
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-component three-bond forming cascade via palladium photoredox catalysis†
Peter
Bellotti
 ,
Maximilian
Koy
,
Christian
Gutheil
,
Steffen
Heuvel
and
Frank
Glorius
*
,
Maximilian
Koy
,
Christian
Gutheil
,
Steffen
Heuvel
and
Frank
Glorius
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
First published on 7th December 2020
Abstract
A highly modular radical cascade strategy based upon radical cyclisation/allylic substitution sequence between alkyl/aryl bromides, 1,3-dienes and nucleophiles ranging from sulfinates to amines, phenols and 1,3-dicarbonyls is described (>80 examples). Palladium phosphine complexes – which merge properties of photo- and cross coupling-catalysts – allow to forge three bonds with complete 1,4-selectivity and stereocontrol, delivering highly value added carbocyclic and heterocyclic motifs that can feature – inter alia – vicinal quaternary centers, free protic groups, gem-difluoro motifs and strained rings. Furthermore, a flow chemistry approach was for the first time applied in palladium–photocatalysed endeavors involving radicals.
Introduction
Cascade strategies have emerged as pivotal transformations that can enable formation of multiple C–C and C–X linkages in a single operational step. Particularly, the rapid generation of intricate chemical architectures and enhanced step-atom economy are among the salient features of domino processes, which are both prevalent in nature and complex total syntheses (Schemes 1A and B).1 In such realm, radical cascade processes2a either driven by single-electron transfer events,2b radical chains or UV-irradiation stand as a synthetic toolbox in natural product synthesis, often complementary to nature's cationic cascade processes (e.g. lanosterol vs. Pattenden's estrone synthesis).2c,d Despite the intriguing bond disconnections that can be envisaged via radical cascades, these methods are often plagued by the use of toxic and hazardous chemicals – for instance organotin reagents and radical initiators.3a–c Seeking for milder and environmentally friendly alternatives of generating radicals, visible light photocatalysis can harness the energy of photons to promote single-electron transfer events by exploiting catalytic amounts of light-absorbing species.4a–e Photocatalysis – often in combination with nickel (synergistic or dual catalysis) – has fostered the rapid development of radical cascade manifolds.5a–f Approaching the problem using a single catalytic species, Gevorgyan,6a–e Shang and Fu,6f,g Glorius6h–k and others6l–n have utilized palladium complexes as single-component7a,b photo- and cross coupling-catalysts for reactions via radical intermediates, obviating the need for a dual catalytic system featuring two species (e.g. synergistic catalysis with Ni and Ir-photocatalyst), therefore simplifying the set up and