
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free alkene carbooxygenation following tandem intramolecular alkoxylation/Claisen rearrangement: stereocontrolled access to bridged [4.2.1] lactones†
Long
Li
a,
Xin-Qi
Zhu
a,
Ying-Qi
Zhang
a,
Hao-Zhen
Bu
a,
Peng
Yuan
a,
Jinyu
Chen
b,
Jingyi
Su
b,
Xianming
Deng
b and
Long-Wu
Ye
 *acd
*acd
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory for Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: longwuye@xmu.edu.cn
bState Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, Fujian 361102, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
dState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 24th January 2019
Abstract
Alkene carbooxygenation has attracted considerable attention over the past few decades as this approach provides an efficient access to various oxygen-containing molecules, especially the valuable O-heterocycles. However, examples of catalytic alkene carbooxygenation via a direct C–O cleavage are quite scarce, and the C–O cleavage in these cases is invariably initiated by transition metal-catalyzed oxidative addition. We report here a novel Brønsted acid-catalyzed intramolecular alkoxylation-initiated tandem sequence, which represents the first metal-free intramolecular alkoxylation/Claisen rearrangement. Significantly, an unprecedented Brønsted acid-catalyzed intramolecular alkene insertion into the C–O bond via a carbocation pathway was discovered. This method allows the stereocontrolled synthesis of valuable indole-fused bridged [4.2.1] lactones, providing ready access to biologically relevant scaffolds in a single synthetic step from an acyclic precursor. Moreover, such an asymmetric cascade cyclization has also been realized by employing a traceless chiral directing group. Control experiments favor the feasibility of a carbocation pathway for the process. In addition, biological tests showed that some of these newly synthesized indole-fused lactones exhibited their bioactivity as antitumor agents against different breast cancer cells, melanoma cells, and esophageal cancer cells.
Introduction
Bridged [4.2.1] lactones are widely distributed heterocycles found in various natural products such as hushinone and citrinovirin, and bioactive compounds (Fig. 1).1 However, such bicyclic frameworks bearing both a medium ring and bridged unit are regarded as difficult skeletons to construct due to entropic effects and the ring strain factor,2 and very few methods have been reported to date.1,3 Hence, novel and stereocontrolled synthesis of bridged [4.2.1] lactone motifs allowing structurally diverse modification is in great demand in both organic and medicinal chemistry.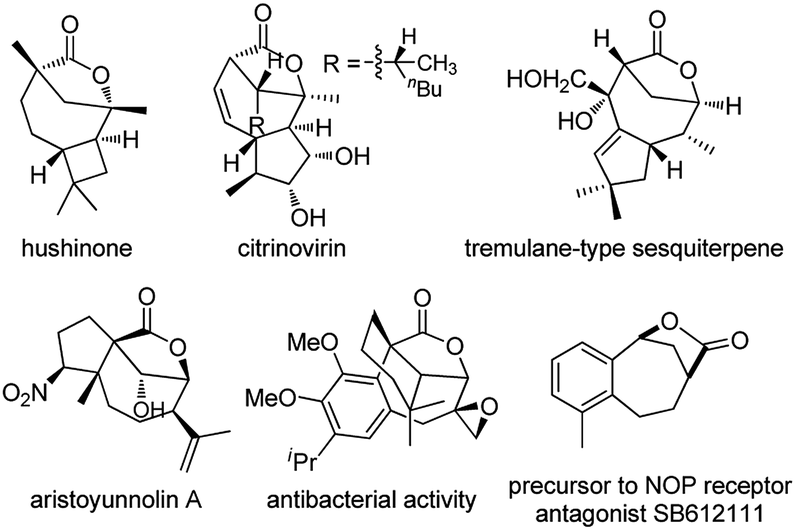 | ||
| Fig. 1 Selected bioactive molecules containing bridged [4.2.1] lactones. | ||
Difunctionalization of unactivated olefins in a single operation is one of the most valuable transformations in organic chemistry.4 Among them, alkene carbooxygenation is particularly attractive as this approach provides an efficient access to various oxygen-containing molecules, especially the valuable O-heterocycles, and various synthetic methods have been developed.4 However, examples of catalytic alkene carbooxygenation via a direct C–O cleavage are quite scarce, and the C–O cleavage in these cases is invariably initiated by transition metal-catalyzed oxidative addition (Scheme 1a).5,6 For example, Douglas et al. reported an elegant protocol for the rhodium-catalyzed intramolecular alkene oxyacylation reaction via an acyl C–O bond activation.6a,b In 2012, Nakao et al. disclosed an intramolecular oxycyanation of alkenes by palladium/BPh3 catalysis.6c Thus, the development of an alternative approach for the catalytic cleavage of the C–O bond and the addition reaction to the alkenes is highly desirable.
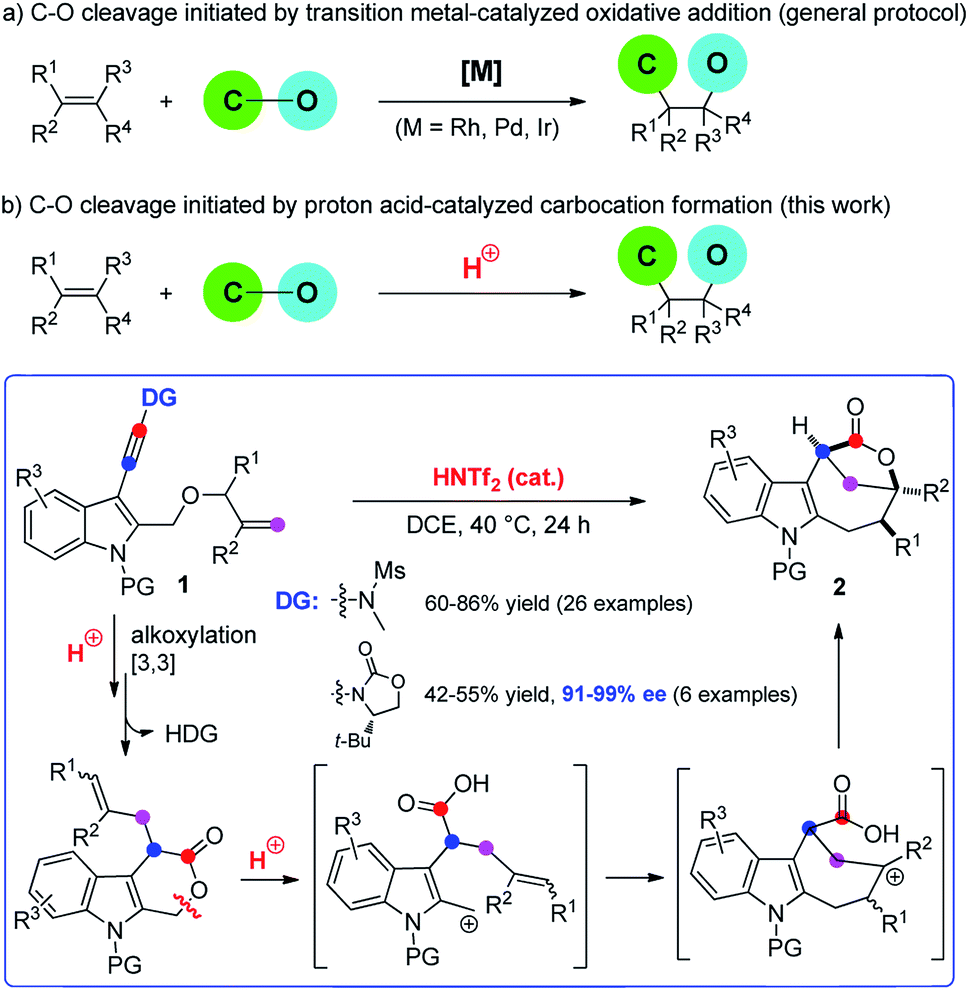 | ||
| Scheme 1 Catalytic alkene carbooxygenation via a C–O cleavage. | ||
Because of their high bond-forming efficiency and atom economy, catalytic tandem intramolecular alkoxylation/Claisen rearrangements have received significant attention.7 In particular, such a cascade cyclization of alkynyl allyl ethers could lead to the formation of various valuable O-heterocycles, which is also established by Hashmi, Liu, Gagosz, Miyata, and others (Scheme 1a).8–10 While these achievements are impressive, almost all of these approaches rely on the use of noble metals such as gold and platinum as catalysts. In our recent study on the catalytic tandem reactions of ynamides for heterocycle synthesis,11,12 we realized the first metal-free intramolecular alkoxylation/Claisen rearrangement of indole-linked ynamide-allyl ethers (Scheme 1b). Interestingly, the resulting six-membered lactone intermediate further underwent an unprecedented Brønsted acid-catalyzed intramolecular carbooxygenation of olefins by C–O bond cleavage involving ring opening, carbocation rearrangement, and then ring closing. This Brønsted acid catalysis led to the highly efficient and stereocontrolled formation of valuable indole-fused bridged [4.2.1] lactones. Furthermore, such an asymmetric cascade cyclization was also realized by employing a traceless chiral directing group. The mechanistic rationale for this cascade reaction is strongly supported by a variety of control experiments. In this paper, we wish to report the results of our detailed investigations of this novel cascade cyclization, including the substrate scope, synthetic applications, biological tests and mechanistic studies.
Results and discussion
Inspired by our previous work on the indolyl ynamide chemistry,12d–f we chose an indole-tethered ynamide 1a as the model substrate for the initial study. As shown in Table 1, indole-fused lactone 2aa was obtained in the presence of most non-noble metals (Table 1, entries 1–4), with Cu(OTf)2 giving the best yield of the desired 2aa (Table 1, entry 4). Different from Hashmi's protocol,8bN-methyl methanesulfonamide here not only serves as the directing group to achieve regioselective attack at the N-terminus of alkyne, but also can be removed spontaneously and regarded as a traceless directing group. Surprisingly, indole-fused bridged [4.2.1] lactone 2a was detected as the main product by employing In(OTf)3 or Fe(OTf)3 as catalysts (Table 1, entries 5–6). Of note, typical gold catalysts, such as Ph3PAuNTf2 and IPrAuNTf2, were not effective in promoting this reaction and the decomposition of 1a was observed in these cases (Table 1, entries 7–8). Various Brønsted acids were also evaluated, but typical organic acids (e.g., TFA, MsOH, and TsOH) were not capable of catalyzing the reaction.13 Gratifyingly, HOTf and HNTf2 (Table 1, entries 9–10) could effectively catalyze this cascade cyclization,14 and the bridged lactone 2a was obtained in 81% yield in the latter case (Table 1, entry 10). The reaction proved to be less efficient when it was performed at 60 °C (Table 1, entry 11) or in other solvents.13 The observed excellent efficiency with HNTf2 as the catalyst can be explained by its high Brønsted acidity combined with the low nucleophilicity of its counterion.15,16|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Reaction conditions | Yieldb (%) | ||
| 2a | 2aa | 1a | |||
| a Reaction conditions: 1a (0.1 mmol), catalyst (0.02 mmol), DCE (2 mL), 40–60 °C, in vials. b Measured by 1H NMR using diethyl phthalate as the internal standard. c 5 mol% of catalyst was used. | |||||
| 1 | Y(OTf)3 | DCE, 40 °C, 48 h | <1 | 20 | 75 |
