
Recent advances in biomaterial-assisted cell therapy
Yu
Chen
abc,
Samira
Pal
a and
Quanyin
Hu
 *abc
*abc
aPharmaceutical Sciences Division, School of Pharmacy, University of Wisconsin-Madison, Madison, WI 53705, USA. E-mail: qhu66@wisc.edu
bCarbone Cancer Center, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI 53705, USA
cWisconsin Center for NanoBioSystems, School of Pharmacy, University of Wisconsin-Madison, Madison, WI 53705, USA
First published on 25th May 2022
Abstract
With the outstanding achievement of chimeric antigen receptor (CAR)-T cell therapy in the clinic, cell-based medicines have attracted considerable attention for biomedical applications and thus generated encouraging progress. As the basic construction unit of organisms, cells harbor low immunogenicity, desirable compatibility, and a strong capability of crossing various biological barriers. However, there is still a long way to go to fix significant bottlenecks for their clinical translation, such as facile preparation, strict stability requirements, scale-up manufacturing, off-target toxicity, and affordability. The rapid development of biotechnology and engineering approaches in materials sciences has provided an ideal platform to assist cell-based therapeutics for wide application in disease treatments by overcoming these issues. Herein, we survey the most recent advances of various cells as bioactive ingredients and outline the roles of biomaterials in developing cell-based therapeutics. Besides, a perspective of cell therapies is offered with a particular focus on biomaterial-involved development of cell-based biopharmaceuticals.
 Yu Chen | Yu Chen received his Master's degree in medicinal chemistry from China Pharmaceutical University in 2017. And he obtained his PhD degree in Pharmaceutics from Fudan University in 2021. He then worked in Dr Quanyin Hu's lab as a postdoctoral fellow at the School of Pharmacy, University of Wisconsin-Madison. His research interest is to leverage chemical biology for cell-based therapeutics. |
 Quanyin Hu | Quanyin Hu is an Assistant Professor at the School of Pharmacy, University of Wisconsin–Madison (UW-Madison). He received his PhD degree in biomedical engineering at the University of North Carolina at Chapel Hill (UNC-CH) and North Carolina State University. Before he joined UW-Madison, he was a postdoc associate at the Koch Institute for Integrative Cancer Research at the Massachusetts Institute of Technology (MIT). His research group focuses on drug delivery, cell therapy, immunotherapy, and personalized therapy. |
1. Introduction
Cell therapy has been receiving increasing attention as a rapidly developing and emerging field in translational medicine.1,2 Cells, the basic functional units of living organisms, are natural producers and carriers of proteins and molecules.3 By exerting complex mechanisms to avoid immune system attack, cells are highly biocompatible with the capability of low toxicity, crossing various biological barriers and homing to specific tissues, thus modulating the pathological environment.4,5 Compared to small molecule drugs or biologics, cell therapies can achieve more integrated and complex functions such as self-proliferation, differentiation, and direct elimination/repair of diseased cells/tissues.6 Also, given the natural tropism and multiple interactions between the cells and diseased microenvironment, cell therapies offer unique advantages in a wide range of diseases.2,7,8In recent years, numerous different types of cell therapies have gradually been developed preclinically or have been approved for clinical trials or even marketing.9 Stem cells and cells derived from their differentiation have become ideal models for guiding the precise treatment of diseases and have shown great potential in various structurally damaging diseases such as spinal cord injury and fibrosis.7,10 The cell research field has been further advanced by the success of chimeric antigen receptor (CAR)-T cell therapy, which is considered a breakthrough in immunotherapy and approved by the U. S. Food and Drug Administration (FDA) to treat hematologic tumors.1 Genetically engineered commensal bacteria have been shown to target tumors and have been applied to treat skin cancer, including melanoma, bringing new ideas to improve cancer research.11,12 The efficacy of cell therapy depends primarily on the rational control and regulation of cell function and behavior in the physiological and pathological environment.13 To obtain therapeutic cell products nowadays, live cells extracted from the donor need to be isolated and purified, genetically modified, and massively amplified before they can be used for subsequent processing, stockpiling, and usage.6 Throughout the therapeutic cycle, maintenance of cell activity, consistency of function, and stability of storage and transportation become significant challenges to meet the clinical demand for cellular products.14
Rapid advances in biotechnologies and engineered materials provide directions to address major challenges in the clinical translation of cell therapies.15 Novel biomaterials can facilitate the clinical translation of cellular products by providing reliable solutions to the obstacles mentioned above.16 With the assistance of engineered biomaterials, it is possible to mimic the in vivo microenvironment and provide a well-defined ecological niche for cells, thus stably producing a cellular phenotype consistent with in vivo function.17 Biomaterials can support specific behaviors such as homing and directed differentiation of certain cells and maintain long-term cellular activity.18 Also, corresponding biomaterials can be constructed to regulate cellular behavior with the help of cellular responsiveness to external specific environments (e.g., composition, charge, stiffness, stress, etc.).19 It is worth emphasizing that the construction of therapeutic cell-biomaterial couples is a promising but challenging approach. During preparation, slight changes in the pH, temperature, ionic strength, and osmotic pressure may significantly affect cell viability and intrinsic function.3 Therefore, by choosing a rational strategy for modifying cells, engineered cells possess new functions conferred by biomaterials and maintain their biological activity to the maximum extent.
This review surveys the latest scientific discoveries and technological innovations in engineering and developing novel biomaterials for adjuvant cell production in various cell therapies such as stem cells, immune cells, and bacteria with detailed examples (Fig. 1). The principles of cell-centered design of adjuvant materials are summarized, including cell surface modification, cell binding, and genetic engineering. Special emphasis is given to applying biomaterials to assist cell therapy in cancer, autoimmune diseases, and tissue regeneration. The prospects and challenges of biomaterial applications in overcoming critical problems in clinical translation of cell therapies under the integration of multidisciplinary theories are also highlighted.
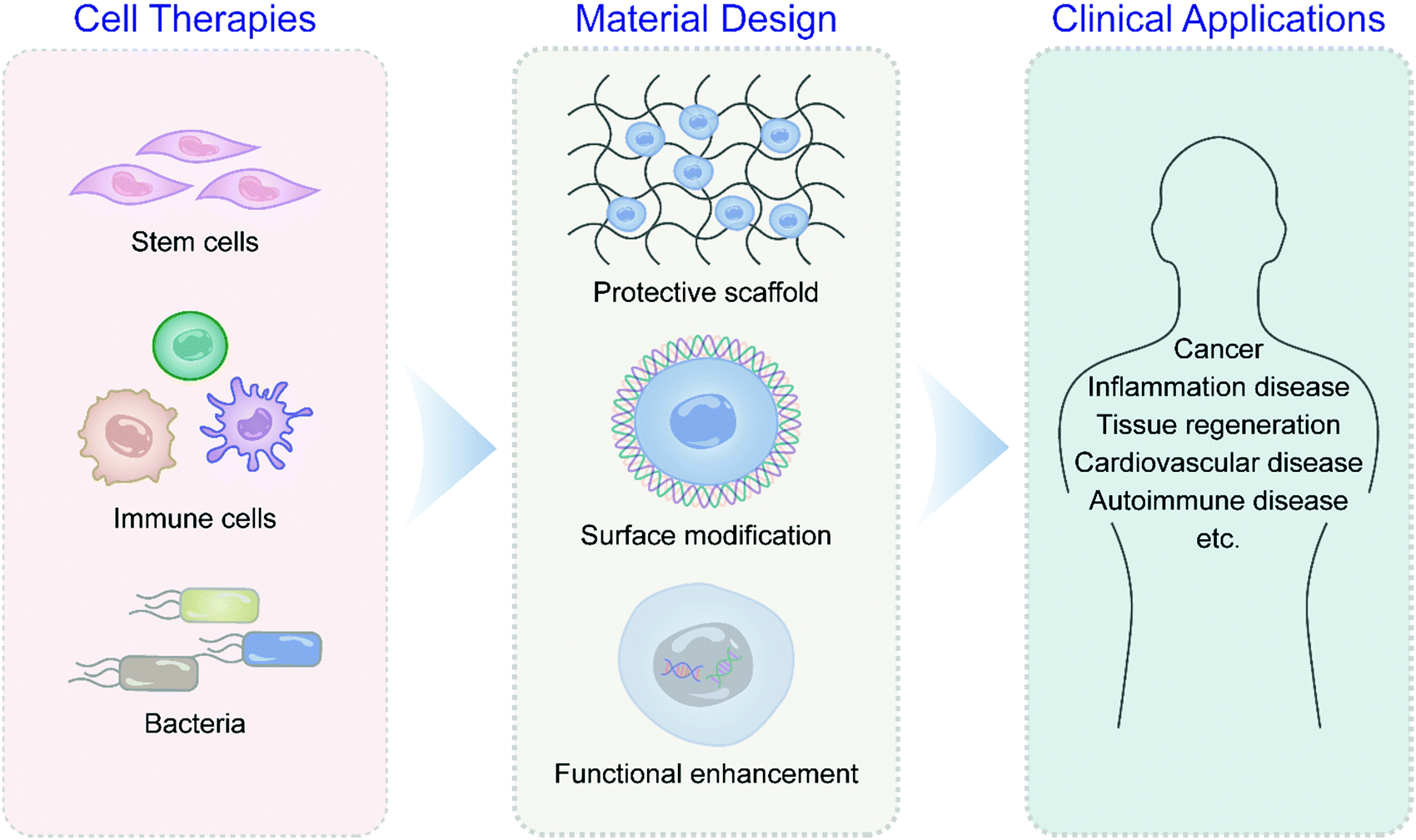 | ||
| Fig. 1 Schematic illustration of biomaterial-assisted immune cell, stem cell, and bacteria therapies for cancers, inflammation diseases, tissue regeneration, cardiovascular diseases and autoimmune diseases. | ||
2. Cell therapies
2.1 Immune cell therapies
2.2 Stem cell therapies
Stem cell therapies hold the potential to revolutionize healthcare by allowing for the regeneration of damaged or dysfunctional tissues, thereby providing treatments for several diseases unsolvable by conventional medicines like muscular dystrophies, diabetes, and neurodegeneration.50–52 Over the last couple of decades, research centered on stem cells has advanced to the use of autologous cells to generate a range of mature cell types and even tiny organs, or organoids ex vivo, by maximizing the regeneration potential of somatic stem cells found in specific tissues, such as bone marrow or skeletal muscle.53 Researchers may currently obtain immature embryonic cells in the terminally differentiated forms by the advent of induced pluripotent stem (iPS) cells.54 These iPS cells can be developed into any cell type found in a given tissue, enabling achievable personalized treatments. These tailored cells can be employed to repair injured tissues and evaluate appropriate medications and treatment recommendations.55 Over the last few years, successful attempts of stem cell therapies have fueled optimism that regenerative medicine tactics would provide a promising treatment regimen for some refractory diseases. It was reported that epidermal stem cells can repair more than 80% of the skin surface area of a young patient with a fatal blistering disorder.56 In other cases, individuals with macular degeneration obtain significantly improved vision after transplanting retinal pigment epithelial cells derived from embryonic stem cells or patient-derived iPS cells into the eye.57 Despite such exciting great achievements, most stem cell clinical trials have failed to receive regulatory approval and commercialization as several challenges preventing the clinical translation of stem cell therapies exist, such as maintenance of the stem cell state, reproducible large-scale expansion of stem cells, uncontrollable in vivo fate, potential tumor formation at the injection site, and harsh storage conditions.532.3 Bacteria therapy
In evolution, microbiota, especially bacteria, have developed a symbiotic relationship with human beings by maintaining the homeostasis of a specific physiological environment. Due to the vital role in immunomodulation and antitumor capacities, bacterial strains involve cancer and several infectious diseases, such as inflammatory bowel disease (IBD), sepsis, and arthritis.11 Besides the direct effects of their structural compositions, the bacteria also affect the pathogenesis, development, and treatment of complicated diseases by producing metabolites.58 These characteristics endow bacteria with incomparable abilities to detect and intervene in the progress of tumors and inflammatory diseases. With a better understanding of bacteria and their habitat, using bacteria in biomedical applications has evolved. Furthermore, with the advancement of genetic engineering technology and microbiology in recent years, the application of engineered bacteria has accelerated.59 In general, bacteria could either directly treat diseases, such as probiotics-mediated treatment against ulcerative colitis, or combine with other therapeutic agents to enhance the treatment efficacy. Bacteria-based therapies are frequently administered orally, which necessitates the protection of bacteria from the stomach's acidic pH and the harsh environment of the gastrointestinal (GI) tract, allowing bacteria to maintain viability and proliferation after reaching the desired site.60 Additionally, mitigating the pathogenicity and immunogenicity of microorganisms is essential in preventing undesirable adverse effects.3. Biomaterial-assisted cell therapies
Cell-based therapeutics have the potential for broad biomedical prospects due to their intrinsic capabilities to flexibly cope with dynamic pathological conditions. However, as “live” medicines, the practical applications of these functional cells are restricted by the challenges of maintaining their viability and functionality, and ensuring their efficacy that is usually restricted by the surrounding environment, which collectively pose additional requirements for designing a favorable harbor to cultivate the cell-based therapeutics at the desired sites.9 Recent years have witnessed explosive growth in materials science, thereby developing various sophisticated biomaterials, both synthetic and natural, that could function as delivery carriers and protection scaffolds to enhance the retention and viability of the therapeutic cells.16,61 Additionally, these biomaterials could endow the cell-based therapeutics with novel functions and thus maximize their potential in disease treatments. Furthermore, with the assistance of the rationally designed biomaterials, the cell behaviors could be more precisely traced and redirected, providing new possibilities for cell therapies.623.1 Biomaterial-assisted immune cell therapies
Despite significant progress in preclinical research of immune cell therapy and the approval of numerous FDA-approved T cell-based medicines, there are still barriers to expanding their clinical applications, such as time-consuming manufacturing methods, off-target toxicity, poor potency in specific diseased conditions, and low cell persistence.1,6 Emerging approaches based on genetic engineering and synthetic biology may provide the solutions to overcome these difficulties, such as removal of checkpoint molecules, upregulation of tumor-homing receptors, and expression of functional secretory proteins to modulate the microenvironment.16 While the genetic modifications facilitate T cells to overcome some challenges, other obstacles, such as expansion difficulties, on-target but off-tumor side effects, and neurotoxicities, have challenged the applications of genetically engineered immune cells.63Biomaterials have been widely used in cancer, cardiovascular disease, metabolic disorders, and tissue engineering for decades by serving as effective components or auxiliary ingredients.9 For example, a biomaterial structure with appropriate stiffness and shape for immune cell culture, expansion, and storage might be built using various compositions and functional motifs. Furthermore, the internal cavity may have a sufficient loading capacity, and the structural framework allows for ligand functionalization to recruit immune cells in situ using recruitment or induction molecules. Immune cell therapies combined with biomaterial platforms can concentrate cells, maintain cell retention, improve pharmacokinetics, and reduce side effects.
