
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGuest recognition enhanced by lateral interactions†
Tianyu
Jiao
 ab,
Kang
Cai
b,
Zhichang
Liu
c,
Guangcheng
Wu
a,
Libo
Shen
a,
Chuyang
Cheng
b,
Yuanning
Feng
b,
Charlotte L.
Stern
b,
J. Fraser
Stoddart
*bde and
Hao
Li
*a
ab,
Kang
Cai
b,
Zhichang
Liu
c,
Guangcheng
Wu
a,
Libo
Shen
a,
Chuyang
Cheng
b,
Yuanning
Feng
b,
Charlotte L.
Stern
b,
J. Fraser
Stoddart
*bde and
Hao
Li
*a
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: lihao2015@zju.edu.cn
bDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: stoddart@northwestern.edu
cSchool of Science, Westlake University, 18 Shilongshan Road, Hangzhou 310024, P. R. China
dInstitute for Molecular Design and Synthesis, Tianjin University, Tianjin 300072, P. R. China
eSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
First published on 23rd April 2019
Abstract
A hexacationic triangular covalent organic cage, AzaEx2Cage6+, has been synthesized by means of a tetrabutylammonium iodide-catalyzed SN2 reaction. The prismatic cage is composed of two triangular 2,4,6-triphenyl-1,3,5-triazine (TPT) platforms bridged face-to-face by three 4,4′-bipyridinium (BIPY2+) spacers. The rigidity of these building blocks leads to a shape-persistent cage cavity with an inter-platform distance of approximately 11.0 Å. This distance allows the cage to accommodate two aromatic guests, each of which is able to undergo π–π interactions with one of the two TPT platform simultaneously, in an A–D–D–A manner. In the previously reported prism-shaped cage, the spacers (pillars) are often considered passive or non-interactive. In the current system, the three BIPY2+ spacers are observed to play an important role in guest recognition. Firstly, the BIPY2+ spacers are able to interact with the carbonyl group in a pyrene-1-carbaldehyde (PCA) guest, by introducing lateral dipole–cation or dipole–dipole interactions. As a consequence, the binding affinity of the cage towards the PCA guest is significantly larger than that of pyrene as the guest, even although the latter is often considered to be a better π-electron donor. Secondly, in the case of the guest 1,5-bis[2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy]naphthalene (BH4EN), the pillars can provide higher binding forces compared to the TPT platform. Hence, peripheral complexation occurs when AzaEx2Cage6+ accommodates BH4EN in MeCN. Thirdly, when both PCA and BH4EN are added into a solution of AzaEx2Cage6+, inclusion and peripheral complexation occur simultaneously to PCA and BH4EN respectively, even though the accommodation of the former guest seems to attenuate the external binding of the latter. This discovery of the importance of lateral interactions highlights the relationship between the electrostatic properties of a highly charged host and its complexation behavior, and as such, provides insight into the design of more complex hosts that bind guests in multiple locations and modes.
Introduction
Cyclophanes, such as calix[n]arenes,1 oxacalixarenes,2 resorcinarenes,3 pillar[n]arene,4 calixpyrroles,5 as well as cationic ones,6 including cyclobis(paraquat-p-phenylene) (CBPQT4+)7 and its extended counterparts,8 represent one of the major focuses in the field of supramolecular chemistry. In the frameworks of these cyclophanes, various arenes are connected by a number of aliphatic linkers such as methylene units, resulting in relatively rigid and preorganized cavities where guest encapsulation could occur without significant entropy loss. For example, in the case of CBPQT4+, π-electron-deficient 4,4′-bipyridinium (BIPY2+) are separated by two p-xylene spacers in a face-to-face manner by approximately 7 Å, which is two times longer than a typical π–π interaction distance. These geometrical properties imply that CBPQT4+ could accommodate a π-conjugated guest within its cavity, where π–π interactions9 could occur between the guest and both the two BIPY2+ platforms in the host.These kind of host–guest recognition features of CBPQT4+ and its homologous cyclophanes can be used to fulfill various tasks, including the construction of molecular switches10 and machines11 in the form of rotaxanes12 and catenanes,13 water purification via the extraction of aromatic compounds into the host,14 and the stabilization of unusual guest conformations.15 There are still several limitations, however, in the design principles of these cyclophanes.
Firstly, the spacers that connect the π-electron-deficient moieties often behave as “passive” or non-interactive building blocks in terms of molecular recognition – except for a few examples where these spacers can take part in relatively weak [C–H⋯π] interactions.16 Secondly, in most cases, host–guest complexation occurs within the host cavity wherein the interactions between the guests and the two platforms occur simultaneously. Furthermore, these interactions are often strengthened by solvophobic effects in polar solvents. As a consequence, most of these cages or rings only have one binding mode or site. There are very few examples17 of guests binding externally to these hosts.
In the current work, we report the design and synthesis of a hexacationic triangular prism, AzaEx2Cage6+, which is composed of two TPT platforms connected face-to-face by three BIPY2+ pillars. The distance between the two platforms is approximately 11.0 Å, which is approximately three-times larger than a typical π–π interaction distance, implying that the cavity in this prism could encapsulate two aromatic guest molecules such as pyrene. Interestingly, in contrast to previously reported hosts that contain phenyl or biphenyl spacers, the BIPY2+ spacers in AzaEx2Cage6+ play a more important role in host–guest complexation.
The host exhibits significantly better binding affinity towards pyrene-1-carbaldehyde (PCA) than towards pyrene in both organic solvents and water because the former guest contains a partially negatively charged carbonyl group that can undergo dipole–cation or dipole–dipole interactions with the positively charged BIPY2+ pillars. Furthermore, the BIPY2+ pillars allow the cage to form an external complex with 1,5-bis[2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy]naphthalene (BH4EN), a guest containing two tetraethylene glycol chains grafted onto a 1,5-dioxynaphthalene (DNP) moiety. This external binding is driven by the donor–acceptor interactions between the DNP unit in the guest and the BIPY2+ pillar in the host, as well as hydrogen bonding between the glycol oxygen atoms and the relatively acidic protons in the BIPY2+ units. The assumption that the DNP unit prefers to interact with the more electron-deficient BIPY2+ pillar instead of the triazine-containing platform inside the cage is supported by both single-crystal X-ray diffraction analysis and theoretical calculations. Furthermore, guest encapsulation and external binding to the cage occur simultaneously in the presence of both PCA and BH4EN, even though binding of the latter slightly decreases upon encapsulation of the former.
We envision that our findings will promote the fundamental understanding, and thus design, of hosts that are able to accommodate guests in multiple modes or sites.
Experimental section
Synthesis and characterization of AzaEx2Cage·6PF6
2,4,6-Tris[4-(bromomethyl)phenyl]-1,3,5-triazine (TBT) – a molecule that comprises three benzyl bromide functions grafted onto a TPT moiety – was synthesized in high yield from the acid-catalyzed trimerization of 4-cyanobenzyl bromide.18TBT was added slowly into a refluxing solution containing a 40-fold excess of 4,4′-bipyridine in MeCN and DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 90 °C in several aliquots over the course of 6 h, yielding the triscationic compound TBTP·3PF6 after counterion exchange. A 1:1 mixture of TBT and TBTP·3PF6 was then combined and heated at 80 °C in MeCN in the presence of 0.2 equiv. of tetrabutylammonium iodide (TBAI) as a catalyst, leading to the formation (Scheme 1) of a substantial amount of yellow precipitate. The 1H NMR spectrum of the precipitate, recorded in CD3SOCD3, indicates that the cage AzaEx2Cage6+, the counterions of which could be either Br−, I− or PF6−, represents the major product in the precipitate, along with a variety of oligomers or polymers as the minor products. Pure AzaEx2Cage·6PF6 was obtained in 16% yield by means of silica gel chromatography (1% NH4PF6 in MeCN (w/v)), before which counterion exchange was performed. Two more water-soluble counterparts, namely AzaEx2Cage·6Cl and AzaEx2Cage·6CF3CO2, were then obtained from AzaEx2Cage·6PF6 by counterion exchange.
