
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly emissive planarized B,N-diarylated benzonaphthoazaborine compounds for narrowband blue fluorescence†
Nhi Ngoc Tuyet Nguyen‡
,
Hanif Mubarok‡,
Taehwan Lee,
Thi Quyen Tran,
Jaehoon Jung * and
Min Hyung Lee*
* and
Min Hyung Lee*
Department of Chemistry, University of Ulsan, Ulsan 44610, Republic of Korea. E-mail: jjung2015@ulsan.ac.kr; lmh74@ulsan.ac.kr
First published on 19th October 2022
Abstract
Highly fluorescent blue emitters with high color purity are of great significance for optical applications. Herein, a series of planarized B,N-diarylated benzonaphthoazaborine compounds, namely, BzNp (1), BuBzNp (2), Bu2BzNp (3), Bu2BzMeNp (4), and Bu2BzBuNp (5), where electron-donating tBu and Me groups are differently introduced into the B-Ph, N-Ph, or benzoazaborine rings, are prepared and characterized. All compounds exhibit low-energy absorptions (λabs = 462–467 nm) and emissions (λPL = 472–478 nm) remarkably red-shifted compared with those found for the pristine dibenzoazaborine compound (404 and 415 nm, respectively). Although the expansion of π-conjugation in the azaborine ring by replacing one phenyl ring with a naphthyl ring is mainly responsible for the redshifts, the emission is also fine-tuned by attached alkyl groups, which have a greater impact on the B-centered LUMO level at the azaborine ring than at the B-Ph ring. The bandgap control and emission tuning are further supported by electrochemical and theoretical studies. Notably, blue to sky-blue fluorescence of all compounds exhibits unitary photoluminescence quantum yields, narrow full width at half maximum values (∼20 nm), and small Stokes shifts (∼11 nm), indicating strong emissions with high color purity.
Introduction
Embedding heteroatoms in polycyclic aromatic hydrocarbons (PAHs) is an effective method for tuning their photophysical properties by modifying their π-orbital interactions, thereby improving the performance of optoelectronic devices, such as organic light-emitting diodes (OLEDs).1–8 The most widely used strategy is the doping of boron (B) and nitrogen (N) atoms into the PAHs because B and N atoms can exert complementary electron-accepting and electron-donating properties, respectively, on the π-conjugated framework, allowing the modulation of the HOMO and LUMO distributions and their energy gap.9–18 Among B,N-doped PAHs, the derivatives of B,N-diphenyl-5,10-dihydro-dibenzo-1,4-azaborine, namely, dibenzoazaborine,19,20 have recently attracted significant attention because the dibenzoazaborine skeleton may constitute the main component of highly efficient emitting materials such as thermally activated delayed fluorescence (TADF) compounds.21,22 More interestingly, B,N-doped PAHs containing multiple azaborine rings have exhibited excellent photophysical properties such as narrowband emissions with high photoluminescence quantum yields (PLQYs) due to the multi-resonance effects of B and N atoms that confine the HOMO and LUMO on different ring atoms.23,24In an effort to extend the feasibility of dibenzoazaborines in optoelectronic applications, Yamaguchi et al. have recently disclosed fully planarized dibenzoazaborine structures in which the B-Ph ring is tethered with the two Ph rings in the dibenzoazaborine moiety through dimethylmethylene bridges (R1 = R2 = H in A in Chart 1).25 It was shown that the chemical and thermal stability of dibenzoazaborines can be improved by structural constraints imposed by a triply-bridged cyclic structure.4 Furthermore, the planarized polycyclic structure featured an expanded π-conjugation through the vacant p(B) orbital, which helped strengthen the B–C bonds.26–28 Interestingly, the planarized dibenzoazaborines exhibited narrowband emissions with full width at half maximum (FWHM) values less than 30 nm.25 However, the compounds fluoresced at the near UV-region (∼415 nm). For practical use in optical applications, emitters should exhibit emissions in the visible region, such as the blue region. For this purpose, we recently reported a series of planarized B,N-diarylated dibenzoazaborine compounds, where various electronic modifications on the B- and/or N-aryl groups were performed to control the HOMO–LUMO bandgap while preserving the dibenzoazaborine ring core (e.g., R1 = H; R2 = aryl in A in Chart 1).29 Although the emission can be effectively controlled to produce deep blue fluorescence at 421–439 nm, we concluded that the bandgap control by modifying the B,N-diaryl groups has a limitation, probably due to the simultaneous electronic effects of the B- or N-aryl substituents on both the HOMO and LUMO levels, partially offsetting the bandgap.
 | ||
| Chart 1 Planarized B,N-diarylated benzonaphthoazaborine derivatives (1–5). | ||
Nonetheless, the preceding results indicate that planarized diarylazaborines are potential candidates for efficient blue emitters with high PLQYs, narrow emission bandwidths, and improved stability compared with typical triarylboryl-based materials. This prompted us to investigate the extension of the emission up to (sky) blue regions. To this end, we decided to modify the central dibenzoazaborine core toward a fused π-conjugated system because the expansion of π-conjugation will have a significant impact on the reduction of the bandgap.30 In this study, we replaced one phenyl ring in dibenzoazaborine with a naphthyl ring to produce a planarized B,N-diphenyl benzonaphthoazaborine compound (R1 = R2 = R3 = H; 1 in Chart 1). Furthermore, for the fine-tuning of the emission, we made additional electronic modifications on the B-Ph, N-Ph, and/or azaborine rings of 1, which afforded compounds 2–5. It is shown that all compounds emit blue to sky-blue fluorescence (λPL = 472–478 nm) in a controlled manner, with unitary PLQYs, very narrow FWHM values (∼20 nm), and small Stokes shifts (∼11 nm). The details of the synthesis and photophysical properties are described, along with theoretical studies.
Experimental
Synthesis of 1a
The mixture of 2,3-dibromonaphthalene (3.00 g, 10.50 mmol), 2-bromoaniline (1.20 g, 7.00 mmol), tris-(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3, 0.19 g, 0.21 mmol), bis[(2-diphenylphosphino)phenyl] ether (DPEPhos, 0.23 g, 0.42 mmol), and sodium tert-butoxide (NaOtBu, 1.00 g, 10.5 mmol) in dry toluene (30 mL) was heated at 80 °C for 12 h. After cooling down, the mixture was diluted with CH2Cl2 (30 mL), filtered through Celite pad, and concentrated under reduced pressure. The crude product was subjected to silica gel column chromatography using CH2Cl2/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v) as an eluent to give 1a as a white solid (yield: 1.40 g, 53%). 1H NMR (CDCl3): δ 8.13 (s, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.64–7.60 (m, 3H), 7.45 (ddd, J = 13.8, 8.2, 6.8 Hz, 2H), 7.36 (dd, J = 8.1, 1.2 Hz, 1H), 7.30 (dd, J = 7.3, 1.4 Hz, 1H), 6.92–6.86 (m, 1H), 6.62 (br, 1H). 13C NMR (CDCl3): δ 140.1, 137.5, 133.4, 133.4, 132.2, 129.9, 128.3, 127.0, 126.8, 126.6, 124.7, 122.9, 118.6, 115.6, 114.7, 112.7.
:10, v/v) as an eluent to give 1a as a white solid (yield: 1.40 g, 53%). 1H NMR (CDCl3): δ 8.13 (s, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.64–7.60 (m, 3H), 7.45 (ddd, J = 13.8, 8.2, 6.8 Hz, 2H), 7.36 (dd, J = 8.1, 1.2 Hz, 1H), 7.30 (dd, J = 7.3, 1.4 Hz, 1H), 6.92–6.86 (m, 1H), 6.62 (br, 1H). 13C NMR (CDCl3): δ 140.1, 137.5, 133.4, 133.4, 132.2, 129.9, 128.3, 127.0, 126.8, 126.6, 124.7, 122.9, 118.6, 115.6, 114.7, 112.7.
Synthesis of 1b
The mixture of 1a (1.40 g, 3.71 mmol), iodobenzene (3.80 g, 18.60 mmol), copper iodide (0.35 g, 1.86 mmol), and potassium carbonate (1.50 g, 11.13 mmol) was refluxed at 200 °C for 24 h. After cooling to room temperature, CH2Cl2 (30 mL) and water (50 mL) were added to the mixture. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 three times (30 mL × 3). The combined organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using CH2Cl2/hexane (1:10, v/v) as an eluent to give 1b as a white solid (yield: 1.45 g, 86%). 1H NMR (CDCl3): δ 8.16 (s, 1H), 7.77–7.71 (m, 1H), 7.64 (d, J = 7.0 Hz, 2H), 7.52 (s, 1H), 7.45 (dd, J = 6.0, 3.2 Hz, 2H), 7.23 (dt, J = 19.4, 7.3 Hz, 4H), 7.07 (t, J = 7.5 Hz, 1H), 7.00 (t, J = 7.2 Hz, 1H), 6.78 (d, J = 7.9 Hz, 2H). 13C NMR (CDCl3): δ 147.8, 146.2, 143.2, 134.8, 133.6, 133.2, 132.2, 129.5, 129.1, 128.5, 127.4, 127.1, 126.8, 126.7, 126.5, 126.4, 122.2, 122.0, 121.4, 121.3.
Synthesis of 1c
To a solution of 1b (1.50 g, 3.31 mmol) in dry THF (30 mL) was added dropwise n-BuLi (2.5 M in hexane, 2.65 mL, 6.62 mmol) at −78 °C. The mixture was stirred at −78 °C for 1 h and then Me2SiCl2 (0.43 g, 3.31 mmol) was slowly added. After stirring at room temperature overnight, the resulting white turbid mixture was quenched by saturated aqueous NH4Cl solution (50 mL) and extracted with diethyl ether (30 mL × 3). The combined organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using CH2Cl2/hexane (1:10, v/v) as an eluent to give 1c as a white powder (yield: 0.97 g, 83%). 1H NMR (CDCl3): δ 8.10 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.70 (dd, J = 10.3, 4.7 Hz, 2H), 7.63–7.54 (m, 2H), 7.43 (d, J = 8.0 Hz, 1H), 7.39–7.35 (m, 2H), 7.33–7.24 (m, 2H), 7.16 (ddd, J = 8.8, 7.2, 1.8 Hz, 1H), 6.99 (dt, J = 7.1, 3.6 Hz, 1H), 6.62 (s, 1H), 6.38 (d, J = 8.6 Hz, 1H), 0.62 (s, 6H). 13C NMR (CDCl3): δ 149.8, 146.9, 144.0, 135.3, 134.8, 134.4, 131.3, 131.2, 130.2, 128.1, 128.0, 127.3, 127.2, 126.7, 123.5, 122.8, 119.8, 119.0, 117.2, 112.1, 0.4.
Synthesis of 1d
Boron tribromide (BBr3, 0.53 g, 2.10 mmol) was added carefully to the flask containing 1c (0.25 g, 0.70 mmol) at room temperature. After stirring at 60 °C for 3 h, the volatiles were removed under reduced pressure at the same temperature for 2 h. The crude mixture was dissolved in anhydrous toluene (10 mL), into which a toluene solution of (2,6-di(prop-1-en-2-yl)phenyl)lithium was added at 0 °C. The latter solution was prepared by the addition of n-BuLi (2.5 M in hexane, 0.4 mL, 1 mmol) into a toluene solution of 2-bromo-1,3-di(prop-1-en-2-yl)benzene (0.20 g, 0.84 mmol) at 0 °C. After stirring at room temperature for 12 h, the mixture was quenched with saturated NH4Cl solution and extracted with diethyl ether. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was subjected to silica gel column chromatography using CH2Cl2/hexane (1:6, v/v) to give 1d as a yellow solid (yield: 0.10 g, 40%). 1H NMR (CD2Cl2): δ 8.47 (s, 1H), 7.84 (d, J = 7.7 Hz, 2H), 7.78 (t, J = 7.3 Hz, 2H), 7.72–7.66 (m, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.45 (m, 7H), 7.31–7.25 (m, 1H), 7.08–7.00 (m, 2H), 6.70 (d, J = 8.7 Hz, 1H), 4.65 (s, 2H), 4.53 (s, 2H), 1.91 (s, 6H). 13C NMR (CD2Cl2): δ 148.2, 148.0, 147.8, 144.5, 142.5, 138.9, 137.6, 136.2, 133.2, 131.5, 129.2, 129.1, 127.9, 127.8, 127.4, 127.3, 125.9, 123.6, 119.4, 117.3, 116.7, 112.3, 112.3, 24.7. 11B NMR (CD2Cl2): δ 55.3.
Synthesis of 1 (BzNp)
The mixture of 1d (0.10 g, 0.22 mmol) and scandium(III) triflate (Sc(OTf)3, 0.22 g, 0.44 mmol) in anhydrous 1,2-dichloroethane (50 mL) was refluxed for 12 h. After cooling down, a saturated aqueous solution of NaHCO3 was added. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified through silica gel chromatography using CH2Cl2/hexane (1:6, v/v) to give 1 as a yellow solid (yield: 0.04 g, 36%). 1H NMR (CD2Cl2): δ 8.75 (m, 1H), 7.77 (m, 4H), 7.72–7.65 (m, 3H), 7.54 (t, J = 8.1 Hz, 1H), 7.47–7.39 (m, 5H), 6.97 (s, 1H), 6.48 (d, J = 8.4 Hz, 1H), 2.23 (s, 6H), 1.83 (s, 6H). 13C NMR (CD2Cl2): δ 160.3, 156.9, 154.8, 154.1, 147.5, 144.0, 142.5, 138.5, 133.8, 132.6, 131.5, 131.0, 130.1, 129.0, 128.5, 126.4, 124.8, 123.9, 122.5, 117.9, 112.6, 111.9, 44.8, 43.0, 35.0, 33.6. 11B NMR (CD2Cl2): δ 43.1. HRMS (EI): m/z [M]+ calcd for C34H28BN: 461.2315; found: 461.2313. Td5 = 320 °C.
Synthesis of 2–5
These compounds were prepared in a manner analogous to the synthesis of 1.Photophysical measurements
UV/Vis absorption and photoluminescence (PL) spectroscopic studies were performed on a Varian Cary 100 and FS5 spectrophotometer (Edinburgh Instruments), respectively. PLQYs (ΦPL) were measured on an absolute PLQY spectrophotometer (Quantaurus-QY C11347-11, Hamamatsu Photonics). Transient PL decay profiles were recorded on a FS5 spectrophotometer using an EPL-375 picosecond pulsed diode laser as an excitation light source.Results and discussion
Synthesis and characterization
Following analogous methods for preparing previous B,N-diarylated dibenzoazaborine compounds (A),25,29 planarized B,N-diarylated benzonaphthoazaborine compounds (1–5) were synthesized, as illustrated in Scheme 1. To fine-tune the HOMO–LUMO bandgap (Eg), which is relevant to emission control, we made electronic modifications at the three positions of the parent B,N-diphenyl benzonaphthoazaborine compound (BzNp, 1), that is, 4-positions of the B-Ph (R3) and N-Ph (R2) rings and the para position to the B atom of the benzoazaborine core (R1). As noted previously, the substituents on the B-Ph and N-Ph rings will mainly affect the LUMO and HOMO, respectively. It is also anticipated that the R1 substituent may have more influence on the B-centered LUMO level owing to its para-connectivity with the B atom. Therefore, for compounds 1–5, the incorporated naphthyl ring will mainly control Eg values, which will be then fine-tuned by the R1−R3 substituents. We chose electron-donating alkyl groups such as tBu and Me groups as the substituents and prepared compounds 1–5, namely, BzNp (1), BuBzNp (2), Bu2BzNp (3), Bu2BzMeNp (4), and Bu2BzBuNp (5), in combination with the different number and position of the substituents. First, 3-bromo-N-(2-bromophenyl)naphthalen-2-amine intermediates (1a, 4a, and 5a) were prepared by the Buchwald–Hartwig amination of 2,3-dibromonaphthalene with 2-bromoanilines. Ullmann coupling of 1a, 4a, and 5a with aryl iodides produced the corresponding 3-bromo-N-(2-bromophenyl)-N-arylnaphthalen-2-amines (1b and 3b–5b), which were then silylated with Me2SiCl2 after dilithiation. | ||
| Scheme 1 Synthetic routes for 1–5. | ||
Note that the use of the THF solvent led to high yields (>73%) of silanes (1c and 3c–5c). However, in the ether solvent, which was used to synthesize previous dibenzoazaborine compounds (A),29 several unknown products, including naphthyl–phenyl coupled species, were formed, consequently resulting in low yields. Next, the silicon–boron exchange reactions of silanes with BBr3 furnished bromo-azaborine intermediates, which were then subject to in situ reactions with 2,6-di(propen-2-yl)phenyllithium26 or 4-tBu-2,6-di(propen-2-yl)phenyllithium to yield arylated benzonaphthoazaborines (1d–5d). The final planarized B,N-diarylated benzonaphthoazaborines 1–5 were obtained via twofold intramolecular Friedel–Crafts cyclization reactions in the presence of Sc(OTf)3.31 All compounds were chemically stable under ambient conditions in solution and solid states. They also exhibited high thermal stability, as judged by high thermal decomposition temperatures (Td5) above 310 °C (Fig. S1†). Notably, Td5 values increased with an increasing number of alkyl substituents, probably due to the increased steric protection of the planarized azaborine core. The chemical structures of the compounds were characterized by multinuclear NMR spectroscopy and high-resolution mass spectrometry. The broad 11B NMR resonances at δ ca. 41–45 ppm, similar to those observed for A, confirmed the presence of a trigonal boron atom of the benzonaphthoazaborine skeleton.25,29
Photophysical and electrochemical properties
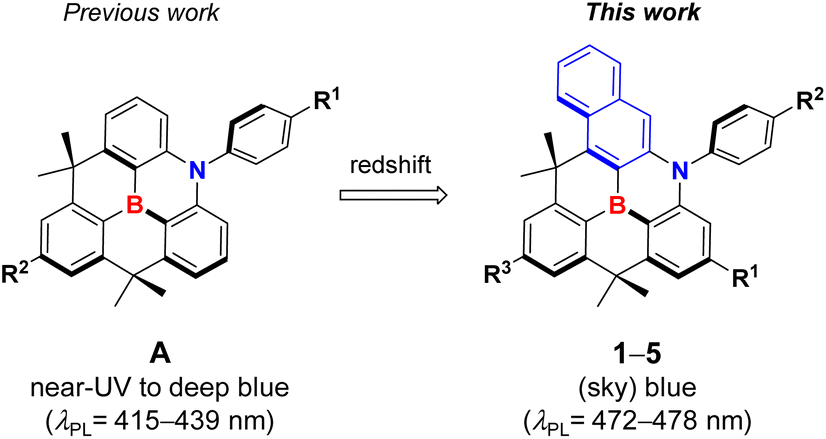
To investigate the photophysical properties of compounds, UV/Vis absorption and photoluminescence (PL) spectra were measured in a toluene solution, and the corresponding spectra are shown in Fig. 1 and S2.† The detailed spectroscopic data are summarized in Table 1. All compounds exhibit similar absorptions in the two regions, that is, the high-energy region at ca. 280−340 nm and the low-energy region at ca. 400–490 nm. The bands are highly structured, indicating the involvement of the ππ* state in the transition. The former bands can be primarily attributed to the local ππ* states of the naphthyl and phenyl rings, whereas the latter is assignable to the ππ* states, corresponding to the HOMO–LUMO transition, centered on the benzonaphthoazaborine core (see the DFT results below). The lowest energy absorption peak for unsubstituted 1 appeared at 467 nm, which was remarkably red-shifted, compared with the peak (404 nm) for the pristine dibenzoazaborine compound (R1 = R2 = H in A).25 This indicates that the incorporation of a naphthyl ring into the azaborine core has a significant impact on the reduction in the bandgap. The introduction of the para alkyl substituents (R3 and R1) with respect to the B-atom resulted in gradual blueshifts of the absorption peaks because of the elevation of the LUMO level. Attaching a tBu group (R2) to the N-Ph ring resulted in a slight redshift (2 vs. 3), but the effect was less than that of the blueshift by a tBu group on the B-Ph ring (1 vs. 2). Although not strong, these results indicate that the low-energy absorption wavelength, that is, the HOMO–LUMO bandgap, can be fine-tuned by introducing alkyl substituents into the B-Ph, N-Ph, and/or benzonaphthoazaborine rings.| Compd | λabsa (nm) (ε × 10−3/M−1 cm−1) | λPLa (nm) | ΦPLa,b (%) | Stokes shift (nm) | FWHMa,c (nm) | τ a,d (ns) | HOMO/LUMOe (eV) | Egf (eV) |
|---|---|---|---|---|---|---|---|---|
| a In oxygen-free toluene at 298 K (2.0 × 10−5 M).b Absolute PLQYs (λexc = 335 nm).c Full width at half maximum.d PL lifetimes.e Estimated from the electrochemical oxidation (HOMO) and reduction (LUMO).f Electrochemical bandgap. | ||||||||
| 1 | 285 (57.82), 442 (7.22), 467 (13.17) | 478 | ∼100 | 11 | 20 | 17.6 | −5.32/−2.32 | 3.00 |
| 2 | 285 (84.32), 440 (15.09), 466 (27.38) | 477 | ∼100 | 11 | 20 | 16.7 | −5.31/−2.31 | 3.00 |
| 3 | 285 (60.19), 441 (9.83), 466 (18.51) | 477 | ∼100 | 11 | 19 | 16.3 | −5.28/−2.30 | 2.98 |
| 4 | 287 (124.95), 437 (18.34), 462 (33.51) | 472 | ∼100 | 10 | 20 | 15.2 | −5.26/−2.21 | 3.05 |
| 5 | 288 (53.18), 438 (12.41), 464 (22.16) | 475 | ∼100 | 11 | 20 | 15.1 | −5.24/−2.18 | 3.06 |
Cyclic voltammetry (CV) was used to examine the electrochemical properties of 1–5 to confirm the bandgap control and HOMO/LUMO energy levels (Fig. 2 and Table 1 and S1†). The oxidation processes revealed that the compounds underwent reversible oxidation centered on benzonaphthoaza moieties (see DFT results below). Compared with the oxidation potential (0.52 V) of unsubstituted 1, the introduction of alkyl groups gradually reduces the oxidation potential by ca. 0.01–0.08 V. The decrease in the potential was the most apparent for compounds with the tBu group on the N-Ph moiety (3–5), which is due to the elevation of the N-centered HOMO level by the electron-donating tBu group. The R1 substituents on the azaborine core also slightly affect the HOMO level (4 and 5). Meanwhile, the compounds underwent reversible (1–3) or quasi-reversible (4 and 5) reductions centered on the boron atom. The reduction potential was very slightly shifted cathodically upon the introduction of the tBu group on the B-Ph moiety (2 and 3 vs. 1). However, relatively large cathodic shifts in the reduction potentials by ca. 0.09–0.12 V occurred for compounds with the R1 substituents on the azaborine core (4 and 5 vs. 3). This implies that the electron-donating effect on the B-centered LUMO level is greater at the azaborine ring than at the B-Ph ring. These electrochemical results indicate that the electronic effects of substituents are more obvious in the azaborine ring core than in the B-Ph and N-Ph moieties, probably due to the localization of frontier molecular orbitals on the conjugated azaborine π-framework. As a result, the electrochemical bandgaps of the compounds were fine-tuned with the greater values for 4 and 5, as similarly observed in the absorptions. It is noteworthy that the bandgap of 1 is largely decreased by 0.48 eV compared with that (3.48 eV) of dibenzoazaborine compound (R1 = R2 = H in A)25 due to the elevated HOMO and lowered LUMO levels (see also the DFT results below).
Next, the emission properties of all compounds (1–5) were investigated in toluene at room temperature (Fig. 1 and Table 1). Unsubstituted 1 exhibited a sky-blue emission at 478 nm, which is significantly red-shifted compared with that (415 nm) of the dibenzoazaborine compound (R1 = R2 = H in A).25 As observed in the absorption, this finding again indicates the impact of the naphthyl ring on the large redshift of the emission. In other words, the near-UV emission can be altered to a blue emission by simply changing the fused phenyl ring to a naphthyl ring. The PL spectrum of 1 underwent gradual blueshifts as the number of alkyl groups increased, exhibiting blue to sky-blue emissions for 2–5 (λPL = 472–477 nm). The trend in the blueshifts is reminiscent of those observed in the absorption and electrochemical bandgap of the compounds, thereby exhibiting more blue-shifted emissions for 4 and 5 containing alkyl groups in the azaborine core. All emissions have weak vibronic shoulders due to the ππ* character of their excited singlet states (S1). Most remarkably, the emission bands are very narrow, with FWHM values of ca. 20 nm. Note that the emission profiles of previous dibenzoazaborine systems exhibited FWHM values greater than 28 nm.25,29 The FWHM being nearly invariant irrespective of alkyl substituents indicates that the rigid benzonaphthoazaborine skeleton is primarily responsible for the narrow emission bandwidths. Moreover, the Stokes shifts of all compounds are small (11 nm), which are comparable to or smaller than those of dibenzoazaborine compounds (>11 nm for A in Chart 1),25,29 although very small values less than 10 nm are known for some azaborine compounds.30,32 This finding indicates that the effect of photoexcitation on structural relaxation is negligibly small. Benefiting from the rigid and planar structures, all compounds also exhibit near-unity PLQYs. Because the electronic transitions are centered on the azaborine ring core, it is likely that the peripheral alkyl substituents barely affect the PLQY. The transient PL decay profiles of all compounds exhibited a single exponential decay in the nanosecond range (τ = 15.1–17.6 ns), confirming the fluorescence nature of the emission (inset in Fig. 1a and S3†. Furthermore, the highly emissive features of 1–5 were retained in the rigid state (PMMA, 1 wt%), showing the near-unity PLQYs (Fig. S4†). These emission properties, including blue to sky-blue fluorescence, high PLQYs, narrow FWHM values, and small Stokes shifts, could be highly desirable for the use of the planarized B,N-diarylated benzonaphthoazaborine compounds as potential candidates for blue fluorescent emitters in OLED devices.
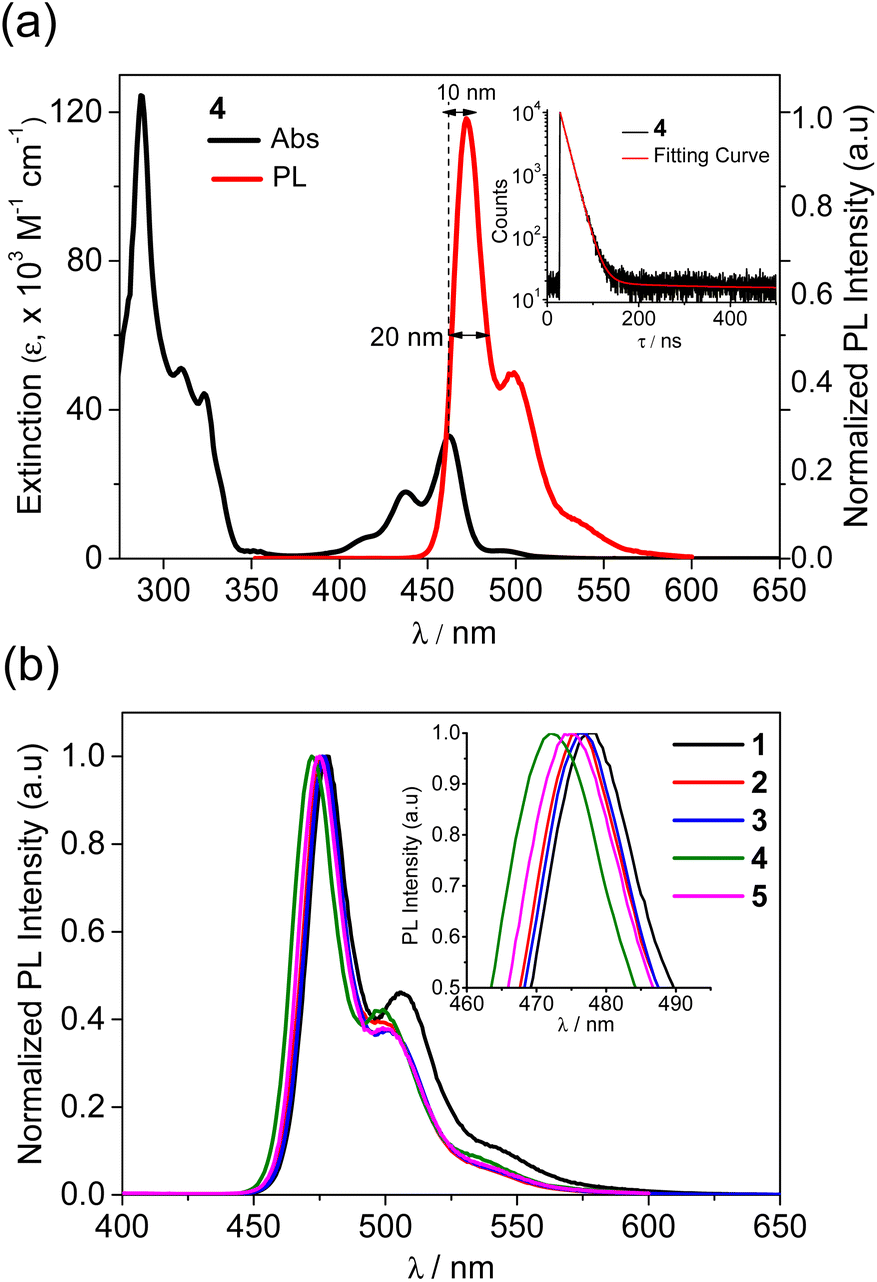 | ||
| Fig. 1 (a) UV/vis absorption and PL spectra of 4 and (b) PL spectra of 1–5 in toluene (2.0 × 10−5 M) at RT. Insets: (a) transient PL decay of 4 and (b) enlarged PL spectra showing FWHM. | ||
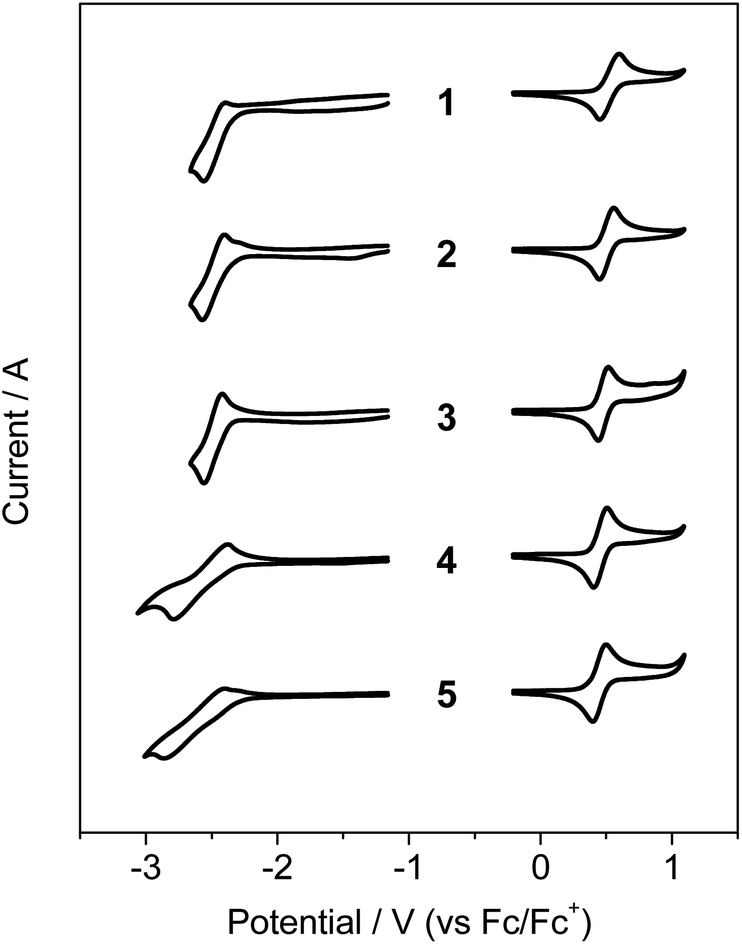 | ||
| Fig. 2 Cyclic voltammograms of 1–5. Solvent: CH2Cl2 for oxidation and THF for reduction (5.0 × 10−4 M). | ||
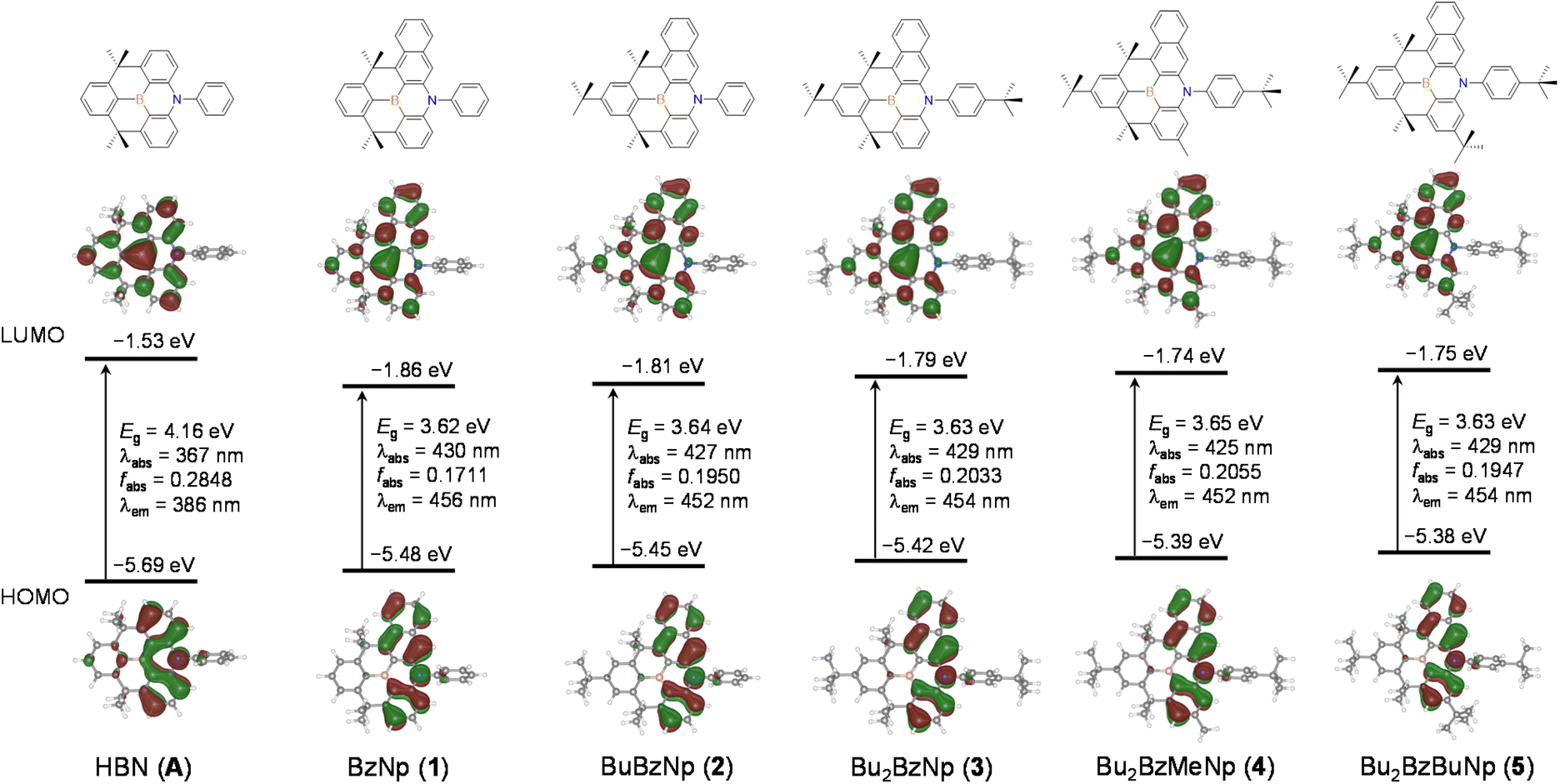 | ||
| Fig. 3 Frontier molecular orbitals of planarized B,N-diphenyl dibenzoazaborine (A) and B,N-diarylated benzonaphthoazaborine (1–5) compounds (isovalue = 0.03) at their ground state (S0) geometries from PBE0/6-31+G(d,p) calculations. Numerical values of MO energies, HOMO–LUMO bandgaps (Eg), oscillator strengths (fabs), and absorption (λabs) and emission (λem) wavelengths are provided. | ||
Finally, we investigated the effect of the naphthyl ring in the azaborine skeleton on aromaticity to confirm the extended π-conjugation effects. To this end, we performed nucleus-independent chemical shift (NICS)36 calculations for compounds 1 and A at the PBE0/6-311++G(d,p) level of theory.36 The NICS(1) values obtained at 1 Å above the ring centers were used to reduce the local effects of sigma bonds.37 As shown in Fig. 4, the central BNC4 cores of the two compounds have weak negative values, indicating a weak aromatic character compared with neighboring phenyl rings. The average NICS(1) value for the outer phenyl ring in 1 exhibits a large negative value (−11.3 ppm) which is greater than that of the phenyl ring in A (−9.8 ppm). Therefore, this finding indicates that the aromaticity of the azaborine skeleton is enhanced by the incorporation of the naphthyl ring, leading to the reduction of energy gap between HOMO and LUMO.
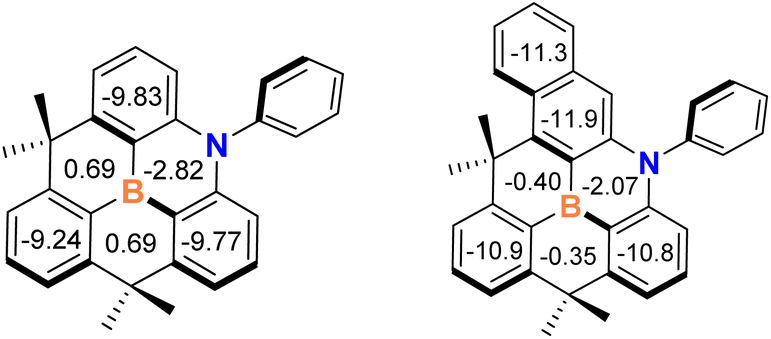 | ||
| Fig. 4 Calculated nucleus-independent chemical shift [NICS(1)] values of planarized B,N-diphenyl dibenzoazaborine (A, left) and B,N-diphenyl benzonaphthoazaborine (1, right) compounds at the PBE0/6-311++G(d,p)//PBE0/6-31+G(d,p) level of theory. | ||
Conclusions
We demonstrated that the expansion of π-conjugation in the azaborine ring has a significant impact on the photophysical properties of planarized dibenzoazaborine compounds. Replacing one phenyl ring in the dibenzoazaborine core with a naphthyl ring significantly reduced the HOMO–LUMO bandgap, resulting in a shift from near-UV or deep blue to (sky) blue fluorescence. The emission was fine-tuned by attaching electron-donating alkyl groups to the B-Ph, N-Ph, and/or azaborine rings. It was shown that the electronic effect on the B-centered LUMO level is greater at the azaborine ring than at the B-Ph ring. These findings were further explained by electrochemical and theoretical studies. Along with emission tuning, all compounds exhibited high PLQYs, very narrow FWHM values, and small Stokes shifts. The findings of this study will be useful for designing highly fluorescent emitters with high color purity for optical applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the 2022 Research Fund of University of Ulsan.Notes and references
- S. Oda and T. Hatakeyama, Bull. Chem. Soc. Jpn., 2021, 94, 950–960 CrossRef CAS.
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 1908677 CrossRef CAS.
- J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 CrossRef PubMed.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS.
- J. Huang and Y. Li, Front. Chem., 2018, 6, 341 CrossRef PubMed.
- S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC.
- J. Wagner, P. Zimmermann Crocomo, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Angew. Chem., Int. Ed., 2022, 61, e202202232 CrossRef CAS PubMed.
- N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS PubMed.
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- A. John, M. Bolte, H.-W. Lerner, G. Meng, S. Wang, T. Peng and M. Wagner, J. Mater. Chem. C, 2018, 6, 10881–10887 RSC.
- D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969–7977 RSC.
- V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 CrossRef CAS PubMed.
- X.-Y. Wang, J.-Y. Wang and J. Pei, Chem.– Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed.
- T. Agou, M. Sekine, J. Kobayashi and T. Kawashima, Chem. Commun., 2009, 1894–1896 RSC.
- T. Agou, T. Kojima, J. Kobayashi and T. Kawashima, Org. Lett., 2009, 11, 3534–3537 CrossRef CAS PubMed.
- T. Agou, J. Kobayashi and T. Kawashima, Chem. Commun., 2007, 3204–3206 RSC.
- Y. Ishikawa, K. Suzuki, K. Hayashi, S.-y. Nema and M. Yamashita, Org. Lett., 2019, 21, 1722–1725 CrossRef CAS PubMed.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- T.-L. Wu, S.-H. Lo, Y.-C. Chang, M.-J. Huang and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 10768–10776 CrossRef CAS PubMed.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- M. Ando, M. Sakai, N. Ando, M. Hirai and S. Yamaguchi, Org. Biomol. Chem., 2019, 17, 5500–5504 RSC.
- V. M. Hertz, N. Ando, M. Hirai, M. Bolte, H.-W. Lerner, S. Yamaguchi and M. Wagner, Organometallics, 2017, 36, 2512–2519 CrossRef CAS.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed.
- I. N. Istiqomah, H. Mubarok, T. Lee, N. T. N. Nguyen, J. Jung and M. H. Lee, Bull. Korean Chem. Soc., 2022, 43, 293–298 CrossRef CAS.
- T. Agou, H. Arai and T. Kawashima, Chem. Lett., 2010, 39, 612–613 CrossRef CAS.
- A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed.
- J.-J. Zhang, L. Yang, F. Liu, Y. Fu, J. Liu, A. A. Popov, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 25695–25700 CrossRef CAS PubMed.
- X. Cai, J. Xue, C. Li, B. Liang, A. Ying, Y. Tan, S. Gong and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202200337 CAS.
- T. Hua, J. Miao, H. Xia, Z. Huang, X. Cao, N. Li and C. Yang, Adv. Funct. Mater., 2022, 32, 2201032 CrossRef CAS.
- G. Meng, H. Dai, T. Huang, J. Wei, J. Zhou, X. Li, X. Wang, X. Hong, C. Yin, X. Zeng, Y. Zhang, D. Yang, D. Ma, G. Li, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, e202207293 CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- P. v. R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. van Eikema Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, photophysical and computational data, and NMR spectra. See DOI: https://doi.org/10.1039/d2ra05163j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |