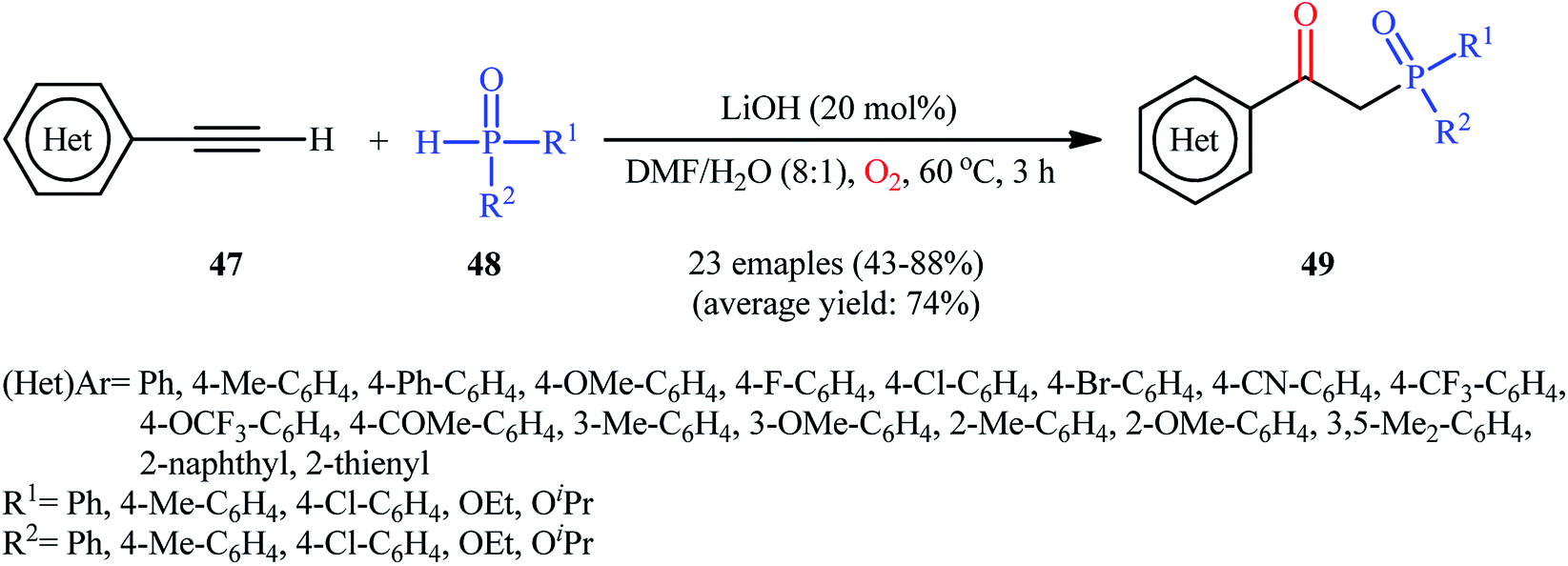DOI:
10.1039/D0RA08074H
(Review Article)
RSC Adv., 2021,
11, 470-483
Recent trends in the direct oxyphosphorylation of C–C multiple bonds
Received
21st September 2020
, Accepted 4th December 2020
First published on 23rd December 2020
Abstract
Due to the wide importance of β-phosphorylated ketones as key building-blocks in the fabrication of various pharmaceutically active organophosphorus compounds, finding new and truly efficient methods for their preparation from simple, low-cost and ubiquitous feedstock materials within a single click is an interesting subject in organic synthesis. Recently, oxyfunctionalization of carbon–carbon multiple bonds has arisen as a straightforward and versatile tool for the synthesis of complex organic molecules from the simple and easily accessible alkenes/alkynes via a single operation. In this context, oxyphosphorylation of alkenes/alkynes with P(O)–H compounds has attracted considerable attention as a unique procedure for the construction of β-phosphorylated ketones. In this review, we outline the recent advances and developments in this fast-growing research field with particular emphasis on the mechanistic aspects of reaction.
 Alireza Bakhtiary | Alireza Bakhtiary was born in Esfahan, Iran, in 1980. He completed his BSc degree in the field of applied chemistry (2007) in Azad University Tehran North Branch, Iran. Shortly after, he continued his education under the supervision of Prof. E. Kianmehr to obtain his MSc degree (2010) in organic chemistry from the University of Tehran, Iran. Later, he went to Canada in order to attend graduate school at the University of Alberta, where he received a PhD degree in chemistry (2018) under the supervision of Prof. J. C. Vederas. Currently, he is hired by pharmaceutical companies as a research scientist. As a scientist, he has broad research interests and experiences which include total synthesis of natural products, solid phase and solution phase peptide synthesis, structural elucidation of peptides using nuclear magnetic resonance spectroscopy and mass spectroscopy, cell culture, protein purification, and computer modeling of protein–ligand interactions. |
 Mohammad Reza Poor Heravi | Mohammad Reza Poor Heravi was born in Seiyahkal-Lahijan, Guilan, Iran in 1965. He received his BSc degree in pure Chemistry from Tabriz University, Iran, in 1987, and MSc degree in Organic Chemistry from Tabriz University, Iran in 1990 with professor A. Shahrisa, and PhD degree from Isfahan University, Iran, with professor H. Loghmanim and carried out visiting research in 2006, The University of Sheffield, UK, with professor Richard F. W. Jackson. He became a sciences faculty member of Payame Noor University in 1990, and associate professor in 2012. His research field includes applications of solvent-free conditions, ionic liquids, fluorination reaction, designing fluorinating reagents and ultrasound irradiation in organic synthesis, and study of methodology in organic chemistry. |
 Akbar Hassanpour | Akbar Hassanpour was born in Tabriz, Iran, in 1978. He received his BS degree in applied chemistry from Tabriz Azad University, Iran, and his MS degree in organic chemistry from Tabriz University, Tabriz, Iran, in 2004 under the supervision of Prof. Kazem D. Safa. He completed his PhD degree in 2008 under the supervision of Prof. Kazem D. Safa. He was a postdoctoral fellow in the lab of Prof. W. A. Stanczyk at the Centre of Molecular and Macromolecular Studies in Łódź, Poland. Now he is working at Azad University as an associate professor. His research interests include organometallic, organic synthesis, and new methodologies in nanomaterial synthesis. |
 Issa Amini | Issa Amini was born in Abhar, Iran, in 1972. He received his BS degree in pure chemistry from the University of Payame Noor, Tehran, Iran, and his MS degree in organic chemistry from Payame Noor University, Tehran, Iran, in 2005 under the supervision of Prof. A. Ramazani. He completed his PhD degree in 2012 under the supervision of Prof. A. Ramazani. Now he is working at Payame Noor University as an assistant professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his BS degree in pure chemistry from the University of Tabriz, Tabriz, Iran, and his MS degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as a professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. He is Editor-in-Chief of two journals including "Chemical Review and Chemistry" and "Journal of Chemistry Letters". |
1. Introduction
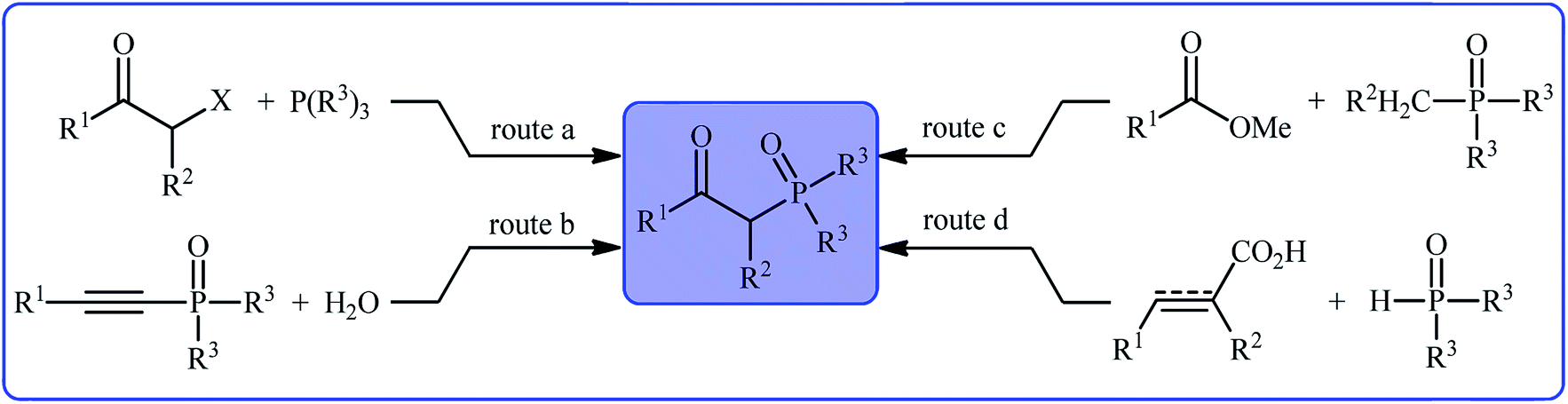
Organophosphorus compounds play a tremendously important role in the fields of organic chemistry,1 medicinal chemistry,2 and agricultural chemistry.3 Among various organophosphorus compounds, β-phosphorylated ketones (e.g., β-ketophosphonates, β-ketophosphine oxides, β-ketophosphinates) are highly valuable building blocks for the synthesis of various value-added chemicals and drug-like molecules.4 Moreover, some β-phosphorylated ketones themselves are endowed with outstanding biological properties.5 Despite wide importance of these motifs in many fields of chemistry, the methods for their preparation are relatively scarce.6 The general protocols for the synthesis of these compounds include the Michaelis–Arbuzov reaction (Scheme 1, route a),7 hydration of alkynyl phosphorus compounds (Scheme 1, route b),8 acylation of alkyl phosphorus compounds (Scheme 1, route c),9 and decarboxylative coupling reaction of alkenyl/alkynyl carboxylic acids with P(O)H compounds (Scheme 1, route d).10 However, most of these methodologies, if not all, suffer from certain disadvantages, such as requiring harsh reaction conditions, requiring pre-functionalized starting materials, and/or expensive catalysts. Therefore, the development of efficient, practical, and environmentally benign methodologies that benefit from simple, inexpensive, and readily available substrates for the construction of this specific class of phosphorus and carbonyl-contain compounds is still highly desirable in synthetic chemistry.
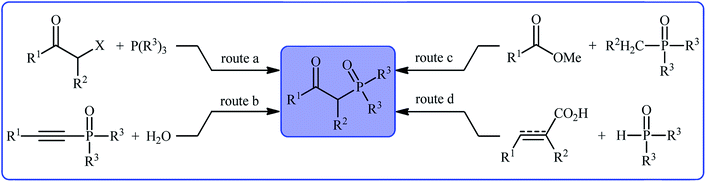 |
| | Scheme 1 Main synthetic routes to β-phosphorylated ketones. | |
Recently, the double functionalization of carbon–carbon multiple bonds in one-pot processes has emerged as a fruitful tool for the rapid synthesis of complex molecular architectures from safe, low-cost, and prevalent alkenes and alkynes.11,12 In this family of reactions, oxyfunctionalization of alkenes/alkynes, which installs a unique functional group and an oxo group in adjacent carbons simultaneously, offers a powerful strategy for the selective synthesis of β-substituted ketones.13 In this context, direct oxyphosphorylation of C–C multiple bonds with P(O)–H compounds has gained a considerable amount of attention in the last couple of years in the preparation of numerous synthetically and biologically important β-phosphorylated ketones (Fig. 1). This protocol features readily accessible starting materials, high atom-, step-, and pot-economy, and high bond-forming efficiency, thus providing a straightforward and highly efficient access to the title compounds. Despite significant advances and developments in this field over the past decade, no comprehensive review can be found in the literature covering this novel and rapidly growing research field. In continuation of our recent published reviews on organophosphorus chemistry10,14 and modern organic synthesis,15 herein, we will highlight the most important developments on the synthesis of β-substituted ketones through the direct oxyphosphorylation of alkenes/alkynes with P(O)–H compounds from 2011 to August 2020. It is worthy of note that a particular emphasis is placed on mechanistic aspects of reactions which may allow possible new insights toward designing more efficient and improved catalytic systems.
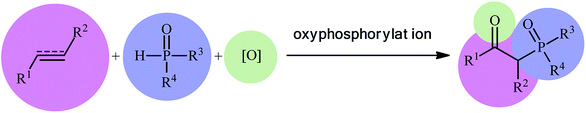 |
| | Fig. 1 Direct oxyphosphorylation of C–C multiple bonds with H–P(O) compounds. | |
2. Oxyphosphorylation of alkenes
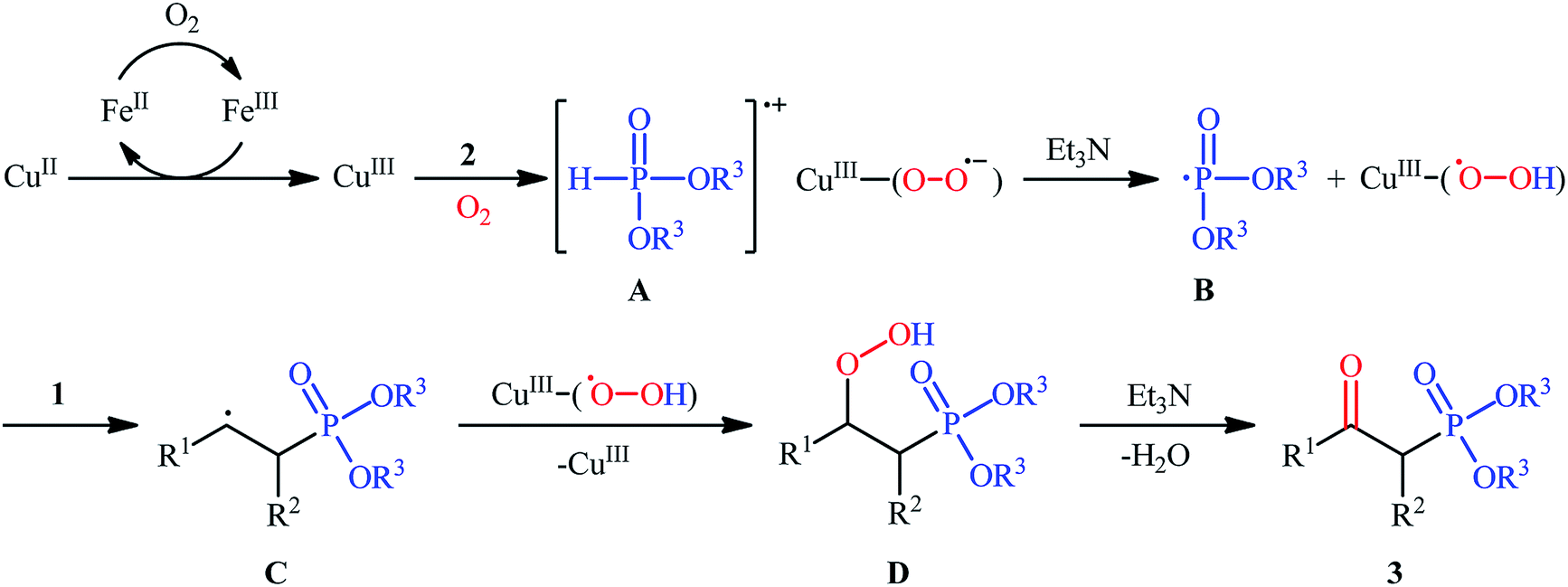
The first mention of the synthesis of β-ketophosphonates through the direct oxyphosphorylation of alkenes with H-phosphonates and dioxygen can be found in a 2011 paper by Ji and Wei.16 To evaluate the catalytic activity of different transition metal complexes, styrene and diisopropyl phosphonate were chosen as the model substrates. They showed that in the presence of a combination of inexpensive and easily accessible CuBr2 and FeBr3 as a catalytic system in DMSO, a variety of electron-rich and electron-deficient styrenes 1 and various H-phosphonates 2 reacted efficiently to give the corresponding β-ketophosphonates 3 in moderate to high yields (Scheme 2). Interestingly, an enamine such as 9-vinylcarbazole was also compatible with this protocol, thus affording the corresponding product in moderate yield. However, 1-octene did not work well in the reaction and therefore no other aliphatic alkenes were examined in this procedure. Of note, the authors demonstrated the scalability of the reaction since diethyl (2-oxo-2-phenylethyl)phosphonate, a key intermediate for the synthesis of various natural products such as (−)-diospongin B, could be obtained in 26 g scale in good yield of 68%. To make the mechanistic course of this conversion clearer, the authors performed 18O isotope labeling and radical trapping experiments. From these results, they speculated that the reaction starts with the formation of dialkyl phosphonate cation radical A and CuIII–(OO˙−) (superoxide) species through the single electron transfer (SET) from H-phosphonate 2 to in situ generated copper(III) species under O2. Next, the abstraction of the H+ by Et3N from the cation radical A leads to phosphonyl radical B and CuIII–(˙OOH) (hydroperoxide) species. Subsequently, the reaction of phosphorus-centered radical B with alkene 1 gives the alkyl radical C, which after interaction with CuIII–(˙OOH) converts to the hydroperoxide intermediate D and regenerates the starting CuIII catalyst. Finally, dehydration this intermediate D with the aim of Et3N affords the desired product 3 (Scheme 3).
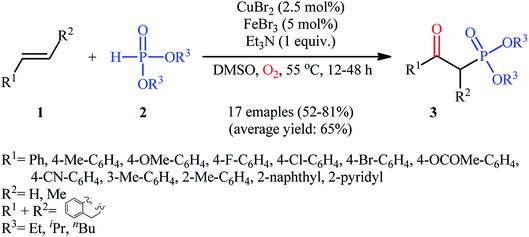 |
| | Scheme 2 Ji's synthesis of β-ketophosphonates 3. | |
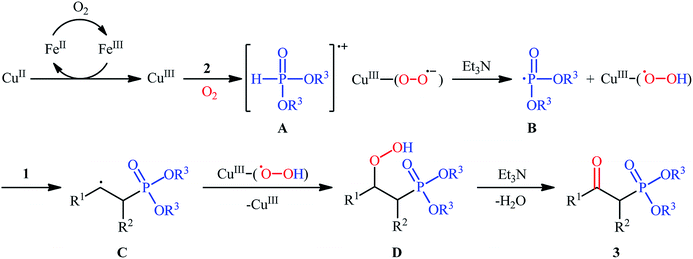 |
| | Scheme 3 Possible mechanism of Cu-catalyzed oxyphosphorylation of alkenes 1 with H-phosphonates 2 and O2. | |
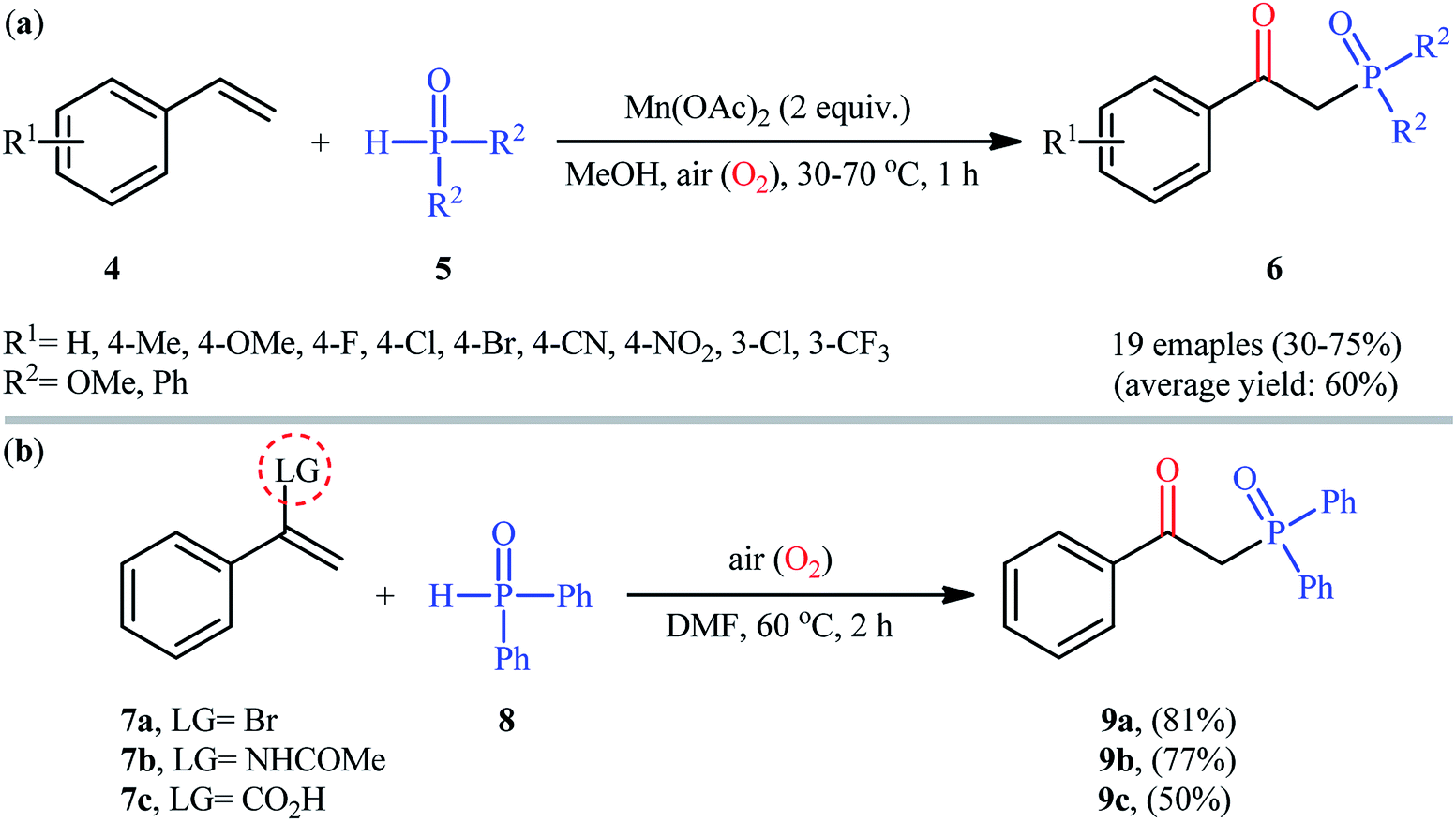
Afterwards, Zeng and Zou along with their co-workers developed a Mn(OAc)2-mediated protocol for the oxyphosphorylation of styrene derivatives 4 with P(O)–H compounds 5 (H-phosphonates and secondary phosphine oxides) under aerobic conditions without the use of any base and additive (Scheme 4a).17 This chemistry showed good functional group tolerance and afforded the target β-ketophosphoryl products 6 in moderate to good yields within 1 h. In this investigation, the authors found some limitation in their methodology, when they attempted to react β-substituted styrenes and non-conjugated terminal alkenes. Unfortunately, in these cases only the hydrophosphorylation products were observed with no formation of the expected β-phosphorylated ketone products. Noteworthy, the reaction of α-substituted styrenes with P(O)–H compounds under the standard conditions also provided the hydroxyphosphorylation products due to the presence of α-substituents. Concurrently, the same strategy was applied by Zhang's group to the synthesis of a diverse range of β-hydroxyphosphine oxides through the reaction of corresponding 1,1-disubstituted alkenes with H-phosphine oxides and O2 under catalyst- and additive-free conditions.18 Following these works, the group of Lei disclosed that if easily removable leaving groups were introduced to α-position of styrenes, β-ketophosphoryl products could be obtained exclusively.19 Thus, a small series of β-ketophosphonates 9 were synthesized in good yields by simple heating of α-substituted styrenes 7 with diphenylphosphine oxide 8 in DMF under air atmosphere and catalyst-free conditions (Scheme 4b).
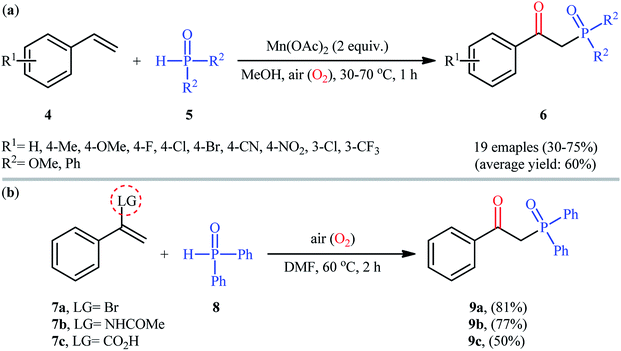 |
| | Scheme 4 (a) Mn-mediated oxyphosphorylation of styrenes 4 with P(O)–H compounds 5; (b) catalyst-free oxyphosphorylation of 1,1-disubstituted alkenes 7 with diphenylphosphine oxide 8. | |
Drawing inspiration from these works, Du–Wei and colleagues were able to demonstrated that a library of β-ketophosphoryl products 12 could be obtained in fair to good yields from the reaction of various terminal and internal alkenes 10 with P(O)–H compounds 11 under an oxygen atmosphere using CuBr2 as a catalyst and Et3N as a base in DCE (Scheme 5).20 Some of the most important information of this C–P bond forming reaction are listed below: (i) all the three kinds of hydrogen phosphoryl compounds (H-phosphonates, H-phosphine oxides, and H-phosphinates) were compatible in this reaction; (ii) the relative reaction rates of P(O)–H compounds followed the order: H-phosphine oxides > H-phosphonates ≅ H-phosphinates; (iii) terminal alkenes afforded higher yields of the expected products compared to the internal alkenes; (iv) the protocol for oxyphosphorylation of aliphatic alkenes was considerably less efficient; and (v) the electronic nature of the substituents on phenyl ring periphery of aromatic alkenes had a little impact on the rate of reaction. The authors suggested reaction mechanism for this transformation is displayed in Scheme 6 and is closely similar to the one proposed by Ji and Wei.
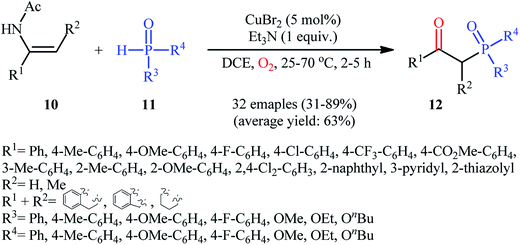 |
| | Scheme 5 Cu-catalyzed oxyphosphorylation of enamides 10 with P(O)–H compounds 11 and O2. | |
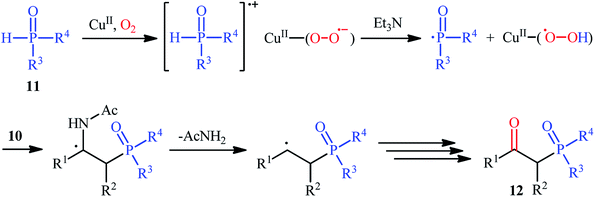 |
| | Scheme 6 Possible reaction pathway for the formation of β-ketophosphoryl compounds 12. | |
In 2017, Chen and co-workers achieved CuSO4·5H2O-catalyzed aerobic oxyphosphorylation of styrenes 13 with various hydrogen phosphoryl compounds 14 under additive-free conditions.21 The reactions were carried out in MeCN at 60 °C, tolerated a wide number of electron-donating and electron-withdrawing functional groups and generally provided the desired β-ketophosphoryl products 15 in modest to excellent yields (Scheme 7a). However, aliphatic alkenes did not work well under standard conditions. It is worthwhile to mention that the optimized reaction condition was also successfully applied in the decarboxylative oxyphosphorylation of alkenyl acids with the same set of P(O)–H compounds. To gain insights into the mechanistic course of this reaction, the authors performed the reaction under an inert (nitrogen) atmosphere and no desired product was observed, indicating that the presence of oxygen is indispensable. Moreover, an isotope labeling study in the presence of H218O was conducted, which revealed that the carbonyl oxygen in the products came from oxygen in air. A similar oxyphosphorylation of (hetero)aromatic alkenes with H-phosphine oxides and O2 was also achieved only using CuCN as the catalyst under additive-free conditions.22 Concurrently, another metal-catalyzed oxyphosphorylation of alkenes was introduced by Gu and Cai.23 In this methodology, styrene derivatives 16 reacted with H-phosphonates 17 in presence of 10 mol% of CuCl as a catalyst, 20 mol% of FeCl3 as a co-catalyst, 1 equiv. of Et3N as a base, and 2 equiv. of DTBP (di-tert-butyl peroxide) as the oxidant in DMF under Ar atmosphere at 90 °C to achieve β-ketophosphonates 18 in 26–62% yields (Scheme 7b). In contrast with previous reports which suggested that carbonyl oxygen in the products should originally be from oxygen (air), the authors proposed that carbonyl oxygen in their products originated from water molecules which are present in DMF solvent. Interestingly, when the same reaction was run at 110 °C, vinylphosphonates were obtained as the sole products. They ascribed this observation by the boiling point of water.
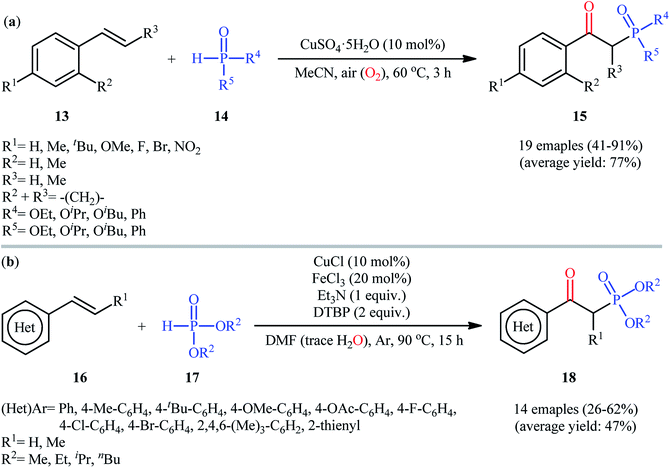 |
| | Scheme 7 (a) Synthesis of β-phosphorylated ketones 15 from styrenes 13 and hydrogen phosphoryl compounds 14 under ambient air; (b) Cai's synthesis of β-ketophosphonates 18. | |
Afterwards, Yamamoto–Makino's group extended the scope of alkenes to vinyl ethers.24 Thus, several alkyl/aryl 2-(diethoxyphosphoryl)acetates 21 were synthesized in moderate yields, ranging from 42% to 57%, via the Mn(acac)3-catalyzed oxyphosphorylation of corresponding vinyl ethers 19 with dimethyl phosphonate 20 and O2 in MeCN at room temperature (Scheme 8). Noteworthy, Mn(acac)2 was also found to promote this transformation; albeit, at lower efficiency. However, other manganese salts such as MnBr2, Mn(OAc)2, Mn(OAc)3·2H2O, and [MnIII(Pc)]Cl proved to be completely ineffective. The outcome of this reaction was strongly depended to the base used, and among the various bases (e.g., Et3N, iPr2Net, Cy2Net, DMAP, DTBMP, TMEDA. DBU, pyridine, 2,2′-bipyridyl, 4-methoxypyridine, 4-(tert-butyl)pyridine, 4-(trifluoromethyl)pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine); pyridine was found to be most effective for this conversion.
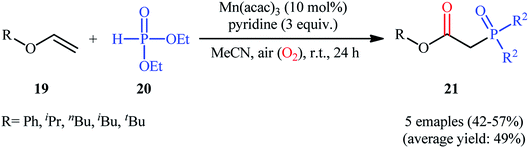 |
| | Scheme 8 Mn-catalyzed direct oxyphosphorylation of vinyl ethers 19 with dimethyl phosphonate 20 developed by Yamamoto and Makino. | |
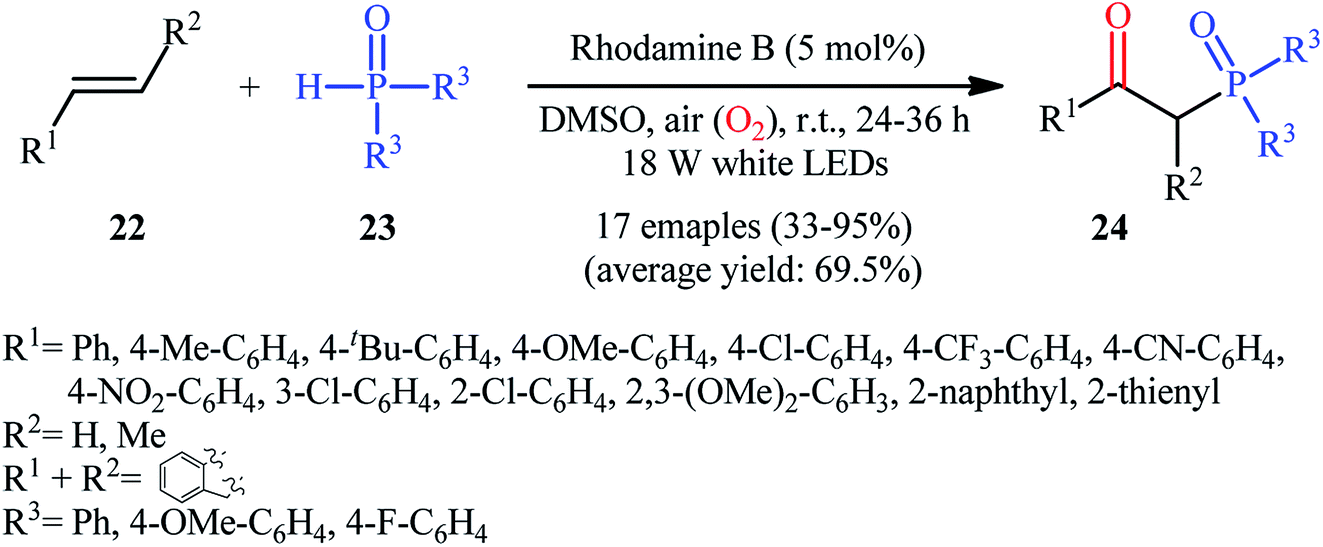
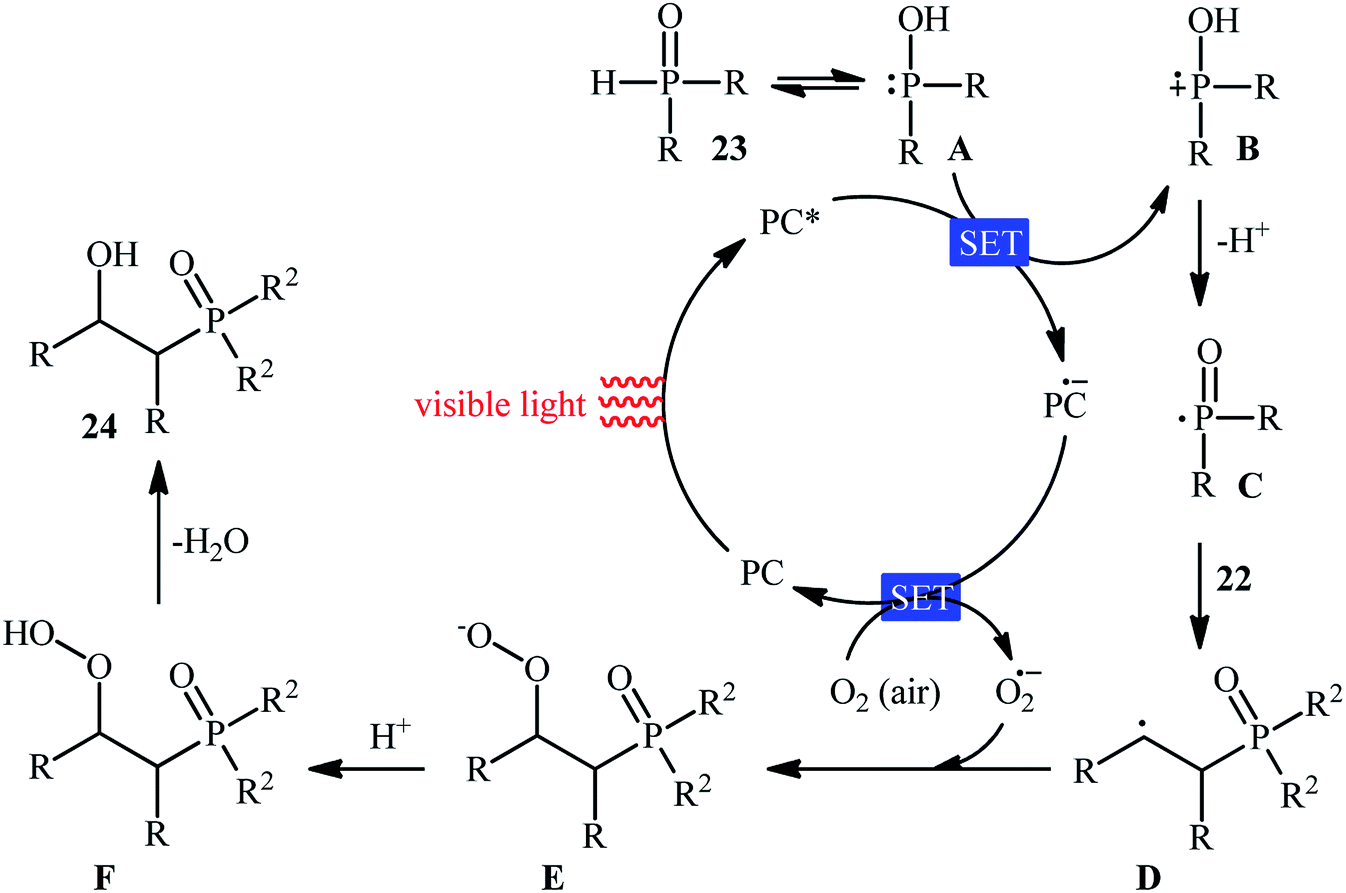
Recently, Zhu et al.25 reported a metal-free, visible light photoredox-catalyzed aerobic oxyphosphorylation of alkenes 22 with H-phosphine oxides 23 at room temperature. No additive or base was used and only 5 mol% of inexpensive rhodamine B catalyst was enough for preparation of a variety of β-ketophosphine oxides 24 in 33–95% yields (Scheme 9). The reaction could also be easily scaled up to the gram-scale as exemplified by the synthesis of 1-(4-chlorophenyl)-2-(diphenylphosphoryl)ethanone on a 1.62 g scale (91%). Remarkably, H-phosphinates were also responded to this reaction. However, H-phosphonates were inert under standard conditions. The radical trapping experiments with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) pointed toward a radical pathway. On the basis of these results and literature reports, the authors suggested that the reaction proceeds through the following key steps (Scheme 10): (i) photoexcitation of the ground state photocatalyst (PC) by visible light to form the excited state photocatalyst (PC*); (ii) single-electron transfer from PC* to phosphinous acid A (the tautomeric form of diarylphosphine oxide 23) to produce the radical cation intermediate B and PC˙− radical anion; (iii) oxidation of PC˙− by O2 to afford 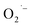 and regenerate the PC; (iv) deprotonation of intermediate B to yield the P-centered radical C; (v) reaction of intermediate C with alkene 22 to give radical D; (vi) the combination of intermediate D with
and regenerate the PC; (iv) deprotonation of intermediate B to yield the P-centered radical C; (v) reaction of intermediate C with alkene 22 to give radical D; (vi) the combination of intermediate D with 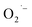 to generate peroxide anion E; (vii) protonation of intermediate E to form intermediate F; and (viii) dehydration of intermediate F to give the desired product 24.
to generate peroxide anion E; (vii) protonation of intermediate E to form intermediate F; and (viii) dehydration of intermediate F to give the desired product 24.
 |
| | Scheme 9 Visible-light-induced direct oxyphosphorylation of alkenes 22 with H-phosphine oxides 23. | |
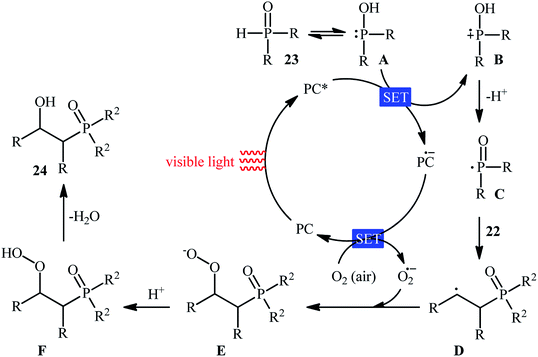 |
| | Scheme 10 Proposed mechanism for the reaction in Scheme 9. | |
Very recently in 2020, the nanocatalyzed version of this reaction was reported by Moghaddam and colleagues, who disclosed that the treatment of various styrenes 25 bearing either electron-donating or electron-withdrawing groups at the phenyl ring periphery with H-phosphonates 26 in the presence of 10 mol% of copper ferrite nanoparticles (CuFe2O4 NPs), 1 equiv. of Et3N, and 2 equiv. of DTBP in MeCN under ambient air afforded the corresponding β-ketophosphonates 27 with yield ranging from 35% to 85% (Scheme 11).26 To evaluate the recyclability and reusability of the catalyst, the oxyphosphorylation of styrene with diethyl phosphonate was performed under the optimized reaction conditions. It was found that the catalyst could be easily recovered by means of a magnetic rod and reused six consecutive cycles without significant decrease in its catalytic activity. Noteworthy, the FE-SEM (field emission scanning electron microscopy) images of the catalysts before and after six cycles of the reaction indicated that the morphology, size, and dispersity of the nanoparticles were not approximately transformed during these runs (Fig. 2).
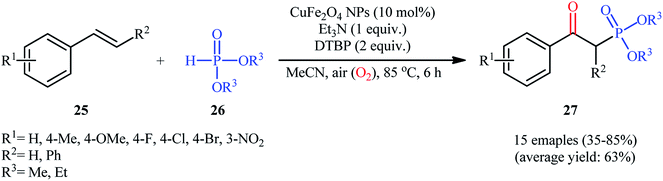 |
| | Scheme 11 Synthesis of β-ketophosphonate derivatives 27 from styrenes 25 and H-phosphonates 26 catalyzed by copper ferrite nanoparticles. | |
 |
| | Fig. 2 The FE-SEM images of CuFe2O4 NPs (a) before and (b) after six times reuse. | |
3. Oxyphosphorylation of alkynes
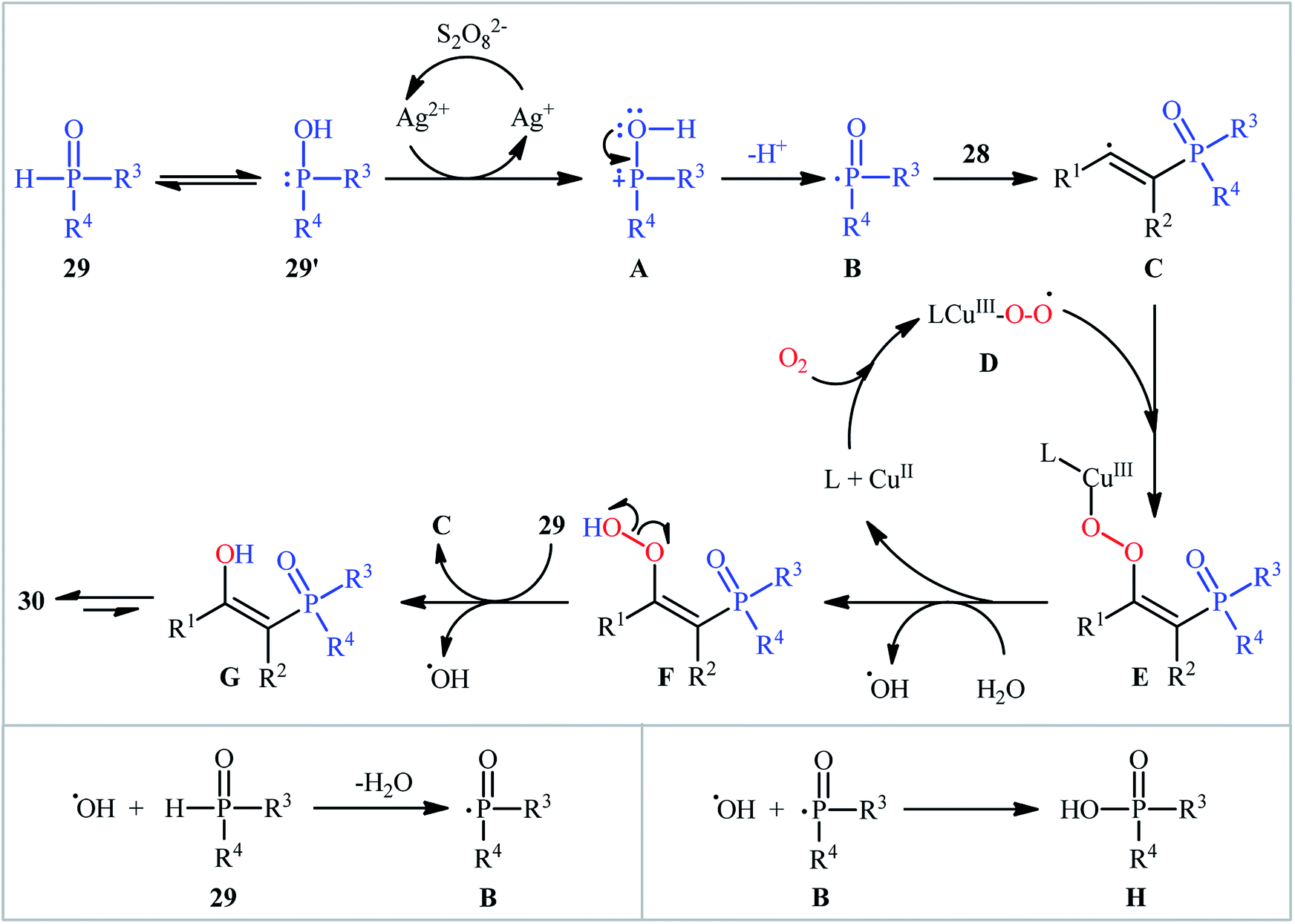
One of the earliest reports on the synthesis of β-phosphorylated ketones via the direct oxyphosphorylation of the corresponding alkynes appeared in 2015, when various available aryl/aliphatic alkynes 28 were converted to β-ketophosphoryl compounds 30 through the silver/copper-catalyzed oxyphosphorylation with P(O)–H compounds 29 and O2 in binary solvent MeCN/H2O with ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at room temperature.27 As shown in Scheme 12, this reaction tolerated all the three kinds of hydrogen phosphoryl compounds and both terminal and internal alkynes, and gave the final products in moderate to excellent yields within 2–3 h. The reactivity order for the P(O)–H compounds under these reaction conditions was H-phosphonates > H-phosphinates ≅ H-phosphine oxides. The results also indicated that terminal alkynes afforded better yields compared to internal alkynes. Beside good yields, broad substrate scope, mild reaction condition, and low reaction times were the advantages, mentioned for this atom economy synthetic strategy. The plausible mechanism for this appealing reaction is outlined based on a series of control experiments (Scheme 13). At the beginning of the reaction, peroxodisulfate oxidize Ag(I) cation to Ag(II) cation by a single-electron transfer process and the generated Ag(II) ion oxidize P(O)–H compound 29 to form cation radical A. Next, deprotonation of this intermediate gives the phosphonyl radical B, which then attacks to the triple bond of alkyne 28 to produce alkenyl radical intermediate C. Subsequently, reaction of this radical with in situ generated active-oxygen copper complex D gives complex E, which in the presence of water quickly converts to hydroperoxide species F. Then, via initial homolysis of the O–O bond, the hydroperoxide F reacts with another molecule of 2 to form a hydroxyl radical, an enol G as well as a phosphonyl radical B. Finally, tautomerization of D affords the observed products 30.
:1 at room temperature.27 As shown in Scheme 12, this reaction tolerated all the three kinds of hydrogen phosphoryl compounds and both terminal and internal alkynes, and gave the final products in moderate to excellent yields within 2–3 h. The reactivity order for the P(O)–H compounds under these reaction conditions was H-phosphonates > H-phosphinates ≅ H-phosphine oxides. The results also indicated that terminal alkynes afforded better yields compared to internal alkynes. Beside good yields, broad substrate scope, mild reaction condition, and low reaction times were the advantages, mentioned for this atom economy synthetic strategy. The plausible mechanism for this appealing reaction is outlined based on a series of control experiments (Scheme 13). At the beginning of the reaction, peroxodisulfate oxidize Ag(I) cation to Ag(II) cation by a single-electron transfer process and the generated Ag(II) ion oxidize P(O)–H compound 29 to form cation radical A. Next, deprotonation of this intermediate gives the phosphonyl radical B, which then attacks to the triple bond of alkyne 28 to produce alkenyl radical intermediate C. Subsequently, reaction of this radical with in situ generated active-oxygen copper complex D gives complex E, which in the presence of water quickly converts to hydroperoxide species F. Then, via initial homolysis of the O–O bond, the hydroperoxide F reacts with another molecule of 2 to form a hydroxyl radical, an enol G as well as a phosphonyl radical B. Finally, tautomerization of D affords the observed products 30.
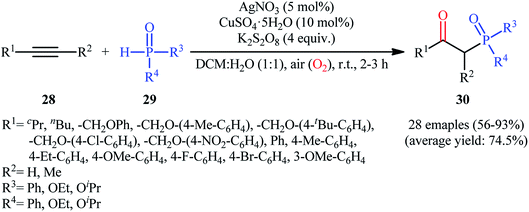 |
| | Scheme 12 Ag/Cu-catalyzed direct oxyphosphorylation of alkynes 28 with P(O)–H compounds 29 and O2. | |
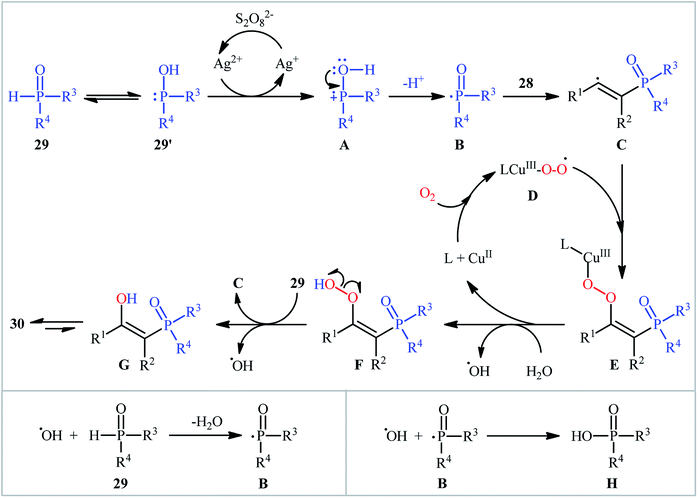 |
| | Scheme 13 Mechanistic proposal from the formation of β-ketophosphoryl compounds 30. | |
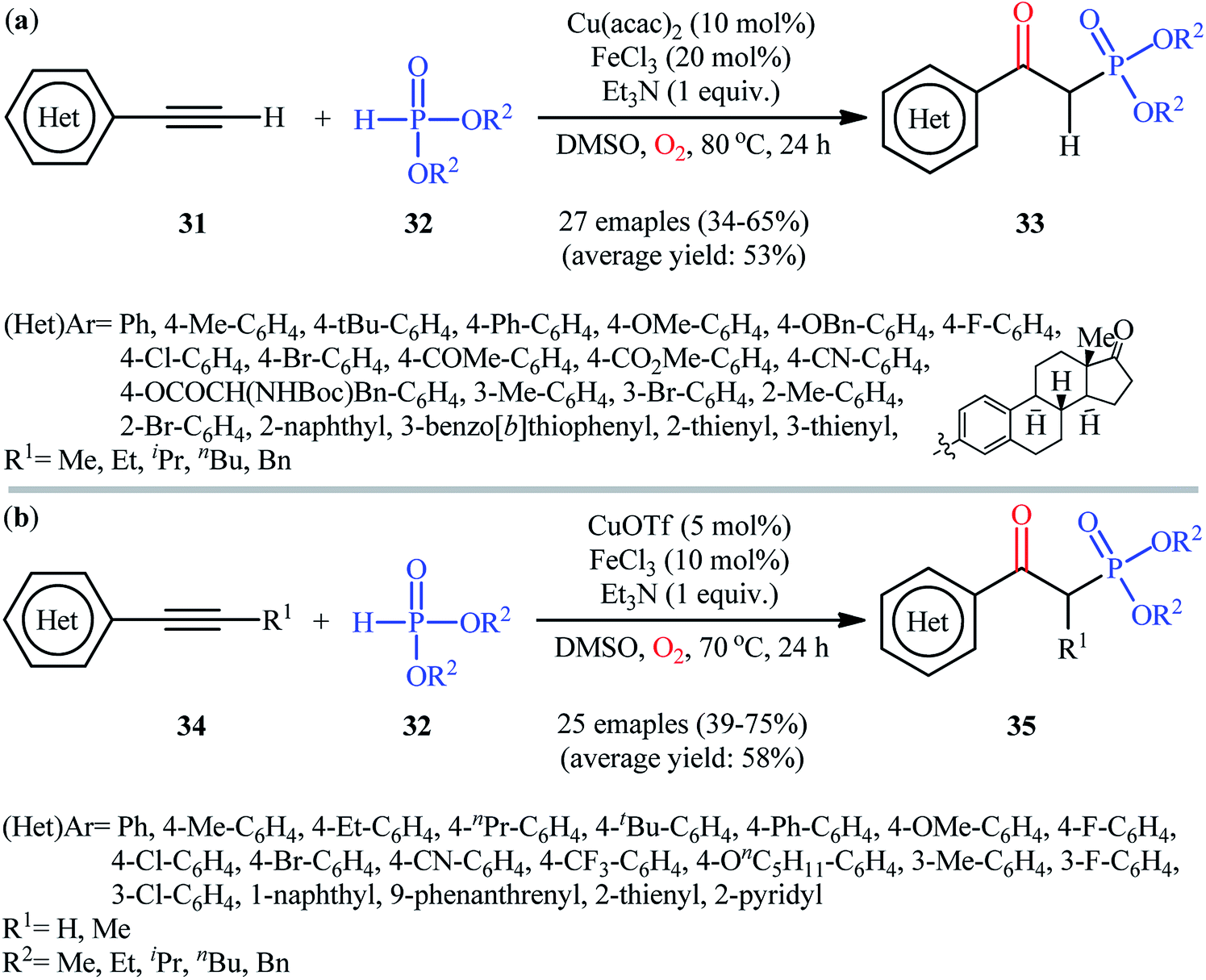
In the same year, He's research team presented an efficient copper/iron-catalyzed oxyphosphorylation of terminal alkynes 31 with H-phosphonates 32 and O2 into β-ketophosphonates 33.28 The results of this investigation indicated that the optimum condition for this transformation was the combination of Cu(acac)2 (10 mol%), FeCl3 (20 mol%), and Et3N (1 equiv.) as a catalytic system using DMSO as the solvent, at 80 °C. Under optimized conditions, the reaction tolerated a diverse set of aromatic and heteroaromatic terminal alkynes and various symmetrical dialkyl H-phosphonates and gave the desired products in moderate to good yields (Scheme 14a). However, both internal and aliphatic terminal alkynes failed to enter into this reaction. Moreover, strongly electron-poor heteroaromatic alkynes, such as 2- and 3-ethynylpyridines were inert under standard conditions. Shortly thereafter, similar work was reported by Song et al.29 Using CuOTf as catalyst and FeCl3 as cocatalyst, they realized the oxyphosphorylation of various terminal and internal alkynes 34 with the same set of H-phosphonates 32 into the corresponding β-phosphorylated ketones 35 (Scheme 14b). Notably, this method allowed for the oxyphosphorylation of 2-ethynylpyridine, which was inert under the He's reaction conditions. This chemistry was also applicable to the coupling of H-phosphine oxides and H-phosphinates with terminal alkynes. However, the reaction did not work well for aliphatic alkynes.
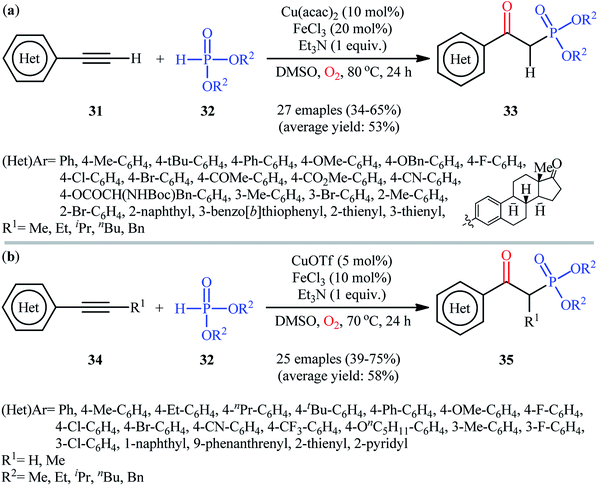 |
| | Scheme 14 (a) Cu/Fe-catalyzed oxidative coupling of aromatic terminal alkynes 31 and with H-phosphonates 32; (b) Song's synthesis of β-phosphorylated ketones 35. | |
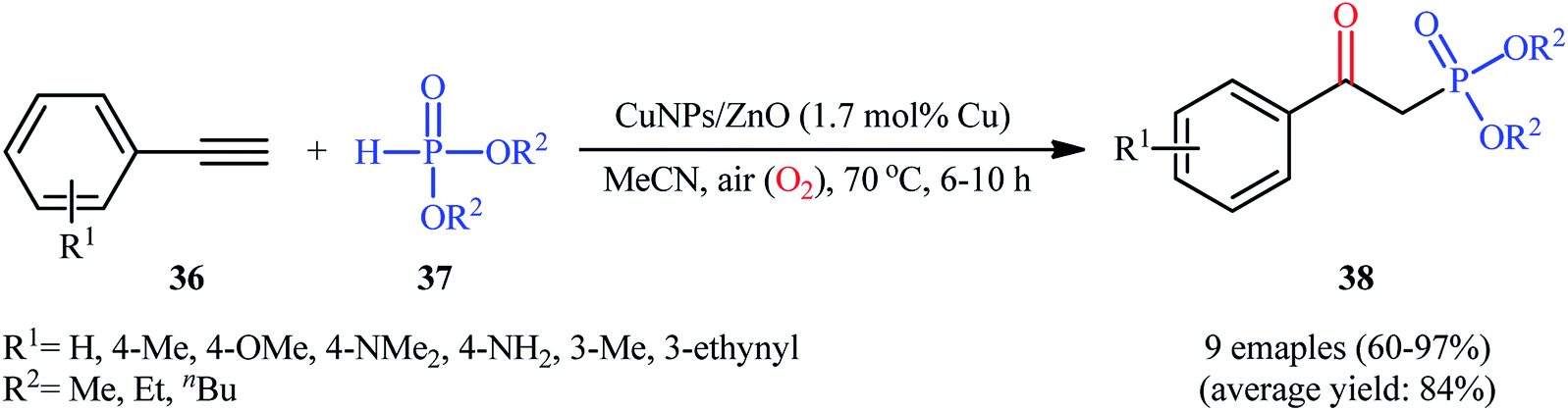
Later, Moglie and Radivoy along with their co-workers introduced the use of copper nanoparticles (CuNPs) supported on ZnO as an efficient catalyst for oxyphosphorylation of terminal alkynes with H-phosphonates under aerobic conditions.30 Thus, a number of β-aryl-β-ketophosphonates 38 were successfully synthesized by treatment of the corresponding aromatic terminal alkynes 36 with dialkyl H-phosphonates 37 in the presence of a catalytic amount of CuNPs@ZnO and in the absence of any additive or ligand (Scheme 15). Interestingly, when aliphatic alkynes were subjected to the same reaction conditions, the anti-Markovnikov vinyl phosphonates were formed as the main reaction products, thus suggesting that the presence of a conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the starting alkyne is mandatory for the formation of the β-ketophosphonate products. The strategy was also successfully applied to the fabrication of β-ketophosphonates from both aromatic and aliphatic terminal alkenes. It should be mentioned that the catalyst demonstrated not to be compatible with the presence of hydroxyl or carboxylic acid groups in the starting alkyne or alkene.
C in the starting alkyne is mandatory for the formation of the β-ketophosphonate products. The strategy was also successfully applied to the fabrication of β-ketophosphonates from both aromatic and aliphatic terminal alkenes. It should be mentioned that the catalyst demonstrated not to be compatible with the presence of hydroxyl or carboxylic acid groups in the starting alkyne or alkene.
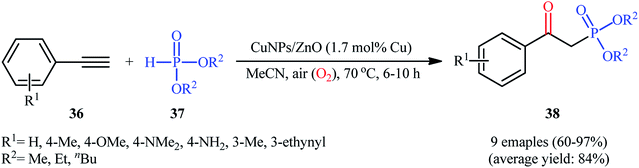 |
| | Scheme 15 CuNPs/ZnO-catalyzed oxyphosphorylation of aromatic terminal alkynes 36 with dialkyl H-phosphonates 37. | |
In 2016, Bu, Lu, and Cai demonstrated an innovative visible-light-induced organocatalytic oxyphosphorylation of alkynes with H-phosphine oxides under mild conditions.31 Thus, in the presence of only 0.5 mol% of rhodamine B as a photocatalyst in iPrOH under an oxygen atmosphere and irradiation with a white LED lamp at room temperature, oxidative coupling of aromatic terminal alkynes 39 and diaryl phosphine oxides 40 furnished the corresponding β-ketophosphine oxide derivatives 41 in good to excellent yields (Scheme 16a). However, the system was unfruitful with aliphatic and nitro-substituted aromatic alkynes, and due to the formation of complex mixtures, the products of internal alkynes could not be separated. It is worthy of note that other photocatalysts such as rose bengal, eosin B, eosin Y, and Ru(bpy)32+ were tested in this reaction and showed lower catalytic efficiency than that of rhodamine B. Of note, no product was observed in the absence of in the absence of a catalyst. The authors proposed mechanism for this transformation is analogous to the one depicted for alkenes in Scheme 10. Almost at the same time, Zhong and co-workers disclosed that the similar reaction could also successfully carried out under conventional heating conditions (55 °C) using 5 mol% of low-cost CuCN as a catalyst.32 Concurrently, in the same paper describing catalyst- and additive-free synthesis of β-ketophosphonates through the oxyphosphorylation of styrene derivatives bearing a leaving group at the α-position and H-phosphine oxides,19 the Lei laboratory described the synthesis of five β-ketophosphine oxides 43 in yield ranging from 48% to 71% via the PhB(OH)2-mediated oxidative coupling of aromatic terminal alkynes 42 with diphenylphosphine oxide 8 in NMP (Scheme 16b).
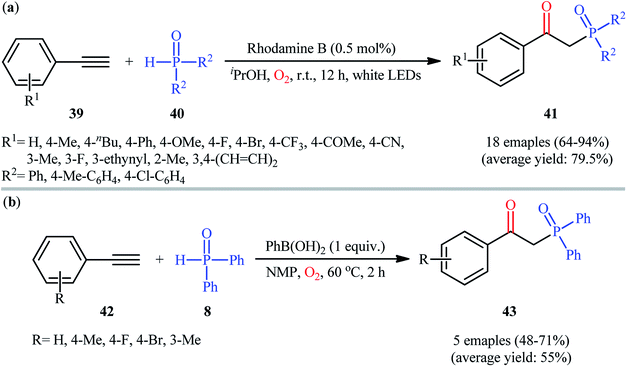 |
| | Scheme 16 (a) Visible-light-mediated oxidative coupling of terminal alkynes 39 and diaryl phosphine oxides 40; (b) PhB(OH)2-mediated oxyphosphorylation of aromatic terminal alkynes 42 with diphenylphosphine oxide 8. | |
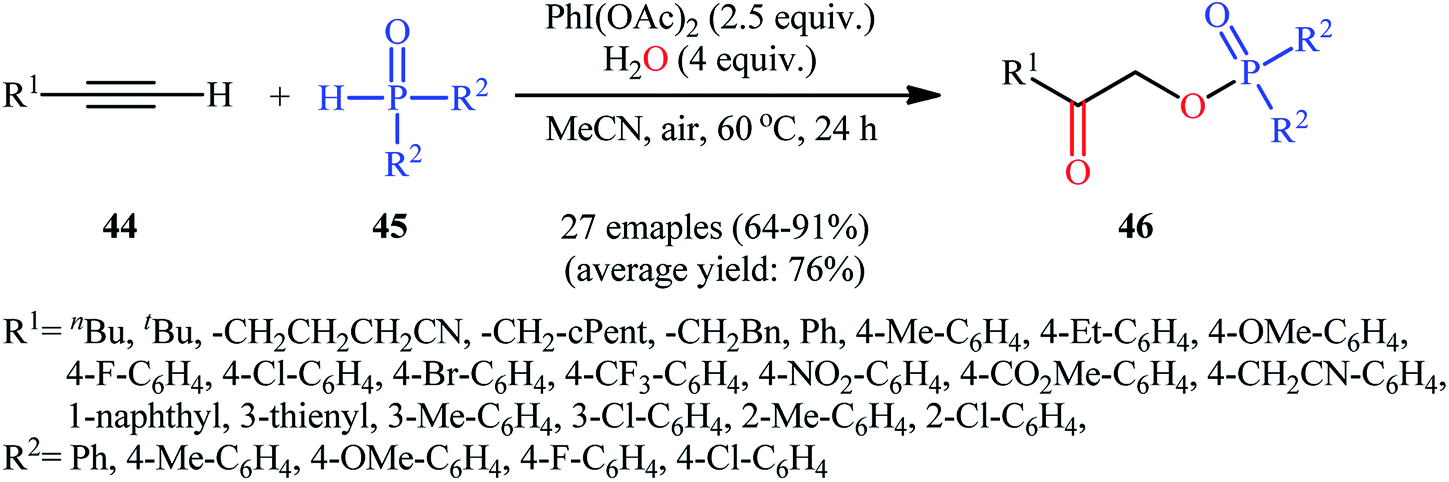
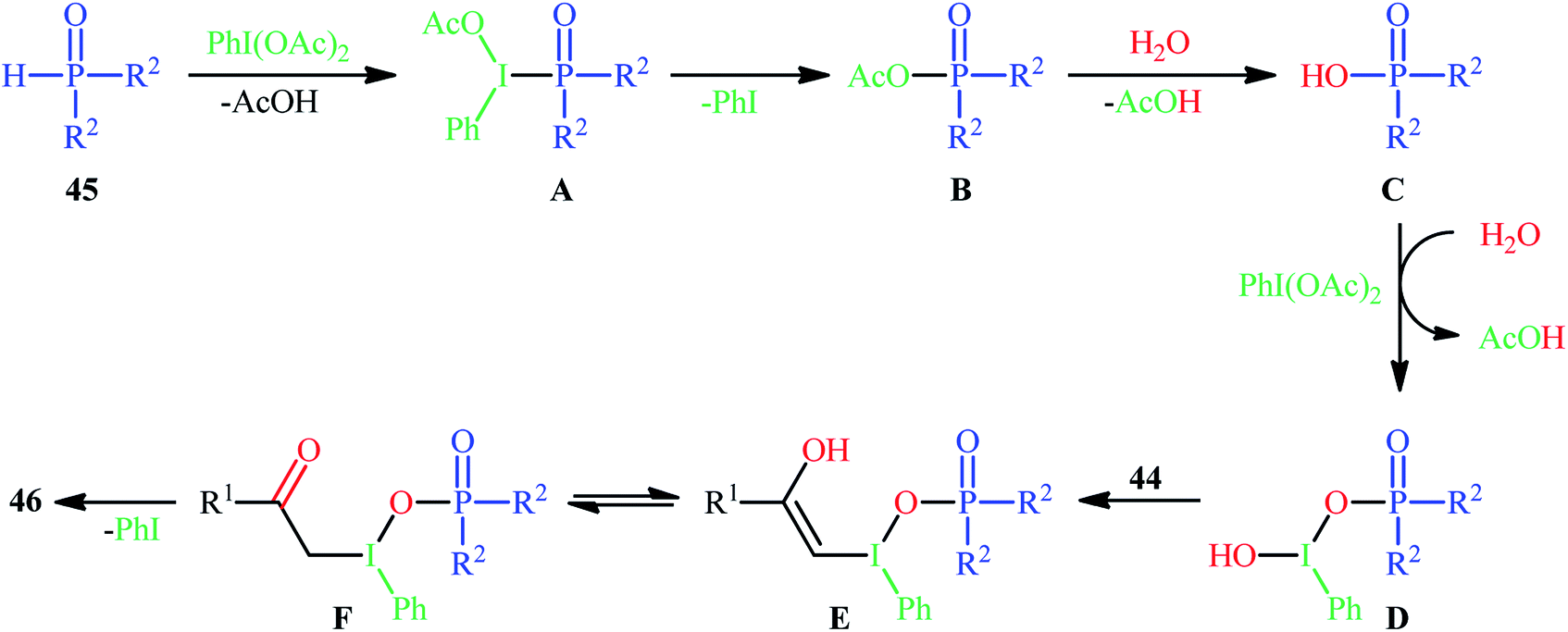
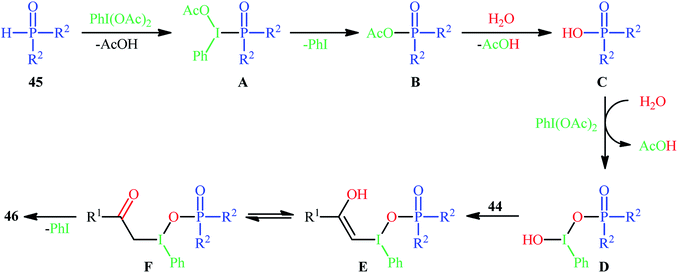
Along this line, Zhang, and Cheng reported a unique and efficient protocol for the metal-free direct synthesis of diaryl (2-oxo-2-alkyl/arylethyl)phosphinates from terminal alkynes, H-phosphine oxides, and water.33 Careful screening of a number of common oxidants such as H2O2, TBHP, PhI(OAc)2, K2S2O8, and m-CPBA; led to PhI(OAc)2 as the most suitable oxidant for this transformation and among the various organic solvents like THF, DCE, EtOH, DMF, MeCN, 1,4-dioxane, and toluene; MeCN proved to be the most efficient solvent. Both aromatic and aliphatic alkynes 44 and a variety of electron-rich and electron-deficient symmetrical diaryl H-phosphine oxides 45 easily took part in the reaction under optimized reaction conditions and provided the desired diaryl (2-oxo-2-alkyl/arylethyl)phosphinates 46 in good to excellent yields within 24 h (Scheme 17). This method was also applicable for gram-scale synthesis without any difficulty. The mechanistic course of this reaction is shown in Scheme 18. Initially, diaryl H-phosphine oxide 45 reacts with the phenyliodine diacetate to give hypervalent iodine intermediate A that, after intermolecular collapse converts to the phosphoric–carboxylic mixed anhydride intermediate B. Thereafter, nucleophilic attack of the water molecule on the anhydride B affords diarylphosphinic acid C that undergoes reaction with phenyliodine diacetate in the presence of water to form the unstable intermediate D. Next, addition of hypervalent iodine reagent D to alkyne 44 leads to intermediate E which undergoes isomerization to give hypervalent iodine intermediate F. Finally, the reductive elimination of this intermediate F affords the target product 46 with the releases of indobenzene.
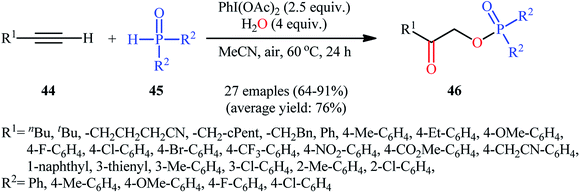 |
| | Scheme 17 Hypervalent iodine-mediated direct synthesis of diaryl (2-oxo-2-alkyl/arylethyl)phosphinates 46 from terminal alkynes 44 and diaryl H-phosphine oxides 45. | |
 |
| | Scheme 18 Mechanism that accounts for the formation of diaryl (2-oxo-2-alkyl/arylethyl)phosphinates 46. | |
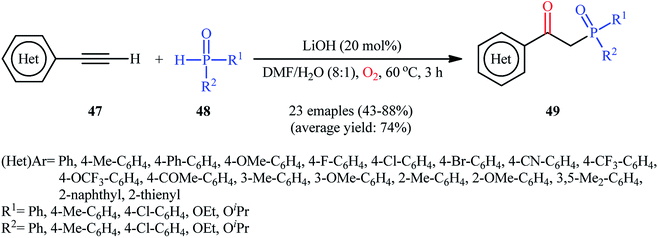
Recently, Shi and Zeng along with their colleagues disclosed the synthesis of β-phosphorylated ketones 49 via base-catalyzed oxyphosphorylation of (hetero)aromatic terminal alkynes 47 with P(O)–H compounds 48 (H-phosphonates, H-phosphine oxides, H-phosphinates) employing 20 mol% of LiOH as the base in an 8:1 mixture of DMF and H2O at 60 °C (Scheme 19).34 A wide panel of important functional groups (e.g., F, Cl, Br, CF3, OCF3, OMe, COMe, CN) at different positions of phenyl rings of phenylacetylenes were well tolerated by this reaction, thus indicating its broad applicability. Unfortunately, no comment was made by the authors regarding the applicability of internal and aliphatic terminal alkynes in this protocol.
 |
| | Scheme 19 Base-promoted direct oxyphosphorylation of (hetero)aromatic terminal alkynes 47 with P(O)–H compounds 48. | |
4. Conclusion
Organophosphorus compounds have aroused a significant interest among synthetic chemists over the past few decades owing to their wide applications in medicinal chemistry, organic synthesis, ligand chemistry, and material science. Among them, β-phosphorylated ketones have attracted significant attention as promising building blocks for the synthesis of various important molecules due to their diverse reaction patterns. In addition to their numerous synthetic utility, some β-phosphorylated ketones themselves are endowed with outstanding biological properties. Therefore, over the years, plenty efforts have been made to identify new and efficient synthetic methods for their preparation from simple and low-cost starting materials. Among the various approaches, oxyphosphorylation of alkenes/alkynes with hydrogen phosphoryl compounds represents an extremely attractive and powerful route for the synthesis of the title compounds which offers several benefits such as high step- and atom-economy as well as the readily available starting materials. As illustrated, both internal and terminals alkenes/alkynes and all the three kinds of P(O)–H compounds (H-phosphonates, H-phosphine oxides, and H-phosphinates) were compatible in this synthetic strategy. Moreover, most of the reactions covered in this review could be easily scaled up to provide multigram quantities of the target β-phosphorylated ketone products without any difficulty. However, despite remarkable developments over past decade on this attractive research arena, many challenges still remain to be overcome: (i) the reported examples on the use of H-phosphinates and internal alkynes as substrates are scarce and therefore, of course, expanding of the substrate scope to these compounds are necessary; (ii) the majority of metal-catalyzed reactions were conducted in the presence of copper catalysts and under relatively harsh conditions. Thus, the development of new metal-based catalytic systems that allow this difunctionalization reaction under milder conditions would be desirable; and (iii) the synthetic utility of these transformations in the preparation of biologically important compounds should be studied. We conclude this review by hoping that it will inspire researchers to make further progress in this appealing research area.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) T. Q. Ly and J. Woollins, Coord. Chem. Rev., 1998, 176, 451–481 CrossRef CAS;
(b) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef;
(c) M.-N. Birkholz, Z. Freixa and P. W. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
-
(a) J. R. Kouvaris, V. E. Kouloulias and L. J. Vlahos, Oncologist, 2007, 12, 738–747 CrossRef CAS;
(b) M. Aapro, A. Carides, B. L. Rapoport, H.-J. Schmoll, L. Zhang and D. Warr, Oncologist, 2015, 20, 450–458 CrossRef CAS;
(c) P. Todd and K. Goa, Drugs, 1992, 43, 346–381 CrossRef CAS;
(d) S. J. Hadziyannis, N. C. Tassopoulos, E. J. Heathcote, T. T. Chang, G. Kitis, M. Rizzetto, P. Marcellin, S. G. Lim, Z. Goodman and J. Ma, Gastroenterol, 2006, 131, 1743–1751 CrossRef CAS;
(e) O. Johnell, B. Jönsson, L. Jönsson and D. Black, Pharmacoeconomics, 2003, 21, 305–314 CrossRef;
(f) S. Shafiei and S. Davaran, Chem. Rev. Lett., 2020, 3, 19–22 Search PubMed;
(g) S. Majedi and S. Majedi, J. Chem. Lett., 2020, 1, 2–8 Search PubMed;
(h) F. Nareetsile, J. T. P. Matshwele, S. Ndlovu and M. Ngaski, Chem. Rev. Lett., 2020, 3, 140–160 Search PubMed.
- M. Kazemi, A. M. Tahmasbi, R. Valizadeh, A. A. Naserian and A. Soni, Agric. Sci. Res. J., 2012, 2, 512–522 Search PubMed.
-
(a) Y. Zhou, S. Yin, Y. Gao, Y. Zhao, M. Goto and L. B. Han, Angew. Chem., Int. Ed. Engl., 2010, 122, 7004–7007 CrossRef;
(b) Y.-L. Liu, Y. Liang, S.-F. Pi and J.-H. Li, J. Org. Chem., 2009, 74, 5691–5694 CrossRef CAS;
(c) Y. Hamashima, T. Suzuki, H. Takano, Y. Shimura, Y. Tsuchiya, K.-i. Moriya, T. Goto and M. Sodeoka, Tetrahedron, 2006, 62, 7168–7179 CrossRef CAS;
(d) G. Lu, B. Lin, Y. Gao, J. Ying, G. Tang and Y. Zhao, Synlett, 2017, 28, 724–728 CAS;
(e) F. A. Davis and Y. Wu, Org. Lett., 2004, 6, 1269–1272 CrossRef CAS.
-
(a) X. Li, A. Bhandari, C. P. Holmes and A. K. Szardenings, Bioorg. Med. Chem. Lett., 2004, 14, 4301–4306 CrossRef CAS;
(b) S. K. Perumal, S. Adediran and R. Pratt, Bioorg. Med. Chem., 2008, 16, 6987–6994 CrossRef CAS;
(c) N. Auberger, B. Al-Dabbagh, A. Bouhss, M. Crouvoisier, C. Gravier-Pelletier and Y. Le Merrer, Org. Biomol. Chem., 2011, 9, 8301–8312 RSC;
(d) V. Cavallaro, Y. F. Moglie, A. P. Murray and G. E. Radivoy, Bioorg. Chem., 2018, 77, 420–428 CrossRef CAS.
- C. Shan, F. Chen, J. Pan, Y. Gao, P. Xu and Y. Zhao, J. Org. Chem., 2017, 82, 11659–11666 CrossRef CAS.
- A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS.
-
(a) X. Li, G. Hu, P. Luo, G. Tang, Y. Gao, P. Xu and Y. Zhao, Adv. Synth. Catal., 2012, 354, 2427–2432 CrossRef CAS;
(b) L. Xie, R. Yuan, R. Wang, Z. Peng, J. Xiang and W. He, Eur. J. Org. Chem., 2014, 2014, 2668–2671 CrossRef CAS.
-
(a) K. M. Maloney and J. Y. Chung, J. Org. Chem., 2009, 74, 7574–7576 CrossRef CAS;
(b) R. R. Milburn, K. McRae, J. Chan, J. Tedrow, R. Larsen and M. Faul, Tetrahedron Lett., 2009, 50, 870–872 CrossRef CAS.
- A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC.
-
(a) E. M. Beccalli, G. Broggini, S. Gazzola and A. Mazza, Org. Biomol. Chem., 2014, 12, 6767–6789 RSC;
(b) J. B. Peng, Adv. Synth. Catal., 2020, 362, 3059–3080 CrossRef CAS;
(c) J. Lin, R. J. Song, M. Hu and J. H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef CAS.
-
(a) N. Yue and F. R. Sheykhahmad, J. Fluorine Chem., 2020, 109629 CrossRef CAS;
(b) L. Feng, X. Li, B. Liu and E. Vessally, J. CO2 Util., 2020, 40, 101220 CrossRef CAS.
- A. Hajra, S. Ghosh, S. Samanta, A. K. Ghosh and S. Neogi, Adv. Synth. Catal., 2020, 362, 4552–4578 CrossRef.
-
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef;
(b) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20 CrossRef;
(c) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(d) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(e) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed;
(f) S. Majedi, S. Majedi and F. Behmagham, Chem. Rev. Lett., 2019, 2, 187–192 Search PubMed;
(g) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett., 2020, 3, 2–8 Search PubMed;
(h) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed.
-
(a) M. Li, S. Abdolmohammadi, M. S. Hoseininezhad-Namin, F. Behmagham and E. Vessally, J. CO2 Util., 2020, 38, 220–231 CrossRef CAS;
(b) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 37 CrossRef CAS;
(c) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef;
(d) J. Wang, P. Su, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 41684–41702 RSC;
(e) Y. Liu, A. G. Ebadi, L. Youseftabar-Miri, A. Hassanpour and E. Vessally, RSC Adv., 2019, 9, 25199–25215 RSC;
(f) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC;
(g) S. Ebrahimiasl, F. Behmagham, S. Abdolmohammadi, R. N. Kojabad and E. Vessally, Curr. Org. Chem., 2019, 23, 2489–2503 CrossRef CAS;
(h) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(i) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(j) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(k) A. Bakhtiary, S. A. Cochrane, P. Mercier, R. T. McKay, M. Miskolzie, C. S. Sit and J. C. Vederas, J. Am. Chem. Soc., 2017, 139, 17803–17810 CrossRef CAS;
(l) S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo, E. M. Rodriguez-Lopez, P. Mercier and J. C. Vederas, PNAS, 2016, 113, 11561–11566 CrossRef CAS;
(m) E. Kianmehr, A. Bakhtiary and C. Zhu, Monatsh. Chem., 2012, 143, 255–262 CrossRef CAS;
(n) S. Mazlom Darbandy, A. Bakhtiary and H. Taheri, IPCBEE, 2011, 23, 125–129 Search PubMed;
(o) A. Amin Abshouri and A. Bakhtiary, J.Commun. Comput., 2012, 9, 387–391 Search PubMed;
(p) A. Amin Abshouri, M. Meybodi and A. Bakhtiary, IEEE proceedings, 2011, 13, 989–993 Search PubMed.
