
Thinking outside the shell: novel sensors designed from plasmon-enhanced fluorescent concentric nanoparticles
Nicolas
Fontaine
 ab,
Audrey
Picard-Lafond
ab,
Jérémie
Asselin
cd and
Denis
Boudreau
*ab
ab,
Audrey
Picard-Lafond
ab,
Jérémie
Asselin
cd and
Denis
Boudreau
*ab
aDepartment of Chemistry, Université Laval, 1045 avenue de la Médecine, Québec, Canada G1V 0A6. E-mail: denis.boudreau@chm.ulaval.ca
bCentre d'Optique, Photonique et Laser, Université Laval, 2375 rue de la Terrasse, Québec, Canada G1V 0A6
cDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge, UK CB3 0FS
dDepartment of Earth Sciences, University of Cambridge, Downing Street, Cambridge, UK CB2 3EQ
First published on 20th August 2020
Abstract
The alteration of photophysical properties of fluorophores in the vicinity of a metallic nanostructure, a phenomenon termed plasmon- or metal-enhanced fluorescence (MEF), has been investigated extensively and used in a variety of proof-of-concept demonstrations over the years. A particularly active area of development in this regard has been the design of nanostructures where fluorophore and metallic core are held in a stable geometry that imparts improved luminosity and photostability to a plethora of organic fluorophores. This minireview presents an overview of MEF-based concentric core–shell sensors developed in the past few years. These architectures expand the range of applications of nanoparticles (NPs) beyond the uses possible with fluorescent molecules. Design aspects that are being described include the influence of the nanocomposite structure on MEF, notably the dependence of fluorescence intensity and lifetime on the distance to the plasmonic core. The chemical composition of nanocomposites as a design feature is also discussed, taking as an example the use of non-noble plasmonic metals such as indium as core materials to enhance multiple fluorophores throughout the UV-Vis range and tune the sensitivity of halide-sensing fluorophores operating on the principle of collisional quenching. Finally, the paper describes how various solid substrates can be functionalized with MEF-based nanosensors to bestow them with intense and photostable pH-sensitive properties for use in fields such as medical therapy and diagnostics, dentistry, biochemistry and microfluidics.
 Nicolas Fontaine | Nicolas Fontaine is a Ph.D. candidate in Chemistry under the supervision of Prof. Denis Boudreau and Dr André Marette at Université Laval in Quebec City, Canada. He received his B.Sc. degree in Chemistry, research mention, at Université Laval in 2017. His research interests include the synthesis of fluorescent probes for ionic and molecular detection and their incorporation to plasmonic-based nanoarchitectures for enhanced fluorometric sensing. |
 Audrey Picard-Lafond | Audrey Picard-Lafond completed her B.Sc. in 2015 and further obtained a M.Sc. degree in organic materials chemistry from Université Laval (J.F. Morin). She is currently pursuing a Ph.D. degree under the supervision of Prof. Denis Boudreau and Dominic Larivière at Université Laval, with research interests including nanomaterial synthesis and characterisation as well as fluorescence- and plasmon-enabled analytical sensing. |
 Jérémie Asselin | Dr Jérémie Asselin is a postdoctoral research fellow in the Optical Nanomaterials group at the University of Cambridge (E. Ringe). He has a Ph.D. in Chemistry from Université Laval (D. Boudreau) in 2018, and a postdoctoral fellowship at University of British Columbia (W. R. Algar). His current research investigates the novel properties of sustainable plasmonic nanoparticles with a combination of optical and electron microscopy techniques. |
 Denis Boudreau | Prof. Denis Boudreau is a member of the Centre for Optics, Photonics and Lasers (COPL) at Université Laval. He has a Ph.D. in Chemistry from Université de Montréal (J. Hubert) and postdoctoral fellowships at University of Gainesville, Florida (J.D. Winefordner) and Chalmers University of Technology in Göteborg, Sweden (O. Axner). His research focuses on optical chemical sensors based on plasmonic nanomaterials. |
1. Introduction
Plasmonic nanomaterials have been used and investigated extensively over the last decades for their unique optical properties. This comes from the collective oscillation of conductive electrons of a metal – the plasmon – when exposed to light, which gives rise to a strong electric field.1–3 The plasmon may then relax through lattice vibrations and induce a high localized temperature that can be transferred into the vicinity, with applications in, e.g., photothermal therapy4–6 and thermo-responsive polymer actuation.7–9 Moreover, while this electromagnetic field enhancement has only recently been applied for photocatalysis,10–12 plasmon coupling with compounds in proximity (proteins, dyes, Raman tags, etc.) has paved the way for a myriad of applications including analytical techniques such as surface-enhanced Raman scattering (SERS)13–15 and metal-enhanced fluorescence (MEF).16–18 Concentrating light energy at the nanoscale with plasmonic materials allows improvements in sensitivity and detection limits without the recourse to the intense light sources or high performance detectors typical of benchtop instruments, which makes plasmonic nanomaterials especially suitable for field use and point-of-care diagnostic platforms.19,20 Sensing techniques benefitting from plasmonics include DNA hybridization,21–23 antigen24 and pathogenic bacteria detection,25 host–guest chemistry with supramolecular receptors such as molecularly imprinted polymers as selective matrices,26 and ion-sensitive dyes,27 to name a few.Since plasmonic materials are sensitive to their dielectric environment, sensing changes in the local refraction index induced by the selective adsorption of biological species can be achieved through different surface chemistries. Localized surface plasmon resonance (LSPR) sensors have been functionalized with molecular probes and antifouling moieties to impart selectivity for selected analytes.28 Several studies have been reported on the control of morphology, i.e. size and geometry of plasmonic substrates at the nanoscale, to tune LSPR frequency.29,30 Exquisite control can be achieved with electron-beam or nanosphere lithography techniques, however these suffer from poor scalability, time-consuming production and thus higher costs.31–33 Colloidal wet syntheses are often used as an alternative to produce plasmonic materials in high volume, using reaction time, reducing agents, and stabilizing ligands as control parameters. Most current synthesis strategies are based on variations of the Turkevich method, which was first demonstrated for the synthesis of gold NPs as a simple one-step reduction reaction involving trisodium citrate in boiling water.34–36 Other strategies, including seeded growth37 and templated synthesis,38,39 have also been devised in recent years with improved control over morphology and colloidal concentration.
Formation of a shell over plasmonic nanoparticles using organic40–42 or inorganic materials43–45 is a popular method to expand the range of their applications. For inorganic shells, silica has been extensively studied due to its chemical stability and well-developed functionalization strategies thanks to a wide variety of commercial silane reagents.46–49 The use of silica-coated plasmonic substrates is particularly popular in MEF applications since a silica matrix allows facile incorporation of fluorophores and can act as a rigid spacer to help improve brightness, photostability, and other luminescence properties.50–52
The use of plasmonics to improve sensing performance has been demonstrated by outstanding research in the field in the last decade, allowing for the simplification or miniaturization of different analytical platforms. This minireview summarizes novel applications of plasmonic NPs used as a versatile material to enhance the analytical performances of optical sensors, with a special emphasis on MEF core–shell nanoparticle designs. First, the influence of composition and morphology on LSPR and their effect on MEF is presented. Experimental trends are then demonstrated using one of the more common core–shell architectures, i.e. Ag cores surrounded by a SiO2 shell (Ag@SiO2). We show how architectural control can be extended to structures based on non-noble metal core–shell (e.g., In@SiO2) and even multiple shell designs encompassing multiple sensing functions (i.e., pH measurements, halide ion sensing) in a single nanoarchitecture. Examples are also given where such MEF-enabled nanosensors are used for real-time mapping of ionic species on surfaces supporting live biochemical systems or in microfluidic devices, and we conclude by presenting perspectives on the next challenges and opportunities for these architectures in the future. For more fundamental studies on the individual components of MEF, we recommend that readers consult recent articles on these particular subjects.53–57
2. Plasmonics and metal-enhanced fluorescence
Metallic NPs can support LSPRs stemming from the collective oscillation of their conduction electrons upon excitation by light. A strong electric field is generated at the interface when the incident light frequency matches that of the LSPR, which allows the NP to act as an antenna for molecules positioned in its near field. Fluorophores will benefit from MEF by a dipole–dipole coupling interaction with the plasmonic NP to enhance both excitation and emission pathways (Fig. 1).58 Since the absorption cross-section of a fluorophore depends on the probability of interaction with a photon, coupling with LSPR leads to larger effective cross-sections. Moreover, this coupled state also improves emissive rates by decreasing the excited state lifetime (τMEF, eqn (2)) compared to that of the free fluorophore (τF, eqn (1)):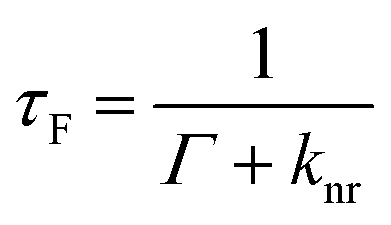 | (1) |
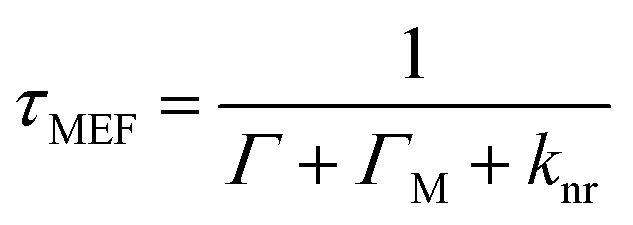 | (2) |
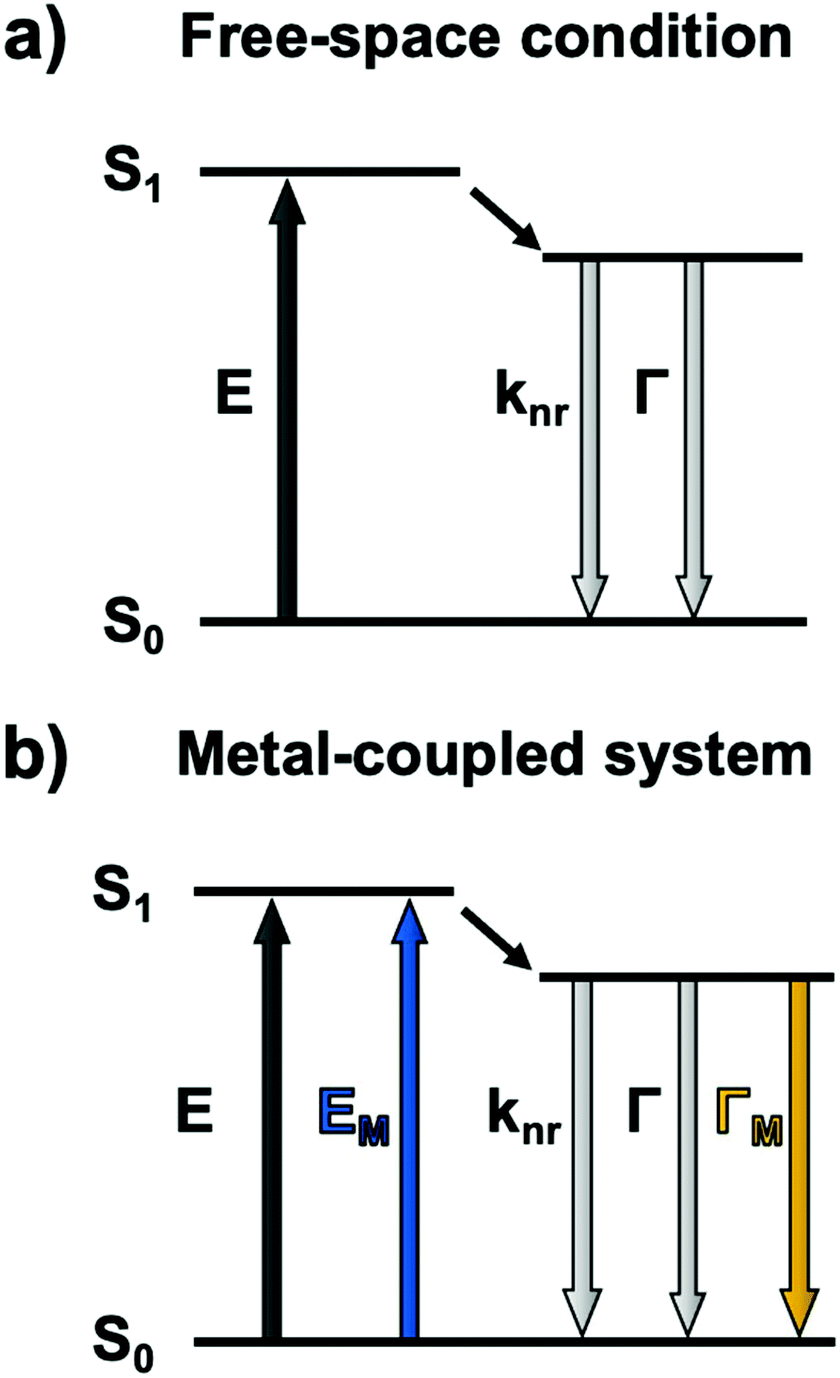 | ||
| Fig. 1 Jablonski diagram for (a) a fluorophore excited in free-space conditions and (b) modified pathways in the vicinity of a plasmonic nanoparticle. E: excitation, knr: cumulative non-radiative decay rate, Γ: radiative decay rate. The M subscript represents metal-enhanced routes. | ||
In the above equations, knr represents the cumulative non-radiative decay rates and Γ and ΓM represent the free-space and metal-coupled radiative rates, respectively.
The reduction of time spent in the excited state also imparts the coupled system with a higher photostability and, in some cases, an improved quantum yield.59 MEF has thus been a useful strategy for several years to counter the low absorptivity and brightness of common organic fluorophores as well as their vulnerability to photobleaching.
The optical absorption and scattering properties of a metal are intrinsically encoded in its dielectric function, which can be used to calculate a plasmonic quality factor.60,61 Since the scattering component generally correlates with fluorescence enhancement, a given fluorophore must spectrally overlap with this part of a plasmon. This interaction is responsible for a coupled system emitting a photon instead of reabsorbing the energy and dissipating it as heat. Although Ag and Au have been used for most plasmonic applications in the last few decades, other metals can sustain high quality LSPRs. The dielectric function of these materials should be composed of a suitably small negative imaginary part (εim), which implies lower losses, and a large real part (εreal). Poor plasmonic metals lose the electric field through damping, a transition to hot electrons relaxing by thermal dissipation; this is the case for Pd and Pt, for instance.62,63 The wavelength-dependent quality factor (QLSPR) can be approximated for different metals as a ratio of the two parts of the dielectric function:
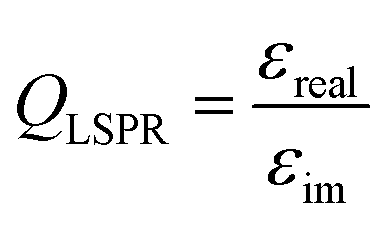 | (3) |
From these theoretical predictions, new elements such as Cu, Al, In and Mg have been investigated experimentally in recent years, which lead to better understanding and control of their colloidal synthesis.64–66 Whereas Ag, Au, Cu, and Al have emerged as powerful antennae for enhanced spectroscopies in narrow ranges of energy in the UV or the visible, the broad plasmonic peaks of Mg and In open exciting new avenues for the development of sensors which emit at different wavelengths throughout the UV, visible, and near-infrared ranges (Fig. 2). The stability of indium in water is of particular interest as it grants good compatibility with applications in biological environments. For example, indium has been shown to improve the low quantum yield of amino acids (tryptophan, tyrosine, phenylalanine) and other high-energy fluorescent emitters.67,68
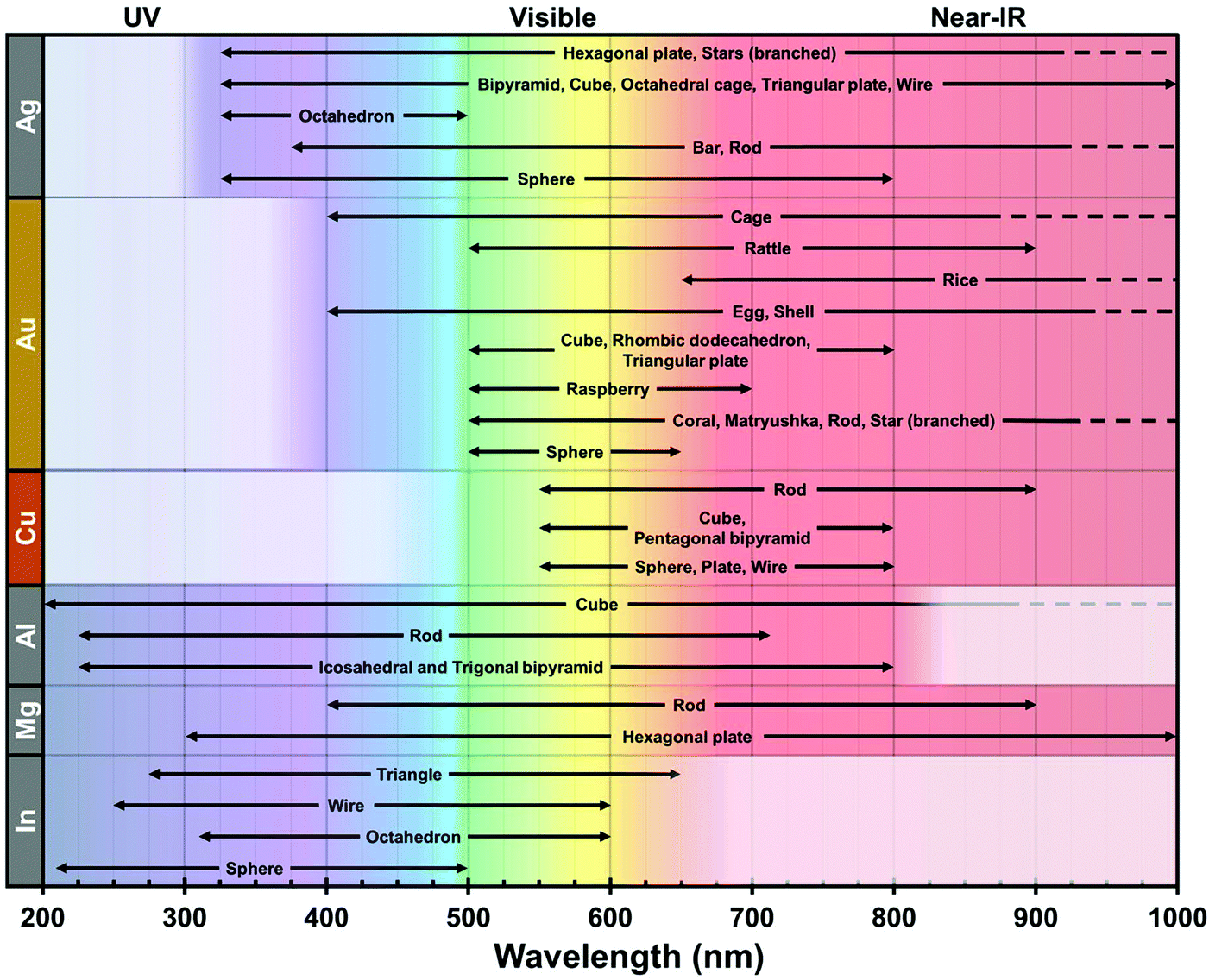 | ||
| Fig. 2 Typical plasmonic spectral range of Ag,69–74 Au,75–78 Cu,79–82 Al,83–86 Mg87,88 and In89–92 over the UV-Vis-NIR spectrum according to their reported geometries. This record is centered on wet chemistry-based products for single metal composition. Other geometries are found in the literature, albeit their experimental spectral properties are unclear. The dashed lines indicate a further extent of the plasmonic region in the NIR. The white box in each row represents the intrinsic limit of the interband transition for each composition. | ||
As much as the quality factor of materials broadly predicts the possible LSPR range, the size and shape of NPs bestow them new plasmonic modes and localized hotspots. Much effort has been put into optimizing syntheses with controlled reaction conditions (reducing agents, temperature, ligands, etc.); however, when developing novel sensor core–shell architectures, this is counterbalanced by taking care to minimize surface chemistry complexity. Condensation of a sol–gel silica shell or a polymer layer on a heterogeneous environment often results in secondary nucleation, uneven surface coatings, and poor reaction repeatability.
3. Experimental study of MEF on intensity and lifetime properties
Comparison of MEF properties in different structures requires well-defined and reproducible synthetic strategies, since their morphology is directly linked with desirable optical properties. When a plasmon and the fluorescence band of a dye overlap significantly, optimal MEF will happen when non-radiative energy transfers are minimized with a thorough control over the dye-metal distance. Several materials have been investigated in the literature for this purpose. The Lakowicz group has previously reported the combination of biotinylated bovine serum albumin (BSA) and avidin monolayers to tune the distance between silver island films and cyanine-dye-labeled oligonucleotides.93 When an optimal spacer thickness was found to be between 5 and 10 nm, discrepancies with other studies were assumed to be arising from the penetration of attached dyes inside the BSA-biotin-avidin layers. In this regard, inorganic materials such as silica shells were largely studied for their enhanced rigidity.52 Moreover, the chemical stability of silica and the versatility of silane chemistry make it a suitable and adaptable shell material to prepare metal core–silica shell nanoarchitectures (M@SiO2) with well-controlled geometry. For example, the MEF effect in relation to metal core size and silica shell thickness as well as the spectral overlap of the LSPR with the fluorescence bands were investigated in Ag@SiO2 architectures. To begin with, a seeded growth method was used to produce Ag NPs of different sizes, this process being a good compromise between reproducibility, control and scale-up capability, followed by a silica coating step with a modified Stöber method.94 Nanoparticles with well-controlled diameters and narrow size distribution were obtained with sizes of (27 ± 4), (40 ± 5), (60 ± 8) and (74 ± 10) nm. The sol–gel silica condensation implied diluting Ag NPs in ethanol with tetraethoxy orthosilicate (TEOS) and dimethylamine (DMA) as basic catalyst. Varying the concentration of TEOS in the reaction medium led to uniform spacers with thicknesses of (2.7 ± 0.3) to (27 ± 2) nm for the (60 ± 8) nm Ag cores (Fig. 3). This upper limit to the thickness range was confirmed to encapsulate the whole LSPR electric field, an observation established from the plateauing wavelength shift at larger thickness values (Fig. 3c).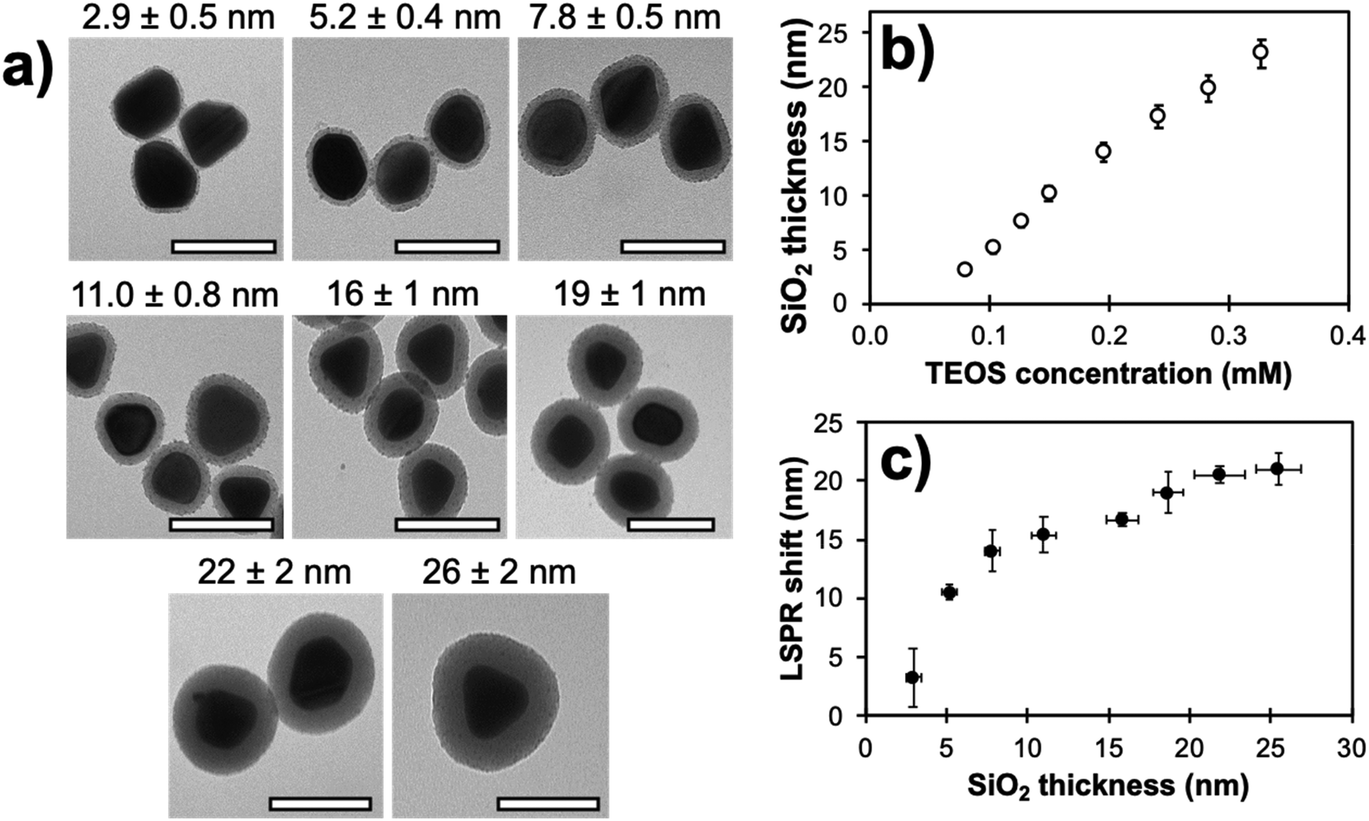 | ||
| Fig. 3 (a) Transmission electron micrographs of (60 ± 8) nm Ag cores with different silica shell thicknesses (scale bar: 100 nm). (b) Experimental SiO2 shell thickness on the (60 ± 8) nm Ag cores presented in (a) in relation to TEOS concentration during synthesis. (c) Corresponding LSPR shift as a function of the shell thickness. Modified from ref. 94 with permission from Springer Nature, Copyright 2016. | ||
The core–shell architectures were functionalized with dyes (fluorescein (FL), eosin Y (EY), or rhodamine B (RB)) by coupling their isothiocyanate derivatives with an aminosilane moiety. Their immobilization inside a (1.0 ± 0.2) nm thin shell (Ag@SiO2@(dye) NPs) was subsequently achieved with a second sol–gel condensation where a limited amount of water was present (coming for the 40% DMA solution) in the ethanol medium.