:1 v/v) at 90 °C in several aliquots over the course of 6 h, yielding the triscationic compound TBTP·3PF6 after counterion exchange. A 1:1 mixture of TBT and TBTP·3PF6 was then combined and heated at 80 °C in MeCN in the presence of 0.2 equiv. of tetrabutylammonium iodide (TBAI) as a catalyst, leading to the formation (Scheme 1) of a substantial amount of yellow precipitate. The 1H NMR spectrum of the precipitate, recorded in CD3SOCD3, indicates that the cage AzaEx2Cage6+, the counterions of which could be either Br−, I− or PF6−, represents the major product in the precipitate, along with a variety of oligomers or polymers as the minor products. Pure AzaEx2Cage·6PF6 was obtained in 16% yield by means of silica gel chromatography (1% NH4PF6 in MeCN (w/v)), before which counterion exchange was performed. Two more water-soluble counterparts, namely AzaEx2Cage·6Cl and AzaEx2Cage·6CF3CO2, were then obtained from AzaEx2Cage·6PF6 by counterion exchange.
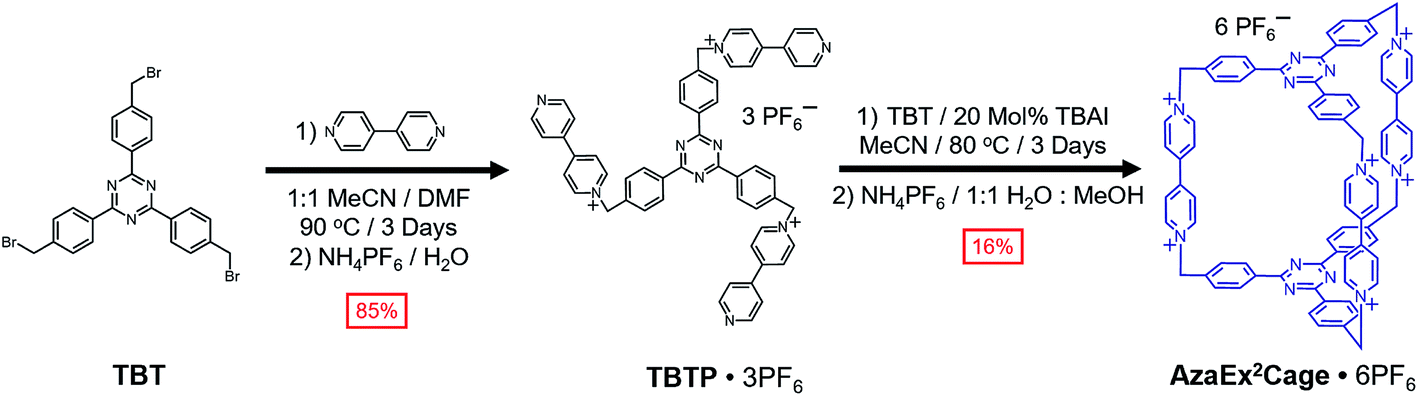 | ||
| Scheme 1 Synthesis of AzaEx2Cage·6PF6 from TBT. | ||
Results and discussion
AzaEx2Cage·6PF6 was fully characterized by NMR spectroscopy, high-resolution mass spectrometry (HRMS), and X-ray diffraction analysis. Its 1H NMR spectrum is remarkably simple (Fig. 1b), indicating that the cage has averaged D3h symmetry in solution.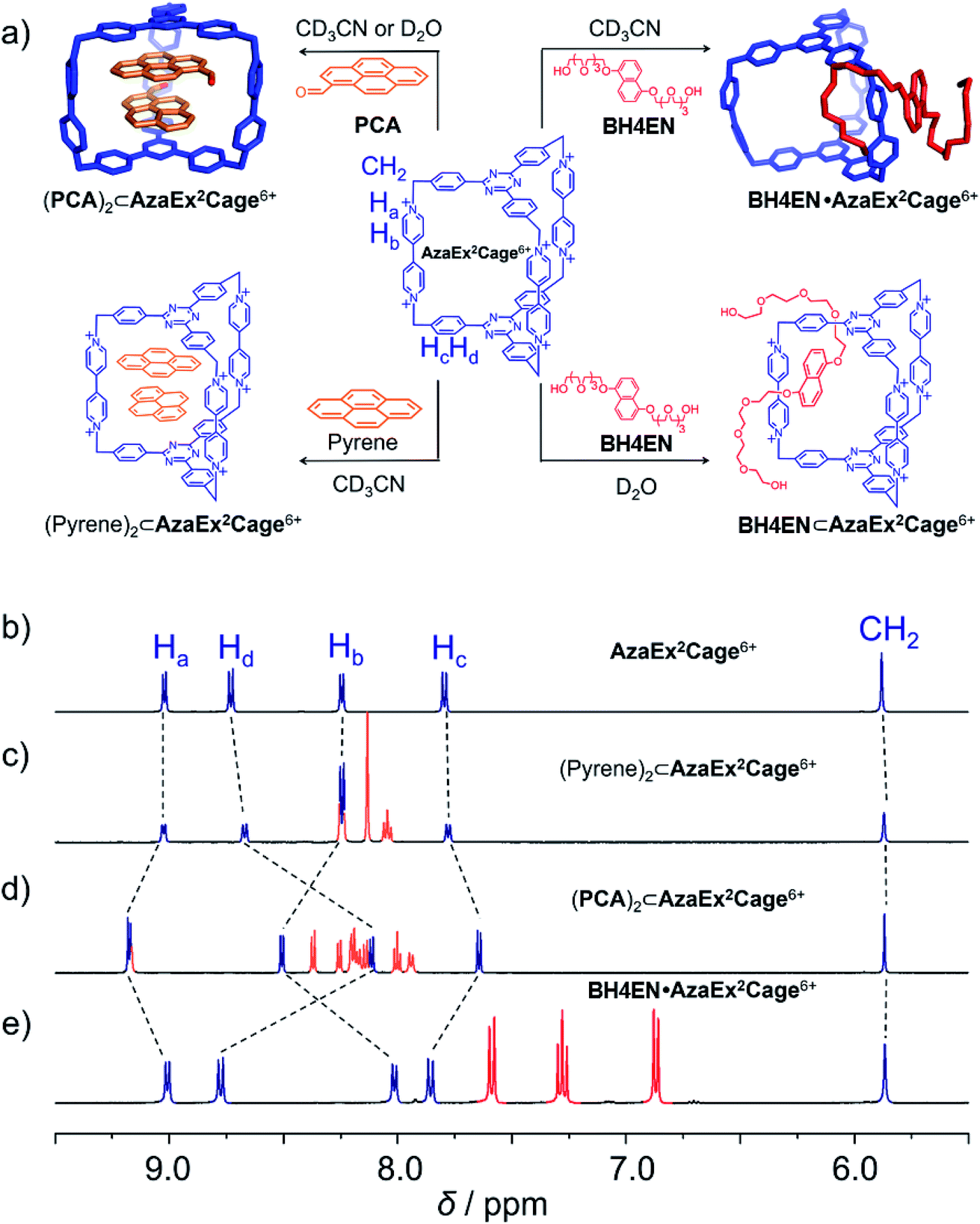 | ||
| Fig. 1 (a) Schematic representation of the ability of AzaEx2Cage6+ to recognize a variety of guests, including pyrene, PCA, and BH4EN. In the cases of pyrene and PCA, 2:1 complexes are formed, even though the ability of the cage to host the former guest is much weaker. In the case of BH4EN, the complex BH4EN·AzaEx2Cage6+ forms in a peripheral and inclusion manner in CD3CN and D2O, respectively. 1H NMR spectra of AzaEx2Cage·6PF6 (500 MHz, 1.0 mM in CD3CN, 298 K) (b) before and after adding (c) pyrene (8.4 equiv.), (d) PCA (10.8 equiv.), and (e) BH4EN (11.4 equiv.). The resonances of the protons in AzaEx2Cage6+ are labeled in the corresponding spectra. Addition of pyrene and PCA results in upfield shifts of the resonances of the phenylene residues (Hc and Hd) in AzaEx2Cage6+, while the presence of BH4EN leads to upfield shifting of the resonances for the protons on BIPY2+ (Ha and Hb). | ||
Single crystals of both AzaEx2Cage·6PF6 and AzaEx2Cage·6CF3CO2 were obtained by vapor diffusion of iPr2O into the corresponding MeCN solutions. Crystal twinning was observed, however, in the case of the former crystals (Fig. S40 and S41†). As expected, in the solid-state structure (Fig. 2) of AzaEx2Cage·6CF3CO2, the two TPT platforms in a cage framework are separated by a distance of approximately 11.0 Å, which is three-times larger than a typical π–π interaction distance,19 indicating that the cage is able to accommodate two aromatic guests in an A–D–D–A (A = acceptor; D = donor) manner. The distance between two adjacent methylene linkers within each TPT platform is around 11.6 Å. As a result, the cage framework has three relatively large (11.0 Å × 11.6 Å) rectangular pore windows. These large windows allow potential guests to undergo relatively fast association/dissociation with the cage. The CF3CO2 counterions are located close to the BIPY2+ pillars. Hydrogen-bonding interactions occur between the CF3CO2− counterions and the protons of the cage in the solid state. Furthermore, pairs of adjacent cage frameworks undergo stacking with each other, driven by π–π interactions between a triazine unit in the platform of one cage and a phenyl moiety of the other.