| 2 | Yb(OTf)3 | DCE, 40 °C, 48 h | <1 | 50 | 30 |
| 3 | Zn(OTf)2 | DCE, 40 °C, 48 h | <1 | 70 | 25 |
| 4 | Cu(OTf)2 | DCE, 40 °C, 24 h | <1 | 84 | <1 |
| 5 | In(OTf)3 | DCE, 40 °C, 48 h | 63 | 15 | <1 |
| 6 | Fe(OTf)3 | DCE, 40 °C, 48 h | 74 | <5 | <1 |
| 7c | Ph3PAuNTf2 | DCE, rt, 10 h | <1 | <1 | <1 |
| 8c | IPrAuNTf2 | DCE, rt, 10 h | <1 | <1 | <1 |
| 9 | HOTf | DCE, 40 °C, 24 h | 72 | <1 | <1 |
| 10 | HNTf 2 | DCE, 40 °C, 24 h | 81 | <1 | <1 |
| 11 | HNTf2 | DCE, 60 °C, 18 h | 67 | <1 | <1 |

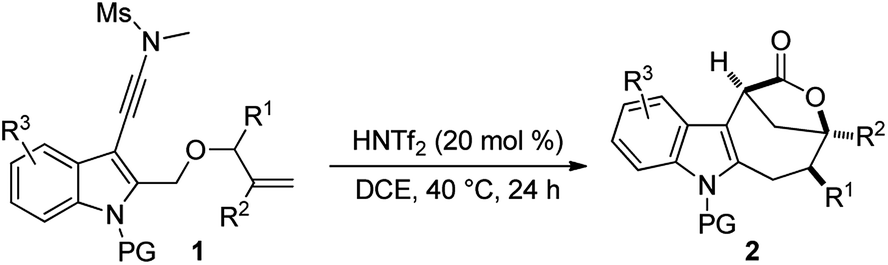
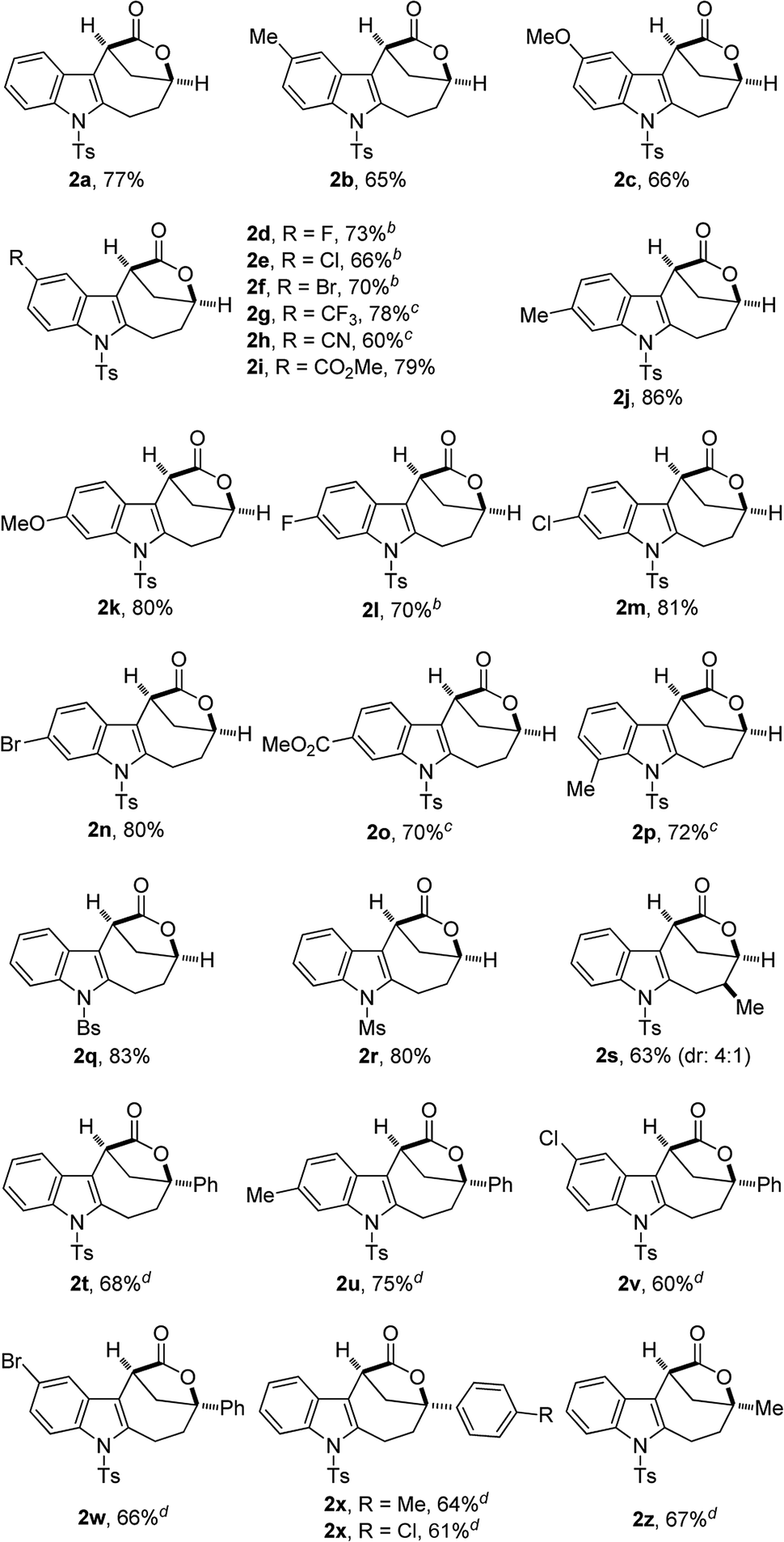
With the optimal reaction conditions in hand (Table 1, entry 10), the scope of this novel tandem reaction was explored (Table 2). This metal-free cascade cyclization proceeded efficiently to furnish a series of indole-fused bridged [4.2.1] lactones in mostly good to excellent yields. For instance, various indolyl-substituted ynamides bearing both electron-donating and -withdrawing groups could be readily converted into the desired indole-fused bicyclic skeletons 2a–2p with yields ranging from 60% to 86%. In particular, the functional groups, such as CF3, CN and CO2Me, were well tolerated under this Brønsted acid catalysis. Further investigation of N-protecting groups demonstrated that the Bs- and Ms-protected substrates 1q–1r gave slightly improved yields. In addition, an ynamide with a methyl group (R1 = Me) was also a suitable substrate for this tandem reaction to afford the corresponding 2s in 63% yield (dr: 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). This result clearly indicated that [3,3] rearrangement, but not [1,3] rearrangement,17 was presumably involved in this multiple cascade sequence.13 Furthermore, the reaction also occurred smoothly with various aryl- or methyl-substituted ynamides (R2 = aryl, Me), and the desired 2t–2z containing a quaternary carbon center could be formed in 60–75% yields by employing 30 mol% of HNTf2 as the catalyst. Importantly, excellent diastereoselectivity (>20:1) was achieved in all cases except for the substrate 1s. The molecular structures of 2a, 2s and 2u were confirmed by X-ray diffraction.18 Thus, this metal-free protocol provides a highly convenient and practical route for the preparation of valuable bridged [4.2.1] lactones.
:1). This result clearly indicated that [3,3] rearrangement, but not [1,3] rearrangement,17 was presumably involved in this multiple cascade sequence.13 Furthermore, the reaction also occurred smoothly with various aryl- or methyl-substituted ynamides (R2 = aryl, Me), and the desired 2t–2z containing a quaternary carbon center could be formed in 60–75% yields by employing 30 mol% of HNTf2 as the catalyst. Importantly, excellent diastereoselectivity (>20:1) was achieved in all cases except for the substrate 1s. The molecular structures of 2a, 2s and 2u were confirmed by X-ray diffraction.18 Thus, this metal-free protocol provides a highly convenient and practical route for the preparation of valuable bridged [4.2.1] lactones.
In addition, this multiple cascade reaction was also applicable to other electron-rich aromatic ring-substituted ynamides such as benzofuran-, pyrrole-, and alkoxy arene-tethered ynamides 3a–3c, delivering the desired bridged [4.2.1] lactones 4a–4c in serviceable yields with excellent dr values (>20:1), as depicted in eqn (1)–(3). Of note, the use of HFIP as an additive led to a significantly improved yield in the case of 3c (eqn (3)).19 Attempts to extend the reaction to non-terminal alkene-substituted ynamide 3d, indole-linked ynamide-allyl amine 3e and sulfide 3f led to the formation of complicated mixtures.

Although our attempts to employ various chiral Brønsted acids such as chiral phosphoric acids and chiral phosphoric amides to catalyze this cascade cyclization failed probably due to the fact that their acidity is not strong enough, the chiral bridged [4.2.1] lactones could be synthesized by employing a chiral oxazolidinone instead of N-methyl methanesulfonamide as the directing group.20 As shown in eqn (4), it was found that the tert-butyl substituted oxazolidinone-derived chiral ynamide 5d gave the best enantioselectivity, and the desired chiral bridged [4.2.1] lactone 2a-ent was formed in 55% yield with 95% ee in the presence of 30 mol% of HNTf2 as the catalyst and 1.5 equiv. of water as the additive. Thus, the chiral auxiliary can be regarded as a traceless directing group to introduce the chirality in a very facile manner. It should be mentioned that significant racemization was observed when prolonging the reaction time.13
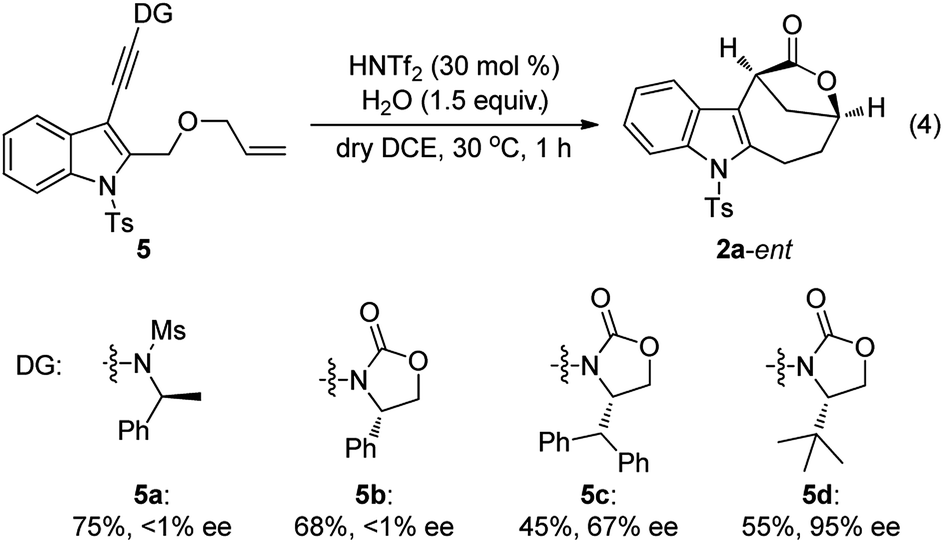
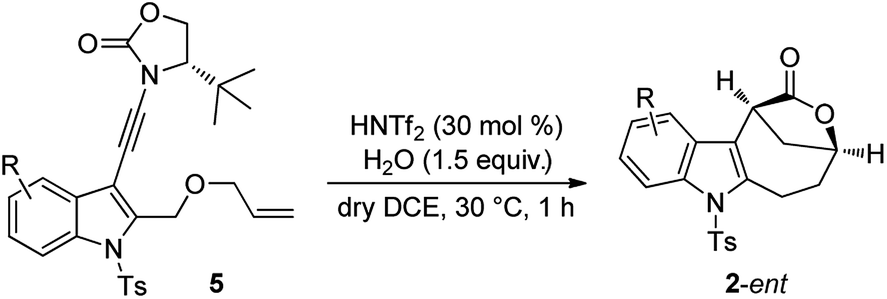
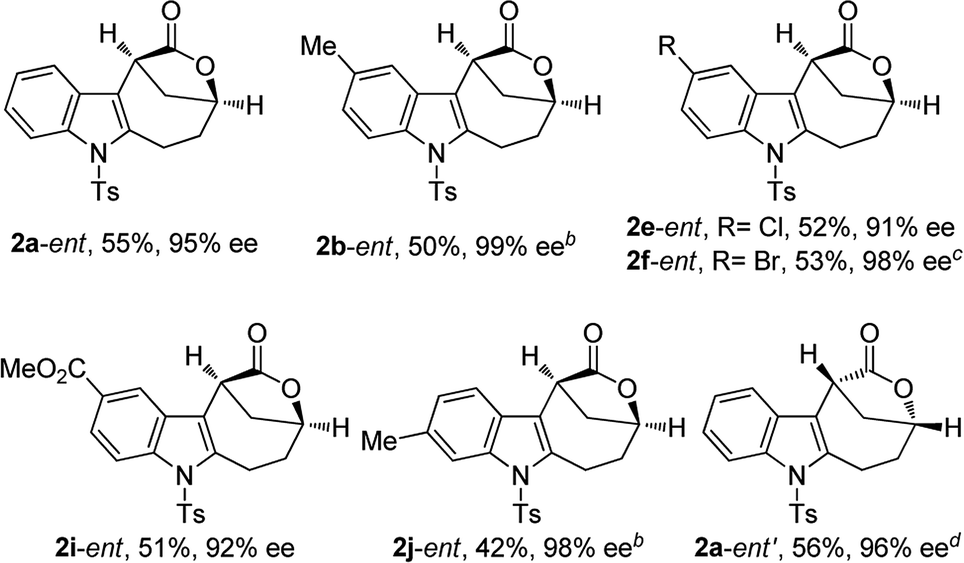
The scope of this asymmetric intramolecular alkoxylation-initiated tandem reaction was further examined by using tert-butyl substituted oxazolidinone-derived chiral ynamides. As depicted in Table 3, the reaction proceeded smoothly with various chiral ynamides 5, allowing the facile synthesis of the corresponding enantioenriched bridged [4.2.1] lactones 2-ent in serviceable yields with excellent ee values (91–99% ee) and excellent dr values (>20:1). Of note, (R)-tert-butyl substituted oxazolidinone-derived chiral ynamide 5d′ could also undergo smooth cascade cyclization to deliver the desired 2a-ent′ with the opposite enantioselectivity.
The potential synthetic utility of this protocol was then demonstrated by the facile diversification of bridged lactone 2a, and importantly, excellent diastereoselectivity was achieved in all cases (Scheme 2). For example, the Ts group in lactone 2a, prepared on a gram scale in 72% yield, was easily removed by treatment with TBAF to afford the corresponding 2ab in 75% yield. In addition, the lactone part of 2a could be selectively reduced to furnish the hemiacetal 2ac (91%, dr > 10:1) with DIBAL-H. By contrast, the use of Et3SiH led to the total reduction of the lactone to produce the desired 2ad in almost quantitative yield. Interestingly, 2a could also be oxidized with DDQ to deliver 2ae in 95% yield. Moreover, the ring of lactone 2a was readily opened by employing PhLi, leading to the stereocontrolled construction of cyclohepta[b]indole scaffold 2af, frequently occurring in natural products and bioactive molecules.21 Finally, the chiral 2a-ent could be further transformed into the indole-fused cycloheptanone 2ah with the ee maintained, and its structure was confirmed by X-ray diffraction,18 which also determined the absolute configuration of 2a-ent.22
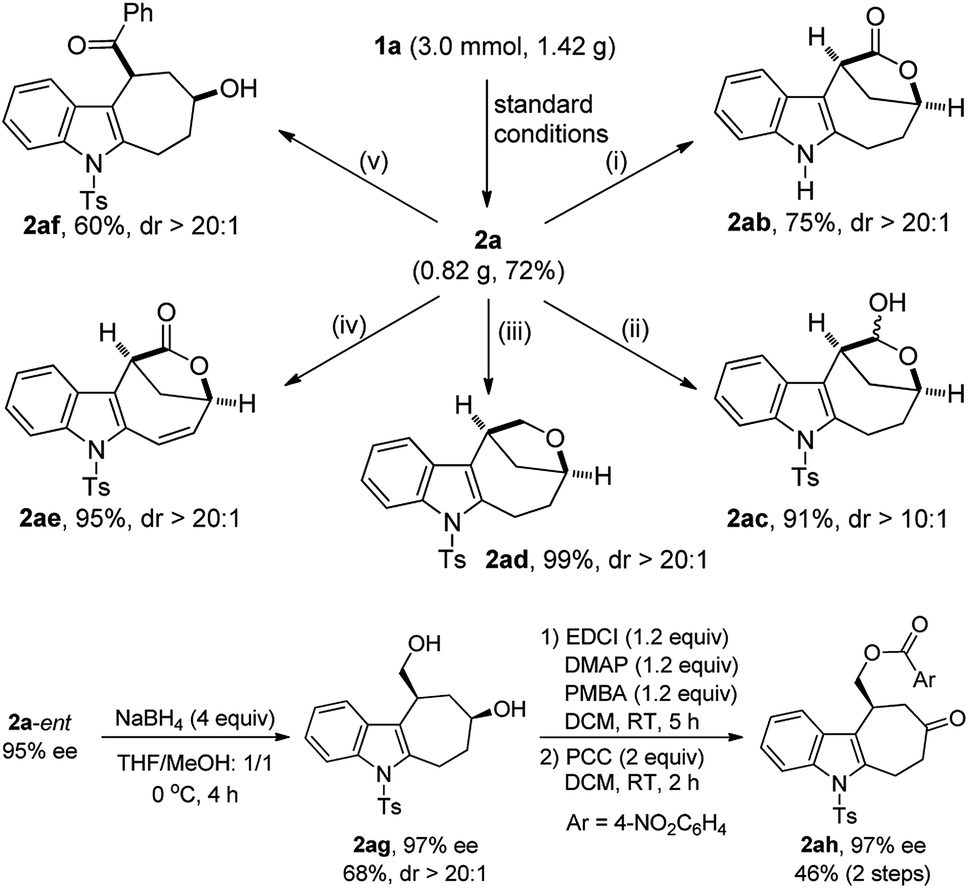 | ||
| Scheme 2 Gram scale reaction and synthetic applications. Reagents and conditions: (i) TBAF (4 equiv.), THF, 65 °C, 6 h; (ii) DIBAL-H (1.5 equiv.), THF, −78 °C, 6 h; (iii) InBr3 (0.5 equiv.), Et3SiH (2.5 equiv.), CHCl3, 60 °C, 10 h; (iv) DDQ (3 equiv.), DCE, 60 °C, 48 h; (v) PhLi (1.2 equiv.), THF, −40 °C, 2 h; −20 °C, 10 h. | ||
Considering the bioactivity reported in the literature for bridged [4.2.1] lactone systems,1 we also tested the above synthesized indole-fused lactones for their biological activity as antitumor agents. The cytotoxic effects of these compounds were evaluated against a panel of cancer cells, including breast cancer cells MDA-MB-231 and MCF-7, melanoma cells A375, and esophageal cancer cells SK-GT-4 and KYSE-450 using cell viability assay.13 The results revealed that these compounds exhibited differential cytotoxicity and selectivity. Compounds 2k, 2l, and 2o selectively inhibited the cell growth of A375 by more than 50% at a concentration of 20 μM, and compound 2af inhibited the cell growth of MCF7 by around 70%. While compound 2ae showed broad activity with a cell viability less than 50% against cancer cells MDA-MB-231, A375 and KYSE-450.
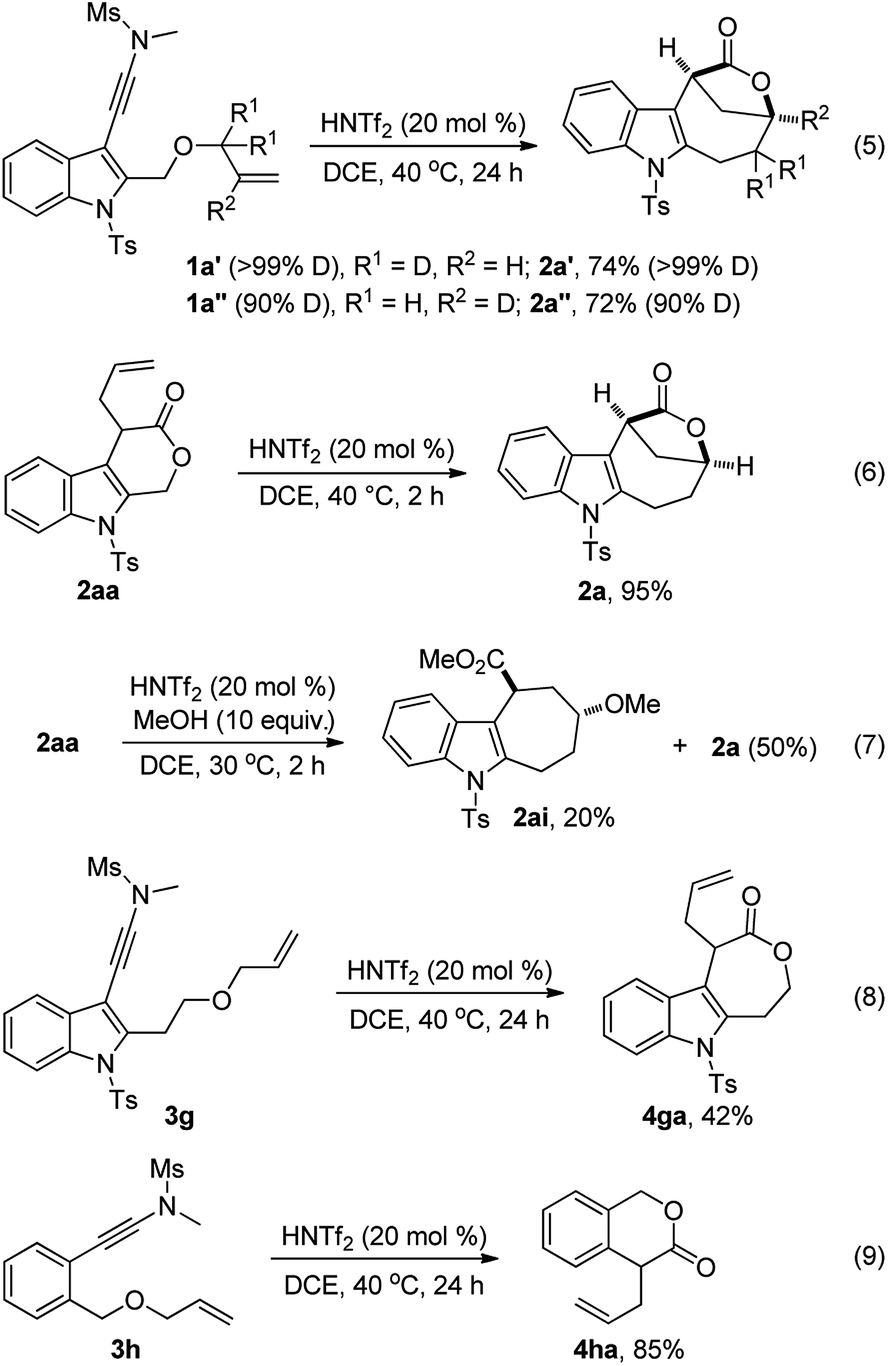
To understand the reaction mechanism, several control experiments were conducted. First, we performed deuterium labeling studies and found that no deuterium loss was observed, thus ruling out any possible reaction pathways involving a hydride shift (eqn (5)). Importantly, 2aa was readily converted into 2a in 95% yield in the presence of HNTf2 while no 2a was formed without an acid catalyst,13 strongly supporting the notion that 2aa is the key intermediate for this tandem reaction (eqn (6)). In addition, the formation of cyclohepta[b]indole 2ai (dr > 20:1) was detected when 2aa was treated with 10 equiv. of MeOH in the presence of HNTf2, which indicates that the cationic intermediate is presumably involved in such a tandem sequence (eqn (7)). Of note, 2a could not be converted into 2ai in the presence of HNTf2 and MeOH.13 Moreover, the cascade cyclization of ynamide 3g or 3h under the standard conditions only led to the formation of the corresponding 4ga (42%) or 4ha (85%), and no desired bridged lactone product was observed (eqn (8) and (9)). These results further suggest that the intramolecular alkene insertion into the C–O bond proceeds via a carbocation pathway, and the generation of a stable electron-rich benzylic carbocation is the key for this lactone expansion process. Finally, it was found that significant incorporation of 18O (>85%) into the product 2a was observed in the presence of 18O-labelled water (10 equiv.), indicating that the oxygen atom on the carbonyl group of 2a originates from water.13
Based on the above experimental observations, a plausible mechanism to rationalize the formation of indole-fused bridged [4.2.1] skeleton 2 is proposed (Scheme 3). Initially, the alkoxy group attacks the acid-activated ynamide 1 to form the Claisen rearrangement precursor Avia a keteniminium intermediate, which undergoes typical [3,3] rearrangement and subsequent trapping by trace water, delivering the indole-fused lactone D23 (that is, 2aa in the case of substrate 1a) along with the generation of sulfonamide. Brønsted acid further promotes the ring opening of lactone D to produce the carbocation intermediate Fvia C–O bond cleavage. Finally, the carbocation of F is trapped by an intramolecular electron-rich alkenyl group to generate another carbocation intermediate G, which is further captured by the intramolecular carboxylic acid group to afford the final product 2.24 Notably, the low nucleophilicity of Tf2N− might be important to maintain the cationic nature and/or reactivity of certain intermediates involved.15
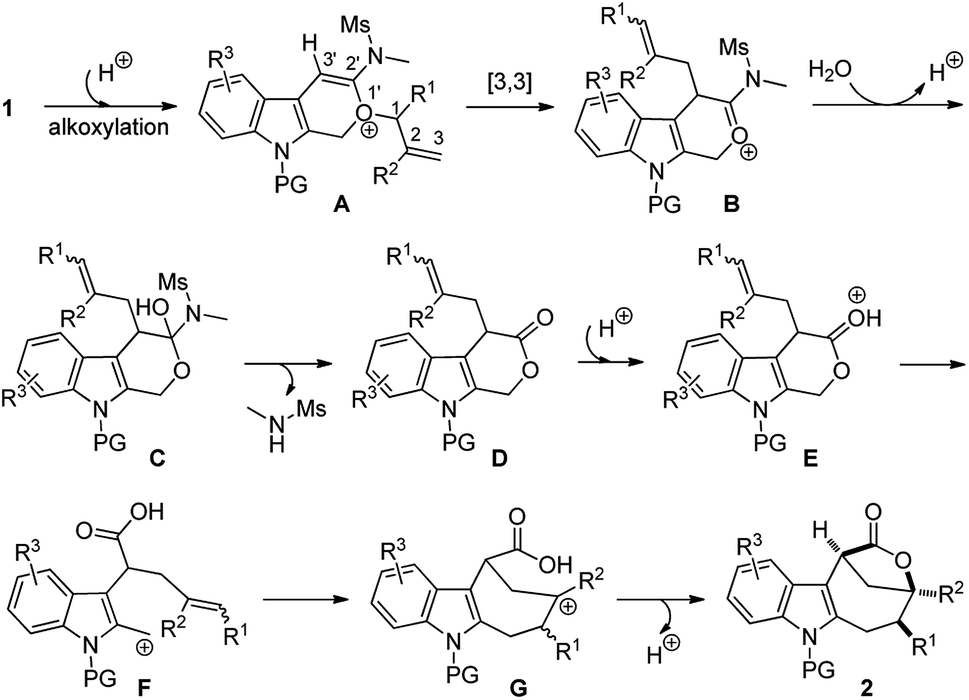 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In summary, we have developed a novel Brønsted acid-catalyzed intramolecular alkoxylation-initiated tandem sequence, which represents the first metal-free intramolecular alkoxylation/Claisen rearrangement to the best of our knowledge. Significantly, an unprecedented Brønsted acid-catalyzed intramolecular alkene insertion into the C–O bond via a carbocation pathway was discovered, which may serve as an alternative approach for alkene carbooxygenation via a direct C–O cleavage. This method enables efficient and stereocontrolled access to valuable indole-fused [4.2.1] lactones under mild reaction conditions, providing ready access to biologically relevant scaffolds in a single synthetic step from an acyclic precursor. Moreover, such an asymmetric cascade cyclization was also realized by employing a traceless chiral directing group. A mechanistic rationale for this novel tandem reaction is well supported by a variety of control experiments. In addition, our preliminary biological tests showed that some of these newly synthesized indole-fused lactones exhibited their bioactivity as antitumor agents against different breast cancer cells, melanoma cells, and esophageal cancer cells. Thus, we believe that this novel multiple cascade reaction will not only inspire chemists to design new rearrangement processes, but also encourage them to find their potential usefulness in organic and medicinal chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21772161 and 21622204), Fundamental Research Funds for the Central Universities (20720180036), PCSIRT, NFFTBS (J1310024), Science & Technology Cooperation Program of Xiamen (3502Z20183015), Young Top-notch Talent Support Program of Fujian Province and XMU Undergraduate Innovation and Entrepreneurship Training Programs. We also thank Prof. Bijie Li (Tsinghua University) and Prof. Zhenghu Xu (Shandong University) for valuable suggestions.Notes and references
- For selected examples, see: (a) D. A. Perrey, J.-X. Li and Y. Zhang, Synthesis, 2017, 49, 1394 CAS; (b) G. Ni, H. Zhang, Y.-Y. Fan, H.-C. Liu, J. Ding and J.-M. Yue, Org. Lett., 2016, 18, 1880 CrossRef CAS PubMed; (c) B. Ranieri, C. Obradors, M. Mato and A. M. Echavarren, Org. Lett., 2016, 18, 1614 CrossRef CAS PubMed; (d) X.-R. Liang, F.-P. Miao, Y.-P. Song, X.-H. Liu and N.-Y. Ji, Bioorg. Med. Chem. Lett., 2016, 26, 5029 CrossRef CAS PubMed; (e) Y.-M. Shi, J. Yang, L. Xu, X.-N. Li, S.-Z. Shang, P. Cao, W.-L. Xiao and H.-D. Sun, Org. Lett., 2014, 16, 1370 CrossRef CAS PubMed; (f) Z.-B. Cheng, W.-W. Shao, Y.-N. Liu, Q. Liao, T.-T. Lin, X.-Y. Shen and S. Yin, J. Nat. Prod., 2013, 76, 664 CrossRef CAS PubMed; (g) T. Hirokawa and S. Kuwahara, Eur. J. Org. Chem., 2013, 2780 CrossRef CAS; (h) Z.-Y. Zhou, J.-G. Tang, F. Wang, Z.-J. Dong and J.-K. Liu, J. Nat. Prod., 2008, 71, 1423 CrossRef CAS PubMed; (i) A. G. González, T. Abad, I. A. Jiménez, A. G. Ravelo, J. G. Luis, Z. Aguiar, L. S. Andrés, M. Plasencia, J. R. Herrera and L. Moujir, Biochem. Syst. Ecol., 1989, 17, 293 CrossRef.
- (a) H. Liu, Y. Wang, X. Guo, L. Huo, Z. Xu, W. Zhang, S. Qiu, B. Yang and H. Tan, Org. Lett., 2018, 20, 546 CrossRef CAS PubMed; (b) W. Zhao, Z. Li and J. Sun, J. Am. Chem. Soc., 2013, 135, 4680 CrossRef CAS PubMed; (c) M. Szostak, L. Yao and J. Aubé, J. Am. Chem. Soc., 2010, 132, 2078 CrossRef CAS PubMed.
- For recent examples, see: (a) Y.-L. Li, M.-C. Liu, Y.-J. Meng and L. Xu, Tetrahedron, 2016, 72, 3171 CrossRef CAS; (b) M. Brambilla, S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron, 2014, 70, 204 CrossRef CAS; (c) F. Grau, A. Heumann and E. Duñach, Angew. Chem., Int. Ed., 2006, 45, 7285 CrossRef CAS PubMed; (d) K. C. Nicolaou, G. E. A. Carenzi and V. Jeso, Angew. Chem., Int. Ed., 2005, 44, 3895 CrossRef CAS PubMed.
- For selected reviews, see: (a) X. Zhu and S. Chiba, Chem. Soc. Rev., 2016, 45, 4504 RSC; (b) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed; (c) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (d) F. Cardona and A. Goti, Nat. Chem., 2009, 1, 269 CrossRef CAS PubMed; (e) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC.
- For a review, see: J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 10224 CrossRef CAS PubMed.
- (a) G. T. Hoang, V. J. Reddy, H. H. K. Nguyen and C. J. Douglas, Angew. Chem., Int. Ed., 2011, 50, 1882 CrossRef CAS PubMed; (b) G. T. Hoang, Z. Pan, J. T. Brethorst and C. J. Douglas, J. Org. Chem., 2014, 79, 11383 CrossRef CAS PubMed; (c) D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544 CrossRef CAS PubMed; (d) M. Arisawa, S. Tanii and M. Yamaguchi, Chem. Commun., 2014, 50, 15267 RSC.
- For reviews, see: (a) H. V. Adcock and P. W. Davies, Synthesis, 2012, 3401 CAS; (b) X. Huang, S. Klimczyk and N. Maulide, Synthesis, 2012, 175 Search PubMed.
- (a) M. Ackermann, J. Bucher, M. Rappold, K. Graf, F. Rominger and A. S. K. Hashmi, Chem.–Asian J., 2013, 8, 1786 CrossRef CAS PubMed; (b) M. C. Blanco Jaimes, V. Weingand, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 12504 CrossRef CAS PubMed; (c) C.-M. Ting, C.-D. Wang, R. Chaudhuri and R.-S. Liu, Org. Lett., 2011, 13, 1702 CrossRef CAS PubMed; (d) F. M. Istrate and F. Gagosz, Beilstein J. Org. Chem., 2011, 7, 878 CrossRef CAS PubMed; (e) M. Ueda, A. Sato, Y. Ikeda, T. Miyoshi, T. Naito and O. Miyata, Org. Lett., 2010, 12, 2594 CrossRef CAS PubMed; (f) S. Sugita, M. Ueda, N. Doi, N. Takeda and O. Miyata, Tetrahedron Lett., 2016, 57, 1786 CrossRef CAS.
- For the relevant Au-catalyzed synthesis of seven-membered carbocycles, see: (a) H. Wu, W. Zi, G. Li, H. Lu and F. D. Toste, Angew. Chem., Int. Ed., 2015, 54, 8529 CrossRef CAS PubMed; (b) H. J. Bae, B. Baskar, S. E. An, J. Y. Cheong, D. T. Thangadurai, I. Hwang and Y. H. Rhee, Angew. Chem., Int. Ed., 2008, 47, 2263 CrossRef CAS PubMed; for the Pt-catalyzed alkoxylation reactions involving the allylic cation, see: (c) A. Fürstner and P. W. Davies, J. Am. Chem. Soc., 2005, 127, 15024 CrossRef PubMed; (d) A. Fürstner, F. Stelzer and H. Szillat, J. Am. Chem. Soc., 2001, 123, 11863 CrossRef.
- For the relevant Au-catalyzed intermolecular hydroalkoxylation/Claisen rearrangement, see: (a) J. Li, L. Lin, B. Hu, P. Zhou, T. Huang, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 885 CrossRef CAS PubMed; (b) M. Egi, K. Shimizu, M. Kamiya, Y. Ota and S. Akai, Chem. Commun., 2015, 51, 380 RSC; (c) A. Gómez-Suárez, D. Gasperini, S. V. C. Vummaleti, A. Poater, L. Cavallo and S. P. Nolan, ACS Catal., 2014, 4, 2701 CrossRef; (d) J. M. Ketcham, B. Biannic and A. Aponick, Chem. Commun., 2013, 49, 4157 RSC; (e) Y. Wang, L. Liu and L. Zhang, Chem. Sci., 2013, 4, 739 RSC; (f) Y. Wang, L. Ye and L. Zhang, Chem. Commun., 2011, 47, 7815 RSC; for the relevant Au-catalyzed intermolecular alkoxylation/Claisen rearrangement, see: (g) S. R. Park, C. Kim, D. Kim, N. Thrimurtulu, H.-S. Yeom, J. Jun, S. Shin and Y. H. Rhee, Org. Lett., 2013, 15, 1166 CrossRef CAS PubMed.
- For recent reviews on ynamide reactivity, see: (a) F. Pan, C. Shu and L.-W. Ye, Org. Biomol. Chem., 2016, 14, 9456 RSC; (b) G. Evano, C. Theunissen and M. Lecomte, Aldrichimica Acta, 2015, 48, 59 CAS; (c) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed; (d) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef CAS PubMed; (e) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS PubMed.
- (a) W.-B. Shen, Q. Sun, L. Li, X. Liu, B. Zhou, J.-Z. Yan, X. Lu and L.-W. Ye, Nat. Commun., 2017, 8, 1748 CrossRef PubMed; (b) B. Zhou, L. Li, X.-Q. Zhu, J.-Z. Yan, Y.-L. Guo and L.-W. Ye, Angew. Chem., Int. Ed., 2017, 56, 4015 CrossRef CAS PubMed; (c) W.-B. Shen, X.-Y. Xiao, Q. Sun, B. Zhou, X.-Q. Zhu, J.-Z. Yan, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2017, 56, 605 CrossRef CAS PubMed; (d) L. Li, X.-M. Chen, Z.-S. Wang, B. Zhou, X. Liu, X. Lu and L.-W. Ye, ACS Catal., 2017, 7, 4004 CrossRef CAS; (e) C. Shu, Y.-H. Wang, B. Zhou, X.-L. Li, Y.-F. Ping, X. Lu and L.-W. Ye, J. Am. Chem. Soc., 2015, 137, 9567 CrossRef CAS PubMed; (f) L. Li, B. Zhou, Y.-H. Wang, C. Shu, Y.-F. Pan, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2015, 54, 8245 CrossRef CAS PubMed; (g) A.-H. Zhou, Q. He, C. Shu, Y.-F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L.-W. Ye, Chem. Sci., 2015, 6, 1265 RSC.
- For details, please see the ESI.†.
- For Brønsted acid-mediated sulfonium [3,3] rearrangement, see: (a) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664 CrossRef CAS PubMed; (b) D. Kaldre, B. Maryasin, D. Kaiser, O. Gajsek, L. González and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 2212 CrossRef CAS PubMed.
- For a review on HNTf2 in organic synthesis, see: (a) W. Zhao and J. Sun, Chem. Rev., 2018, 118, 10349 CrossRef CAS PubMed; (b) K. Takasu, Synlett, 2009, 1905 CrossRef CAS; (c) J. Sun, Triflimide in Encyclopedia of Reagents for Organic Synthesis, Wiley, Hoboken, 2010, DOI:10.1002/047084289X.rn01222; for selected examples involving the crucial role of Tf2N−, see: (d) W. Zhao, H. Qian, Z. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 10005 CrossRef CAS PubMed; (e) W. Zhao, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 6209 CrossRef CAS PubMed; (f) J. Sun and S. A. Kozmin, J. Am. Chem. Soc., 2005, 127, 13512 CrossRef CAS PubMed; (g) K. Ishihara, Y. Hiraiwa and H. Yamamoto, Chem. Commun., 2002, 1564 RSC.
- For a review on the Tf2NH-catalyzed reaction of ynamides, see: (a) G. Evano, M. Lecomte, P. Thilmany and C. Theunissen, Synthesis, 2017, 49, 3183 CrossRef CAS; for recent leading examples, see: (b) Y. Zhao, C. Wang, Y. Hu and B. Wan, Chem. Commun., 2018, 54, 3963 RSC; (c) Y. Wang, L.-J. Song, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 9704 CrossRef CAS PubMed; (d) C. Theunissen, B. Métayer, N. Henry, G. Compain, J. Marrot, A. Martin-Mingot, S. Thibaudeau and G. Evano, J. Am. Chem. Soc., 2014, 136, 12528 CrossRef CAS PubMed.
- For a recent review, see: C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240 RSC.
- CCDC 1831051 (2a), 1831053 (2s), 1831054 (2u) and 1864037 (2ah) contains the supplementary crystallographic data for this paper.†.
- G. Alachouzos and A. J. Frontier, Angew. Chem., Int. Ed., 2017, 56, 15030 CrossRef CAS PubMed.
- J. A. Mulder, R. P. Hsung, M. O. Frederick, M. R. Tracey and C. A. Zificsak, Org. Lett., 2002, 4, 1383 CrossRef CAS PubMed.
- E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390 CrossRef CAS PubMed.
- For a possible molecular model of transition state to explain the chirality induction, see the ESI.†.
- For the relevant Brønsted acid-promoted hydrolysis, see: (a) D. Fujino, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2014, 136, 6255 CrossRef CAS PubMed; (b) S. K. Pawar, D. Vasu and R.-S. Liu, Adv. Synth. Catal., 2014, 356, 2411 CrossRef CAS; (c) S. N. Karad, S. Bhunia and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8722 CrossRef CAS PubMed.
- For the relevant proton acid-mediated cationic polyene cyclization, see: S. Kano, T. Yokomatsu, Y. Yuasa and S. Shibuya, J. Org. Chem., 1985, 50, 3449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1831051, 1831053, 1831054 and 1864037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00079h |
| This journal is © The Royal Society of Chemistry 2019 |