Steady-state and time-resolved fluorescence measurements were used to evaluate the distance-dependence behaviour of MEF for each core size and shell thicknesses. The fluorescence enhancement was measured by comparing signal intensity of core–shell NPs (FNP) with that of dyes without the core's influence (F0), which corresponds to the intensity of the same number of molecules in the same chemical environment where only the plasmonic influence is absent. The latter was indirectly determined from Ftotal, referring to the signal of reaction blanks where the same chemical conditions are met (ethanol, water, DMA, TEOS), except for the core–shell NPs, and Fresidual, i.e. the fluorescence of the reaction supernatants after centrifugation of the Ag@SiO2@(dye) NPs. As such, enhancement factors (EF) were basically determined from:
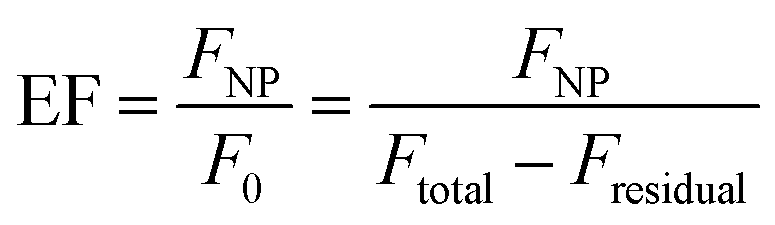 | (4) |
This method benefits from not requiring the dissolution of either metal cores or shells, which may lead to experimental losses, or to the imprecise determination of the concentration of doped silica NPs.
Fig. 4a compares the distance-dependence of the enhancement factor for Ag@SiO2@(FL) with different core sizes. As previously reported,95 larger NPs with higher scattering cross-sections support an electric field which reaches farther into the shell leading to increased EF values for all silica thicknesses. Thinner shells show the onset of non-radiative quenching by energy transfer towards the metal; this has also been demonstrated with shorter spacing molecules or polymer layers.96,97 Fluorophores 5–10 nm away from the plasmonic cores were more strongly enhanced for all sizes, and EFs decreased for distances over 10 nm as the LSPR electric field's influence diminished.
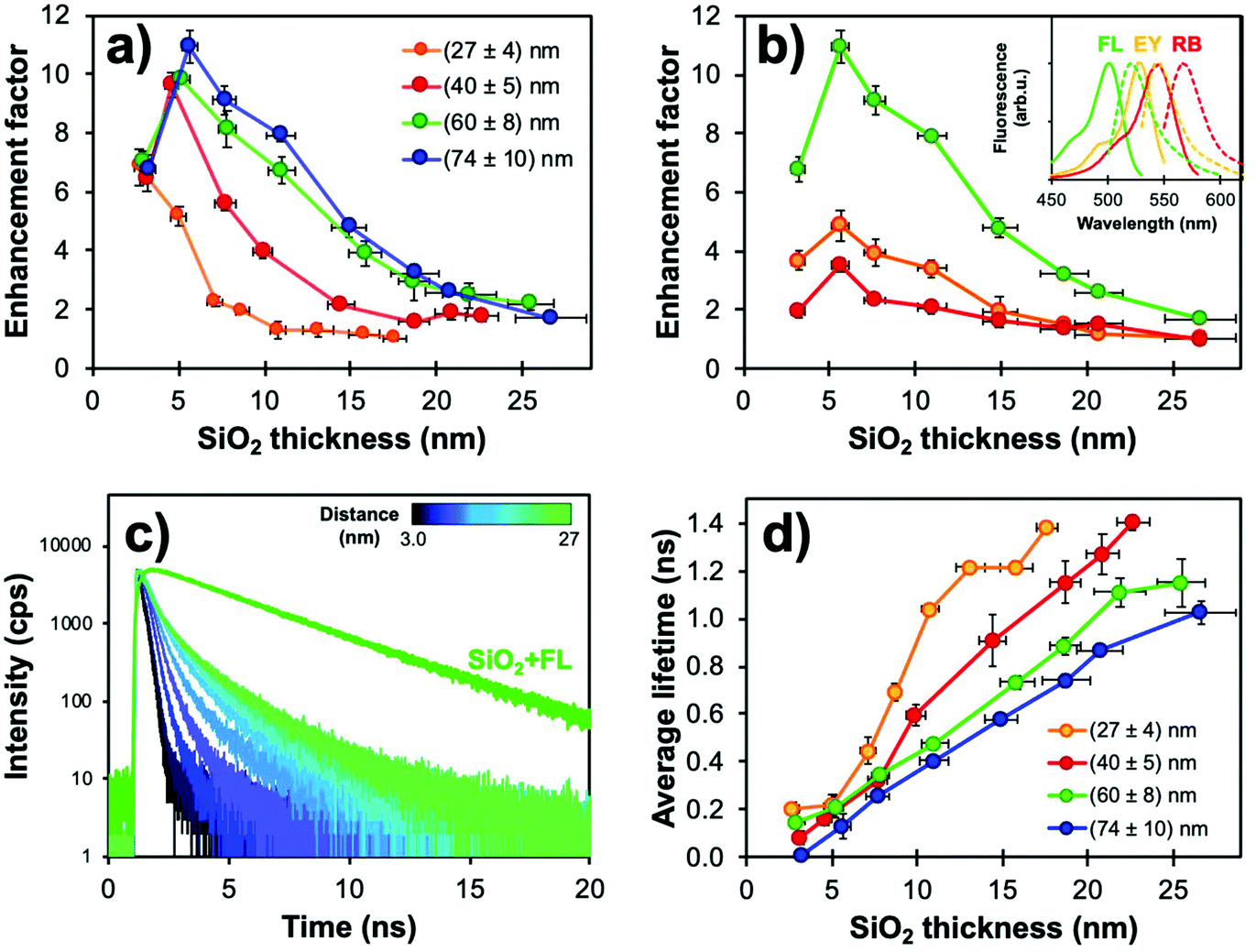 | ||
| Fig. 4 (a) Fluorescence enhancement factors (EF) determined from steady-state measurements for Ag@SiO2@(FL) (λexc = 485 nm, λem = 520 nm) with varying core sizes and silica spacer thickness (N = 3). (b) EF values for (74 ± 10) nm cores with changing fluorophores and silica spacer thickness. Parameters used for the acquisitions of FL, EY and RB were λexc = 485, 520, 550 nm and λem = 520, 550, 575 nm, respectively. Inset: Normalized fluorescence spectra for each dye. (c) Corresponding fluorescence decay curves obtained from the TCSPC measurements for (74 ± 10) nm Ag@SiO2@(FL) with shell thickness variation. (d) Average fluorescence lifetimes of Ag@SiO2(FL) for different core sizes (λexc = 485 nm, λem = 520 nm). Modified from ref. 94 with permission from Springer Nature, Copyright 2016. | ||
Moreover, there is a strong influence of the spectral overlaps on experimental EF values. This phenomenon can best be compared with two different silane-derived xanthene dyes (EY and RB) with poorer overlap of the excitation and emission bands with the Ag extinction. Higher EF values were observed for FL, which has a better overlap with Ag NPs; the “sweet spot” between metal quenching and enhancement was unaffected for all dyes in the 5–10 nm range (Fig. 4b), which is consistent with other studies in the literature.52,93,96
Core–shell NPs were further investigated using lifetime measurements by time-correlated single-photon counting (TCSPC) to confirm whether the steady-state results were indeed caused by MEF. Decay curves and fluorescence lifetimes for Ag@SiO2@(FL) are shown in Fig. 4c and d. The fluorescence lifetimes were well under that for FL encapsulated in solid silica NPs (SiO2@(FL); 3.03 ns). The fluorescence quenching by energy transfer to the metal for small metal-dye distances, as determined from steady state measurements, is not observed in TCSPC as the method relies solely on fluorescence decay curves instead of signal intensity. This emphasizes the fact that both intensity and lifetime measurements are essential to confirm MEF. Since larger plasmonic cores have higher scattering efficiency, lifetimes were shorter for the (60 ± 8) and (74 ± 10) nm cores, which is consistent with higher EF values. Interestingly, even for thicker spacers, which should encapsulate the full LSPR electric field, fluorescence lifetimes remained shorter than the value for FL in silica; this could indicate that the plasmonic influence on excitation and emission pathways follows different distance dependencies.
The Chen group reported a similar study on the interaction of silica-coated gold nanoparticles and rose Bengal dye.50 Despite using a different plasmonic metal than silver, similar results were obtained based on thorough characterization of the MEF effect. For instance, increasing the metal core size improved the fluorescence enhancement factor and optimal metal-dye distance was established at 10 nm. Results also showed that while the electric field enhancement drops with increasing distance from the particle, shorter distances also decrease the fluorescence intensity. This observation was supported by TCSPC measurements where the transition from near-single exponential to biexponential decay rate models with decreasing shell thickness suggested distinct relaxation pathways for the excited dye. These findings are of significant relevance as design guidelines for the development of highly luminescent probes based on plasmonic materials.
Less commonly reported in the literature, fluorescence anisotropy measurements can be particularly useful to probe the close vicinity of a luminescent molecule.98 This instrumental method compares the parallel (I∥) and orthogonal (I⊥) polarization emission intensities to calculate an anisotropy (r) parameter:
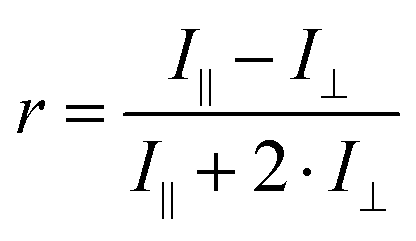 | (5) |
The r value is correlated with the rotational mobility of fluorescent molecules, where an unrestricted moiety would be able to reorient freely while in the excited state (r = 0). As such, Fig. 5 shows that functionalizing silica NPs with FL increases the anisotropy significantly compared to the free molecule, from 0.01 to 0.07. While still encapsulating FL in a sol–gel silica environment, MEF-based core–shell architectures further increase anisotropy values by allowing less time in the excited state. This decreased fluorescence lifetime (0.6 ns with MEF, and 3.0 ns without) would then decrease the potential rotation of the already-restricted molecule, which leads here to an anisotropy of 0.17. Anisotropy measurements have been used extensively to determine shape and size of macromolecules, binding constants, reaction kinetics as well as medium viscosity and composition. More recently, the Qiu group reviewed several nanomaterials-based fluorescence anisotropy strategies for applications ranging from immunoassays to ion detection.99 The results above regarding amplified anisotropy in MEF conditions could further improve these types of fluorescence polarization analyses.100
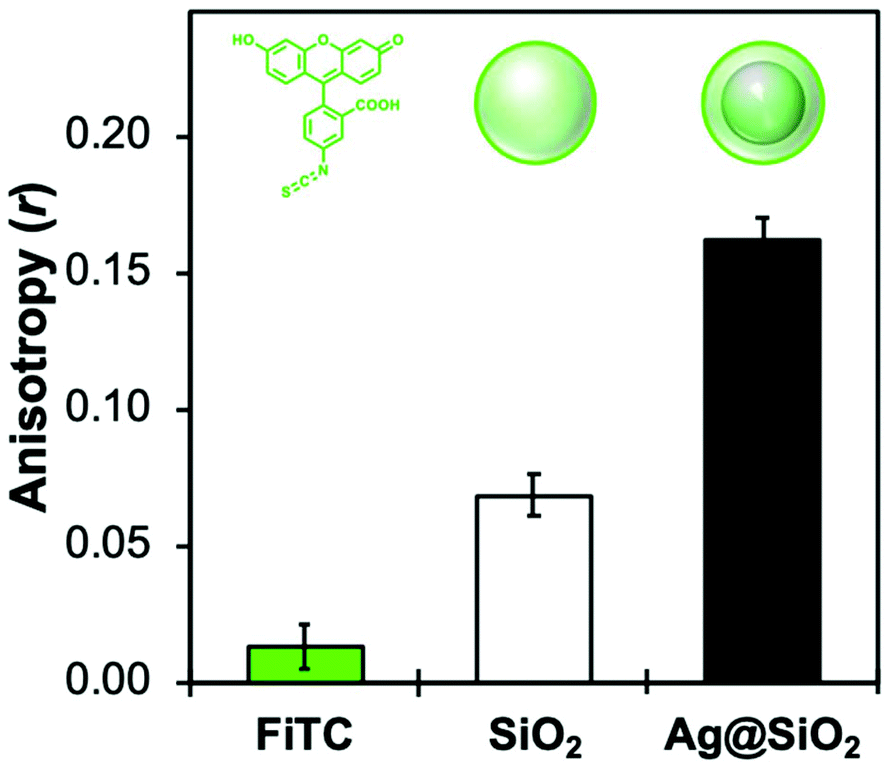 | ||
| Fig. 5 Fluorescence anisotropy values measured at 20 °C for molecular fluorescein isothiocyanate (FiTC), encapsulated SiO2 NPs (80 nm), and 80 nm Ag core-15 nm silica shell MEF architectures functionalized with a FL derivative (λexc = 485 nm, λem = 520 nm). | ||
4. Applications
The wavelength-dependence of MEF allows for the development of novel sensors when a fluorescent moiety responds to a change of local environment (pH, temperature, ionic concentrations, etc.) with a change in excitation and/or emission spectrum.101 For example, FL is an inexpensive pH-sensitive fluorophore for which the quantum yield varies with the protonation states of the molecule.102 In the pH range of ∼6–8, the stoichiometry of the monoanionic and dianionic species changes and a net increase of fluorescence can be measured (Fig. 6). In Ag@SiO2 MEF-based architectures, this intrinsic variation also improves the excitation of monoanionic fluorescein and thus adds a slight shoulder at 440 nm. Ratiometric normalization of the signal becomes possible by ratioing the fluorescence signal intensities measured while exciting at 440 and 490 nm. This powerful technique allows quantitative pH measurements from fluorescein-functionalized MEF systems, correcting for source fluctuations, photobleaching, quenching and other changes in the local environment of the fluorophore.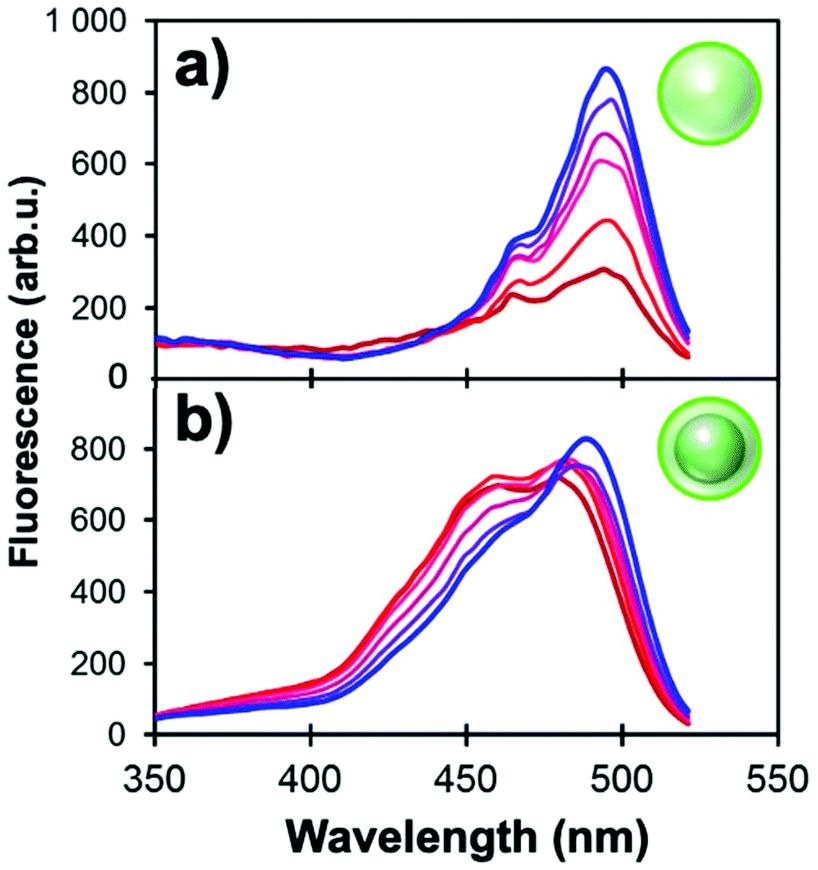 | ||
| Fig. 6 Excitation spectra of (a) 80 nm SiO2 NPs and (b) Ag@SiO2 NPs (60 nm core and 12 nm shell) functionalized with FL derivatives (λem = 540 nm). pH values of the phosphate buffers range incrementally from 5.9 (red) to 7.6 (blue). | ||
4.1 Multiplex sensors using indium plasmonics
Most applications of MEF in literature take advantage of the “turn-on” effect on signal to improve analytical parameters such as limits of detection and sensitivity. In such cases TCSPC will usually reveal a shorter fluorescence lifetime (Fig. 4) indicative of an interaction of the plasmon with the excited-state molecule. On the other hand, this effect is rarely used to improve the analytical properties of a nanosensor. A recent example of this strategy is an architecture in which the increase in emissive relaxation rates is used to tune the dynamic range of quenching-sensitive fluorescent halide ion indicators, quinolinium for instance.103 As a proof-of-concept, a MEF-based multiplex sensor using two sensitive dyes was designed to measure simultaneously pH104 and halide ions (Cl−, Br−, I−),105 two relevant physiological parameters among others such as protein content, ionic species (Na+, K+) involved in neuronal action potentials, and Ca2+ in cell signaling.106 A third fluorophore was also incorporated in the concentric nanoarchitecture to act as an internal reference.Quinolinium and its derivatives are reported for their halide sensing capabilities through dynamic quenching,107,108 meaning collisional interactions with a quenching species that induce external conversion of the excited dye and its return to the fundamental electronic state without emitting a photon. This process can be described by the Stern–Volmer equation:
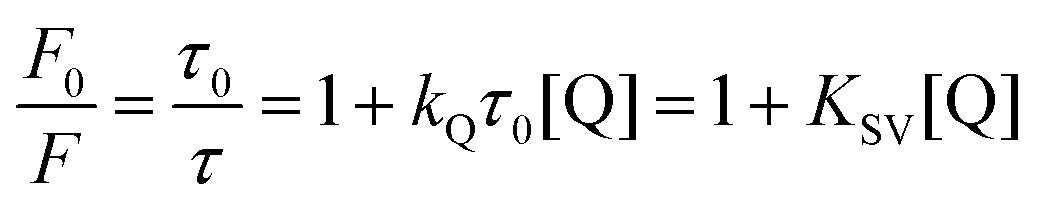 | (6) |
In the above equation, F0 and F represent the fluorescence without and with quencher (Q), respectively. τ0 and τ are the corresponding fluorescence lifetimes. KQ is the quenching constant of Q on the studied system and KSV relates to the Stern–Volmer constant (KQτ0).109Eqn (6) thus predicts a linear relationship for dynamic quenching events, in which the slope corresponds to the Stern–Volmer constant. For a given probe with a lifetime τ0, the Stern–Volmer constant is expected to increase with the polarizability of a quenching ion. This trend can be observed experimentally for MQ with KSV of 109, 280 and 488 M−1 for Cl−, Br−, and I−, respectively.
This high sensitivity of MQ to halide ions, however, can hinder applications at high salt concentrations since most of the fluorescence loss has already happened. Because of the dependence of KSV on τ0, MEF and concentric plasmonic NPs can be used to decrease the fluorescence lifetime of MQ and thus adjust the ionophore's response to the halide concentration range of the system being measured. This concept was implemented using a nanoarchitecture based on silica-coated indium nanoparticles previously investigated for the enhancement of luminescence of amino acids and a quinolinium dye (Fig. 7).103
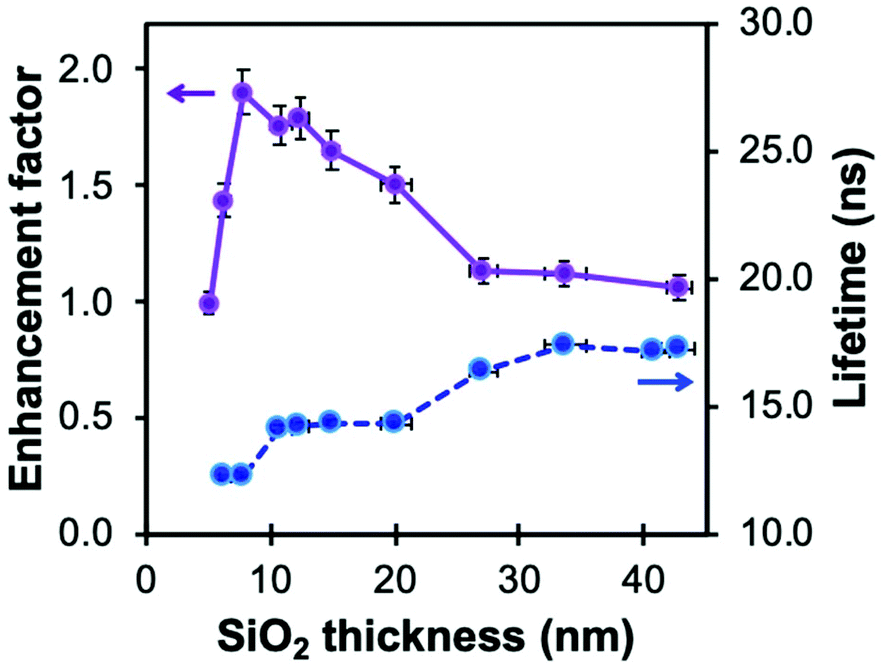 | ||
| Fig. 7 Fluorescence enhancement factor of silanized methoxyquinolinium (MQ, λex = 340 nm, λem = 440 nm) and related lifetime (λex = 308 nm) in In@(RB)@SiO2@MQ architectures as a function of spacer thickness. Reproduced from ref. 103 with permission from the Royal Society of Chemistry, Copyright 2017. | ||
Despite the lower enhancement factors reported for indium than for silver or gold, due to a lower quality factor and the presence of a 5–8 nm thick In2O3 passivation layer, indium NPs have been investigated by several groups for sensing purposes in the UV region. For instance, the Kawata group reported the use of indium-coated fused silica glass substrate for SERS in the deep UV.110 An indium coating produced by thermal vapor deposition increased the SERS signal 11-fold from thin adenine films. Geddes et al. reported the detection by metal-enhanced fluorescence of tyrosine and tryptophan, residues contained in BSA.68 Using a “sandwich” sample geometry for the solution loading, a four-fold enhancement factor was reported for an indium-coated substrate compared to bare glass. Hence, indium metal offers interesting attributes for enhancing the signal from UV-excited dyes. Moreover, the broad plasmon of In NPs overlaps exceedingly well with the excitation and emission bands of MQ (Fig. 8a). Analogously to Ag@SiO2@(FL) NPs, silica coating of this material can be achieved with a modified Stöber process to form a spacer between the metal and the dye. Moreover, successive coatings can be performed to tune the shell thickness while varying the shell dye doping, a useful property for a multiplex core–shell design.
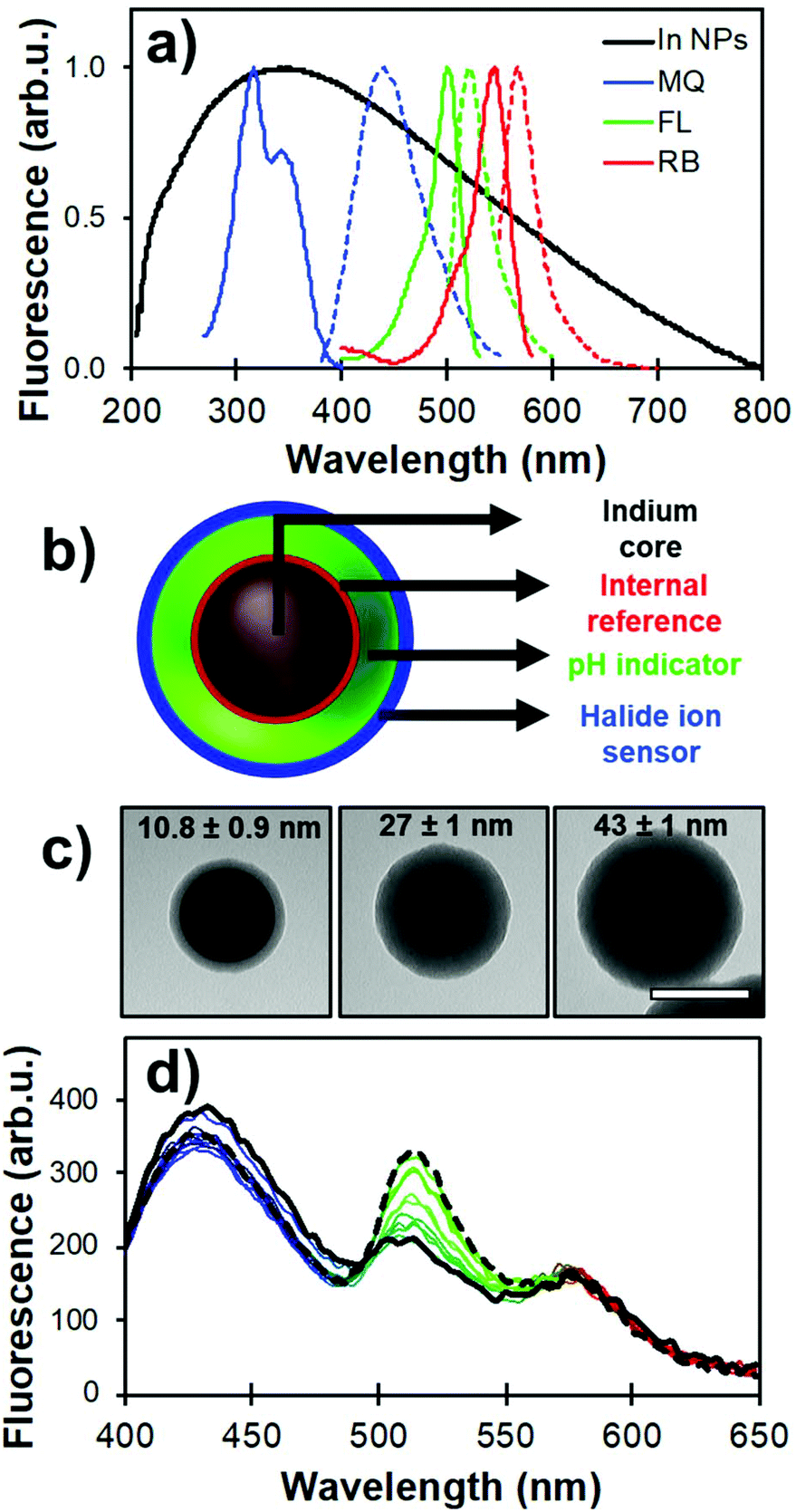 | ||
| Fig. 8 (a) LSPR of (70 ± 20) nm indium NPs (black) and its overlap with the fluorescence spectra of MQ (blue), FL (green), and RB (red). (b) Schematic representation of the core-multishell architecture with dyes positions relative to the indium core (In@(RB)@(FL)@MQ). (c) TEM images of (70 ± 20) nm In@(RB)@SiO2 with different silica spacer thicknesses. Scale bar is 100 nm. (d) Raw fluorescence spectra from a pH calibration where FL emission (λem = 540 nm) increases in phosphate buffer from pH 5.5 (solid black line) to 8.0 (dashed line). Adapted from ref. 103 with permission from the Royal Society of Chemistry, Copyright 2017. | ||
Strategic positioning of the dyes – modified to form silane-bearing compounds – relative to the core in the nanoarchitecture design helped to improve the photophysical behavior of each probe as well as maintain their accessibility towards the analytes of interest. In the present case, RB was incorporated in the inner silica shell for MQ signal normalization. A second shell was then formed with FL for pH sensing. Finally, MQ molecules were grafted on the outer surface for increased accessibility towards the chemical environment. Fig. 8b and c illustrate the nanoarchitecture design and show representative examples of silica shell thickness modulation over indium plasmonic cores. The analytical properties of the designed core–shell architecture were assessed using steady-state and time-resolved fluorescence measurements.
