
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3,4,3′-Tri-O-methylellagic acid as an anticancer agent: in vitro and in silico studies†
Andika Pramudya Wardanaab,
Muhammad Ikhlas Abdjanb,
Nanik Siti Aminah *bc,
Mochamad Zakki Fahmib,
Imam Siswantobd,
Alfinda Novi Kristantibc,
Mirza Ardella Saputrae and
Yoshiaki Takayaf
*bc,
Mochamad Zakki Fahmib,
Imam Siswantobd,
Alfinda Novi Kristantibc,
Mirza Ardella Saputrae and
Yoshiaki Takayaf
aPhD Student of Mathematics and Natural Sciences, Faculty of Science and Technology, Universitas Airlangga, Surabaya 60115, Indonesia
bDepartment of Chemistry, Faculty of Science and Technology, Universitas Airlangga, Surabaya 60115, Indonesia. E-mail: nanik-s-a@fst.unair.ac.id; Fax: +62-31-5936502; Tel: +62-31-5936501
cBiotechnology of Tropical Medicinal Plants Research Group, Universitas Airlangga, Indonesia
dBioinformatic Laboratory, UCoE Research Center for Bio-Molecule Engineering, Universitas Airlangga, Surabaya, Indonesia
eNanotechnology Engineering, Faculty of Advanced Technology and Multidiscipline, Universitas Airlangga, Surabaya 60115, Indonesia
fFaculty of Pharmacy, Meijo University, 150 Yagotoyama, Tempaku, Nagoya, 468-8503, Japan
First published on 19th October 2022
Abstract
We report a natural product compound isolated from Syzygium polycephalum known as 3,4,3′-tri-O-methylellagic acid (T-EA) as a candidate drug for cancer treatment. The characterization of the isolated T-EA compound was carried out using various spectroscopic methods. The in vitro evaluation showcased the inhibition activity of T-EA towards the T47D and HeLa cell lines with EC50 values of 55.35 ± 6.28 μg mL−1 and 12.57 ± 2.22 μg mL−1, respectively. Meanwhile, the in silico evaluation aimed to understand the interaction of T-EA with enzymes responsible for cancer regulation at the molecular level by targeting the hindrance of cyclin-dependent kinase 9 (CDK9) and sirtuin 1 (SIRT1) enzymes. T-EA showed a binding free energy towards the SIRT1 protein of ΔGbind (MM-GBSA): −30.98 ± 0.25 kcal mol−1 and ΔGbind (MM-PBSA): −24.07 ± 0.30 kcal mol−1, while that of CDK9 was ΔGbind (MM-GBSA): −29.50 ± 0.22 kcal mol−1 and ΔGbind (MM-PBSA): −25.87 ± 0.40 kcal mol−1. The obtained results from this research could be considered as important information on 3,4,3′-tri-O-methylellagic acid as a drug to treat cervical and breast cancers.
Introduction
The exact cause of cancer is still difficult to explain with certainty although many hypotheses have been reported. For example, this disease is assumed to be caused by various factors in the environment, such as pollution, chemicals, and viruses, as well as carcinogenic substances in food.1 GLOBOCAN 2020 data estimated around 19.3 million new cancer cases and 10 million cancer deaths worldwide. Amongst those cases, in women, breast cancer is recorded to be as high as 11.7% of all cancer cases worldwide and 6.9% of all death cases in 2020,2 while cervical cancer constituted 3.1% of new cases and 3.4% of death cases. Therefore, treating these two types of cancer using drugs is of great concern.The development of cancer drugs is currently one of the major efforts being made to overcome the high death rate caused by cancer. One of the target proteins of these drugs is known as cyclin-dependent kinase 9 (CDK9), a major transcriptional regulator and a promising subject for developing a cancer cure.3 The CDK9 enzyme is present in almost all types of human cancer, and this enzyme promotes genome integrity to prevent replication stress and DNA damage.4 In cervical cancer, the CDK9 enzyme is upregulated during cervical lesions. This enzyme acts as a proto-oncogene in cervical cancer, modulating cell proliferation and apoptosis through the AKT2/p53 pathway.5 Additionally, the development of cancer drugs using the sirtuin inhibitory mechanism is also an alternative. Sirtuin1 (SIRT1) is an isoform of the sirtuin enzyme bunch (SIRT1–SIRT7) that indicates cancer growth.6 Overexpression of SIRT1 promotes the development of cancer cells in breast cancer.7 Furthermore, this enzyme can deacetylate different proteins to intervene in cell development through cell cycle pathways (FOXO3a, RB1, KU70, and E2F1).8 Moreover, the SIRT1 enzyme can intervene in cancer growth through the apoptotic pathway by restraining p53 action.9 Therefore, inhibition of the SIRT1 enzyme is promising for study as an inhibitory target in suppressing cancer growth.
Several ellagic acid derivatives have been reported to have the capability to inhibit the growth of HepG2 cancer cells, such as 3,3′-di-O-methylelaga-4′-O-β-D-silopiranoside acid.10 Meanwhile, the 4,4′-di-O-methylellagic acid compound can hinder the spread of colon cancer cells.11 Moreover, 3,4,3′-tri-O-methylellagic acid compounds have been reported to be cytotoxic against two cancer cell lines (RBL2H3 and RAW264.7).12 Therefore, ellagic acid derivatives have potential to be developed as cancer drugs. In a previous study, we used Syzygium polycephalum extract and the nanoencapsulation form of the extract for an anti-cancer assay using the T47D and HeLa cell lines, and they showed good extract potency.13 Based on these considerations, we tried to isolate the ellagic acid derivative from the stem bark of S. polycephalum and carried out activity tests using the T47D and HeLa cell lines. Specifically, we report an examination of the ellagic acid derivative from S. polycephalum, which was assessed for anticancer action in vitro and in silico.
Methodology
Materials
S. polycephalum bark, silica gel TLC 60 F254 (Merck), silica gel 60 (0.063–0.200 mm) (Merck), methanol, dichloromethane, n-hexanes, phosphate buffered saline, doxorubicin, sodium dodecyl sulfate, and 3-(4,5′-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium (MTT).Isolation and elucidation
3,4,3′-Tri-O-methylellagic acid (T-EA) was isolated from S. polycephalum following previously reported methods.13 The isolated compound was characterized using UV-Vis spectrometry (Shimadzu UV-1800), FTIR spectroscopy (Shimadzu IRTracer-100), DART-MS (Thermo Scientific Inc.), 1H-NMR spectroscopy (Bruker 600 MHz, in DMSO-d6), 13C-NMR spectroscopy (Bruker 150 MHz, in DMSO-d6), HSQC, and HMBC.In vitro anticancer assay
Cancer cells (T47D and HeLa cell lines) from cell culture were put into 96-well plates with 80% confluence and incubated at 37 °C with 5% CO2 for 24 hours. After incubation, the medium was discarded, and 100 μL of T-EA solution was added at different concentrations. Cells were then re-incubated for 24 hours and 100 μL of MTT solution was added to each well. After more incubation for 3 hours, 100 μL of sodium dodecyl sulfate was added to each well. The resultant formazan-based absorbance was determined using an ELISA reader at λmax 560 nm and the following equation was used to calculate the cell viability (eqn (1)).
 | (1) |
Molecular docking and MD simulation
Investigation of molecular docking and molecular dynamics (MD) simulation of the T-EA compound used the NAD-dependent protein deacetylase sirtuin-1/SIRT1 (PDB code 4I5I)14 and the cyclin-dependent kinase 9/CDK9 (PDB code 3TNH) enzymes.14,15 SIRT1 was used as a promoter in breast cancer (T47D cell line)16 and CDK9 enzyme as a promotor in cervical cancer (HeLa cell line).17 Selected protein codes have been reported as potential targets for studying the inhibition mechanism of small organic molecules at the molecular level through in silico studies.18,19 (6S)-2-Chloro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-6 carboxamide (PDB code 4I5) was used as an inhibitor of the SIRT1 enzyme14 and 4-[(E)-(3,5-diamino-1h-pyrazol-4-yl)diazenyl]phenol (PDB code F18) was used as an inhibitor of the CDK9 enzyme 15. The electrostatic potential (ESP) charge of T-EA was calculated using the density functional theory (DFT/B3LYP/6-31G(d,p)) method with a Gaussian 16W package.20 The redocking process used the native ligand on each target protein's active site as a reference. Furthermore, the AMBER force field (FF14SB) and the Austin Model 1-Bond Charge Correction (AM1-BCC) were utilized to obtain ligand and receptor boundaries, and bonded, nonbonded, and charge data.Molecular docking examination utilized the depiction of the Dock6 program bundle, in which the group circle determination employed a range of 10.0 Å from the local ligand's directions on the receptor's dynamic site. Flexible conformation with energy calculation used a functional grid score to determine ligand–receptor interactions, with a validation criterion using ligand root-mean-square displacement (RMSD) value of 2.0 Å.21 The score function integrated molecular docking and the MD simulation used the General AMBER Force Field (GAFF).22 The solvent model utilized the TIP3P water solvent model with a base distance of 12 Å. Then, sodium ions (Na+) were randomly added to neutralise the recreated framework.23 The additional hydrogen atoms and water molecules were minimized by 500 stages of steepest descent and 3000 stages of conjugated gradient, although the remainder of the atoms were restrained. Finally, the whole framework was completely minimized by similar techniques. The heating stage for each system was run for 200 ps in stages from 0 K to 298 K. The density (300 ps) and equilibrium (1000 ps) stages were carried out with harmonic restraints of 30, 20, 10, and 5 kcal mol−1 Å−2. Thus, the entire simulation process for each system was carried out under the NPT (310 K, 1 atm) ensemble to 100 ns to generate trajectories.
System stability analyses, such as total energy, temperature, and RMSD, were performed using all trajectories with a simulation time of 100 ns. On the other hand, analysis of hydrogen bond and binding affinity (ΔGbind) was calculated using the last 20 ns of the trajectory. Calculation of binding free energy (ΔG) and decomposition free energy (ΔGresiduebind) used the MMPBSA.py tool of the AMBER18 package.24 In addition, the binding free energy (ΔGbind) determination was achieved using the Molecular Mechanics-Poisson Boltzmann/Generalized Born Surface Area (MM-PB/GBSA) method. Methodically, binding free energy (ΔGbind) can be calculated using eqn (2). The entropy change variable (−TΔS) was neglected due to its high computational cost and low prediction accuracy.25 In particular, the energy components that involve the bond free energy can be represented by the gas (eqn (3)) and the solvation state (eqn (4)). The gaseous state indicated the component comprising bonded energy (ΔEbonded), van der Waals energy (ΔEvdW), and electrostatic energy (ΔEele). Bonded energy (ΔEbonded) represents the bond, angle, and torsion energies. Therefore, the conformational energy value for that parameter is zero. Moreover, the solvation state includes the total Poisson Boltzmann/Generalized Born models (ΔGelesolv) and solvent-accessible surface area energy (ΔGnonpolarsolv). Finally, binding free energy can be determined utilizing the four energy variables shown in eqn (5).
| ΔGbind = ΔGgas + ΔGsolv − TΔS | (2) |
| ΔGgas = ΔEbonded + ΔEvdW + ΔEele | (3) |
| ΔGsolv = ΔGelesolv + ΔGnonpolarsolv | (4) |
| ΔGbind = ΔEbonded + ΔEvdW + ΔGelesolv + ΔGnonpolarsolv | (5) |
Drug-likeness, bioavailability, and pharmacokinetics
Drug criterion prediction of T-EA, such as drug-likeness and bioavailability, was studied using the SwissADME web service (http://www.swissadme.ch/index.php).26 The study included physicochemical properties, lipophilicity, water-solubility, drug-likeness, and pharmacokinetics. All calculations were carried out using the T-EA 2D structure model (SMILES format).Results and discussion
Structure elucidation
The yellowish solid of the isolated T-EA (15.2 mg) was obtained from S. polycephalum following a previously reported method.13 Characterization using UV-Vis spectra (CH3OH, λmax) showed that T-EA had two absorption bands at 372.3 and 247.6 nm (Fig. S1†), which illustrated the existence of carbonyl and benzene groups. The IR spectra (Fig. S2†) showed the presence of –OH stretch groups (3423 cm−1), –CH stretch groups (2963, 2914, and 2853 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch groups (1751 and 1726 cm−1), C–C benzene groups (1607, 1573, and 1490 cm−1), –CH3 groups (1367 cm−1), and –O-aryl and –OCH3 groups (1242, 1121, and 1092 cm−1).
O stretch groups (1751 and 1726 cm−1), C–C benzene groups (1607, 1573, and 1490 cm−1), –CH3 groups (1367 cm−1), and –O-aryl and –OCH3 groups (1242, 1121, and 1092 cm−1).
The following 1H-NMR studies in DMSO-d6 suggested that the isolated T-EA had three sharp singlets at δH 4,05, 4.04, and 3.99 (3H, s), indicating the presence of three methoxy groups. Additionally, two singlet signals indicated two protons in the aromatic ring at δH 7.62 and 7.51 (1H, s), respectively. The 13C-NMR spectra showed twelve carbon signals for two aromatic rings, two carbon signals for two carbonyls, and three carbon signals accounting for three methoxy groups, as detailed in Table 1. The twelve aromatic carbons indicated two –CH aromatic carbons at δC 107.5 (C, C-5) and 111.8 (C, C-5′), two quaternary carbons attached to the O of the lactones at δC 141.5 (C, C-2) and 141.0 (C, C-2′), and three aromatic carbons attached to three methoxy groups at δC 140.8 (C, C-3), 153.4 (C, C-4), and 140.4 (C, C-3′), while the signal at δC 153.7 (C, C-4′) represented one aromatic carbon adjacent to the hydroxyl group. The remainder of the aromatic carbons appearing at δC 111.9 (C, C-1) and 110.7 (C, C-1′) were two quaternary carbons connecting the two aromatic rings, while signals at δC 113.6 (C, C-6) and 112.5 (C, C6′) represented quaternary carbons next to two carbonyls, with signals at δC 158.6 (C, C-7) and 158.4 (C, C-7′). Lastly, carbon signals of the three methoxy group carbons were observed at δC 61.3 (–OMe, C-3), 56.7 (–OMe, C-4), and 60.9 (–OMe, C-3′). Chemical shift analysis data are provided in the ESI (Fig. S3–S6).† These results as well as data comparison with the literature supported the conclusion that the isolated compound was indeed 3,4,3′-tri-O-methylellagic acid (Fig. 1).
| Position | T-EA | Reference 13 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 1 | 111.9 (C) | 112.0 (C) | ||
| 2 | 141.5 (C) | 141.5 (C) | ||
| 3 | 140.8 (C) | 140.9 (C) | ||
| 4 | 153.4 (C) | 152.8 (C) | ||
| 5 | 107.5 (CH) | 7.62 (s) | 107.5 (CH) | 7.63 (s) |
| 6 | 113.6 (C) | 112.6 (C) | ||
| 7 | 158.6 (CO) |
158.6 (CO) |
||
| 1′ | 110.7 (C) | 111.1 (C) | ||
| 2′ | 141.0 (C) | 141.0 (C) | ||
| 3′ | 140.4 (C) | 140.3 (C) | ||
| 4′ | 153.7 (C) | 153.8 (C) | ||
| 5′ | 111.8 (CH) | 7.51 (s) | 111.7 (CH) | 7.52 (s) |
| 6′ | 112.5 (C) | 112.0 (C) | ||
| 7′ | 158.4 (CO) |
158.4 (CO) |
||
| 3-OCH3 | 61.3 (CH3) | 4.04 (s) | 61.3 (CH3) | 4.03 (s) |
| 4-OCH3 | 56.7 (CH3) | 3.99 (s) | 56.7 (CH3) | 3.99 (s) |
| 3′-OCH3 | 60.9 (CH3) | 4.05 (s) | 61.0 (CH3) | 4.04 (s) |
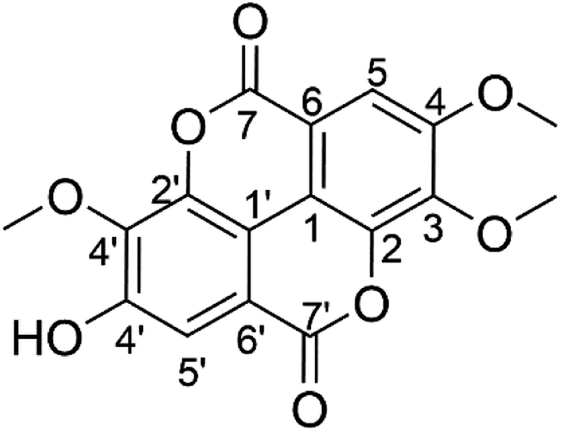 | ||
| Fig. 1 The structure of 3,4,3′-tri-O-methylellagic acid. | ||
In vitro assay: T47D and HeLa cell lines
In order to analyse the potential anticancer activity of T-EA, an assay using the MTT method was conducted measuring the growth inhibition ability of the HeLa and T47D cell lines (Fig. 2). This isolated T-EA compound showed HeLa cell line inhibitory activity better than that against T47D the cell line, in which EC50 ± SE for the HeLa cell was 12.57 ± 2.22 μg mL−1 and it was 55.35 ± 6.28 μg mL−1 for the T47D cell line. In comparison, the positive control of doxorubicin had EC50 ± SE values of 2.66 ± 0.27 μg mL−1 for the HeLa cell line and 0.03 ± 0.01 μg mL−1 for the T47D cell line (Fig. S7†). In addition to these experiments, molecular docking and MD simulations were conducted to study the interaction of T-EA and target proteins at the molecular level, which could be very useful when studying the activity of T-EA as a potential cancer drug candidate through the SIRT1 and CDK9 enzyme inhibitory mechanisms. These two enzymes were chosen as they were presumed to be crucial in cancer cell development.16,17 | ||
| Fig. 2 The T-EA concentration-effect: the percentage of cell viability of the HeLa and T47D cell lines. | ||
Molecular docking analysis
The receptor's active site was calculated based on the primary coordinates of the native ligand.27 The selection of the cluster sphere indicated the coordinates of the native ligand binding to the receptor's active site (Fig. 3). Comparing poses on each native ligand (4I5 and F18) resulted in good root-mean-square displacement (RMSD) values of 0.449 Å and 0.519 Å. It indicates the coordinates of the redocking native ligands (pose) had good coordinates with the crystal native ligands (reference). Next, analysis of ligand–receptor binding affinity was performed using the grid score function.28 Energy component calculations, such as grid score, van der Waals energy (EvdW), electrostatic energy (Eele), and internal repulsive energy (Eint), were performed in the gas phase. The redocking process used a flexible conformation with a grid score of each native ligand of 4I5: −59.10 kcal mol−1 and F18: −49.10 kcal mol−1 (Table S1†). The grid score results for each native ligand were used as comparison data with the T-EA ligand as a candidate. Furthermore, hydrogen bond interaction is known as an important parameter responsible for ligand–receptor interactions.29 Three amino acid residues were required in the H-bond interaction at 4I5-4I5I and five amino acid residues were required at F18-3TNH. Such information was essential in evaluating H-bond interactions between the candidate ligand and each receptor.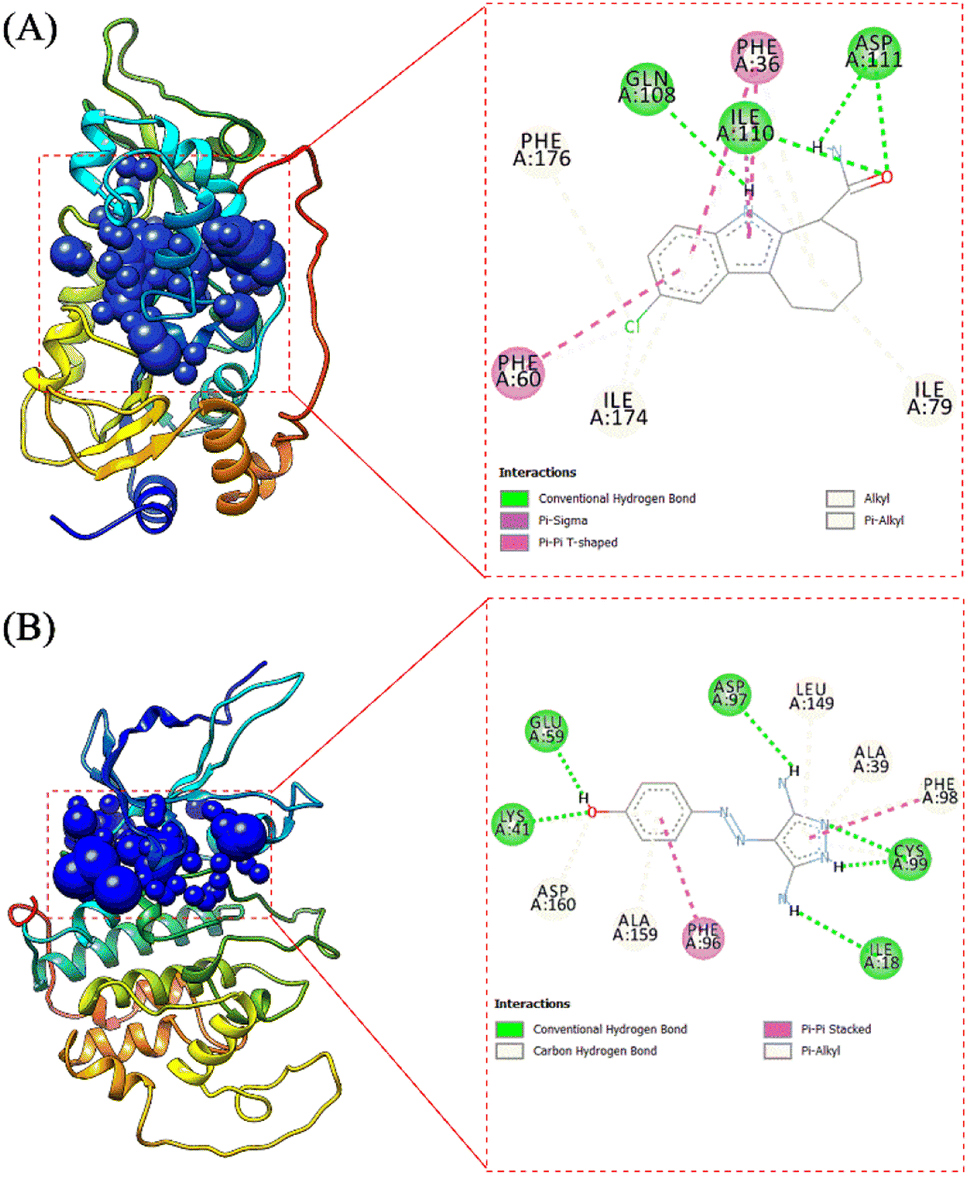
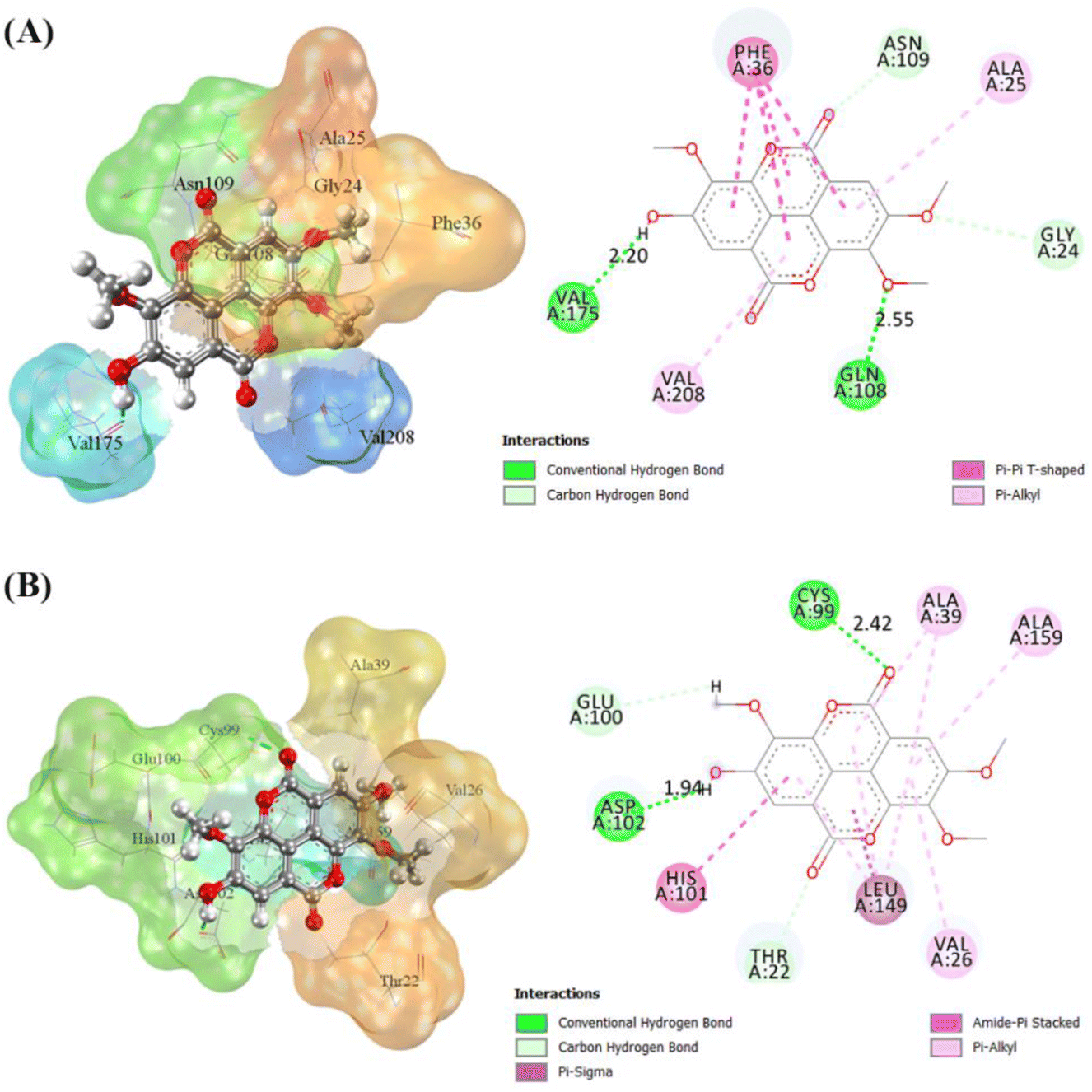
The coordinates from the redocking results were used to dock the T-EA ligand at each receptor (4I5I and 3TNH), and the results showed a good conformation of T-EA with each receptor. This was because the grid score (kcal mol−1) of T-EA was less than that of the native ligand (Table S2†). Moreover, molecular docking studies could also be used to evaluate the inhibitor activity of T-EA with the SIRT1 and CDK9 enzymes. The interaction of T-EA on the active site of the SIRT1 enzyme involved seven amino acid residues (GLY24, ALA25, PHE36, GLN108, ASN109, VAL175, and VAL208) and two residues (GLN108 and VAL175) that were responsible for H-bond interactions (Fig. 4A). Meanwhile, the T-EA interaction on the active site of the CDK9 enzyme involved nine amino acid residues (THR22, VAL26, ALA39, CYS99, GLU100, HIS101, ASP102, LEU149, and ALA159) and two residues (GLN108 and VAL175) that were responsible for H-bond interactions (Fig. 4B). In particular, the residues involved in the H-bond interactions were chosen to evaluate MD simulation usage.
MD simulation: system stability, binding free energy, decomposition free energy, and hydrogen bonding
MD simulation was used to investigate the interaction dynamics between the T-EA ligand and SIRT1 and CDK9 enzymes during the simulation time. System stability analysis of each complex was used to observe the stability of interaction between ligand and receptor.23,30 Several crucial parameters were measured in the system analysis using trajectories (100 ns), including total energy (kcal mol−1), temperature (K), and complex RMSD (nm). Analyses of total energy and temperature used the process_mdout.perl tool contained in the AMBER18 package (Fig. 5). The results showed average total energy and temperature values of T-EA–4I5I (−111.836 ± 1236.402 kcal mol−1; 297.966 ± 4.081 K) and T-EA–3TNH (−143.934 ± 1558.157 kcal mol−1; 297.942 ± 4.043 K). Overall, each system displayed good stability, which indicated no significant fluctuations in the total energy and temperature parameters. | ||
| Fig. 5 mdout analysis plotted along 100 ns of MD simulation. | ||
Next, a crucial complex RMSD analysis was performed by using the cpptraj tool included in the AMBER18 package to assess the system stability.30,31 The results revealed that the stability of each system was good, with no significant fluctuations occurring during the simulation time (Fig. 6). Specifically, the T-EA–3TNH system (RMSD: 0.347 ± 0.072) showed better stability than the T-EA–4I5I system (RMSD: 0.383 ± 0.110). This indicated that the T-EA–3TNH system at the start of 20 ns fluctuated and did not deliver great fluctuations until 100 ns. Meanwhile, the T-EA–4I5I system fluctuated at 0–60 ns and did not experience significant fluctuations up to 100 ns. However, each system showed good RMSD stability in the last 20 ns of simulation time (80–100 ns). These trajectories were then used for further analysis of binding free energy (ΔGbind), decomposition free energy (ΔGresiduebind), and hydrogen bonding.30
 | ||
| Fig. 6 Trajectory analysis of root-mean-square displacement during the 100 ns of simulation: T-EA–4I5I (top) and T-EA–3TNH (bottom). | ||
The binding free energy of the T-EA–SIRT1 and T-EA–CDK9 complexes was calculated using the MM-PB/GBSA technique,24 in which ΔGbind comprised contributions from gas and solvation terms to each complex (Table 2). The energy improvement in the gas term, such as EvdW and Eelec, resulted in an excellent contribution to the ΔGbind of each complex. However, the energy commitment to the solvation term (precisely: Eelesolv (GBSA) and Eelesolv (PBSA)) made an unfavourable contribution to ΔGbind of each complex respectively. The prediction results of ΔGbind showed that the candidate T-EA had good interaction with the SIRT1 and CDK9 enzymes. The promising binding free energy indicated a more negative value in thermodynamics. The negative value of (ΔGbind) is expected to provide strong binding on the receptor active site. Its interaction would likely change the conformational structure and inactivate the SIRT1 and CDK9 enzymes' ability to respond to cancer cell division. Additionally, the MM-GBSA approach was utilized to calculate the decomposition energy, aiming to determine the contribution of amino acid residues in the receptor's active site (Fig. S8†). A good residue contribution shown by an amino acid residue is ΔGresiduebind ≤ −1.0 kcal mol−1.30 The T-EA–3TNH complex involved nine residues, and the T-EA–4I5I complex involved eight residues with good energy contributions. Notably, these outcomes depicted a decent connection with molecular docking (Fig. 4), with some identical amino acid residues being recorded in the final trajectories (20 ns of MD simulation). Several amino acid residues common to both molecular docking and MD simulation affected the T-EA–4I5I complex (PHE36, GLN108, ASN109, VAL175, and VAL208) and the T-EA–3TNH complex (VAL26, CYS99, HIS101, ASP102, and LEU149).
| Energy component (kcal mol−1) | T-EA–4I5I | T-EA–3TNH |
|---|---|---|
| Gas term | ||
| EvdW | −42.79 ± 0.23 | −41.30 ± 0.26 |
| Eelec | −27.84 ± 0.33 | −26.58 ± 0.59 |
| ΔGgas | −70.63 ± 0.36 | −67.89 ± 0.53 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Solvation term | ||
| Eelesolv (GBSA) | 44.64 ± 0.26 | 43.19 ± 0.45 |
| Enonpolarsolv (GBSA) | −4.99 ± 0.01 | −4.81 ± 0.01 |
| ΔGsolv (GBSA) | 39.65 ± 0.26 | 38.38 ± 0.45 |
| Eelesolv (PBSA) | 52.10 ± 0.40 | 47.73 ± 0.60 |
| Enonpolarsolv (PBSA) | −5.54 ± 0.01 | −5.71 ± 0.01 |
| ΔGsolv (PBSA) | 46.55 ± 0.40 | 42.01 ± 0.60 |
|
||
| Binding free | ||
| ΔGbind (MM-GBSA) | −30.98 ± 0.25 | −29.50 ± 0.22 |
| ΔGbind (MM-PBSA) | −24.07 ± 0.30 | −25.87 ± 0.40 |
Lastly, analysis of hydrogen bond interaction was also conducted to uncover the significant part of the ligand–receptor interactions. The cpptraj tool was used for each complex in the final 20 ns of trajectories of MD simulation (Fig. S9†); hydrogen bonding with an occupation percentage of 70% represented a strong H-bond.31 The results indicated that there were two H-bonds within the T-EA–4I5I complex at the residues GLN108 (62.3%) and VAL175 (99.7%), while two H-bonds at CYS99 (88.5%) and ASP102 (57.2%) were observed in the T-EA–3TNH complex. In summary, the results of molecular docking and MD simulations show a strong correlation in the determination of the hydrogen bond. It indicates each residue on each complex well measured in molecular docking studies and MD simulations.
Drug-likeness, bioavailability, and pharmacokinetics prediction
Predictions of drug-likeness and bioavailability could provide data for predicting the capacity of T-EA to be a decent medication candidate dependent on its design. The drug-likeness analysis of T-EA was performed based on several rules, such as Lipinski's rule, Ghose's rule, Veber's rule, Egan's rule, and Muegge's rule.32 The criteria for acceptance of T-EA as a drug based on several rules was as follows: Lipinski's rule (MlogP ≤ 4.15, MW ≤ 500 Da, ∑HBD ≤ 5, and ∑O + N ≤ 10); Ghose's rule (160 ≤ MW ≤ 480 Da, −0.4 ≤ WlogP ≤ 5.6, 40 ≤ MR ≤ 130, and 20 ≤ atoms ≤ 70); Veber's rule (number of rotatable bonds ≤ 10 and TPSA ≤ 140 Å2); Egan's rule (WlogP ≤ 5.88 and TPSA ≤ 131.6 Å2); and Muegge's rule (200 ≤ MW ≤ 600 Da, −2 ≤ XlogP3 ≤ 5, TPSA ≤ 150 Å2, number of rings ≤ 7, number of carbon > 4, number of heteroatoms > 1, number of rotatable bonds ≤ 15, ∑O + N ≤ 10, and ∑HBD ≤ 15). These results suggested that T-EA has good drug-likeness criteria and could potentially be a promising drug candidate because several aspects of the existing rules have been met (Table S3†).
The oral bioavailability prediction aimed to provide theoretical information on the physicochemical properties of T-EA. In general, the criteria for a drug candidate to have good oral bioavailability include lipophilicity (−0.7 < XlogP3 < 5.0), size (150 D < MW < 500 D), polarity (20 Å2 < TPSA < 130 Å2), insolubility (0 < ESOL < 6), insaturation (0.25 < Csp3 < 1), and flexibility (0 < number of rotatable bonds < 9). Based on these results, T-EA only violated one criterion, namely insaturation (Csp3: 0.18) (Fig. 7), meaning it could still be considered to have good potential as an oral drug as a drug candidate is usually described as having low appropriateness as an oral medication if it does not meet multiple standards.33 This theoretical information could then be used as initial data for experimental testing considerations.
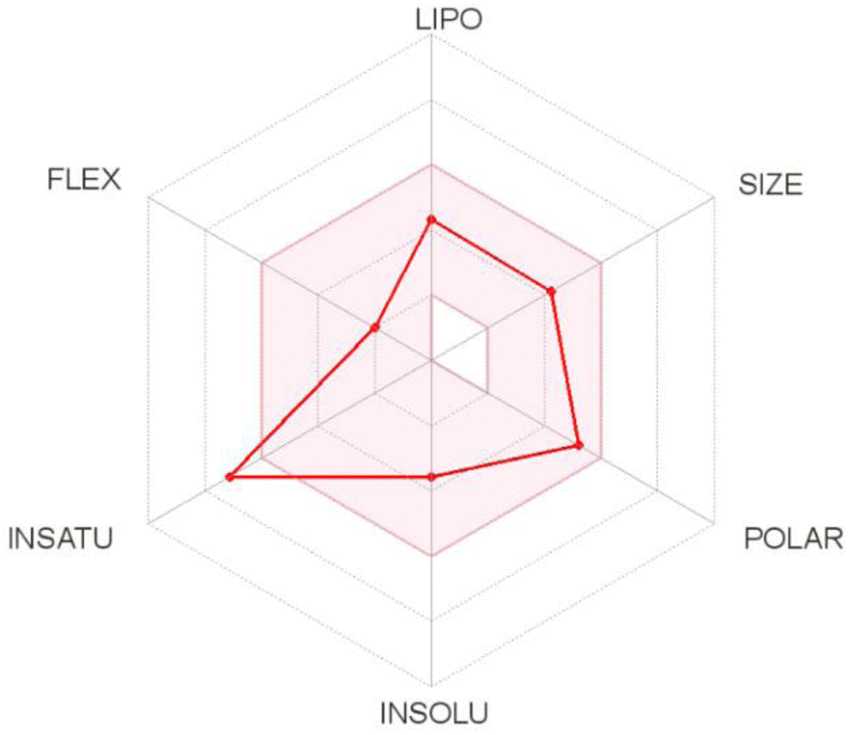 | ||
| Fig. 7 The suitable physicochemical space for oral bioavailability prediction of T-EA. | ||
Prediction of the gastrointestinal (GI) absorption parameter showed that T-EA possessed high GI absorption, which meant that T-EA can pass through the GI membrane well. Another parameter, blood–brain barrier (BBB) permeability, was also evaluated to describe the capacity of the medication to pass the BBB. However, T-EA did not distribute well across the BBB, which identified this compound as having good potential as a drug candidate because it does not influence the function of the central nervous system. The effect of T-EA on cytochrome isoenzymes (CYPs), like CYP2C19 and CYP2D6, was considered to be an urgent subject of study in the body's metabolism.34 In conclusion, T-EA showed potential as a drug candidate due a lack of both interference with enzyme activities and unwanted side effects on the body when the metabolic process takes place.35
Conclusions
3,4,3′-Tri-O-methylellagic acid (T-EA) was successfully isolated from S. polycephalum and its structure was confirmed through experimental data obtained using various spectroscopic methods. While in vitro evaluation of the T47D and HeLa cell lines showed that T-EA could inhibit the growth of these two cells, the isolated T-EA exhibited better cell growth inhibitory activity towards the HeLa cell line compared to the T47D cell line. Moreover, in silico studies of the SIRT1 and CDK9 enzymes as target proteins suggested that T-EA could have potential as a cancer drug that could repress these two protein's statements. Additionally, the interaction of T-EA with each receptor showed that several amino acid residues contributed to the H-bond interaction, T-EA–4I5I (GLN108: 62.3% and VAL175: 99.7%) and T-EA–3TNH (CYS99: 88.5% and ASP102: 57.2%). T-EA also showed good drug-likeness, oral bioavailability, and pharmacokinetic properties as a drug candidate. Overall, both in vitro and in silico studies showcased that T-EA had promising potential anticancer activity.Conflicts of interest
The authors have no competing interests.Acknowledgements
The authors would like to thank the Parasitology Laboratory, Department of Parasitology, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada for assisting in providing the T47D and HeLa cell lines. We are grateful for the computational resources provided by UCoE Research Center for Bio-Molecule Engineering, Universitas Airlangga (BIOME-UNAIR) for this work. This research was supported by the Universitas Airlangga “Hibah Riset Mandat” scheme in 2022 (contract number: 214/UN3.15/PT/2022).References
- E. A. Waters, J. Muff and J. G. Hamilton, Genet. Med., 2014, 16, 913–921 CrossRef
.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed
- M. H. Rahaman, M. Kumarasiri, L. B. Mekonnen, M. Yu, S. Diab, H. Albrecht, R. W. Milne and S. Wang, Endocr.-Relat. Cancer, 2016, 23, T211–T226 CAS
- E. Cerami, J. Gao, U. Dogrusoz, B. E. Gross, S. O. Sumer, B. A. Aksoy, A. Jacobsen, C. J. Byrne, M. L. Heuer, E. Larsson, Y. Antipin, B. Reva, A. P. Goldberg, C. Sander and N. Schultz, Cancer Discov., 2012, 2, 401–404 CrossRef PubMed
- J. Xu, S. Xu, Y. Fang, T. Chen, X. Xie and W. Lu, IUBMB Life, 2019, 71, 347–356 CAS
- Z. Lin and D. Fang, Genes Cancer, 2013, 4, 97–104 CrossRef CAS
- V. Carafa, L. Altucci and A. Nebbioso, Front. Pharmacol., 2019, 9, 1–14 Search PubMed
- E. Zhao, J. Hou, X. Ke, M. N. Abbas, S. Kausar, L. Zhang and H. Cui, Cancers, 2019, 11, 1–22 Search PubMed
- J. T. Lee and W. Gu, Genes Cancer, 2013, 4, 112–117 CrossRef CAS
- H. Zhang, Z. J. Guo, W. M. Xu, X. J. You, L. Han, Y. X. Han and L. J. Dai, Oncol. Lett., 2014, 7, 525–530 CrossRef PubMed
- A. R. De Molina, T. Vargas, S. Molina, J. Sánchez, J. Martínez-Romero, M. González-Vallinas, R. Martín-Hernández, R. Sánchez-Martínez, M. G. De Cedrón, A. Dávalos, L. Calani, D. Del Rio, A. González-Sarrías, J. C. Espín, F. A. Tomás-Barberán and G. Reglero, J. Pharmacol. Exp. Ther., 2015, 353, 433–444 CrossRef PubMed
- X. D. Su, R. H. Guo, H. X. Li, J. Y. Ma, Y. R. Kim, Y. H. Kim and S. Y. Yang, Bioorg. Med. Chem. Lett., 2018, 28, 2210–2216 CrossRef CAS
- A. P. Wardana, N. S. Aminah, M. Z. Fahmi, A. N. Kristanti, H. I. Zahrah, Y. Takaya and M. I. Choudhary, Trop. J. Nat. Prod. Res., 2020, 4, 945–952 CAS
- X. Zhao, D. Allison, B. Condon, F. Zhang, T. Gheyi, A. Zhang, S. Ashok, M. Russell, I. MacEwan, Y. Qian, J. A. Jamison and J. G. Luz, J. Med. Chem., 2013, 56, 963–969 CrossRef CAS
- S. Baumli, A. J. Hole, M. E. M. Noble and J. A. Endicott, ACS Chem. Biol., 2012, 7, 811–816 CrossRef CAS
- Y. W. Yi, H. J. Kang, H. J. Kim, Y. Kong, M. L. Brown and I. Bae, Oncotarget, 2013, 4, 984–994 CrossRef
- F. Morales and A. Giordano, Cell Cycle, 2016, 15, 519–527 CrossRef CAS
- D. Kesuma, B. T. Purwanto and M. Rudyanto, J. Chin. Pharm. Sci., 2020, 29, 123–129 CrossRef
- Y. Nkizinkiko, J. Desantis, J. Koivunen, T. Haikarainen, S. Murthy, L. Sancineto, S. Massari, F. Ianni, E. Obaji, M. I. Loza, T. Pihlajaniemi, J. Brea, O. Tabarrini and L. Lehtiö, Sci. Rep., 2018, 8, 1–10 CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- K. Liu and H. Kokubo, J. Comput.-Aided Mol. Des., 2020, 34, 1195–1205 CrossRef CAS
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed
- P. Mahalapbutr, N. Darai, W. Panman, A. Opasmahakul, N. Kungwan, S. Hannongbua and T. Rungrotmongkol, Sci. Rep., 2019, 9, 1–11 CrossRef PubMed
- B. R. Miller, T. D. Mcgee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS
- H. Luo, D. F. Liang, M. Y. Bao, R. Sun, Y. Y. Li, J. Z. Li, X. Wang, K. M. Lu and J. K. Bao, Int. J. Oral Sci., 2017, 9, 53–62 CrossRef CAS
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef
- L. G. Ferreira, R. N. Dos Santos, G. Oliva and A. D. Andricopulo, Molecules, 2015, 20, 13384–13421 CrossRef CAS
- N. S. Pagadala, K. Syed and J. Tuszynski, Biophys. Rev., 2017, 9, 91–102 CrossRef CAS
- A. K. Varma, R. Patil, S. Das, A. Stanley, L. Yadav and A. Sudhakar, PLoS One, 2010, 5, 1–10 Search PubMed
- B. Nutho, S. Pengthaisong, A. Tankrathok, V. S. Lee, J. R. K. Cairns, T. Rungrotmongkol and S. Hannongbua, Biomolecules, 2020, 10, 1–19 CrossRef
- P. Mahalapbutr, K. Thitinanthavet, T. Kedkham, H. Nguyen, L. thi ha Theu, S. Dokmaisrijan, L. Huynh, N. Kungwan and T. Rungrotmongkol, J. Mol. Struct., 2019, 1180, 480–490 CrossRef CAS
- F. T. Ndombera, G. K. K. Maiyoh and V. C. Tuei, J. Pharm. Pharmacol., 2019, 7, 165–176 Search PubMed
- S. Yasmeen and P. Gupta, Bioinform. Biol. Insights, 2019, 13, 1–11 Search PubMed
- J. D. Tyzack and J. Kirchmair, Chem. Biol. Drug Des., 2019, 93, 377–386 CrossRef CAS
- Y. Han, J. Zhang, C. Q. Hu, X. Zhang, B. Ma and P. Zhang, Front. Pharmacol., 2019, 10, 1–12 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Additional XRD and FT-IR analytical data. See DOI: https://doi.org/10.1039/d2ra02922g |
| This journal is © The Royal Society of Chemistry 2022 |