DOI:
10.1039/D1NR06131C
(Minireview)
Nanoscale, 2022,
14, 4908-4921
Emerging low-molecular weight nucleopeptide-based hydrogels: state of the art, applications, challenges and perspectives
Received
17th September 2021
, Accepted 15th March 2022
First published on 16th March 2022
Abstract
Over the last twenty years, low-molecular weight gelators and, in particular, peptide-based hydrogels, have drawn great attention from scientists thanks to both their inherent advantages in terms of properties and their high modularity (e.g., number and nature of the amino acids). These supramolecular hydrogels originate from specific peptide self-assembly processes that can be driven, modulated and optimized via specific chemical modifications brought to the peptide sequence. Among them, the incorporation of nucleobases, another class of biomolecules well-known for their abilities to self-assemble, has recently appeared as a new promising and burgeoning approach to finely design supramolecular hydrogels. In this minireview, we would like to highlight the interest, high potential, applications and perspectives of these innovative and emerging low-molecular weight nucleopeptide-based hydrogels.
 Loic Stefan | Dr Loic Stefan received his Ph.D in chemistry from Université de Bourgogne (Dijon, France) in 2013, focusing on the nucleobase self-assemblies for the development of supramolecular tools with applications in biology and biotechnologies (group of Dr D. Monchaud). Then, he joined Politecnico di Milano (Milan, Italy) to work on peptide and protein self-assemblies, studying the impact of halogenation on their organization process and properties (group of Pr. P. Metrangolo). Since autumn 2016, he is a CNRS researcher working at the Laboratory of Macromolecular Physical Chemistry (Nancy, France). His current research interests are the developments of both innovative soft materials based on peptide and nucleobase derivatives, and metal-binding peptide derivatives exhibiting antioxidant properties. |
Introduction
Gradually becoming major in the realm of both soft matter and bioinspired materials, low-molecular weight peptide hydrogels have been highly studied for more than two decades and are still the focus of intense research interest.1,2 These supramolecular hydrogels hinge on the natural ability of protein structures (i.e., by extension, peptides and amino acids) to self-assemble thanks to specific non-covalent interactions (e.g., hydrogen bonds, van der Waals and electrostatic forces, π–π interactions). However, this self-organisation process relies on peculiar peptide sequences, and only few supramolecular assemblies are able to subsequently form self-supporting materials. Indeed, the development of such low-molecular weight hydrogels requires a tailored peptide design, as discussed in several authoritative and recent reviews.2–5 Depending on the intended applications, several properties can be expected, such as modularity, mechanical tunability, responsiveness to physical (e.g., temperature, light), mechanical (e.g., stress, pressure), chemical (e.g., pH, ionic strength) or biological (e.g., enzyme) stimuli, (bio)degradability or biomimicry.6–8 In the latter, key structural characteristics such as pore size, solvent diffusivity, density of the network, swelling and viscoelasticity of the hydrogels are key parameters to be controlled.9,10 In particular, the storage modulus (also termed elastic modulus) G′, corresponding to the solid-state behaviour of a soft material, is a pivotal property which has to be finely tuned: for instance, for tissue engineering, the storage modulus should match the stiffness of the natural extracellular matrix of the cell phenotype (from G′ < 1 kPa to >50 kPa for neural and bone cells, respectively), and can be harnessed to direct the stem cell differentiation.10–12 In this context, thanks to their exceptional inherent qualities of modularity, biocompatibility, biodegradability, atom economy and generally non-immunogenicity,9,13 low-molecular weight peptide-based hydrogels have demonstrated their potential and have opened new horizons in terms of applications, mainly in the biomedical and biotechnological domains7,8,14,15 (e.g., for regenerative medicine, drug delivery, bioprinting, or as media for cell culture for which commercial products16 are available) and in the nanotechnological field.17,18 Interestingly, while only a handful of native peptides exclusively comprised of proteinogenic amino acids have been reported, chemical modifications and/or functionalisations have been overwhelmingly brought to peptide sequences to improve the self-assembly and, subsequently, to improve the mechanical and physicochemical hydrogel properties.5,18,19 Thus, the incorporation of large aromatic moieties (e.g., Fmoc, naphthyl),20,21 aliphatic chains,2,22 non-natural amino acids,23 halogen atoms24,25 or pseudopeptide bonds26–28 are among the most popular approaches. Concomitantly, several research groups, including ours, have recently concentrated their efforts on the modification of the peptide moiety by the addition of another class of biomolecules, namely the DNA-nucleobases, which emerges as an efficient and highly promising approach.29 Thus, the design, properties, relevance, interests and applications of these new types of innovative low-molecular weight nucleopeptide-based hydrogels are discussed all along this minireview.
Nucleopeptide self-assemblies
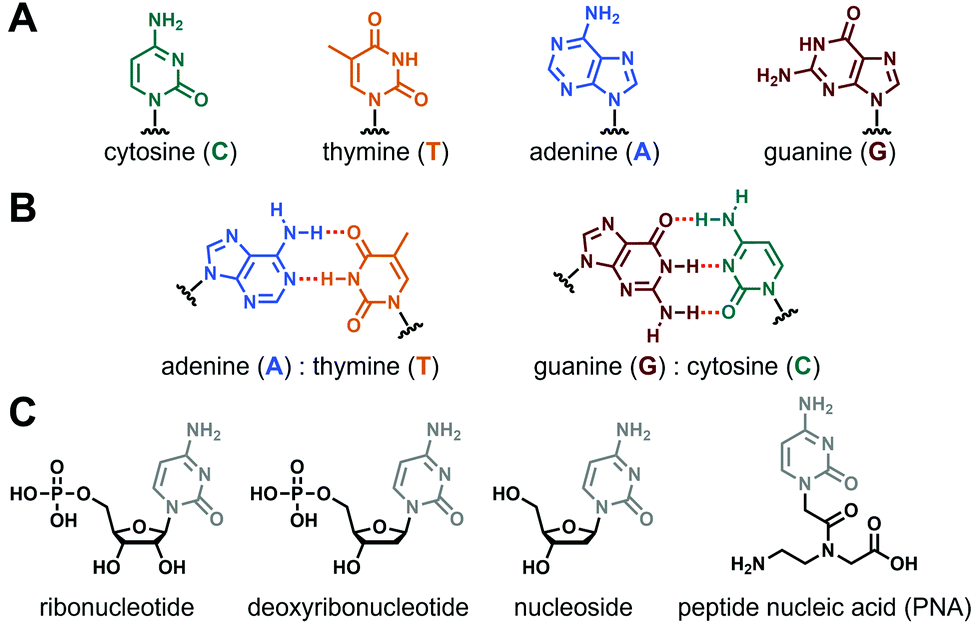
Nucleopeptides are natural or synthetic chemical compounds in which a peptide sequence is functionalised by one or several nucleobases or nucleobase-incorporating moieties (vide infra). Nucleobases are aromatic structures found in DNA, RNA and other biomolecules (e.g., ATP, coenzyme A) and which, such as peptide and proteins, play pivotal biological roles in living organisms. The main nucleobases (Fig. 1A) are the two purines guanine (G) and adenine (A), and the three pyrimidines cytosine (C), thymine (T, in DNA) and uracil (U, in RNA), even if other natural (e.g., inosine, hypoxanthine), fully synthetic and functionalized ones exist.30–32 Thanks to their inherent abilities to self-assemble mainly via hydrogen bonds and/or π-stacking interactions forming canonical Watson–Crick base pairs (Fig. 1B) or other structures (e.g., Hoogsteen or wobble base pairs, triplets, tetrads), they are ideal tools to construct a myriad of complex and responsive supramolecular systems,33–38 including hydrogels.39–42
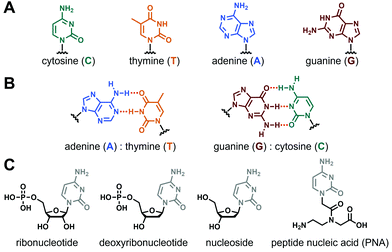 |
| | Fig. 1 (A) Chemical structures of nucleobases and (B) the two canonical Watson–Crick base pairing. (C) Chemical structures of nucleobase-containing moieties. | |
The incorporation of nucleobase(s) into peptide sequences can follow several strategies considering different derivatives, from the nucleobase alone to the biologically-relevant nucleosides or nucleotides (in their ribo- or deoxyribo- forms, see Fig. 1C), to the non-natural peptide nucleic acids (e.g., aegPNA, Fig. 1C) and other synthetic derivatives (LNA, etc.).43,44 Thus, thanks to the wealth of possibilities for the modulation of the two constitutive parts, i.e., the peptide (e.g., length, sequence, nature of the amino acids) and the nucleobase-containing (e.g., number, sequence, position in the peptide, nature of the base, type of derivative considered) moieties, nucleopeptides appear as a highly versatile class of molecules with almost virtually infinite possibilities in terms of design and molecular structures, offering to chemists a fascinating field to explore and to understand.29
While not found in Nature (except for the nucleoamino acid willardiine) and that their prebiotic role has been hypothesized,45–47 nucleopeptides are mainly synthetic compounds developed for biological applications47 thanks to their ability to interact with proteins or enzymes such as serum albumin48 or the reverse-transcriptase of HIV.49 Harnessing the abilities of peptides and nucleobases to self-assemble in specific conditions, several research groups reported on the use of nucleopeptides to study their self-organisation and to develop new properties and applications29 such as tuneable or functional supramolecular architectures,50–53 fluorescence,54–57 to selectively sequestrate ATP in cells58 or to deliver RNA into cells,59 to name a few. In some instances and depending on both the design and the mode of preparation (i.e., the formulation), nucleopeptides are able to form low-molecular weight gels, an emerging and promising approach to develop innovative soft materials.