To obtain the effective and safe improvement of T cell therapy against solid tumors, Hao et al. attached a liposome containing the metabolism-modulating drug avasimibe onto the surface of the T cells without an obvious effect on their abilities through a lipid-inserting manner (Fig. 2(A)).66 While liposomes could stably encapsulate avasimibe during circulation and extravasation, the metabolic modulator would undergo controllable release at the tumor site to upregulate the cholesterol levels of the T cell membrane, causing T cell receptor clustering long-term activation. By combining T cell therapy with metabolic intervention, this immunotherapy strategy exhibited improved efficacy against tumor resistance in mice with melanoma and glioblastoma. Furthermore, there were no clear systemic negative effects from the infusion of modified T cells. Given the ease of production and desirable safety profile, these cell-surface anchor-engineered T cells provide a promising strategy to improve the treatment efficacy of T cell therapy against solid tumors. Tang et al. proposed another “backpack” strategy to effectively increase the utility and functionality of T cell therapy towards solid tumors (Fig. 2(B)).67 In this study, protein nanogels encapsulating the interleukin-15 super-agonist complex, which could sense membrane reduction potential changes after T cell activation and release the supporting protein drugs, were constructed and conjugated onto the surface of engineered T cells. By covalently coupling the nanogels to the surface of the proteins, the T cells were endowed with more potency and had enhanced function and persistence without noticeable toxicity after intravenous administration. The efficacy and safety of adoptive T cell therapy were increased by using and creating these “chemical backpacks”. Furthermore, the backpacks possess favorable biodegradability, an easy preparation process, and desirable storage stability, providing these T cell therapies with translational promises.
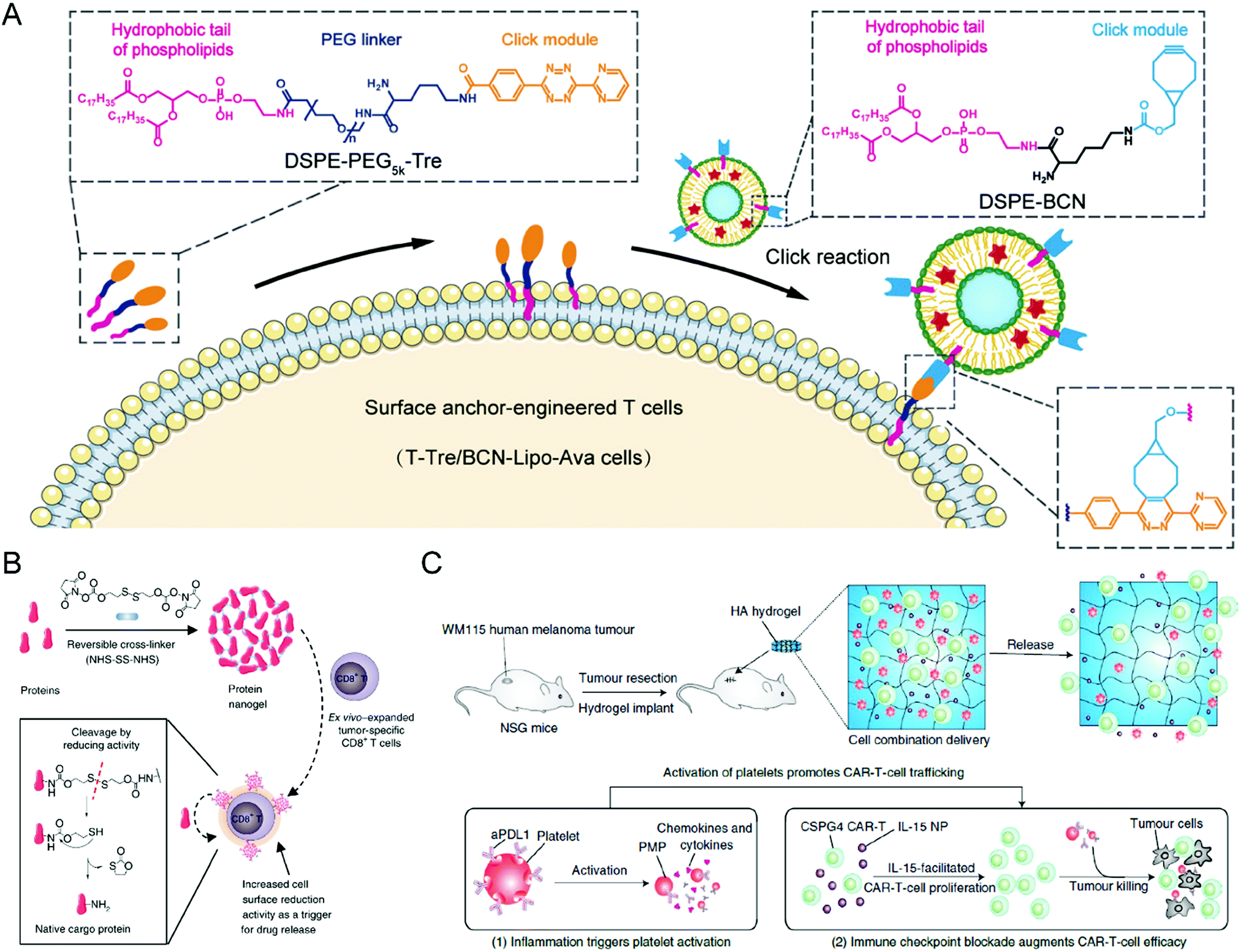 | ||
| Fig. 2 (A) Schematic illustration of preparing liposome-anchored T cells. (B) Schematic illustration of protein nanogel preparation and the relevant release mechanism in response to the local reducing microenvironment. (C) Schematic illustration of the hydrogel loading with CAR-T cells and anti-PDL1-conjugated platelets inhibiting post-surgery tumor recurrence. (A) was reproduced from ref. 66 with permission from the American Association for the Advancement of Science, Copyright 2020. (B) was reproduced from ref. 67 with permission from Nature Publishing Group, Copyright 2018. (C) was reproduced from ref. 69 with permission from Nature Publishing Group, Copyright 2021. | ||
Compared to intravenous injection, regional administration of effector T cells could circumvent the physiologic barriers and tumor-infiltrating obstacles. Coon et al. proposed nitinol thin films as the micromesh implants to load CAR-T cells, realizing enhanced T cell treatment against solid tumors.68 The resultant T cell-loaded micromesh films spatially conformed to the implantation site by which effector cells then underwent rapid proliferation, leading to abundant effector cells directly to the TME, and dramatically enhanced animal survival of mice bearing non-resectable ovarian cancer. Additionally, coating self-expandable stents with these T-cell-loaded films and placing them into subcutaneous tumors could extend the potency of the stents by delaying tumor ingrowth. Collectively, CAR-T-loaded micropatterned nitinol thin films can boost the benefits of T cell-based therapies by allowing direct access to tumors. Moreover, the stability of the film and the reproducibility of T-cell loading could be rationally adjusted by changing the pore geometry and mechanical properties of the micromeshes. Additionally, the thinness of micromeshes renders nutrients available to the effector cells, increasing their duration at the tumor location.
The development of novel biomaterials also provides more possibilities combined with other standard treatment modalities for T cell therapies, improving their treatment efficacy and expanding their applications. For example, Hu et al. designed a hydrogel containing CAR-T cells with the chondroitin sulfate proteoglycan 4 (CSPG4) antigens, which are abundantly expressed in human melanoma cells yet restricted in normal cells (Fig. 2(C)).69 The hydrogel creates an environment so that the CAR-T cells can suppress any residual tumor cells post-surgery and prevent any future regrowth or recurrence of the tumor. Given that the inflammatory conditions can trigger the platelet activation to release platelet-derived microparticles (PMPs), the surface of platelets was conjugated with anti-PDL1 (engineered monoclonal antibodies against programmed-death ligand 1) to prepare P-aPDL1 that release aPDL1 antibodies to block PDL1 of cancer cells under an inflammatory environment secondary to the cancer surgery, further preventing the T cell exhaustion. To promote the proliferation of the CAR-T cells, IL-15 NPs were loaded into the hydrogel and P-aPDL1. The coculture assay showed that CAR-T cells loaded in the hydrogel could undergo rapid expansion and effectively damage WM115 melanoma cells. The hydrogel demonstrated notable efficacy in controlling local tumor regrowth. Besides, Ogunnaike et al. proposed fibrin gels loaded with CAR-T cells to achieve slow release of the effector cells to the target region and promote higher antitumor activity than directly injecting CAR-T cells to the site.70 A porous gel generated from human fibrinogen through thrombin-mediated enzymatic polymerization was designed to encapsulate T cells, which could gradually release the effector cells at the tumor resection cavity after in situ gel formation. The CAR-T cells that were deposited by the fibrin gel demonstrated effective antitumor activity in glioblastoma resection mouse models. This study is particularly advantageous for targeting tumor recurrence, especially for brain tumors, compared to the direct injection of CAR-T cells. Ultimately, the fibrin gel loaded with CAR-T cells may prove incredibly effective when used over a long period post-surgery.
Recently, sophisticated biomaterial-assisted techniques have been developed to improve the efficacy, specificity, and safety of T cell treatments. To combat the shortcomings of T cell therapies, material-based platforms are being developed to concentrate cells, maintain cell retention, optimize pharmacokinetics, and prevent adverse effects. Co-loading T cells and other functional agents in integrated formulations gives effector cells exhaustion resistance, potentially boosting efficacy and durability in solid tumors. Furthermore, materials with TME-regulating abilities may help T cells expand or persist, resist the suppressive microenvironment, or act as a tumor-infiltrating promoter.
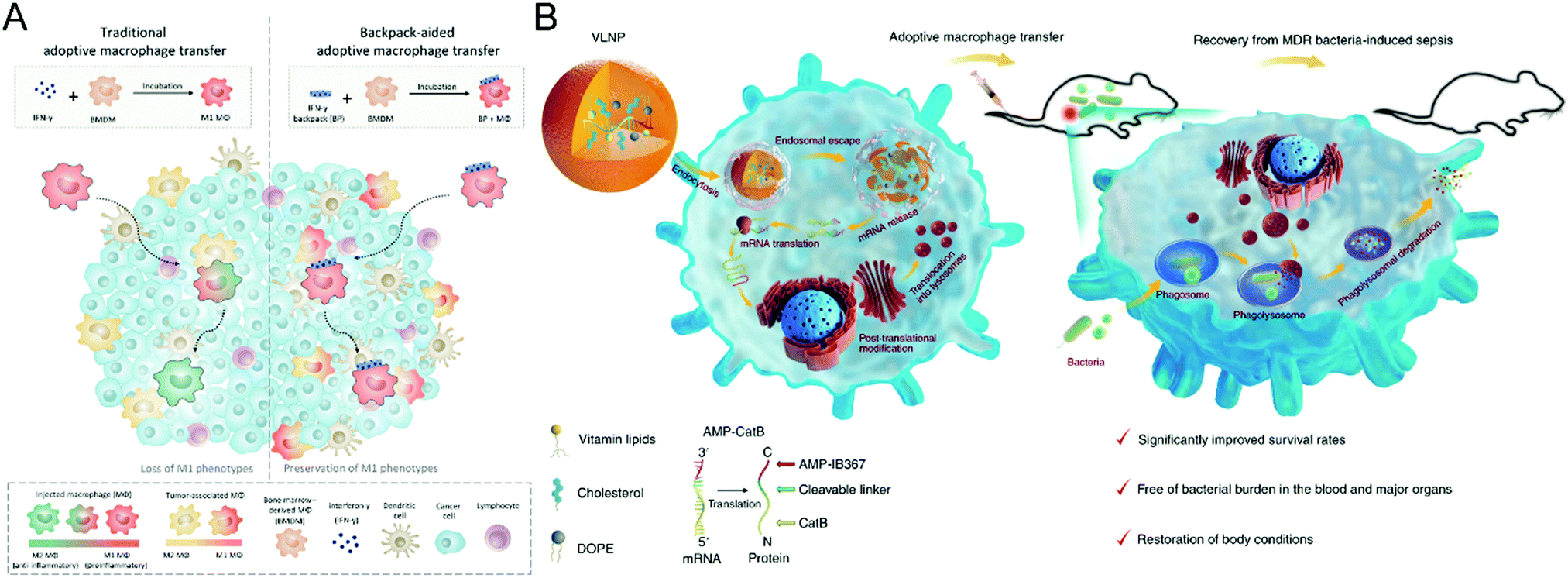 | ||
| Fig. 3 (A) Schematic illustration of cellular backpack-based macrophage therapies. (B) Schematic illustration of preparing the vitamin-derived lipid nanoparticles and the action mechanism of adoptive macrophage-mediated sepsis treatment. (A) was reproduced from ref. 71 with permission from the American Association for the Advancement of Science, Copyright 2020. (B) was reproduced from ref. 72 with permission from Nature Publishing Group, Copyright 2020. | ||
Besides induction of phenotype polarization, biomaterials could extend the functions of macrophages, leading to a synergistic effect on refractory diseases. Hou et al. propose a macrophage-based cell therapy that overcomes sepsis-causing bacteria resistance (Fig. 3(B)).72 The method employs vitamin C lipid nanoparticles to transfect macrophages with antimicrobial peptides and cathepsin B (AMP-CatB) mRNA. Since the lysosome of macrophages is the targeting site for bactericidal activities, the vitamin C lipid nanoparticles enable the specific accumulation of AMP-CatB lysosomes. After transfer, the adoptive macrophages could eliminate multidrug-resistant pathogens such as Staphylococcus aureus and Escherichia coli, resulting in immunocompromised septic mice recovering completely. This study paves the way for developing nanomaterial-assisted macrophage therapy for infectious disorders and an alternate technique for combating multidrug-resistant bacteria-induced sepsis.
Material-engineered macrophages can also be used as an adjuvant treatment to combine chemotherapy, radiotherapy, and phototherapy to generate a synergistic effect, in addition to direct polarization of phenotypes and improvement of abilities. Unfortunately, the insufficient tumor-targeting capability poses challenges to macrophages to target malignant tumors, particularly metastatic cancers. However, chemically or genetically coupling targeting ligands is expected to endow engineered macrophages with metastatic tumor-targeting abilities.3 Furthermore, material-modified macrophages also serve as drug delivery systems to transport functional agents to the specific diseased site through their inherent homing effect.
As for the difficulties in ex vivo expansion, Ahn et al. developed a 3D hyaluronic acid-based niche, termed 3D-ENHANCE, to culture NK cells (Fig. 4(A)).73 Other than commonly used 2D methods, 3D-ENHANCE enables NK-92 cell lines and human epidermal growth factor receptor (EGFR)-specific CAR-NK cells with upregulated mRNA expression, increased cytokine secretion, rapid growth proliferation, and enhanced cancer cell-killing capacities. Moreover, the controllable biodegradability renders 3D-ENHANCE an implantable cell reservoir in the post-surgery cavity. The in vivo evaluation demonstrated that implanting 3D-ENHANCE into the surgical sites could reduce relapse and metastases; thus, this strategy provides a cytokine-free niche for ex vivo proliferation and postsurgical therapy of NK cells, which improves their low therapeutic efficiency.
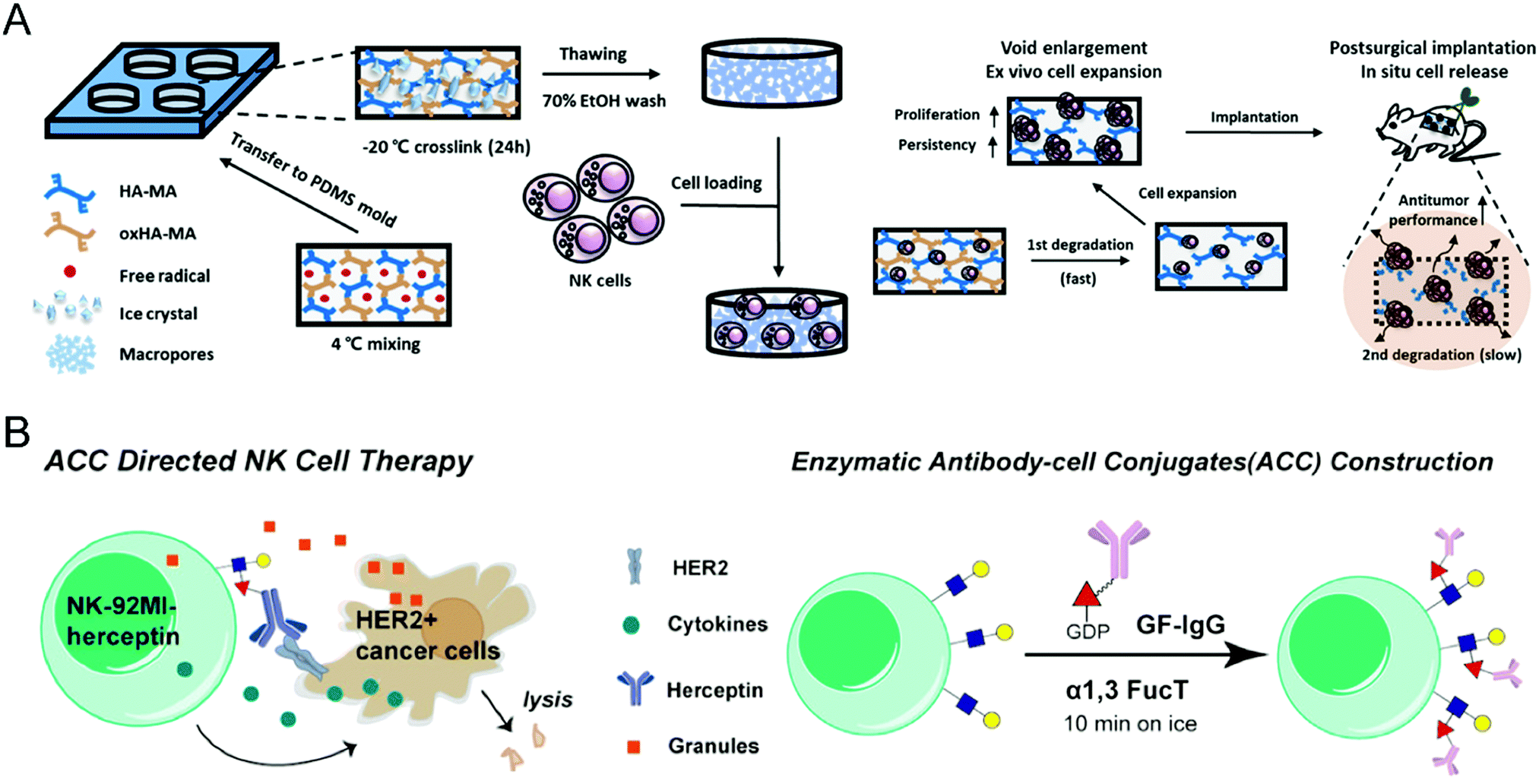 | ||
| Fig. 4 (A) Schematic illustration of preparing NK cell-loaded 3D-ENHANCE and its relevant action mechanism. (B) Schematic illustration of enzymatic surface antibody modification to prepare Herceptin-conjugated NK cells. (A) was reproduced from ref. 73 with permission from Elsevier, Copyright 2020. (B) was reproduced from ref. 75 with permission from the American Chemical Society, Copyright 2018. | ||
To enhance the specificity of effector NK cells against cancer cells, modification with targeting ligand on their surface serves as a feasible option. Yang et al. incorporated the synthetic CD30-specific aptamers into the membrane of NK cells to produce aptamer-engineered NK cells (ApEn-NK) with an unnoticeable effect on genome and cell viability.74 The resultant NK cells selectively bind to CD30-overexpressing lymphoma cells and effectively eliminate the malignant cells. Besides, based on the chemoenzymatic approach, Li et al. constructed antibody-modified NK-92MI cells through fucosyltransferase-mediated biomacromolecule transfer (Fig. 4(B)).75 The Herceptin-engineered NK cells demonstrated increased abilities to eliminate HER2+ cancer cells, leading to a significantly improved anticancer effect on human tumor xenograft models.
A rapidly increasing number of biomaterial-based strategies have been designed for NK cell therapies. Especially, the adoptive transfer of NK cells provides a desirable option for cancer management due to the advances in in vitro expansion and activation approaches. However, overcoming the immunosuppressive microenvironment remains an urgent issue, which might compromise the potential of NK cell therapy for further applications.76
Recently, many researchers have attempted to employ various biomaterials, such as scaffolds, polymeric particles, or hydrogels, to promote the viabilities and abilities of DCs. Srinivasan et al. reported a multicomponent immunomodulatory scaffold prepared by incorporating glutaraldehyde-crosslinked gelatin microparticles into an agarose matrix.77 The resultant scaffold could create a suitable microenvironment for re-educating and generating DCs with a tolerogenic or regulatory phenotype through controllable release of granulocyte monocyte colony-stimulating factor (GM-CSF), dexamethasone (DEX), or peptidoglycan (PGN). The scaffold-treated DCs were demonstrated to suppress the proliferation of T cells. Moreover, Thomas et al. developed a poly(ethylene glycol) (PEG)-based hydrogel for local transplantation of IL-10-treated DCs.78 The DC-containing hydrogel could reprogram the immune microenvironment of the injection site, realizing prevention from the progression of sclerosis.
Surface engineering is also a practical approach to promoting the maturation and functionality of DCs. Liu et al. decorated the surface of DCs with polydopamine through a calcium-mediated process, thereby achieving controllable regulation of the DC maturation status.79 While polydopamine serves as a ROS scavenger to inhibit DC maturation, the photothermal effect after irradiation with an 808 nm laser could initiate the maturation process, leading to effective activation. Yu et al. provided another surface modification approach to enhance the antigen presentation of DCs (Fig. 5(A)).80 The synthetic glycopolymer modified onto the DC membrane could enhance the frequency and duration of their interaction with T cells, resulting in promoted T cell activation and increased anticancer efficacy. Collectively, biomaterial-based surface engineering provides a promising strategy for more efficient DC therapies.
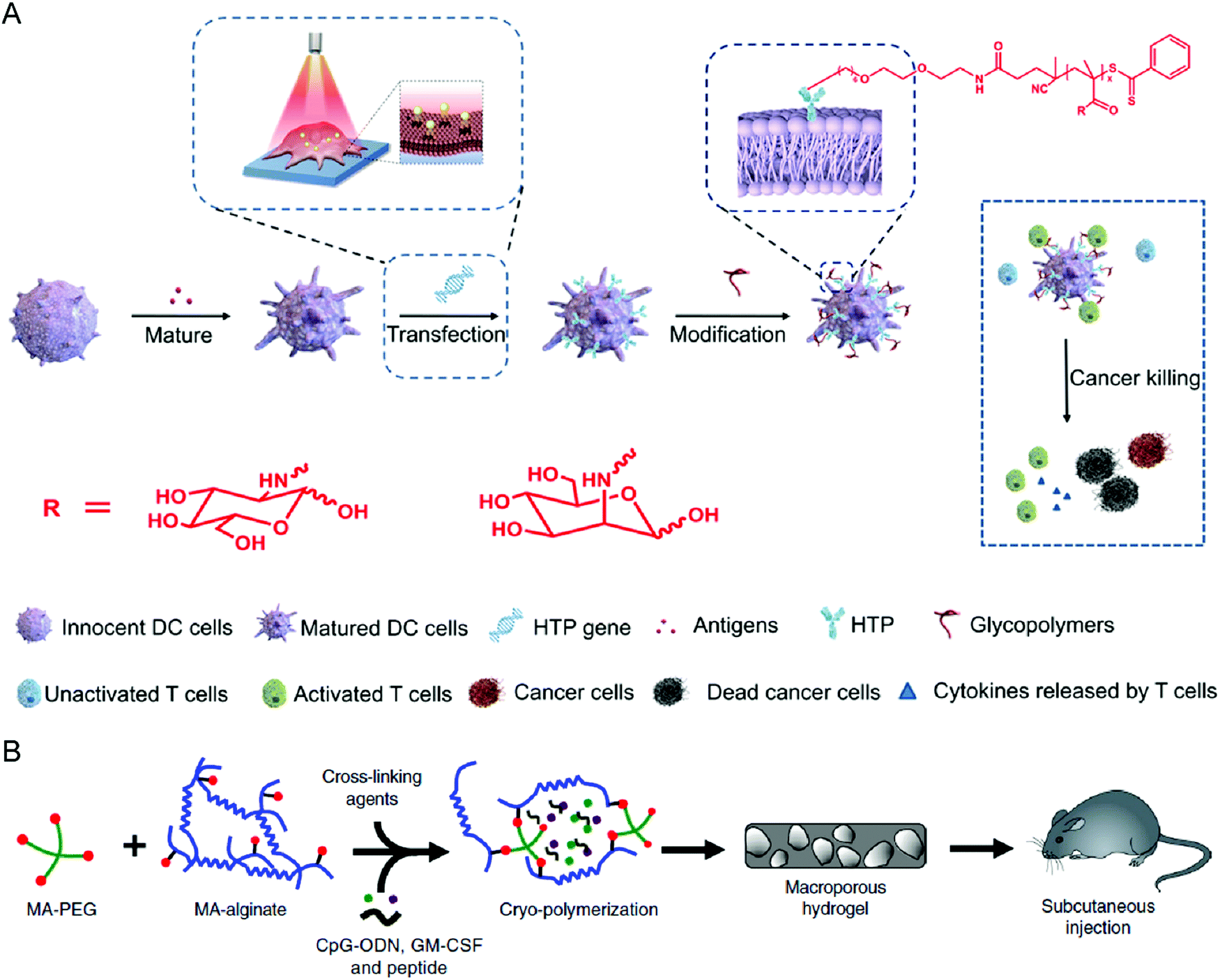 | ||
| Fig. 5 (A) Schematic illustration of modifying DCs with glycopolymers. (B) Schematic illustration of preparing a cryogel loaded with immunomodulatory agents. (A) was reproduced from ref. 80 with permission from the American Association for the Advancement of Science, Copyright 2020. (B) was reproduced from ref. 82 with permission from Nature Publishing Group, Copyright 2020. | ||
Given their specific functions of antigen presentation and T cell activation, in vivo education of DCs is a simple method to exert the treatment efficacy of DC therapies. Kim et al. designed an implantable microporous poly(lactide-co-glycolide) (PLG) scaffold encapsulating GM-CSF to in vivo recruit and enrich DCs to the pore of PLG scaffolds.81 From the same group, Shah et al. showed another macroporous biomaterial formed by covalent cross-linking between PEG and alginate, termed cryogel, to encapsulate acute myeloid leukemia (AML)-associated antigens, cytosine–guanosine oligodeoxynucleotide (CpG-ODN), and GM-CSF (Fig. 5(B)).82 After subcutaneous injection, the cyrogel promotes in situ DC maturation and local T cell infiltration, leading to a potent anti-AML immune response. Other than the commonly used ex vivo education of DCs, these studies provide a new direction for DC-based treatments.
For DC therapies, many studies have been performed to maximize the therapeutic potentials of DCs through designing new biomaterials. Moreover, benefiting from the understanding of materials science, rational combination therapies can be developed to improve the treatment efficacy of DCs with other standard therapies.
Liu et al. reported an in situ Treg cell reprogramming strategy, employing poly(L-lactic acid) (PLLA) nanofibrous spongy microspheres as the injectable scaffold that contains PLLA/PEG-functionalized mesoporous silica nanoparticles (MSNs) and PLG microspheres (Fig. 6).83 This system could enrich, expand, and educate Treg cells and thus create a local immunotolerant microenvironment. Finally, the multifunctional microspheres are demonstrated to relieve bone loss in a murine periodontitis model. In another study, Fisher et al. synthesized chemokine (C–C motif) ligand 22 (CCL22)-encapsulated PLG microparticles to realize immune tolerance and minimize rejection after limb transplantation.84 After intragraft administration, the microparticle systems could recruit Treg cells in allograft skin and promote their immunosuppressive functions with unnoticeable effects on conventional T cells. This study provides a practical approach to reducing the immunological rejection caused by transplantation.
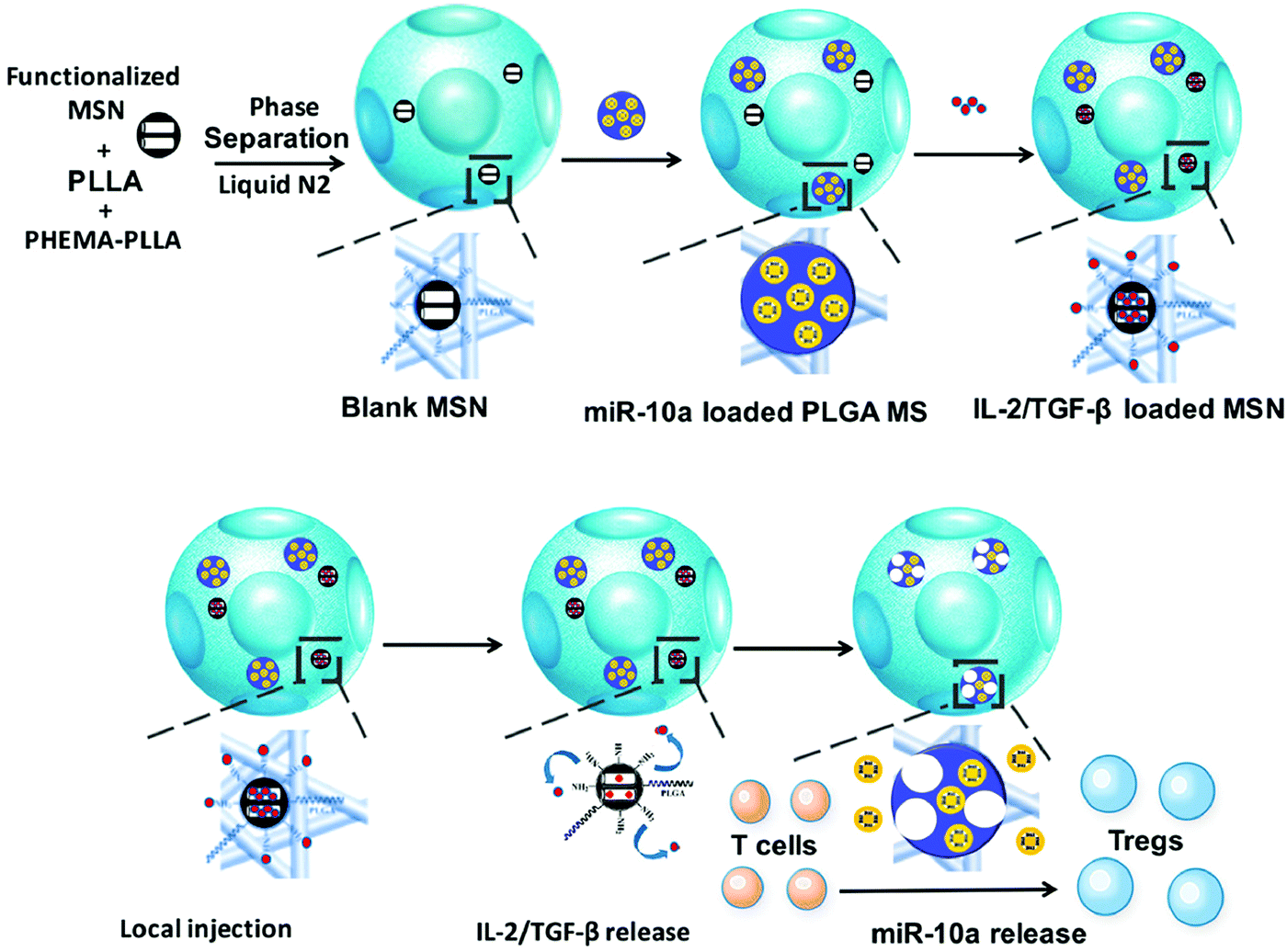 | ||
| Fig. 6 Schematic illustration of preparing the multifunctional microspheres and their relevant action mechanism. Fig. 6 was reproduced from ref. 83 with permission from the American Chemical Society, Copyright 2018. | ||
Besides the above-mentioned in vivo recruitment of Treg cells, co-transplantation of organs with Treg cells is another feasible application for Treg cell therapies. Graham et al. employed a PLG scaffold to co-transplant Treg cells and islet grafts for protecting islets from rejection in diabetic models.85 The resultant PLG scaffold could recruit and educate host-derived Treg cells to co-localize inside grafts, leading to long-term and systemic immunological tolerance.
With the explosive growth in biomaterials, researchers would develop increasing strategies to expand the applications and accelerate the clinical translation of cell therapies.17 As for Treg cell-based therapies, expansion and function persistence remain the major issues requiring much more effort.
3.2 Biomaterial-assisted stem cell therapies
Stem cell treatment holds great potential in tissue regeneration and functional recovery. Stem cell therapeutics exert functions after intravenous or local administration through cell replacement, cytokine secretion, and dying-cell clearance.52 Despite their enormous abilities, stem cell therapies suffer from several difficulties in clinical translation, including low viability in the hostile diseased microenvironment and unfavorable biodistribution, immune rejection, and uncontrollable differentiation.53 As a result, developing a biomaterial-based bioengineering strategy to address the limitations is a desirable option for enhancing the clinical translation potential of stem cell therapy.Given that multiple factors compromise the treatment efficacy of mesenchymal stromal cells (MSCs), Gonzalez-Pujana et al. developed a multifunctional hydrogel system with tunable mechanical properties to maximize their immunomodulatory properties (Fig. 7(A)).86 Immunomodulatory extracellular matrix hydrogels (iECM) generated from a click functionalized-alginate and fibrillar collagen incorporated by interferon γ (IFN-γ)-loaded heparin-coated beads, which increased the expression of a wide panel of key immunomodulatory and regulatory genes in bone marrow-derived primary human MSCs (hMSCs) and enhanced the secretion of indoleamine 2,3-dioxygenase 1 (IDO1) and galectin-9 (GAL9). Furthermore, the coculturing assay demonstrated that iECM-encapsulated hMSCs could significantly suppress the proliferation of activated human T cells, indicating enhanced immunomodulatory capabilities. Sultan et al. reported another kind of hydrogel system based on silk fibroin (SF) for brain injury treatment (Fig. 7(B)), which could encapsulate engineered hMSCs and facilitate the secretion of brain-derived neurotrophic factor (BDNF).87 The SF that serves as the base for the proposed hydrogel systems is collected from the cocoons of a particular species of silk moths, Bombyx mori. The authors prepared multiple hydrogels with differing concentrations of SF and tested the relevant tensile strength and gel point. The results showed that SF-based hydrogels could increase the neurological function of specific brain-damaged areas in the rat model, serving as a minimally invasive alternative for treating brain injury.
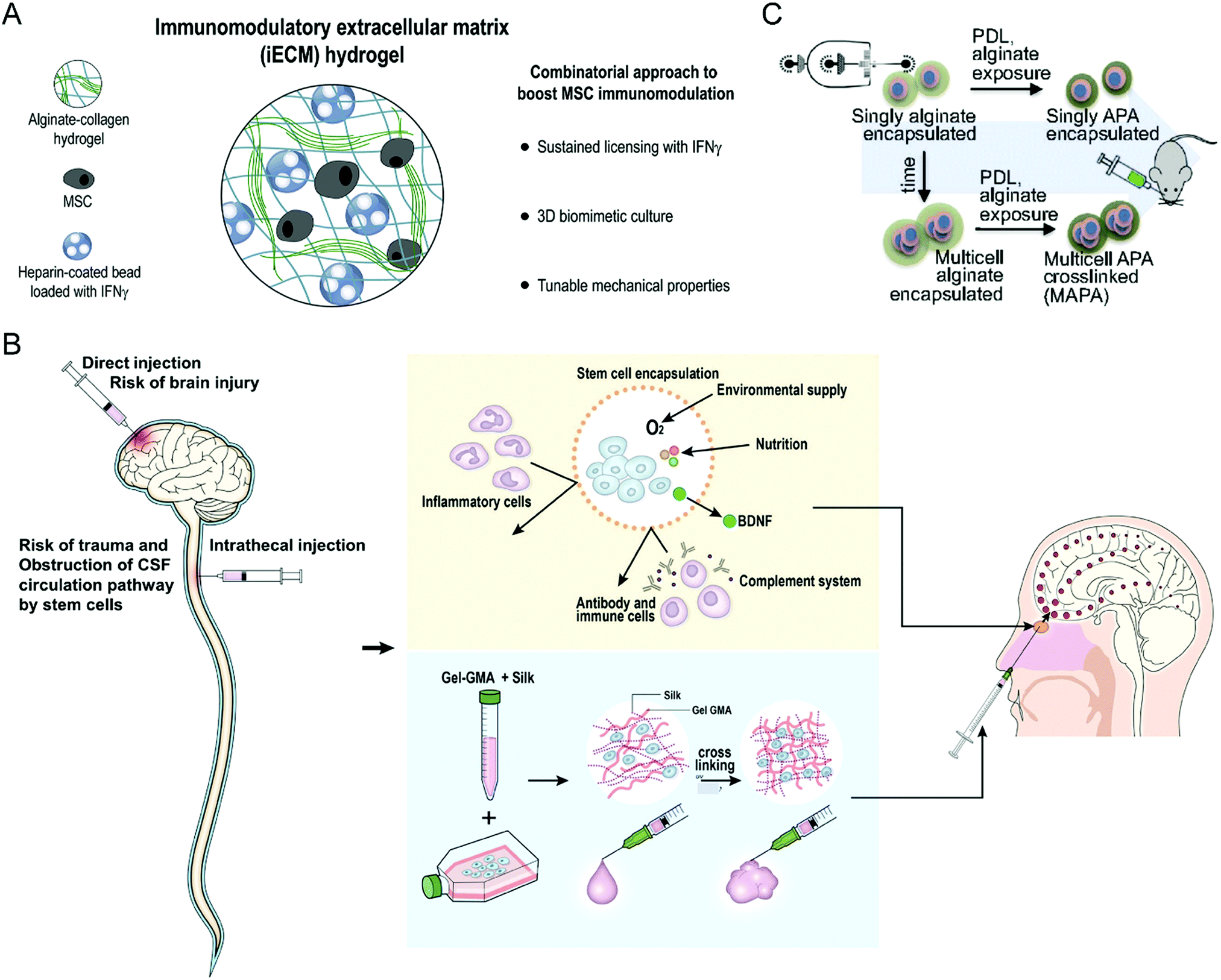 | ||
| Fig. 7 (A) Schematic illustration of an iECM hydrogel for improving the immunomodulatory ability of MSCs. (B) Schematic illustration of silk fibroin-based hydrogel encapsulating hMSCs for brain injury treatment. (C) Schematic illustration of encapsulating mMSCs into alginate microgels. (A) was reproduced from ref. 86 with permission from Elsevier, Copyright 2020. (B) was reproduced from ref. 87 with permission from Elsevier, Copyright 2020. (C) was reproduced from ref. 88 with permission from the National Academy of Sciences of the United States of America, Copyright 2019. | ||
Besides the hydrogel scaffold systems, Mao et al. encapsulated the single MSC into an alginate microgel through the microfluidic device to maximize the therapeutic potential and overcome the short in vivo cell persistence and phenotype inconsistencies of stem cells (Fig. 7(C)).88 Prior to administration, exposure to inflammatory cytokines upregulates the immunomodulatory genes in encapsulated cells. This biomaterial encapsulation strategy could significantly improve the half-life of MSCs after intravenous injection due to alginate-mediated cell cluster formation and subsequent cross-linking with polylysine. Furthermore, these surface modifications are conducive to mitigating the clearance risks caused by innate and adaptive immune responses. The encapsulated stem cells finally improve the engraftment of major histocompatibility complex (MHC)-mismatched donor cells in a bone marrow transplant model, indicating that this microgel encapsulation approach is practical to prolong stem cell survival and boost their overall immunomodulatory potency.
Moreover, stem cells have been widely used to treat cardiovascular diseases. Given the poor retention of the transplanted cells and inevitable entrapment in the lungs after intravenous injection, the Cheng group has developed various biomaterials, such as polymeric nanogels,89 platelet nanovesicles,90 ferumoxytol nanoparticles,91 and chemically engineered antibodies,92 to enhance the treatment efficacy of the stem cell-mediated heart repair. As for stem cell transplantation, Tang et al. attempted to encapsulate human cardiac stem cells into a thermosensitive nanogel with a porous and convoluted inner structure,89 which protects the stems cells from the attack by immune cells but permits nutrient, oxygen, and secretion diffusion. Therefore, the resulting nanogel facilitates the repair of the heart and reduces scar sizes without systemic inflammation and local T-cell infiltration. Furthermore, given the low retention and engraftment of the transplanted cells, Tang et al. developed a fast, straightforward and safe engineering approach for cardiac stem cells to improve their vascular delivery (Fig. 8(A)).90 Through fusion with platelet nanovesicles, the modified cells could express a platelet surface maker and maintain their tissue regeneration abilities, leading to selective accumulation at the injured site for exerting a repairing function. Also, Vandergriff et al. employed FDA-approved ferumoxytol nanoparticles to label stem cells, realizing enhanced cell engraftment and treatment efficacy through magnetically-targeted cell delivery.91 Besides, Li et al. proposed the biorthogonal click chemistry-mediated pretargeting strategy to redirect endogenous circulating stem cells to the injured heart (Fig. 8(B)).92 Especially, the affinity between platelets and endothelial progenitor cells (EPCs) could promote the binding of EPCs to the injured blood vessels, leading to effective stem cell-based heart repair. Benefiting from these tailored biomaterials, they have provided a promising prospect for stem cell-based treatments of cardiovascular disease.
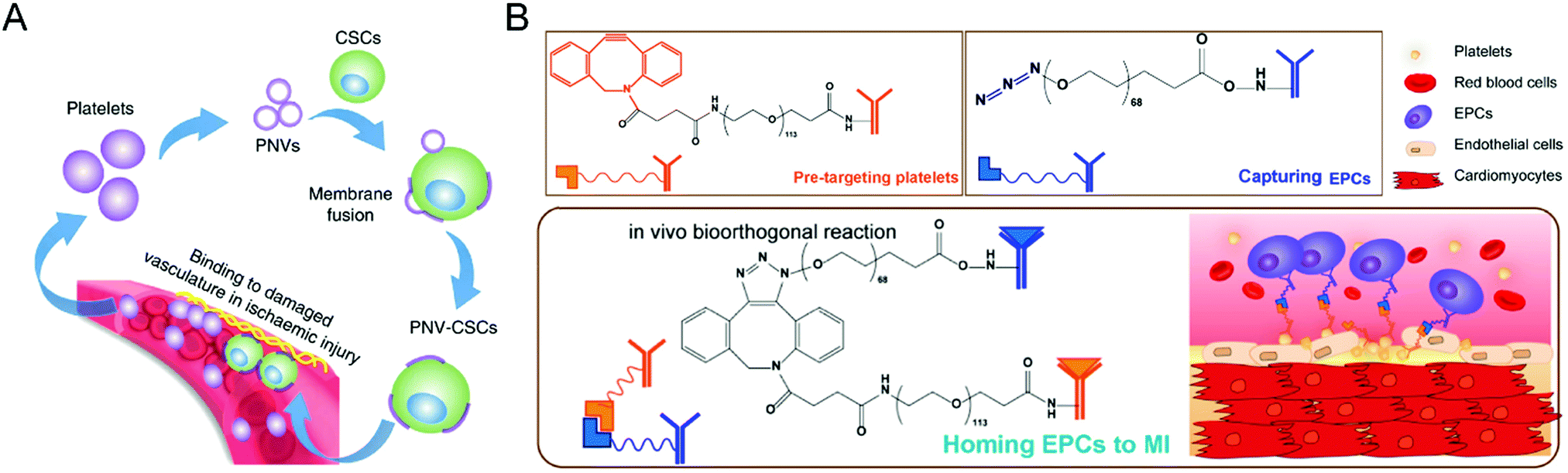 | ||
| Fig. 8 (A) Schematic illustration of targeted heart injury repair mediated by stem cells decorated with platelet nanovesicles for improving the repairing capability of MSCs. (B) Schematic illustration of redirecting endogenous circulating stem cells to the injured heart through a biorthogonal click chemistry-mediated pretargeting strategy. (A) was reproduced from ref. 90 with permission from Nature Publishing Group, Copyright 2018. (B) was reproduced from ref. 92 with permission from the American Chemical Society, Copyright 2018. | ||
Stem cell therapy has advanced at an unparalleled rate in transplantation and regenerative medicine due to its self-renewal capabilities. The versatility of stem cells enables researchers to choose matched stem cell types according to the needs of the specific disease treatment. Due to the advances in materials science, both scaffold and capsule systems can not only provide stem cells with protection and maintenance but also regulate their development to increase therapeutic benefits. Despite their enormous potential, the immaturity of stem cell expansion and differentiation poses a big challenge to their clinical translation. In addition, carcinogenicity and mutagenicity remain the major issues to be addressed.
3.3 Biomaterial-assisted bacteria therapies
Bacteria have the potential to selectively target the diseased site by sensing the pathological cues, leading to either suppression or acceleration of the disease progression. For example, probiotics could reprogram microbial homeostasis and promote the repairment of the mucosal barrier in the GI tract to improve the IBD treatment.93 In contrast, certain bacteria would deteriorate the pathological processes by blocking the immune response-mediated clearance. These essential properties render bacteria a promising therapeutic modality for specific disorders from the perspective of healthcare. However, the clinical translation of bacterium therapies is hampered by their possible toxicity and immunogenicity, indicating the requirement of biomaterials engineering. The design principles for bacteria-based therapeutics are centered on minimizing toxicity, increasing in vivo survivability and proliferation capabilities, and endowing bacteria with extra functionalities for improved treatment potency. The convergence of bacteria and materials science will pave the way for novel therapeutic possibilities involving engineered microbes.Despite the extensive exploration of bacterial-mediated biotherapies for cancer treatments, the poor potency hinders their clinical application, mainly due to the low tumor-specific accumulation after administration. Geng et al. conjugated aptamers onto the surface of bacteria through a cytocompatible amidation approach,94 leading to significantly improved tumor localization following systemic injection (Fig. 9(A)). Compared to unmodified counterparts, bacteria with an optimal conjugation density of aptamers realize high accumulation at the tumor site and remarkable selectivity against tumor cells. Furthermore, the engineered Salmonella triggered the anticancer immune response inside the tumor and thus exhibited improved treatment efficacy in both 4T1 and H22 murine tumor models. This study demonstrated the feasibility of surface conjugation to change the bacterial behavior and increase antitumor biotherapy.
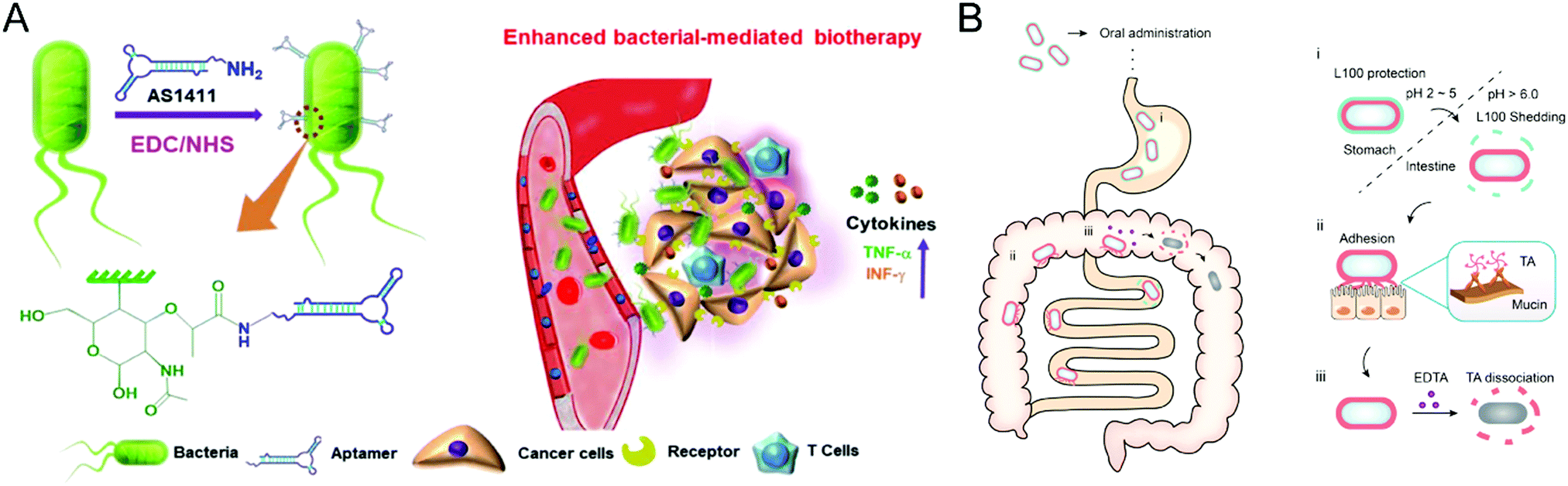 | ||
| Fig. 9 (A) Schematic illustration of aptamer-modified bacteria for enhanced biotherapy. (B) Schematic illustration of the action mechanism of the double layer-coating bacteria in the GI tract. (A) was reproduced from ref. 94 with permission from Nature Publishing Group, Copyright 2021. (B) was reproduced from ref. 95 with permission from Elsevier, Copyright 2021. | ||
Moreover, the surface coating serves as another common material-based engineering technique for bacteria. Considering the promise of probiotics in treating IBD by modulating gut microbiota homeostasis, Liu et al. developed a double-layer coating method to facilitate the resistance of probiotics against the harsh conditions in the GI tract and thus enhance the retention time of probiotics (Fig. 9(B)).95 The coating strategy employed tannic acid (TA) and enteric L100 to encapsulate Escherichia coli Nissle 1917 (EcN) strain, increasing availability and persistence after oral administration. Then, the engineered EcN systems selectively released TA-EcN in the intestine through pH-responsive degradation of the outer L100 layer. The powerful mucoadhesive abilities of the TA layer prolonged the EcN retention time without a noticeable effect on the viability and proliferation of EcN, achieving potent prophylactic and treatment efficacy in murine IBD models. Furthermore, the TA layer can be eliminated by adding ethylenediaminetetraacetic acid to reduce the possible adverse reactions caused by long-term TA mucoadhesion. This coating strategy provides a practical approach for bacteria therapies to treat GI tract disorders.
Due to the interactions with intestinal microbiota and many disorders, bacterial therapies provide a novel strategy for complex diseases. However, many bacteria possess a pathogenicity and strong colonization ability. Furthermore, the immunogenicity of most bacteria would cause undesirable adverse reactions like systemic inflammatory response syndrome and the risk of sepsis. Additionally, bacteria are sensitive to hostile physiological environments, including gastric acid, which may compromise their availability after oral administration. Biomaterial-assisted techniques can facilitate bacteria to fully exert their therapeutic functions and improve in vivo biosafety. Synthetic polymers or biofilms can be coated on the surface of bacteria to protect them from a harsh environment. Surface modification with antibodies or targeting ligands could also promote selective accumulation in the diseased site and treatment efficacy. Moreover, advanced material-based engineering methodologies can endow bacteria with novel functionalities, extending their clinical applications.
3.4 Biomaterial-assisted other cell therapies
The past decade has witnessed the important role of biomaterials in promoting the efficiency of immune cell, stem cell, and bacteria therapies, inspiring the applications of various biomaterials to other cell therapies. For example, Nash et al. developed an alginate capsule to load human ARPE-19 cells as a cytokine-producing factory (Fig. 10(A)).96 After tumor-adjacent administration, the capsules could controllably release effector cytokines and trigger the anticancer immune response, eradicating intraperitoneal tumors without systemic toxicities. As for the treatment of diabetes, Bose et al. reported a retrievable and implantable microdevice constructed by a polydimethylsiloxane reservoir and polycarbonate track-etched membrane to protect xenogeneic cells from the destruction of host immune systems without affecting their therapeutic functions (Fig. 10(B)).97 Vegas et al. encapsulated human embryonic stem cell-derived β cells into alginate derivatives for glycemic correction after intraperitoneal administration.98 In another study, Bochenek et al. tested the immune protection of seven alginate hydrogel spheres for allogeneic pancreatic islet cells.99 They found that a chemically modified alginate derivate, Z1-Y25, and the relevant formulation enabled long-time transplantation of pancreatic islets without the need for immunosuppression. Wang et al. employed a nanofiber-integrated cell encapsulation device to protect human pluripotent stem cell-derived functional β cells from immune rejection and thus realize diabetes reversal within one week (Fig. 10(C)).100 Moreover, the device possesses desirable scalability and retrievability with an unnoticeable fibrotic response. Besides hydrogels, the microneedle is an emerging device for transdermal drug delivery, which could also assist cell therapies in a painless, low-invasive, and long-time manner.101 Ye et al. integrated microneedles with pancreatic-cell capsules that respond to changes in blood glucose and produce insulin (Fig. 10(D)).102 Then, the microneedles were incorporated with glucose signal amplifiers, which were constructed by synthetic nanovesicles containing glucose oxidase, α-amylase, and glucoamylase and could trigger insulin release from pancreatic-cell capsules through the glucose-involved enzymatic cascade. Finally, the microneedle patch rapidly reduced blood glycemic levels in type 1 diabetic mice. Collectively, as the therapeutic cell species increase, the demands for novel biomaterials will rise; in other words, as materials science advances, more cell therapies will be developed and improved. | ||
| Fig. 10 (A) Schematic illustration of preparing the cytokine factory and its relevant action mechanism. (B) Schematic illustration of the device design and its relevant action mechanism. (C) Schematic illustration of the device design and structure. (D) Schematic illustration of the design of the functionalized microneedle path and its relevant action mechanism. (A) was reproduced from ref. 96 with permission from the American Association for the Advancement of Science, Copyright 2022. (B) was reproduced from ref. 97 with permission from Nature Publishing Group, Copyright 2020. (C) was reproduced from ref. 100 with permission from the American Association for the Advancement of Science, Copyright 2021. (D) was reproduced from ref. 102 with permission from Wiley-VCH Verlag GmbH & Co. KgaA, Copyright 2016. | ||
4. Conclusions and perspectives
The inherent ability to interact with and adapt to specific physiological processes renders cells an emerging treatment modality widely investigated for various diseases. The proliferation and differentiation potentials could be applied to tissue repair and regeneration. Due to the capacities of stimulus-responsive protein secretion, cell therapies are suitable for dynamic, long-term therapeutic administration. Biomaterials play a key role in supporting, delivering, and functionalizing the therapeutic cells to enhance their biomedical applications. Collectively, biomaterials can serve as supportive scaffolds or protective coatings for cell-based therapeutics to maintain their viability and intrinsic functionality and induce specific cell phenotypes through educating, training, or redirecting the target cells. Also, the biomaterials frequently serve as the delivery vehicles to transport abundantly available cells to the diseased sites and increase their retention by improving the targeting effect or protecting them from environmental damages. Moreover, given the strong therapeutic abilities of “live” medicine, versatile biomaterials could equip these therapeutic cells with additional functions and thus synergistically exert treatment efficacy. Biomaterial engineering techniques have been demonstrated to accelerate the translation of therapeutic cells. However, the costs and difficulties of cell collection, expansion, and engineering may compromise applicability in the clinic. The stringent quality control and safety standards for the materials also pose additional challenges to the approval of material-based cell therapeutics. Apart from the materials already approved for the clinic, new material modification and characterization approaches and the relevant mechanisms usually require extensive evaluations, leading to high time and manufacturing costs. Overall, with the increasingly rapid growth in developing sophisticated and smart biomaterials, material-modified cells would possess various new therapeutic potentials and extensive application prospects.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Q. H. acknowledges the start-up package support from the University of Wisconsin-Madison.References
- H. J. Jackson, S. Rafiq and R. J. Brentjens, Nat. Rev. Clin. Oncol., 2016, 13, 370–383 CrossRef CAS PubMed.
- N. Shimasaki, A. Jain and D. Campana, Nat. Rev. Drug Discovery, 2020, 19, 200–218 CrossRef CAS PubMed.
- Z. Li, Y. Wang, Y. Ding, L. Repp, G. S. Kwon and Q. Hu, Adv. Funct. Mater., 2021, 31, 2100088 CrossRef CAS.
- J. Xue, Z. Zhao, L. Zhang, L. Xue, S. Shen, Y. Wen, Z. Wei, L. Wang, L. Kong, H. Sun, Q. Ping, R. Mo and C. Zhang, Nat. Nanotechnol., 2017, 12, 692–700 CrossRef CAS PubMed.
- Q. Hu, W. Sun, J. Wang, H. Ruan, X. Zhang, Y. Ye, S. Shen, C. Wang, W. Lu, K. Cheng, G. Dotti, J. F. Zeidner, J. Wang and Z. Gu, Nat. Biomed. Eng., 2018, 2, 831–840 CrossRef CAS PubMed.
- C. M. Britten, A. Shalabi and A. Hoos, Nat. Rev. Drug Discovery, 2021, 20, 476–488 CrossRef CAS PubMed.
- S. Yamanaka, Cell Stem Cell, 2020, 27, 523–531 CrossRef CAS PubMed.
- L. Bagno, K. E. Hatzistergos, W. Balkan and J. M. Hare, Mol. Ther., 2018, 26, 1610–1623 CrossRef CAS PubMed.
- A. L. Facklam, L. R. Volpatti and D. G. Anderson, Adv. Mater., 2020, 32, e1902005 CrossRef PubMed.
- J. Wu, D. Song, Z. Li, B. Guo, Y. Xiao, W. Liu, L. Liang, C. Feng, T. Gao, Y. Chen, Y. Li, Z. Wang, J. Wen, S. Yang, P. Liu, L. Wang, Y. Wang, L. Peng, G. N. Stacey, Z. Hu, G. Feng, W. Li, Y. Huo, R. Jin, N. Shyh-Chang, Q. Zhou, L. Wang, B. Hu, H. Dai and J. Hao, Cell Res., 2020, 30, 794–809 CrossRef CAS PubMed.
- Z. Li, Y. Wang, J. Liu, P. Rawding, J. Bu, S. Hong and Q. Hu, Adv. Mater., 2021, 33, 2102580 CrossRef CAS PubMed.
- S. Patyar, R. Joshi, D. S. Byrav, A. Prakash, B. Medhi and B. K. Das, J. Biomed. Sci., 2010, 17, 21 CrossRef CAS PubMed.
- E. W. Weber, M. V. Maus and C. L. Mackall, Cell, 2020, 181, 46–62 CrossRef CAS PubMed.
- C. Raffin, L. T. Vo and J. A. Bluestone, Nat. Rev. Immunol., 2020, 20, 158–172 CrossRef CAS PubMed.
- R. Chen, L. Li, L. Feng, Y. Luo, M. Xu, K. W. Leong and R. Yao, Biomaterials, 2020, 230, 119627 CrossRef CAS PubMed.
- Y. Xue, J. Che, X. Ji, Y. Li, J. Xie and X. Chen, Chem. Soc. Rev., 2022, 51, 1766–1794 RSC.
- Z. Liao, W. Zhang, H. Zheng, Y. Wang, J. Yu, H. Li and Z. Gu, J. Controlled Release, 2022, 344, 272–288 CrossRef CAS PubMed.
- X. Hu, Z. Xia and K. Cai, J. Mater. Chem. B, 2022, 10, 1486–1507 RSC.
- A. Isser, N. K. Livingston and J. P. Schneck, Biomaterials, 2021, 268, 120584 CrossRef CAS PubMed.
- J. H. Esensten, J. A. Bluestone and W. A. Lim, Annu. Rev. Phytopathol., 2017, 12, 305–330 CrossRef CAS PubMed.
- N. Zacharakis, H. Chinnasamy, M. Black, H. Xu, Y. C. Lu, Z. Zheng, A. Pasetto, M. Langhan, T. Shelton, T. Prickett, J. Gartner, L. Jia, K. Trebska-McGowan, R. P. Somerville, P. F. Robbins, S. A. Rosenberg, S. L. Goff and S. A. Feldman, Nat. Med., 2018, 24, 724–730 CrossRef CAS PubMed.
- S. J. Schuster, J. Svoboda, E. A. Chong, S. D. Nasta, A. R. Mato, O. Anak, J. L. Brogdon, I. Pruteanu-Malinici, V. Bhoj, D. Landsburg, M. Wasik, B. L. Levine, S. F. Lacey, J. J. Melenhorst, D. L. Porter and C. H. June, N. Engl. J. Med., 2017, 377, 2545–2554 CrossRef CAS PubMed.
- A. P. Rapoport, E. A. Stadtmauer, G. K. Binder-Scholl, O. Goloubeva, D. T. Vogl, S. F. Lacey, A. Z. Badros, A. Garfall, B. Weiss, J. Finklestein, I. Kulikovskaya, S. K. Sinha, S. Kronsberg, M. Gupta, S. Bond, L. Melchiori, J. E. Brewer, A. D. Bennett, A. B. Gerry, N. J. Pumphrey, D. Williams, H. K. Tayton-Martin, L. Ribeiro, T. Holdich, S. Yanovich, N. Hardy, J. Yared, N. Kerr, S. Philip, S. Westphal, D. L. Siegel, B. L. Levine, B. K. Jakobsen, M. Kalos and C. H. June, Nat. Med., 2015, 21, 914–921 CrossRef CAS PubMed.
- M. Martino, C. Alati, F. A. Canale, G. Musuraca, G. Martinelli and C. Cerchione, Int. J. Mol. Sci., 2021, 22, 2150 CrossRef CAS PubMed.
- S. Srivastava, S. N. Furlan, C. A. Jaeger-Ruckstuhl, M. Sarvothama, C. Berger, K. S. Smythe, S. M. Garrison, J. M. Specht, S. M. Lee, R. A. Amezquita, V. Voillet, V. Muhunthan, S. Yechan-Gunja, S. P.-S. Pillai, C. Rader, A. M. Houghton, R. H. Pierce, R. Gottardo, D. G. Maloney and S. R. Riddell, Cancer Cell, 2021, 39, 193–208e110 CrossRef CAS PubMed.
- A. Sarivalasis, M. Morotti, A. Mulvey, M. Imbimbo and G. Coukos, Ther. Adv. Med. Oncol., 2021, 13, 1–17 Search PubMed.
- B. Sun, D. Yang, H. Dai, X. Liu, R. Jia, X. Cui, W. Li, C. Cai, J. Xu and X. Zhao, Cancer Immunol. Res., 2019, 7, 1813–1823 CrossRef CAS PubMed.
- K. Pilipow, A. Darwich and A. Losurdo, Semin. Cancer Biol., 2021, 72, 90–101 CrossRef CAS PubMed.
- R. C. Larson and M. V. Maus, Nat. Rev. Cancer, 2021, 21, 145–161 CrossRef CAS PubMed.
- F. Moroni, B. J. Dwyer, C. Graham, C. Pass, L. Bailey, L. Ritchie, D. Mitchell, A. Glover, A. Laurie, S. Doig, E. Hargreaves, A. R. Fraser, M. L. Turner, J. D.-M. Campbell, N. W.-A. McGowan, J. Barry, J. K. Moore, P. C. Hayes, D. J. Leeming, M. J. Nielsen, K. Musa, J. A. Fallowfield and S. J. Forbes, Nat. Med., 2019, 25, 1560–1565 CrossRef CAS PubMed.
- N. R. Anderson, N. G. Minutolo, S. Gill and M. Klichinsky, Cancer Res., 2021, 81, 1201–1208 CrossRef CAS PubMed.
- Y. Xia, L. Rao, H. Yao, Z. Wang, P. Ning and X. Chen, Adv. Mater., 2020, 32, e2002054 CrossRef PubMed.
- M. Najafi, N. Hashemi Goradel, B. Farhood, E. Salehi, M. S. Nashtaei, N. Khanlarkhani, Z. Khezri, J. Majidpoor, M. Abouzaripour, M. Habibi, I. R. Kashani and K. Mortezaee, J. Cell. Biochem., 2019, 120, 2756–2765 CrossRef CAS PubMed.
- C. D. Mills, L. L. Lenz and R. A. Harris, Cancer Res., 2016, 76, 513–516 CrossRef CAS PubMed.
- K. Wu, K. Lin, X. Li, X. Yuan, P. Xu, P. Ni and D. Xu, Front. Immunol., 2020, 11, 1731 CrossRef CAS PubMed.
- M. Martinez and E. K. Moon, Front. Immunol., 2019, 10, 128 CrossRef CAS PubMed.
- C. Varol, A. Mildner and S. Jung, Annu. Rev. Immunol., 2015, 33, 643–675 CrossRef CAS PubMed.
- F. J. van Dalen, M. van Stevendaal, F. L. Fennemann, M. Verdoes and O. Ilina, Molecules, 2018, 24, 9 CrossRef PubMed.
- Y. Gong, R. G.-J. Klein Wolterink, J. Wang, G. M.-J. Bos and W. T.-V. Germeraad, J. Hematol. Oncol., 2021, 14, 73 CrossRef CAS PubMed.
- M. A. Morgan, H. Buning, M. Sauer and A. Schambach, Front. Immunol., 2020, 11, 1965 CrossRef CAS PubMed.
- D. Bollino and T. J. Webb, Transl. Res., 2017, 187, 32–43 CrossRef CAS PubMed.
- E. Liu, D. Marin, P. Banerjee, H. A. Macapinlac, P. Thompson, R. Basar, L. Nassif Kerbauy, B. Overman, P. Thall, M. Kaplan, V. Nandivada, I. Kaur, A. Nunez Cortes, K. Cao, M. Daher, C. Hosing, E. N. Cohen, P. Kebriaei, R. Mehta, S. Neelapu, Y. Nieto, M. Wang, W. Wierda, M. Keating, R. Champlin, E. J. Shpall and K. Rezvani, N. Engl. J. Med., 2020, 382, 545–553 CrossRef CAS PubMed.
- A. Cerwenka and L. L. Lanier, Nat. Rev. Immunol., 2016, 16, 112–123 CrossRef CAS PubMed.
- H. Klingemann, OncoImmunology, 2014, 3, e28147 CrossRef PubMed.
- G. M. Mantia-Smaldone and C. S. Chu, BioDrugs, 2013, 27, 453–468 CrossRef CAS PubMed.
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Nat. Rev. Immunol., 2020, 20, 7–24 CrossRef CAS PubMed.
- L. E. Fisher, L. Kammerling, M. R. Alexander and A. M. Ghaemmaghami, Curr. Opin. Biotechnol, 2021, 74, 194–203 CrossRef PubMed.
- J. A. Bluestone, Nat. Rev. Immunol., 2005, 5, 343–349 CrossRef CAS PubMed.
- M. Romano, G. Fanelli, C. J. Albany, G. Giganti and G. Lombardi, Front. Immunol., 2019, 10, 43 CrossRef CAS PubMed.
- J. Li, Q. Zhang, W. Wang, F. Lin, S. Wang and J. Zhao, J. Neurol., 2020, 268, 4095–4107 CrossRef PubMed.
- O. Lindvall and Z. Kokaia, Nature, 2006, 441, 1094–1096 CrossRef CAS PubMed.
- S. Chari, A. Nguyen and J. Saxe, Cell Stem Cell, 2018, 22, 781–782 CrossRef CAS PubMed.
- J. Galipeau and L. Sensebe, Cell Stem Cell, 2018, 22, 824–833 CrossRef CAS PubMed.
- G. Q. Daley, M. W. Lensch, R. Jaenisch, A. Meissner, K. Plath and S. Yamanaka, Cell Stem Cell, 2009, 4, 200–201 CrossRef CAS PubMed ; author reply 202.
- W. Chen, P. H. Tsai, Y. Hung, S. H. Chiou and C. Y. Mou, ACS Nano, 2013, 7, 8423–8440 CrossRef CAS PubMed.
- R. F. Aten, A. J. Eisenfeld, N. J. MacLusky and R. B. Hochberg, J. Steroid Biochem., 1982, 16, 447–449 CrossRef CAS PubMed.
- M. Mandai, A. Watanabe, Y. Kurimoto, Y. Hirami, C. Morinaga, T. Daimon, M. Fujihara, H. Akimaru, N. Sakai, Y. Shibata, M. Terada, Y. Nomiya, S. Tanishima, M. Nakamura, H. Kamao, S. Sugita, A. Onishi, T. Ito, K. Fujita, S. Kawamata, M. J. Go, C. Shinohara, K. I. Hata, M. Sawada, M. Yamamoto, S. Ohta, Y. Ohara, K. Yoshida, J. Kuwahara, Y. Kitano, N. Amano, M. Umekage, F. Kitaoka, A. Tanaka, C. Okada, N. Takasu, S. Ogawa, S. Yamanaka and M. Takahashi, N. Engl. J. Med., 2017, 376, 1038–1046 CrossRef CAS PubMed.
- C. D. Mohan, S. Rangappa, S. C. Nayak, R. Jadimurthy, L. Wang, G. Sethi, M. Garg and K. S. Rangappa, Semin. Cancer Biol., 2021 DOI:10.1016/j.semcancer.2021.05.006.
- S. Zhou, Nature, 2016, 536, 33–34 CrossRef CAS PubMed.
- S. Li, W. Jiang, C. Zheng, D. Shao, Y. Liu, S. Huang, J. Han, J. Ding, Y. Tao and M. Li, J. Controlled Release, 2020, 327, 801–833 CrossRef CAS PubMed.
- Z. Li, Y. Ding, J. Liu, J. Wang, F. Mo, Y. Wang, T. J. Chen-Mayfield, P. M. Sondel, S. Hong and Q. Hu, Nat. Commun., 2022, 13, 1845 CrossRef CAS PubMed.
- A. A. Abdeen and K. Saha, Trends Biotechnol., 2017, 35, 971–982 CrossRef CAS PubMed.
- D. Li, X. Li, W. L. Zhou, Y. Huang, X. Liang, L. Jiang, X. Yang, J. Sun, Z. Li, W. D. Han and W. Wang, Signal Transduction Targeted Ther., 2019, 4, 35 CrossRef PubMed.
- M. O. Li, N. Wolf, D. H. Raulet, L. Akkari, M. J. Pittet, P. C. Rodriguez, R. N. Kaplan, A. Munitz, Z. Zhang, S. Cheng and N. Bhardwaj, Cancer Cell, 2021, 39, 725–729 CrossRef CAS PubMed.
- D. C. Singleton, A. Macann and W. R. Wilson, Nat. Rev. Clin. Oncol., 2021, 18, 751–772 CrossRef PubMed.
- M. Hao, S. Hou, W. Li, K. Li, L. Xue, Q. Hu, L. Zhu, Y. Chen, H. Sun, C. Ju and C. Zhang, Sci. Transl. Med., 2020, 12, eaaz6667 CrossRef CAS PubMed.
- L. Tang, Y. Zheng, M. B. Melo, L. Mabardi, A. P. Castano, Y. Q. Xie, N. Li, S. B. Kudchodkar, H. C. Wong, E. K. Jeng, M. V. Maus and D. J. Irvine, Nat. Biotechnol., 2018, 36, 707–716 CrossRef CAS PubMed.
- M. E. Coon, S. B. Stephan, V. Gupta, C. P. Kealey and M. T. Stephan, Nat. Biomed. Eng., 2020, 4, 195–206 CrossRef CAS PubMed.
- Q. Hu, H. Li, E. Archibong, Q. Chen, H. Ruan, S. Ahn, E. Dukhovlinova, Y. Kang, D. Wen, G. Dotti and Z. Gu, Nat. Biomed. Eng., 2021, 5, 1038–1047 CrossRef CAS PubMed.
- E. A. Ogunnaike, A. Valdivia, M. Yazdimamaghani, E. Leon, S. Nandi, H. Hudson, H. Du, S. Khagi, Z. Gu, B. Savoldo, F. S. Ligler, S. Hingtgen and G. Dotti, Sci. Adv., 2021, 7, eabg5841 CrossRef CAS PubMed.
- C. W.-t Shields, M. A. Evans, L. L. Wang, N. Baugh, S. Iyer, D. Wu, Z. Zhao, A. Pusuluri, A. Ukidve, D. C. Pan and S. Mitragotri, Sci. Adv., 2020, 6, eaaz6579 CrossRef CAS PubMed.
- X. Hou, X. Zhang, W. Zhao, C. Zeng, B. Deng, D. W. McComb, S. Du, C. Zhang, W. Li and Y. Dong, Nat. Nanotechnol., 2020, 15, 41–46 CrossRef CAS PubMed.
- Y. H. Ahn, L. Ren, S. M. Kim, S. H. Seo, C. R. Jung, D. S. Kim, J. Y. Noh, S. Y. Lee, H. Lee, M. Y. Cho, H. Jung, S. R. Yoon, J. E. Kim, S. N. Lee, S. Kim, I. W. Shin, H. S. Shin, K. S. Hong, Y. T. Lim, I. Choi and T. D. Kim, Biomaterials, 2020, 247, 119960 CrossRef CAS PubMed.
- S. Yang, J. Wen, H. Li, L. Xu, Y. Liu, N. Zhao, Z. Zeng, J. Qi, W. Jiang, W. Han and Y. Zu, Small, 2019, 15, e1900903 CrossRef PubMed.
- J. Li, M. Chen, Z. Liu, L. Zhang, B. H. Felding, K. W. Moremen, G. Lauvau, M. Abadier, K. Ley and P. Wu, ACS Cent. Sci., 2018, 4, 1633–1641 CrossRef CAS PubMed.
- S. Im, D. Jang, G. Saravanakumar, J. Lee, Y. Kang, Y. M. Lee, J. Lee, J. Doh, Z. Y. Yang, M. H. Jang and W. J. Kim, Adv. Mater., 2020, 32, e2000020 CrossRef PubMed.
- S. Srinivasan and J. E. Babensee, ACS Biomater. Sci. Eng., 2020, 6, 4062–4076 CrossRef CAS PubMed.
- A. M. Thomas, N. M. Beskid, J. L. Blanchfield, A. M. Rosado, A. J. Garcia, B. D. Evavold and J. E. Babensee, J. Biomed. Mater. Res., Part A, 2021, 109, 1247–1255 CrossRef CAS PubMed.
- Y. Liu, Y. Han, H. Dong, X. Wei, D. Shi and Y. Li, ACS Appl. Mater. Interfaces, 2020, 12, 4163–4173 CrossRef CAS PubMed.
- L. Yu, R. Feng, L. Zhu, Q. Hao, J. Chu, Y. Gu, Y. Luo, Z. Zhang, G. Chen and H. Chen, Sci. Adv., 2020, 6, eabb6595 CrossRef PubMed.
- J. Kim, W. A. Li, W. Sands and D. J. Mooney, ACS Appl. Mater. Interfaces, 2014, 6, 8505–8512 CrossRef CAS PubMed.
- N. J. Shah, A. J. Najibi, T. Y. Shih, A. S. Mao, A. Sharda, D. T. Scadden and D. J. Mooney, Nat. Biomed. Eng., 2020, 4, 40–51 CrossRef CAS PubMed.
- Z. Liu, X. Chen, Z. Zhang, X. Zhang, L. Saunders, Y. Zhou and P. X. Ma, ACS Nano, 2018, 12, 9785–9799 CrossRef CAS PubMed.
- J. D. Fisher, W. Zhang, S. C. Balmert, A. M. Aral, A. P. Acharya, Y. Kulahci, J. Li, H. R. Turnquist, A. W. Thomson, M. G. Solari, V. S. Gorantla and S. R. Little, Sci. Adv., 2020, 6, eaax8429 CrossRef CAS PubMed.
- J. G. Graham, X. Zhang, A. Goodman, K. Pothoven, J. Houlihan, S. Wang, R. M. Gower, X. Luo and L. D. Shea, Tissue Eng., Part A, 2013, 19, 1465–1475 CrossRef CAS PubMed.
- A. Gonzalez-Pujana, K. H. Vining, D. K.-Y. Zhang, E. Santos-Vizcaino, M. Igartua, R. M. Hernandez and D. J. Mooney, Biomaterials, 2020, 257, 120266 CrossRef CAS PubMed.
- M. T. Sultan, B. Y. Choi, O. Ajiteru, D. K. Hong, S. M. Lee, H. J. Kim, J. S. Ryu, J. S. Lee, H. Hong, Y. J. Lee, H. Lee, Y. J. Suh, O. J. Lee, S. H. Kim, S. W. Suh and C. H. Park, Biomaterials, 2021, 266, 120413 CrossRef CAS PubMed.
- A. S. Mao, B. Ozkale, N. J. Shah, K. H. Vining, T. Descombes, L. Zhang, C. M. Tringides, S. W. Wong, J. W. Shin, D. T. Scadden, D. A. Weitz and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15392–15397 CrossRef CAS PubMed.
- J. Tang, X. Cui, T. G. Caranasos, M. T. Hensley, A. C. Vandergriff, Y. Hartanto, D. Shen, H. Zhang, J. Zhang and K. Cheng, ACS Nano, 2017, 11, 9738–9749 CrossRef CAS PubMed.
- J. Tang, T. Su, K. Huang, P. U. Dinh, Z. Wang, A. Vandergriff, M. T. Hensley, J. Cores, T. Allen, T. Li, E. Sproul, E. Mihalko, L. J. Lobo, L. Ruterbories, A. Lynch, A. Brown, T. G. Caranasos, D. Shen, G. A. Stouffer, Z. Gu, J. Zhang and K. Cheng, Nat. Biomed. Eng., 2018, 2, 17–26 CrossRef CAS PubMed.
- A. C. Vandergriff, T. M. Hensley, E. T. Henry, D. Shen, S. Anthony, J. Zhang and K. Cheng, Biomaterials, 2014, 35, 8528–8539 CrossRef CAS PubMed.
- Z. Li, D. Shen, S. Hu, T. Su, K. Huang, F. Liu, L. Hou and K. Cheng, ACS Nano, 2018, 12, 12193–12200 CrossRef CAS PubMed.
- D. T. Riglar and P. A. Silver, Nat. Rev. Microbiol., 2018, 16, 214–225 CrossRef CAS PubMed.
- Z. Geng, Z. Cao, R. Liu, K. Liu, J. Liu and W. Tan, Nat. Commun., 2021, 12, 6584 CrossRef CAS PubMed.
- J. Liu, W. Li, Y. Wang, Y. Ding, A. Lee and Q. Hu, Nano Today, 2021, 41, 101291 CrossRef CAS.
- A. M. Nash, M. I. Jarvis, S. Aghlara-Fotovat, S. Mukherjee, A. Hernandez, A. D. Hecht, P. D. Rios, S. Ghani, I. Joshi, D. Isa, Y. Cui, S. Nouraein, J. Z. Lee, C. Xu, D. Y. Zhang, R. A. Sheth, W. Peng, J. Oberholzer, O. A. Igoshin, A. A. Jazaeri and O. Veiseh, Sci. Adv., 2022, 8, eabm1032 CrossRef CAS PubMed.
- S. Bose, L. R. Volpatti, D. Thiono, V. Yesilyurt, C. McGladrigan, Y. Tang, A. Facklam, A. Wang, S. Jhunjhunwala, O. Veiseh, J. Hollister-Lock, C. Bhattacharya, G. C. Weir, D. L. Greiner, R. Langer and D. G. Anderson, Nat. Biomed. Eng., 2020, 4, 814–826 CrossRef CAS PubMed.
- A. J. Vegas, O. Veiseh, M. Gurtler, J. R. Millman, F. W. Pagliuca, A. R. Bader, J. C. Doloff, J. Li, M. Chen, K. Olejnik, H. H. Tam, S. Jhunjhunwala, E. Langan, S. Aresta-Dasilva, S. Gandham, J. J. McGarrigle, M. A. Bochenek, J. Hollister-Lock, J. Oberholzer, D. L. Greiner, G. C. Weir, D. A. Melton, R. Langer and D. G. Anderson, Nat. Med., 2016, 22, 306–311 CrossRef CAS PubMed.
- M. A. Bochenek, O. Veiseh, A. J. Vegas, J. J. McGarrigle, M. Qi, E. Marchese, M. Omami, J. C. Doloff, J. Mendoza-Elias, M. Nourmohammadzadeh, A. Khan, C. C. Yeh, Y. Xing, D. Isa, S. Ghani, J. Li, C. Landry, A. R. Bader, K. Olejnik, M. Chen, J. Hollister-Lock, Y. Wang, D. L. Greiner, G. C. Weir, B. L. Strand, A. M.-A. Rokstad, I. Lacik, R. Langer, D. G. Anderson and J. Oberholzer, Nat. Biomed. Eng., 2018, 2, 810–821 CrossRef CAS PubMed.
- X. Wang, K. G. Maxwell, K. Wang, D. T. Bowers, J. A. Flanders, W. Liu, L. H. Wang, Q. Liu, C. Liu, A. Naji, Y. Wang, B. Wang, J. Chen, A. U. Ernst, J. M. Melero-Martin, J. R. Millman and M. Ma, Sci. Transl. Med., 2021, 13, eabb4601 CrossRef CAS PubMed.
- J. Yu, Y. Zhang, Y. Ye, R. DiSanto, W. Sun, D. Ranson, F. S. Ligler, J. B. Buse and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8260–8265 CrossRef CAS PubMed.
- Y. Ye, J. Yu, C. Wang, N. Y. Nguyen, G. M. Walker, J. B. Buse and Z. Gu, Adv. Mater., 2016, 28, 3115–3121 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |