PubMed.
- C. H. Sohn, S. Yin, I. Peng, J. A. Loo and J. L. Beauchamp, Investigation of the mechanism of electron capture and electron transfer dissociation of peptides with a covalently attached free radical hydrogen atom scavenger, Int. J. Mass Spectrom., 2015, 390, 49–55 CrossRef PubMed.
- D. Nardiello, C. Palermo, A. Natale, M. Quinto and D. Centonze, Strategies in protein sequencing and characterization: multi-enzyme digestion coupled with alternate CID/ETD tandem mass spectrometry, Anal. Chim. Acta, 2015, 854, 106–117 CrossRef PubMed.
- J. Heyda, J. C. Vincent, D. J. Tobias, J. Dzubiella and P. Jungwirth, Ion Specificity at the Peptide Bond: Molecular Dynamics Simulations of N-Methylacetamide in Aqueous Salt Solutions, J. Phys. Chem. B, 2010, 114(2), 1213–1220 CrossRef PubMed.
- R. C. Dunbar, J. D. Steill, N. C. Polfer, G. Berden and J. Oomens, Peptide Bond Tautomerization Induced by Divalent Metal Ions: Characterization of the Iminol Configuration, Angew. Chem., Int. Ed., 2012, 51(19), 4591–4593 CrossRef PubMed.
- R. C. Dunbar, G. Berden and J. Oomens, How does a small peptide choose how to bind a metal ion? IRMPD and computational survey of CS versus iminol binding preferences, Int. J. Mass Spectrom., 2013, 354, 356–364 CrossRef.
- R. C. Dunbar, J. Martens, G. Berden and J. Oomens, Complexes of Ni(II) and Cu(II) with small peptides: deciding whether to deprotonate, Phys. Chem. Chem. Phys., 2016, 18(38), 26923–26932 RSC.
- V. G. Voinov, P. D. Hoffman, S. E. Bennett, J. S. Beckman and D. F. Barofsky, Electron Capture Dissociation of Sodium-Adducted Peptides on a Modified Quadrupole/Time-of-Flight Mass Spectrometer, J. Am. Soc. Mass Spectrom., 2015, 26(12), 2096–2104 CrossRef PubMed.
- D. Asakawa, T. Takeuchi, A. Yamashita and Y. Wada, Influence of Metal–Peptide Complexation on Fragmentation and Inter-Fragment Hydrogen Migration in Electron Transfer Dissociation, J. Am. Soc. Mass Spectrom., 2014, 25(6), 1029–1039 CrossRef PubMed.
- H. I. Okur, J. Hladilkova, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and their Biological Functions, J. Phys. Chem. B, 2017, 121(9), 1997–2014 CrossRef PubMed.
- P. Jungwirth and P. S. Cremer, Beyond Hofmeister, Nat. Chem., 2014, 6(4), 261–263 CrossRef PubMed.
- W. Sang-Aroon and V. Ruangpornvisuti, Conformational analysis of alkali metal complexes of anionic species of aspartic acid, their interconversion and deprotonation: A DFT investigation, J. Mol. Graphics Modell., 2008, 26(6), 982–990 CrossRef PubMed.
- M. Z. Steinberg, R. Elber, F. W. McLafferty, R. B. Gerber and K. Breuker, Early Structural Evolution of Native Cytochrome c after Solvent Removal, ChemBioChem, 2008, 9(15), 2417–2423 CrossRef PubMed.
- K. Breuker and F. W. McLafferty, Stepwise evolution of protein native structure with electrospray into the gas phase, 10(−12) to 10(2) s, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(47), 18145–18152 CrossRef PubMed.
- K. Breuker, S. Brüschweiler and M. Tollinger, Electrostatic stabilization of a native protein structure in the gas phase, Angew. Chem., Int. Ed., 2011, 50(4), 873–877 CrossRef PubMed.
- O. S. Skinner, F. W. McLafferty and K. Breuker, How Ubiquitin Unfolds after Transfer into the Gas Phase, J. Am. Soc. Mass Spectrom., 2012, 23(6), 1011–1014 CrossRef PubMed.
- L. Konermann, Molecular Dynamics Simulations on Gas-Phase Proteins with Mobile Protons: Inclusion of All-Atom Charge Solvation, J. Phys. Chem. B, 2017, 121(34), 8102–8112 CrossRef PubMed.
- J. Bonner, Y. A. Lyon, C. Neilessen and R. R. Julian, Photoelectron Transfer Dissociation Reveals Surprising Favorability of Zwitterionic States in Large Gaseous Peptides and Proteins, J. Am. Chem. Soc., 2017, 139(30), 10286–10293 CrossRef PubMed.
- D. Kim, P. J. Pai, A. J. Creese, A. W. Jones, D. H. Russell and H. J. Cooper, Probing the Electron Capture Dissociation Mass Spectrometry of Phosphopeptides with Traveling Wave Ion Mobility Spectrometry and Molecular Dynamics Simulations, J. Am. Soc. Mass Spectrom., 2015, 26(6), 1004–1013 CrossRef PubMed.
- Z. Boughlala, C. F. Guerra and F. M. Bickelhaupt, Alkali Metal Cation Affinities of Anionic Main Group-Element Hydrides Across the Periodic Table, Chem.–Asian J., 2017, 12(19), 2604–2611 CrossRef PubMed.
- M. J. Stevens and S. L. B. Rempe, Ion-Specific Effects in Carboxylate Binding Sites, J. Phys. Chem. B, 2016, 120(49), 12519–12530 CrossRef PubMed.
- S. W. Park, C. W. Kim, J. H. Lee, G. Shim and K. S. Kim, Comparison of Arsenic Acid with Phosphoric Acid in the Interaction with a Water Molecule and an Alkali/Alkaline-Earth Metal Cation, J. Phys. Chem. A, 2011, 115(41), 11355–11361 CrossRef PubMed.
- K. Breuker, H. Oh, D. M. Horn, B. A. Cerda and F. W. McLafferty, Detailed unfolding and folding of gaseous ubiquitin ions characterized by electron capture dissociation, J. Am. Chem. Soc., 2002, 124(22), 6407–6420 CrossRef PubMed.
- M. Schennach, E. M. Schneeberger and K. Breuker, Unfolding and Folding of the Three-Helix Bundle Protein KIX in the Absence of Solvent, J. Am. Soc. Mass Spectrom., 2016, 27(6), 1079–1088 CrossRef PubMed.
- M. Schennach and K. Breuker, Probing Protein Structure and Folding in the Gas Phase by Electron Capture Dissociation, J. Am. Soc. Mass Spectrom., 2015, 26(7), 1059–1067 CrossRef PubMed.
- M. Schennach and K. Breuker, Proteins with Highly Similar Native Folds Can Show Vastly Dissimilar Folding Behavior When Desolvated, Angew. Chem., Int. Ed., 2014, 53(1), 164–168 CrossRef PubMed.
- F. Lermyte, D. Valkenborg, J. A. Loo and F. Sobott, Radical solutions: principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure, Mass Spectrom. Rev., 2018, 1–22 Search PubMed.
- B. Ganisl, M. Taucher, C. Riml and K. Breuker, Charge as you like! Efficient manipulation of negative ion net charge in electrospray ionization of proteins and nucleic acids, Eur. J. Mass Spectrom., 2011, 17(4), 333–343 CrossRef PubMed.
- M. Taucher, U. Rieder and K. Breuker, Minimizing Base Loss and Internal Fragmentation in Collisionally Activated Dissociation of Multiply Deprotonated RNA, J. Am. Soc. Mass Spectrom., 2010, 21, 278–285 CrossRef PubMed.
- E. W. Robinson, R. D. Leib and E. R. Williams, The role of conformation on electron capture dissociation of ubiquitin, J. Am. Soc. Mass Spectrom., 2006, 17(10), 1469–1479 CrossRef PubMed.
- A. I. S. Holm, P. Hvelplund, U. Kadhane, M. K. Larsen, B. Liu, S. B. Nielsen, S. Panja, J. M. Pedersen, T. Skrydstrup, K. Stochkel, E. R. Williams and E. S. Worm, On the mechanism of electron-capture-induced dissociation of peptide dications from N-15-labeling and crown-ether complexation, J. Phys. Chem. A, 2007, 111(39), 9641–9643 CrossRef PubMed.
- M. Schneider, C. Masellis, T. Rizzo and C. Baldauf, Kinetically Trapped Liquid-State Conformers of a Sodiated Model Peptide Observed in the Gas Phase, J. Phys. Chem. A, 2017, 121(36), 6838–6844 CrossRef PubMed.
- J. A. Silveira, K. L. Fort, D. Kim, K. A. Servage, N. A. Pierson, D. E. Clemmer and D. H. Russell, From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations, J. Am. Chem. Soc., 2013, 135(51), 19147–19153 CrossRef PubMed.
- N. A. Pierson and D. E. Clemmer, An IMS–IMS threshold method for semi-quantitative determination of activation barriers: interconversion of proline cis ↔ trans forms in triply protonated bradykinin, Int. J. Mass Spectrom., 2015, 377, 646–654 CrossRef PubMed.
- A. E. Counterman and D. E. Clemmer, Large anhydrous polyalanine ions: evidence for extended helices and onset of a more compact state, J. Am. Chem. Soc., 2001, 123, 1490–1498 CrossRef PubMed.
- A. E. Counterman and D. E. Clemmer, Compact → extended helix transitions of polyalanine in vacuo, J. Phys. Chem. B, 2003, 107(9), 2111–2117 CrossRef.
- R. Pepin, A. Petrone, K. J. Laszlo, M. F. Bush, X. S. Li and F. Turecek, Does Thermal Breathing Affect Collision Cross Sections of Gas-Phase Peptide Ions? An Ab Initio Molecular Dynamics Study, J. Phys. Chem. Lett., 2016, 7(14), 2765–2771 CrossRef PubMed.
- N. A. Pierson, S. J. Valentine and D. E. Clemmer, Evidence for a Quasi-Equilibrium Distribution of States for Bradykinin [M + 3H](3+) Ions in the Gas Phase, J. Phys. Chem. B, 2010, 114(23), 7777–7783 CrossRef PubMed.
- A. J. Soulby, J. W. Heal, M. P. Barrow, R. A. Roemer and P. B. O'Connor, Does deamidation cause protein unfolding? A top-down tandem mass spectrometry study, Protein Sci., 2015, 24(5), 850–860 CrossRef PubMed.
- A. T. Iavarone, K. Paech and E. R. Williams, Effects of charge state and cationizing agent on the electron capture dissociation of a peptide, Anal. Chem., 2004, 76(8), 2231–2238 CrossRef PubMed.
- R. Jertz, J. Friedrich, C. Kriete, E. N. Nikolaev and G. Baykut, Tracking the Magnetron Motion in FT-ICR Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2015, 26(8), 1349–1366 CrossRef PubMed.
- G. R. Grimsley, J. M. Scholtz and C. N. Pace, A summary of the measured pK values of the ionizable groups in folded proteins, Protein Sci., 2009, 18(1), 247–251 Search PubMed.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, pK values of the ionizable groups of proteins, Protein Sci., 2006, 15(5), 1214–1218 CrossRef PubMed.
- E. P. Hunter and S. G. Lias, Gas Phase Ion Energetics Data, In NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, retrieved July 21, 2017, http://webbook.nist.gov Search PubMed.
- V. Addario, Y. Z. Guo, I. K. Chu, Y. Ling, G. Ruggerio, C. F. Rodriquez, A. C. Hopkinson and K. W. M. Siu, Proton affinities of methyl esters of N-acetylated amino acids, Int. J. Mass Spectrom., 2002, 219(1), 101–114 CrossRef.
- M. M. Kish, G. Ohanessian and C. Wesdemiotis, The Na+ affinities of alpha-amino acids: side-chain substituent effects, Int. J. Mass Spectrom., 2003, 227(3), 509–524 CrossRef.
- C. H. Ruan and M. T. Rodgers, Cation–pi interactions: structures and energetics of complexation of Na+ and K+ with the aromatic amino acids, phenylalanine, tyrosine, and tryptophan, J. Am. Chem. Soc., 2004, 126(44), 14600–14610 CrossRef PubMed.
- A. Gapeev and R. C. Dunbar, Na+ affinities of gas-phase amino acids by ligand exchange equilibrium, Int. J. Mass Spectrom., 2003, 228(2–3), 825–839 CrossRef.
- S. J. Ye, A. A. Clark and P. B. Armentrout, Experimental and theoretical investigation of alkali metal cation interactions with hydroxyl side-chain amino acids, J. Phys. Chem. B, 2008, 112(33), 10291–10302 CrossRef PubMed.
- A. L. Heaton, R. M. Moision and P. B. Armentrout, Experimental and theoretical studies of sodium cation interactions with the acidic amino acids and their amide derivatives, J. Phys. Chem. A, 2008, 112(15), 3319–3327 CrossRef PubMed.
- M. F. Bush, J. Oomens and E. R. Williams, Proton Affinity and Zwitterion Stability: New Results from Infrared Spectroscopy and Theory of Cationized Lysine and Analogues in the Gas Phase, J. Phys. Chem. A, 2009, 113(2), 431–438 CrossRef PubMed.
- M. L. Stover, C. E. Plummer, S. R. Miller, C. J. Cassady and D. A. Dixon, Gas-Phase Acidities of Phosphorylated Amino Acids, J. Phys. Chem. B, 2015, 119(46), 14604–14621 CrossRef PubMed.
- M. Remko, P. T. Van Duijnen and C. W. von der Lieth, Structure and stability of Li(I) and Na(I) – Carboxylate, sulfate and phosphate complexes, J. Mol. Struct.: THEOCHEM, 2007, 814(1–3), 119–125 CrossRef.
- C. E. Bartman, H. Metwally and L. Konermann, Effects of Multidentate Metal Interactions on the Structure of Collisionally Activated Proteins: Insights from Ion Mobility Spectrometry and Molecular Dynamics Simulations, Anal. Chem., 2016, 88(13), 6905–6913 CrossRef PubMed.
- K. Breuker and F. W. McLafferty, The thermal unfolding of native cytochrome c in the transition from solution to gas phase probed by native electron capture dissociation, Angew. Chem., Int. Ed., 2005, 44(31), 4911–4914 CrossRef PubMed.
- E. F. Strittmatter and E. R. Williams, The role of proton affinity, acidity, and electrostatics on the stability of neutral versus ion-pair forms of molecular dimers, Int. J. Mass Spectrom., 2001, 212(1–3), 287–300 CrossRef.
- S. R. Harvey, M. Porrini, R. C. Tyler, C. E. MacPhee, B. F. Volkman and P. E. Barran, Electron capture dissociation and drift tube ion mobility-mass spectrometry coupled with site directed mutations provide insights into the conformational diversity of a metamorphic protein, Phys. Chem. Chem. Phys., 2015, 17(16), 10538–10550 RSC.
- S. R. Harvey, M. Porrini, A. Konijnenberg, D. J. Clarke, R. C. Tyler, P. R. R. Langridge-Smith, C. E. MacPhee, B. F. Volkman and P. E. Barran, Dissecting the Dynamic Conformations of the Metamorphic Protein Lymphotactin, J. Phys. Chem. B, 2014, 118(43), 12348–12359 CrossRef PubMed.
- M. Meot-Ner, The ionic hydrogen bond, Chem. Rev., 2005, 105(1), 213–284 CrossRef PubMed.
- L. Vasicek and J. S. Brodbelt, Enhanced Electron Transfer Dissociation through Fixed Charge Derivatization of Cysteines, Anal. Chem., 2009, 81(19), 7876–7884 CrossRef PubMed.
- X. J. Li, J. J. Cournoyer, C. Lin and P. B. O'Connor, The Effect of Fixed Charge Modifications on Electron Capture Dissociation, J. Am. Soc. Mass Spectrom., 2008, 19(10), 1514–1526 CrossRef PubMed.
- L. Vrbka, J. Vondrasek, B. Jagoda-Cwiklik, R. Vacha and P. Jungwirth, Quantification and rationalization of the higher affinity of sodium over potassium to protein surfaces, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(42), 15440–15444 CrossRef PubMed.
- S. Warnke, G. von Helden and K. Pagel, Protein Structure in the Gas Phase: The Influence of Side-Chain Microsolvation, J. Am. Chem. Soc., 2013, 135(4), 1177–1180 CrossRef PubMed.
- R. A. Zubarev, Reactions of polypeptide ions with electrons in the gas phase, Mass Spectrom. Rev., 2003, 22, 57–77 CrossRef PubMed.
- B. A. Budnik, Y. O. Tsybin, P. Håkansson and R. A. Zubarev, Ionization energies of multiply protonated polypeptides obtained by tandem ionization in Fourier transform mass spectrometers, J. Mass Spectrom., 2002, 37(11), 1141–1144 CrossRef PubMed.
- S. L. Zhou, B. S. Prebyl and K. D. Cook, Profiling pH changes in the electrospray plume, Anal. Chem., 2002, 74(19), 4885–4888 CrossRef PubMed.
- M. Girod, X. Dagany, R. Antoine and P. Dugourd, Relation between charge state distributions of peptide anions and pH changes in the electrospray plume. A mass spectrometry and optical spectroscopy investigation, Int. J. Mass Spectrom., 2011, 308(1), 41–48 CrossRef.
- P. Liigand, A. Heering, K. Kaupmees, I. Leito, M. Girod, R. Antoine and A. Kruve, The Evolution of Electrospray Generated Droplets is Not Affected by Ionization Mode, J. Am. Soc. Mass Spectrom., 2017, 28(10), 2124–2131 CrossRef PubMed.
- M. Zachariou, I. Traverso, L. Spiccia and M. T. W. Hearn, Potentiometric investigations into the acid–base and metal ion binding properties of immobilized metal ion affinity chromatographic (IMAC) adsorbents, J. Phys. Chem., 1996, 100(30), 12680–12690 CrossRef.
- C. S. Hoaglund-Hyzer, Y. J. Lee, A. E. Counterman and D. E. Clemmer, Coupling ion mobility separations, collisional activation techniques, and multiple stages of MS for analysis of complex peptide mixtures, Anal. Chem., 2002, 74(5), 992–1006 CrossRef PubMed.
- H. J. Cooper, R. R. Hudgins, K. Håkansson and A. G. Marshall, Characterization of amino acid side chain losses in electron capture dissociation, J. Am. Soc. Mass Spectrom., 2002, 13(3), 241–249 CrossRef PubMed.
- S. A. McLuckey and D. E. Goeringer, Slow heating methods in tandem mass spectrometry, J. Mass Spectrom., 1997, 32, 461–474 CrossRef.
- J. Vusurovic, E. M. Schneeberger and K. Breuker, Interactions of Protonated Guanidine and Guanidine Derivatives with Multiply Deprotonated RNA Probed by Electrospray Ionization and Collisionally Activated Dissociation, ChemistryOpen, 2017, 6(6), 739–750 CrossRef PubMed.
- E. M. Schneeberger and K. Breuker, Native Top-Down Mass Spectrometry of TAR RNA in Complexes with a Wild-Type tat Peptide for Binding Site Mapping, Angew. Chem., Int. Ed., 2017, 56(5), 1254–1258 CrossRef PubMed.
- B. Ganisl and K. Breuker, Does Electron Capture Dissociation Cleave Protein Disulfide Bonds?, ChemistryOpen, 2012, 1(6), 260–268 CrossRef PubMed.
- S. J. Ye and P. B. Armentrout, Absolute thermodynamic measurements of alkali metal cation interactions with a simple dipeptide and tripeptide, J. Phys. Chem. A, 2008, 112(16), 3587–3596 CrossRef PubMed.
- D. Semrouni, O. P. Balaj, F. Calvo, C. F. Correia, C. Clavaguera and G. Ohanessian, Structure of Sodiated Octa-Glycine: IRMPD Spectroscopy and Molecular Modeling, J. Am. Soc. Mass Spectrom., 2010, 21(5), 728–738 CrossRef PubMed.
- O. P. Balaj, C. Kapota, J. Lemaire and G. Ohanessian, Vibrational signatures of sodiated oligopeptides (GG–Na+, GGG–Na+, AA–Na+ and AAA–Na+) in the gas phase, Int. J. Mass Spectrom., 2008, 269(3), 196–209 CrossRef.
