
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbocation catalysis in confined space: activation of trityl chloride inside the hexameric resorcinarene capsule†
Margherita
De Rosa
 *a,
Stefania
Gambaro
a,
Annunziata
Soriente
a,
Paolo
Della Sala
a,
Veronica
Iuliano
a,
Carmen
Talotta
a,
Carmine
Gaeta
a,
Antonio
Rescifina
*b and
Placido
Neri
*a
*a,
Stefania
Gambaro
a,
Annunziata
Soriente
a,
Paolo
Della Sala
a,
Veronica
Iuliano
a,
Carmen
Talotta
a,
Carmine
Gaeta
a,
Antonio
Rescifina
*b and
Placido
Neri
*a
aLaboratory of Supramolecular Chemistry, Dipartimento di Chimica e Biologia “A. Zambelli”, Università di Salerno, Via Giovanni Paolo II, I-84084, Fisciano (SALERNO), Italy. E-mail: maderosa@unisa.it; neri@unisa.it
bDipartimento di Scienze del Farmaco e della Salute, Università di Catania, viale Andrea Doria, 6, 95125 Catania, Italy. E-mail: arescifina@unict.it
First published on 7th July 2022
Abstract
Carbocation catalysis can be performed inside the confined space of the hexameric resorcinarene capsule. The inner cavity of the capsule can host the trityl carbocation, which catalyses the Diels–Alder reaction between dienes and unsaturated aldehydes. Experimental results and in silico calculations show that the hexameric resorcinarene capsule C6 can promote the formation of the trityl carbocation from trityl chloride through the cleavage of the carbon–halogen bond promoted by OH⋯X− hydrogen bonding. Here it is shown that the combination of the nanoconfined space and the latent carbocation catalysis provides a convenient complementary strategy for the typical carbocation catalysis. The latent strategy bypasses the typical pitfalls associated with active carbocations and provides control of the reaction efficiency in terms of reaction rate, conversion, and selectivity.
1. Introduction
Carbocations are relatively common species, generally unstable and non-isolable intermediates in several fundamental organic transformations and relevant industrial processes.1 However, stable enough carbocations can also be obtained by an appropriate substitution around the carbocationic center with suitable stabilizing groups.2 Classical carbocations are trivalent, planar species also called carbenium ions, whereas non-classical carbonium ions contain carbon centres with tetra-, penta-, or even higher coordination.3Because of their positive charge, both carbenium and carbonium ions have been involved in catalytic systems.4,5 In particular, carbenium ions (e.g., triarylmethylium or trityl cations) have been used as highly efficient and highly versatile Lewis acid catalysts in a range of transformations, including Diels–Alder cycloaddition,6 Mukaiyama aldol reaction,7 Sakurai allylation,8 Michael reaction,9 halogenation,10 epoxide rearrangement,4b hetero-ene cyclization,4b and oxo-metathesis.11
In recent years, several research groups have focused their attention on the hexameric capsule (1)6·8H2O (C6), initially reported by Atwood,12 which is obtained by self-assembly of six resorcinarene 1 units and eight water molecules (Fig. 1). C6 has been largely exploited as a nanoreactor13 thanks to its ability to selectively host a wide range of substrates and to accelerate some organic reactions with excellent chemo-, regio-, and stereoselectivity. One advantage of using C6 as a nanocontainer is that its large cavity (internal volume of 1375 Å3) can be easily engineered by introducing specific artificial cofactors.14 In addition, the π-electron-rich cavity of C6 shows a high affinity for cationic species, which are stabilized by cation–π interactions.13 For this reason, the C6 capsule behaves as a good catalyst for organic reactions involving cationic intermediates and/or transition states, which are shielded from chemical quenching. In this way, interesting reactions involving cyclase mimics,15 iminium catalysis,14 Brønsted acid catalysis,16 H-bond catalysis,17 and halogen-bond catalysis18 have been performed in the confined space of a C6 capsule, frequently giving different outcomes with respect to the bulk medium. Another interesting catalysis performed inside the hexameric capsule was a mild Friedel–Crafts alkylation of (hetero)arenes in which C6 acts as a Lewis acid catalyst by activating the C–Cl bond of benzyl chloride through H-bonding interactions.19
 | ||
| Fig. 1 Chemical drawing of C-undecylresorcin[4]arene 1 (left) and a reduced model (R = Me) of the hexameric capsule (1R)6·(H2O)8 (C6) (right). | ||
Based on these considerations, the question arises as to whether the hexameric C6 capsule is also capable of carbocation catalysis with stable carbenium ions such as trityl cations: can trityl cations be hosted inside C6 to achieve catalysis?
Can C6 act as a Lewis acid catalyst by activating the less expensive trityl chloride (TrCl, 2a, Scheme 1) as the carbocation source?
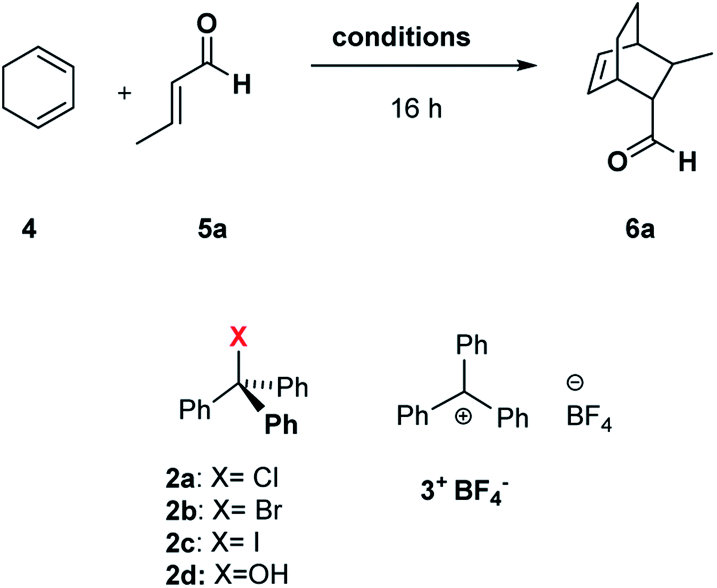 | ||
| Scheme 1 Diels–Alder reaction between 1,3-cyclohexadiene 4 and crotonaldehyde 5a in the presence of trityl chloride 2a and the C6 capsule. | ||
2. Results and discussion
Initially, we investigated the uptake of trityl chloride 2a by C6, in water-saturated CDCl3 as the solvent, in order to form the capsule. Interestingly, upon adding 2a to a C6 solution, a red-orange color developed (see ESI Fig. S1†), indicative of the formation of the trityl cation (Tr+, 3+). This was confirmed by an absorption band at 424 nm in the UV-vis spectrum (Fig. S2 and S3†). The occurrence of such a combination band instead of the typical twin absorption band reported for the carbocation20a could be attributed to the electronic interactions between the phenyl rings of trityl cation and the aromatic walls of the capsule.20b The formation and encapsulation of 3+ by C6 was also demonstrated by 1D and 2D NMR studies (see the ESI†). An HSQC spectrum provided compelling evidence for the up-field shifted resonances of o-, m-, and p-H of the encapsulated Tr+ cation (Fig. S14–S16†). In detail, the 2D HSQC spectrum of the mixture TrCl/C6 in CDCl3 showed relevant 1J correlations at 7.12/128.9, 6.85/142.2, and 4.70/140.9 ppm attributable to encapsulated 3+. Independent proof of the capability to encapsulate Tr+ by C6 was obtained by adding the trityl tetrafluoroborate salt 3+·BF4− to its water-saturated CDCl3 solution. Also in this case, a similar red-orange color developed, and 1D and 2D NMR studies clearly showed the encapsulation of the Tr+ cation inside the hexameric capsule C6 (ESI, see Fig. S18–S22†). In detail, the 1D and 2D NMR spectra of the C6/3+·BF4− mixture (ESI†) showed a set of up-field shifted aromatic resonances (o-, m-, and p-H) of the encapsulated Tr+ cation very similar to the signals observed upon mixing TrCl with C6. Thus, the 2D HSQC experiment indicated the presence of 1J correlations at 7.13/129.3, 6.83/141.4, and 4.75/141.0 ppm attributable to the encapsulated 3+. Differently, the 1H NMR spectrum of 3+·BF4− in dry CDCl3 showed signals at 8.20 (p-H), 7.86 (m-H), and 7.70 (o-H). Another evidence came from the 13C NMR spectrum of the mixture C6/2a (Fig. S12 and S13†) which shows the presence of a characteristic up-field shifted (Ph)3C+ signal at 207.5 ppm. A very similar value (206.8 ppm) was observed in the 13C NMR spectrum of the mixture C6/3+·BF4− (Fig. S20†), whereas the free salt 3·BF4− gives an unshielded resonance at 211.3 ppm (Fig. S20†). Furthermore, DOSY NMR experiments performed on the mixtures C6/2a (Fig. S17†) and C6/3+·BF4− (Fig. S22†), showed that the signals of encapsulated 3+ cation diffuse at the same rate as the capsule thus probing its internalization. In summary, all these data confirm that the C6 capsule can act as a Lewis acid catalyst by activating trityl chloride 2a to give the encapsulated carbocation 3+@C6. At this point, we investigated the catalytic ability of the encapsulated triphenylmethylium 3+@C6, obtained by mixing trityl chloride 2a with C6, in the Diels–Alder (DA) reaction between 1,3-cyclohexadiene 4 and crotonaldehyde 5a (Scheme 1) in water-saturated CDCl3 as the solvent. When the reaction mixture (Table 1, entry 1) was stirred at 30 °C for 16 h, adduct 6a was obtained in 35% yield, whereas no product was obtained in the absence of 2a (entry 2), C6 (entry 3), or both (entry 4), thus confirming the crucial role played by the C6 capsule as catalyst. The yield of 6a was then increased to 92% by raising the temperature to 50 °C and using a 4/5a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 6). With decreasing amount of 2a, the reactivity slightly slowed down, showing a lower conversion to product 6a after 16 h (Table 1, entry 7).
:1 (Table 1, entry 6). With decreasing amount of 2a, the reactivity slightly slowed down, showing a lower conversion to product 6a after 16 h (Table 1, entry 7).
| Entrya | T (°C) | 2a (mol%) | Capsule (mol%) |
4:5a |
6a yieldb (%) |
|---|---|---|---|---|---|
| a Reactions were performed on a 0.16 mmol scale using 4 (from 1 to 3 equiv.), 5a (1 equiv.), 2a (0.26 equiv.), and C6 (0.26 equiv.) in 1.1 mL of water-saturated CDCl3, for 16 h. b Isolated yield. | |||||
| 1 | 30 | 26 | 26 | 1:1 |
35 |
| 2 | 30 | — | 26 | 1:1 |
— |
| 3 | 30 | 26 | — | 1:1 |
— |
| 4 | 30 | — | — | 1:1 |
— |
| 5 | 50 | 26 | 26 | 1:1 |
50 |
| 6 | 50 | 26 | 26 | 3:1 |
92 |
| 7 | 50 | 10 | 26 | 3:1 |
80 |
As concerns the stereochemistry of the DA reaction, in all instances, a high selectivity for endo-6a over exo-6a was observed with a ratio >99:1.
The catalytic role played by the self-assembled resorcinarene capsule C6 was corroborated by a series of control experiments (Table 2) in accord with a standard protocol previously described by us and others.13–17 In particular, the DA reaction in Scheme 1 was performed in the presence of Et4N+·BF4− as a competitive guest for C6,13–17 and under these conditions, no hint of product 6a was detected in the reaction mixture (Table 2, entry 1). Interestingly, under such conditions no red-orange colour develops (Fig. S36 and S37†) indicating the absence of encapsulated Tr+ cation. On the other hand, in the presence of a very large quaternary ammonium cation (salt 14, reported at pages S55 and S66†), too bulky to be accommodated within the cavity of C6(Fig. S23 and S24†), both the reaction (Fig. S39†) and the colour development (Fig. S38†) proceeded regularly. Furthermore, no hint of product 6a was detected when the reaction in Scheme 1 was performed in the presence of hydrogen-bond competitor solvents (DMSO, Table 2, entry 2) able to disaggregate the capsule. These data strongly indicate that the reaction reported in Scheme 1 occurs in the nanoconfined space inside the resorcinarene capsule C6.
| Entrya | TrX | Additiveb (equiv.) | 6a yieldc (%) |
endo:exod |
|---|---|---|---|---|
| a Reaction conditions: 4 (0.45 M), 5a (0.15 M), C6 and TrX (0.039 M) in 1.1 mL of water-saturated CDCl3, at 50 °C for 16 h. b Amount of additive with respect to C6. c Isolated yield. d Determined by 1H NMR analysis of the crude reaction mixture according to literature data.21 e The reaction was performed in the absence of C6. | ||||
| 1 | –Cl | Et4NBF4 (10) | — | — |
| 2 | –Cl | DMSO (30) | — | — |
| 3 | BF4− | — | 90 | >99:1 |
| 4e | BF4− | — | 43 | >99:1 |
| 5 | –Br | — | 68 | >99:1 |
| 6 | –I | — | — | — |
| 7 | –OH | — | — | — |
Previously,19 we showed that the hexameric capsule C6 was able to promote a Friedel–Crafts benzylation by a catalytically relevant H-bonding interaction between the bridged water molecules of the capsule and benzyl chloride, which was fundamental for the activation of the C–Cl bond in the substrate.19 Based on these findings, we postulate that the bridged water molecules of the capsule C6 establish an analogous H-bonding interaction, Tr–Cl⋯H–OH (C6), that is fundamental for the activation of the C–Cl bond of 2a and the formation of the trityl cation (Tr+, 3+) (vide infra, Fig. S78†).
To corroborate the catalytic role of this H-bonding interaction, we studied the reaction in Scheme 1 using trityl halides (TrX, X = Br and I) with halogen atoms of different H-bonding acceptor abilities. Interestingly, when 4 was reacted with 5a in the presence of hexameric capsule C6 and trityl bromide 2b, product 6a was obtained with a lower yield (68% yield, Table 2, entry 5), while no conversion to 6a was observed in the presence of trityl iodide 2c because the iodine atom in 2c is a weaker H-bond acceptor.22 Analogously, no hint of product 6a was observed performing the reaction in Scheme 1 in the presence of trityl alcohol 2d as the cofactor (Table 2, entry 7). These results clearly indicated that the C6 capsule is more effective in abstracting the chloride anion over the bromide and iodide anions from the corresponding trityl halide, whereas it is incapable of detaching the OH group. Entries 3 and 4 in Table 2 clearly show that C6 is catalytically active also in the presence of a preformed trityl cation, such as in salt 3+·BF4−. In fact, this salt was an active catalyst by itself (43% yield, Table 2, entry 4), but gave superior performances in the presence of the C6 capsule (90% yield, Table 2, entry 3).
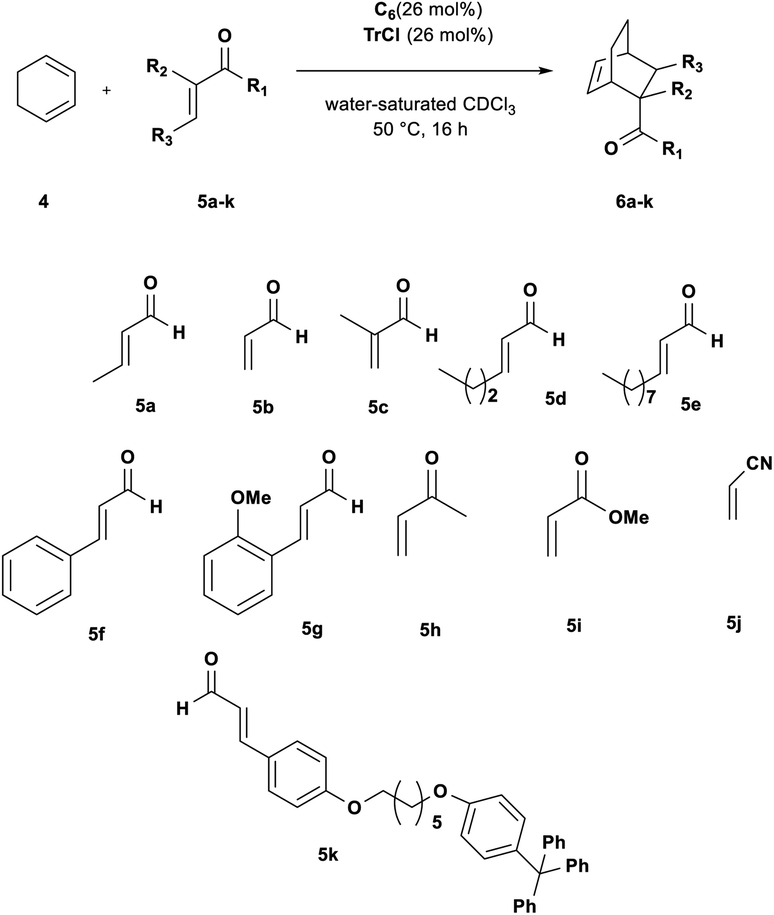
At this point, we investigated the substrate scope of the DA reaction promoted by C6 exploring a variety of carbonyl compounds (Table 3). Under the standard reaction conditions, the procedure proved to be compatible with most structurally distinct substrates, affording the corresponding products in moderate to high yields. The reaction of both aldehydes 5a and 5c with diene 4 to give the products 6a and 6c in the presence of the capsule was slower than the reaction of acrolein 5b (see also Fig. 2), but at prolonged reaction time, all of them reached full conversion and no evidence of diene decomposition was detected.4a
|
|
||||
|---|---|---|---|---|
| Entrya | 5 | Conv.b (%) | 6 yieldc (%) |
endo:exod |
| a Reaction conditions: 4 (0.45 M), 5 (0.15 M), C6 and TrCl (0.039 M) in 1.1 mL of water-saturated CDCl3, at 50 °C for 16 h. All aldehydes showed no background reaction in the absence of C6 and TrCl under reaction conditions. b Determined by 1H NMR analysis of the crude reaction mixture. c Isolated yield. d Determined by 1H NMR analysis of the crude reaction mixture according to literature data.21 | ||||
| 1 | 5a | 100 | 92 | >99:1 |
| 2 | 5b | 100 | 85 | >99:1 |
| 3 | 5c | 100 | 88 | >99:1 |
| 4 | 5d | 78 | 72 | >99:1 |
| 5 | 5e | 47 | 46 | >99:1 |
| 6 | 5f | 55 | 47 | >99:1 |
| 7 | 5g | 67 | 67 | >99:1 |
| 8 | 5h | 100 | 87 | >99:1 |
| 9 | 5i | — | — | — |
| 10 | 5j | — | — | — |
| 11 | 5k | — | — | — |
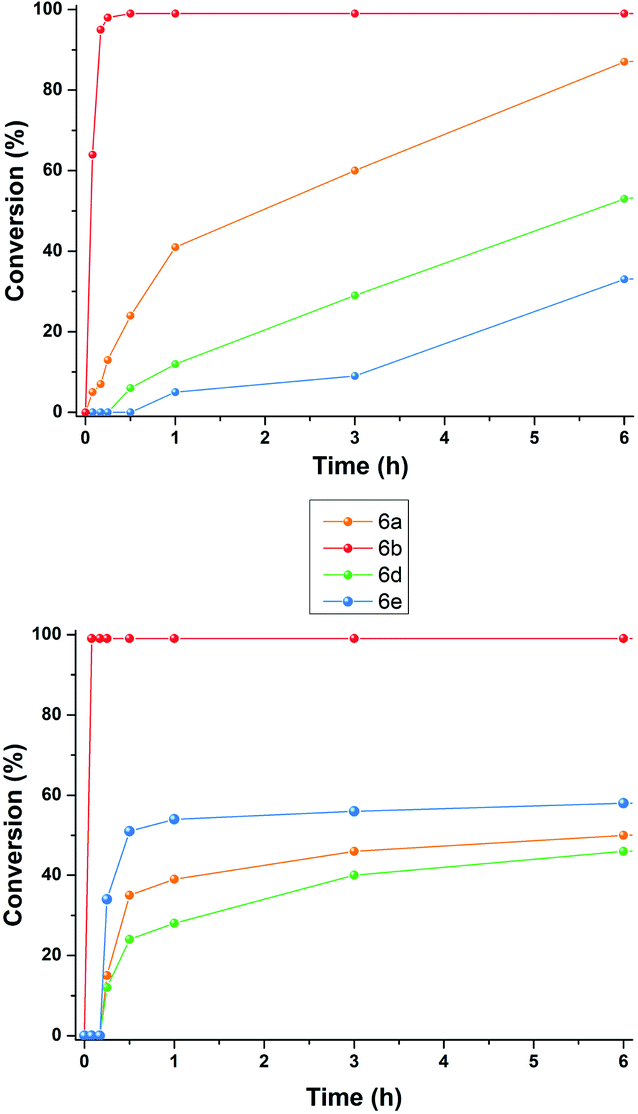 | ||
| Fig. 2 Reaction progress for DA reaction between 4 and aldehydes 5a–e in the presence of: (top) 2a (26 mol%) and (26 mol%); (bottom) 3+·BF4−. | ||
Interestingly, the data reported in Table 3 clearly showed that the efficiency of the DA reaction between 4 and enals 5a–j in the presence of C6 depended on the length of the R3 alkyl chain in their β-position.
In particular, as chain length increased from 5a (R3 = Me, Table 3, entry 1) to 5d (R3 = n-C3H9, Table 3, entry 4) and 5e (R3 = n-C8H17, Table 3, entry 5), the conversion to 6x greatly decreased from 92 to 46%.
With these results in hand, we studied and monitored the DA reaction between cyclohexadiene 4 with aldehydes 5a, 5d, and 5e in the presence of preformed trityl fluoroborate salt 3+·BF4− without capsule C6 (Fig. 2, bottom). 1H NMR analysis of DA reactions over time showed that the reactions catalyzed by the preformed trityl fluoroborate salt and in the absence of C6 led to the DA adducts 6a, 6d, and 6e, in moderate and comparable yields after 16 h, regardless of the alkyl chain length of the R3 substituent. On the other hand, in the presence of 2a and C6, the adducts 6a and 6d were formed faster than 6e (Fig. 2, top). These results confirm that the reaction occurs within the cavity of C6 and the nano-environment of the capsule is sensitive to the different steric dimensions of the aldehydes.
Probably, C6 imposes a barrier for the entrance of sterically hindered aldehydes, thus making their reaction relatively slow compared to the other ones. These results indicate that the capsule C6 is able to exert a substrate selectivity toward the substrates 5a, b, d, and e.13–17
In fact, in agreement with these considerations, and with the results previously reported by Franzén and coworkers,4 trityl cation is temperature, air-, and moisture-sensitive, consequently, when the DA reaction was performed in the presence of cyclohexadiene and larger aldehyde such as 5d and 5e, which show slower kinetic of complexation inside C6, then the diene decomposition is favoured thus stopping the DA reaction and resulting in lower conversions. In fact, when only cyclohexadiene is added to a solution of trityl cation in dichloromethane complete decomposition or polymerization of the diene is observed.4 In summary, under our reaction conditions, once trityl cation is formed from TrCl inside C6, if the aldehyde had kinetic difficulty getting into the capsule, then after prolonged reaction times we observed full consumption of the diene by its polymerization and the appearance of trityl alcohol. In addition, it is interesting to highlight that the encapsulated carbocation derived from 2a@C6 was also effective for less-activated cinnamic aldehydes 5f and 5g, under the general reaction conditions, affording the corresponding cycloadducts in moderate yields (Table 3, entries 6 and 7). The reaction of cinnamic aldehydes with cyclohexadiene 4 is not easy; different unsuccessful attempts have been reported in the literature characterized by the formation of complex mixtures of by-products due to the diene decomposition or polymerization.23,24 Furthermore, Franzén and coworkers reported that the reaction does not work in the presence of preformed trityl fluoroborate salt 3+·BF4−,4a but it is necessary to use a less active and more stable trityl cation, such as (p-MeOPh)3CBF4, in order to obtain the adducts in moderate yield.
In our case, the C6 capsule can gradually provide the trityl carbocation in the reaction mixture in situ, thus avoiding diene side-reactions due to too long reaction times with less reactive dienophiles and tuning the Lewis acid properties of the trityl cation catalyst according to the reactivity of substrates. However, in accordance with literature data,4a the reaction failed with dienophiles such as acrylonitrile and methyl acrylate.
To extend the scope of our system, we carried out further studies with two other dienes with different reactivity,25 such as open diene 2,3-dimethylbutadiene 7 and cyclopentadiene 9.
The reactions with diene 7 proved to be more sensitive than those with diene 4 to the size of aldehydes.
The DA reactions with aldehydes 5a–c afforded the corresponding adducts in high yields and selectivity just running at room temperature (Table 4, entries 1–3). A significant drop in reactivity was observed with sterically hindered aldehydes accompanied by a total consumption of more reactive diene 7 through polymerization (Table 4, entries 4–8). Increasing the reaction temperature and prolonging the reaction time did not improve the reaction efficiency. Interestingly, the corresponding reaction with OMe ortho-substituted cinnamic aldehyde 5g led to better conversion, probably because it is preferentially encapsulated inside C6. Uptake NMR studies clearly showed that 5g is encapsulated inside C6 to a greater extent than 5f, and consequently, higher efficiency was observed for the conversion of 5g in the product (see Fig. S25–S32†). We also examined the reaction with α,β-unsaturated ketone 5h. Methyl vinyl ketone 5h showed a reactivity comparable with acrolein, and the desired cycloadduct was afforded quantitatively (Table 4, entry 9).
|
|
|||
|---|---|---|---|
| Entrya | 5 | Conv.b (%) | 8 yieldc (%) |
| a Reaction conditions: 7 (0.45 M), 5 (0.15 M), C6 and TrX (0.039 M) in 1.1 mL of water-saturated CDCl3, at rt for 16 h. b Determined by 1H NMR analysis of the crude reaction mixture according to literature data.21a,b,26 c Isolated yield. d The reaction was carried out at 50 °C. | |||
| 1 | 5a | 82 | 75 |
| 2 | 5b | 100 | 82 |
| 3 | 5c | 80 | 73 |
| 4 | 5d | 32 | 27 |
| 5 | 5e | 7 | 5 |
| 6 | 5f | 16 | 16 |
| 7d | 5f | 34 | 27 |
| 8 | 5g | 61 | 60 |
| 9 | 5h | 100 | 92 |
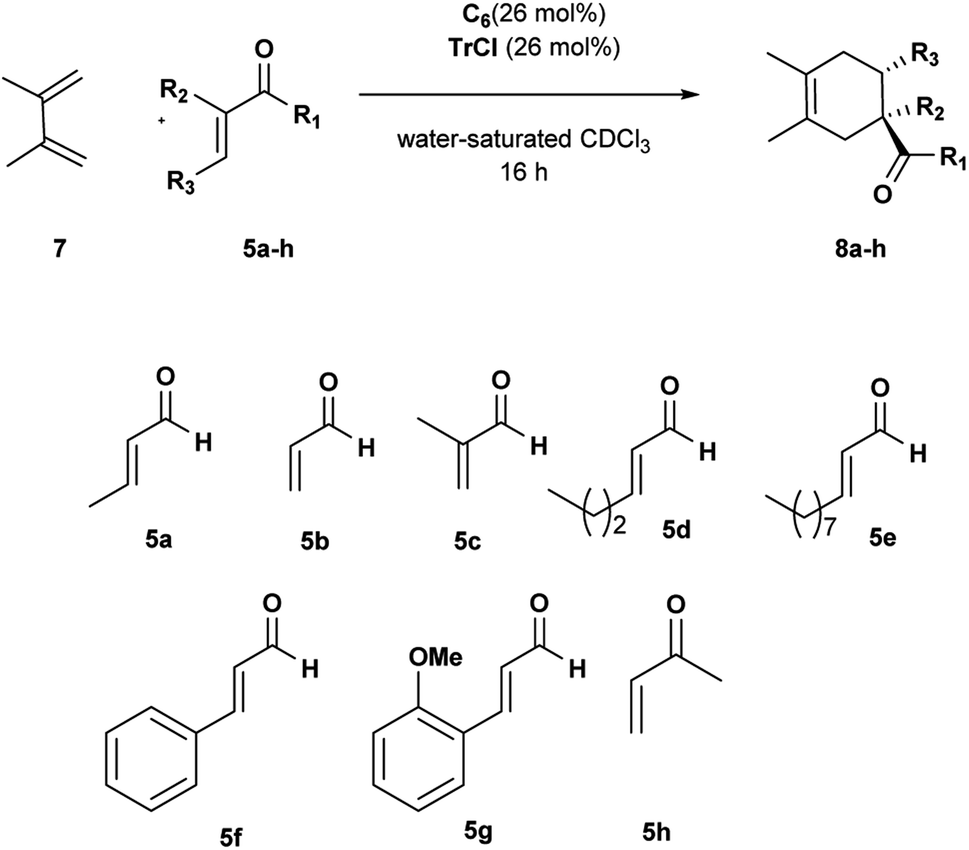
Changing the diene to more reactive cyclopentadiene 9 had a significant influence on the rate and efficiency of the reaction. Aldehydes 5a–c all reached full conversion in 1 h (Table 5, entries 1, 3 and 6); by increasing the size of the R3 group in aldehydes 5d and 5e a size selectivity could be initially detected but extending the reaction time to 16 h the adducts were obtained in high conversion for both aldehydes (Table 5, entries 7 and 8).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | 5 | t (h) | Conv.b (%) | 10 yieldc (%) |
endo:exod |
| a Reaction conditions: 9 (0.45 M), 5 (0.15 M), C6 and TrCl (0.039 M.) in 1.1 mL of water-saturated CDCl3, at rt. b Determined by 1H NMR analysis of the crude reaction mixture. c Isolated yield. d Determined by 1H NMR analysis of the crude reaction mixture according to literature data.21b,21d,27 e The reaction was performed in the absence of C6 and TrCl. | |||||
| 1 | 5a | 1 | 100 | 90 | 86:14 |
| 2e | 5a | 1 | — | — | — |
| 3 | 5b | 1 | 100 | Quant | 86:14 |
| 4e | 5b | 1 | 9 | — | — |
| 5e | 5b | 3 | 33 | 27 | 77:23 |
| 6 | 5c | 1 | 100 | Quant | 2:98 |
| 7 | 5d | 4 | 94 | 90 | 69:25 |
| 16 | 100 | 96 | 77:23 |
||
| 8 | 5e | 4 | 78 | 72 | 84:16 |
| 16 | 95 | 91 | 84:16 |
||
| 9 | 5f | 4 | 54 | 50 | 91:9 |
| 16 | 72 | 68 | 89:11 |
||
| 10 | 5g | 4 | 37 | 34 | 84:16 |
| 16 | 61 | 60 | 80:20 |
||
| 11 | 5h | 2 | Quant | 93 | >99:1 |
Thus, we subsequently evaluated the reaction with cinnamic aldehydes 5f and 5g. In a similar fashion to the reaction with 4, both substrates gave the target products in satisfactory yields confirming the feasibility of the reaction (Table 5, entries 9 and 10). Finally, also ketone 5h was found to be a very good dienophile for this reaction giving cycloadduct 8h in high yield (Table 5, entry 11).
To further investigate the reaction mechanism that leads to the peculiar catalytic role played by the hexameric capsule C6 (Scheme 1), we conducted an in silico study by employing both molecular dynamics (MD) simulations and quantum-mechanical (QM) investigations.
In this last case, we employed a reduced model capsule CM assembled with the C-methylresorcinarene and the ONIOM method. Initially, to corroborate our findings on the catalytic role of the capsule as a Lewis acid, we studied the encapsulation of trityl chloride 2a and its ionized form to give the (Tr+·Cl−@CM) complex. Interestingly, in the most stable structure of 2a@CM (Fig. S78†), the chlorine atom of 2a engages a hydrogen bond with a structural water molecule, which weakens the C–Cl bond making it significantly longer.
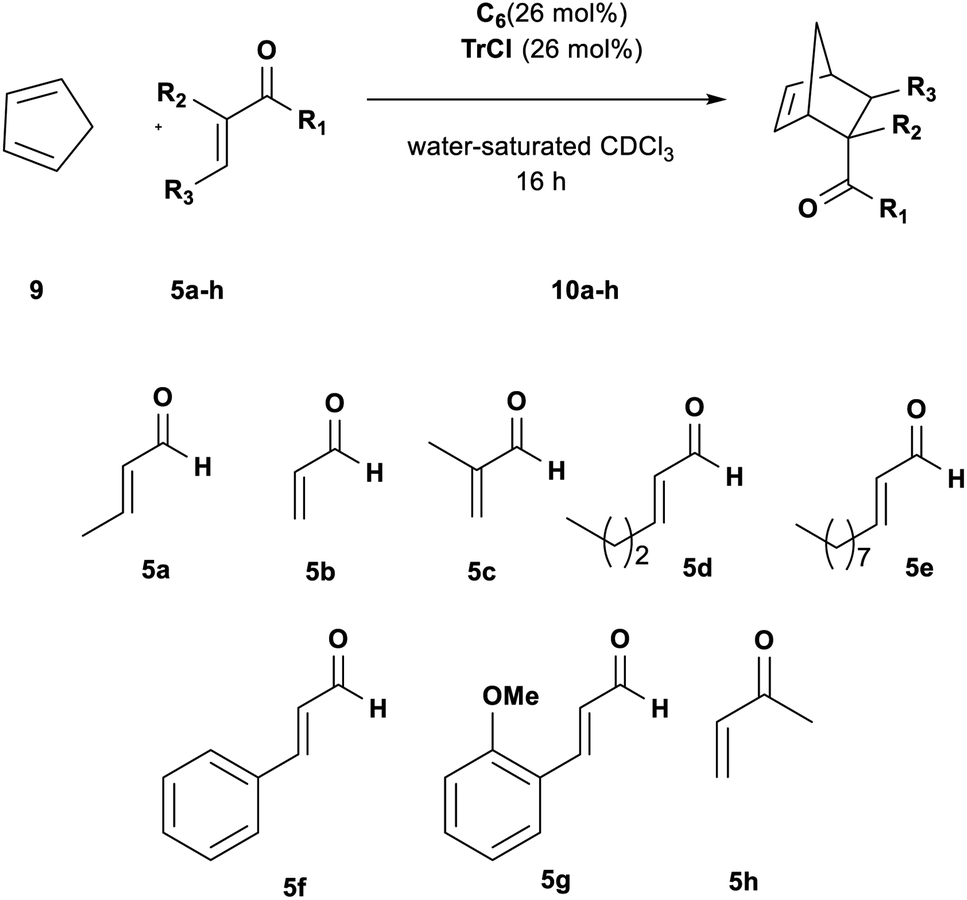
The most stable structure of the Tr+·Cl−@CM complex was found through an MD study of 2 μs, at the molecular mechanics (MM) level with chloroform as the explicit solvent, starting from the structure with the trityl carbocation inside the capsule and the chloride anion outside, near a structural water molecule. Finally, the most stable structure obtained by analyzing the MD trajectory was more stable than the reagents (ΔG = −1.14 kcal mol−1), showing the peculiarity of possessing the chloride anion replacing a structural water molecule.28 The replaced H2O molecule was anchored to the internal wall of the capsule through two hydrogen bonds with another structural water molecule and an oxygen atom of a resorcinol moiety (Fig. 3, top).
 | ||
| Fig. 3 The most stable structure of ionized trityl chloride inside the hexameric resorcinarene capsule (Tr+·Cl−@CM). (Top) Balls and sticks refer to the QM level. (Bottom) Bottom view regarding the plane that cuts the capsule parallel to the trityl cation longitudinal axes; the dots and dashed-lines in purple refer to CH–π interactions. | ||
In this structure, the trityl cation is in its catalytically active form and is nested in one side of the capsule with its three phenyl rings pointing towards the inner cavity of three resorcin[4]arene macrocycles (Fig. 3, bottom) and establishing stabilizing CH–π and π–π interactions.
MD simulations with the other trityl halides and TrBF4 brought approximatively the same structure obtained with the chloride anion.
The Gibbs free energy of inclusion (ΔG) and the electrophilicity index (ω) of the investigated compounds are reported in Table 6. Except for TrF, all other trityls easily enter into the capsule, and the electrophilicity indexes of their complexes are comparable to that of free TrBF4, indicating that they can behave as Lewis acids (in contrast, the electrophilicity indexes of all free trityl halides are in the range of 1.11–1.52, indicating that they are poor electrophiles).
The explanation of the different reactivity observed for TrX (X = Cl, Br, I, BF4) comes from the MD studies. All TrX@CM complexes, starting with the counterion of the trityl outside the capsule, incorporate the anion within 2 ns in the position previously occupied by a structural water molecule, with the exception of iodide that employs about 80 ns. Interestingly, TrCl@CM and TrBF4@CM complexes remain stable for the entire simulation period (2 μs), whereas TrBr@CM and TrI@CM become unstable at around 50 and 82 ns (just 2 ns after acquiring the adequate catalytic power), respectively, and then the network of hydrogen bonds begins to falter, leading to the opening of the capsule. This is ideally in accordance with the high yields observed for X = Cl and BF4, and for the moderate yield obtained with X = Br. In the case of X = I, the complex collapses very quickly and does not give rise to the cycloaddition reaction. Subsequently, we studied, at the QM/SE level, the DA reaction inside the capsule using as a model reaction the TrCl@CM complex, cyclohexa-1,3-diene 4, and (E)-crotonaldehyde 5a. At the same time, we investigated the same reaction using TrBF4 in the absence of the capsule (Scheme 2). The relative Gibbs free energies and the transferred charges for the species depicted in Scheme 2 are reported in Table 6, whereas all energetic parameters are given in Table S1.† The endo/exo TS energy difference in both reactions is high (2.59 and 2.63 kcal mol−1) and in accordance with the experimentally observed endo/exo ratio.
 | ||
| Scheme 2 Endo and exo routes for the in silico studies of the model DA reactions and nomenclature adopted: TS-endo and TS-exo refer to the reaction with TrBF4, while TS-endo@CM and TS-exo@CM refer to the reaction with TrCl and the capsule. | ||
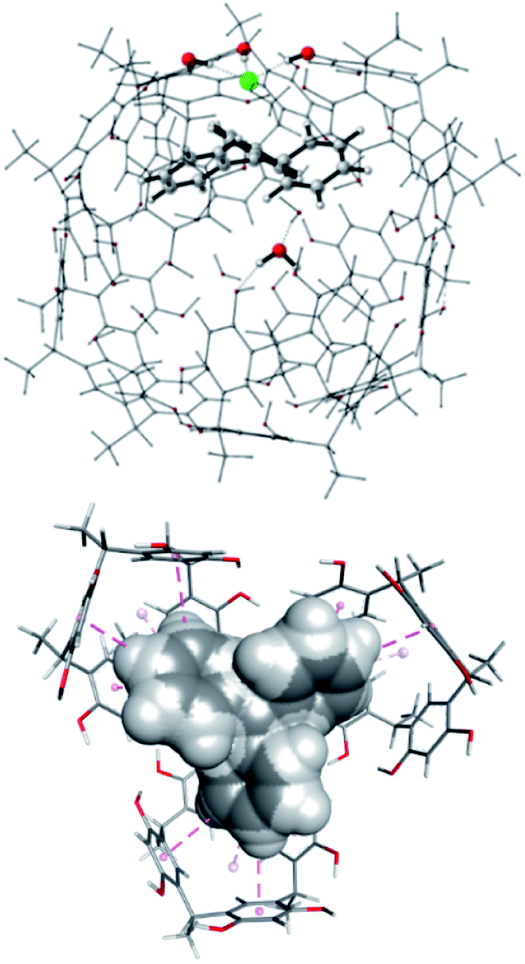
Although the TS energy barrier is highest for the reaction inside the capsule (and this justifies the improvement of the yield observed at 50 °C), the yield and the course of the reaction worsen in its absence (vide infra for a possible explanation). In both the model reactions, we were unable to locate a concerted transition state due to the high positive charge that develops on the carbon beta to the dienophile during its complexation with the trityl carbocation. Effectively, the DA reactions at hand proceed with a stepwise mechanism in which the first step consists of a Michael-type addition of the diene to the β-carbon of the dienophile, whereas in the second one, barrierless in a vacuum, the reaction proceeds by joining the two carbon atoms bearing the developing opposite partial charges.
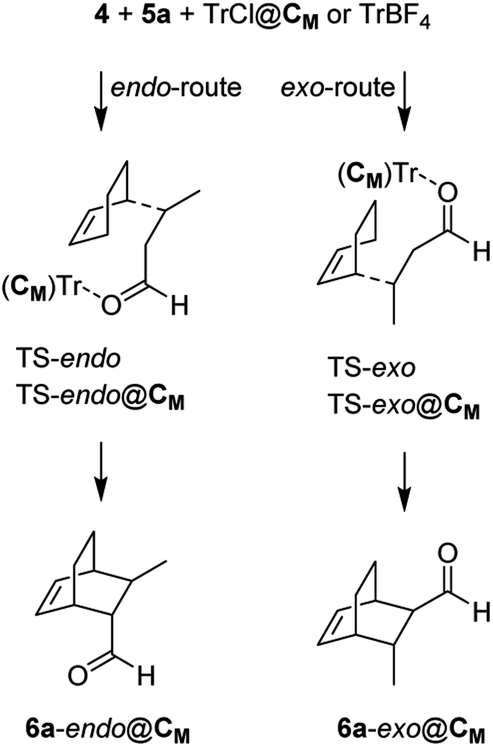
Interestingly, the two reactions proceed with a different geometry arrangement with respect to the trityl carbocation and the respective counteranions, as depicted in Fig. 4 that report the 3D structures of TS-endo and TS-endo@CM.
 | ||
| Fig. 4 Geometries for the most energetically favored TS-endo (DA with TrBF4 without the capsule, left) and TS-endo@CM (DA with TrCl and the capsule, right) routes (only at the QM level). Lengths are in Å. Carried out with CYLview.29 | ||
This behavior could justify the differences in the efficiency of the reactions with TrBF4 without and with the capsule. When BF4− is from the same side of the diene and the dienophile probably hinders the progress of the reaction. In contrast, the presence of the capsule precludes the possibility of making the reaction take place from the same side of the anion, as the space is not enough. Moreover, in this last case, the trityl cation is firmly anchored thanks to the phenyls that point inside the cavity of the resorcinarenes and, under these conditions, its electrophilicity index increase by 0.21 eV (Table 6). Noteworthily, the structural water molecule that in TrCl@CM was anchored to the capsule wall now has moved to the trityl cation and establishes two OH–π interactions with two phenyls, and in all TSs, the (E)-crotonaldehyde is in s-cis conformation (Fig. 4, right). Finally, both the TS-endo and TS-exo are more advanced with respect to the corresponding TS-endo@CM and TS-exo@CM, which is in accord with the lower charge transfer found in the latter (Table 7).
Presumably, the presence of the capsule and the displacement of the structural water molecule anchored to its internal wall towards the phenyls of trityl mitigates the ionicity of the transition state while failing to make the DA reaction concerted.
Conclusions
In conclusion, we have herein reported that the DA reaction can occur in the nanoconfined space of the hexameric resorcinarene capsule C6, and the capsule is able to promote the reaction through the activation of the dienophile by a carbocation catalyst generated in situ. C6 can form an active trityl ion from TrCl as a carbocation precursor through the cleavage of the carbon–halogen bond promoted by OH⋯X− hydrogen bonding. We believe that the combination of the nanoconfined space and the latent carbocation catalysis would provide a complementary strategy to the typical carbocation catalysis. The latent strategy bypasses the typical pitfalls associated with active carbocations and provides control of the reaction efficiency in terms of reaction rate, conversion, and selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Regione Campania (POR CAMPANIA FESR 2007/2013 O.O.2.1, CUP B46D14002660009) for the FT-ICR mass spectrometer facilities and the “Centro di Tecnologie Integrate per la Salute” (CITIS) (project PONa3_00138) for the 600 MHz NMR facilities. Financial support from the University of Salerno (FARB) is acknowledged.Notes and references
- G. A. Olah, Angew. Chem., Int. Ed., 1995, 34, 1393–1405 CrossRef CAS.
- M. Horn and H. Mayr, J. Phys. Org. Chem., 2012, 25, 979–988 CrossRef CAS , and references therein..
- G. A. Olah, J. Am. Chem. Soc., 1972, 94, 808–820 CrossRef CAS.
- (a) J. Bah and J. Franzén, Chem.–Eur. J., 2014, 20, 1066–1072 CrossRef CAS PubMed; (b) J. Bah, V. R. Naidu, J. Teske and J. Franzén, Adv. Synth. Catal., 2015, 357, 148–158 CrossRef CAS; (c) V. R. Naidu, J. Bah and J. Franzén, Eur. J. Org. Chem., 2015, 2015, 1834–1839 CrossRef; (d) V. R. Naidu, S. Ni and J. Franzén, Chem. Cat. Chem., 2015, 7, 1896–1905 CAS.
- (a) R. Properzi, P. S. J. Kaib, M. Leutzsch, P. Gabriele, M. Raja, C. K. De, L. Song, P. R. Schreiner and B. List, Nat. Chem., 2020, 12, 1174–1179 CrossRef CAS PubMed; (b) X. Tang, W. Chen, X. Yi, Z. Liu, Y. Xiao, Z. Chen and A. Zheng, Angew. Chem., Int. Ed., 2021, 60, 4581–4587 CrossRef CAS PubMed.
- (a) A. Brunner, S. Taudien, O. Riant and H. B. Kagan, Chirality, 1997, 9, 478–486 CrossRef CAS; (b) S. Taudien, O. Riant and H. B. Kagan, Tetrahedron Lett., 1995, 36, 3513–3582 CrossRef CAS; (c) S. Ni, V. R. Naidu and J. Franzén, Eur. J. Org. Chem., 2016, 9, 1708–1713 CrossRef; (d) Q. Zhang, J. Lv, S. Li and S. Luo, Org. Lett., 2018, 20, 2269–2272 CrossRef CAS PubMed.
- (a) S. Kobayashi, S. Murakami and T. A. Mukaiyama, Chem. Lett., 1985, 14, 447–450 CrossRef; (b) S. Kobayashi, M. Murakami and T. Mukaiyama, Chem. Lett., 1985, 14, 1535–1538 CrossRef; (c) S. E. Denmark and C.-T. Chen, Tetrahedron Lett, 1994, 35, 4327–4330 CrossRef CAS.
- T. Mukaiyama, H. Nagaoka, M. Murakami and M. Ohshima, Chem. Lett., 1985, 14, 977–980 CrossRef.
- S. Kobayashi, M. Murakami and T. Mukaiyama, Chem. Lett., 1985, 14, 953–956 CrossRef.
- S. Ni, M. A. A. El Remaily and J. Franzén, Adv. Synth. Catal., 2018, 360, 4197–4204 CrossRef CAS.
- S. Ni and J. Franzén, Chem. Commun., 2018, 54, 12982–12985 RSC.
- (a) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS; (b) A. Shivanyuk and J. Rebek, J. Am. Chem. Soc., 2003, 125, 3432–3433 CrossRef CAS PubMed; (c) L. Avram and Y. Cohen, Org. Lett., 2002, 4, 4365–4368 CrossRef CAS PubMed.
- (a) D. Ajami and J. Rebek Jr, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed; (b) L. Catti, Q. Zhang and K. Tiefenbacher, Chem.–Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed; (c) G. Borsato, J. Rebek Jr and A. Scarso, in Selective Nanocatalysts and Nanoscience Concepts for Heterogeneous and Homogeneous Catalysis, ed. A. Zecchina, S. Bordiga and E. E. Groppo, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, pp. 105–168 Search PubMed; (d) L. Avram, Y. Cohen and J. Rebek Jr, Chem. Commun., 2011, 47, 5368–5375 RSC; (e) G. Borsato and A. Scarso, in Organic Nanoreactors, ed. S. Sadjadi, Academic Press, 2016, ch. 7, pp. 203–234 Search PubMed; (f) L. Catti, T. Brauer, Z. Qi and K. Tiefenbacher, Chimia, 2016, 70, 810–814 CrossRef CAS PubMed; (g) Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS PubMed; (h) C. Gaeta, C. Talotta, M. De Rosa, P. La Manna, A. Soriente and P. Neri, Chem.–Eur. J., 2019, 25, 4899–4913 CrossRef CAS PubMed; (i) C. Gaeta, P. La Manna, M. De Rosa, A. Soriente, C. Talotta and P. Neri, Chem. Cat. Chem., 2020, 13, 1638–1658 Search PubMed; (j) C. Gaeta, C. Talotta, M. De Rosa, P. La Manna, A. Soriente and P. Neri, in Reactivity in Confined Spaces, ed. R. S. Forgan and G. O. Lloyd, The Royal Society of Chemistry, 2021, ch. 5, pp. 133–166 Search PubMed.
- (a) T. M. Bräuer, Q. Zhang and K. Tiefenbacher, Angew. Chem., Int. Ed., 2016, 55, 7698–7701 CrossRef PubMed; (b) P. La Manna, M. De Rosa, C. Talotta, C. Gaeta, A. Soriente, G. Floresta, A. Rescifina and P. Neri, Org. Chem. Front., 2018, 5, 827–837 RSC; (c) S. Gambaro, C. Talotta, P. Della Sala, A. Soriente, M. De Rosa, C. Gaeta and P. Neri, J. Am. Chem. Soc., 2020, 142, 14914–14923 CrossRef CAS PubMed.
- (a) Q. Zhang, J. Rinkel, B. Goldfuss, J. S. Dickschat and K. Tiefenbacher, Nat. Catal., 2018, 1, 609–615 CrossRef CAS PubMed; (b) Q. Zhang and K. Tiefenbacher, Angew. Chem., Int. Ed., 2019, 58, 12688–12695 CrossRef CAS PubMed.
- (a) Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2013, 135, 16213–16219 CrossRef CAS PubMed; (b) J. M. Köster and K. Tiefenbacher, Chem. Cat. Chem., 2018, 10, 2941–2944 Search PubMed.
- (a) S. Gambaro, M. De Rosa, A. Soriente, C. Talotta, G. Floresta, A. Rescifina, C. Gaeta and P. Neri, Org. Chem. Front., 2019, 6, 2339–2347 RSC; (b) P. La Manna, C. Talotta, M. De Rosa, A. Soriente, C. Gaeta and P. Neri, Org. Lett., 2020, 22, 2590–2594 CrossRef CAS PubMed.
- P. La Manna, M. De Rosa, C. Talotta, A. Rescifina, G. Floresta, A. Soriente, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2020, 59, 811–818 CrossRef CAS PubMed.
- P. La Manna, C. Talotta, G. Floresta, M. De Rosa, A. Soriente, A. Rescifina, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2018, 57, 5423–5428 CrossRef CAS PubMed.
- (a) T. Shida in Electronic Absorption Spectra of radical Ions, Elsevier, Amsterdam, The Netherlands, 1988 Search PubMed; (b) R. Rathore, C. L. Burns and I. A. Guzei, J. Org. Chem., 2004, 69, 1524–1530 CrossRef CAS PubMed.
- (a) Z. Zhu and J. H. Espenson, J. Am. Chem. Soc., 1997, 119, 3507–3512 CrossRef CAS; (b) H. F. T. Klare, K. Bergander and M. Oestreich, Angew. Chem., Int. Ed., 2009, 48, 9077–9079 CrossRef CAS PubMed; (c) R. K. Schmidt, K. Müther, C. Mück-Lichtenfeld, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2012, 134, 4421–4428 CrossRef CAS PubMed; (d) E. Gould, T. Lebl, A. M. Z. Slawin, M. Reid, T. Daviesb and A. D. Smith, Org. Biomol. Chem., 2013, 11, 7877–7892 RSC.
- G. R. Desiraju and T. Steiner, The weak hydrogen bond: in structural chemistry and biology, Oxford University Press, Oxford; New York, 1999 Search PubMed.
- A. C. Kinsman and M. Kerr, Org. Lett., 2000, 2, 3515–3520 CrossRef PubMed.
- T. Arndt, P. K. Wagner, J. J. Koenig and M. Breugst, Chem. Cat. Chem., 2021, 13, 2922–2930 CAS.
- (a) R. S. Paton, S. Kim, A. G. Ross, S. J. Danishefsky and K. N. Houk, Angew. Chem., Int. Ed., 2011, 50, 10366–10368 CrossRef CAS PubMed; (b) B. J. Levandowski and K. N. Houk, J. Org. Chem., 2015, 80, 3530–3537 CrossRef CAS PubMed.
- (a) F. Fu, Y.-C. Teo and T.-P. Loh, Org. Lett., 2006, 8, 5999–6001 CrossRef CAS PubMed; (b) E. Taarning and R. Madsen, Chem.–Eur. J., 2008, 14, 5638–5644 CrossRef CAS PubMed.
- (a) K. Ishihara, H. Kurihara, M. Matsumoto and H. Yamamoto, J. Am. Chem. Soc., 1998, 120, 6920–6930 CrossRef CAS; (b) H. Gotoh and Y. Hayashi, Org. Lett., 2007, 9, 2859–2862 CrossRef CAS PubMed; (c) H. He, B.-J. Pei, H.-H. Chou, T. Tian, W.-H. Chan and A. W. M. Lee, Org. Lett., 2008, 10, 2421–2424 CrossRef CAS PubMed.
- S. Merget, L. Catti, G. M. Piccini and K. Tiefenbacher, J. Am. Chem. Soc., 2020, 142, 4400–4410 CrossRef CAS PubMed.
- C. Y. Legault, CYLview20, Université de Sherbrooke, 2020, http://www.cylview.org Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, in silico study, and characterisation for all compounds. See https://doi.org/10.1039/d2sc02901d |
| This journal is © The Royal Society of Chemistry 2022 |