- W. Wei and J. X. Ji, Angew. Chem., Int. Ed. Engl., 2011, 123, 9263–9265 CrossRef.
- G.-Y. Zhang, C.-K. Li, D.-P. Li, R.-S. Zeng, A. Shoberu and J.-P. Zou, Tetrahedron, 2016, 72, 2972–2978 CrossRef CAS.
- M.-S. Li, Q. Zhang, D.-Y. Hu, W.-W. Zhong, M. Cheng, J.-X. Ji and W. Wei, Tetrahedron Lett., 2016, 57, 2642–2646 CrossRef CAS.
- P. Peng, Q. Lu, L. Peng, C. Liu, G. Wang and A. Lei, ChemComm, 2016, 52, 12338–12341 RSC.
- W. Liang, Z. Zhang, D. Yi, Q. Fu, S. Chen, L. Yang, F. Du, J. Ji and W. Wei, Chin. J. Chem., 2017, 35, 1378–1382 CrossRef CAS.
- X. Chen, X. Chen, X. Li, C. Qu, L. Qu, W. Bi, K. Sun and Y. Zhao, Tetrahedron, 2017, 73, 2439–2446 CrossRef CAS.
- G. Nan and H. Yue, Tetrahedron Lett., 2018, 59, 2071–2074 CrossRef CAS.
- J. Gu and C. Cai, Org. Biomol. Chem., 2017, 15, 4226–4230 RSC.
- D. Yamamoto, H. Ansai, J. Hoshino and K. Makino, Chem. Pharm. Bull., 2018, 66, 873–879 CrossRef CAS.
- Y. Shi, R. Chen, K. Guo, F. Meng, S. Cao, C. Gu and Y. Zhu, Tetrahedron Lett., 2018, 59, 2062–2065 CrossRef CAS.
- F. M. Moghaddam, M. Daneshfar, R. Azaryan and J.-L. Pirat, Catal. Commun., 2020, 106015 CrossRef CAS.
- X. Chen, X. Li, X.-L. Chen, L.-B. Qu, J.-Y. Chen, K. Sun, Z.-D. Liu, W.-Z. Bi, Y.-Y. Xia and H.-T. Wu, ChemComm, 2015, 51, 3846–3849 RSC.
- N. Yi, R. Wang, H. Zou, W. He, W. Fu and W. He, J. Org. Chem., 2015, 80, 5023–5029 CrossRef CAS.
- M. Zhou, M. Chen, Y. Zhou, K. Yang, J. Su, J. Du and Q. Song, Org. Lett., 2015, 17, 1786–1789 CrossRef CAS.
- V. Gutierrez, E. Mascaró, F. Alonso, Y. Moglie and G. Radivoy, RSC Adv., 2015, 5, 65739–65744 RSC.
- M.-j. Bu, G.-p. Lu and C. Cai, Catal. Sci. Technol., 2016, 6, 413–416 RSC.
- W.-W. Zhong, Q. Zhang, M.-S. Li, D.-Y. Hu, M. Cheng, F.-T. Du, J.-X. Ji and W. Wei, Synth. Commun., 2016, 46, 1377–1385 CrossRef CAS.
- D.-Y. Hu, M.-S. Li, W.-W. Zhong, J.-X. Ji, J. Zhu, W. Wei, Q. Zhang and M. Cheng, Chin. Chem. Lett., 2016, 27, 1691–1695 CrossRef CAS.
- W. Zhong, T. Tan, L. Shi and X. Zeng, Synlett, 2018, 29, 1379–1384 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b






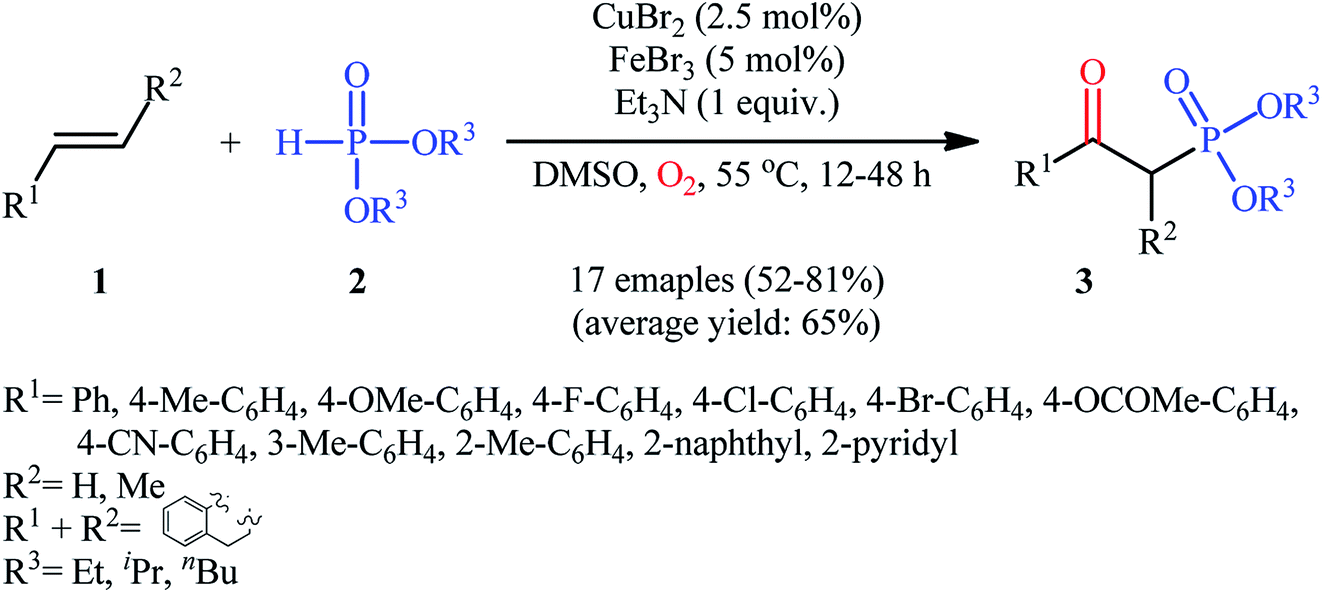
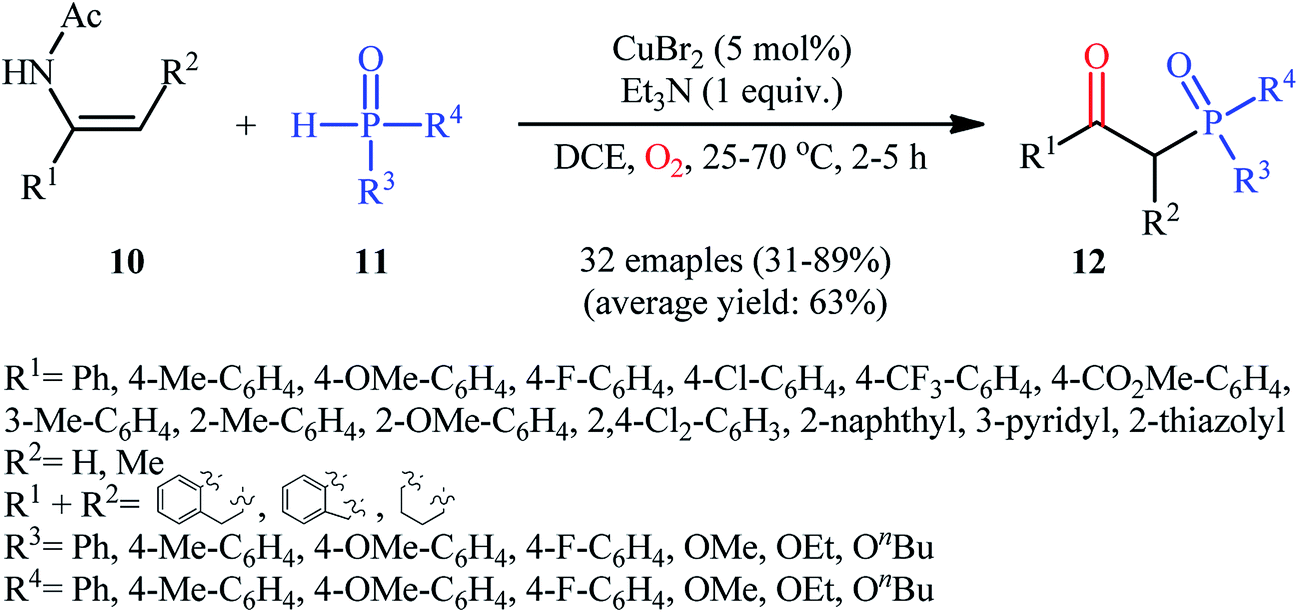


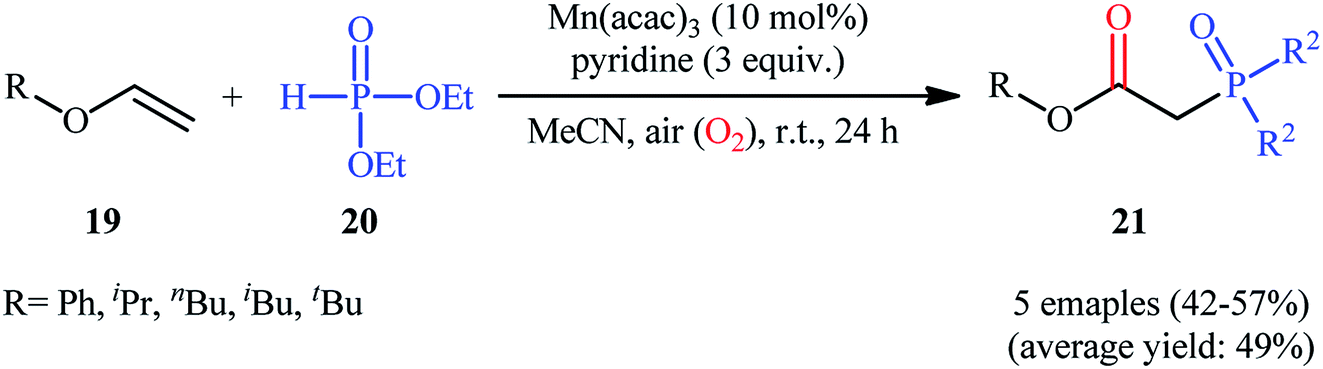
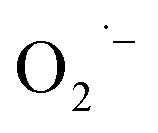 and regenerate the PC; (iv) deprotonation of intermediate B to yield the P-centered radical C; (v) reaction of intermediate C with alkene 22 to give radical D; (vi) the combination of intermediate D with
and regenerate the PC; (iv) deprotonation of intermediate B to yield the P-centered radical C; (v) reaction of intermediate C with alkene 22 to give radical D; (vi) the combination of intermediate D with 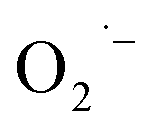 to generate peroxide anion E; (vii) protonation of intermediate E to form intermediate F; and (viii) dehydration of intermediate F to give the desired product 24.
to generate peroxide anion E; (vii) protonation of intermediate E to form intermediate F; and (viii) dehydration of intermediate F to give the desired product 24.