- J. Dzubiella, Salt-specific stability and denaturation of a short salt-bridge-forming alpha-helix, J. Am. Chem. Soc., 2008, 130(42), 14000–14007 CrossRef PubMed.
- C. Lin, J. J. Cournoyer and P. B. O'Connor, Probing the gas-phase folding kinetics of peptide ions by IR activated DR-ECD, J. Am. Soc. Mass Spectrom., 2008, 19(6), 780–789 CrossRef PubMed.
- A. R. Ledvina, G. C. McAlister, M. W. Gardner, S. I. Smith, J. A. Madsen, J. C. Schwartz, G. C. Stafford, J. E. P. Syka, J. S. Brodbelt and J. J. Coon, Infrared Photoactivation Reduces Peptide Folding and Hydrogen-Atom Migration following ETD Tandem Mass Spectrometry, Angew. Chem., Int. Ed., 2009, 48(45), 8526–8528 CrossRef PubMed.
- C. Lin, J. J. Cournoyer and P. B. O'Connor, Use of a double resonance electron capture dissociation experiment to probe fragment intermediate lifetimes, J. Am. Soc. Mass Spectrom., 2006, 17(11), 1605–1615 CrossRef PubMed.
- H. Takahashi, S. Sekiya, T. Nishikaze, K. Kodera, S. Iwamoto, M. Wada and K. Tanaka, Hydrogen Attachment/Abstraction Dissociation (HAD) of Gas-Phase Peptide Ions for Tandem Mass Spectrometry, Anal. Chem., 2016, 88(7), 3810–3816 CrossRef PubMed.
- S. G. Lias, Ionization Energy Evaluation, In NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, retrieved December 31, 2017, http://webbook.nist.gov Search PubMed.
- X. F. Chen, G. Q. Liu, Y. L. E. Wong, L. L. Deng, Z. Wang, W. Li and T. W. D. Chan, Dissociation of trivalent metal ion (Al3+, Ga3+, In3+ and Rh3+)–peptide complexes under electron capture dissociation conditions, Rapid Commun. Mass Spectrom., 2016, 30(6), 705–710 CrossRef PubMed.
- H. C. Liu and K. Håkansson, Divalent metal ion-peptide interactions probed by electron capture dissociation of trications, J. Am. Soc. Mass Spectrom., 2006, 17(12), 1731–1741 CrossRef PubMed.
- D. Asakawa and E. De Pauw, Difference of Electron Capture and Transfer Dissociation Mass Spectrometry on Ni2+-, Cu2+-, and Zn2+-Polyhistidine Complexes in the Absence of Remote Protons, J. Am. Soc. Mass Spectrom., 2016, 27(7), 1165–1175 CrossRef PubMed.
- J. Dong and R. W. Vachet, Coordination Sphere Tuning of the Electron Transfer Dissociation Behavior of Cu(II)–Peptide Complexes, J. Am. Soc. Mass Spectrom., 2012, 23(2), 321–329 CrossRef PubMed.
- T. G. Flick, W. A. Donald and E. R. Williams, Electron Capture Dissociation of Trivalent Metal Ion–Peptide Complexes, J. Am. Soc. Mass Spectrom., 2013, 24(2), 193–201 CrossRef PubMed.
- J. J. Commodore and C. J. Cassady, Effects of acidic peptide size and sequence on trivalent praseodymium adduction and electron transfer dissociation mass spectrometry, J. Mass Spectrom., 2017, 52(4), 218–229 CrossRef PubMed.
- A. Kramida, Y. Ralchenko, J. Reader and N. A. Team, NIST Atomic Spectra Database (Version 5.5.3), [Online] [Wed Mar 28 2018], National Institute of Standards and Technology, Gaithersburg, MD, 2018, Available: https://physics.nist.gov/asd Search PubMed.
- T. Dudev and C. Lim, Competition among Metal Ions for Protein Binding Sites: Determinants of Metal Ion Selectivity in Proteins, Chem. Rev., 2014, 114(1), 538–556 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02470g |
| This journal is © The Royal Society of Chemistry 2018 |