RB was incorporated in the first inner silica shell to limit its interaction with the probed medium. Despite being located in the immediate vicinity of the plasmonic core, loss of brightness to quenching by energy transfer to the metal is limited due to the suboptimal spectral overlap (Fig. 8a). This was confirmed by fluorescence lifetime measurements showing a minimal decrease in excited state lifetime of RB observed in the nanoarchitecture compared to that of free RB. The RB signal was also shown to be insensitive to pH in the range 6 to 8, and the Stern–Volmer constant of molecular RB against chlorine ions is nearly two orders of magnitude lower than molecular MQ.
As mentioned previously, FL exhibits intrinsic excitation ratiometry where signal normalization using two excitation bands allows for a linear measurement range over pH 6.5 to 8.0 (Fig. 8d), which is in the physiological range. The slight shift towards higher pH compared to fluorescein embedded in silver-based core–shell architecture (5.5 to 7.5) is attributable to a variation in the fluorescence enhancement of the monoanionic form of the dye, causing a shift of spectral overlap to higher energies.
The halide sensing ability of MQ normalized with the RB signal was evaluated for Cl− by comparing several shell thicknesses, i.e. as a function of distance of the MQ surface layer relative to the plasmonic core. The shortest lifetime measured for MQ was obtained for the 10 nm spacer where the probe is subjected to a more intense electric field (Fig. 9a). Longer fluorescence lifetimes were measured for thicker spacers. The 40 nm silica shell yielded a shorter excited state lifetime than that free MQ molecules, indicating that dipole–dipole coupling can still occur between the metal and the dye at this distance. Moreover, when the free MQ is almost fully quenched in 50 mM KCl, the MQ-appended plasmonic architectures of various shell thicknesses show a significantly lower sensitivity towards halide ions as they still exhibit strong residual fluorescence signal relative to the values without quencher.
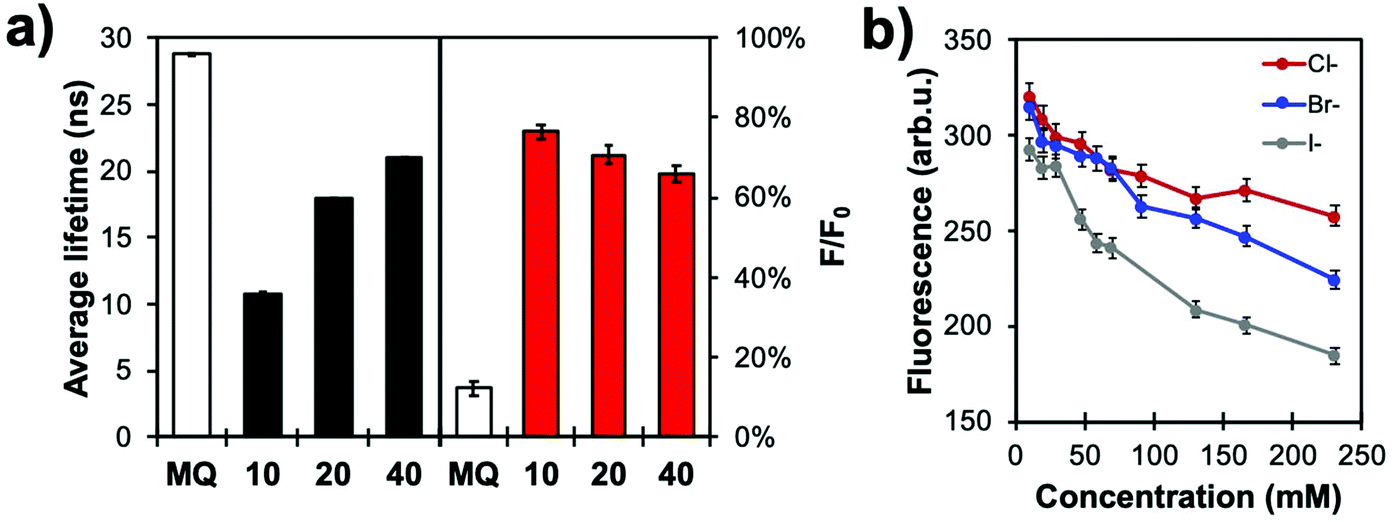 | ||
| Fig. 9 (a) Fluorescence lifetime (left) and relative intensities (F/F0, right) of MQ and In@(RB)@(FL)@MQ architectures with different silica thicknesses (in nanometres) in a 50 mM KCl solution in water (pH 6.4; N = 3). (b) Calibration curves of In@(RB)@(FL)@MQ (spacer thickness: 20 nm) with different halides (Cl−, Br−, I−) in aqueous solution (0.1 M phosphate buffer, pH 7.2; N = 3). Reproduced from ref. 103 with permission from the Royal Society of Chemistry, Copyright 2017. | ||
Normalizing the MQ signal with RB results in quasi-linear calibration curves (Fig. 9b). The higher quenching efficiency of high atomic mass ions is also observed, as the sensor shows greatest response for iodide, with KSV values of 11, 17 and 35 M−1 for Cl−, Br− and I−, respectively, for the 20 nm spacer architecture. This data illustrates how modulating the core-MQ distance affects the excited state lifetime of MQ, decreasing the sensitivity of the probe and moving its dynamic range to higher concentrations. This feature can be used to adapt the sensing range to the application at hand, or for species with different quenching efficiencies.
4.2 Immobilization of nanoprobe sensors on solid substrates via click chemistry
Material-embedded dyes can improve analytical applications by offering benefits such as higher fluorophore density, higher brightness and better protection against their chemical environment. As a result, many biomedical studies have performed proof-of-concept experiments where fluorescent nanoarchitectures are internalized in vitro or in vivo and monitored.111 However, in most biological applications, the colloidal NPs are still affected by the ion- and protein-rich environment of the cell, and the invasiveness of the method can limit their application as free-floating probes.112,113 Hence, supporting the fluorescent nanosensors on substrates is an efficient means to study biological interactions via the extracellular space, e.g. near biological membranes, in a non-invasive manner while also granting good compatibility with spatially resolved imaging techniques.Glass coverslips are planar, transparent, uniform and easy-to-handle materials adapted to surface characterization techniques such as dark field, fluorescence and scanning electron microscopies. Numerous functionalization routes can be used to bind fluorescent molecules or NPs to silica surfaces, whether physically adsorbed or covalently attached. Amongst these methods, the CuI-catalyzed 1,3-dipolar cycloaddition of alkynes and azides is generally known for its selectivity, substrate versatility and high efficiency at ambient conditions.114 As such, this click chemistry route has been demonstrated in a study where FL-doped Ag core–silica shell NPs (Ag@(FL) NPs) were functionalized with alkyne-terminated silanes and subsequently coupled to an azide-bearing glass coverslip (Fig. 10a).115 Coupling was achieved through a combination of copper sulfate and ascorbic acid in a water/ethanol mixture to generate the CuI ions in situ to activate the alkynes and begin the cycloaddition process.116 It should be noted that although the azide and alkyne silanes are permutable, the grafting of alkynes on the colloidal material led to an improved accessibility to catalytic CuI, thus increasing reaction kinetics and enabling a better grafting density. This methodology yields a uniform and aggregation-free substrate as confirmed both by scanning electron and fluorescence microscopies (Fig. 10b). This strategy was efficiently employed with other NP compositions such as Au (Au@SiO2), In (In@SiO2) and SiO2 NPs. Likewise, the method has been applied to EY-doped Ag@SiO2 NPs, thus confirming that the choice for the incorporated dye does not compromise the coupling efficiency at the interface. This versatility of using different architectures allowed the grafting of distinct emitters on the same substrate without observing unsought resonant energy transfers between different dyes, often expected in molecular mixtures.
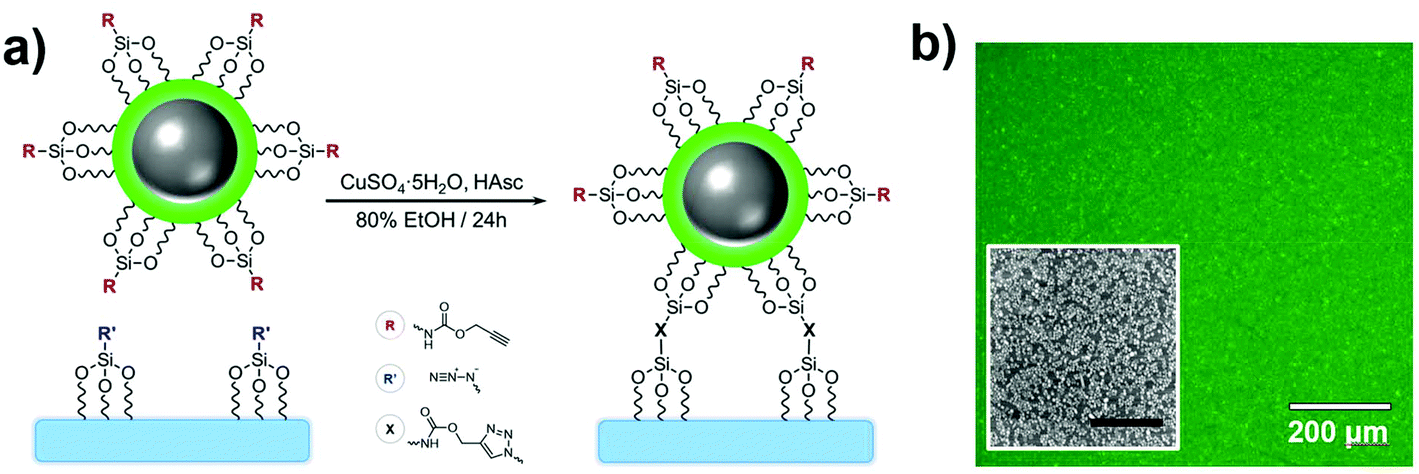 | ||
| Fig. 10 (a) Grafting scheme of Ag@(FL) NPs to a SiO2 substrate through CuI-catalyzed 1,3-dipolar cycloaddition. (b) Fluorescence image of the substrate grafted with Ag@(FL) NPs (λexc = 489 ± 6 nm, λem = 536 ± 20 nm). Inset: SEM image of a region the substrate (scale bar: 1 μm). Adapted from ref. 115 with permission from the Royal Society of Chemistry, Copyright 2014. | ||
Photostability has been characterized through different ways in literature; as the total amount of emitted photons, the duration of single-molecule fluorescence events, and ensemble decay measurements. Here, because NPs are functionalized with many FL moieties, single-NP decays are compared between different Ag core-SiO2 shell architectures while immobilized on a substrate. Fig. 11 shows a photobleaching experiment where single NPs were immobilized on a glass substrate and their punctual intensity signature was traced over 60 minutes. Fluorescence decay (I(t)) predicts a single-exponential relationship following:
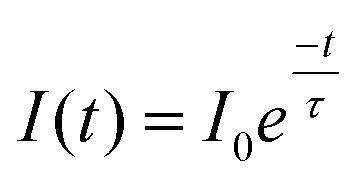 | (7) |
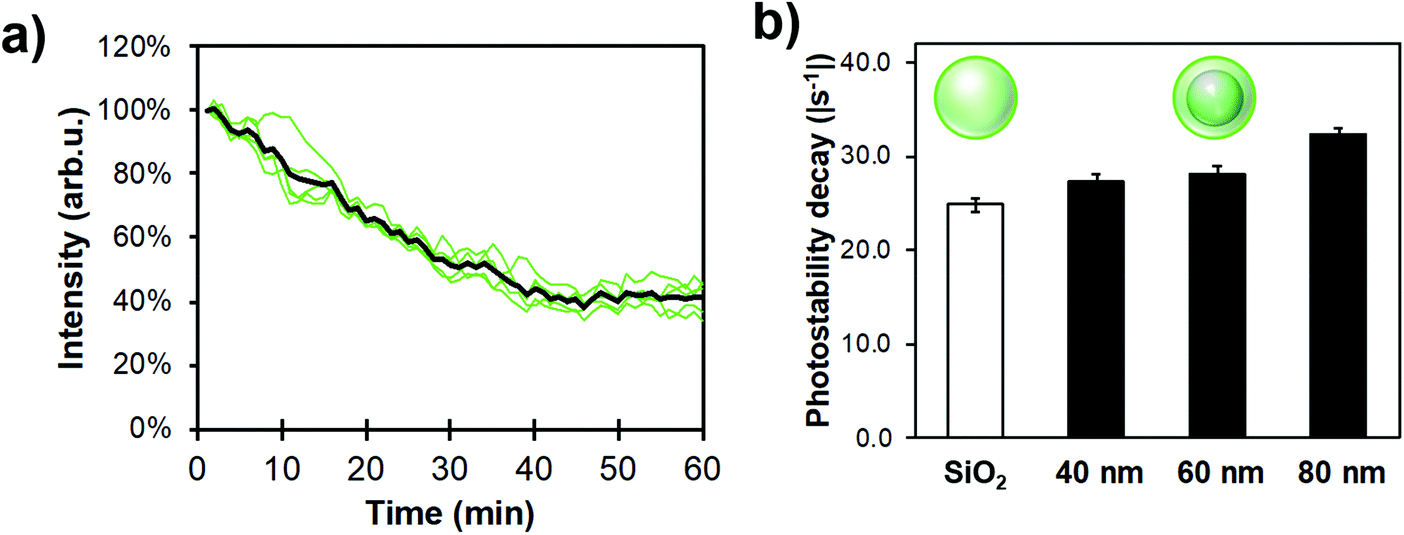 | ||
| Fig. 11 Experimental single-NP photostability measurements for different FL-functionalized architectures; (a) 5 single-particle exponential decays of a 80 nm Ag core-15 nm SiO2 shell architecture (green) and their average trace (black), (b) Photostability decay for different core–shell MEF systems and 80 nm silica NPs as reference. Samples were dispersed in phosphate buffer (pH 7.3, 0.010 M) and measurements were performed using a 488 nm excitation and a 506 nm highpass emission filter. For all datasets N > 100 NPs, error bars represent the standard error to the mean. | ||
Ag cores improved the photostability compared to FL-doped silica NPs, albeit not drastically; this trend still increased with bigger NP sizes (i.e. higher scattering cross-section). Therefore, this result helps to design sensing conditions appropriately, meaning that illumination intensity could be decreased for MEF samples where longer exposure experiments are beneficial.
A relevant application of immobilized Ag@(FL) NPs is the 2D spatial pH mapping of the extracellular environment since the quantification of physiological ions in the vicinity of biological membranes is invariably connected to most intracellular metabolic pathways. For instance, as cancer cells induce significant acidification of the extracellular medium,117 mapping proton activity is invaluable to distinguish healthy from unhealthy cells. When using a coverslip grafted with nanosensors, the fluorophores and NPs are unable to detach and migrate through the phospholipid bilayer, thus measuring the extracellular pH values specifically.
Human cardiac fibroblasts, known for their good adhesive properties,118 were grown on the pH-sensitive substrate without noticeable cytotoxicity (Fig. 12a); in fact, the increased surface roughness improved cell viability on NP-grafted surfaces. Carbonic anhydrase enzymes (CA12) are expressed by fibroblast cells to facilitate the solubilisation of aqueous carbon dioxide119 and were chosen to demonstrate the pH mapping of the coverslip sensors. Ratiometric imaging confirmed a significant pH increase correlated with line profiles from the center of the cell (Fig. 12b), an observation ascribed to proton diffusion from the CA12 enzymes bound to the cell membrane. The resolution was estimated at 1 μm, mostly limited by the numerical aperture of the objective and the NP grafting density. Good temporal resolution could also be expected for metabolic pathways, since fluorescence lifetimes are orders of magnitude faster than most biological mechanisms. This decreased fluorescence lifetime by MEF further improves photostability, allowing for prolonged analyses of cell samples.
 | ||
| Fig. 12 (a) 2D pH cartography of a GFP (green fluorescent protein) + CA12 transfected cardiac fibroblast on the pH-responsive surface with λexc = 489 ± 6 nm/436 ± 5 nm and λem = 536 ± 20 nm. (b) pH measured as a function of the distance from the cell center (arrows in c) for an unaltered cell (control with GFP) and the GFP + CA12 transfected fibroblast. Adapted from ref. 115 with permission from the Royal Society of Chemistry, Copyright 2014. | ||
This analytical tool could be adapted for other biologically relevant targets. For example, the covalent immobilisation of the In@(RB)@(FL)@MQ NPs described in section 4.1 would allow for simultaneous, ratiometric pH and Cl− mapping, as long as appropriate excitation and emission filters are used. Moreover, the possibility of immobilizing a variety of ionophores on a single substrate in different NPs could expand its potential as a multiplex platform with minimal interference on the analytical responses.
4.3 Integration of MEF nanoprobes in microfluidic devices
Perhaps the most enticing aspect of microfluidics is that the ability to manipulate small volumes of liquids reliably offers robust control over chemical conditions and kinetics, and imparts predictable laminar flows to induce precise and real-time perturbations to a closed system.120–122 However, performing spatially and temporally resolved chemical measurements in constricted volumes of liquid remains a challenge. As a transparent substrate well adapted to optical measurements, Ag@(FL)-appended coverslips are well suited for integration in microfluidic devices, thus opening possibilities regarding highly-resolved luminescent sensing of confined systems under dynamic flux.The microfluidic device fabricated is a polydimethylsiloxane (PDMS)-casted Y-channel bound to a glass coverslip supporting a patterned coverage of immobilized NPs (Fig. 13a).123 This selective disposition of NPs inside the channel was achieved by a stamping process applied to a fully covered Ag@(FL) coverslip pre-assembled by the above-described click chemistry route. The NP-coated surface was put in contact with a PDMS stamp activated by oxygen plasma to leave with an imprint identical to the inside of the microfluidic channel, due to a strong binding of the Ag@(FL) NPs with the activated PDMS stamp. The final PDMS microfluidic device was then bound to the exposed glass of the coverslip to generate the pH-sensitive channel (Fig. 13b).
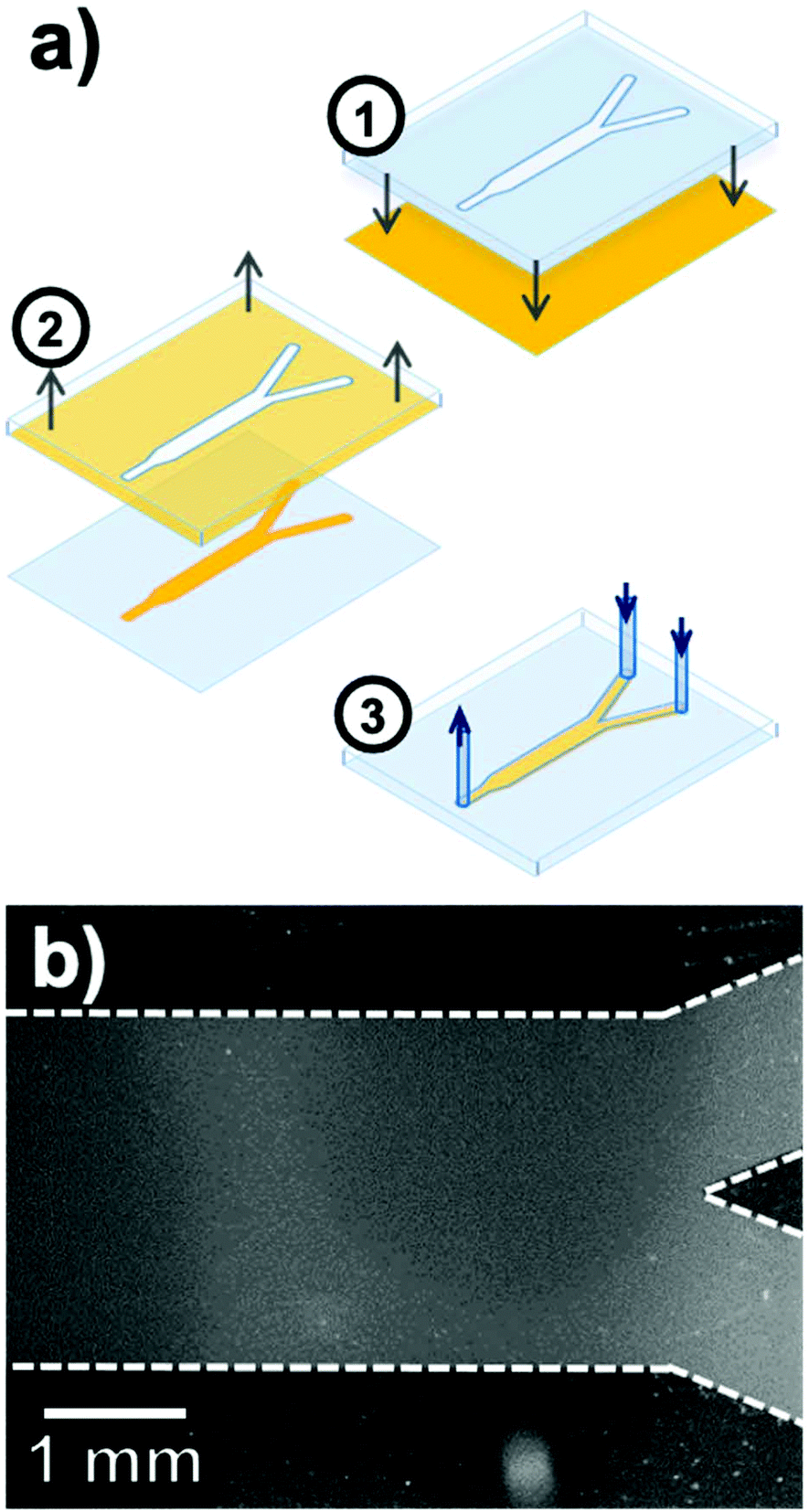 | ||
| Fig. 13 (a) Fabrication steps for the pH-sensitive microfluidic Y-channel including (1) contact of the plasma-activated PDMS stamp with the Ag@(FL) coated glass coverslip, (2) removal of the nanoparticle-bound stamp and (3) deposition of the PDMS microfluidic channel on the patterned glass surface. (b) Fluorescence image of the pH-sensitive channel after the stamping process. Adapted from ref. 123 with permission from the Royal Society of Chemistry, Copyright 2016. | ||
The analytical capabilities of this device were evaluated by eluting buffer solutions in the adjoining channels and acquiring ratiometric fluorescence images. First, as eluting liquids in microchannels induces mechanical shear forces, the robustness of the Ag@(FL) NPs to mechanical detachment was tested by flowing a pH 5.5 buffer at rates ranging from 0 mL h−1 to 5 mL h−1, representing shear stress values between 0 Pa and 0.15 Pa. The pH at various locations in the channel remained accurate with an average reading of 5.6 ± 0.2. Moreover, the raw fluorescence intensity (λexc = 489 ± 6 nm) was also stable, implying that the NPs remained immobilized on the surface. This stability under a dynamic flow is indispensable for sensing applications inside fluidic devices. The pH-sensitive microchannel also showed good contrast for distinct buffer solutions flowing alongside in the Y microchannel. Solutions of pH 5.0 and 7.0 were flowed concomitantly in the channel with flow rate ratios varying between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and 3:1, revealing by ratiometric pH imaging that the interface of the buffer solutions shifted towards the channel wall adjoining the slower flowing stream (Fig. 14a). Moreover, since proton diffusion across the interface and turbulence occurs over long distances, the cross-sectional gradients could be reported along the channel length. For identical buffer solution rates (1:1 ratio), the well-defined solution interface near the initial Y-junction inlets gave a resolution of 0.08 unit of pH per pixel. This parameter improved to 0.025 unit of pH per pixel further downstream due to the anticipated reduction in the slope of the pH gradient (Fig. 14b). Temporal resolution of the microfluidic system developed was also assessed by evaluating the effect of disturbances in the co-flowing streams of pH 5.0 and 7.0 buffer solutions. Short flow imbalances were manually initiated in the flow rates by applying a momentary force on the inlet pump piston. This disruption, recorded in a 4 frames per second video, led to the appearance of sinusoidal oscillations propagating at the interface through the channel length (Fig. 14c).
:3 and 3:1, revealing by ratiometric pH imaging that the interface of the buffer solutions shifted towards the channel wall adjoining the slower flowing stream (Fig. 14a). Moreover, since proton diffusion across the interface and turbulence occurs over long distances, the cross-sectional gradients could be reported along the channel length. For identical buffer solution rates (1:1 ratio), the well-defined solution interface near the initial Y-junction inlets gave a resolution of 0.08 unit of pH per pixel. This parameter improved to 0.025 unit of pH per pixel further downstream due to the anticipated reduction in the slope of the pH gradient (Fig. 14b). Temporal resolution of the microfluidic system developed was also assessed by evaluating the effect of disturbances in the co-flowing streams of pH 5.0 and 7.0 buffer solutions. Short flow imbalances were manually initiated in the flow rates by applying a momentary force on the inlet pump piston. This disruption, recorded in a 4 frames per second video, led to the appearance of sinusoidal oscillations propagating at the interface through the channel length (Fig. 14c).