Low-molecular weight nucleopeptide-based hydrogels
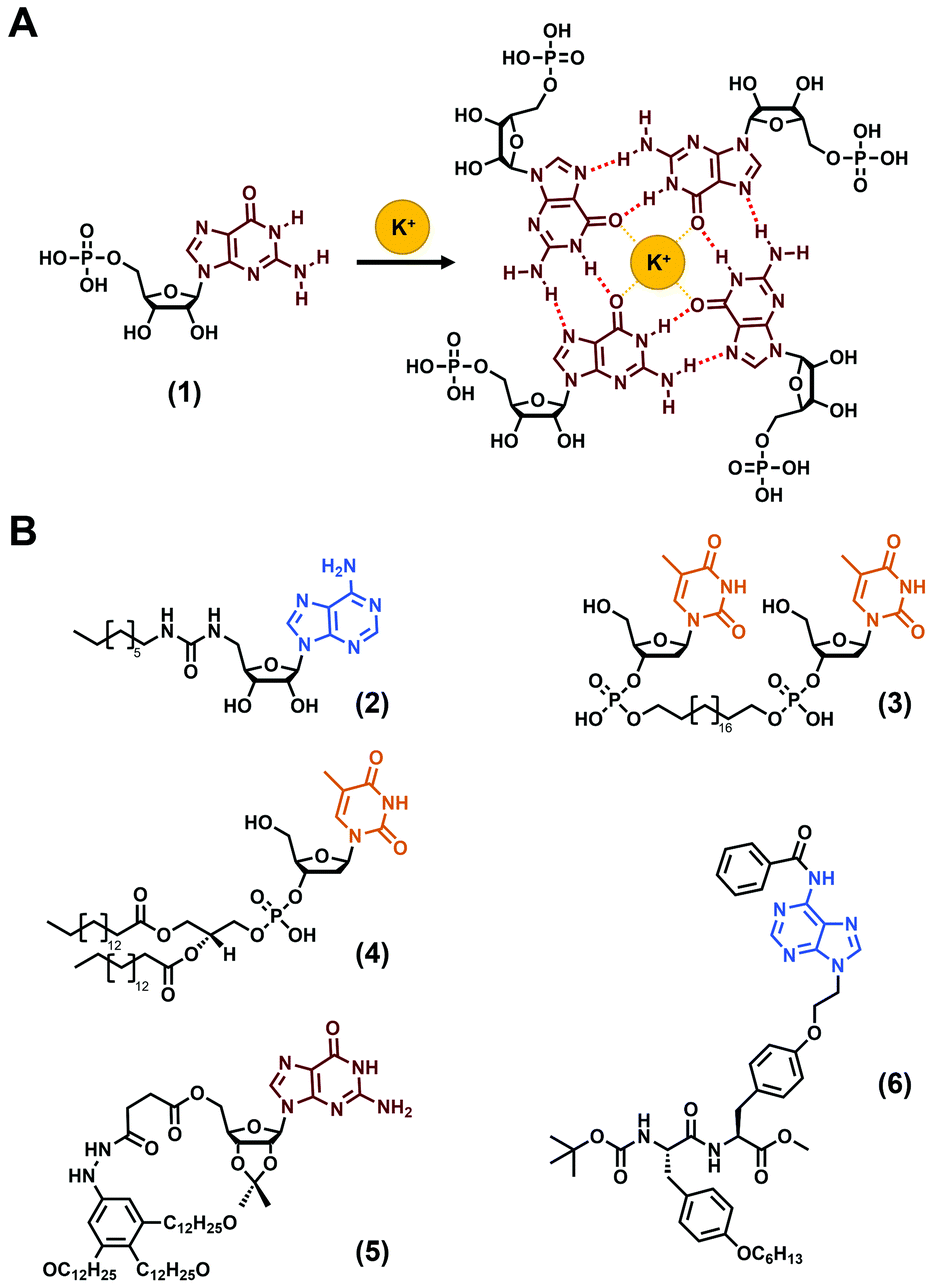
The use of nucleobases to develop hydrogels is a time-tested approach firstly reported in the first decades of the 1900s with the observation that concentrated solutions of guanylic acids (also termed guanosine-5′-monophosphate (1), Fig. 2A) are able to gelify.60–62 Understood later, this phenomenon relies on the fascinating propensity of guanines to self-organise into G-quartets (Fig. 2A), high ordered supramolecular structures that have been harnessed in the field of soft matter, including hydrogels, since then.68–70 While long DNA strands have also been reported to form hydrogels, they suffer, inter alia, from their high cost.39,40,71,72 Thus, low-molecular weight gelators incorporating nucleobases have been favoured and several molecular structures have been reported,19,39,41 such as derivatized nucleosides, sugar and lipids, or via the synthesis of bolaamphiphile compounds (see (2)–(4)Fig. 2B for some examples) or, more recently, via the functionalization of peptide sequences. Thus, over the last 20 years, the development of low-molecular weight nucleopeptide-based gels has been gradually growing, with a large majority of efforts focused on hydrogels, even if few organogels have been reported (e.g., (5), (6), Fig. 2B).67 The following subsections are dedicated to the discussion on the designs, properties and applications of the nucleopeptide-based hydrogels, based on the nature of the nucleobase moieties incorporated.
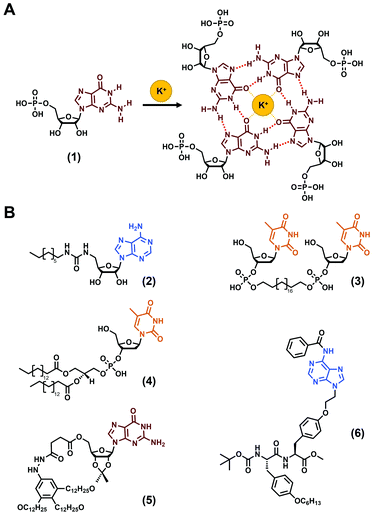 |
| | Fig. 2 (A) G-quartet formation from the guanosine-5′-monophosphate (1). (B) Chemical structures of nucleobase-containing compounds able to form hydrogels ((2),63(3)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 64 and (4)65) or organogels ((5)66 and (6)67). 64 and (4)65) or organogels ((5)66 and (6)67). | |
Nucleopeptides incorporating nucleotide derivatives
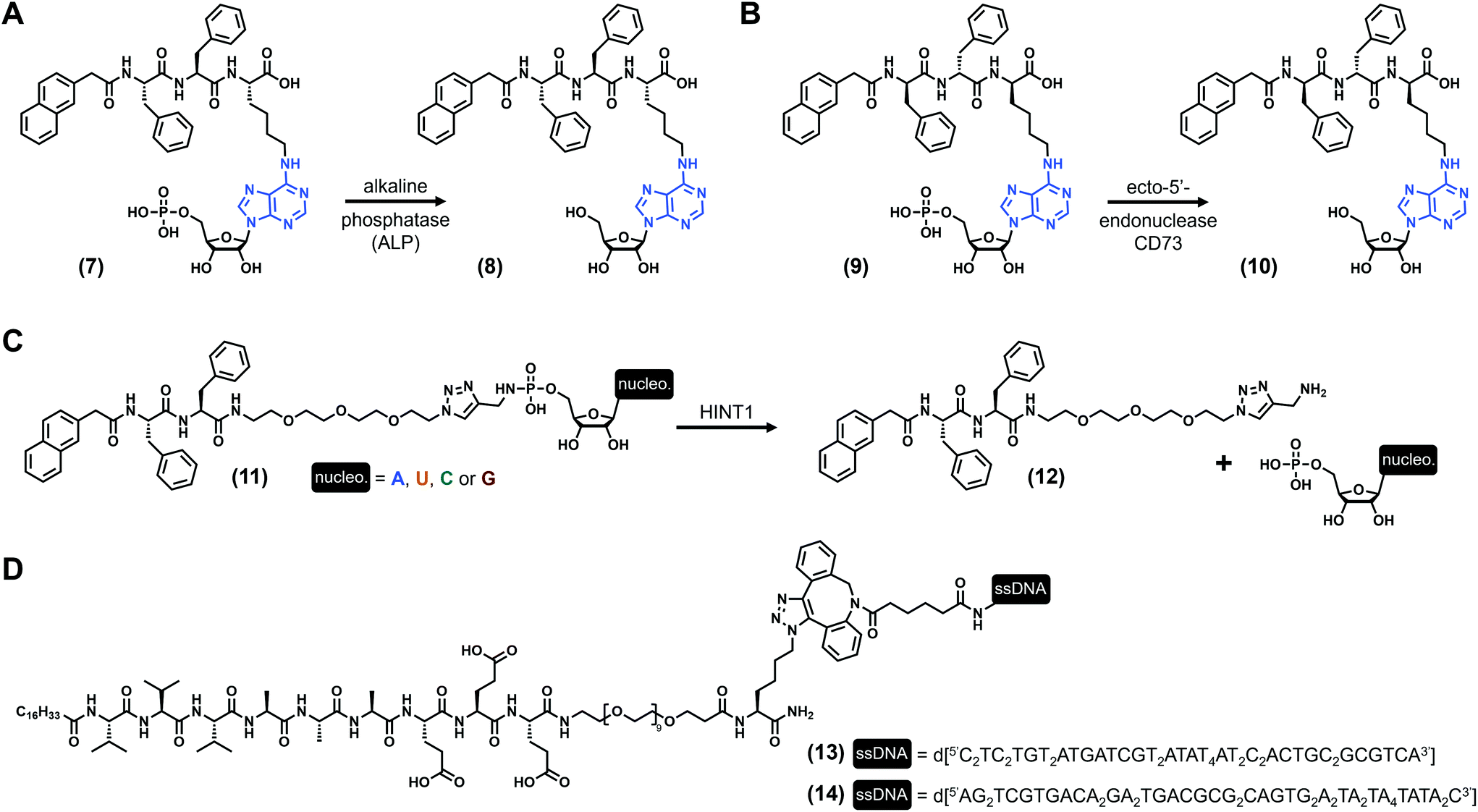
Nucleotides (Fig. 1C) are comprised of a (deoxy)ribose functionalized by both a nucleobase and a phosphate moiety (up to three phosphate groups) at its C1′ and C5′ positions, respectively. Monophosphate nucleotides are the key constituents of DNA and RNA, while monomeric nucleotides play pivotal biochemical roles (e.g., AMP, ATP). Using these types of derivatives, B. Xu and coworkers73 synthesized the tripeptide Phe–Phe–Lys, protected at its N-term by a naphthyl group, and on which the lysine has been functionalized at its N-ε by an adenosine monophosphate via its amine in position 6 ((7), Fig. 3A). Upon the addition of alkaline phosphatase ALP (10 units), a clear solution of (7) (3.5 wt%) turns into a transparent hydrogel with a storage modulus G′ ∼ 100 Pa (pH 7.4). This gelification process is triggered by the dephosphorylation of the starting compound (giving (8), Fig. 3A), allowing the self-assembly of the nucleopeptides which subsequently form long nanofibers with diameters of 11–17 nm. Other experiments using the dephosphorylated compound (8) in the presence of the 10-mer nucleic acid d[5′(T)3′10] show an increase of the elasticity of the hydrogels, suggesting additional cross-linking based on specific Watson–Crick interactions. Few years later, the same compound in which the L-tripeptide was replaced by the enantiomeric D equivalent ((9), Fig. 3B) was studied in the presence of another enzyme, the ecto-5′-nucleotidase CD73.74 Once again, the dephosphorylation leads to the formation of weak hydrogels after three days at a concentration of 2.0 wt% (pH 7.0), with G′ ∼ 25 Pa. Additionally, these compounds exhibit interesting cell biocompatibility, especially with the MCF-7 cells. Three years later, West et al. reported75 on a series of nucleopeptides comprised of a short Phe–Phe dipeptide functionalized at its N-term by a naphthyl group and, at its C-term, by a short polyethylene glycol linked to a nucleoside monophosphate via a phosphoramidate function. The four synthesized derivatives (with nucleobase = A, U, C, G, see (11), Fig. 3C), soluble in Dulbecco's phosphate-buffered saline (DPBS) (0.9 wt/vol), are mixed with the histidine triad nucleotide binding protein 1 HINT1, leading to the release of the nucleotide monophosphates from the pro-gelators and, subsequently, to the formation of hydrogels for all the compounds. Rheological investigations on the adenine and uracil derivatives show maximum storage moduli of ∼7 kPa and ∼6 kPa, respectively, obtained after less than 7 minutes for both. For these three examples, nucleopeptides are comprised of a nucleoside monophosphate, and the gelification occurs once the phosphate73,74 or the nucleoside75 is released from the molecule upon addition of a specific enzyme acting as a trigger of the gelification process, making these nucleopeptides pro-gelators instead of real gelators. Concomitantly, the Luijten and Stupp groups published the same year in Science76 reversible nucleopeptide-based self-assembling systems. Thus, two 9-mer peptides (Val3–Ala3–Glu3) were functionalized by an alkyl chain on their N-terminal and by complementary oligonucleotides (from 10 to 45 nucleotides) on their C-term (through a short PEG–lysine linker, (13) and (14), Fig. 3D. Note: due to their length, these molecules are not stricto sensu low-molecular weight compounds). When mixed together, the peptide parts self-assemble into fibres, cross-linked by double helix formation thanks to complementary base pairing. At the macroscopic level, the mixture forms stiff hydrogels with tuneable mechanical properties: the storage modulus (G′ ∼ 18 kPa) can be modulated via the introduction of an extra single-stranded DNA termed “invader”. Indeed, increasing the concentration of the invader strand is deleterious for the inter-fibre DNA duplex due to a strand-displacement mechanism, decreasing the size of the fibre bundles (as observed by electron microscopy) and subsequently weakening the mechanical properties of the hydrogel in a concentration-dependent manner (down to less than 1 kPa). To counteract this effect, the addition of anti-invader single-stranded DNA (designed to interact with the invader thanks to a complementary DNA sequence) allows cross-linking duplex to reform, leading to an increase of the hydrogel stiffness. Then, the authors have extrapolated these results to develop another series of peptide amphiphile sequences (without nucleobases) exhibiting extracellular matrix biomimetic features.
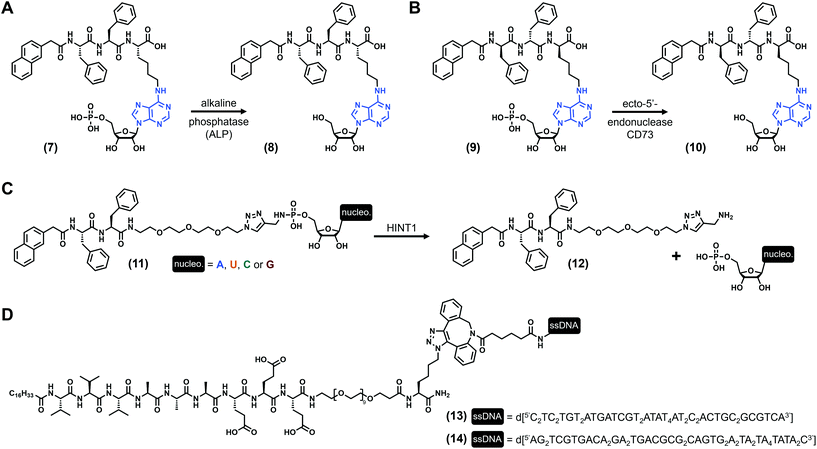 |
| | Fig. 3 Chemical structures of low-molecular weight supramolecular hydrogel-forming nucleopeptides, incorporating nucleotide derivatives. | |
Nucleopeptides incorporating nucleoside derivatives
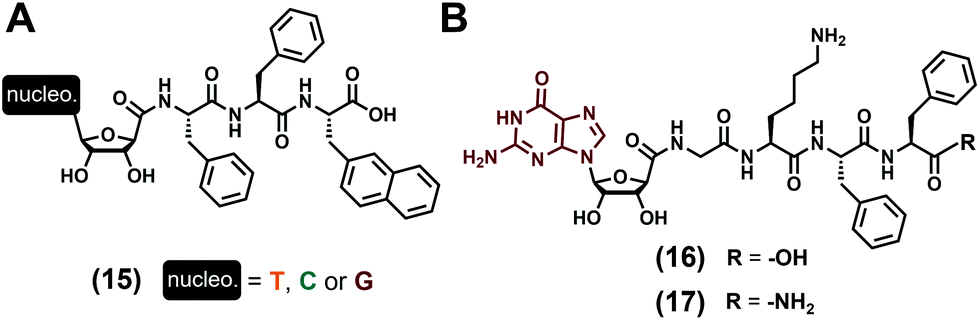
Lacking the phosphate groups, nucleosides (Fig. 1C) are only comprised of the nucleobase and the (deoxy)ribose and have been considered for the development of two distinct series of low-molecular weight hydrogelators. In the first instance, D. Wu et al. focused in 201477 on the tripeptide Phe–Phe–2Nal (2Nal stands for the 2-naphthyl-L-alanine) grafted on its N-term to the nucleoside derivative (with nucleobase = T, C, G, see (15), Fig. 4A) via the C5′ position of the ribose, thanks to an amide bond. All the three compounds have been formulated in water (pH 7.0) at 1 wt%, sonicated and heated up, and are able to form hydrogels with very significant differences in terms of mechanical properties. Indeed, while the presence of thymine leads to a relatively strong gel (G′ = 762 Pa) in which two sets of fibres (diameters of 5 and 28 nm) are observed in TEM, the two others incorporating cytosine and guanine are really weak, with storage moduli G′ of 5 Pa and 1 Pa, respectively, which can be explained by the presence of shorter fibres for the former, and of fibres and nanoparticles (diameters ∼39 nm) for the latter. The cell compatibility of such compounds has been evaluated in diluted conditions (without the formation of gel) and do not exhibit significant toxicity up to 500 μmol L−1. Only considering guanosine as the nucleoside derivative and substituting the previous tripeptide sequence by the 4-mer Gly–Lys–Phe–Phe ((16) and (17), Fig. 4B), the J.-E. Smith-Carpenter group78 has focused its work on the impact of the nature of the C-term function on the gelification process. Based on visual macroscopic investigations, they observed that the carboxylic acid derivative (16) is able to form a self-supporting hydrogel after 48 hours (0.2 wt%) in a 80/20 water/acetonitrile mixture (pH 4.5), while the amide derivative fails to gelify even after weeks. These differences can be explained by the morphology of the nanoobjects they form, with twisted nanofibers and nanosheets observed for the self-assembly of (16) and for the amide compound (17), respectively. In terms of supramolecular organisation, the authors proposed a main β-sheet formation for the peptide parts, and they speculate the role of the guanosine moiety which can assemble as G-quartets or G-ribbon structures for the carboxylic and amide derivatives, respectively. Previously considered as nucleotide derivatives, compounds (7)73 and (9)74 (Fig. 3A) can also be categorized as nucleoside-containing low-molecular weight hydrogelators because they require a dephosphorylation to trigger the gelification process, i.e., the hydrogel formation stems from the nucleoside derivative (i.e., (8) and (10), respectively) assembly abilities.
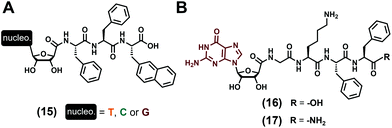 |
| | Fig. 4 Chemical structures of low-molecular weight supramolecular hydrogel-forming nucleopeptides, incorporating nucleoside derivatives. | |
Nucleopeptides incorporating peptide nucleic acids
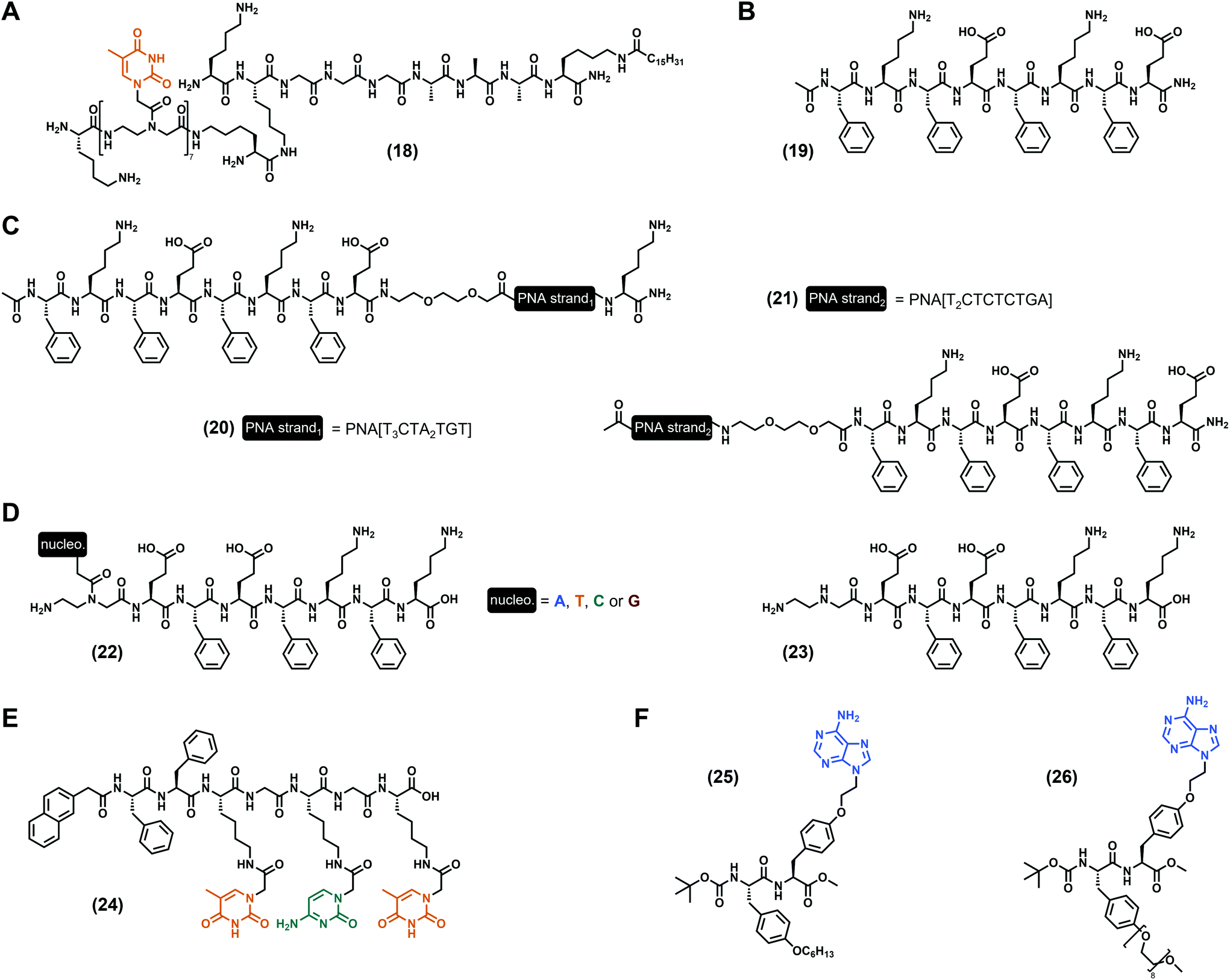
As discussed hereinbefore, the incorporation of nucleotide and nucleoside derivatives into peptide sequences is probably the more Nature-inspired approach since these moieties are found in biological media, including in the DNA structure for the former. However, peptide nucleic acids, also termed PNA (Fig. 1C), are synthetic DNA equivalents in which the phosphate–ribose moiety is substituted by a peptide backbone, with the N-(2-aminoethyl)glycine as the most commonly used,43 even if other PNA exist.44,79 PNA predominantly found applications in biological contexts thanks to their high stability, including as antigens, as progenes, as anti-microRNA, in antisense strategies to inhibit protein expression, or for gene editing.80 Moreover, PNA are viewed as a relevant and promising substitute for DNA in bio- and nano-technologies, or even in material science thanks to, inter alia, their higher chemical stability compared to DNA.81 Thus, PNA have been considered for the development of low-molecular weight nucleopeptide-based hydrogels with, for the best of our knowledge, the first example reported in 2005 by the Stupp group. In their work,82 they designed a PNA/peptide amphiphile conjugate ((18), Fig. 5A) bearing seven thymines in a row, grafted on the N-ε of the second lysine (from the N-term). Interestingly, at acidic pH (pH < 4) 100 nm-long nanofibers have been observed, confirming the ability of nucleopeptides to self-assemble and to form hydrogels. As used by Freeman et al. more than a decade later (with the invader strand, vide supra),76 the addition of the complementary single-stranded DNA containing seven adenines in a row modifies the self-assembly as observed by electron microscopy, even if the impact of the nucleobase hybridization on the gel properties is not discussed. Based on the octapeptide (Phe–Glu)2–(Phe–Lys)2 and derivatives alternating charged and aromatic amino acids firstly reported by the Saiani group,83,84 B. L. Nilsson and co-workers85 developed a complex multicomponent system using three peptides, namely the Ac–(Phe–Lys–Phe–Glu)2–NH2 ((19), Fig. 5B) and two other analogues on which two different 10-mer PNA sequences were added either at the N-term, or at the C-term ((20) and (21), Fig. 5C). While the mixture of these three compounds at a 5:1:1 ratio, respectively, leads to the hydrogel formation with a storage modulus of 148 Pa (at 4.2 mM total peptide concentration), the addition of an extra 27-mer single-stranded DNA (0.6 mM), designed to improve the fibril–fibril cross-linking, increases the mechanical properties with G′ = 245 Pa. Such a system confirms the relevance of multicomponent hydrogels86–88 and highlights the high potential of peptide fibril hybridization on the modulation of the material behaviour. Inspired by the same peptide sequences, our group reported in 2020 a series of four nucleopeptides based on the (Phe–Glu)2–(Phe–Lys)2 on which the N-term Phe was substituted by one PNA (nucleobase = A, T, C, G, see (22), Fig. 5D).89 Working under controlled conditions in the presence of the Tris.HCl (for tris(hydroxymethyl)aminomethane hydrochloride) buffer (pH 7.4) and at 15 mM of nucleopeptide, we first showed that the mechanical properties of the hydrogels can be finely tuned depending on the nature of the nucleobase. Indeed, while the presence of a pyrimidine (i.e., cytosine or thymine) leads to close storage moduli compared to the unmodified octapeptide (G′ ∼ 300 Pa), purines drastically improve the stiffness with an increase of circa 20-fold (G′ = 6.5 kPa) and 70-fold (G′ = 21.6 kPa) for adenine and guanine, respectively. Interestingly, all the nucleobases enhance the resistance to mechanical stress (up to 40-fold) and bring thermoreversible properties to the hydrogels. Then, we determined that these variations in terms of mechanical properties can be, inter alia, attributed to the formation of different nanoobjects which constrained molecular water (from the solvent) in different ways. Also, the additional experiments (e.g., FTIR, CD, NMR, ThT assays and fluorescence) highlight the ability of such nucleopeptides to self-assemble into β-sheet structures influenced by the nucleobases which play a pivotal role, mainly via intermolecular π-stacking interactions. Interestingly, the latter lead to the generation of uncommon red-edge excitation shift (REES) effects.90–92 Thus, more than the proof that incorporation of PNA can improve the mechanical properties of peptide hydrogels, we highlighted that a multiscale approach is a relevant strategy to study these innovative and complex systems, in order to further understand and decipher the influence of nucleobases on the supramolecular assembly processes. On the strength of these results, we reported one year later93 on the mixture of these different compounds and, in particular, on the mix of nucleopeptides bearing complementary nucleobases, i.e., adenine with thymine, guanine with cytosine. As reference systems, we studied each nucleobase-containing compound in the presence of the nucleobase-lacking equivalent ((23), Fig. 5D), corresponding to the heptapeptide with an aminoethyl glycine moiety at its N-term. Interestingly, the best mechanical properties have been obtained for the A + T and G + C-containing-nucleopeptides mixtures (G′ = 5.8 kPa and 6.8 kPa, respectively), which also have the highest resistances to stress (τy = 131 Pa and 59 Pa, respectively) and thermoreversibility properties. Thanks to a multiscale analysis, we highlighted that the stiffness of the hydrogels is linked to the compactness of the network (determined by SAXS) and to the ratio of constrained water molecules (determined by relaxometry). Preserving the REES properties, the A + T and the G + C-containing-nucleopeptides mixtures co-assemble via the formation of β-sheets, leading to fibrillar networks in which π-stacking interactions between nucleobases have been confirmed by fluorescence spectroscopy. Using other original PNA on which the nucleobase (cytosine or thymine) is grafted on the N-ε of a lysine residue, X. Du et al.94 synthesized a heptapeptide ((24), Fig. 5E), protected at its N-term by a naphthyl group. While formulated in phosphate-buffered saline (PBS, pH 7.4) the nucleopeptide alone results in a hydrogel with a storage modulus of G′ ∼ 500 Pa (at 1.0 wt%), the addition of 1.75 mM of a complementary single-stranded DNA d[5′(ACA)3′4] or d[5′(A2CA)4A3′] leads to stiffer gels with G′ = 1.1 kPa and 1.8 kPa, respectively. Interestingly, this improvement of the mechanical properties can originate from the formation of clusters of nanofibers in the presence of single-stranded DNA, thanks to hybridization with the nucleopeptides. A supplementation of plasmid DNA also increases the stiffness of the gel to G′ ∼ 1.5 kPa. Thanks to its ability to interact with DNA, the nucleopeptide, which is not cytotoxic against HeLa cells, is able to deliver labelled hairpin DNA into the cytosol after 1 hour of incubation. Functionalizing the hydroxyl group of a tyrosine by a N9-ethyl-adenine, Serpell et al. reported67 on the Boc N-protected dipeptide derivative ((25), Fig. 5F) which requires the presence of 5% DMSO to form a gel in aqueous solution. Mixing the latter in the presence of an equimolar quantity of its PEG analogue (26) leads to a stable hydrogel with thicker and less branched ribbons compared to (25) alone, which assembles into fibres. Studied in diluted conditions, this binary system also shows good biocompatibility, offering possibilities for future biological applications.
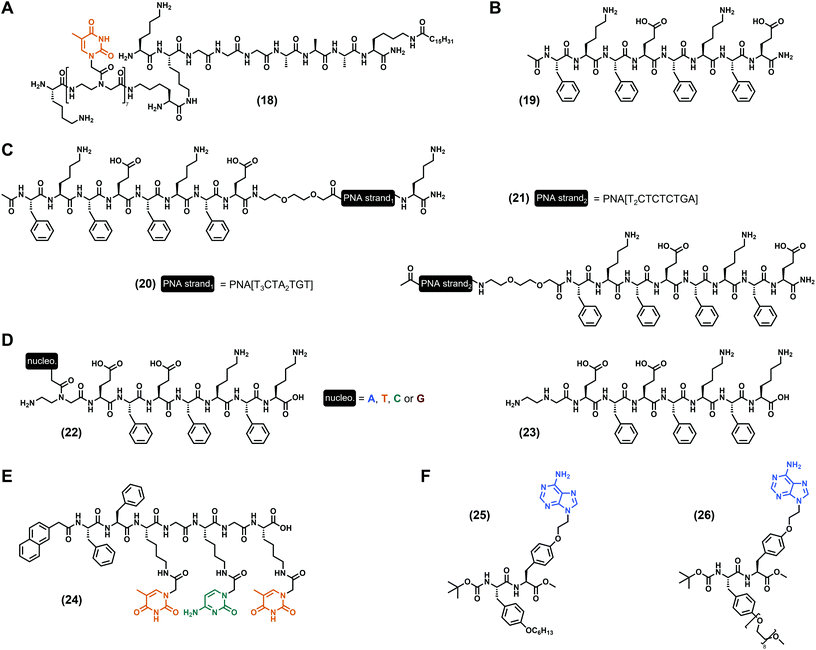 |
| | Fig. 5 Chemical structures of low-molecular weight supramolecular hydrogel-forming nucleopeptides, incorporating peptide–nucleic acid (PNA) derivatives. | |
Nucleopeptides incorporating nucleobases directly covalently linked
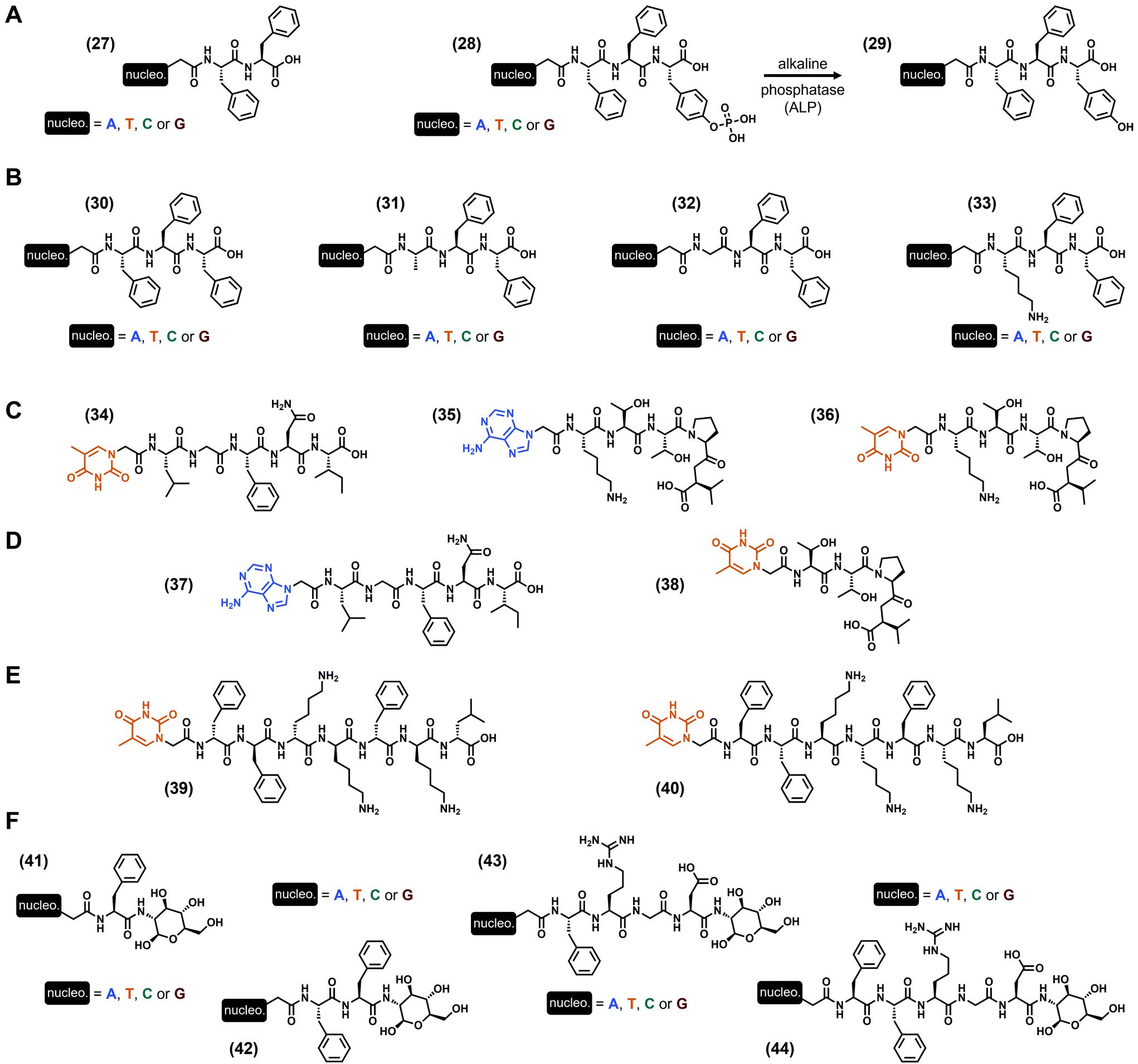
In all the previous examples, nucleobases were incorporated via a scaffold on which they are grafted, namely a phosphate-(deoxy)ribose (i.e., nucleotide), a deoxyribose (i.e., nucleoside) or a peptide derivative (i.e., peptide nucleic acids). A more minimalist approach discussed in this subsection is the direct incorporation of the nucleobase into the peptide sequence, via a functionalization at the N-term of the peptide sequence. Thus, the first example of this strategy to develop hydrogels has been reported by the B. Xu group in 201195 with the study of two different series of nucleopeptides. The shorter one is based on the dipeptide Phe–Phe96 with one nucleobase at the N-term (A, T, C or G, see (27), Fig. 6A), and all these compounds form hydrogels in slightly acidic conditions (pH 5.0) at 2 wt%, with higher storage moduli for the purine-containing nucleopeptides (G′ = 12.6 kPa and 8.1 kPa for the guanine and adenine derivatives, respectively). Interestingly, the mixture of the adenine and thymine derivatives leads to an increase of the modulus to G′ = 18.0 kPa, better than each compound alone, suggesting a cooperative synergistic effect between the complementary nucleobases. Then, a second series comprised of a phosphotyrosine at the C-term ((28), Fig. 6A) has been synthesized for which gelification occurs upon dephosphorylation by the alkaline phosphatase (i.e., the resulting tripeptide sequence is Phe–Phe–Tyr, see (29), Fig. 6A). At physiological pH (pH 7.4), hydrogelation is observed for three compounds (i.e., except for the cytosine derivative) with the adenine-containing nucleopeptide being the stiffest (G′ = 2.1 kPa). However, when the latter is mixed with the thymine derivative, the resulting storage modulus drops to 150 Pa. Additional experiments using extra oligomeric deoxyadenosine d[5′(A)3′10] in the presence of the thymine-incorporating nucleopeptides (27) or (28) exhibit an improvement of the mechanical properties, which can originate from specific interactions between complementary nucleobases. This early study on the ability of nucleopeptides to form hydrogels also confirms their potential use for biological applications, thanks to their good biocompatibility and stability, depending on the nature of the nucleobase. Also focusing on the use of phenylalanines to design the peptide part, L. J. Suggs and coworkers97 reported in 2019 on a series of four Phe–Phe–Phe tripeptides each one functionalized by one nucleobase (i.e., A, T, C or G, see (30), Fig. 6B). Using the pH switch (from basic to more acidic) to formulate the corresponding hydrogels in water, they highlighted better gelification properties for the thymine and adenine derivatives with G′ = 1.6 and 1.4 kPa (at 1 wt%) and critical gelification concentrations of 0.4 wt% and 0.45 wt%, respectively, at pH 7.5. Interestingly, when nucleopeptides containing complementary nucleobases were mixed, the resulting storage moduli increased, in contrast with the mixtures in the presence of non-complementary nucleobases. These observations, supported by molecular dynamics simulations, suggest the presence of Watson–Crick interactions, in addition to π–π stacking interactions. These latter, predominant for the adenine-incorporating (30) derivative which is also able to form hydrogels at physiological pH, have been harnessed to entrap the anticancer drug doxorubicin, an aromatic compound known for its ability to interact with DNA by intercalation.98 Firstly, the presence of the drug has an effect on the mechanical properties of the gel with an increase of its storage modulus from G′ ∼ 1 kPa (at 15 mM) to G′ ∼ 10 kPa in the presence of 1 mM doxorubicin. Thanks to the long release of the drug from the gel, in cellulo experiments show a significant and progressive death of the 4T1 mice breast cancer cells after 5 days, while the direct use of doxorubicin in solution kills all the cells in less than 3 days. These long release properties were confirmed by in vivo experiments in mice with lower tumor volume increase with the loaded gel compared to the drug in solution after 12 days. These experiments confirm once more the interest of low-molecular weight hydrogels to develop biocompatible soft materials. Still working with the Phe triad, they recently considered99 the thymine-incorporating (30)-based hydrogel as a scaffold for cell culture, with similar fibroblast cell metabolism (up to 4 days) compared to the commercially available PuraMatrix™ peptide hydrogel, but with more moderate proliferation rate. Moreover, their study highlights the impact of the formulation (e.g., type of buffer, presence of NaCl, CaCl2 or MgCl2) on, inter alia, the mechanical properties of the resulting hydrogels. Indeed, while gels are obtained at pH 6.8 in MES (for 2-(N-morpholino)ethanesulfonic acid) and HEPES (for N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)) buffers with G′ = 869 Pa and 232 Pa, respectively, the presence of PIPES (for piperazine-N,N′-bis(2-ethanesulfonic acid) or PBS buffer is deleterious. Not only considering the Phe amino acid in their design, the same group developed three other series of nucleopeptides97 on which the N-term amino acid Phe has been substituted by the uncharged alanine or glycine, or by the positively charged lysine (see (31)–(33), Fig. 6B, respectively). Depending on the nature of both the nucleobase and this N-term amino acid, a wide range of mechanical properties can be obtained with critical gelification properties spanning from 0.4 wt% to 1.43 wt%, at required pH from 4.0 to 7.5, and storage moduli from less than 10 Pa to more than 90 kPa. In particular, the two stiffest hydrogels are obtained with the Lys–Phe–Phe (33) derivatives functionalized with thymine and adenine, with G′ = 94.5 kPa and 25.5 kPa, respectively, at 1 wt%. Moreover, all the nucleopeptides show excellent biocompatibility in dilute conditions. Inspired by Nature and mainly by the ability of some different proteins to interact each other via specific fragments, D. Yuan et al. reported in 2015 on the use of two peptide sequences, namely the pentapeptides Leu–Gly–Phe–Asn–Ile from the PDZ domain and Lys–Thr–Thr–Pro–Val known to interact with.100 Functionalizing the former with a thymine and the latter with a complementary adenine or a thymine (as a control), three nucleopeptides were obtained ((34)–(36), Fig. 6C)). Interestingly, while all the compounds alone remain soluble after 48 hours (16.4 mM) in PBS (pH 6.2), the two mixtures in which the peptide sequences are different (i.e., (34) + (35) and (34) + (36)) form transparent hydrogels. However, the nature of the nucleobase drastically impacts the mechanical properties, with G′ = 8.9 kPa in the presence of the thymine (34) + adenine (35) derivatives, dropping around 10-fold lower with the two thymine (34) and (36) derivatives, with G′ = 0.9 kPa. In parallel, the mixture of compounds (35) + (36) for which the peptide sequences are identical fails to gelify. Thus, these experiments highlight the role of both the presence of the two heterodimeric peptide sequences and the complementary nucleobases on the resulting abilities to self-assemble and to form hydrogels. In these conditions, nanofibers were observed with more entanglements than for the mixture of the two thymine-containing nucleopeptides. Additionally, the two hydrogels show excellent stability (>98%) against proteinase K after 24 hours, and are cell-compatible as confirmed by the cell-viability assays on the HeLa and PC12 cells. The same year, the same group synthesized another series of nucleopeptides with analogue peptide sequences (and some compounds with an additional saccharide on the C-term).101 Among all the different gelification assays, only the mixture of (37) and (38) (Fig. 6D) bearing the Leu–Gly–Phe–Asn–Ile and the Thr–Thr–Pro–Val
sequences and the complementary thymine and adenine, respectively, is effective even if the rigidity of the resulting gel is low with G′ = 3.7 Pa. In 2018, working on the development of nucleopeptides able to selectively sequester ATP (instead of ADP) in cancer cells, the B. Xu group reported58 on two heptapeptides with the same peptide sequence Phe2–Lys2–Phe–Lys–Leu either in its L- or D-enantiomeric form, and functionalized at their N-term with a thymine. Thus, both compounds (39) and (40) (Fig. 6E) exhibit selective hydrogelation upon the addition of one equivalent of ATP in PBS and human serum, while they remain liquid alone or precipitate with ADP. In terms of stiffness, the equimolar (39) or (40) (0.4 wt% in PBS) and ATP mixtures lead to hydrogels with G′ ∼ 15 kPa and 20 kPa, respectively. These examples confirm the interests of nucleopeptides for applications in biological contexts, and show that specific designs can lead to complex hydrogels with unusual triggers, like ATP. To end this subsection, we now discuss nucleopeptides on which an additional saccharide has been added on the C-term. Previously mentioned but failing to form soft materials (vide supra), these types of compounds have been first developed by X. Li et al. in 2011 as hydrogelators.102 The first series, comprised of the single amino acid Phe twice functionalized by a nucleobase (A, T, C or G) and a glycosamide at its N-term and C-term, respectively ((41), Fig. 6F), can be formulated as hydrogels using the pH switch method to reach final pH between 4.0 (G derivative) and 7.0 (T and C derivatives), and storage moduli from G′ = 139 kPa (G derivative) to 6 kPa (A derivative) at 3 wt%. Interestingly, the thymine derivative has been used in addition with the oligomeric deoxyadenosine d[5′(A)3′10] (which increases its stiffness) to deliver the latter into the cytosol and nuclei of HeLa cells. The presence of a supplementary phenylalanine (compounds (42), Fig. 6F) leads to a modification of these gelification abilities, with a pH range spanning from 4.0 to 8.5 (T derivative) and the stiffest hydrogel obtained for the cytosine derivative (G′ = 220 kPa at 3 wt%). Such compounds also exhibit a good stability against proteinase K, thanks to the presence of the glycoside moiety. Then, other glyco-nucleopeptide analogues incorporating the well-known Arg–Gly–Asp (i.e., RGD) tripeptide have been reported (compounds (43) and (44), Fig. 6F).103 In the presence of one Phe in the sequence (nucleopeptides (43)), hydrogels have been obtained for all the nucleobases, and with highest stiffness for the guanine derivative (G′ = 35 kPa, 3 wt%, pH 4.0); in contrast, when two Phe are present in the sequence (nucleopeptides (44)), only the adenine and cytosine derivatives are able to form a gel at pH 4.0, with storage moduli of 2.4 to 6 kPa, respectively, at 3 wt%. As the previous series, the stability of these compounds against proteolysis has been improved thanks to the presence of the glycoside moiety at the C-term. Finally, the ability of adenine–Phe–Arg–Gly–Asp–glycosamide to self-assemble has been harnessed for promoting both the proliferation of murine embryonic cells (mES) without compromising their pluripotency, and the development of zygotes of mice into blastocysts.104
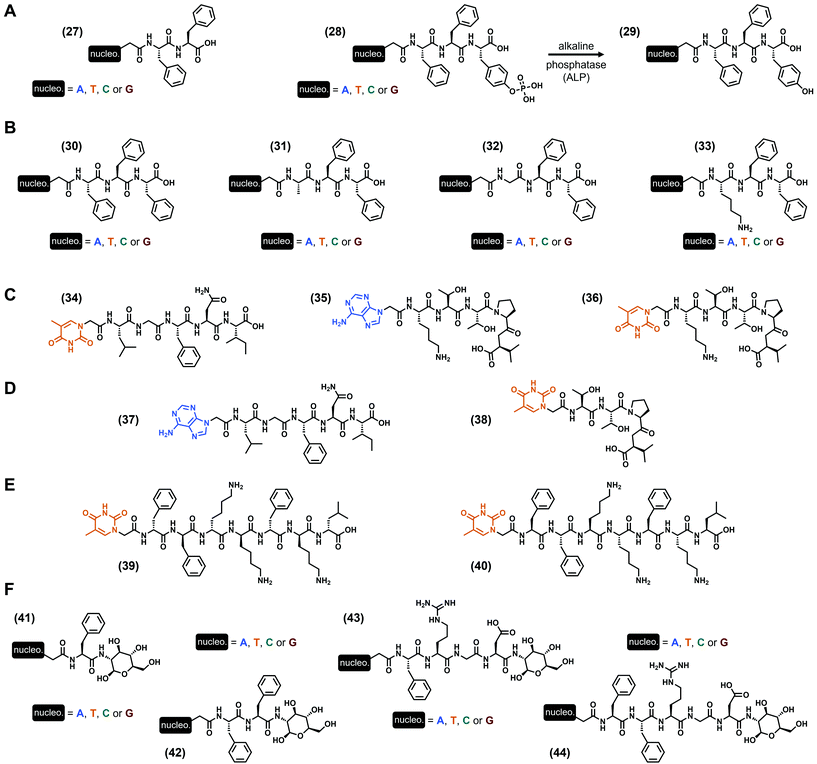 |
| | Fig. 6 Chemical structures of low-molecular weight supramolecular hydrogel-forming nucleopeptides, incorporating nucleobases directly covalently linked. | |
Summary, conclusions and perspectives of low-molecular weight nucleopeptide-based hydrogels
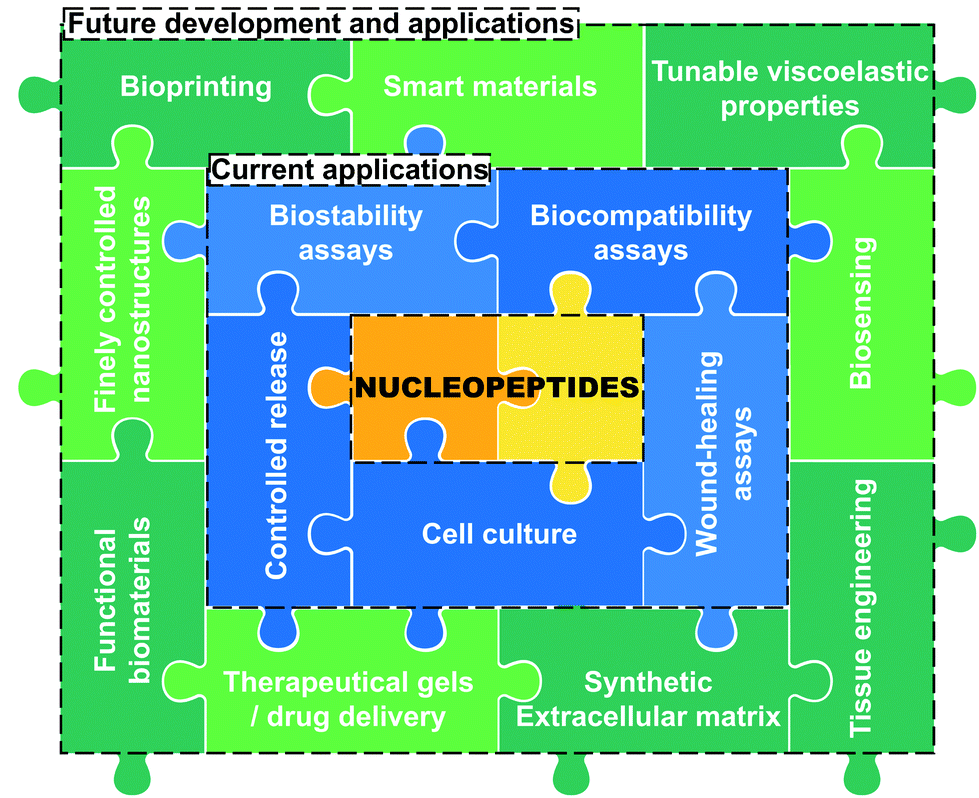
As discussed all along this minireview, the incorporation of nucleobase(s) into peptide sequences through different approaches in terms of structure and design leads to nucleopeptides exhibiting a panel of physicochemical and mechanical properties we summarized in Tables 1 and 2. These data highlight the efficiency of low-molecular weight nucleopeptide-based hydrogels in different experimental conditions, including the formulation, the pH or the nature of the buffer if used, offering several possibilities in terms of applications, from cell culture to long release of anticancer drugs. Nucleopeptides have already been reported for various other applications (vide supra)29,47,105,106 thanks to their inherent properties derived from their two main constituents, i.e., the peptide and the nucleobase-moiety parts, which can be harnessed for the development of soft materials. While the pros and cons of the former have been discussed hereinbefore (see introduction),8,14,15,17,18 the latter exhibit different advantages and drawbacks depending on the nature of the derivative. Indeed, the choice of nucleotide(s) will improve the solubility of the nucleopeptides because of the presence of negatively charged phosphate groups, whereas the incorporation of nucleobases directly grafted, nucleosides and especially PNA, which are exempt of charge, will have the opposite effect with a higher propensity to aggregation and/or precipitation.29,43,44 All these derivatives have a good biocompatibility,40,107–109 as confirmed by several examples (e.g., (15),77(25),67(30),97–99(41)102), though with better chemical (i.e., against hydrolysis and depurination) and biological (i.e., against nucleases) stability for nucleobase(s) directly grafted and PNA.43,44,80,81 Also, nucleotides and PNA offer the possibility for incorporating several nucleobases in a row (i.e., strands), even if such an approach presents challenges in terms of chemical synthesis.29,106,110 Concerning the impact of the type of derivative on the viscoelastic properties, it appears that the incorporation of nucleosides leads to relatively weak hydrogels (up to <0.8 kPa) compared to the other nucleopeptides which exhibit higher storage moduli up to hundreds of kPa (see Tables 1 and 2). For all these soft materials, the resulting properties drastically depend on the type of the nucleobase derivative incorporated and, also, on the nature of the nucleobase (i.e., A, T, C or G), and it is still difficult and premature to discern a clear trend from the current data. For these reasons, further research on the role(s) of nucleobases in the self-assembly and hydrogel formation processes of nucleopeptides are required even if challenging. Indeed, the precise role(s) and impact(s) of the nucleobases on the supramolecular assembly process and on the hydrogel formation is complex to understand because nucleobases affect the supramolecular organisation via hydrophobic interactions, but also via more specific ones such as π-stacking89,93,97 and hydrogen bonds,85,95,97 including Watson–Crick pairings. However, for the latter, no formal proof (via, e.g., X-ray diffraction or NMR spectroscopy111) has been provided, even if indirect evidences support their presence.76,85,95,97,100,102 Such differences at the supramolecular scale feed through to the nano- and microscopic scales, with the formation of different objects, depending on the nature of the nucleobase, as observed inter alia by electron microscopy. Interestingly, this difficulty to understand these nucleopeptide-based systems and the precise impact of each constituent at all scales are similar to what is reported for peptide-based hydrogels. For these reasons, we consider that to further comprehend them, a multiscale analysis is a relevant approach trying to decipher the role of nucleobases on the self-assembly process. Interestingly, this minireview highlights the different strategies scientists have used to develop nucleopeptide-based hydrogels, with diversity in terms of both the peptide sequence (even if phenylalanine, one of the most used amino acids to develop low-molecular weight peptide-based hydrogels thanks to, inter alia, its π-stacking ability,5,96,112 is often present) and the nucleobase-incorporating moieties (e.g., nucleotide, nucleoside, PNA, direct functionalization) grafted. However, due to the burgeoning aspect of this research topic, this diversity complicates the rationalisation of the impact of nucleobases on the structural, physicochemical and mechanical properties of the assemblies, with studies at different concentrations, pH, conditions, method of formulation, etc. Moreover, only few works89,93 report on the comparison of such nucleopeptides with the starting peptide sequence or a derivative, which can help to decipher both the roles and benefits of the nucleobase(s) compared to a reference compound. While nucleobases are perfect systems to design supramolecular assemblies based on their inherent abilities to interact each other,29,33,35,37,40 the formulation of mono-component hydrogels has been more often considered than the mixture of nucleopeptides to form multicomponent hydrogels, which has recently appeared as a challenging but promising and relevant strategy to generate sophisticated next-generation materials.86–88,113 Altogether, the data discussed herein highlight the high potential of this new class of compounds able to form hydrogels in precise conditions and for which the resulting properties are sensitive to and dependent on the nature of the incorporated nucleobase(s). Thus, it is safe to bet that the interest for these systems will increase in the next years, with the development of innovative low-molecular weight nucleopeptide-based hydrogels which will open the way to new applications (Fig. 7), thanks to a precise control of the properties via the presence of the nucleobase(s).
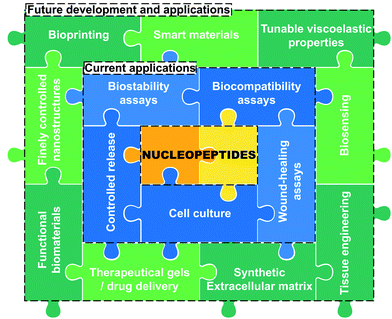 |
| | Fig. 7 Overview of the current (in blue) and future (in green) development and applications of nucleopeptide-based hydrogels. | |
Table 1 Selected key properties of reported low-molecular weight nucleopeptide-based hydrogels
| Compound |
Nucleobase moiety |
Experimental conditions |
Gelification conditions |
Gelification time |
CCG |
Mechanical properties |
Other characterizations |
Biological applications |
Ref. |
|
n/s = not specified, n/a = not applicable. Diluted conditions. CCG for critical concentration of gelation, Tris.HCl = tris(hydroxymethyl)aminomethane hydrochloride, PBS = phosphate-buffered saline, DPBS = Dulbecco's PBS. |
|
(7)/(8) |
Nucleotide |
Buffer (n/s), pH 7.4 |
Enzyme (ALP) addition |
n/s |
0.8 wt% |
G′ = 100 Pa (3.5 wt%) |
TEM |
n/a |
73
|
|
(9)/(10) |
Nucleotide |
Water, pH 7.0 |
Enzyme (CD73) addition |
∼2 days |
n/s |
n/s
|
TEM |
Biostabilitya (serum), biocompatibility assaysa |
74
|
|
(11)/(12) |
Nucleotide |
DPBS, pH 7.3 |
Enzyme (HINT1) addition, mix, vortex |
<5 min |
n/s |
G′ = 6 kPa (U) to 7 kPa (A) (0.9 wt% + 6 μM enzyme). |
TEM, cryo-TEM |
n/a |
75
|
|
(13) + (14) |
Nucleotide |
Water (with NaCl, KCl, CaCl2) |
Mix |
n/s |
n/s |
G′ ∼ 18 kPa (adjustable upon addition of ssDNA) |
Coarse-grained molecular dynamics, SAXS, TEM, cryo-TEM, SEM, AFM |
Cell culture |
76
|
|
(15)
|
Nucleoside |
Water, pH 7.0 |
Sonication, heating |
n/s |
n/s |
G′ = 1 Pa (G) to 762 Pa (T) (1 wt%) |
CD, TEM |
Biocompatibility assaysa |
77
|
|
(16)
|
Nucleoside |
Water/acetonitrile (80/20), pH 4.5, ±KCl |
Vortex, sonication |
∼2 days |
<0.2 wt% |
n/s |
FTIR, PXRD, TEM |
n/a |
78
|
|
(18)
|
aegPNA |
Water, pH < 7 |
pH switch |
n/s |
<1.0 wt% |
n/s |
CD |
n/a |
82
|
|
(19) + (20) + (21) |
aegPNA |
Water |
Vortex |
n/s |
n/s |
G′ = 148 Pa (4.2 mM peptide) to 245 Pa (+ssDNA). |
CD, TEM |
n/a |
85
|
|
(22)
|
aegPNA |
Tris.HCl, pH 7.4 |
Heating |
From <5 min to <6 hours |
n/s |
G′ = 201 Pa (C) to 21.6 kPa (G) (15 mM) |
NMR, FTIR, CD, fluorescence, ThT assays, TEM, NMR relaxometry |
n/a |
89
|
|
(22) + (23) |
aegPNA |
Tris.HCl, pH 7.4 |
Heating |
From 2 min to >4 days |
n/s |
G′ = 28 Pa (C + (23)) to 6.8 kPa (C + G) (7.5 mM + 7.5 mM) |
FTIR, CD, fluorescence, ThT assays, SAXS, TEM, NMR relaxometry |
n/a |
93
|
|
(24)
|
Other PNA |
PBS, pH 7.4 |
Mix |
n/s |
0.07 wt% |
G′ ∼ 500 Pa (1 wt%) to 1800 Pa (+ssDNA) |
CD, TEM, DLS |
Biostabilitya (proteinase K), biocompatibility assaysa, hairpin DNA deliverya |
94
|
|
(25)
|
Other PNA |
Water/DMSO (95/5) |
Mix, heating |
n/s |
0.5 wt% |
n/s |
CD, TEM, AFM |
n/a |
67
|
|
(25) + (26) |
Other PNA |
Water |
Mix, heating |
n/s |
0.5 wt% |
n/s |
CD, TEM, cryo-TEM, AFM |
Biocompatibility assaysa |
67
|
Table 2 Selected key properties of reported low-molecular weight nucleopeptide-based hydrogels
| Compound |
Nucleobase moiety |
Experimental conditions |
Gelification conditions |
Gelification time |
CCG |
Mechanical properties |
Other characterizations |
Biological applications |
Ref. |
|
n/s = not specified, n/a = not applicable. Diluted conditions. CCG for critical concentration of gelation, Tris.HCl = tris(hydroxymethyl)aminomethane hydrochloride, PBS = phosphate-buffered saline, DPBS = Dulbecco's PBS. |
|
(27)
|
Directly grafted |
Water, pH 5.0 |
Mix |
n/s |
n/s |
G′ = 26 Pa (C) to 12.6 kPa (G) (2 wt%) |
MM calculation, CD, TEM |
Biocompatibility assaysa |
95
|
|
(28)/(29) |
Directly grafted |
Water, pH 7.4 |
Enzyme (ALP) addition |
n/s |
n/s |
G′ = from 2.9 Pa (T) to 2.1 kPa (A) (2 wt%) |
CD, TEM |
Biostabilitya (proteinase K), biocompatibility assaysa, in vitro wound-healing assay |
95
|
|
(30)
|
Directly grafted |
Water, pH 6.5 to 7.5, different buffers for (T) |
pH switch |
<1 min |
From 0.4 wt% (T) to 1.0 wt% (C) |
G′ = 154 Pa (G, 1.5 wt%) to 1.6 kPa (T, 1.0 wt%). |
MD simulation, CD, TEM, |
Biocompatibility assaysa, for (A): release of doxorubicin (in vitro, in vivo). |
97–99
|
|
(31)
|
Directly grafted |
Water, pH 4.0 to 6.0 |
pH switch |
<1 min |
From 0.52 wt% (T) to 1.34 wt% (C) |
G′ = 5.6 Pa (G, 1.5 wt%) to 1.9 kPa (T, 1.0 wt%) |
TEM |
n/a |
97
|
|
(32)
|
Directly grafted |
Water, pH 4.0 to 5.5 |
pH switch |
<3 min |
From 0.5 wt% (T) to 1.43 wt% (G) |
G′ = 189 Pa (G, 1.5 wt%) to 1.5 kPa (A, 1.0 wt%) |
TEM |
n/a |
97
|
|
(33)
|
Directly grafted |
Water, pH 6.5 to 7.5 |
pH switch |
From <5 min to <10 min |
From 0.5 wt% (T, A) to 1.25 wt% (C) |
G′ = 1.4 kPa (C, 1.5 wt%) to 94.5 kPa (T, 1.0 wt%) |
TEM |
n/a |
97
|
|
(34) + (35) |
Directly grafted |
PBS, pH 6.2 |
Mix |
∼2 days |
n/s |
G′ = 8.9 kPa (8.2 mM + 8.2 mM) |
FTIR, CD, TEM |
Biostabilitya (proteinase K), biocompatibility assaysa |
100
|
|
(37) + (38) |
Directly grafted |
PBS, pH 6.2 |
Mix |
Overnight |
n/s |
G′ = 3.7 Pa (8.3 mM + 8.3 mM) |
TEM |
Biostabilitya (proteinase K), biocompatibility assaysa |
101
|
|
(39)/(40) |
Directly grafted |
PBS, pH 7.4 |
ATP addition (1eq.), mix |
n/s |
n/s |
G′ = ∼15 kPa ((39)) to 20 kPa ((40)) |
n/a |
Biocompatibility assaysa |
58
|
|
(41)
|
Directly grafted |
Water, pH 4.0 to 7.0 |
pH switch |
n/s |
n/s |
G′ = 6 kPa (A) to 139 kPa (G) (3 wt%) |
CD, TEM |
Biostabilitya (proteinase K), biocompatibility assaysa |
102
|
|
(42)
|
Directly grafted |
Water, pH 4.0 to 8.5 |
pH switch |
n/s |
n/s |
G′ = 32 kPa (T) to 220 kPa (C) (3 wt%) |
CD, TEM |
Biostabilitya (proteinase K), biocompatibility assaysa |
102
|
|
(43)
|
Directly grafted |
Water, pH 4.0 to 7.4 |
pH switch |
n/s |
n/s |
G′ = 2.1 kPa (G) to 35 kPa (T) (3 wt%) |
TEM |
Biostabilitya (proteinase K) |
102 and 103 |
|
(44)
|
Directly grafted |
Water, pH 4.0 to 7.4 |
pH switch |
n/s |
n/s |
G′ = 2.4 kPa (C) to 6 kPa (A) (3 wt%) |
TEM |
Biostabilitya (proteinase K) |
103
|
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
L. S. and M.-C. A.-P acknowledge the Centre National de la Recherche Scientifique (CNRS) for funding. L. S. and P. H. thank the Agence Nationale de la Recherche (ANR-20-CE06-0010-01 MUNCH) for funding. T. G. thanks the Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation (MESRI) for his Ph.D. grant.
Notes and references
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC
 .
.
- L. M. De Leon Rodriguez, Y. Hemar, J. Cornish and M. A. Brimble, Chem. Soc. Rev., 2016, 45, 4797–4824 RSC .
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS .
- S. Mondal, S. Das and A. K. Nandi, Soft Matter, 2020, 16, 1404–1454 RSC .
- R. Das, B. Gayakvad, S. D. Shinde, J. Rani, A. Jain and B. Sahu, ACS Appl. Bio Mater., 2020, 3, 5474–5499 CrossRef CAS .
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef PubMed .
- X. Q. Dou and C. L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed .
- A. K. Das and P. K. Gavel, Soft Matter, 2020, 16, 10065–10095 RSC .
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed .
- X. Ding, H. Zhao, Y. Li, A. L. Lee, Z. Li, M. Fu, C. Li, Y. Y. Yang and P. Yuan, Adv. Drug Delivery Rev., 2020, 160, 78–104 CrossRef CAS PubMed .
- K. Liang, K. H. Bae and M. Kurisawa, J. Mater. Chem. B, 2019, 7, 3775–3791 RSC .
- Z. Yang, H. Xu and X. Zhao, Adv. Sci., 2020, 7, 1903718 CrossRef CAS PubMed .
- W. Y. Seow and C. A. E. Hauser, Mater. Today, 2014, 17, 381–388 CrossRef CAS .
- J. Li, R. Xing, S. Bai and X. Yan, Soft Matter, 2019, 15, 1704–1715 RSC .
- K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539–7551 RSC .
- Among the commercially-available peptide-based hydrogels, we can mention Hydromatix™ (Sigma-Aldrich, Saint-Louis, USA), PuraMatrix™ (Corning®, Corning, USA), PeptiGels® (Manchester Biogel, Manchester, England), PGmatrix™ (PepGel, Manhattan, USA), Biogelx™ (Biogelx Ltd, Motherwell, Scotland).
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC .
-
L. Stefan, in Amino Acid - New Insights and Roles in Plant and Animal, ed. T. Asao and M. Asaduzzaman, IntechOpen, 2017, pp. 31–73 Search PubMed .
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed .
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC .
- C. Diaferia, G. Morelli and A. Accardo, J. Mater. Chem. B, 2019, 7, 5142–5155 RSC .
- H. Chang, C. Li, R. Huang, R. Su, W. Qi and Z. He, J. Mater. Chem. B, 2019, 7, 2899–2910 RSC .
- H. Arakawa, K. Takeda, S. L. Higashi, A. Shibata, Y. Kitamura and M. Ikeda, Polym. J., 2020, 52, 923–930 CrossRef CAS .
- A. Pizzi, C. Pigliacelli, A. Gori, Nonappa, O. Ikkala, N. Demitri, G. Terraneo, V. Castelletto, I. W. Hamley, F. Baldelli Bombelli and P. Metrangolo, Nanoscale, 2017, 9, 9805–9810 RSC .
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 7574 CrossRef PubMed .
- M. M. Nguyen, K. M. Eckes and L. J. Suggs, Soft Matter, 2014, 10, 2693–2702 RSC .
- A. Rajbhandary and B. L. Nilsson, Pept. Sci., 2017, 108, e22994 Search PubMed .
- M. I. A. Ibrahim, G. Pickaert, L. Stefan, B. Jamart-Grégoire, J. Bodiguel and M. C. Averlant-Petit, RSC Adv., 2020, 10, 43859–43869 RSC .
- T. Macculloch, A. Buchberger and N. Stephanopoulos, Org. Biomol. Chem., 2019, 17, 1668–1682 RSC .
- D. H. Appella, Curr. Opin. Chem. Biol., 2009, 13, 687–696 CrossRef CAS PubMed .
- A. Marx and K. Betz, Chem. – Eur. J., 2020, 26, 3446–3463 CrossRef CAS PubMed .
- K. Duffy, S. Arangundy-Franklin and P. Holliger, BMC Biol., 2020, 18, 112 CrossRef PubMed .
- A. del Prado, D. González-Rodríguez and Y. L. Wu, ChemistryOpen, 2020, 9, 409–430 CrossRef CAS PubMed .
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC .
- F. Pu, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 1285–1306 RSC .
- Y. Zhao, X. Dai, F. Wang, X. Zhang, C. Fan and X. Liu, Nano Today, 2019, 26, 123–148 CrossRef CAS .
- M. Surin, Polym. Chem., 2016, 7, 4137–4150 RSC .
- P. Barthélemy, C. R. Chim., 2009, 12, 171–179 CrossRef .
- J. Gačanin, C. V. Synatschke and T. Weil, Adv. Funct. Mater., 2020, 30, 1906253 CrossRef .
- Y. Shao, H. Jia, T. Cao and D. Liu, Acc. Chem. Res., 2017, 50, 659–668 CrossRef CAS PubMed .
- G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 RSC .
- V. Morya, S. Walia, B. B. Mandal, C. Ghoroi and D. Bhatia, ACS Biomater. Sci. Eng., 2020, 6, 6021–6035 CrossRef CAS PubMed .
- E. Uhlmann, A. Peyman, G. Breipohl and D. W. Will, Angew. Chem., Int. Ed., 1998, 37, 2796–2823 CrossRef CAS PubMed .
- C. Sharma and S. K. Awasthi, Chem. Biol. Drug Des., 2017, 89, 16–37 CrossRef CAS PubMed .
- B. M. A. G. Piette and J. G. Heddle, Trends Ecol. Evol., 2020, 35, 397–406 CrossRef PubMed .
- A. K. Bandela, N. Wagner, H. Sadihov, S. Morales-Reina, A. Chotera-Ouda, K. Basu, R. Cohen-Luria, A. de la Escosura and G. Ashkenasy, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2015285118 CrossRef CAS PubMed .
- P. L. Scognamiglio, C. Platella, E. Napolitano, D. Musumeci and G. N. Roviello, Molecules, 2021, 26, 3558 CrossRef CAS PubMed .
- G. N. Roviello, G. Oliviero, A. Di Napoli, N. Borbone and G. Piccialli, Arabian J. Chem., 2020, 13, 1966–1974 CrossRef CAS .
- G. N. Roviello, S. Di Gaetano, D. Capasso, S. Franco, C. Crescenzo, E. M. Bucci and C. Pedone, J. Med. Chem., 2011, 54, 2095–2101 CrossRef CAS PubMed .
- M. L. Daly, Y. Gao and R. Freeman, Bioconjugate Chem., 2019, 30, 1864–1869 CrossRef CAS PubMed .
- N. Gour, D. Kedracki, I. Safir, K. X. Ngo and C. Vebert-Nardin, Chem. Commun., 2012, 48, 5440–5442 RSC .
- A. Chotera, H. Sadihov, R. Cohen-Luria, P. A. Monnard and G. Ashkenasy, Chem. – Eur. J., 2018, 24, 10128–10135 CrossRef CAS PubMed .
- A. Buchberger, C. R. Simmons, N. E. Fahmi, R. Freeman and N. Stephanopoulos, J. Am. Chem. Soc., 2020, 142, 1406–1416 CrossRef CAS PubMed .
- C. Avitabile, C. Diaferia, V. Roviello, D. Altamura, C. Giannini, L. Vitagliano, A. Accardo and A. Romanelli, Chem. – Eur. J., 2019, 25, 14850–14857 CrossRef CAS PubMed .
- C. Diaferia, C. Avitabile, M. Leone, E. Gallo, M. Saviano, A. Accardo and A. Romanelli, Chem. – Eur. J., 2021, 27, 14307–14316 CrossRef CAS PubMed .
- N. Gour, J. Nixon Abraham, M. Chami, A. Castillo, S. Verma and C. Vebert-Nardin, Chem. Commun., 2014, 50, 6863–6865 RSC .
- R. Bucci, A. Bossi, E. Erba, F. Vaghi, A. Saha, S. Yuran, D. Maggioni, M. L. Gelmi, M. Reches and S. Pellegrino, Sci. Rep., 2020, 10, 19331 CrossRef CAS PubMed .
- H. Wang, Z. Feng, Y. Qin, J. Wang and B. Xu, Angew. Chem., Int. Ed., 2018, 57, 4931–4935 CrossRef CAS PubMed .
- S. Tomassi, C. Ieranò, M. E. Mercurio, E. Nigro, A. Daniele, R. Russo, A. Chambery, I. Baglivo, P. V. Pedone, G. Rea, M. Napolitano, S. Scala, S. Cosconati, L. Marinelli, E. Novellino, A. Messere and S. Di Maro, Bioorg. Med. Chem., 2018, 26, 2539–2550 CrossRef CAS PubMed .
- P. A. Levene and W. A. Jacobs, Ber. Dtsch. Chem. Ges., 1909, 42, 2469–2473 CrossRef CAS .
- I. Bang, Biochem. Z., 1910, 26, 293–311 CAS .
- M. V. Buell and M. E. Perkins, J. Biol. Chem., 1927, 72, 21–26 CrossRef CAS .
- S. M. Park and B. H. Kim, Soft Matter, 2008, 4, 1995–1997 RSC .
- R. Iwaura, K. Yoshida, M. Masuda, K. Yase and T. Shimizu, Chem. Mater., 2002, 14, 3047–3053 CrossRef CAS .
- B. Dessane, R. Smirani, G. Bouguéon, T. Kauss, E. Ribot, R. Devillard, P. Barthélémy, A. Naveau and S. Crauste-Manciet, Sci. Rep., 2020, 10, 2850 CrossRef CAS PubMed .
- X. Wang, L. Zhou, H. Wang, Q. Luo, J. Xu and J. Liu, J. Colloid Interface Sci., 2011, 353, 412–419 CrossRef CAS PubMed .
- C. J. Serpell, M. Barłóg, K. Basu, J. F. Fakhoury, H. S. Bazzi and H. F. Sleiman, Mater. Horiz., 2014, 1, 348–354 RSC .
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123–136 CrossRef CAS PubMed .
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650–668 CrossRef .
- T. Bhattacharyya, P. Saha and J. Dash, ACS Omega, 2018, 3, 2230–2241 CrossRef CAS PubMed .
- J. Chu, C. Chen, X. Li, L. Yu, W. Li, M. Cheng, W. Tang and Z. Xiong, Anal. Chim. Acta, 2021, 1157, 338400 CrossRef CAS PubMed .
- J. Bush, C. H. Hu and R. Veneziano, Appl. Sci., 2021, 11, 1–20 Search PubMed .
- X. Du, J. Li, Y. Gao, Y. Kuang and B. Xu, Chem. Commun., 2012, 48, 2098–2100 RSC .
- D. Wu, X. Du, J. Shi, J. Zhou, N. Zhou and B. Xu, J. Colloid Interface Sci., 2014, 447, 269–272 CrossRef PubMed .
- H. T. West, C. M. Csizmar and C. R. Wagner, Biomacromolecules, 2018, 19, 2650–2656 CrossRef CAS PubMed .
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS PubMed .
- D. Wu, X. Du, J. Shi, J. Zhou and B. Xu, Chin. J. Chem., 2014, 32, 313–318 CrossRef CAS .
- K. Boback, K. Bacchi, S. O'neill, S. Brown, J. Dorsainvil and J. E. Smith-Carpenter, Molecules, 2020, 25, 5493 CrossRef CAS PubMed .
- T. Sugiyama and A. Kittaka, Molecules, 2012, 18, 287–310 CrossRef PubMed .
- E. Quijano, R. Bahal, A. Ricciardi, W. M. Saltzman and P. M. Glazer, Yale J. Biol. Med., 2017, 90, 583–598 CAS .
- D. Bonifazi, L. E. Carloni, V. Corvaglia and A. Delforge, Artif. DNA PNA XNA, 2012, 3, 112–122 CrossRef PubMed .
- M. O. Guler, J. K. Pokorski, D. H. Appella and S. I. Stupp, Bioconjugate Chem., 2005, 16, 501–503 CrossRef CAS PubMed .
- A. Mohammed, A. F. Miller and A. Saiani, Macromol. Symp., 2007, 251, 88–95 CrossRef CAS .
- J. Gao, C. Tang, M. A. Elsawy, A. M. Smith, A. F. Miller and A. Saiani, Biomacromolecules, 2017, 18, 826–834 CrossRef CAS PubMed .
- J. T. M. DiMaio, T. M. Doran, D. M. Ryan, D. M. Raymond and B. L. Nilsson, Biomacromolecules, 2017, 18, 3591–3599 CrossRef CAS PubMed .
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC .
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC .
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC .
- T. Giraud, S. Bouguet-Bonnet, P. Marchal, G. Pickaert, M. C. Averlant-Petit and L. Stefan, Nanoscale, 2020, 12, 19905–19917 RSC .
- O. Berger, L. Adler-Abramovich, M. Levy-Sakin, A. Grunwald, Y. Liebes-Peer, M. Bachar, L. Buzhansky, E. Mossou, V. T. Forsyth, T. Schwartz, Y. Ebenstein, F. Frolow, L. J. W. Shimon, F. Patolsky and E. Gazit, Nat. Nanotechnol., 2015, 10, 353–360 CrossRef CAS PubMed .
- A. P. Demchenko, Luminescence, 2002, 17, 19–42 CrossRef CAS PubMed .
- S. Haldar, A. Chaudhuri and A. Chattopadhyay, J. Phys. Chem. B, 2011, 115, 5693–5706 CrossRef CAS PubMed .
- T. Giraud, S. Bouguet-Bonnet, M. J. Stébé, L. Richaudeau, G. Pickaert, M. C. Averlant-Petit and L. Stefan, Nanoscale, 2021, 13, 10566–10578 RSC .
- X. Du, J. Zhou, X. Li and B. Xu, Interface Focus, 2017, 7, 20160116 CrossRef PubMed .
- X. Li, Y. Kuang, H. C. Lin, Y. Gao, J. Shi and B. Xu, Angew. Chem., Int. Ed., 2011, 50, 9365–9369 CrossRef CAS PubMed .
- S. Marchesan, A. V. Vargiu and K. E. Styan, Molecules, 2015, 20, 19775–19788 CrossRef CAS PubMed .
- K. Baek, A. D. Noblett, P. Ren and L. J. Suggs, ACS Appl. Bio Mater., 2019, 2, 2812–2821 CrossRef CAS PubMed .
- K. Baek, A. D. Noblett, P. Ren and L. J. Suggs, Biomater. Sci., 2020, 8, 3130–3137 RSC .
- A. D. Noblett, K. Baek and L. J. Suggs, ACS Biomater. Sci. Eng., 2021, 7, 2605–2614 CrossRef CAS PubMed .
- D. Yuan, X. Du, J. Shi, N. Zhou, J. Zhou and B. Xu, Angew. Chem., Int. Ed., 2015, 54, 5705–5708 CrossRef CAS PubMed .
- D. Yuan, X. Du, J. Shi, N. Zhou, A. A. Baoum, K. O. Al Footy, K. O. Badahdah and B. Xu, Beilstein J. Org. Chem., 2015, 11, 1352–1359 CrossRef CAS PubMed .
- X. Li, Y. Kuang, J. Shi, Y. Gao, H. C. Lin and B. Xu, J. Am. Chem. Soc., 2011, 133, 17513–17518 CrossRef CAS PubMed .
- X. Li, X. Du, Y. Gao, J. Shi, Y. Kuang and B. Xu, Soft Matter, 2012, 8, 7402–7407 RSC .
- X. Du, J. Zhou, O. Guvench, F. O. Sangiorgi, X. Li, N. Zhou and B. Xu, Bioconjugate Chem., 2014, 25, 1031–1035 CrossRef CAS PubMed .
- H. Wang, Z. Feng and B. Xu, Theranostics, 2019, 9, 3213–3222 CrossRef CAS PubMed .
- N. Stephanopoulos, Bioconjugate Chem., 2019, 30, 1915–1922 CrossRef CAS PubMed .
- J. Li, L. Mo, C. H. Lu, T. Fu, H. H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC .
- K. R. Singh, P. Sridevi and R. P. Singh, Eng. Rep., 2020, 2, e12238 CAS .
- L. Chen, J. Zhang, Z. Lin, Z. Zhang, M. Mao, J. Wu, Q. Li, Y. Zhang and C. Fan, Acta Pharm. Sin. B, 2022, 12, 76–91 CrossRef CAS PubMed .
- N. Venkatesan and B. H. Kim, Chem. Rev., 2006, 106, 3712–3761 CrossRef CAS PubMed .
-
N. Greenbaum and R. Ghose, Nuclear Magnetic Resonance (NMR) Spectroscopy: Structure Determination of Proteins and Nucleic Acids, in Encyclopedia of Life Sciences (ELS), John Wiley & Sons, Ltd, Chichester, 2010 ( DOI:10.1002/9780470015902.a0003100.pub2) .
- T. Das, M. Häring, D. Haldar and D. Díaz Díaz, Biomater. Sci., 2018, 6, 38–59 RSC .
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC .
Footnote |
| † These authors contribute equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Paul
Hoschtettler†
,
Paul
Hoschtettler†