reducing operational costs.4a–e Furthermore, previously inaccessible transformations – which significantly complement well-established means of generating radicals in synthetic endeavours – have been devised.7a–e Particularly, the plethora of substrates amenable for palladium–catalysed coupling reactions extends well-beyond sp2-hybridised species,8a,b thus offering a gateway towards increasingly complex scaffolds to «Escape from Flatland» (Lovering et al.).9 In this regard, radical species prove highly suitable to generate congested quaternary centres, thus overcoming typical limitations of ionic pathways. In 2020, our group6j,k and Gevorgyan6e independently reported a radical trapping/allylic substitution sequence under palladium–photocatalysis, affording decorated alkenes: in the latter case, aryl-substituted dienes controlled the 1,2-selective difunctionalisation process, while in the former a sterically driven 1,4-substitution has been observed. Given the scarcity of palladium–photocatalysed three-bond three-component cascades10 and perpetuating our interest in the development of metallaphotoredox strategies towards highly congested carbon architectures,6h–k we wondered whether visible light could enable a domino reaction between olefin-tethered alkyl/aryl bromides, 1,3-dienes and nucleophiles. Building on the aforementioned radical palladium π-allyl generation approaches,6e–k the perspective strategy would involve the following steps (Scheme 1C): (I) excitation of palladium(0) complex A through visible light; (II) SET to generate an alkyl/aryl radical D; (III) 5-exo trig cyclisation to generate intermediate E;11 (IV) radical trapping with dienes to generate a stabilised allyl radical F and the second bond; (V) Pd(I)-radical-to-polar crossover towards Pd(II)-allyl intermediate G; (VI) allylic substitution to yield the product H and the catalytically active palladium(0) species.12 A careful tuning of the catalytic manifold is needed to prevent the following potential pitfalls: (I) unproductive protodehalogenation; (II) early Pd(I)-radical recombination, followed by fast β-H elimination;7d (III) elimination processes from G; (IV) scarce control of regio- (1,2/1,4) and E-selectivity due to η3–η1–η3 equilibria.13a–c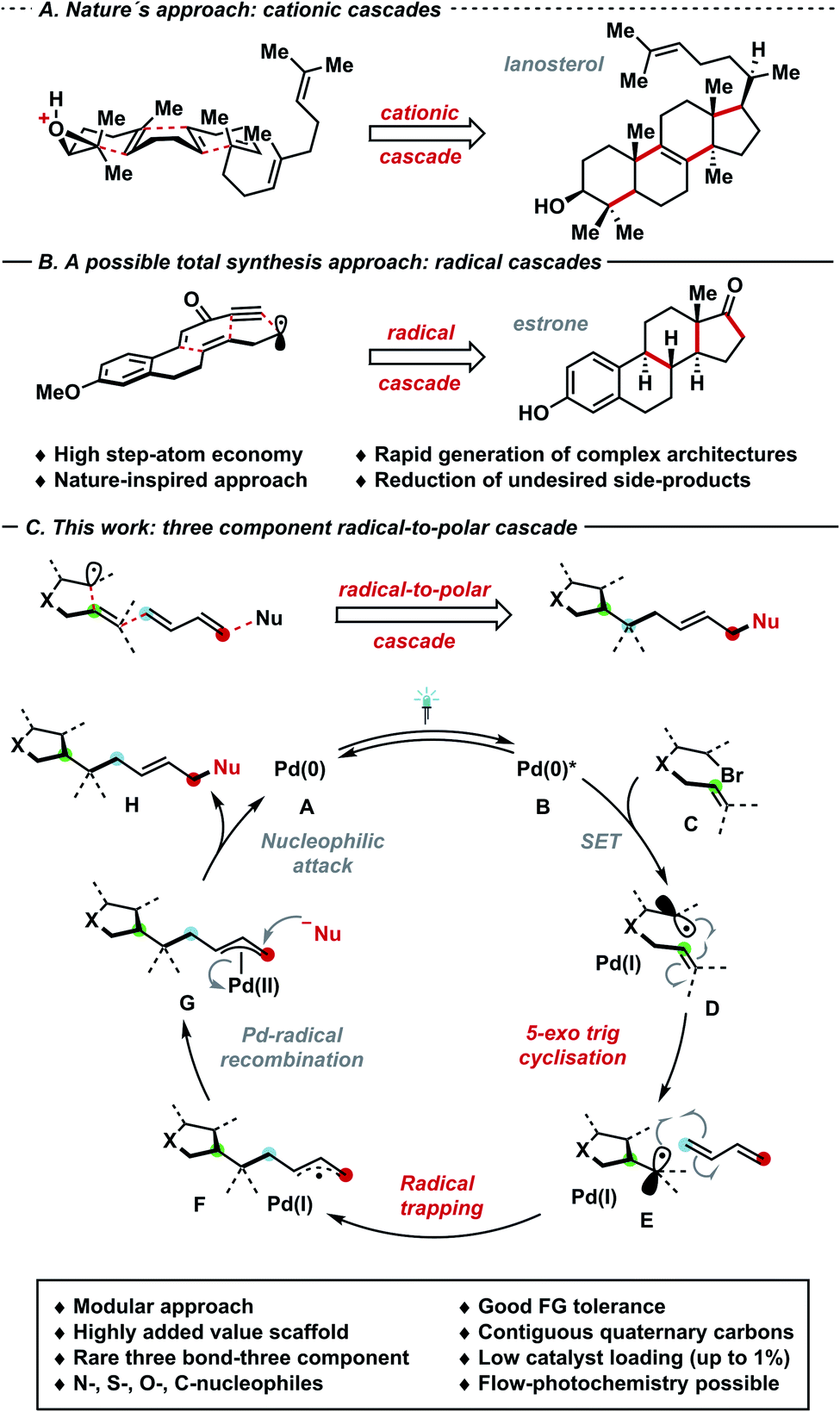 | ||
| Scheme 1 (A) Cationic cascades: lanosterol biosynthesis; (B) radical cascades: total synthesis of estrone; (C) three-component three-bond cascade strategy. | ||
Herein, we report the palladium–photocatalysed three-component three-bond cascade featuring a powerful 5-exo trig cyclisation, radical trapping and allylic functionalisation sequence to generate highly complex motifs bearing one or more contiguous quaternary centres with excellent 1,4-selectivity and stereocontrol (E/Z ratio). Remarkably, this process can tolerate both (hetero-)aryl and alkyl bromides bearing different functional groups, a vast array of nitrogen, sulfur, carbon and oxygen nucleophiles and variously substituted dienes, allowing the modification of the core structure at will in a highly modular fashion. Furthermore, the newly developed palladium–photocatalysed cross coupling process was performed in flow, thus enabling the streamlined product formation with higher output thanks to reduced reaction times.14a,b
Results and discussion
In line with our previous results on palladium–photocatalysed processes,6h–k we investigated a set of bidentate ligands (Fig. 1C) and conditions (Fig. 1A) in the three-component coupling of bromide 1a with butadiene 2a and sodium p-toluenesulfinate 3a to yield dihydrobenzofuran 4a. Interestingly, DPEPhos (L1) outperformed rac-BINAP (L2) and XantPhos (L3) as ligand, while moderate conversion (45%) could be detected even in the absence of added ligand (entry 2). While the presence of L2 caused a diminished initial cyclisation rate (C to E), the ligand was successfully used upon longer reaction times when the allylic substitution step proved troublesome (Scheme 2C, 3C and D).15 Furthermore, ethereal solvents – in particular 1,4-dioxane – afforded better yields in comparison with other polar aprotic solvents (entry 3).16 Both inorganic and organic bases (entry 4) proved compatible with the catalytic manifold, as well as reduced catalyst loading (2 or 1 mol%), albeit with a moderate reduction of yield of 4a (77 and 62%, respectively). In the case of sulfinate nucleophiles – while not strictly needed to observe reactivity – stoichiometric amounts of base proved to be beneficial for the conversion (entry 8).15 In contrast, both light irradiation and palladium catalyst are required to observe any product formation. Furthermore, heating at reflux in absence of light (entry 7) did not afford 4a, reinforcing the notion that the process is photon-driven rather than thermally triggered. | ||
| Fig. 1 (A) Optimisation of the three-component coupling. aConditions: 1a (0.12 mmol), 2a (0.15 mmol, 2 M in THF), 3a (0.10 mmol), K2CO3 (0.15 mmol), Pd(PPh3)4 (5 mol%), DPEPhos (10 mol%), 1,4-dioxane (0.1 M), 30 W 450 nm LEDs, 25–30 °C, 24 h. bYields were determined using 1H NMR spectroscopy with CH2Br2 as internal standard; cReaction time increased to 48 h. (B) Sensitivity assessment radar diagram. (C) Ligands screened in the catalytic reaction. p-Tol = para-tolyl. DPEPhos = (oxydi-2,1-phenylene)bis(diphenylphosphine); BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; XantPhos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene. | ||
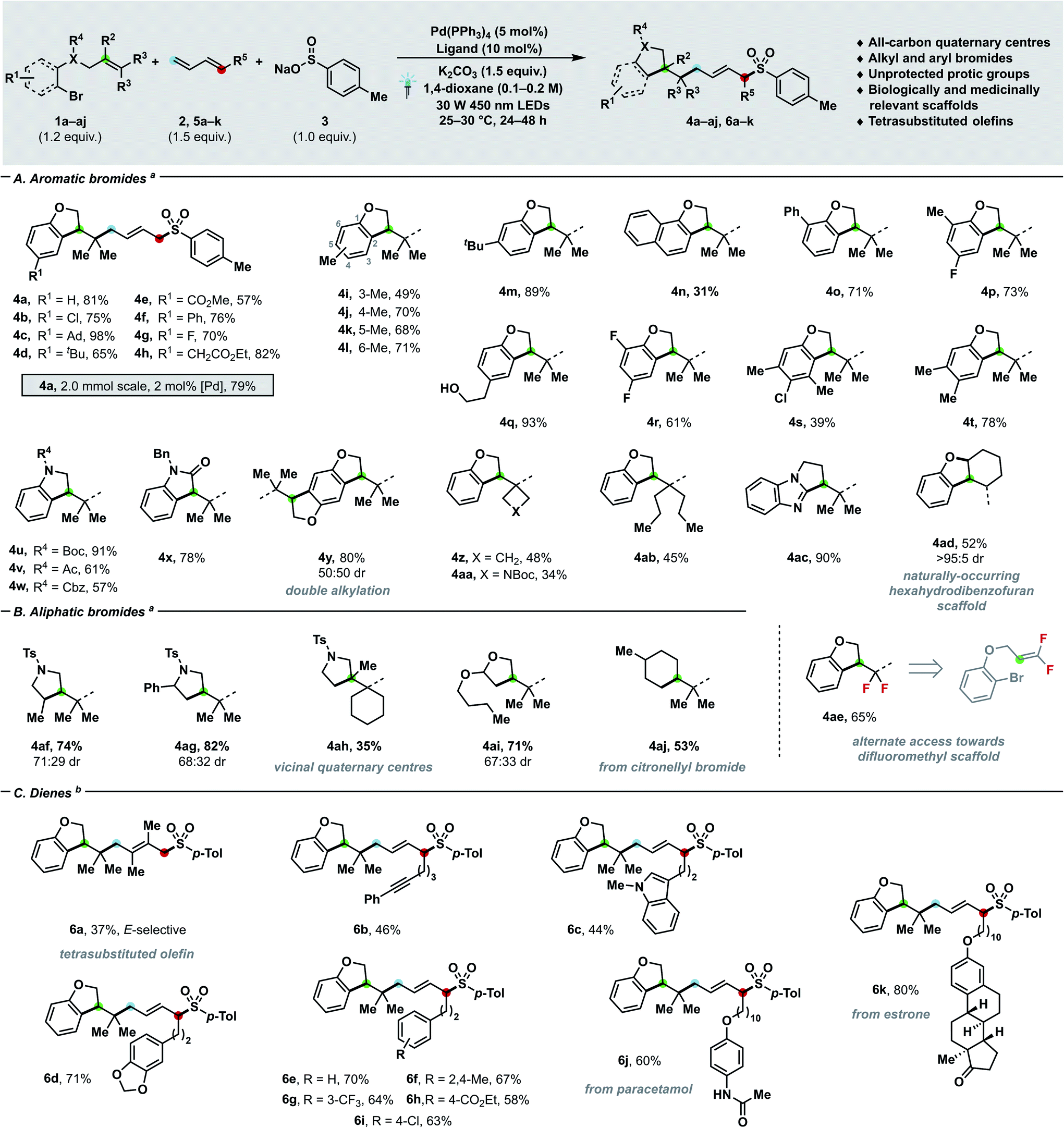 | ||
| Scheme 2 Substrate scope with regards to bromides (A and B) and dienes (C). Yields of isolated products are given. For experimental details, see ESI.†aConditions: 1a–aj (0.24 mmol), 2a (0.30 mmol, 2 M in THF), 3a (0.20 mmol), K2CO3 (0.30 mmol), Pd(PPh3)4 (5 mol%), DPEPhos (10 mol%), 1,4-dioxane (0.1 M), 30 W 450 nm LEDs, 25–30 °C. bConditions: 1a (0.24 mmol), 5a–k (0.30 mmol), 3a (0.20 mmol), K2CO3 (0.30 mmol), Pd(PPh3)4 (5 mol%), rac-BINAP (10 mol%), 1,4-dioxane (0.2 M), 30 W 450 nm LEDs, 25–30 °C. Ad = 1-adamantyl, Ph = phenyl, Boc = tert-butoxycarbonyl, Cbz = carboxybenzyl, Ac = acetyl, Bn = benzyl, Ts = para-toluenesulfonyl. | ||
Under the optimized conditions, a sensitivity screening approach (Fig. 1B) assessed the inertness of the reaction towards concentration fluctuations, different light intensities, presence of trace water and even upon scaling-up the process. Conversely, high oxygen content, significantly elevated or reduced temperatures proved detrimental to the reaction yield.17
Having assessed the feasibility of the cascade process, we investigated the generality of our newly developed manifold with respect of bromides (Schemes 2A and B), dienes (Scheme 2C) and nucleophilic partners (Scheme 3). When olefin-tethered bromoaryl ethers were used as radical precursors (1a–aj), the corresponding dihydrobenzofurans arising from the three-bond three-component coupling were obtained with excellent E- (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) and 1,4-selectivity (>20:1). Furthermore – when the catalyst loading was reduced to 2 mol% and the process scaled-up to 2 mmol – product 4a was afforded in 79% yield. A plethora of different functional groups, spanning from aliphatic (4c–d, 4i–m) to halide substituents such as chlorides (4b, 4s), fluorides (4g, 4p, 4r), as well as aryls (4f, 4o), aryl/alkyl esters (4e, 4h) and condensed aromatics (4n) were tolerated. As shown by examples 4i–l, substitution can be accommodated at any available ring position, while yield differences can be ascribed to the interplay between steric and electronic effects. Notably, protic groups like alcohols (4q) did not interfere with the catalytic process, thus enabling to tune the substitution of coupling partners almost at will. By reacting a doubly O-alkylated hydroquinone substrate, the tetrahydrobenzodifuran 4y arising from the two-fold three-component coupling could be obtained in 80% yield. Interestingly, the catalytic manifold can generate highly decorated heterocyclic systems other than dihydrobenzofurans – namely indolines (4u–w), oxindole (4x) and dihydropyrrolbenzimidazole (4ac) – which are prevalent nitrogen containing scaffolds in medicinal chemistry.18 The tethered olefin substitution could be tuned (4z–ab, 4ad, 4ae) to access different highly congested quaternary carbon centres such as cyclobutane (4z) and Boc-azetidine (4aa). By employing 1,1-difluorosubstituted olefins, a gem-difluoro moiety (4ae) could be installed in good yield (65%), thus offering a mild alternative mean19 of generating such MedChem-relevant building blocks to nucleophilic fluorination.20 When cyclic disubstituted olefins were employed, the synthesis of hexahydrodibenzofuran 4ad – a prevalent motif in natural products (e.g. cannabielsoic acids and bisabosquals)21a,b – can be achieved with complete diastereocontrol. Delightfully, aliphatic bromides successfully delivered both carbocyclic (4aj) and heterocyclic (4af–4ai) coupling products in moderate to good yields (35–81%) and even congested vicinal quaternary centres (4ah) could be forged in a single step.22 The coupling of a secondary alkyl bromide (4af) further testifies the generality of the strategy with respect to the electrophilic partner.
:5) and 1,4-selectivity (>20:1). Furthermore – when the catalyst loading was reduced to 2 mol% and the process scaled-up to 2 mmol – product 4a was afforded in 79% yield. A plethora of different functional groups, spanning from aliphatic (4c–d, 4i–m) to halide substituents such as chlorides (4b, 4s), fluorides (4g, 4p, 4r), as well as aryls (4f, 4o), aryl/alkyl esters (4e, 4h) and condensed aromatics (4n) were tolerated. As shown by examples 4i–l, substitution can be accommodated at any available ring position, while yield differences can be ascribed to the interplay between steric and electronic effects. Notably, protic groups like alcohols (4q) did not interfere with the catalytic process, thus enabling to tune the substitution of coupling partners almost at will. By reacting a doubly O-alkylated hydroquinone substrate, the tetrahydrobenzodifuran 4y arising from the two-fold three-component coupling could be obtained in 80% yield. Interestingly, the catalytic manifold can generate highly decorated heterocyclic systems other than dihydrobenzofurans – namely indolines (4u–w), oxindole (4x) and dihydropyrrolbenzimidazole (4ac) – which are prevalent nitrogen containing scaffolds in medicinal chemistry.18 The tethered olefin substitution could be tuned (4z–ab, 4ad, 4ae) to access different highly congested quaternary carbon centres such as cyclobutane (4z) and Boc-azetidine (4aa). By employing 1,1-difluorosubstituted olefins, a gem-difluoro moiety (4ae) could be installed in good yield (65%), thus offering a mild alternative mean19 of generating such MedChem-relevant building blocks to nucleophilic fluorination.20 When cyclic disubstituted olefins were employed, the synthesis of hexahydrodibenzofuran 4ad – a prevalent motif in natural products (e.g. cannabielsoic acids and bisabosquals)21a,b – can be achieved with complete diastereocontrol. Delightfully, aliphatic bromides successfully delivered both carbocyclic (4aj) and heterocyclic (4af–4ai) coupling products in moderate to good yields (35–81%) and even congested vicinal quaternary centres (4ah) could be forged in a single step.22 The coupling of a secondary alkyl bromide (4af) further testifies the generality of the strategy with respect to the electrophilic partner.
 | ||
| Scheme 3 Substrate scope with regards to nucleophiles (A–D). Yields of the corresponding isolated products are given. For experimental details, see ESI.†aConditions: 1b (0.20 mmol), 2a (0.30 mmol, 2 M in THF), 7a–i (0.20 mmol), K2CO3 (0.30 mmol), Pd(PPh3)4 (5 mol%), DPEPhos (10 mol%), 1,4-dioxane (0.1 M), 25–30 °C. bProduct 8f underwent spontaneous partial dechlorination upon purification. cConditions: 1b (0.20 mmol), 2a (0.60 mmol), 9a–l (0.30 mmol), Et3N (0.60 mmol), Pd(PPh3)4 (5 mol%), DPEPhos (10 mol%), 1,4-dioxane (0.1 M), 30 W 450 nm LEDs, 25–30 °C. dKOH (0.6 mmol) was used as base. e93:7 E/Z. f94:6 E/Z. gConditions: 1a (0.30 mmol), 2a (0.60 mmol), 11a–k (0.20 mmol), K2CO3 (0.60 mmol), Pd(PPh3)4 (5 mol%), rac-BINAP (10 mol%), 1,4-dioxane (0.2 M), 30 W 450 nm LEDs, 25–30 °C. hConditions: 1a (0.20 mmol), 2a (0.30 mmol), 13a–d (0.60 mmol), KOAc (0.30 mmol), Pd(PPh3)4 (5 mol%), rac-BINAP (10 mol%), 1,4-dioxane (0.2 M), 30 W 450 nm LEDs, 25–30 °C. i1h (0.20 mmol), 13b (0.20 mmol) and K2CO3 (0.30 mmol) were used. Cy = cyclohexyl. | ||
When the challenging 2,3-dimethylbutadiene was used as coupling partner, the corresponding E-configured tetrasubstituted alkene (6a) was obtained in synthetically useful yield (37%).23 Then, we sought to investigate monosubstituted dienes (6b–k) in the three-component coupling. Pleasingly, a wide array of functional groups comprising alkynes (6b), indoles (6c), variously decorated arenes (6d–i), pharmaceuticals (6j) and natural products (6k) can be incorporated with exquisite site- and E-selectivity in moderate to excellent yields (44–80%).
To further prove the generality of our protocol with regards of coupling partners, a plethora of different S- (Scheme 3A), N- (Scheme 3B), O- (Scheme 3C) and C-nucleophiles (Scheme 3D) were successfully employed. Both aryl (8a–g) and alkyl sulfinates (8h–i) delivered the desired allyl sulfones, which are relevant scaffolds both in medicinal and synthetic chemistry.24a,b Alkyl (8a, 8c–e), fluorine (8b, 8g), and chlorine (8f) substitutions were well-tolerated and even sterically demanding ortho-substituted (8c–f) aryl sulfinates afforded the coupling products with complete 1,4- and E-selectivity. Thanks to the high prevalence of nitrogen in nature and drugs, the generation of a new C–N linkage via our protocol was highly desirable. Delightfully, both aliphatic amines (10e–f), heterocyclic amines (10a–d, 10g–i) and electron-rich N-containing aromatics (10j–l) can deliver highly sought-after tertiary allyl amines which are widely encountered in drugs (e.g. cinnarizine or azaprocin) or N-alkylated heterocyles in good yields (57–80%). Piperidine (10c), piperazine (10i), pyrrolidine (10b), indole (10j, 10k), morpholine (10a) and tetrahydroisoquinoline (10h) – which all rank among the top 20 of most frequently encountered N-heterocycles in FDA-approved drugs25 – can be successfully coupled. Pleasingly, variously decorated phenols spanning from guaiacol (12a), 3-methoxyphenol (12b), mequinol (12c), hydroxyaniline (12d) to paracetamol (12g), ethynylestradiol (12h) and estrone (12i) afforded the corresponding aryl ethers in moderate (64%) to quantitative yields. Furthermore, we tested the compatibility of our protocol towards both electron-rich and -poor heteroaromatics tethered to the phenol such as indole (12e), pyrrole (12f), pyridine (12j) and tetrazole (12k): in either case, products were afforded in moderate to excellent yields (31–93%). Delightfully, product 10a could still be obtained in 63% yield at 1 mol% catalyst loading, which has limited precedence in palladium–photocatalysis (common loading: 5–10 mol%).7c,d Carbon-based nucleophiles such as 1,3-diketones (14a, d), C2-substituted acetoacetates (14c) and diethyl malonate (14b) effectively performed as coupling partners to provide the allylated products (50–72%). The presented three-bond three-component protocol proved compatible with photo-flow conditions (Fig. 2A) which have never been applied – to the best of our knowledge – in palladium–photoredox endeavours involving radicals.26 In line with the notion of a more efficient illumination and mass transfer, remarkably improved results with a residence time of 30 minutes (flow rate: 4 mL h−1) were obtained in comparison with batch reaction (0.31 mmol h−1vs. 0.14 mmol h−1).14 To highlight the potential follow-up chemistry and the highly value added motifs that the present project enables to achieve, we performed the following selected transformations. Allyl amine 10a underwent diastereoselective Lewis acid-catalysed Belluš–Claisen rearrangement to generate α,β-disubstituted-γ,δ-unsaturated amide 15.27 Furthermore, we identified the allyl sulfone 4a as possible precursor of the highly decorated internal olefin 16, which was obtained via reductive desulfonylation with excellent regio- and stereocontrol (Fig. 2B).28
 | ||
| Fig. 2 (A) Reaction under flow conditions, comparison with batch process and synthetic application of the product. Flow reaction: 1b (0.20 mmol), 2a (0.60 mmol), morpholine (0.30 mmol), Et3N (0.60 mmol), 1,4-dioxane (0.1 M), 30 W 450 nm LEDs, 25–30 °C, flow rate: 4 mL h−1. (B) Reductive desulfonylation. (C) Radical probe experiment. (D) TEMPO trapping experiments. (E) UV-vis spectroscopic analysis. (F) In situ formation of catalytically active species from Pd(DPEPhos)Cl2. asyn:anti nomenclature in accordance to ref. 27. bSee Fig. 1 – entry 1. c1a (0.12 mmol), 2a (0.15 mmol, 2 M in THF), 3a (0.10 mmol), K2CO3 (0.15 mmol), 1,4-dioxane (0.1 M), 30 W 450 nm LEDs, 25–30 °C, 24 h. dYields were determined using 1H NMR spectroscopy with CH2Br2 as internal standard. eIsolated yield. DIPEA = diisopropylethylamine; dppe = 1,2-bis(diphenylphosphino)ethane; DIBAL-H = diisobutylaluminium hydride. | ||
In line with previous investigations from our and other groups that shed light on the palladium–photoredox system,6a–n we performed selected experiments16 to corroborate our mechanistic proposal (Scheme 1C). Reinforcing the hypothesised intermediacy of radicals during the initial dicarbofunctionalisation cascade, the ring-opening coupling process of methylcyclopropyl radical A to release primary radical B was observed under standard reaction conditions (Fig. 2C). To support the envisaged radical-to-polar crossover mechanism, intermediate G could be detected by means of accurate mass spectrometry on the reaction.16 While TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) inhibited the catalytic turnover, trace amount of the adduct 19 arising from trapping of the initially cyclised radical could be detected. When the Pd(PPh3)4/L1 system was used in stoichiometric fashion, the cyclized TEMPO-adduct 19 could be isolated in 68% yield – with no residual bromide 1a – thus testifying that radical inhibition cannot interfere with the initial radical formation and intramolecular 5-exo trig cyclization, but exclusively with the subsequent steps (Fig. 2D). By UV-visible analysis of the reaction components, we proved that only Pd(PPh3)4 or Pd(PPh3)4/DPEPhos were absorbing blue light, thus suggesting that the transition metal enables the photocatalytic process to occur (Fig. 2E, see also Fig. 1 – entry 7). Upon addition of 1a, the fluorescence attributable to the Pd(PPh3)4/DPEPhos system changed, hinting that aryl bromides can interact with the catalytically competent mixture. As suggested by 31P NMR spectroscopy, facile displacement of PPh3 by bidentate DPEPhos occurs under the experimental conditions. By testing Pd(DPEPhos)Cl2 in combination with various reductants, we observed a considerable degree of catalytic activity in presence of phosphines (Fig. 2F).16 This latter finding – in combination with the observed fast ligand displacement by 31P NMR – corroborates the notion of DPEPhos-ligated species to be active under the reaction conditions.
Conclusions
In conclusion, we have developed a highly modular three-component dicarbofunctionalisation/allylic substitution sequence that can generate three C–C and C–Nu linkages in sequence by exploiting palladium as dual catalyst. The catalytic manifold tolerates a variety of functional groups – including unprotected protic groups and small ring systems – thus allowing to generate highly relevant motifs (e.g. allyl sulfones, allyl amines) with remarkable levels of structural flexibility. Moreover, the amenable coupling partners span from aryl to alkyl bromides and allow the generation of sterically congested quaternary centres and even vicinal quaternary carbons (4ah), which are challenging scaffolds to be accessed. For the first time, a palladium–catalysed photoredox reaction involving radical species was performed in flow, enabling the reduction of reaction times and the potential streamlined utilization of the process. Mechanistic experiments are supportive of a radical-to-polar crossover mechanism, whereby palladium acts – upon visible light irradiation – both as SET- and transition metal-catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (SFB 858, Leibniz Award) and the Friedrich-Ebert-Stiftung (C. G.) is gratefully acknowledged. The authors acknowledge Dr Matthias Letzel for mass spectrometry assistance, Frederik Sandfort, Dr Huan-Ming Huang (all WWU Münster) and Dr Stefano Crespi (University of Groningen) for the helpful discussion. We acknowledge Ley and co-workers for the flow chemistry icon template29 in Fig. 2A, distributed under the Creative Commons License-CC-CY-4.0.Notes and references
- For selected reviews on cascade reactions, with emphasis on total syntheses:
(a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed
; (b) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC
- For selected reviews and examples on radical cascade reactions:
(a) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC
- For selected reviews on radical reactions:
(a) D. P. Curran, Synthesis, 1988, 6, 417–439 CrossRef
- For excellent reviews on visible light photocatalysis:
(a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS
- For selected reports on visible light-enabled radical cascades:
(a) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed
- For selected examples on Pd-photoredox C–C bond forming reactions:
(a) D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212–14216 CrossRef CAS PubMed
- For an excerpt on thermally induced Pd-catalysed radical processes:
(a) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111–6137 CrossRef CAS
-
(a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS
- A single example featuring CO2 as coupling partner has been reported: L. Sun, J.-H. He, W.-J. Zhou, X. Zeng and D.-G. Yu, Org. Lett., 2018, 20, 3049–3052 CrossRef CAS PubMed
- J.-W. Ma, X. Chen, Z.-Z. Zhou and Y.-M. Liang, J. Org. Chem., 2020, 85, 9301–9312 CrossRef CAS
- For a review concerning allylic functionalisation involving radicals: H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC
- For selected reports dealing with regio- and stereoselectivity issues in Tsuji-Trost reactions:
(a) B. M. Trost and P. E. Strege, J. Am. Chem. Soc., 1975, 97, 2534–2535 CrossRef CAS
- Selected reviews on photo-flow chemistry:
(a) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed
- Remaining bromide 1a could be detected after 24 hours, hinting that the initial step occurs at a reduced rate.
- See ESI† for details.
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS PubMed
- For selected reviews and examples on such heterocyclic motifs in medicinal chemistry:
(a) Y. Wang, W.-J. Liu, L. Yin, H. Li, Z.-H. Chen, D.-X. Zhu, X.-Q. Song, Z.-Z. Cheng, P. Song, Z. Wang and Z.-G. Li, Bioorg. Med. Chem. Lett., 2018, 28, 974–978 CrossRef CAS PubMed
- Standard routes towards electrophilic fluorination, see: K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed
- For a review on fluorine utilization in MedChem: E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS
-
(a) A. Shani and R. Mechoulam, Tetrahedron, 1974, 30, 2437–2446 CrossRef CAS
- E. A. Peterson and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943–11948 CrossRef CAS PubMed
- For a review on tetrasubstituted olefins: A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698–4745 CrossRef CAS PubMed
- For selected reviews on sulfones:
(a)
K. A. Scott and J. T. Njardarson, in, Sulfur Chemistry, Topics in Current Chemistry Collections, ed. X. Jiang, Springer, Cham, 2018, pp. 1–34 Search PubMed
- The above-mentioned scaffolds rank 1st, 3rd, 5th, 9th, 17th and 19th, respectively. See: E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed
- Single example of facilitation of Pd-photoflow chemistry via Pd(0)-Pd(II): I. Abdiaj, L. Huck, J. M. Mateo, A. de la Hoz, M. V. Gomez, A. Díaz-Ortiz and J. Alcázar, Angew. Chem., Int. Ed., 2018, 57, 13231–13236 CrossRef CAS PubMed
- T. P. Yoon, V. M. Dong and D. W. C. MacMillan, J. Am. Chem. Soc., 1999, 121, 9726–9727 CrossRef CAS
- R. O. Hutchins and K. Learn, J. Org. Chem., 1982, 47, 4380–4382 CrossRef CAS
- B. Deadman, R. Ingham, D. L. Browne, R. Turner, I. R. Baxendale, S. V Ley, 2014, DOI:10.6084/m9.figshare.1170073.v3.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05551d |
| This journal is © The Royal Society of Chemistry 2021 |