
A dual responsive photonic liquid for independent modulation of color brightness and hue†
Yun
Liu
 ac,
Qingsong
Fan
c,
Guanghao
Zhu
b,
Gongpu
Shi
a,
Huiru
Ma
*b,
Wei
Li
a,
Tianlong
Wu
a,
Jitao
Chen
a,
Yadong
Yin
*c and
Jianguo
Guan
*a
ac,
Qingsong
Fan
c,
Guanghao
Zhu
b,
Gongpu
Shi
a,
Huiru
Ma
*b,
Wei
Li
a,
Tianlong
Wu
a,
Jitao
Chen
a,
Yadong
Yin
*c and
Jianguo
Guan
*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, International School of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China. E-mail: guanjg@whut.edu.cn
bDepartment of Chemistry, School of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, Wuhan 430070, China. E-mail: mahr@whut.edu.cn
cDepartment of Chemistry, University of California, Riverside, CA, 92521, USA. E-mail: yadong.yin@ucr.edu
First published on 8th May 2021
Abstract
Responsive chromic materials are highly desirable in the fields of displays, anti-counterfeiting, and camouflage, but their advanced applications are usually limited by the unrealized delicate and independent tunability of their three intrinsic attributes of color. This work achieves the separate, continuous, and reversible modulation of structural color brightness and hue with an aqueous suspension of dual-responsive Fe3O4@polyvinylpyrrolidone (PVP)@poly(N-isopropyl acrylamide) (PNIPAM) flexible photonic nanochains. The underlying modulation mechanism of color brightness was experimentally and numerically deciphered by analyzing the morphological responses to stimuli. When an increasing magnetic field was applied, the random worm-like flexible photonic nanochains gradually orientated along the field direction, due to the dominant magnetic dipole interaction over the thermal motion, lengthening the orientation segment length up to the whole of the nanochains. Consequently, the suspension displays increased color brightness (characterized by diffraction intensity). Meanwhile, the color hue (characterized by diffraction frequency) could be controlled by temperature, due to the volume changes of the interparticle PNIPAM. The achieved diverse color modulation advances the next-generation responsive chromic materials and enriches the basic understanding of the color tuning mechanisms. With versatile and facile color tunability and shape patterning, the developed responsive chromic liquid promises to have attractive potential in full-color displays and in adaptive camouflages.
New conceptsThe developed responsive chromic materials mainly carry out on–off switch or overall changes in color when stimulated by single or multiple stimuli. Limited by the current color modulation methods, the independent delicate modulation of the three intrinsic attributes of color remains a challenge. We demonstrate a structural color brightness modulation principle that relies on the dynamic controllability of the periodic range of the photonic crystalline structures while keeping their lattice constant the same, in order to achieve tunable structural colors with changeable brightness and maintained hue in an aqueous suspension of Fe3O4@polyvinylpyrrolidone (PVP)@poly(N-isopropyl acrylamide) (PNIPAM) flexible photonic nanochains, by changing the external magnetic field (H). The flexible photonic nanochains with random worm-like conformations exhibit gradually increasing periodic ranges along the field direction with increasing field strength, due to the dominant magnetic dipole interaction over the thermal motion. Macroscopically, the suspension displays a continuous and rapid response of color brightness to the strength of H. Furthermore, the color hue can be adaptively tuned by altering the temperature when needed. The achievement of a versatile and respective tunability of the color brightness and hue by modulating different external stimuli creates an exciting new avenue for the development of advanced responsive chromic materials. |
Introduction
Color plays an essential role in carrying and covering visual information.1–3 In nature, some living creatures, such as chameleons, turtle beetles, seahorses, and certain fish, can utilize their body color changes for communication of information, temperature regulation, warning or as camouflage, boosting their adaptability to the surrounding environment and survival ability from predators.4–6 Inspired by these phenomena, a large number of responsive chromic materials that change color in response to external fields such as light, electric field, magnetic field, temperature, pressure, and chemical agents, have been developed for the growing demands within self-adaptive camouflages, intelligent textiles, sensing, smart displays, energy-saving building materials and in fascinating decorations over the past decades.7–20 However, most of the materials can only carry out the general control of color, especially as an on–off switch or in broad frequency ranges.7–9,15 In order to meet the critical requirements for their application in full-color displays, high-security anti-counterfeiting and environmentally adaptive camouflage, developing next-generation responsive chromic materials that can adaptively modulate their color hue, saturation, and brightness (HSB) in a respective, continuous, and reversible manner by two or multiple independent external stimuli, is essential.4,21 So far, such responsive chromic materials are not available, due to the limitations of the existing color modulation mechanisms or the basic materials. Herein, we report a versatile color-changing aqueous liquid that can independently, continuously, and reversibly tune the color brightness and hue by modulating the external magnetic field (H) and surrounding temperature (T), based on the dual responsive individual photonic nanochains (PNCs) of thermosensitive polymer encapsulating superparamagnetic colloidal nanocrystal clusters (CNCs).While colors generally stem from chemical colorants, brilliant structural colors can be generated through light-matter interactions such as diffraction, scattering, and interference.22–24 These are environmentally friendly, energy-saving, and anti-photobleaching, demonstrating their increasing importance for future color materials.25,26 As typical materials exhibiting structural colors, photonic crystals (PCs) are periodically arranged dielectric materials. The photonic band gaps are in the range of the visible spectra when the lattice constants are comparable to the wavelength of visible light.27 In this case, their diffraction wavelength, diffraction intensity, and peak width at half height correspond to the hue, brightness, and saturation, respectively. When stimuli-responsive materials are used as building blocks or are incorporated into the PCs as the surrounding matrix, the band gaps may be tuned by changing the lattice constant, refractive index, or orientation state, endowing them with dynamic tunability of the structural colors in response to external stimuli, such as pH, T, humidity, electric field and H.28–34 Among the materials, magnetically responsive PCs have the advantages of having a simple assembly process, being reversible and having a rapid response to H, as well as bright structural colors.31,35–37 They are based on establishing a balance between the magnetic attraction and the electrostatic or steric repulsion, thus they display tunable color hues that cover the entire visible spectra.38–40 The one-dimensional periodically arrayed structures of superparamagnetic CNCs can be fixed by inorganic materials or polymers, obtaining various forms of one-dimensional magnetic PCs including films,41 balls,30,42–44 fibers,45 and individual PNCs.46–50 They can display either constant structural colors or tunable colors with overall changes in HSB when stimulated by a single external field.
In this paper, the independent, continuous, and reversible modulation of structural colors for their brightness and hue has been demonstrated using an aqueous suspension of magnetically and thermally responsive Fe3O4@polyvinylpyrrolidone (PVP)@poly(N-isopropyl acrylamide) (PNIPAM) flexible PNCs. As indicated by both the experimental and numerical simulation results, the continuous and reversible tunability of the color brightness at a constant color hue benefits from the flexibility of encapsulated PNIPAM and the large chain length, and this guarantees the gradual domination of magnetic interaction over thermal motion with increasing H and enables the orientation and stretch of the chains along the field direction. Consequently, the flexible PNCs exhibit better periodic structures along the field direction under stronger H, leading to enhanced color brightness. Meanwhile, the versatile controllability of the color hue was achieved due to the thermally responsive property of the linking PNIPAM, which adjusts the interparticle distance by volume changes with T. The realization of the diverse tunability of structural color in both brightness and hue can be expected to help develop next-generation chromic materials and enrich the basic understanding of structural color modulation mechanisms. The obtained dual responsive liquid shows great potential for smart structural color materials.
Results and discussion
A dual-responsive aqueous dispersion of Fe3O4@polyvinylpyrrolidone (PVP)@poly(N-isopropyl acrylamide) (PNIPAM) flexible photonic nanochains (PNCs) was used to investigate the independent, continuous, and reversible modulation of color brightness and hue. The monodisperse Fe3O4@PVP colloid nanocrystal clusters (CNCs) with an average diameter of 140 nm (Fig. S1, ESI†) were used as building blocks for the Fe3O4@PVP@PNIPAM flexible PNCs, which were fabricated using a hydrogen bond-guided template polymerization method.49 As shown in Fig. 1a, the formation procedures of the Fe3O4@PVP@PNIPAM flexible PNCs include the self-assembly of Fe3O4@PVP CNCs induced by external H, and the subsequent in situ polymerization of NIPAM initiated by UV light. In the reaction system, PAA functions as a bridge between PVP and NIPAM by forming hydrogen bonds.49 The formation procedure can be illustrated as follows: first, PAA forms hydrogen bonds with PVP and NIPAM, leading to a much higher NIPAM concentration within the PVP brush shells of Fe3O4@PVP CNCs than that outside in the solution. Then, the CNCs with NIPAM within the PVP shells aligned along H with a nearly equal interparticle distance when an external field H was applied. Finally, the polymerization of NIPAM on the Fe3O4 particle surface and the interparticle regions was initiated by UV light, resulting in individual one-dimensional periodical nanochains linked by polymers. The addition and amount of PAA significantly influenced the formation of the flexible PNCs, because it determines the amount of NIPAM that is adsorbed onto the surface of the Fe3O4@PVP CNCs. The product obtained without the addition of PAA (Fig. S2a, ESI†) exhibits a monodisperse sphere morphology with a rough surface, and this shows no obvious difference compared to the initial Fe3O4@PVP CNCs (Fig. S1, ESI†), indicating the poor interaction between PVP and NIPAM. When the PAA concentration was 0.31 mg mL−1, nanochains with a perfect chain structure could still not be obtained due to the insufficient surface coating of PNIPAM (Fig. S2b, ESI†). When the PAA concentration was further increased to 1.67 mg mL−1, flexible PNCs could be obtained, but they tended to form bundle chains with thick polymer shells (Fig. S2c, ESI†). The slightly decreased interparticle distance within the nanochains that was obtained at a high PAA concentration was caused by the compression of the PVP brush shells, due to their strong interaction with PAA.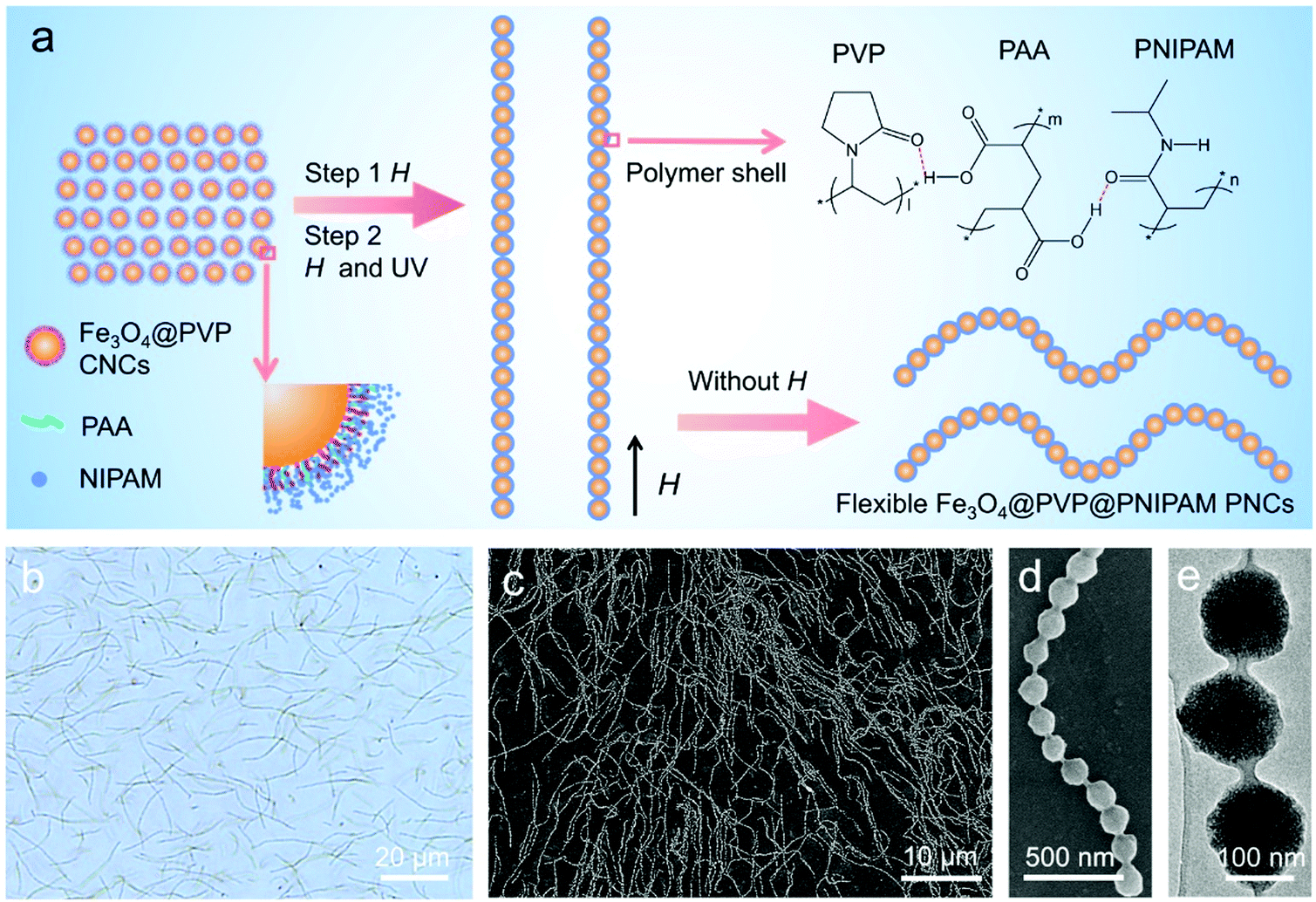 | ||
| Fig. 1 The preparation and characterization of the typical Fe3O4@PVP@PNIPAM flexible PNCs. (a) Schematic illustration showing the formation process of the Fe3O4@PVP@PNIPAM flexible PNCs; (b) bright-field optical microscopy image; (c and d) SEM images; (e) TEM image. | ||
The bright-field optical microscopy image and scanning electron microscopy (SEM) images (Fig. 1b and c, and Fig. S3a in the ESI†) clearly show that the products of the individual nanochains exhibit a randomly natural bending state with an average end-to-end distance (D) of about 13 μm. When an external magnetic field (H) was applied, the nanochains aligned along the field direction with an average chain length (L) of up to about 15 μm, which is larger than D (Fig. S3b and c in the ESI†). This confirms the polymer-analogous flexibility of the nanochains. The magnified SEM image (Fig. 1d), TEM image (Fig. 1e), and the FT-IR spectrum (Fig. S4a, ESI†) reveal that the nanochains are pearl necklace-like structures, in which the particle cores are coated and separated by a PNIPAM layer. The exact mass percentage of polymers was calculated as 34.37%, based on the TG-DSC data (Fig. S4b, ESI†). Furthermore, the superparamagnetic property of the obtained Fe3O4@PVP@PNIPAM flexible PNCs, endowing them with the reversible magnetically responsive property, was confirmed by the magnetic hysteresis loop (Fig. S4c, ESI†), which showed negligible remanent magnetization (Mr) and coercivity (Hc) with a mass saturation magnetization (Ms) value of 33.09 emu·g−1.
The aqueous suspension of the obtained Fe3O4@PVP@PNIPAM flexible PNCs exhibits independent, continuous, and reversible tunability of diffraction intensity and diffraction wavelength under different H and temperature (T) values. As seen in Fig. 2a, the aqueous suspension of the typical Fe3O4@PVP@PNIPAM flexible PNCs at 6 °C exhibited a yellow color, but had enhanced brightness with increasing H. At H = 10 Gs, only a dark yellow color appeared. When H was increased to 50 Gs, a homogeneous color could be observed. The color became much brighter when H was further enhanced to 100 and 400 Gs. The 2D contour map of the reflection spectra (Fig. 2b) confirms that the flexible PNCs under different H values show the same diffraction peak at around 600 nm, but the reflectance (R) increases with the increase in H, and this is consistent with the dark-field optical microscopy observations. This completely distinguishes it from that of the rigid straight magnetic counterparts, of which the structural color brightness is almost independent of the strength of H. According to Bragg's law:
λ = 2nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ, sinθ, | (1) |
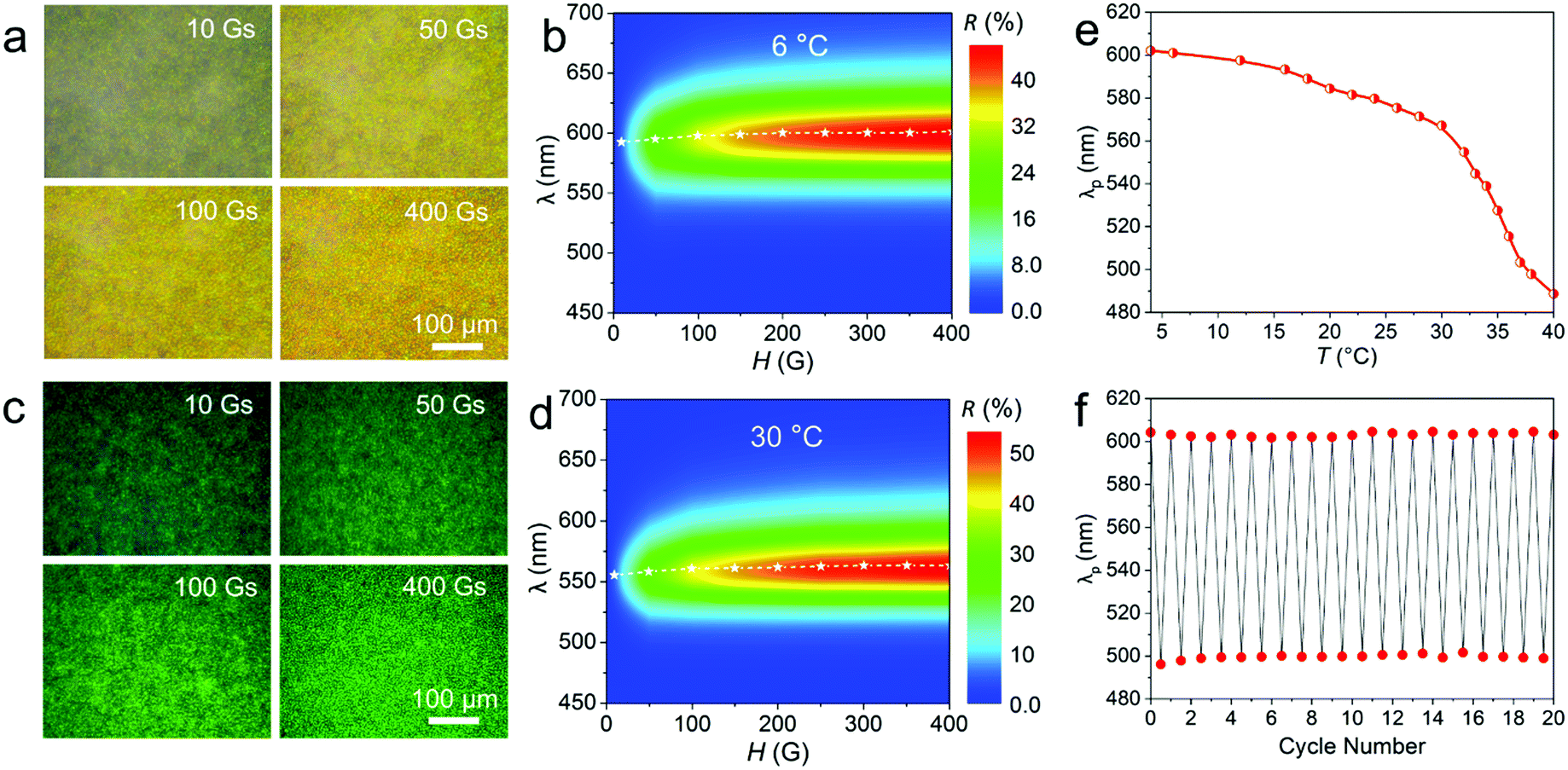 | ||
| Fig. 2 The dual responsive optical properties of the typical Fe3O4@PVP@PNIPAM flexible PNCs. (a and c) Dark-field optical microscopy images, and (b and d) 2D contour maps of the reflection spectra obtained under different H and T values. (a and b) 6 °C; (c and d) 30 °C. (e) Diffraction wavelength as a function of T. (f) Cycling performance showing the variation in diffraction peaks between 4 and 38 °C over 20 cycles. The magnetic field direction in (a and c) is parallel to the observation direction. | ||
The dynamic changes of color hue with T are shown in Movies S2 and S3 (ESI†). When the sample was stimulated by a drop of ice water (4 °C), the diffraction color changed from green to yellow (Movie S2, ESI†). When it was stimulated by a drop of warm water at 40 °C, the diffraction color rapidly changed from green to blue and then recovered to green (Movie S3, ESI†), showing the reversible tunability of the color hue. The shift in diffraction color was attributed to the thermosensitive property of PNIPAM, as this exhibits a reversible transition between the hydrophilic state and the hydrophobic state as T changes.52 When stimulated by warm water, the PNIPAM in the interlayer and in the shell of the nanochains experienced rapid shrinking by expelling water out, leading to a decrease in the interparticle distance and thus a blue shift in the diffraction color. When T was recovered to room temperature, PNIPAM returned to a swollen state, leading to increased interparticle distance and the corresponding recovery of the structural color. The as-obtained flexible PNCs showed a more rapid speed of color change than the corresponding thermochromic films and balls,44,53 and this can be illustrated by their reduced volume change relaxation time:54
| τ = A2π−2S−1, | (2) |
In order to understand the magnetic field strength-dependent optical properties of the flexible PNCs, we recorded R versus the magnetic field induction time (t) at different H values. As shown in Fig. 3a, with an increase of t, R quickly increases before reaching a constant maximum value (Rmax). Increasing H enhances Rmax but reduces the shortest time needed for the corresponding Rmax (tmin). The value of tmin is almost on the second scale if H is stronger than 10 Gs and is typically less than 1 s at a H value stronger than 100 Gs. This confirms the swift response of the PNCs to H. As depicted in Fig. 3b, the correlation of tmin and Rmax with H can be well fitted by the Boltzmann function model, implying that the incorporation of magnetic energy balances the thermal motion of the flexible PNCs, leading to the improvement of Rmax. The diffraction intensity tunability of the flexible PNCs with H may benefit from their polymer-like flexibility and superparamagnetic property, enabling them to become ordered structures segment by segment with increasing H.
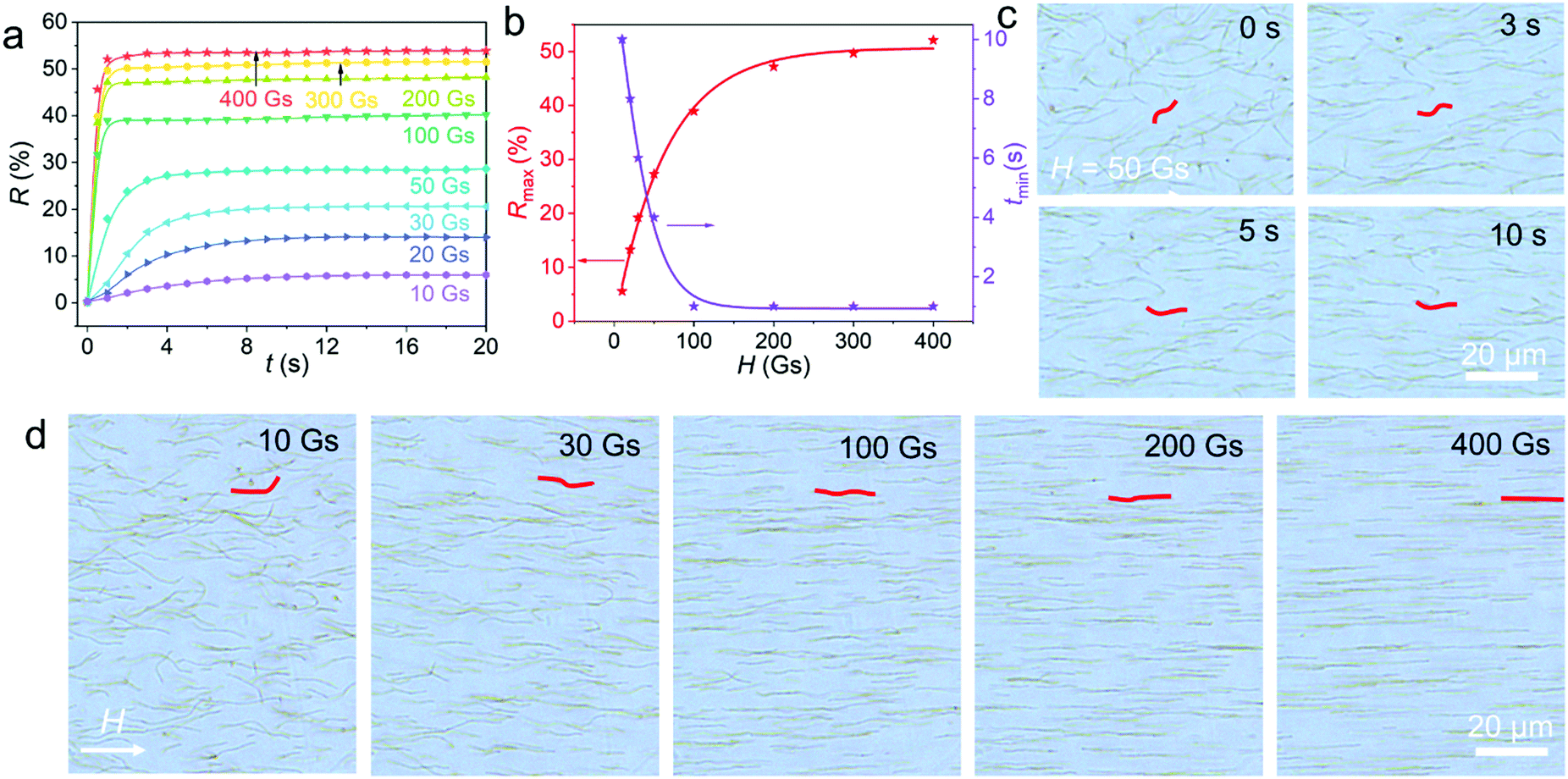 | ||
| Fig. 3 The influence of H and t on the reflectance and orientation of the typical Fe3O4@PVP@PNIPAM flexible PNCs. (a) R, as a function of t at different H values. (b) The influence of H on the corresponding maximum reflectance (Rmax) and the shortest time needed for Rmax (tmin). The solid lines are the fitted results of the Rmax and tmin data as a function of H based on the Boltzmann model. (c) Bright-field optical microscopy images obtained under 50 Gs with different t values. (d) Bright-field optical microscopy images obtained at different H values with t = 10 s. Typical nanochains are highlighted in red in (c) and in (d) to ease observation. | ||
To intuitively substantiate the above assumption, we used optical microscopy to directly observe the orientation process of the flexible PNCs. As shown in Fig. 3c and in Movie S4 (ESI†), when a weak H value of 50 Gs was applied, the randomly dispersed flexible PNCs partly orientated along the field direction as t was increased from 0 to 5 s, but they still exhibited bent segments that could not be stretched into a straight state. When t was extended to 10 s, the PNCs did not show any clear changes in conformation, except for small fluctuations, indicating that the complete stretch orientation could not be achieved at low H by prolonging t, in accordance with the optical property results (Fig. 3a). Fig. 3d shows the balance conformations of the flexible PNCs under different H values. Most of the PNCs did not achieve the deflection orientation along H under 10 Gs. When H was increased to 20 and 30 Gs, the flexible PNCs started to orientate along H, but they existed in an obvious bending state, indicating that the energy supplied by the weak magnetic field was insufficient to balance the thermal motion of most segments and stretch the bent chains. When H was further enhanced to 100 and 200 Gs, most of the flexible long PNCs carried out stretch orientation with a few bent segments. All the PNCs were stretched entirely into a straight state at H = 400 Gs. The small movement towards the field direction under strong H is caused by the magnetic packing force induced by the inhomogeneous magnetic field supplied by a magnet during the observation process. Careful observation indicated that short PNCs are easier for facilitating the complete orientation along H than the long ones, suggesting stronger H dependence of the optical properties for the long flexible PNCs than for the short ones. Movies S5 and S6 (ESI†) show the dynamic orientation process of the flexible PNCs under 400 Gs and when varying H from 0 to 400 to 0 Gs, respectively. The movies confirm the rapid orientation under strong H and a continuous, reversible, and controllable orientation transformation with H, resulting in the tunable periodicity and optical properties of the long flexible PNCs with H.
Based on the microscopy observations, we assumed a schematic illustration for the orientation process of the flexible PNC under H (Fig. 4a). Flexible long PNCs exhibit random worm-like conformations due to the large entropic elasticity endowed by the thermal motion of the flexible PNIPAM gels, and this is similar to in the case of flexible polymer chains associated with the rotation of C–C bonds caused by thermal disturbance.55,56 When an external field H is applied, the superparamagnetic Fe3O4 CNCs in the chain acquire an induced dipole moment:
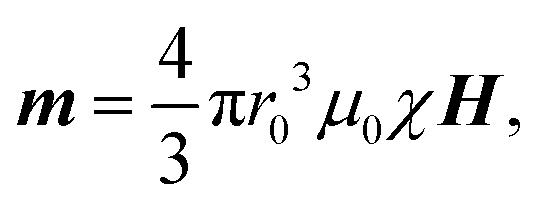 | (3) |
| Fd = 3m2(1 − 3cos2α)rd−4 | (4) |
| Fp = ∇(mH) | (5) |
 | ||
| Fig. 4 (a) Schematic illustration for the orientation process of a flexible PNC under H. (b–e) The calculated probabilities of different conformations shown by 50000 flexible PNCs under different H values. | ||
The relationship between the magnetic field strength and the conformation of the flexible PNCs was qualitatively illustrated using a quasi-3D simulation experiment, which gives the probabilities of different chain conformations based on their total energy. The total energy of each flexible PNC at a certain H was calculated by considering the Weeks–Chandler–Andersen (WCA) interaction, the bonding interaction, and the magnetic dipole–dipole interaction.58,59 With the calculated energy of each conformation, their probabilities were evaluated from the partition functions. A detailed description of the model (Fig. S6, ESI†) and the conformation probability calculation are shown in the ESI.† Fig. S7 (ESI†) shows that most conformations exhibit similar probabilities without H, and this is consistent with the randomly dispersed bending states of the flexible PNCs. The few blue dots in the central area indicate that the probabilities of the corresponding conformations are close to 0, due to the high repulsive WCA potential caused by the overlapping of spheres in those conformations. When a weak magnetic field (50 Gs) is applied along the positive x-direction, the chains with ends at the right side have higher probabilities than those with ends at the top or bottom sides (Fig. 4b). The change in the conformation probabilities with H (Fig. 4b–e) reveals that it becomes more obvious for the PNCs to align themselves along the direction of H to minimize the total energy of the system. When H = 400 Gs, there are much larger probabilities for the PNCs to have ends at the most right-hand side than at the other positions, indicating the most stable conformation of the self-aligned straight state under strong H, and this is consistent with the experimental observations and with the assumption illustrated in Fig. 4a. This reveals that due to the superparamagnetic property of the CNCs, the flexibility of the PNIPAM shells and linkers, and the characteristic large chain length, the flexible PNCs could exhibit balanced states with aligned segments of different lengths along H, showing their unique H-dependent diffraction intensity and color brightness.
As observed from the optical images (Fig. 3d), the smaller the chain lengths, the faster the total orientation occurs along H. Thus, for the flexible PNCs with different average chain lengths (Fig. S8, ESI†), the short PNCs show a more rapid increase in the relative reflectance (R/Rmax) values with an increase in H than the long ones (Fig. 5a). The higher R/Rmax values for the short PNCs under weak H can be attributed to their limited disordered segments and rapid total orientation along H, showing the limited tuning ability of the diffraction intensity, and this is consistent with the optical observations. In contrast, long flexible PNCs possess much more disordered segments, which can be stretched to an orientated state gradually by increasing H, guaranteeing their long-range periodic structure under strong H, which is beneficial to obtaining high reflectance and strong brightness. The results further prove that the large chain length is essential for the flexible PNCs to obtain magnetic field strength-dependent tunability for color brightness.
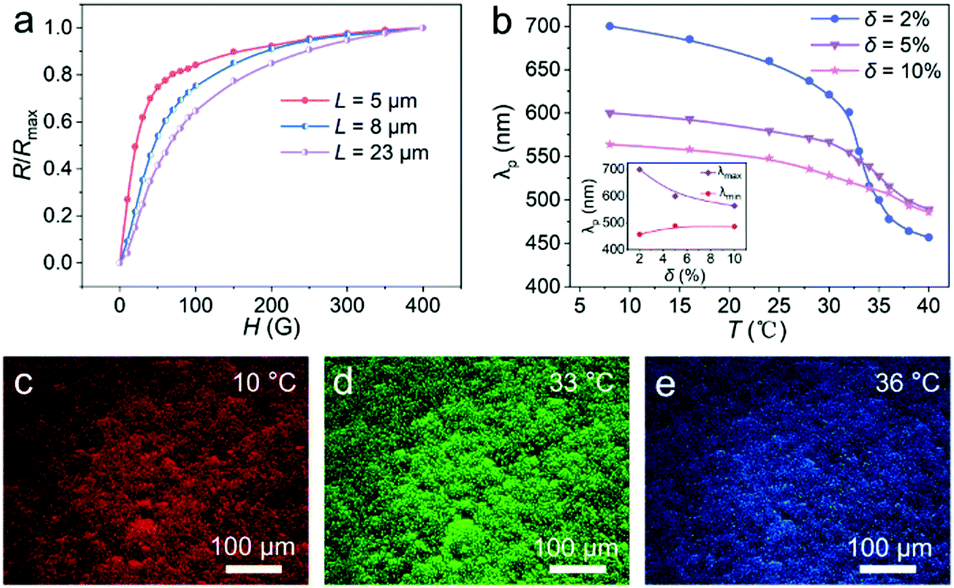 | ||
| Fig. 5 The influence of L and δ on the optical properties of the Fe3O4@PVP@PNIPAM flexible PNCs. (a) Plots showing the magnetic field strength responsive optical properties of the flexible PNCs with different L values. (b) Plots showing the temperature-responsive optical properties of the flexible PNCs with different δ values. The inset in (b) shows the maximum and minimum diffraction wavelengths of the flexible PNCs with different δ values. (c–e) Dark-field optical microscopy images of the flexible PNCs with δ = 2% at different temperatures. | ||
In addition to the large chain length, the magnetic field strength-dependent diffraction intensity of the Fe3O4@PVP@PNIPAM flexible PNCs also benefits from their flexibility that originates from the soft property of the linking PNIPAM. The flexible PNCs with different cross-linking degrees were obtained by tuning the molar ratio of BIS and NIPAM (δ) during the preparation procedure (Fig. S9, ESI†). As seen from Fig. S10 (ESI†), the flexible Fe3O4@PVP@PNIPAM PNCs with δ = 2%, 5%, and 10% all exhibit a magnetic field tunable diffraction intensity due to the intrinsic soft property of PNIPAM. When H = 0–50 Gs, the PNCs with δ = 10% exhibit larger R/Rmax values than those with δ = 2% and 5%, which only show negligible differences with irregular fluctuations. When H = 50–400 Gs, the R/Rmax values of the PNCs at a certain H value increase as δ increases from 2% to 5%, but they decrease as δ further increases from 5% to 10%. The rapid increase of the R/Rmax values for the PNCs with δ = 10% at weak H can be attributed to their rapid rotation orientation along the field direction, and this benefits from their less bending original conformations. With an increase in H, the orientation of the flexible PNCs is dominated by the segment stretch orientation along H, and the PNCs with lower δ possess more disordered segments, resulting in a slower increase in the R/Rmax values. However, flexible PNCs with δ = 10% exhibit a decreased sensitivity of magnetic field strength due to their reduced flexibility and the high magnetic energy needed for the stretch of segments with enhanced mechanical strength. The flexible nanochains with different cross-linking degrees at a certain H value slightly differ in their R/Rmax values from an overall perspective. This phenomenon could be reasonably explained by the full consideration that the PNCs obtained with different δ values have different intrinsic soft attributes of PNIPAM, and different lattice distances and polymer content of PNCs.
While the crosslinking degree of the flexible Fe3O4@PVP@PNIPAM PNCs does not significantly influence their magnetic field strength-responsive optical property, their temperature-responsive optical property is clearly affected. Fig. 5b shows that the tuning range of the diffraction wavelength decreases from 243 to 78 nm as δ increases from 2% to 10%. The maximum diffraction wavelength (λmax) for the flexible PNCs of different δ values decreases with increasing δ. In contrast, the minimum diffraction wavelength (λmin) first increases as δ increases from 2% to 5%, and then remains almost constant as δ further increases from 5% to 10%. The higher λmax and lower λmin values for flexible PNCs with δ = 2% indicate their larger and smaller interparticle distance at low and high temperatures. This confirms that the low crosslinked flexible PNCs show strong swelling or shrinking ability due to the loose structure of the polymer networks, guaranteeing their good temperature responsiveness. The flexible PNCs with δ = 2% achieved tunable hues ranging from red to blue (Fig. 5c–e), and their continuous and reversible temperature-responsive color change progress is shown in Movie S7 (ESI†). The results demonstrate that the tunability of the color hue covering the entire visible light spectrum can be achieved by simply adjusting the crosslinking degree of the linking PNIPAM. Furthermore, the Fe3O4@PVP@PNIPAM flexible PNCs with original diffraction colors ranging from red to blue could be easily obtained by tuning the reaction parameters during the sample fabrication process (Fig. S11, ESI†), which could be utilized to control the structural parameters of the flexible PNCs and thus their dual-responsive properties. By combining the delicate fabrication process and the respective or combined stimuli of magnetic field and temperature, targeted structural colors with independent, continuous, and reversible modulation in brightness and hue could be achieved by the developed aqueous suspension of Fe3O4@PVP@PNIPAM flexible PNCs.
Conclusions
In summary, the independent, continuous, and reversible modulation of structural colors in brightness and hue has been achieved by an aqueous suspension of dual responsive Fe3O4@PVP@PNIPAM flexible PNCs. Benefiting from the superparamagnetic property of Fe3O4@PVP CNCs, the soft nature of the linking PNIPAM, and the large chain length, the as-obtained flexible PNCs show gradual stretching orientation along the field direction, due to the domination of their magnetic potential energy over their entropic elasticity in an increasing external magnetic field, ensuring the enhancement of diffraction intensity and structural color brightness. The tunability of the structural color brightness of the flexible PNCs relies on their large chain-length and flexibility. Meanwhile, the hue of the flexible PNCs can be easily tuned by changing the surrounding temperature due to the temperature-dependent volume shrinking or swelling behavior of PNIPAM. Furthermore, the delicate fabrication of the flexible PNCs with different original colors and the single or combined stimuli of magnetic field and temperature could be utilized to achieve target colors with predesigned brightness and hue. The independent, continuous, and reversible tunability of structural colors in brightness and hue of the developed photonic liquid promises the next generation of adaptive chromic materials and enriches the basic understanding of color modulation mechanisms.Author contributions
J. G. and H. M. conceived the idea and designed the experiments. H. M., Y. Y. and J. G. supervised the project. Y. L., G. Z., G. S. and J. C. conducted the experiments and analyzed the results. Y. L., Q. F., W. L. and T. W. designed and conducted the simulation calculations. Y. L. and Q. F. wrote the manuscript. H. M., Y. Y. and J. G. revised the manuscript. All authors contributed to the discussion and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2020YFA0710100), the National Natural Science Foundation of China (51521001), the Natural Science Foundation of Hubei Province (2019CFB572), the Fundamental Research Funds for the Central Universities (WUT: 2017-YB-003), and the China Scholarship Council (CSC). Yin is grateful for the financial support from the US National Science Foundation (DMR-1810485).References
- P. Wu, J. Wang and L. Jiang, Mater. Horiz., 2020, 7, 338–365 RSC.
- C. Fenzl, T. Hirsch and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2014, 53, 3318–3335 CrossRef CAS PubMed.
- E. S. A. Goerlitzer, R. N. K. Taylor and N. Vogel, Adv. Mater., 2018, 30, 1706654 CrossRef PubMed.
- L. Shang, W. Zhang, K. Xu and Y. Zhao, Mater. Horiz., 2019, 6, 945–958 RSC.
- J. Teyssier, S. V. Saenko, D. van der Marel and M. C. Milinkovitch, Nat. Commun., 2015, 6, 6368 CrossRef CAS PubMed.
- Y. Zhao, Z. Xie, H. Gu, C. Zhu and Z. Gu, Chem. Soc. Rev., 2012, 41, 3297–3317 RSC.
- A. Abdollahi, H. Roghani-Mamaqani and B. Razavi, Prog. Polym. Sci., 2019, 98, 101149 CrossRef CAS.
- A. Abdollahi, H. Roghani-Mamaqani, B. Razavi and M. Salami-Kalajahi, ACS Nano, 2020, 14, 14417–14492 CrossRef PubMed.
- Y. Wang, Q. Zhao and X. Du, Mater. Horiz., 2020, 7, 1341–1347 RSC.
- T. H. Park, H. Eoh, Y. Jung, G. W. Lee, C. E. Lee, H. S. Kang, J. Lee, K. B. Kim, D. Y. Ryu, S. Yu and C. Park, Adv. Funct. Mater., 2021, 31, 2008548 CrossRef CAS.
- S. Li, Y. Zeng, W. Hou, W. Wan, J. Zhang, Y. Wang, X. Du and Z. Gu, Mater. Horiz., 2020, 7, 2944–2950 RSC.
- Y. Wang, Y. Yu, J. Guo, Z. Zhang, X. Zhang and Y. Zhao, Adv. Funct. Mater., 2020, 30, 2000151 CrossRef CAS.
- Y. Yang, Y. Chen, Z. Hou, F. Li, M. Xu, Y. Liu, D. Tian, L. Zhang, J. Xu and J. Zhu, ACS Nano, 2020, 14, 16057–16064 CrossRef PubMed.
- S. U. Kim, S. H. Lee, I. H. Lee, B. Y. Lee, J. H. Na and S. D. Lee, Opt. Express, 2018, 26, 13561 CrossRef CAS PubMed.
- A. C. Arsenault, D. P. Puzzoo, I. Manners and G. A. Ozin, Nat. Photonics, 2007, 1, 468–472 CrossRef CAS.
- H. Fudouzi and Y. N. Xia, Langmuir, 2003, 19, 9653–9660 CrossRef CAS.
- J. Hou, M. Li and Y. Song, Angew. Chem., Int. Ed., 2018, 57, 2544–2553 CrossRef CAS PubMed.
- Y. Cui, Y. Ke, C. Liu, Z. Chen, N. Wang, L. Zhang, Y. Zhou, S. Wang, Y. Gao and Y. Long, Joule, 2018, 2, 1707–1746 CrossRef CAS.
- C. Xiong, J. Zhao, L. Wang, H. Geng, H. Xu and Y. Li, Mater. Horiz., 2017, 4, 862–868 RSC.
- Z. Li, J. Jin, F. Yang, N. Song and Y. Yin, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- Y. Bao, Y. Yu, H. Xu, C. Guo, J. Li, S. Sun, Z. Zhou, C. Qiu and X. Wang, Light: Sci. Appl., 2019, 8, 95 CrossRef PubMed.
- B. M. Boyle, T. A. French, R. M. Pearson, B. G. McCarthy and G. M. Miyake, ACS Nano, 2017, 11, 3052–3058 CrossRef CAS PubMed.
- J. Hou, M. Li and Y. Song, Nano Today, 2018, 22, 132–144 CrossRef CAS.
- J. Chen, J. Feng, Z. Li, P. Xu, X. Wang, W. Yin, M. Wang, X. Ge and Y. Yin, Nano Lett., 2019, 19, 400–407 CrossRef CAS PubMed.
- A. G. Dumanlia and T. Savin, Chem. Soc. Rev., 2016, 45, 6698–6724 RSC.
- Y. Li, Q. Fan, X. Wang, G. Liu, L. Chai, L. Zhou, J. Shao and Y. Yin, Adv. Funct. Mater., 2021, 2010746 CrossRef CAS.
- E. Yablonovitch, Phys. Rev. Lett., 1987, 58, 2059–2062 CrossRef CAS PubMed.
- Z. Li and Y. Yin, Adv. Mater., 2019, 1807061 CrossRef PubMed.
- J. Ge and Y. Yin, Angew. Chem., Int. Ed., 2011, 50, 1492–1522 CrossRef CAS PubMed.
- H. Ma, Y. Tan, J. Cao, S. C. Lian, K. Chen, W. Luo and J. Guan, J. Mater. Chem. C, 2018, 6, 4531–4540 RSC.
- L. He, M. Wang, J. Ge and Y. Yin, Acc. Chem. Res., 2012, 45, 1431–1440 CrossRef CAS PubMed.
- F. Yang, F. Mou, Y. Jiang, M. Luo, L. Xu, H. Ma and J. Guan, ACS Nano, 2018, 12, 6668–6676 CrossRef CAS PubMed.
- L. Bai, Z. Xie, W. Wang, C. Yuan, Y. Zhao, Z. Mu, Q. Zhong and Z. Gu, ACS Nano, 2014, 8, 11094–11100 CrossRef CAS PubMed.
- X. Jia, T. Zhang, J. Wang, K. Wang, H. Tan, Y. Hu, L. Zhang and J. Zhu, Langmuir, 2018, 34, 3987–3992 CrossRef CAS PubMed.
- M. Wang and Y. Yin, J. Am. Chem. Soc., 2016, 138, 6315–6323 CrossRef CAS PubMed.
- Z. Li, M. Wang, X. Zhang, D. Wang, W. Xu and Y. Yin, Nano Lett., 2019, 19, 6673–6680 CrossRef CAS PubMed.
- Z. Li, F. Yang and Y. Yin, Adv. Funct. Mater., 2019, 1903467 Search PubMed.
- W. Luo, H. Ma, F. Mou, M. Zhu, J. Yan and J. Guan, Adv. Mater., 2014, 26, 1058–1064 CrossRef CAS PubMed.
- H. Wang, Q. Chen, Y. Yu, K. Cheng and Y. Sun, J. Phys. Chem. C, 2011, 115, 11427–11434 CrossRef CAS.
- J. Ge, L. He, J. Goebl and Y. Yin, J. Am. Chem. Soc., 2009, 131, 3484–3486 CrossRef CAS PubMed.
- C. Li, X. Zhou, K. Wang, K. Li, M. Li and Y. Song, Compos. Commun., 2019, 12, 47–53 CrossRef.
- Y. Zhao, L. Shang, Y. Cheng and Z. Gu, Acc. Chem. Res., 2014, 47, 3632–3642 CrossRef CAS PubMed.
- J. Ge, H. Lee, L. He, J. Kim, Z. Lu, H. Kim, J. Goebl, S. Kwon and Y. Yin, J. Am. Chem. Soc., 2009, 131, 15687–15694 CrossRef CAS PubMed.
- W. Luo, J. Yan, Y. Tan, H. Ma and J. Guan, Nanoscale, 2017, 9, 9548–9555 RSC.
- S. Shang, Z. Liu, Q. Zhang, H. Wang and Y. Li, J. Mater. Chem. A, 2015, 3, 11093–11097 RSC.
- H. Wang, Q. Chen, Y. Sun and M. He, J. Phys. Chem. C, 2010, 114, 19660–19666 CrossRef CAS.
- Y. Hu, L. He and Y. Yin, Angew. Chem., Int. Ed., 2011, 50, 3747–3750 CrossRef CAS PubMed.
- H. Ma, K. Tang, W. Luo, L. Ma, Q. Cui, W. Li and J. Guan, Nanoscale, 2017, 9, 3105–3113 RSC.
- W. Luo, Q. Cui, K. Fang, K. Chen, H. Ma and J. Guan, Nano Lett., 2020, 20, 803–811 CrossRef CAS PubMed.
- L. Kong, Y. Feng, W. Luo, F. Mou, K. Ying, Y. Pu, M. You, K. Fang, H. Ma and J. Guan, Adv. Funct. Mater., 2020, 2005243 CrossRef CAS.
- J. M. Weissman, H. B. Sunkara, A. S. Tse and S. A. Asher, Science, 1996, 274, 959–963 CrossRef CAS PubMed.
- F. Mou, C. Chen, Q. Zhong, Y. Yin, H. Ma and J. Guan, ACS Appl. Mater. Interfaces, 2014, 6, 9897–9903 CrossRef CAS PubMed.
- H. Ma, M. Zhu, W. Luo, W. Li, K. Fang, F. Mou and J. Guan, J. Mater. Chem. C, 2015, 3, 2848–2855 RSC.
- K. Matsubara, M. Watanabe and Y. Takeoka, Angew. Chem., Int. Ed., 2007, 46, 1688–1692 CrossRef CAS PubMed.
- T. T. Perkins, D. E. Smith and S. Chu, Science, 1997, 276, 2016–2021 CrossRef CAS.
- D. E. Smith and S. Chu, Science, 1998, 281, 1335–1340 CrossRef CAS PubMed.
- D. Lisjak and A. Mertelj, Prog. Mater. Sci., 2018, 95, 286–328 CrossRef CAS.
- J. Wei, F. Song and J. Dobnikar, Langmuir, 2016, 32, 9321–9328 CrossRef CAS PubMed.
- X. Xue and E. P. Furlania, Phys. Chem. Chem. Phys., 2014, 16, 13306–13317 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data. See DOI: 10.1039/d1mh00556a |
| This journal is © The Royal Society of Chemistry 2021 |