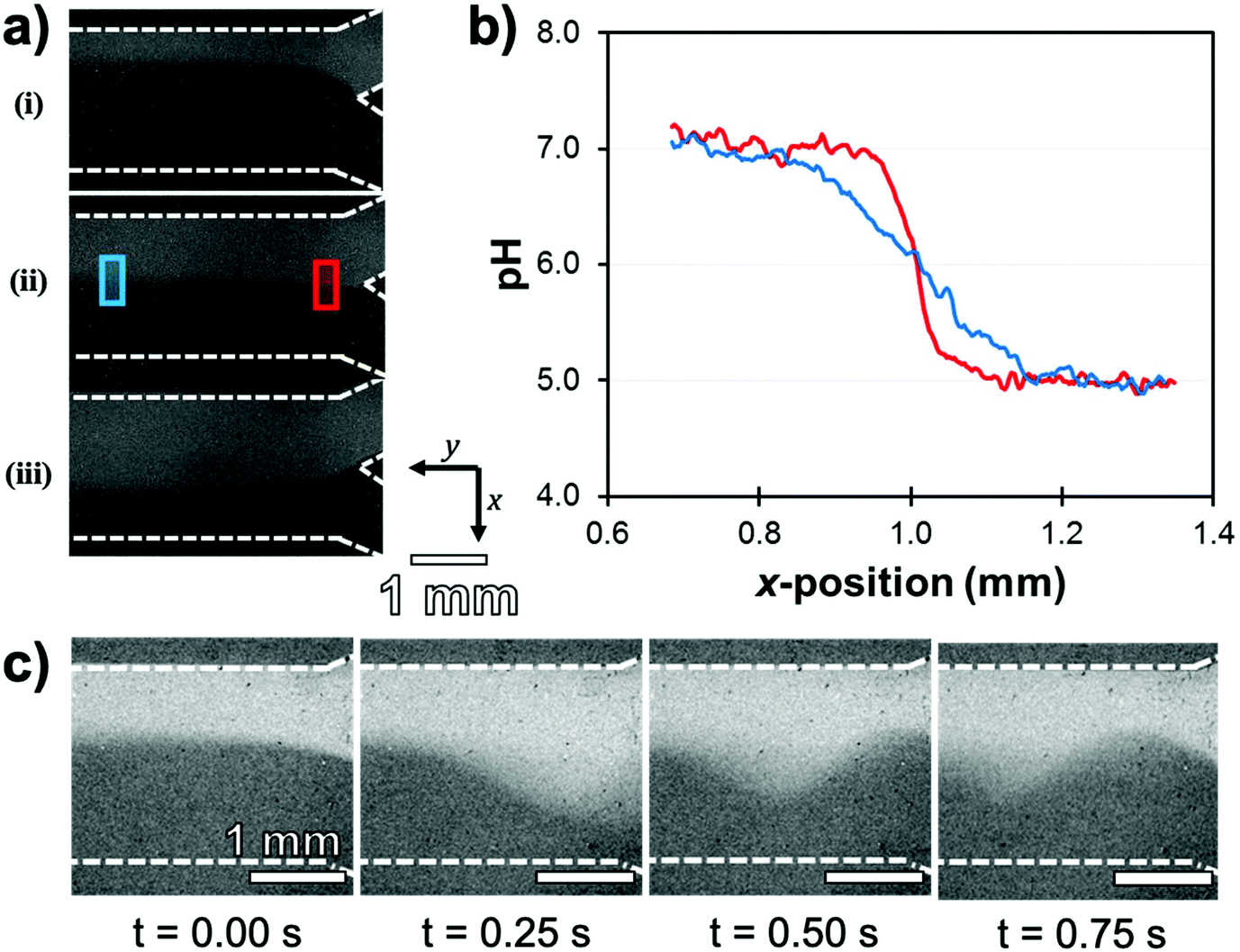 | ||
| Fig. 14 (a) Ratiometric pH images of the microfluidic channel with streams of pH 7.0 (upper channel) and pH 5.0 (lower channel) buffer solutions flowing at a total rate of 2.0 mL h−1. Relative flow rates (pH 7:pH 5) are (i) 1:3, (ii) 1:1 and (iii) 3:1. (b) Cross-sectional pH values (x-position) at longitudinal y positions of 0.5 mm (red) and 3.5 mm (blue). (c) Consecutive frames from a movie showing a pressure oscillation propagating downstream as a result of flow instabilities in pH 7 (3.0 mL h−1) and pH 5 (1.6 mL h−1) solutions inputs. Adapted from ref. 123 with permission from the Royal Society of Chemistry, Copyright 2016. | ||
The evaluation of the pH-sensitive microfluidic device validates some important analytical characteristics such as stability through time and mechanical stress, and high temporal and spatial resolutions. These benefits were further tested by studying the behaviour of live oral biofilms grown from Streptococcus salivarius. In human oral cavities, lactic acid bacterial biofilms are responsible for tooth decay through local acidification and fermentation at the gum line interface.124 The fermentation of carbohydrates such as glucose by these biofilms is classically reported with a Stephan curve plotting the pH of the biofilm upon nutrient ingestion.125 Upon growth of the biofilm inside the pH-sensitive microfluidic channel, pH fluctuations were monitored by addition of either glucose-containing or glucose-free nutrient solutions. Real-time measurements showed that removing 0.1% wt glucose from the flowing growth solution resulted in a return from pH 5.4 to 7.2 in 30 min. However, resuming the flowing of 0.1% wt glucose-containing nutrient fluid induced a rapid acidification. This behaviour is typical of a Stephan curve, where an increase in nutrient causes a rapid production of lactic acid at the interface with the biofilm, and the following recovery to ambient conditions occurs in 15–40 min.126 Whereas traditional methods to perform Stephan curve measure an average pH value for the whole biofilm and its environment,127 the spatial and temporal resolutions of this microfluidic device allow identifying microheterogeneity and transport pathways under and outside of the live biofilm.
5. Conclusions and perspectives
The use of fluorescence detection lives on in most fields of analytical science as it requires simple instrumentation and offers high sensitivity. Furthermore, a plethora of commercial and custom dyes are available with distinct absorptive and emissive properties tunable by trivial parameters such as functional groups and solvent conditions. Regardless, for certain applications, dye sensitivity is insufficient or some other photophysical characteristic needs to be improved. Undeniably, MEF allows to overcome these limitations by increasing the overall luminosity and robustness. Then again, the ability to control the photophysical state of the dye is also a genuine enabling feature offered by MEF. This minireview has highlighted that the fluorescence lifetime, photostability, anisotropy and susceptibility to dynamic quenching of a NP-appended dye can be controlled by careful consideration of NP composition, geometry, dye positioning, spectral overlap, and so on. With this information, targeted properties of a system can be altered predictably and even modulated to fit the requirements of an application at hand. For example, the effect of MEF on excited state lifetimes could be harnessed to modulate the extent of changes in fluorescence anisotropy in sensing strategies based on this principle.128,129In addition to quantitative, robust and reproducible sensing performance, the plasmonic core–shell architectures described in this paper are also capable of multiplex detection. The incorporation of distinct dyes within a single architecture enables ratiometric correction with an internal reference and concomitant sensing of different analytes. This is an unmatched advantage compared to free-state fluorophore systems. The scope of applicability of MEF concentric NPs is further stressed by addressing the sensing of biological pathways using non-invasive fluorescence microscopy, with high spatial and temporal resolution, and even in confined volumes such as microfluidic channels.
In light of these advances, it is clear that one of the next steps in applied research is to extend the use of MEF concentric core–shell structures to other biologically and environmentally relevant ions and molecules. The development of ionophores and molecular-sensitive fluorophores is a blossoming field of research that would benefit from the fluorescence enhancement, increased photostability and easy implementation on substrates. In fact, numerous analyte-sensitive molecules are immobilized on surfaces for similar motives as those stated throughout this review and they would benefit from the advantageous effects of MEF on their sensing process.130–133 Furthermore, substrate-based platforms should not be limited to planar surfaces; non-planar materials such as paper and optical fibers offer advantages such as higher surface area, flexibility and even the possibility to perform sensing by inserting a nanosensor-coated fiber within a sample.134,135 These aforementioned structural developments all tend to extend the diversity of applications of metal-enhanced fluorescence-based systems.
Combining fluorescent inorganic materials with plasmonic NPs is another avenue worth researching to yield advantages distinct from those imparted by molecular dyes. For instance, the narrow fluorescence bands and photostability of quantum dots136 or the upconversion capabilities of lanthanide-based NPs137,138 may generate MEF systems with outstanding luminescence properties. Exploring NP-coating beyond silica is also a stimulating possibility; zinc oxide is reported for its fluorescence-enhancing capabilities, and investigating its use as a shell-spacing material can allow the latter to participate in the overall optical properties of the system.139,140
Lastly, while this review discussed mostly silver and indium NPs, other non-centrosymmetric morphologies, such as those disclosed in Fig. 2, could extend applications in the NIR biological window and provide other plasmonic modes to benefit from. Yet, the surface chemistry of these nanomaterials challenges the control of shell formation and fluorophore immobilization at relevant positions, a feature which will require refining to expand application-oriented outcomes.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada, the “Fonds de la Recherche du Québec – Nature et Technologies” and the Canadian Foundation for Innovation.References
- P. A. D. Gonçalves, T. Christensen, N. Rivera, A. P. Jauho, N. A. Mortensen and M. Soljačić, Nat. Commun., 2020, 11, 1–13 Search PubMed.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- C. Vilches and R. Quidant, in Colloids for Nanobiotechnology: Synthesis, Characterization and Potential Applications, ed. W. Parak and N. Feliu, Elsevier, Amsterdam, 1st edn, 2020, vol. 16, ch. 12, pp. 307–352 Search PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- X. Huang and M. A. El-Sayed, Alexandria J. Med., 2011, 47, 1–9 CrossRef.
- L. Jauffred, A. Samadi, H. Klingberg, P. M. Bendix and L. B. Oddershede, Chem. Rev., 2019, 119, 8087–8130 CrossRef CAS PubMed.
- M. Nguyen, N. Felidj and C. Mangeney, Chem. Mater., 2016, 28, 3564–3577 CrossRef CAS.
- A. Barhoumi, W. Wang, D. Zurakowski, R. S. Langer and D. S. Kohane, Nano Lett., 2014, 14, 3697–3701 CrossRef CAS PubMed.
- A. Gellé and A. Moores, Curr. Opin. Green Sustainable Chem., 2019, 15, 60–66 CrossRef.
- N. Wu, Nanoscale, 2018, 10, 2679–2696 RSC.
- K. Li, N. J. Hogan, M. J. Kale, N. J. Halas, P. Nordlander and P. Christopher, Nano Lett., 2017, 17, 3710–3717 CrossRef CAS PubMed.
- J. H. Lee, H. Y. Cho, H. K. Choi, J. Y. Lee and J. W. Choi, Int. J. Mol. Sci., 2018, 19, 2021 CrossRef CAS PubMed.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- Y. Jeong, Y. M. Kook, K. Lee and W. G. Koh, Biosens. Bioelectron., 2018, 111, 102–116 CrossRef CAS PubMed.
- J. F. Li, C. Y. Li and R. F. Aroca, Chem. Soc. Rev., 2017, 46, 3962–3979 RSC.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- Z. Li, L. Leustean, F. Inci, M. Zheng, U. Demirci and S. Wang, Biotechnol. Adv., 2019, 37, 107440 CrossRef PubMed.
- E. Mauriz, P. Dey and L. M. Lechuga, Analyst, 2019, 144, 7105–7129 RSC.
- F. Jin, H. Li and D. Xu, Anal. Chim. Acta, 2019, 1077, 297–304 CrossRef CAS PubMed.
- L. D. S. Lapitan Jr., Y. Guo and D. Zhou, Analyst, 2015, 140, 3872–3887 RSC.
- D. Brouard, M. L. Viger, A. G. Bracamonte and D. Boudreau, ACS Nano, 2011, 5, 1888–1896 CrossRef CAS PubMed.
- W. Xiang, Q. Lv, H. Shi, B. Xie and L. Gao, Talanta, 2020, 214, 120716 CrossRef CAS PubMed.
- D. C. Vanegas, C. L. Gomes, N. D. Cavallaro, D. Giraldo-Escobar and E. S. McLamore, Compr. Rev. Food Sci. Food Saf., 2017, 16, 1188–1205 CrossRef CAS.
- Y. Lv, Y. Qin, F. Svec and T. Tan, Biosens. Bioelectron., 2016, 80, 433–441 CrossRef CAS PubMed.
- D. D. Xu, B. Zheng, C. Y. Song, Y. Lin, D. W. Pang and H. W. Tang, Sens. Actuators, B, 2019, 282, 650–658 CrossRef CAS.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- N. G. Khlebtsov, L. A. Trachuk and A. G. Mel'nikov, Opt. Spectrosc., 2005, 98, 77–83 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- Z. Xie, W. Yu, T. Wang, H. Zhang, Y. Fu, H. Liu, F. Li, Z. Lu and Q. Sun, Plasmonics, 2011, 6, 565–580 CrossRef.
- S. W. Lee, K. S. Lee, J. Ahn, J. J. Lee, M. G. Kim and Y. B. Shin, ACS Nano, 2011, 5, 897–904 CrossRef CAS PubMed.
- X. Zhang, C. R. Yonzon and R. P. Van Duyne, J. Mater. Res., 2006, 21, 1083–1092 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052–7071 CrossRef CAS PubMed.
- Y. Wan, Z. Guo, X. Jiang, K. Fang, X. Lu, Y. Zhang and N. Gu, J. Colloid Interface Sci., 2013, 394, 263–268 CrossRef CAS PubMed.
- Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- F. Kretschmer, S. Mühlig, S. Hoeppener, A. Winter, M. D. Hager, C. Rockstuhl, T. Pertsch and U. S. Schubert, Part. Part. Syst. Charact., 2014, 31, 721–744 CrossRef.
- J. Schubert and M. Chanana, Curr. Med. Chem., 2018, 25, 4553–4586 CrossRef CAS PubMed.
- M. Potara, M. Focsan, A. M. Craciun, I. Botiz and S. Astilean, in New Polymer Nanocomposites for Environmental Remediation, ed. C. M. Hussain and A. K. Mishra, Elsevier, Amsterdam, 2018, ch. 15, pp. 361–387 Search PubMed.
- R. Contreras-Cáceres, J. Pacifico, I. Pastoriza-Santos, J. Pérez-Juste, A. Fernández-Barbero and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 3070–3076 CrossRef.
- M. Khatami, H. Q. Alijani, M. S. Nejad and R. S. Varma, Appl. Sci., 2018, 8, 411 CrossRef.
- A. Mirzaei, K. Janghorban, B. Hashemi and G. Neri, J. Nanopart. Res., 2015, 17, 1–36 CrossRef CAS.
- P. Mélinon, S. Begin-Colin, J. L. Duvail, F. Gauffre, N. H. Boime, G. Ledoux, J. Plain, P. Reiss, F. Silly and B. Warot-Fonrose, Phys. Rep., 2014, 543, 163–197 CrossRef.
- S. Talebzadeh, C. Queffélec and D. A. Knight, Nanoscale Adv., 2019, 1, 4578–4591 RSC.
- Q. Chen, Y. Ge, H. Granbohm and S. P. Hannula, Nanomaterials, 2018, 8, 362 CrossRef PubMed.
- M. Ma, H. Chen, Y. Chen, X. Wang, F. Chen, X. Cui and J. Shi, Biomaterials, 2012, 33, 989–998 CrossRef CAS PubMed.
- Y. Kobayashi, H. Katakami, E. Mine, D. Nagao, M. Konno and L. M. Liz-Marzán, J. Colloid Interface Sci., 2005, 283, 392–396 CrossRef CAS PubMed.
- H. H. Lin and I. C. Chen, J. Phys. Chem. C, 2015, 119, 26663–26671 CrossRef CAS.
- K. Aslan, M. Wu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2007, 129, 1524–1525 CrossRef CAS PubMed.
- O. G. Tovmachenko, C. Graf, D. J. van den Heuvel, A. van Blaaderen and H. C. Gerritsen, Adv. Mater., 2006, 18, 91–95 CrossRef CAS.
- C. R. Simovski, Opt. Lett., 2019, 44, 2697–2700 CrossRef CAS.
- S. Sun, I. L. Rasskazov, P. S. Carney, T. Zhang and A. Moroz, J. Phys. Chem. C, 2020, 124, 13365–13373 CrossRef CAS.
- H. Zong, X. Wang, X. Mu, J. Wang and M. Sun, Chem. Rec., 2019, 19, 818–842 CrossRef CAS PubMed.
- J. Dong, Z. Zhang, H. Zheng and M. Sun, Nanophotonics, 2015, 4, 472–490 CAS.
- R. Knoblauch, H. Ben Hamo, R. Marks and C. D. Geddes, J. Phys. Chem. C, 2020, 124, 4723–4737 CrossRef CAS.
- J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308–1346 RSC.
- C. D. Geddes, Reviews in Plasmonics 2017, Springer, Cham, Switzerland, 2019 Search PubMed.
- Y. Gutiérrez, M. Losurdo, F. González, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2020, 124, 7386–7395 CrossRef.
- M. G. Blaber, M. D. Arnold and M. J. Ford, J. Phys.: Condens. Matter, 2010, 22, 143201 CrossRef CAS PubMed.
- E. González, F. Merkoçi, R. Arenal, J. Arbiol, J. Esteve, N. G. Bastús and V. Puntes, J. Mater. Chem. A, 2016, 4, 200–208 RSC.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS PubMed.
- Y. Gutierrez, D. Ortiz, J. M. Sanz, J. M. Saiz, F. Gonzalez, H. O. Everitt and F. Moreno, Opt. Express, 2016, 24, 20621–20631 CrossRef PubMed.
- J. M. Sanz, D. Ortiz, R. Alcaraz De La Osa, J. M. Saiz, F. González, A. S. Brown, M. Losurdo, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2013, 117, 19606–19615 CrossRef CAS.
- J. M. McMahon, G. C. Schatz and S. K. Gray, Phys. Chem. Chem. Phys., 2013, 15, 5415–5423 RSC.
- F. Magnan, J. Gagnon, F. G. Fontaine and D. Boudreau, Chem. Commun., 2013, 49, 9299–9301 RSC.
- A. I. Dragan and C. D. Geddes, J. Appl. Phys., 2010, 108, 094701 CrossRef.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836–2846 CrossRef.
- Q. Zhang, N. Li, J. Goebl, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 18931–18939 CrossRef CAS PubMed.
- H. Tanimoto, K. Hashiguchi and S. Ohmura, J. Phys. Chem. C, 2015, 119, 19318–19325 CrossRef CAS.
- B. J. Wiley, S. H. Im, Z. Y. Li, J. McLellan, A. Siekkinen and Y. Xia, J. Phys. Chem. B, 2006, 110, 15666–15675 CrossRef CAS PubMed.
- F. Huang, G. Ma, J. Liu, J. Lin, X. Wang and L. Guo, Small, 2016, 12, 5442–5448 CrossRef CAS PubMed.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- H. Yockell-Lelièvre, F. Lussier and J. F. Masson, J. Phys. Chem. C, 2015, 119, 28577–28585 CrossRef.
- H. Wang, D. W. Brandl, P. Nordlander and N. J. Halas, Acc. Chem. Res., 2007, 40, 53–62 CrossRef CAS PubMed.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS PubMed.
- Q. C. Sun, Y. Ding, S. M. Goodman, H. H. Funke and P. Nagpal, Nanoscale, 2014, 6, 12450–12457 RSC.
- T. M. D. Dang, T. T. T. Le, E. Fribourg-Blanc and M. C. Dang, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2011, 2, 015009 Search PubMed.
- M. Jin, G. He, H. Zhang, J. Zeng, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 10560–10564 CrossRef CAS PubMed.
- I. Pastoriza-Santos, A. Sánchez-Iglesias, B. Rodríguez-González and L. M. Liz-Marzán, Small, 2009, 5, 440–443 CrossRef CAS PubMed.
- S. Tian, O. Neumann, M. J. McClain, X. Yang, L. Zhou, C. Zhang, P. Nordlander and N. J. Halas, Nano Lett., 2017, 17, 5071–5077 CrossRef CAS PubMed.
- M. J. McClain, A. E. Schlather, E. Ringe, N. S. King, L. Liu, A. Manjavacas, M. W. Knight, I. Kumar, K. H. Whitmire, H. O. Everitt, P. Nordlander and N. J. Halas, Nano Lett., 2015, 15, 2751–2755 CrossRef CAS PubMed.
- B. D. Clark, C. R. Jacobson, M. Lou, J. Yang, L. Zhou, S. Gottheim, C. J. DeSantis, P. Nordlander and N. J. Halas, Nano Lett., 2018, 18, 1234–1240 CrossRef CAS PubMed.
- B. D. Clark, C. R. Jacobson, M. Lou, D. Renard, G. Wu, L. Bursi, A. S. Ali, D. F. Swearer, A. L. Tsai, P. Nordlander and N. J. Halas, ACS Nano, 2019, 13, 9682–9691 CrossRef CAS PubMed.
- J. Asselin, C. Boukouvala, E. R. Hopper, Q. M. Ramasse, J. S. Biggins and E. Ringe, ACS Nano, 2020, 14, 5968–5980 CrossRef CAS PubMed.
- J. S. Biggins, S. Yazdi and E. Ringe, Nano Lett., 2018, 18, 3752–3758 CrossRef CAS PubMed.
- R. Das and R. K. Soni, Appl. Surf. Sci., 2019, 489, 755–765 CrossRef CAS.
- R. Das and R. K. Soni, RSC Adv., 2017, 7, 32255–32263 RSC.
- F. Magnan, J. Gagnon, F. G. Fontaine and D. Boudreau, Chem. Commun., 2013, 49, 9299–9301 RSC.
- N. H. Chou, X. Ke, P. Schiffer and R. E. Schaak, J. Am. Chem. Soc., 2008, 130, 8140–8141 CrossRef CAS PubMed.
- J. Malicka, I. Gryczynski, Z. Gryczynski and J. R. Lakowicz, Anal. Biochem., 2003, 315, 57–66 CrossRef CAS PubMed.
- J. Asselin, P. Legros, A. Grégoire and D. Boudreau, Plasmonics, 2016, 11, 1369–1376 CrossRef CAS.
- D. D. Evanoff and G. Chumanov, J. Phys. Chem. B, 2004, 108, 13957–13962 CrossRef CAS.
- J. Zhang, N. Ma, F. Tang, Q. Cui, F. He and L. Li, ACS Appl. Mater. Interfaces, 2012, 4, 1747–1751 CrossRef CAS PubMed.
- R. Chhabra, J. Sharma, H. Wang, S. Zou, S. Lin, H. Yan, S. Lindsay and Y. Liu, Nanotechnology, 2009, 20, 485201 CrossRef PubMed.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, Boston, MA, 3rd edn, 2006, ch. 10, pp. 353–382 Search PubMed.
- J. Chen, J. Liu, X. Chen and H. Qiu, Chin. Chem. Lett., 2019, 30, 1575–1580 CrossRef CAS.
- H. Szmacinski and J. R. Lakowicz, Anal. Chem., 2008, 80, 6260–6266 CrossRef CAS PubMed.
- S. Wu, H. Min, W. Shi and P. Cheng, Adv. Mater., 2020, 32, 1–14 Search PubMed.
- N. Klonis and W. H. Sawyer, J. Fluoresc., 1996, 6, 147–157 CrossRef CAS PubMed.
- J. Asselin, M. P. Lambert, N. Fontaine and D. Boudreau, Chem. Commun., 2017, 53, 755–758 RSC.
- G. Miesenböck, D. A. De Angelis and J. E. Rothman, Nature, 1998, 394, 192–195 CrossRef PubMed.
- R. K. Skitchenko, D. Usoltsev, M. Uspenskaya, A. V. Kajava and A. Guskov, Bioinformatics, 2020, 36, 3064–3071 CrossRef PubMed.
- H. Bischof, S. Burgstaller, M. Waldeck-Weiermair, T. Rauter, M. Schinagl, J. Ramadani-Muja, W. F. Graier and R. Malli, Cells, 2019, 8, 492 CrossRef CAS PubMed.
- C. D. Geddes, Sens. Actuators, B, 2001, 72, 188–195 CrossRef CAS.
- M. S. Mehata and H. B. Tripathi, J. Lumin., 2002, 99, 47–52 CrossRef CAS.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, Boston, MA, 3rd edn, 2006, ch. 8, pp. 277–330 Search PubMed.
- Y. Kumamoto, A. Taguchi, M. Honda, K. Watanabe, Y. Saito and S. Kawata, ACS Photonics, 2014, 1, 598–603 CrossRef CAS.
- B. Korzeniowska, R. Nooney, D. Wencel and C. McDonagh, Nanotechnology, 2013, 24, 442002 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- J. Asselin, C. Roy, D. Boudreau, Y. Messaddeq, R. Bouchareb and P. Mathieu, Chem. Commun., 2014, 50, 13746–13749 RSC.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
- F. Grinnell and M. H. Bennett, J. Cell Sci., 1981, 48, 19–34 CAS.
- C. P. S. Potter and A. L. Harris, Br. J. Cancer, 2003, 89, 2–7 CrossRef CAS PubMed.
- A. J. DeMello, Nature, 2006, 442, 394–402 CrossRef CAS PubMed.
- T. A. Duncombe, A. M. Tentori and A. E. Herr, Nat. Rev. Mol. Cell Biol., 2015, 16, 554–567 CrossRef CAS PubMed.
- A. Karimi, D. Karig, A. Kumar and A. M. Ardekani, Lab Chip, 2015, 15, 23–42 RSC.
- M. Parvinzadeh Gashti, J. Asselin, J. Barbeau, D. Boudreau and J. Greener, Lab Chip, 2016, 16, 1412–1419 RSC.
- P. W. Caufield, C. N. Schön, P. Saraithong, Y. Li and S. Argimón, J. Dent. Res., 2015, 94, 110S–118S CrossRef CAS PubMed.
- R. M. Stephan and B. F. Miller, J. Dent. Res., 1943, 22, 45–51 CrossRef.
- I. Kleinberg, J. Dent. Res., 1961, 40, 1087–1111 CrossRef.
- W. M. Edgar, Caries Res., 1976, 10, 241–254 CrossRef CAS PubMed.
- L. Wang, H. Ye, H. Q. Sang and D. D. Wang, Chin. J. Anal. Chem., 2016, 44, 799–803 CAS.
- N. Duan, S. J. Wu and Z. P. Wang, Chin. J. Anal. Chem., 2011, 39, 300–304 CAS.
- K. Kołątaj, J. Krajczewski and A. Kudelski, Environ. Chem. Lett., 2020, 18, 529–542 CrossRef.
- J. Luan, A. Seth, R. Gupta, Z. Wang, P. Rathi, S. Cao, H. Gholami Derami, R. Tang, B. Xu, S. Achilefu, J. J. Morrissey and S. Singamaneni, Nat. Biomed. Eng., 2020, 4, 518–530 CAS.
- M. Zheng, Y. Kang, D. Liu, C. Li, B. Zheng and H. Tang, Sens. Actuators, B, 2020, 319, 128263 CrossRef CAS.
- K. Girigoswami and N. Akhtar, Int. J. Nano Dimens., 2019, 10, 1–17 CAS.
- X. A. Ton, V. Acha, P. Bonomi, B. Tse Sum Bui and K. Haupt, Biosens. Bioelectron., 2015, 64, 359–366 CrossRef CAS PubMed.
- B. J. Yun, J. E. Kwon, K. Lee and W. G. Koh, Sens. Actuators, B, 2019, 284, 140–147 CrossRef CAS.
- A. K. Tobias and M. Jones, J. Phys. Chem. C, 2019, 123, 1389–1397 CrossRef CAS.
- Y. Yang, P. Su and Y. Tang, ChemNanoMat, 2018, 4, 1097–1120 CrossRef CAS.
- C. D. S. Brites, S. Balabhadra and L. D. Carlos, Adv. Opt. Mater., 2019, 7, 1801239 CrossRef.
- N. R. Nirala and G. Shtenberg, Front. Chem., 2019, 7, 1–8 CrossRef PubMed.
- N. R. Nirala and G. Shtenberg, Nanomaterials, 2020, 10, 1–12 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |