
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast screening of illicit drugs in beverages and biological fluids by direct coupling of thin film microextraction to dielectric barrier discharge ionization-mass spectrometry
Mario F.
Mirabelli†
a,
Emanuela
Gionfriddo‡†
b,
Janusz
Pawliszyn
b and
Renato
Zenobi
 *a
*a
aETH Zurich, Department of Chemistry and Applied Biosciences, 8093 Zurich, Switzerland. E-mail: zenobi@org.chem.ethz.ch
bDepartment of Chemistry, University of Waterloo, Waterloo, Ontario, Canada
First published on 8th March 2019
Abstract
A direct and fast method for screening and quantification of illicit drugs in beverages and biological fluids was developed by using dielectric barrier discharge (DBD) as ionization technique, in combination with high-resolution mass spectrometry. Extraction of the targeted analytes was carried out with thin film microextraction (TFME) using ultrasound as agitation method. The targeted analytes were then thermally desorbed and introduced into the source without the need for any cryofocusing apparatus. The performance of this new analytical set up were compared to conventional TFME-TDU-GC/MS, showing enhanced linear dynamic range and lower limits of quantitation (low pg ml−1) achievable at the same extraction conditions (5 min extraction time). The performance of the method was tested in different beverages and body fluids, confirming its applicability for quantitative analysis of the targeted drugs in complex samples.
Introduction
The identification and quantification of illicit drugs usually involve the analysis of blood, urine or hair samples, often using time-consuming and labor-intensive techniques. An alternative approach for screening, prevention or investigative purposes can also include analysis of beverages used as media for voluntary or involuntary drug intake. Moreover, beverages are samples that can provide direct on-site proof of illegal drug sale, intended application and/or consumption. The ideal sampling scenario for this type of application would consist of minimum sample handling, using small sample volumes and very short extraction times, followed by fast data acquisition and processing. Solid-phase microextraction (SPME) satisfies these requirements and enables a simultaneous sampling/preconcentration step. Among the various geometries of SPME, thin film microextraction (TFME) provides a viable alternative to the classical fiber, with a higher surface area and coating volume. These characteristics of TFME translate into enhanced extraction efficiency, without sacrificing extraction kinetics. TFME devices were successfully used both for gas- and liquid-chromatography applications.1–3 Generally, polydimethylsiloxane (PDMS)-based TFME devices are preferred for thermal desorption due to the thermal stability of the polymer, while polyacrylonitrile (PAN)-based thin films are better for solvent desorption. The devices used in this work are made of PDMS loaded with divinylbenzene (DVB) particles, a combination that demonstrated enhanced extraction efficiency with a broader range of analytes extracted, when compared to pure PDMS thin films.4 The flexibility of this type of self-supported TFME device allows for easy handling and introduction into desorption liners used for thermal desorption. In previous studies, the PDMS/DVB TFME devices were directly interfaced to mass spectrometry (MS), through an electron ionization (EI) source using a single quadruple mass spectrometer.5 The coupling was carried out by connecting a thermal desorption unit (TDU) to the mass spectrometer through a deactivated fused silica column. This approach employed a sample introduction system that involves two thermal desorption stages (the first to desorb the analytes from the TFME device, and the second to release the analytes into the chromatographic column) with a liquid nitrogen cryofocusing step in between, which yields narrow chromatographic peaks. The analytical set-up described above was necessary to enable ionization in vacuum conditions in the EI source.Recently, the rapid development of various ambient mass spectrometry technologies opened the door to quick and easy direct introduction of samples or extracts into mass spectrometers.6,7 In particular, the dielectric barrier discharge ionization (DBDI) source was already successfully used for the analysis of analytes collected by SPME devices in the fiber8,9 and arrow10 geometries. More recently, capillary atmospheric pressure photoionization (cAPPI) was successfully employed for the direct SPME analysis of nonpolar compounds like PAHs.11
A DBD ionization source consists of a low-temperature plasma, which is ignited between two electrodes separated by a dielectric material.12–15 In recent years, DBDI has been increasingly used for analysis of various sample matrices for direct mass spectrometric16–19 or chromatographic analyses.20–23
The source employed in this work uses an “active capillary” sampling geometry, meaning that the source is directly connected to the mass spectrometer, and the flow of gas and analytes through the source is determined by the vacuum inside the mass spectrometer. This enhances the sensitivity, due to a loss-less transmission of the ions formed into the mass spectrometer. Interfacing of this DBDI source to liquid and gas chromatography was also recently reported.23,24 In addition to its simple design, our DBDI source can also operate with different gases for the plasma ignition, depending on the specific analytical needs. Finally, in contrast to hard ionization methods such as EI, DBDI is a soft ionization method that generates little to no fragmentation, mostly yielding protonated molecules.
In this work, we propose the direct coupling of a self-supported TFME device to a DBD ion source and high-resolution mass spectrometry, as a convenient and rapid analytical setup for quantification of drugs in beverages and biofluids. No cryofocusing system was required since the gas flow rate around the TFME device was sufficiently high to allow for rapid thermal desorption of the analytes. Compared to other approaches,5 the desorption chamber used was directly connected to the ionization source, minimizing desorption time and consequently potential thermal degradation of compounds on their way into the ionization source. This SPME extraction/desorption strategy ensures that most of the matrix components and molecules with very low volatility are either not extracted from the sample or not desorbed from the SPME thin film, thus giving minimal sample carry-over. The method proposed herein also provides the advantage of fast sampling and data acquisition without sacrificing the sensitivity of the analysis.
Experimental
Materials
HPLC-grade water and acetonitrile were obtained from Sigma-Aldrich (Buchs, Switzerland). Analytical-grade drug standards (Cocaine, Diazepam, Desipramine, Imipramine, MDMA, MDEA, Methadone, and Fentanyl), human plasma (containing 4% trisodium citrate as anticoagulant) and dimethyldichlorosilane (DMDCS, 5% in toluene) were purchased from Sigma-Aldrich. Urine samples were obtained from a healthy male volunteer employed at ETH Zurich, and were not treated further before spiking and extraction.For the preparation of the TFME devices, a SYLGARD184 silicone elastomer kit was purchased from Dow Corning (Midland, MI, USA). DVB particles with a diameter of 3–5 μm were kindly provided by Millipore Sigma. The TFME devices were prepared according to the procedure proposed by Jiang et al.4 The dimension and shape of the TFME device were customized as a 20 × 3 mm rectangle with a film thickness of 250 ± 10 μm.
Sample preparation
Drug mixtures were prepared from individual standards at a concentration of 50 μg mL−1 in acetonitrile, and stored at −20 °C. Diluted solutions used for SPME analysis were prepared in ACN from the mixture and were stored at a temperature of 4 °C. Aqueous dilutions were prepared with HPLC-grade water, keeping the total ACN concentration below 1% (v/v). Beverages (vodka, wine, and cola) and biological fluids (human urine and blood plasma) were spiked with drugs at different concentration levels, keeping the total ACN concentration below 1% (v/v), and equilibrated at room temperature for 6 hours prior to analysis.The PDMS/DVB TFME devices were supported by a stainless-steel wire (0.125 mm diameter, 60 mm length) and were fully immersed in the sample matrix during extraction from 1.5 mL glass vials. Samples were extracted for 5 minutes at 40 °C. Instead of a classic stirring/shaking approach, an ultrasound-assisted extraction was used since the thin film devices were found to be stable to ultrasound. For this purpose, a lab sonicator was used, and the sonication bath was kept at a constant temperature of 40 °C (±1 °C).
Thermal desorption
Thin film SPME devices were thermally desorbed in a lab-built desorption chamber directly connected to the DBD ionization source (Fig. 1). The desorption chamber was made from aluminum and a fine temperature control (±0.5 °C) was achieved with a PID temperature controller. Inside the aluminum body, a glass liner (I.D. 4 mm) chemically deactivated by silanization (using 5% DMDCS in toluene) was used to reduce carryover due to adsorption of compounds to the transfer line. The liner was connected to the ionization source using high-temperature O-rings.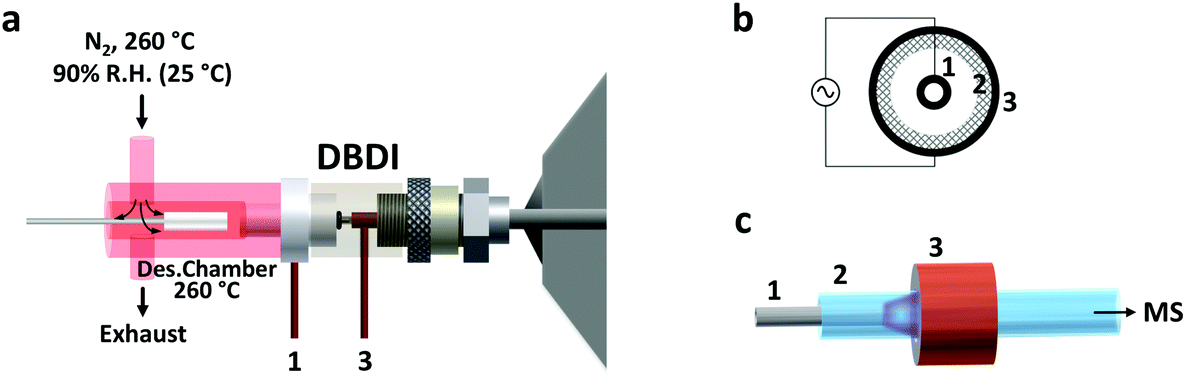 | ||
| Fig. 1 Schematic drawing of the setup used in this study. (a) The entire setup (desorption chamber and ionization source) is shown. Pre-heated and humidified nitrogen (at 90% relative humidity, R.H., at 25 °C) gas was introduced in the desorption chamber from the top. The nitrogen gas flow rate was higher than the flow rate through the DBDI source. Therefore, the incoming nitrogen was split between an exhaust line and the ionization source. This prevented any air (and oxygen) from the laboratory room to enter the setup. The DBDI source, which was directly connected to the inlet of the mass spectrometer, was operated by applying alternating voltages to an inner electrode (1) and an outer electrode (3), separated by a dielectric material (2). In the right part of the figure, the details of the DBDI source inner core are shown: (b) axial view of the ionization source core: (c) three-dimensional view of the electrode and dielectric arrangement. The low temperature plasma forming upon application of high voltage is shown in purple color. | ||
N2 was used as desorption gas, at a temperature of 260 °C and a relative humidity of 90% (at 25 °C). The carrier gas flow was maintained constant at 3 L min−1 by means of a mass-flow controller (Bronkhorst High Tech B.V. Ruurlo, Netherlands). Of the total 3.0 L min−1, only 1.0 L min−1 entered the ionization source. This overflow of nitrogen was used to prevent room air from entering the setup. This prevented the reproducibility of the results from being negatively impacted, considering that the desorption chamber has an open design (no seals/septa used). In addition, the presence of oxygen from the air during desorption should be avoided since it could oxidize the SPME coating at high temperatures.
DBD ionization source
The active capillary plasma source was operated by applying an AC voltage to two concentric electrodes, separated by a dielectric capillary connected to the MS inlet8 (Fig. 1). A 1.6 kVp–p potential was used, with an operating frequency of 5.75 kHz. Under these conditions mostly protonated molecules were generated, with very little to no fragmentation.Mass spectrometry
A high-resolution LTQ Orbitrap mass spectrometer (Thermo Fischer Scientific, San José, USA) was used. The total gas flow entering the source was constant and dictated by the inlet of the mass spectrometer, i.e., the metal transfer capillary dimensions, and measured to be 1.0 L min−1. Quantification was performed in full scan mode, with centroid acquisition and a mass resolution of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FWHM at 400 m/z. The MS interface parameters were as follows: capillary voltage, 4 V; tube lens voltage, 65 V; capillary temperature, 275 °C. The acquisition was performed with a mass window of 150 to 500 m/z, 1 micro scan, and with a maximum injection time of 150 ms. Automatic gain control was used. The mass accuracy achieved was below 1 ppm with the use of lock masses. A mass tolerance of 2 ppm was used for creation of extracted ion chromatograms and signal integration.
000 FWHM at 400 m/z. The MS interface parameters were as follows: capillary voltage, 4 V; tube lens voltage, 65 V; capillary temperature, 275 °C. The acquisition was performed with a mass window of 150 to 500 m/z, 1 micro scan, and with a maximum injection time of 150 ms. Automatic gain control was used. The mass accuracy achieved was below 1 ppm with the use of lock masses. A mass tolerance of 2 ppm was used for creation of extracted ion chromatograms and signal integration.
Complementary TDU-GC-MS analyses
Complementary Thermal Desortion Unit-Gas-Chromatography/Mass Spectrometry (TDU-GC/MS) analyses of the target analytes were performed by a Agilent GC 6890N coupled to a 5973N Inert MSD detector (Agilent Technologies, CA, USA). Thermal desorption of the target analytes from the TFME devices was performed in a cooling injection system 3 (CIS 3) equipped with a thermal desorption unit (GERSTEL Inc., Linthicum, MD, USA). The TDU desorption liners containing the TFME devices were transferred into the TDU unit using the GERSTEL multipurpose system 2 (MPS2) autosampler. Chromatographic separation of the selected analytes was carried out with a DB-5MS-UI capillary column (length of 30 m, 0.25 mm ID, 0.25 μm film thickness) from Agilent J&W (Santa Clara, USA). Helium at 99.999% purity from Praxair (Kitchener, ON, Canada) was used as carrier gas, at a flow of 1.2 mL min−1. The oven temperature program was started at an initial temperature of 40 °C held for 2 min, subsequently a temperature of 180 °C was reached using 10 °C min−1 ramp and held for 2 min, and a final temperature of 300 °C was reached using a 10 °C min−1 ramp, and held for 5 min. Desorption from the TFME devices was carried out at 250 °C for 10 min. The temperatures of the MS transfer line, ion source, and quadrupole were 280 °C, 150 °C and 230 °C, respectively. Ionization of the target molecules was by electron ionization (EI) at 70 eV.Results and discussion
The use of DBD ionization for the analysis of thin films produced clean mass spectra, with little to no fragmentation for the compounds of interest. An example is shown in Fig. 2, where a spectrum obtained after extraction of the target drugs at a concentration of 10 ng mL−1 from 1.5 mL of spiked blood plasma is displayed.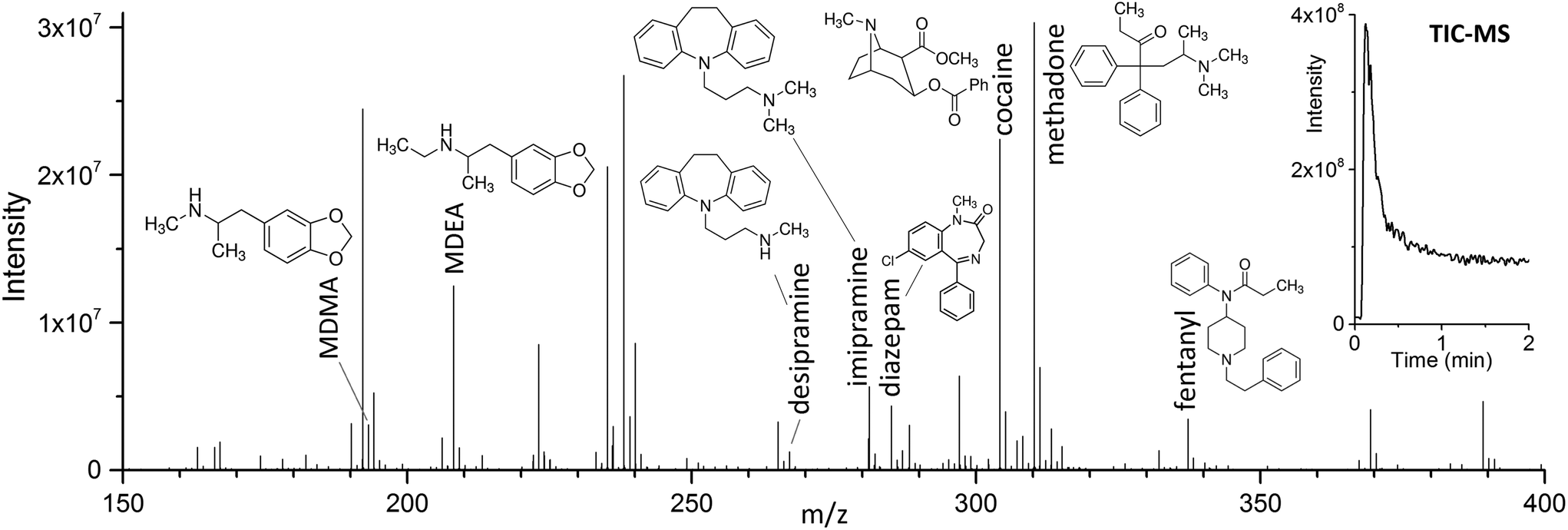 | ||
| Fig. 2 Direct TFME-DBDI full scan mass spectrum of a 10 ng mL−1 mix of drugs in blood plasma (containing 1% ACN), extracted for 5 minutes with ultrasound. The data shows the simultaneous ionization of the compounds and the low level of background signals. On the top right corner, the TIC for the desorption (260 °C) is shown. | ||
As previously shown for SPME fibers, a fast desorption is obtained with the use of small I.D. desorption chambers, in order to ensure a quick displacement of the desorbed compounds from the proximity of the fiber.25 We have recently shown the direct coupling of SPME to MS with the use of an open system,8 where the best results were obtained using a desorption chamber with a narrow internal diameter (I.D.) even when employing a high desorption gas flow rate. Since TFME devices can be manufactured in shapes significantly bigger than fibers, a desorption chamber with a proper I.D., able to accommodate the devices, had to be used. In our case, we tried to minimize the dimensions of the thin films, without compromising their extraction capacity deriving from a geometry with higher surface area and coating volume. TFME devices having a width of 3 mm were found to be easy to manufacture and handle for this application. A length of 20 mm was chosen to be compatible with the 1.5 mL glass vials. Shorter films did not offer advantages during desorption. The desorption chamber was equipped with 4 mm I.D. glass liners, passivated by silanization, and capable of accommodating the thin films without bending the devices.
Considering the larger volume of the 4 mm I.D. desorption chamber, and the different geometry of the thin films compared to SPME fibers, it was necessary to verify that the linear gas velocity applied would guarantee efficient desorption of the analytes. Therefore, the desorption kinetics for the target drugs was investigated using different temperatures, ranging from 200 °C to 260 °C (Fig. 3). Considering previous findings on the thermal stability of SPME matrices, higher temperatures were not tested.2,4 Most of the target analytes showed a fast desorption kinetics, with a complete desorption in less than 2 min at 260 °C. Diazepam was found to be strongly retained by the film, resulting in a slower desorption, which was complete only after 6 min at 260 °C.
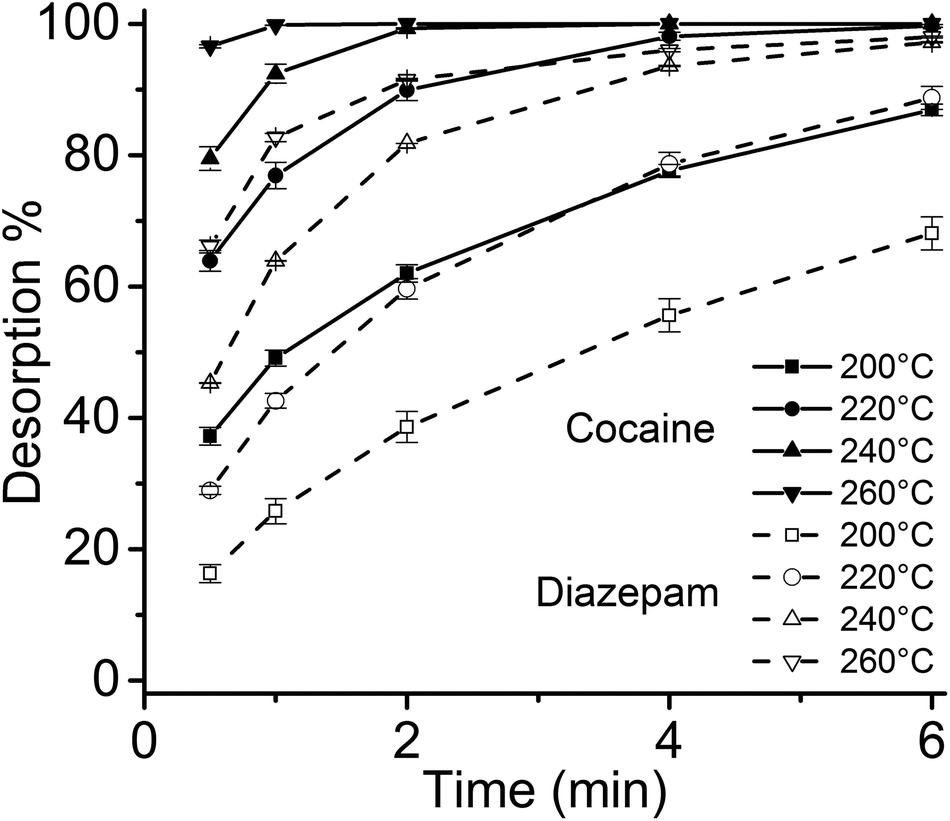 | ||
| Fig. 3 SPME-DBDI-MS desorption time profiles of cocaine and diazepam at different temperatures (200 to 260 °C) for the 3 × 20 mm thin film microextraction device (n = 3). A 4 mm I.D. glass liner desorption chamber (chemically deactivated by silanization) was used. | ||
Considering the flexibility of the self-supported TFME device used, two different sample agitation modes were tested to optimize the extraction conditions: agitation by stir bar and sonication. Generally, the use of sonication is not widespread for SPME, due to concerns regarding the SPME devices’ stability toward ultrasound energy and high variability of the sonication bath temperature during continuous and extensive sonication procedures. However, in this work, we found that the self-supported TFME devices provided enough stability to endure sonication for up to 120 min. We also carefully controlled the temperature of the sonication bath to ensure constant extraction temperature (within ±1 °C). The extraction efficiency and kinetics of the TFME devices were evaluated using both agitation modes, setting the extraction temperature to 40 °C and determining extraction time profiles from 1 to 120 min in ultrapure water. As shown in Fig. 4, for the representative analyte fentanyl, sonication is able to provide a more efficient agitation method with faster extraction kinetics. In light of the results obtained, sonication was selected as the agitation method for this study and an extraction time of 5 min was selected as a good compromise between sensitivity and throughput for the method developed.
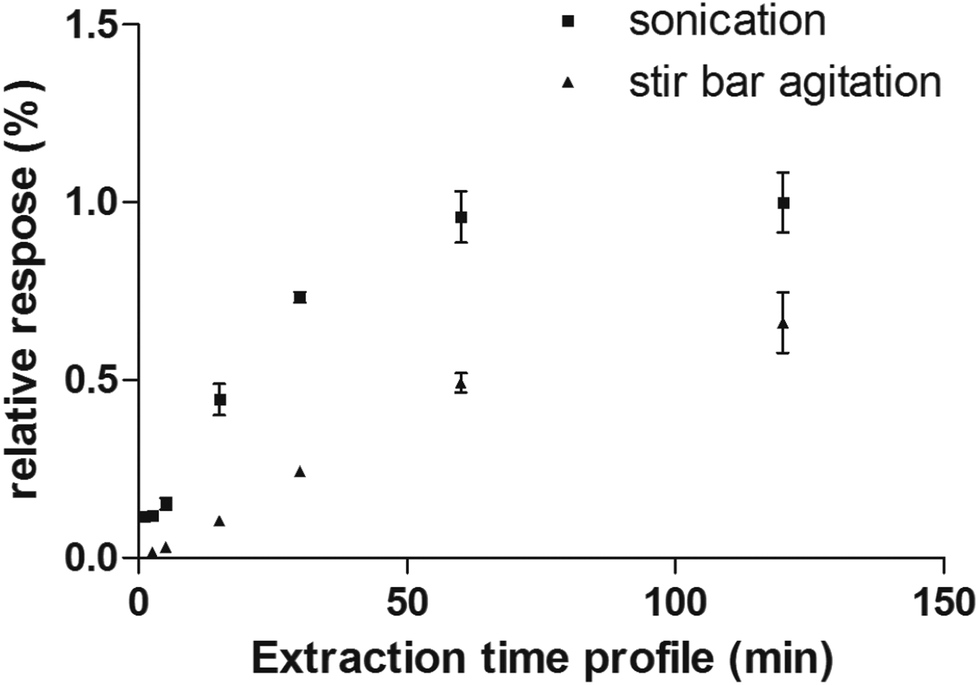 | ||
| Fig. 4 Extraction time profile of fentanyl extracted from water obtained by sonication and stir bar agitation methods. | ||
In addition to extraction of drugs from water samples, real sample matrices were used to simulate cases of drug analysis in forensic and medical applications, i.e., vodka, wine, cola, human urine, and blood plasma. The beverages chosen for this study present different challenges due to their composition: in particular, the ethanol content of vodka and wine, which constitutes a competing phase for the partitioning of the analytes into the TFME device, and/or the presence of interfering molecules that can affect the quantification of drugs at ultra-trace level. In the case of cola, sugars are present at very high concentrations (ca. 10%). In wine, several polyphenolic compounds with low volatility are present, and the EtOH content ranges usually from 12% to 14% (v/v). In the vodka sample, the concentration of EtOH was even higher (37.5%).
The influence of the amount of alcohol in the sample matrix on the extraction efficiency was investigated by extracting the target analytes from water/ethanol mixtures of various compositions (EtOH concentrations of 0, 10, 20, 40 and 60% (v/v)). The ultrasound-assisted extractions were performed in pre-equilibrium conditions (5 min) at 40 °C. The resulting extracted ion chromatograms were integrated over a 0.5 min time period. The results are reported in Fig. 5.
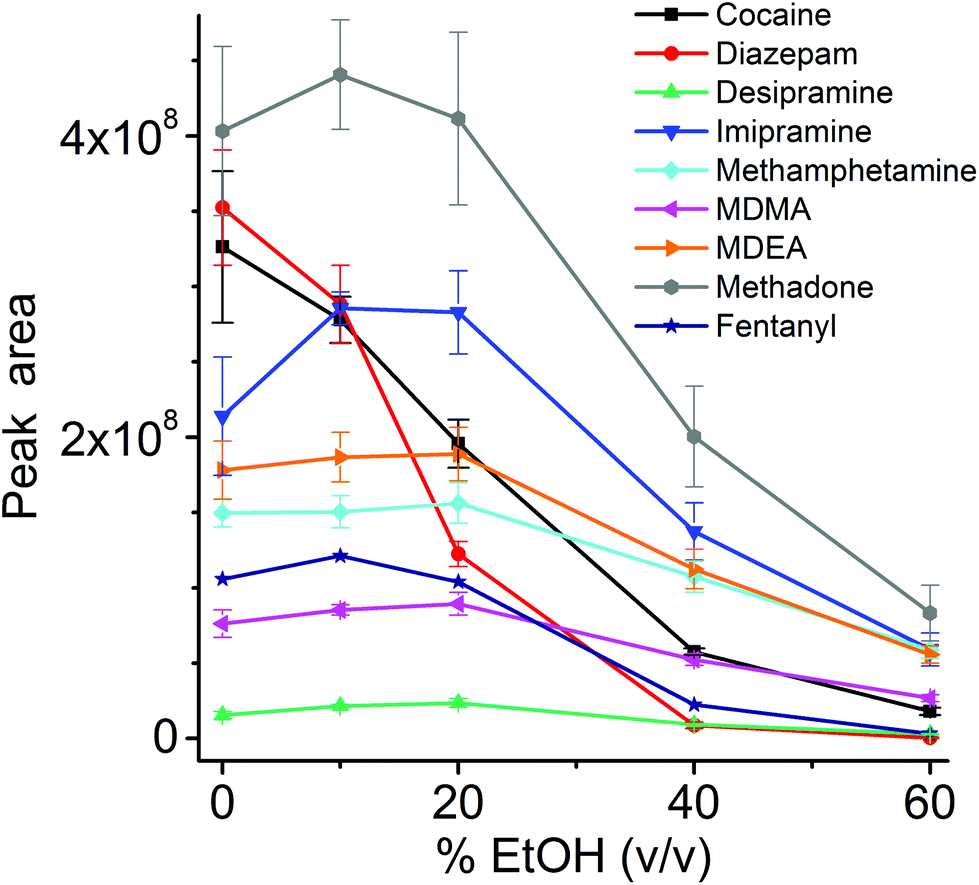 | ||
| Fig. 5 Extraction efficiency (n = 4) for the considered drugs from aqueous solutions at different concentration of EtOH. The ultrasound-assisted extractions were performed in pre-equilibrium (5 min) at 40 °C using 3 × 20 mm thin films immersed in 1.5 mL liquid (in 1.5 mL vials). Each extracted ion chromatogram was integrated for 0.5 min. | ||
Most analytes were efficiently extracted up to EtOH concentration of 20%. The extraction efficiency from alcoholic solutions is compound-dependent, and is a function of the partitioning of the analytes between the sample solution and the TFME device, which is influenced both by the analytes’ hydrophobicity and π–π interactions with the extraction phase. In most cases, more than 50% of the signal was still present at 40% EtOH content, when compared to extraction from pure water, with the exception of cocaine and diazepam, which showed a lower extraction efficiency at high alcohol concentration. Since we employed a sensitive DBDI source coupled to a high capacity SPME device, the reduction in the extraction efficiency did not jeopardize the quantification of target analytes at the pg mL−1 level in vodka.
In forensic applications, e.g., a drink spiked with illicit substances, a variable concentration of drugs is expected, depending on the potency of the drug. Potent drugs like fentanyl are effective even at μg levels. In these cases, the drug concentration would be high enough to allow shorter extraction times to be used. To demonstrate the potential of a faster approach, extractions of drinks spiked at 1 μg mL−1 were performed at room temperature without stirring, by inserting the thin film in the liquid samples for only 5 seconds. After extraction, a quick (3 seconds) manual rinsing step in LC-MS grade water was performed to remove potential residues of matrix components, and afterward the thin films were directly desorbed. This approach allows for a quasi-real-time analysis (Fig. 6).
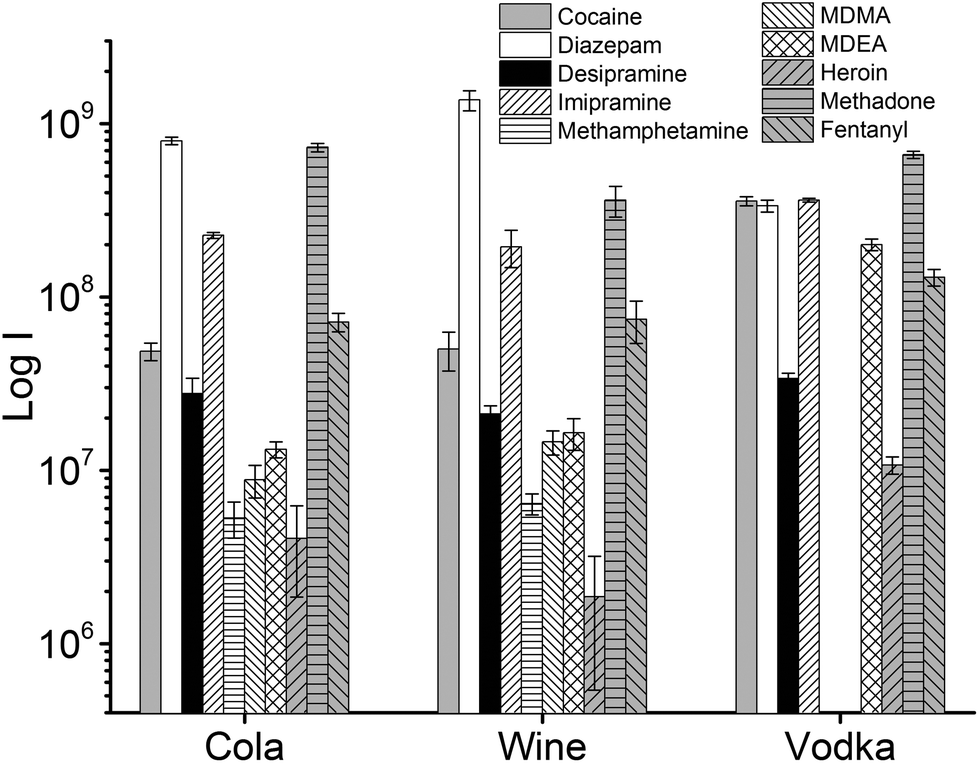 | ||
| Fig. 6 TFME-DBDI-MS ion signals for fast extractions (n = 3, 5 seconds) of beverages (1.5 mL) spiked with a mix of drugs at 1 μg mL−1. No agitation of the sample was performed. A quick (3 s) rinse of the TFME device by immersion in LC-MS grade water was performed prior to thermal desorption and DBDI-MS analysis. Each extracted ion chromatogram was integrated for 0.5 min. | ||
An extraction time of 5 min, together with the accelerated extraction kinetics obtained with the ultrasound-assisted extraction, was sufficient for the quantification of the considered drugs at ultra-trace levels. Calibration curves (1/x weighted) for the drugs in vodka, urine and blood plasma are reported in Table 1.
| Compound | I S | Equation | LDR, pg mL−1 | R 2 | LOD, pg mL−1 | |
|---|---|---|---|---|---|---|
| Vodka | Cocaine | Cocaine-d3 | y = 1.11 × 10−3x − 6.93 × 10−2 | 30–30k | 0.9961 | 10 |
| Diazepam | Diazepam-d5 | y = 1.12 × 10−3x − 1.64 × 10−1 | 100–30k | 0.9923 | 30 | |
| Desipramine | Fentanyl-d5 | y = 3.49 × 10−4x − 1.07 × 10−2 | 100–30k | 0.9978 | 30 | |
| Imipramine | Diazepam-d5 | y = 1.26 × 10−2x + 3.72 × 10−1 | 100–30k | 0.9950 | 10 | |
| MDMA | MDMA-d5 | y = 1.07 × 10−3x − 2.06 × 10−2 | 100–30k | 0.9929 | 10 | |
| MDEA | MDEA-d5 | y = 9.05 × 10−4x + 4.00 × 10−3 | 100–30k | 0.9998 | 10 | |
| Methadone | Methadone-d3 | y = 9.27 × 10−4x − 1.26 × 10−2 | 30–30k | 0.9992 | 10 | |
| Fentanyl | Fentanyl-d5 | y = 8.59 × 10−4x − 1.39 × 10−2 | 100–30k | 0.9994 | 30 | |
| Urine | Cocaine | Cocaine-d3 | y = 1.18 × 10−3x − 3.58 × 10−2 | 30–10k | 0.9978 | 30 |
| Diazepam | Diazepam-d5 | y = 9.79 × 10−4x − 1.45 × 10−3 | 10–10k | 0.9982 | 3 | |
| Desipramine | Cocaine-d3 | y = 1.04 × 10−4x − 1.02 × 10−2 | 100–10k | 0.9931 | 100 | |
| Imipramine | Cocaine-d3 | y = 2.75 × 10−3x − 6.67 × 10−3 | 10–10k | 0.9998 | 3 | |
| MDMA | Diazepam-d5 | y = 1.23 × 10−6x + 1.99 × 10−5 | 100–10k | 0.9882 | 30 | |
| MDEA | Diazepam-d5 | y = 5.21 × 10−6x − 3.01 × 10−4 | 300–10k | 0.9982 | 30 | |
| Methadone | Methadone-d3 | y = 9.83 × 10−4x − 7.44 × 10−3 | 10–10k | 0.9982 | 3 | |
| Fentanyl | Fentanyl-d5 | y = 1.05 × 10−3x − 8.66 × 10−3 | 10–10k | 0.9985 | 10 | |
| Blood plasma | Cocaine | Cocaine-d3 | y = 1.18 × 10−3x − 7.45 × 10−2 | 100–30k | 0.9996 | 100 |
| Diazepam | Diazepam-d5 | y = 1.17 × 10−3x − 1.69 × 10−1 | 300–30k | 0.9961 | 300 | |
| Desipramine | Fentanyl-d5 | y = 1.89 × 10−4x − 5.43 × 10−2 | 300–30k | 0.9986 | 300 | |
| Imipramine | Methadone-d3 | y = 2.07 × 10−4x − 2.17 × 10−2 | 300–30k | 0.9993 | 100 | |
| MDMA | MDMA-d5 | y = 1.36 × 10−3x + 5.78 × 10−1 | 300–30k | 0.9993 | 300 | |
| MDEA | MDEA-d5 | y = 1.27 × 10−3x − 9.79 × 10−3 | 100–30k | 0.9996 | 100 | |
| Methadone | Methadone-d3 | y = 1.07 × 10−3x − 2.98 × 10−2 | 30–30k | 0.9998 | 30 | |
| Fentanyl | Fentanyl-d5 | y = 1.18 × 10−3x − 1.39 × 10−1 | 300–30k | 0.9997 | 300 |
The results obtained by the TFME-DBDI setup are even more rewarding when compared to what can be obtained by a conventional TDU-GC/MS system with the same extraction conditions. In general, when calibration was attempted in urine, cola, wine, and vodka, using the same extraction conditions, only in a few cases (Table 2) were consistent results obtained. In all cases, LOQs were above 10 ng mL−1. Moreover, compounds like MDMA and MDEA cannot be analyzed by gas-chromatography without a derivatization step, which would add extra effort to the analytical workflow.
| Compound | Equation | LDR, ng mL−1 | R 2 | LOQ, ng mL−1 | |
|---|---|---|---|---|---|
| Cola | Cocaine | — | — | — | — |
| Diazepam | y = 0.0106x − 0.0368 | 10–1000 | 0.9993 | 10 | |
| Desipramine | — | — | — | — | |
| Imipramine | y = 0.00156x − 0.0974 | 100–1000 | 0.9923 | 100 | |
| MDMA | — | — | — | — | |
| MDEA | — | — | — | — | |
| Methadone | — | — | — | — | |
| Fentanyl | — | — | — | — | |
| Urine | Cocaine | y = 0.0103x + 0.0127 | 10–500 | 0.9998 | 10 |
| Diazepam | y = 0.0003x − 0.0324 | 10–1000 | 0.9993 | 10 | |
| Desipramine | — | — | — | — | |
| Imipramine | y = 0.0198x − 0.444 | 10–1000 | 0.9974 | 10 | |
| MDMA | — | — | — | — | |
| MDEA | — | — | — | — | |
| Methadone | y = 0.0113x − 0.444 | 10–1000 | 0.9912 | 10 | |
| Fentanyl | y = 0.0106x − 0.0368 | 10–1000 | 0.9993 | 10 |
The reported direct coupling of TFME with DBDI showed good compatibility with the sample matrices investigated. In particular, the reproducibility data reported in Table 3 show an average intra-day RSD of 6% both for 1 ng mL−1 and 10 ng mL−1 samples, with all values below 21%.
| 1 ng mL−1 | 10 ng mL−1 | ||
|---|---|---|---|
| Vodka | Cocaine | 2.0 | 7.1 |
| Diazepam | 1.9 | 6.6 | |
| Desipramine | 9.7 | 13 | |
| Imipramine | 3.5 | 4.8 | |
| MDMA | 7.4 | 7.2 | |
| MDEA | 3.8 | 8.6 | |
| Methadone | 0.1 | 8.1 | |
| Fentanyl | 9.7 | 10 | |
| Urine | Cocaine | 7.6 | 2.1 |
| Diazepam | 2.9 | 1.8 | |
| Desipramine | 5.7 | 8.3 | |
| Imipramine | 14 | 13 | |
| MDMA | 21 | 14 | |
| MDEA | 17 | 9.8 | |
| Methadone | 3.6 | 1.9 | |
| Fentanyl | 7.1 | 2.4 | |
| Blood plasma | Cocaine | 1.0 | 1.2 |
| Diazepam | 5.7 | 2.3 | |
| Desipramine | 11 | 9.8 | |
| Imipramine | 4.9 | 8.5 | |
| MDMA | 3.6 | 1.8 | |
| MDEA | 1.8 | 1.9 | |
| Methadone | 1.4 | 1.1 | |
| Fentanyl | 1.4 | 2.8 |
Compared to other direct approaches where no sample pretreatment is performed,18,19,26 the use of TFME as a fast and simple technique for liquid sample cleanup/up-concentration is very practical, because it minimizes the amount of interfering compounds ionized together with the analytes of interest (with possible suppression effects) and results in lower LODs due to the pre-concentration provided by the thin film. Although waiving the sample preparation completely would reduce the analysis time to just a few seconds per sample, it appears necessary to perform at least a simple sample pretreatment in order to be able to analyze complex matrices.
The method reported in this work can be fairly directly compared with other SPME-MS direct coupling strategies.27 For example, a method based on the thermal desorption of the analyte prior or during ionization using a DART source was used by Vasiljevic et al. for the analysis of drugs of abuse in oral fluids and urine.28 A sensitivity in the ng mL−1 range was achieved by extracting samples with a coated plastic mesh SPME device, analyzed in transmission mode. In this study, the LODs in urine for cocaine, diazepam, MDMA, methadone and fentany were 0.1, 0.5, 2.5, 1 and 0.5 ng mL−1, respectively. The values reported in this study for the same analytes are, 0.03, 0.003, 0.03, 0.003 and 0.01 ng mL−1, respectively, i.e., more than one order of magnitude lower. Evans-Nguyen et al. also reported the analysis of benzodiazepines in human plasma by gold-wire SPME fibers,29 obtaining a sensitivity in the sub-ppb range.
Solvent-based SPME desorption methods were also successfully used. These methods are generally less comparable to the one described here, because of the different ionization mechanisms and sample desorption. Gómez-Ríos et al. reported the analysis of drugs of abuse in human urine and plasma at the sub-ppb concentration level by using an open port probe (OPP) ionization source.30 Tascon et al. quantified immunosuppressants in whole blood at the sub-ppb level with a microfluidic open interface (MOI) ionization source.31 Coated blade spray32 (CBS) was also used for the quantification od drugs of abuse in human urine and plasma. These techniques, namely OPP, MOI, and CBS, represent a viable alternative to DBDI when an ESI-like ionization is preferred. The use of the abovementioned approaches can be advantageous when only a minor modification of an ESI source is desired (this is the case for OPP or MOI). CBS, although being a more flexible technique, still poses some challenges in terms of automation. The same applies to nano electrospray-based direct SPME-MS couplings,33 where the amount of manual operation still strongly limits the analytical throughput.
Matrix effects in TFME-DBDI
As always when dealing with complex samples, matrix effects can arise and compromise the analytical results. In our case, the extraction of drugs from alcoholic beverages was dependent on the ethanol concentration in the beverage itself. This is obvious from Fig. 5, and is a well-known phenomenon due to the competition between the organic solvent and the thin film extraction phase for the partitioning of the drugs. Interestingly, a small amount of alcohol increased the extraction efficiency for some of the drugs (Fig. 5), probably due to a faster extraction kinetics. For the other two beverages (wine and coke) the extraction efficiency was also affected by interfering compounds, i.e., mainly sugars in coke and polyphenolic compounds in wine. The presence of interfering compounds not only affects the extraction, but it can also cause suppression effects inside the ionization source, due to competition phenomena.The strongest matrix effects were observed in blood plasma, and to a lesser extent, in urine (Fig. 7). In fact, both of these matrices contain proteins, hormones, metabolites and salts. Some of their constituents can bind some of the analytes, reducing the effective amount that the thin film can extract (via free concentration).
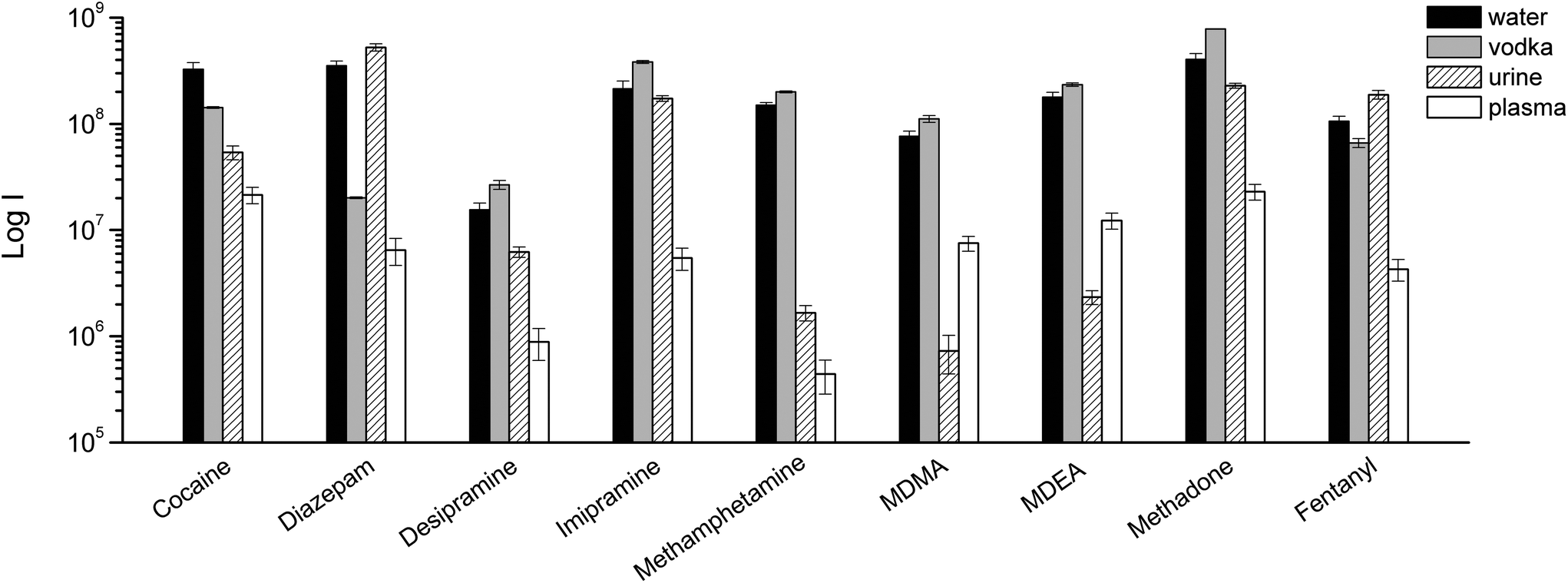 | ||
| Fig. 7 TFME-DBDI-MS ion signals for different drugs at 1 ng mL−1 extracted from vodka, urine, and plasma (1.5 mL, 5 min extraction, n = 4). | ||
Despite the presence of almost 40% ethanol, vodka showed the lowest matrix effect when compared to urine and plasma. For plasma and urine, which are more complex matrices and contain a greater variety of interfering compounds, ionization suppression effects were observed in addition to competition effects during extraction, as indicated by the higher level of MS signal background from blank extractions. The different response obtained for urine and blood samples also depends on the highest degree of binding of the analytes to matrix components.
Conclusions
We present the development of a direct interface between thin film microextraction (TFME) and dielectric barrier discharge ionization (DBDI)-mass spectrometry. Eight drugs were extracted from biological fluids (urine, blood plasma) and beverages (vodka, cola, and wine) using fast extractions, from 5 min to 5 s. Ultrasound-assisted extraction was also employed to accelerate the extraction kinetics. The proposed method was shown to have enhanced performances when compared to a conventional TDU-GC-EI-MS approach, with limits of detection between 3 and 100 pg mL−1 in urine, 10 and 30 pg mL−1 in vodka, and 30 and 300 pg mL−1 in blood plasma. These values are below the range of concentrations investigated in cases of drug intoxication or therapeutic drug monitoring, which are usually in the ng mL−1 range.34 The high sensitivity of the method is particularly helpful when trying to identify illicit drugs in urine or blood plasma, even several days following their consumption, when most of them are already metabolized by the body.The approach was found to be very robust, with an average intra-day RSD of 6% both for 1 ng mL−1 and 10 ng mL−1 samples, with all values below 21%. The figures of merit were obtained with an extraction time of only 5 minutes, although currently with manual handling of the TFME during extraction and desorption. Therefore, room for further improvements in terms of throughput and reproducibility is possible with automation of the analytical workflow. This would also enable the proposed approach to be used in many analytical laboratories, for research and routine measurements.
As future perspectives, the bio-compatible nature of the thin films could also allow for direct sampling of saliva or sweat in a non-invasive way, simplifying sample collection procedures and allowing for more rapid results to be obtained. This might, for example, be helpful for anti-doping investigations, where speed is extremely important to rapidly assess the compliance of the athlete to regulations. In such cases, the extraction time can even be reduced to below 1 minute, allowing for on-site/real time sampling.
Notes
The original data used in this publication are made available in a curated data archive at ETH Zurich (https://www.researchcollection.ethz.ch) under the DOI: 10.3929/ethz-b-000329059.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Novartis AG (Dr Juan Zhang) for the generous donation of the high resolution Orbitrap-MS system used within this study. They also thank Christian Marro from the technical workshop of ETH for the valuable support and the construction of the experimental setup. The authors are grateful to Dominika Gruszecka from the University of Waterloo for her assistance with experiments and data processing.References
- H. Piri-Moghadam, E. Gionfriddo, A. Rodriguez-Lafuente, J. J. Grandy, H. L. Lord, T. Obal and J. Pawliszyn, Anal. Chim. Acta, 2017, 964, 74–84 CrossRef CAS PubMed.
- J. J. Grandy, E. Boyacı and J. Pawliszyn, Anal. Chem., 2016, 88, 1760–1767 CrossRef CAS PubMed.
- R. Jiang, E. Cudjoe, B. Bojko, T. Abaffy and J. Pawliszyn, Anal. Chim. Acta, 2013, 804, 111–119 CrossRef CAS PubMed.
- R. Jiang and J. Pawliszyn, Anal. Chem., 2014, 86, 403–410 CrossRef CAS PubMed.
- E. Boyacı, K. Goryński, C. R. Viteri and J. Pawliszyn, J. Chromatogr. A, 2016, 1436, 51–58 CrossRef PubMed.
- X. Li, W. Ma, H. Li, W. Ai, Y. Bai and H. Liu, Anal. Bioanal. Chem., 2017, 1–10 Search PubMed.
- R. Javanshad and A. R. Venter, Anal. Methods, 2017, 9, 4896–4907 RSC.
- M. F. Mirabelli, J.-C. Wolf and R. Zenobi, Anal. Chem., 2016, 88, 7252–7258 CrossRef CAS PubMed.
- M. F. Mirabelli, E. Gionfriddo, J. Pawliszyn and R. Zenobi, Analyst, 2018, 143, 891–899 RSC.
- A. K. Huba, M. F. Mirabelli and R. Zenobi, Anal. Chim. Acta, 2018, 1030, 125–132 CrossRef CAS PubMed.
- M. F. Mirabelli and R. Zenobi, Anal. Chem., 2018, 90, 5015–5022 CrossRef CAS PubMed.
- C. C. Guo, F. Tang, J. Chen, X. Wang, S. Zhang and X. Zhang, Anal. Bioanal. Chem., 2015, 407, 2345–2364 CrossRef CAS PubMed.
- C. Meyer, S. Müller, E. L. Gurevich and J. Franzke, Analyst, 2011, 136, 2427–2440 RSC.
- J. D. Harper, N. A. Charipar, C. C. Mulligan, X. Zhang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2008, 80, 9097–9104 CrossRef CAS PubMed.
- U. Kogelschatz, Plasma Chem. Plasma Process., 2003, 23, 1–46 CrossRef CAS.
- S. Martínez-Jarquín and R. Winkler, TrAC, Trends Anal. Chem., 2017, 89, 133–145 CrossRef.
- M. Benassi, A. Berisha, W. Romão, E. Babayev, A. Römpp and B. Spengler, Rapid Commun. Mass Spectrom., 2013, 27, 825–834 CrossRef CAS PubMed.
- A. U. Jackson, J. F. Garcia-Reyes, J. D. Harper, J. S. Wiley, A. Molina-Díaz, Z. Ouyang and R. Graham Cooks, Analyst, 2010, 135, 927 Search PubMed.
- M. Smoluch, M. Babij, D. Zuba, G. Schroeder, T. Gotszalk and J. Silberring, Int. J. Mass Spectrom., 2015, 386, 32–36 CrossRef CAS.
- H. Hayen, A. Michels and J. Franzke, Anal. Chem., 2009, 81, 10239–10245 CrossRef CAS PubMed.
- B. Gilbert-López, H. Geltenpoth, C. Meyer, A. Michels, H. Hayen, A. Molina-Díaz, J. F. García-Reyes and J. Franzke, Rapid Commun. Mass Spectrom., 2013, 27, 419–429 CrossRef PubMed.
- B. Gilbert-López, J. F. García-Reyes, C. Meyer, A. Michels, J. Franzke, A. Molina-Díaz and H. Hayen, Analyst, 2012, 137, 5403–5410 RSC.
- M. F. Mirabelli, J.-C. Wolf and R. Zenobi, Anal. Bioanal. Chem., 2016, 408, 3425–3434 CrossRef CAS.
- M. F. Mirabelli, J.-C. Wolf and R. Zenobi, Analyst, 2017, 142, 1909–1915 RSC.
- J. Pawliszyn, Handbook of Solid Phase Microextraction, Elsevier, Waltham, 2012 Search PubMed.
- L. C. Chen, Y. Hashimoto, H. Furuya, K. Takekawa, T. Kubota and K. Hiraoka, Rapid Commun. Mass Spectrom., 2009, 23, 333–339 CrossRef CAS PubMed.
- G. A. Gómez-Ríos and M. F. Mirabelli, TrAC, Trends Anal. Chem., 2019, 112, 201–211 CrossRef.
- T. Vasiljevic, G. A. Gómez-Ríos and J. Pawliszyn, Anal. Chem., 2018, 90, 952–960 CrossRef CAS PubMed.
- K. M. Evans-Nguyen, T. L. Hargraves and A. N. Quinto, Anal. Methods, 2017, 9, 4954–4957 RSC.
- G. A. Gómez-Ríos, C. Liu, M. Tascon, N. Reyes-Garcés, D. W. Arnold, T. R. Covey and J. Pawliszyn, Anal. Chem., 2017, 89, 3805–3809 CrossRef PubMed.
- M. Tascon, M. N. Alam, G. A. Gómez-Ríos and J. Pawliszyn, Anal. Chem., 2018, 90, 2631–2638 CrossRef CAS PubMed.
- M. Tascon, G. A. Gómez-Ríos, N. Reyes-Garcés, J. Poole, E. Boyacl and J. Pawliszyn, Anal. Chem., 2017, 89, 8421–8428 CrossRef CAS PubMed.
- G. A. Gómez-Ríos, N. Reyes-Garcés, B. Bojko and J. Pawliszyn, Anal. Chem., 2016, 88, 1259–1265 CrossRef PubMed.
- Clinical Laboratory Reference, Cutoff and Toxicity Levels for Drugs-of-Abuse Testing http://www.clr-online.com/CLR2017-13_Table-of-Cutoff-Toxicity-DOA.pdf.
Footnotes |
| † These authors contributed equally. |
| ‡ Current address: Department of Chemistry and Biochemistry, The University of Toledo, Toledo, OH, USA. |
| This journal is © The Royal Society of Chemistry 2019 |