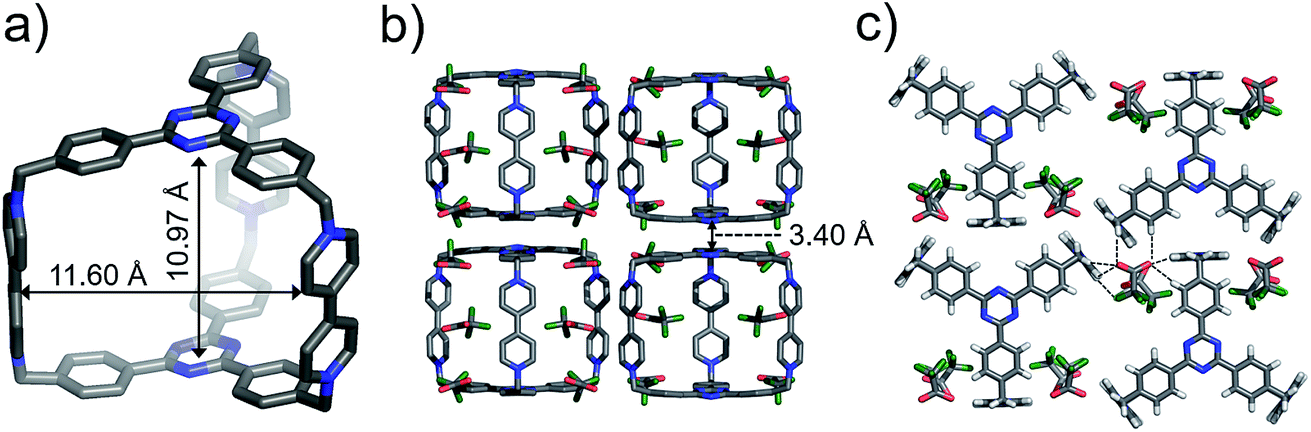 | ||
| Fig. 2 Solid-state (super)structures of AzaEx2Cage·6CF3CO2 obtained by single-crystal X-ray crystallography. (a) Stick representation of AzaEx2Cage6+, CF3CO2− counterions are omitted for the sake of clarity. (b and c) Side-on views of the packing structure revealing that π–π and hydrogen-bonding interactions play important roles in the crystal packing. The close contacts in the range of 2.22–2.46 Å between CF3CO2− counteranions and the protons in the cage indicate the occurrence of Hydrogen-bonding interactions, which are labeled with dashed lines. Hydrogen atoms and disordered solvent molecules are omitted for the sake of clarity. C, gray; N, blue; F, green; O, red. | ||
Cyclic voltammetry (CV) analysis (Fig. S36a†) of AzaEx2Cage·6PF6 in degassed DMF reveals two consecutive reversible redox processes. The reduction processes at −0.264 and −0.662 V can be assigned to two consecutive three-electron reductions, i.e., BIPY2+/BIPY+˙ and BIPY+˙/BIPY0, by three identical BIPY2+ pillars in a cage. The simultaneous reduction of the three pillars indicates the absence of electron communication between the BIPY2+ units within the cage framework.
The UV-Vis-NIR absorption spectrum (Fig. S36b†) of AzaEx2Cage3(+˙) was obtained by adding Zn dust as a reductant to a solution of AzaEx2Cage·6PF6 in MeCN. The spectrum is similar to that of 4,4′-dimethylviologen radical cations, further demonstrating that neither radical pairing nor dimerization occurs between any two of the three BIPY+˙ pillars of the cage AzaEx2Cage3(+˙). The absence of intramolecular radical pairing interactions20 is not surprising because the three BIPY+˙ units are separated by approximately 11.6 Å within the rigid cage framework.
Host–guest recognition
We first investigated the ability of AzaEx2Cage6+ to accommodate two π-electron-rich guests, since the two TPT platforms are π-electron acceptors. The 1H NMR resonances for AzaEx2Cage·6PF6 recorded in CD3CN exhibit only relatively small shifts upon addition of different guests, including pyrene, triphenylene, and perylene. For example, upon addition of 8.37 equiv. of pyrene, the resonances of the Hc and Hd protons in the phenylene residues of AzaEx2Cage·6PF6 undergo (Fig. 1c) upfield shifts of just −0.02 and −0.06 ppm, respectively. These findings indicate that the ability of AzaEx2Cage6+ to accommodate pyrene within its cavity is remarkably small. Side-on interactions between the host platforms and the guests in a external manner may also contribute to the upfield shifts of the cage proton resonances in the 1H NMR spectra, at least partially. For example, after adding 30 equiv. of pyrene into a solution of TBT in CD3CN, the resonances of the corresponding phenylene protons undergo upfield shifts of −0.02 and −0.04 ppm, respectively. In fact, the binding constants of AzaEx2Cage6+ to two pyrene molecules in CD3CN, i.e., K1 and K2, are too low to be determined accurately by means of 1H NMR titration experiments. A 1:1 binding model was employed to fit the NMR titration data, for which Keq = 11.1 ± 0.2 M−1 was determined (Fig. S22 and S23†). Several attempts were made to obtain single crystals of the complex (pyrene)2⊂AzaEx2Cage6+ by vapor diffusion of iPr2O into MeCN solutions containing both pyrene and AzaEx2Cage·6PF6 at different guest-to-host ratios ranging from 5:1 to 40:1. All of these attempts, however, yielded only a few single crystals corresponding to “empty” AzaEx2Cage·6PF6 cages, further supporting our conclusion that the binding affinity of (pyrene)2⊂AzaEx2Cage6+ is at best very weak.
We also envisioned that AzaEx2Cage·6Cl might be capable of accommodating pyrene in water by taking advantage of the hydrophobic effect. After sonicating suspensions of pyrene in D2O solutions of AzaEx2Cage·6Cl (1.0 mM) at both room temperature and 80 °C for no less than 5 h, however, no resonances corresponding to pyrene were observed in the 1H NMR spectra. In addition, the resonances of the cage protons remained almost completely unshifted. These results are in contrast to our previously reported findings, i.e., that the two smaller cages, namely ExCage6+ (ref. 14) and AzaExCage6+,21 can encapsulate a variety of polycyclic aromatic hydrocarbons with remarkably high binding constants in both organic solvents and water. For example, the Ka values for pyrene⊂ExCage6+ and pyrene⊂AzaExCage6+ are 6.77 × 105 (ref. 14) and 4.93 × 105 M−1,21 respectively, in MeCN. The lower binding affinity in the case of AzaEx2Cage6+ could be explained by the facts that (i) the TPT platform of AzaEx2Cage6+ is a weaker π-electron acceptor than the more electron-deficient triscationic counterparts such as 1,3,5-pyridinium-phenyl in ExCage6+ and 2,4,6-pyridinium-1,3,5-triazine in AzaExCage6+, (ii) the formation the 2:1 complex (pyrene)2⊂AzaEx2Cage6+ is less entropically favored than formation of the 1:1 complexes pyrene⊂ExCage6+ and pyrene⊂AzaExCage6+ (i.e., ExCage6+ and AzaExCage6+ have more preorganized cavities for encapsulating pyrene guests) and (iii) the A–D–D–A binding mode in the case of (pyrene)2⊂AzaEx2Cage6+ involves a less favored D–D interaction, making its formation more energetically demanding than that of the A–D–A mode that occurs in both pyrene⊂ExCage6+ and pyrene⊂AzaExCage6+.
Pyrene-1-carbaldehyde (PCA) is considered to be a weaker π-electron donor than pyrene, since it contains an electron-withdrawing formyl group. Surprisingly, PCA undergoes significantly stronger binding within the cavity of AzaEx2Cage·6PF6 compared to that of pyrene. For example, upon addition of 10.8 equiv. of PCA to a CD3CN solution of AzaEx2Cage·6PF6 (Fig. 1d), the resonances of the Hc and Hd protons in the platforms of the cage undergo much more significant upfield shifts, i.e., Δδ for Hc and Hd are −0.154 and −0.618 ppm, respectively, compared with −0.02 and −0.06 ppm, respectively, when 8.37 equiv. of pyrene is added (Fig. 1c). The upfield shifts reveal the presence of π–π interactions between the TPT platform and the pyrene moiety in the guest, providing the phenylene units in the platform with a magnetically shielded environment. Only one set of sharp resonances corresponding to either the host or the guest is observed in the 1H NMR spectra (Fig. 1d and 3b), regardless of the host-to-guest ratio, indicating that host–guest association/dissociation occurs relatively rapidly on the 1H NMR timescale. This observation is consistent with the solid-state structure of AzaEx2Cage6+ – i.e., each cage contains three large pore windows allowing for rapid guest exchange.
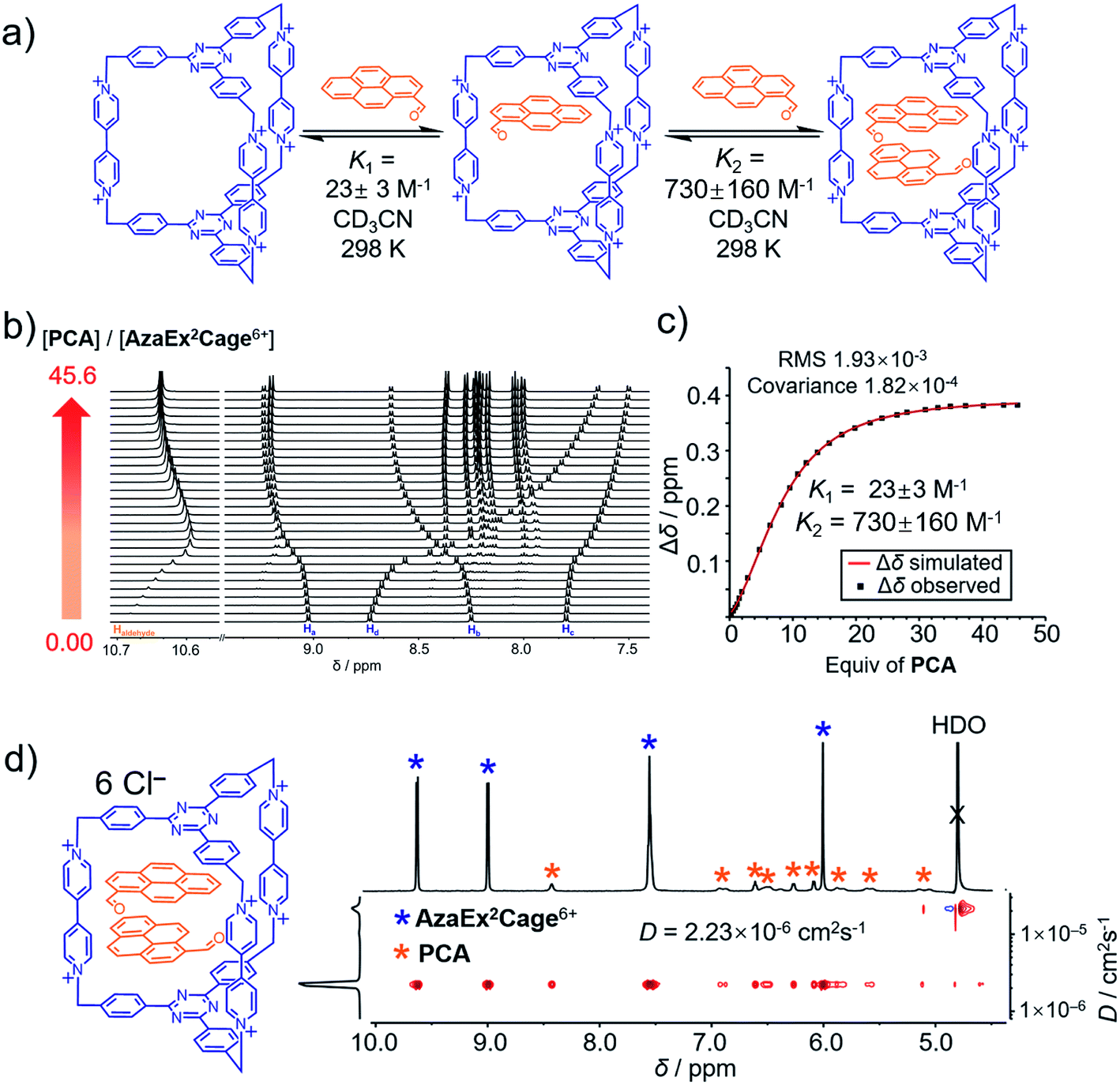 | ||
| Fig. 3 (a) Schematic representation of the ability of AzaEx2Cage6+ to accommodate two PCA guests, in which the corresponding binding constants K1 and K2 determined by 1H NMR spectroscopy in CD3CN at room temperature are shown. (b) Stacked 1H NMR spectra of AzaEx2Cage·6PF6 in CD3CN upon addition of 0 to 45.6 equiv. of PCA relative to AzaEx2Cage6+. (c) Plot of the downfield resonance shifts of Hb protons in AzaEx2Cage6+ (Δδ) versus the amount of PCA guest added relative to AzaEx2Cage6+. [AzaEx2Cage·6PF6] = 0.904 mM for all spectra. (d) 2D DOSY spectrum of the (PCA)2⊂AzaEx2Cage6+ recorded in D2O. | ||
1H NMR titration experiments (Fig. 3b and c) revealed that the binding constants for the formation of (PCA)2⊂AzaEx2Cage6+ in CD3CN, i.e., K1 and K2, are 23 ± 3 and 730 ± 160 M−1, respectively. The positive cooperativity effect is not surprising, given that the cage cavity with a length of 11 Å is too large and less preorganized to accommodate the first guest, which is able to undergo π–π interactions with only one of the two platforms in the host. After accommodation of the first guest, the cavity becomes smaller in size and therefore more complementary towards binding of the second guest molecule. In addition, the encapsulation of the second guest is driven by an extra guest–guest π–π interaction, which does not occur in the case of the first guest. The 2:1 complex (PCA)2⊂AzaEx2Cage6+ forms exclusively in D2O upon sonicating PCA solid in a D2O solution of AzaEx2Cage·6Cl (1.0 mM) at room temperature. Measuring the integration of the resonances for the protons corresponding to both the host and guest in the 1H NMR spectrum (Fig. 3d) confirms the 1:2 host-to-guest stoichiometry of the complex, which is largely a result of the low solubility of the unbound guest in water. The 2D DOSY spectrum (Fig. 3d) further confirms the formation of the complex (PCA)2⊂AzaEx2Cage6+. In addition, in water, the resonance for the formyl proton in PCA undergoes a remarkable upfield shift, i.e., δ = 8.41 ppm; Δδ ≈ 2 ppm. This observation indicates that a short contact occurs between the aldehyde and one of the viologen pillars in the host. The latter unit thus provides a magnetically shielded chemical environment for the former group, which supports our assumption that the formyl group can undergo cation–dipole interactions with one of the viologen pillars.
Single crystals of (PCA)2⊂AzaEx2Cage·6PF6 suitable for X-ray diffraction were obtained by vapor diffusion of iPr2O into its MeCN solution at 4 °C over 3 days, providing unambiguous evidence for the formation of a 2:1 complex (Fig. 4). As expected, two guest molecules are accommodated within the cavity of the cage framework. π–π Stacking interactions occur between the two platforms in the host and the two guests in an A–D–D–A manner, as inferred from the observation that the distances between triazine/pyrene, pyrene/pyrene, and pyrene/triazine are 3.42, 3.46, and 3.46 Å, respectively. The closest contacts between the two carbonyl oxygen atoms and the corresponding BIPY2+ pillars of the host are 2.78 and 3.01 Å, indicating the occurrence of dipole–cation or dipole–dipole interactions. This secondary interaction is responsible for the enhanced binding affinities of PCA with the cage cavity relative to those of pyrene. It is noteworthy that, in the solid state, the two carbonyl groups of the two guests interact with two different BIPY2+ pillars in the cage in order to minimize the repulsion between the partially anionic carbonyl oxygen atoms. Thus, (PCA)2⊂AzaEx2Cage·6PF6 exhibits axial chirality in the solid state, even though the two enantiomers co-crystallize in a racemic crystal lattice.
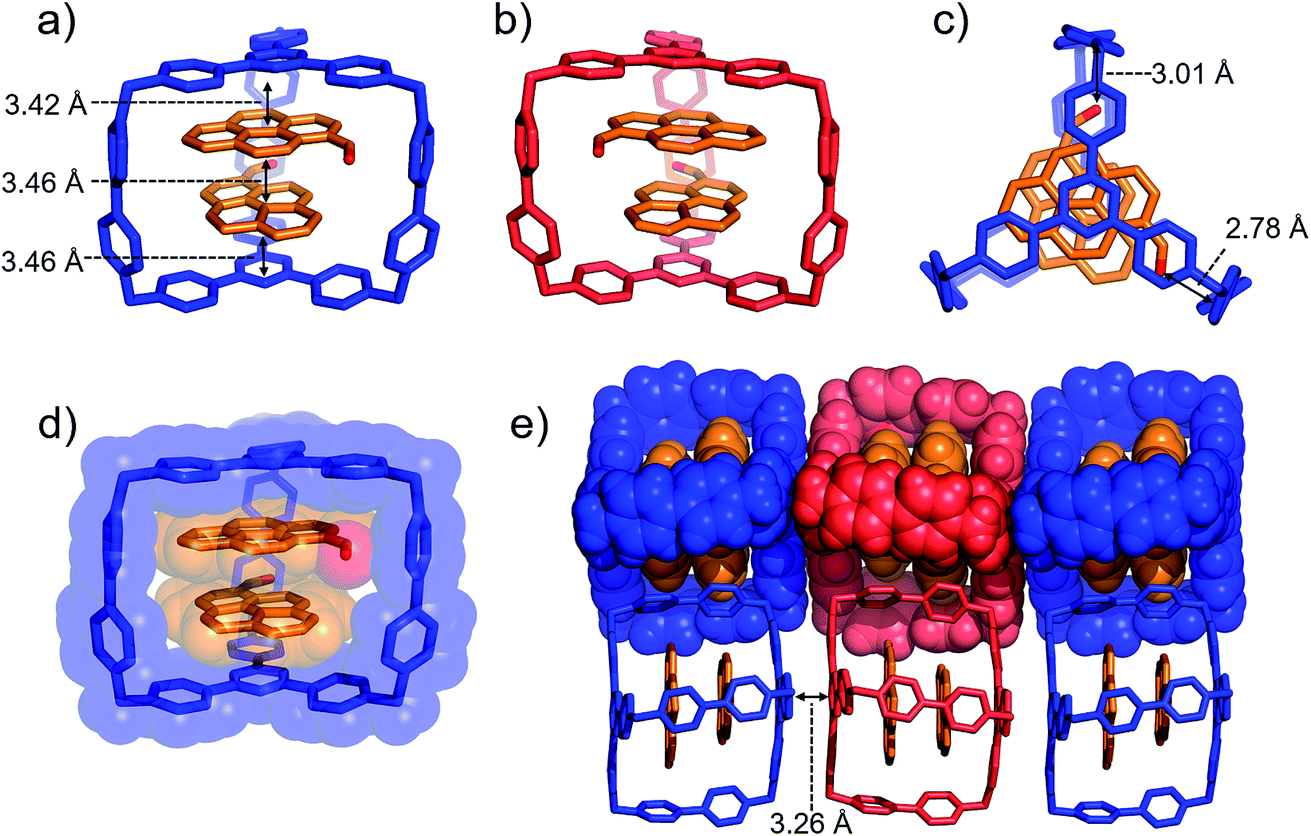 | ||
| Fig. 4 (a) Side-on stick diagrams view of the crystal structures of (PCA)2⊂AzaEx2Cage6+ forms with different chirality including (a) R (blue) and (b) S (red). The axial chirality of the complex results from the different relative rotation directions of the carbonyl groups in the two PCA guests within the cage cavity. The interplane distances between the platforms in the host and the two guests are 3.42, 3.46, and 3.46 Å, indicating the occurrence of π–π stacking interactions. (c) Top view of the X-ray crystal structure of (PCA)2⊂AzaEx2Cage6+. The distances between the two carbonyl oxygen atoms and the corresponding BIPY2+ pillars are 2.78 and 3.01 Å, respectively, indicating the occurrence of dipole–cation or dipole–dipole interactions. (d) Side-on stick diagram view overlaid with a space-filling representation of the crystal structure of (PCA)2⊂AzaEx2Cage6+. (e) Side view of the crystal lattice of (PCA)2⊂AzaEx2Cage·6PF6 showing that complexes of different chirality undergo stacking with each other, making the crystal lattice a racemic mixture. PF6− counterions are omitted for the sake of clarity. | ||
BH4EN, which is another π-electron-rich compound that bears two tetraethylene glycol chains grafted onto a 1,5-dioxynaphthalene (DNP) moiety, was also added into a CD3CN solution of AzaEx2Cage·6PF6 in order to investigate their recognition behavior. Interestingly, instead of forming an inclusion complex in either a 1:1 or 1:2 manner, it seems that the external complex BH4EN·AzaEx2Cage6+ is formed. This assumption was supported initially by the 1H NMR spectrum (Fig. 1e). Upon addition of 11.4 equiv. of BH4EN, the resonance of the Hb proton in BIPY2+ undergoes a significant upfield shift (Δδ ≈ −0.229 ppm), while those of Hc and Hd in the platforms are barely shifted. This observation indicates that, instead of undergoing π–π interactions with the cage platform, the DNP moiety prefers to interact with the BIPY2+ pillars in the host, which therefore experiences a magnetically shielded environment. No NOESY cross-peaks between the phenylene units in the host platforms and DNP protons in the guest are observed in the NOESY spectrum (Fig. S27†) of the complex BH4EN·AzaEx2Cage6+, indicating that the interactions between DNP and BIPY2+ occur on the periphery of the cage molecule. The formation of external complex was unambiguously confirmed by single-crystal X-ray diffraction. In the solid state (Fig. 5), the face-to-face distance between DNP and BIPY2+ is around 3.46 Å, confirming the occurrence of π-electron donor–acceptor interactions. The two glycol chains of the guest penetrate into the cavities of two adjacent cage molecules. The shortest contact between an oxygen atom in the glycol chain and one of the Hb protons in BIPY2+ is around 2.24 Å, indicating the existence of [C–H⋯O] interactions. The combination of donor–acceptor and hydrogen-bonding interactions make the formation of the external complex more thermodynamically favorable than formation of the inclusion complex wherein the DNP guest undergoes π–π interactions with the triazine residue in the platforms. In water, the recognition mode for BH4EN and AzaEx2Cage6+ is similar, i.e., adding BH4EN to a solution (1.0 mM) of AzaEx2Cage·6Cl in D2O also results in an upfield shift of the resonances for the BIPY2+ protons. However, an interesting difference is that strong NOESY cross-peaks between the DNP protons (Hy and/or Hz) and the phenylene protons (Hc and Hd) in the host platforms are observed (Fig. 6). These observations indicate that, in water, the complex BH4EN⊂AzaEx2Cage6+ may exist in the form of an inclusion complex instead of the external complex observed in CD3CN. This observation could be explained by the fact, that in water where the hydrophobic effect becomes predominant, the formation of the inclusion complex is more thermodynamically favored than formation of the external complex. Our attempts to obtain single crystals of the complex BH4EN⊂AzaEx2Cage6+ from water proved unsuccessful, probably because of the relatively high aqueous solubilities of both the host AzaEx2Cage·6Cl and the guest BH4EN.
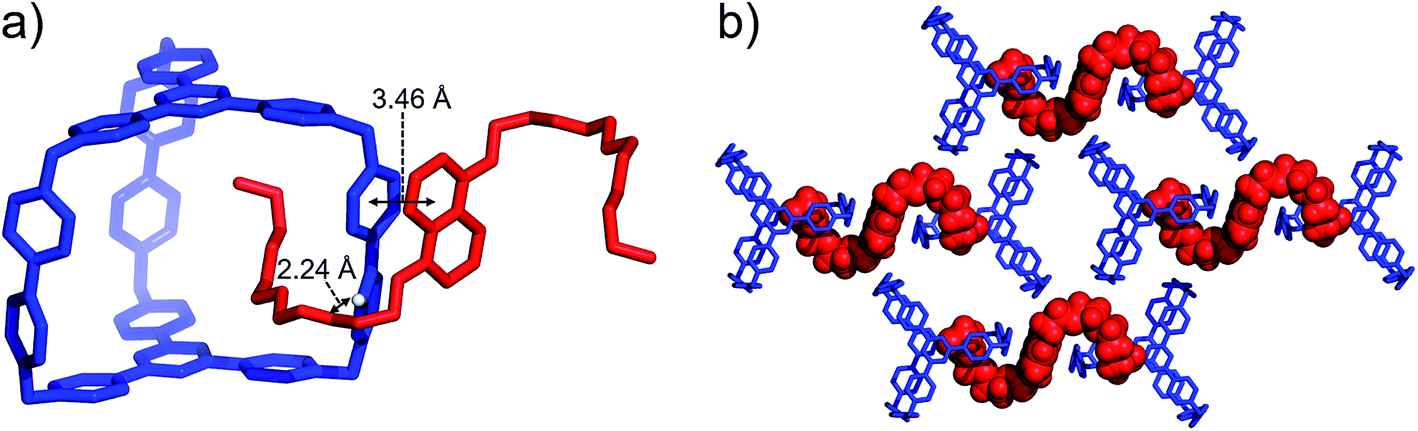 | ||
| Fig. 5 (a) Side view of the crystal structure of the complex BH4EN·AzaEx2Cage·6PF6 in the form of a stick diagram. (b) Top view of the complex BH4EN·AzaEx2Cage·6PF6, showing that each BH4EN molecule interacts with two BIPY2+ pillars in two adjacent cages. PF6− counterions are omitted for the sake of clarity. | ||
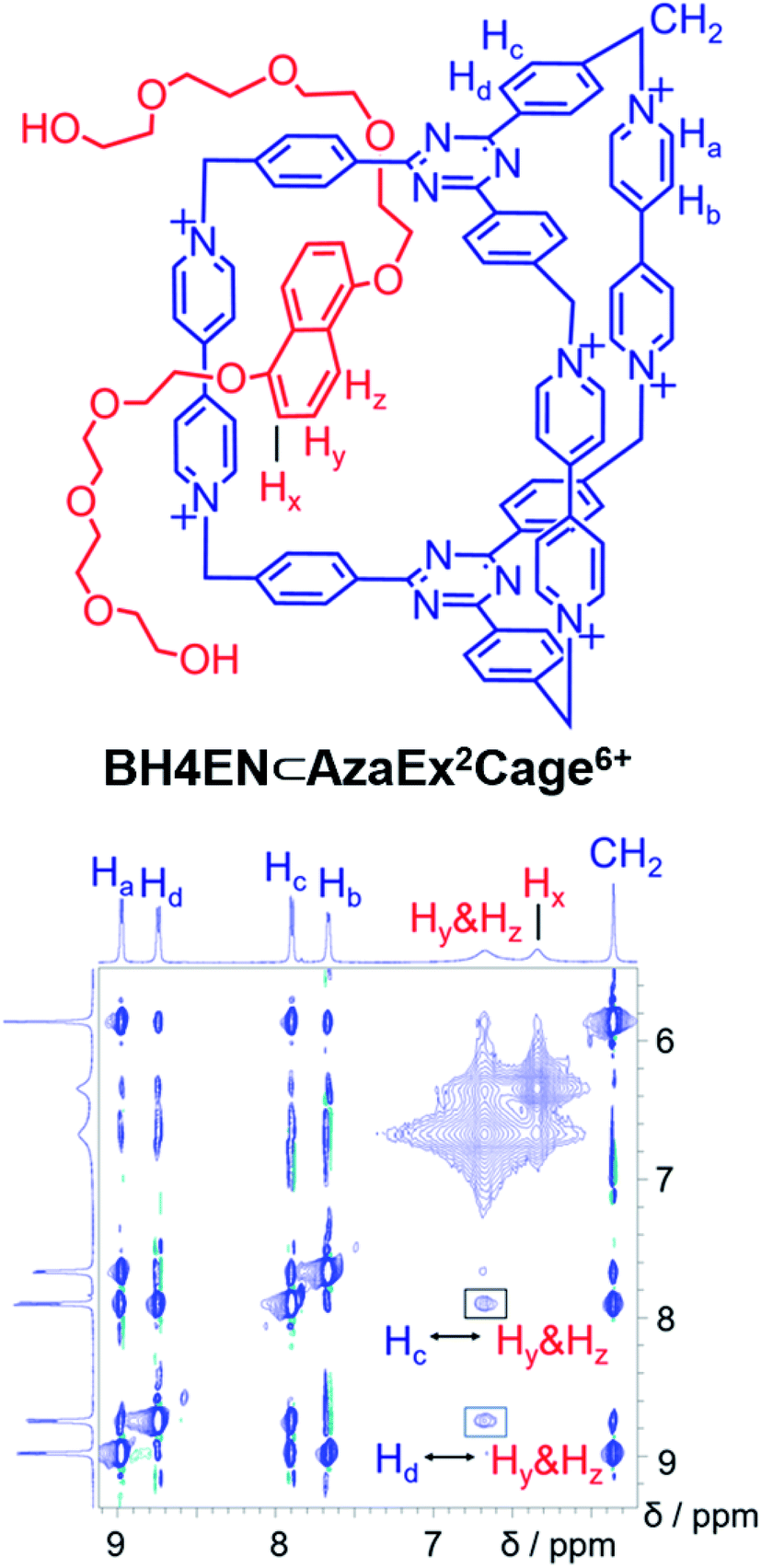 | ||
| Fig. 6
1H–1H NOESY spectrum of a 1:6 mixture of AzaEx2Cage·6Cl and BH4EN (500 MHz, D2O, 298 K). Through-space proton couplings between the Hc and Hd protons in the phenylene groups of the cage and the Hy and/or Hz protons in the guests are labeled in the spectrum, demonstrating that BH4EN·AzaEx2Cage6+ may exist in the form of an inclusion complex in water. | ||
When both PCA and BH4EN are added to a CD3CN solution of AzaEx2Cage·6PF6, inclusion and external complexation occur simultaneously, as inferred from the corresponding 1H NMR spectroscopic analyses (Fig. S30–S33†), in which the proton resonances of both the platforms and pillars undergo shifts upon addition of PCA and BH4EN, respectively. Diffraction-grade single-crystals of BH4EN·(PCA)2⊂AzaEx2Cage·6PF6 were obtained by slow vapor diffusion of iPr2O into a three-component 10:4:1 mixture of PCA, BH4EN and AzaEx2Cage·6PF6 in MeCN (2.0 mM) at 4 °C over a period of 3 days. In the solid state (Fig. 7), the cavity of a cage is occupied by two PCA guests, while a BH4EN guest resides on the external surface of a BIPY2+ pillar in the host. The glycol chains of the BH4EN guest are excluded from the cage cavity by the two PCA guests. This observation implies that hydrogen bonding between the glycol chains and the BIPY2+ units in the host might be attenuated to some extent in the solid state.
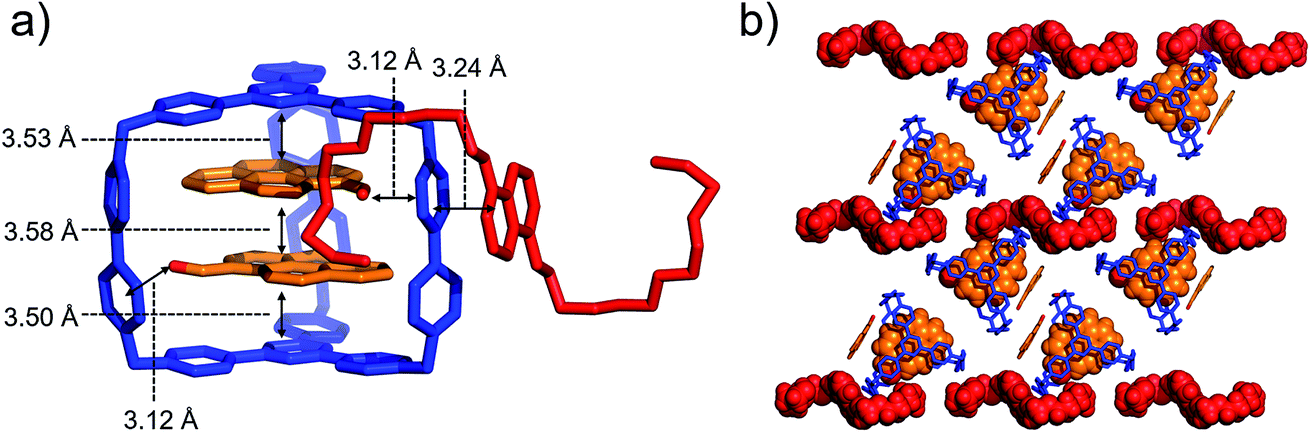 | ||
| Fig. 7 (a) Side view of the crystal structure of the complex BH4EN·(PCA)2⊂AzaEx2Cage·6PF6 in the form of a stick diagram. (b) Top view of the complex BH4EN·(PCA)2⊂AzaEx2Cage·6PF6. PF6− counterions are omitted for the sake of clarity. | ||
A 1H NMR titration experiment was performed to evaluate the impact of PCA on the binding affinity of between AzaEx2Cage·6PF6 and BH4EN. In the absence of PCA, the binding constant (Ka) for the complex BH4EN·AzaEx2Cage6+ (Fig. S26 and S33†) is approximately around 36.7 ± 0.4 M−1 in CD3CN, while upon adding 26.6 equiv. of PCA, the Ka value decreases to 4.9 ± 0.1 M−1. The decrease of the binding constant in solution is consistent with the solid-state observation that encapsulation of PCA guests expels the glycol chains of BH4EN from the cage cavity and helps to decrease the strength of hydrogen bonding. Interestingly, in the presence of 7.8 equiv. of BH4EN, the binding constants between the cage and two PCA guests (Fig. S31†) seem to be barely changed as compared with those in the absence of BH4EN. We interprete this observation to mean that the impact of external binding on the environment inside the cage is of less importance.
Quantum mechanical calculations
In order to gain a better understanding of the recognition characteristics of AzaEx2Cage6+ in terms of both inclusion and peripheral complexation, we have performed density functional theory (DFT) calculations to investigate the electronic properties AzaEx2Cage6+. Geometry optimization of AzaEx2Cage6+, BH4EN⊂AzaEx2Cage6+, (pyrene)2⊂AzaEx2Cage6+, and (PCA)2⊂AzaEx2Cage6+ were performed at the M06-2X level of theory22 with the 6-31G(d) basis set.23 The single-point energies and solvent effects in MeCN were computed at the M06-2X level of theory with the 6-311++G(d,p) basis set for all the atoms, based on the optimized gas-phase structures. Solvation energies were evaluated by a self-consistent reaction field (SCRF) using the SMD model.24 Natural population analysis25 of AzaEx2Cage6+ demonstrates that the bipyridinium, methylene and TPT units take +1.301, +0.327 and +0.074 (see the ESI†), respectively, implying that the BIPY2+ pillars of the cage, including both pyridinium moieties and the methylene linkers, takes up most of the positive charge on the cage. Expressed another way, the BIPY2+ pillars are more electron-deficient than the TPT platforms (Fig. 8a). On one hand, since the DNP unit in a BH4EN guest represents a π-electron-rich donor, it is not surprising that DNP prefers to interact with BIPY2+, forming a BH4EN·AzaEx2Cage6+ complex (Fig. 8b) whose formation is also strengthened by hydrogen-bonding interactions. On the other hand, the guests pyrene and PCA have larger π-electron surfaces. As a consequence, they prefer to interact with the TPT platform driven by π–π stacking interactions as well as solvophobic effects. To visualize the cation–dipole interactions, we performed reduced density gradient (RDG) analysis26 on the (PCA)2⊂AzaEx2Cage6+. The analysis shows clearly the occurrence of noncovalent bonding interactions (Fig. S50†) between the formyl group on PCA and the viologen pillars in AzaEx2Cage6+. | ||
| Fig. 8 Electrostatic potential maps for (a) AzaEx2Cage6+ and (b) BH4EN⊂AzaEx2Cage6+ obtained by using DFT calculations. Light-yellow and deep-blue colors in the maps represent negative and positive electrostatic potentials, respectively, demonstrating that the BIPY2+ units represent better π-electron acceptors on account of their stronger π-electron deficiency. DFT-optimized structures of (c) (PCA)2⊂AzaEx2Cage6+ and (d) (pyrene)2⊂AzaEx2Cage6+. The free energy changes (ΔG) for the formation of (PCA)2⊂AzaEx2Cage6+ and (pyrene)2⊂AzaEx2Cage6+ complexes are calculated to be −7.5 and −3.9 kcal mol−1, respectively. | ||
DFT calculations were also performed to evaluate the free energy change (ΔG), which is defined as the free energy difference between the complex and the corresponding unbound guest and host (Fig. 8c and d), for the formation of (pyrene)2⊂AzaEx2Cage6+ and (PCA)2⊂AzaEx2Cage6+. The ΔG values for the formation of (pyrene)2⊂AzaEx2Cage6+ and (PCA)2⊂AzaEx2Cage6+ were calculated to be −3.9 and −7.5 kcal mol−1, respectively. The more negative ΔG for (PCA)2⊂AzaEx2Cage6+ indicates that its formation is more thermodynamically favored than that of (pyrene)2⊂AzaEx2Cage6+. These results are consistent with the observations that the formation of the former complex is enhanced by dipole–cation or dipole–dipole interactions between the guest carbonyl oxygen atoms, which bear partially negative charges, and the dicationic BIPY2+ units in the host.
Conclusion
In summary, we have introduced a hexacationic triangular prismatic cage that is composed of two TPT platforms connected by three BIPY2+ pillar-shaped spacers. Both the platforms and the pillars play important roles in host–guest recognition. The cage can form 2:1 inclusion complexes with polycyclic aromatic hydrocarbons, including pyrene and PCA. The latter guest exhibits remarkably enhanced binding affinities within the cage cavity compared with those of the former, owing to dipole–cation and dipole–dipole interactions between the carbonyl groups and the laterally pillar-shaped spacers in the host. In addition, because the three BIPY2+ pillars represent both an efficient π-electron acceptor and hydrogen bonding donor, one of them can interact with a BH4EN guest, which contains a π-electron-donating DNP unit and two hydrogen-bond-accepting glycol chains. External complexation occurs in organic solvents, while inclusion complexation can occur in water. When both PCA and BH4EN are present, both inclusion and peripheral complexation occur simultaneously, even though accommodation of the former guests seems to suppress the external binding of the latter.
These findings improve our fundamental understanding of the relationship between the electrostatic properties of the building blocks of supramolecular systems and their host–guest recognition properties. Specifically, laterally charged moieties within host molecules could supply effective intermolecular interaction to drive the inclusion and peripheral complexations. Furthermore, this work will inform the development of a design principle for more complex cage molecules that can bind guests in multiple modes and sites.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Chinese “Thousand Youth Talents Plan”, the National Natural Science Foundation of China (No. 21772173), the Natural Science Foundation of Zhejiang Province (No. LR18B020001). H. L. and T. J. also wish to express their appreciation for the financial support from Zhejiang University. Z. L. thanks Westlake University for the startup funds. J. F. S. expresses appreciation to King Abdulaziz City for Science and Technology (KACST) and Northwestern University (NU) for support of this work. We thank Prof. Xin Hong (Department of Chemistry, Zhejiang University) for his help with the computational resources used in this investigation.References
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS PubMed.
- W. Maes and W. Dehaen, Chem. Soc. Rev., 2008, 37, 2393–2402 RSC.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS.
- T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- P. A. Gale, P. Anzenbacher and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57–102 CrossRef CAS.
- (a) F. Diederich, Angew. Chem., Int. Ed. Engl., 1988, 27, 362–386 CrossRef; (b) M. Buhner, W. Geuder, W. K. Gries, S. Hunig, M. Koch and T. Poll, Angew. Chem., Int. Ed. Engl., 1988, 27, 1553–1556 CrossRef.
- (a) B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547–1550 CrossRef; (b) P. R. Ashton, B. Odell, M. V. Reddington, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1550–1553 CrossRef.
- (a) M. Asakawa, P. R. Ashton, S. Menzer, F. M. Raymo, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 1996, 2, 877–893 CrossRef CAS; (b) P. R. Ashton, S. E. Boyd, A. Brindle, S. J. Langford, S. Menzer, L. Perez-Garcia, J. A. Preece, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, New J. Chem., 1999, 23, 587–602 RSC; (c) H. Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed; (d) E. J. Dale, N. A. Vermeulen, M. Juríček, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Acc. Chem. Res., 2016, 49, 262–273 CrossRef CAS PubMed; (e) E. J. Dale, D. P. Ferris, N. A. Vermeulen, J. J. Henkelis, I. Popovs, M. Juríček, J. C. Barnes, S. T. Schneebeli and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 3667–3670 CrossRef CAS PubMed; (f) X. Gong, R. M. Young, K. J. Hartlieb, C. Miller, Y. Wu, H. Xiao, P. Li, N. Hafezi, J. Zhou, L. Ma, T. Cheng, W. A. Goddard III, O. K. Farha, J. T. Hupp, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 4107–4116 CrossRef CAS PubMed; (g) I. Roy, S. Bobbala, J. Zhou, M. T. Nguyen, S. K. M. Nalluri, Y. Wu, D. P. Ferris, E. A. Scott, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 7206–7212 CrossRef CAS PubMed; (h) Y. Shi, K. Cai, H. Xiao, Z. C. Liu, J. W. Zhou, D. K. Shen, Y. Y. Qiu, Q. H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS PubMed.
- (a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS; (b) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- (a) Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) M. A. Olson, Y. Y. Botros and J. F. Stoddart, Pure Appl. Chem., 2010, 82, 1569–1574 CAS; (c) R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC; (d) Y. R. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS PubMed; (e) A. I. Share, K. Parimal and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 1665–1675 CrossRef CAS PubMed.
- (a) B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed; (b) R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445–455 CrossRef CAS PubMed; (c) A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed; (d) C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS PubMed; (e) J. P. Collin, C. Dietrich-Buchecker, P. Gavina, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed; (f) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines: A Journey into the Nanoworld, 2nd edn, Wiley-VCH, Weinheim, 2006 Search PubMed; (g) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed; (h) J. F. Stoddart, Nat. Chem., 2009, 1, 14–15 CrossRef CAS PubMed; (i) M. M. Boyle, R. A. Smaldone, A. C. Whalley, M. W. Ambrogio, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2011, 2, 204–210 RSC; (j) C. Y. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. F. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- (a) D. H. Qu, Q. C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296–5299 CrossRef CAS PubMed; (b) H. Zheng, W. Zhou, J. Lv, X. Yin, Y. Li, H. Liu and Y. Li, Chem.–Eur. J., 2009, 15, 13253–13262 CrossRef CAS PubMed; (c) Q. Jiang, H. Y. Zhang, M. Han, Z. J. Ding and Y. Liu, Org. Lett., 2010, 12, 1728–1731 CrossRef CAS PubMed; (d) C. Romuald, E. Busseron and F. Coutrot, J. Org. Chem., 2010, 75, 6516–6531 CrossRef CAS PubMed.
- (a) A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399–9400 CrossRef CAS PubMed; (b) M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS; (c) D. Cao, M. Amelia, L. M. Klivansky, G. Koshkakaryan, S. I. Khan, M. Semeraro, S. Silvi, M. Venturi, A. Credi and Y. Liu, J. Am. Chem. Soc., 2010, 132, 1110–1122 CrossRef CAS PubMed; (d) J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. C. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS PubMed.
- E. J. Dale, N. A. Vermeulen, A. A. Thomas, J. C. Barnes, M. Juríček, A. K. Blackburn, N. L. Strutt, A. A. Sarjeant, C. L. Stern, S. E. Denmark and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 10669–10682 CrossRef CAS PubMed.
- (a) M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222–228 CrossRef PubMed; (b) B. M. Schmidt, T. Osuga, T. Sawada, M. Hoshino and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 1561–1564 CrossRef CAS PubMed.
- (a) P. R. Ashton, C. L. Brown, E. J. T. Chrystal, T. T. Goodnow, A. E. Kaifer, K. P. Parry, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 634–639 RSC; (b) M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 630–634 RSC.
- (a) B. E. Tiedemann and K. N. Raymond, Angew. Chem., Int. Ed., 2005, 45, 83–86 CrossRef PubMed; (b) C. Sgarlata, J. S. Mugridge, M. D. Pluth, B. E. Tiedemann, V. Zito, G. Arena and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 1005–1009 CrossRef CAS PubMed; (c) T. Sawada and M. Fujita, J. Am. Chem. Soc., 2010, 132, 7194–7201 CrossRef CAS PubMed; (d) W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 7038–7043 CrossRef CAS PubMed; (e) F. J. Rizzuto, W. Y. Wu, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2016, 55, 7958–7962 CrossRef CAS PubMed; (f) D. Preston, J. E. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed; (g) X. Bai, C. Jia, Y. Zhao, D. Yang, S. C. Wang, A. Li, Y. T. Chan, Y. Y. Wang, X. J. Yang and B. Wu, Angew. Chem., Int. Ed., 2018, 57, 1851–1855 CrossRef CAS PubMed.
- J. Samanta and R. Natarajan, Org. Lett., 2016, 18, 3394–3397 CrossRef CAS PubMed.
- (a) M. Yoshizawa, J. Nakagawa, K. Kumazawa, M. Nagao, M. Kawano, T. Ozeki and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 1810–1813 CrossRef CAS PubMed; (b) M. M. Zhang, M. L. Saha, M. Wang, Z. X. Zhou, B. Song, C. J. Lu, X. Z. Yan, X. P. Li, F. H. Huang, S. C. Yin and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 5067–5074 CrossRef CAS PubMed.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Coti, D. Benítez, E. Tkatchouk, J. C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42–49 CrossRef CAS PubMed.
- N. Hafezi, J. M. Holcroft, K. J. Hartlieb, E. J. Dale, N. A. Vermeulen, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, Angew. Chem., Int. Ed., 2015, 54, 456–461 CAS.
- (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1858887–1858890 and 1858892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00591a |
| This journal is © The Royal Society of Chemistry 2019 |