
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Olefin hydroboration catalyzed by an iron-borane complex†
Laura A.
Grose
and
Darren
Willcox
 *
*
Department of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: darren.willcox@manchester.ac.uk
First published on 23rd May 2023
Abstract
The well-defined iron(0) complex [(iPrDPBPh)Fe2-(μ-1,2-N2)] (A) reacts with HBpin to afford the complex [(η3-H2iPrDPB)Fe(η3-H2Bpin)] (B) via oxidative addition of the H–B bond. Complex A is an effective pre-catalyst for the hydroboration of a range of olefins in synthetically useful yields (typically >80%) under neat conditions.
Reduction of unsaturated carbon-carbon and carbon-heteroatom bonds by hydrofunctionalization is of paramount importance to the synthesis of pharmaceuticals and agrochemicals.1 Amongst the hydrofunctionalization reactions, hydroboration has proven to be a powerful reaction in the generation of value-added reagents for organic synthesis.2 This is in part due to the wide availability of boranes coupled with their high reactivity and selectivity towards unsaturated substrates. To date, many catalytic systems have been reported for hydroboration, with a large proportion being based on transition metals.2,3 However, it is Earth-abundant catalysts which are taking centre stage, exhibiting high levels of activity in the hydroboration of unsaturated C–C bonds.4–8 Of these metals, iron has proved to be popular in novel catalytic systems.9,10
Arguably, the most common approach for iron-catalyzed olefin hydroboration is via the use of a neutral ligand in the presence of Fe(II) salts, and a catalytic amount of an activator, namely Grignard reagents or hydride additives.11 However, there are a limited number of reports which use molecular-defined low valent iron pre-catalysts. The Chirik group have reported that a bis(imino)pyridine iron dinitrogen complex (Fe1) was efficient for the hydroboration of terminal and disubstituted olefins with 1 mol% catalyst loading under neat conditions (Scheme 1a).12 The group of Darcel reported an efficient NHC-iron complex (Fe2) which facilitated the hydroboration of functionalized terminal olefins using HBpin with 5 mol% catalyst loading under photochemical conditions (Scheme 1b).13
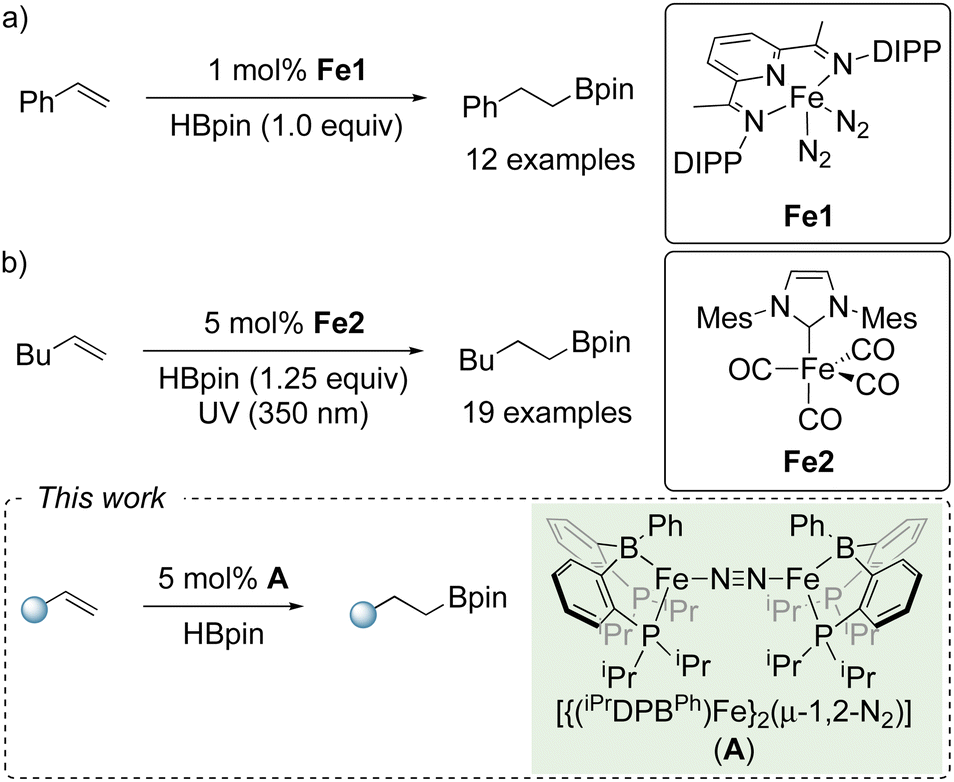 | ||
| Scheme 1 (a) and (b) Previous studies using well-defined iron complexes as catalysts for olefin hydroboration; (c) this work. DIPP = 2,6-diisopropylphenyl and Mes = 2,4,6-trimethylphenyl. | ||
Metal-borane complexes are emerging as promising systems for cooperative catalysis. It has been recently demonstrated that late first-row transition metal-borane complexes can readily facilitate well-defined two-electron chemistry. To date, catalysis using these systems is limited to a handful of examples, such as olefin hydrogenation (using Ni), and hydrosilylation of aldehydes and ketones (Co, Fe and Ni) all proceeding via addition of E–H (E = H or Ph2SiH) across the metal-borane interaction leading to the formation of a borohydrido-hydride species.14
Motivated by this bifunctional reactivity and its untapped potential for realizing well-defined two-electron catalytic processes with iron, we sought to broaden the scope of catalytic transformations facilitated by iron-borane complexes, namely [{(iPrDPBPh)Fe}2(μ-1,2-N2)] (A), which was first developed by Peters.14f Herein, we report the activation of the B–H bond in HBpin across the Fe–B core in complex A, and subsequent application in olefin hydroboration (Scheme 1c).
Firstly, a stoichiometric reaction between complex A and HBpin was conducted. Treatment of A with an excess of HBpin (6 equivalents) at 50 °C in benzene-d6 for 24 h afforded diamagnetic (S = 0) complex B in 85% isolated yield (Scheme 2a). When 20 equivalents of HBpin were added no rate enhancement was observed, with the same yield obtained after 24 hours. Complex B could be purified by silica gel column chromatography eluting with hexanes and exhibits good air-stability, with no decomposition observed after 7 days. The 1H NMR spectrum of complex B features two upfield proton resonances (δ = −16.71 ppm and −20.68 ppm) corresponding to iron-hydride signals, which integrate in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with respect to each other (Fig. 1a). The hydride signal at −16.71 ppm is comparable with reports by Wang and Song for η3-H2Bpin complexes.15 The 31P NMR spectrum displays a broad triplet at 98.1 ppm with 2JPH = 17.8 Hz. The 11B NMR spectrum displays two resonances at δ = 36.3 ppm and δ = 47.9 ppm, diagnostic of two chemically inequivalent boron centres. From 1H-11B correlation spectroscopy, the proton resonance at δ = −16.71 ppm interacts only with the boron resonance at δ = 47.9 ppm, whereas the resonance at δ = −20.68 ppm interacts exclusively with the boron resonance at δ = 36.3 ppm, indicating two discrete borohydride-type fragments. The 11B NMR resonance at δ = 47.9 ppm is in good agreement with that reported by Lin and Peters for a η3-H2BR2 hydroborate complex,16 and the δ = 36.3 resonance is in good agreement with a recently reported η3-H2Bpin complex.15b Isotopic labelling studies using DBpin showed the corresponding deuterides in the 2H NMR spectrum at the same positions as hydride resonances in the 1H NMR spectrum. The infrared spectrum of complex B contains a broad and intense stretch at ca. 1900 cm-1 corresponding to a Fe–H–B fragment. As expected, when using DBpin, this stretching frequency is sensitive to the isotopic labelling (νFe-D-Bca. 1300 cm-1, see ESI†).14f
:1 ratio with respect to each other (Fig. 1a). The hydride signal at −16.71 ppm is comparable with reports by Wang and Song for η3-H2Bpin complexes.15 The 31P NMR spectrum displays a broad triplet at 98.1 ppm with 2JPH = 17.8 Hz. The 11B NMR spectrum displays two resonances at δ = 36.3 ppm and δ = 47.9 ppm, diagnostic of two chemically inequivalent boron centres. From 1H-11B correlation spectroscopy, the proton resonance at δ = −16.71 ppm interacts only with the boron resonance at δ = 47.9 ppm, whereas the resonance at δ = −20.68 ppm interacts exclusively with the boron resonance at δ = 36.3 ppm, indicating two discrete borohydride-type fragments. The 11B NMR resonance at δ = 47.9 ppm is in good agreement with that reported by Lin and Peters for a η3-H2BR2 hydroborate complex,16 and the δ = 36.3 resonance is in good agreement with a recently reported η3-H2Bpin complex.15b Isotopic labelling studies using DBpin showed the corresponding deuterides in the 2H NMR spectrum at the same positions as hydride resonances in the 1H NMR spectrum. The infrared spectrum of complex B contains a broad and intense stretch at ca. 1900 cm-1 corresponding to a Fe–H–B fragment. As expected, when using DBpin, this stretching frequency is sensitive to the isotopic labelling (νFe-D-Bca. 1300 cm-1, see ESI†).14f
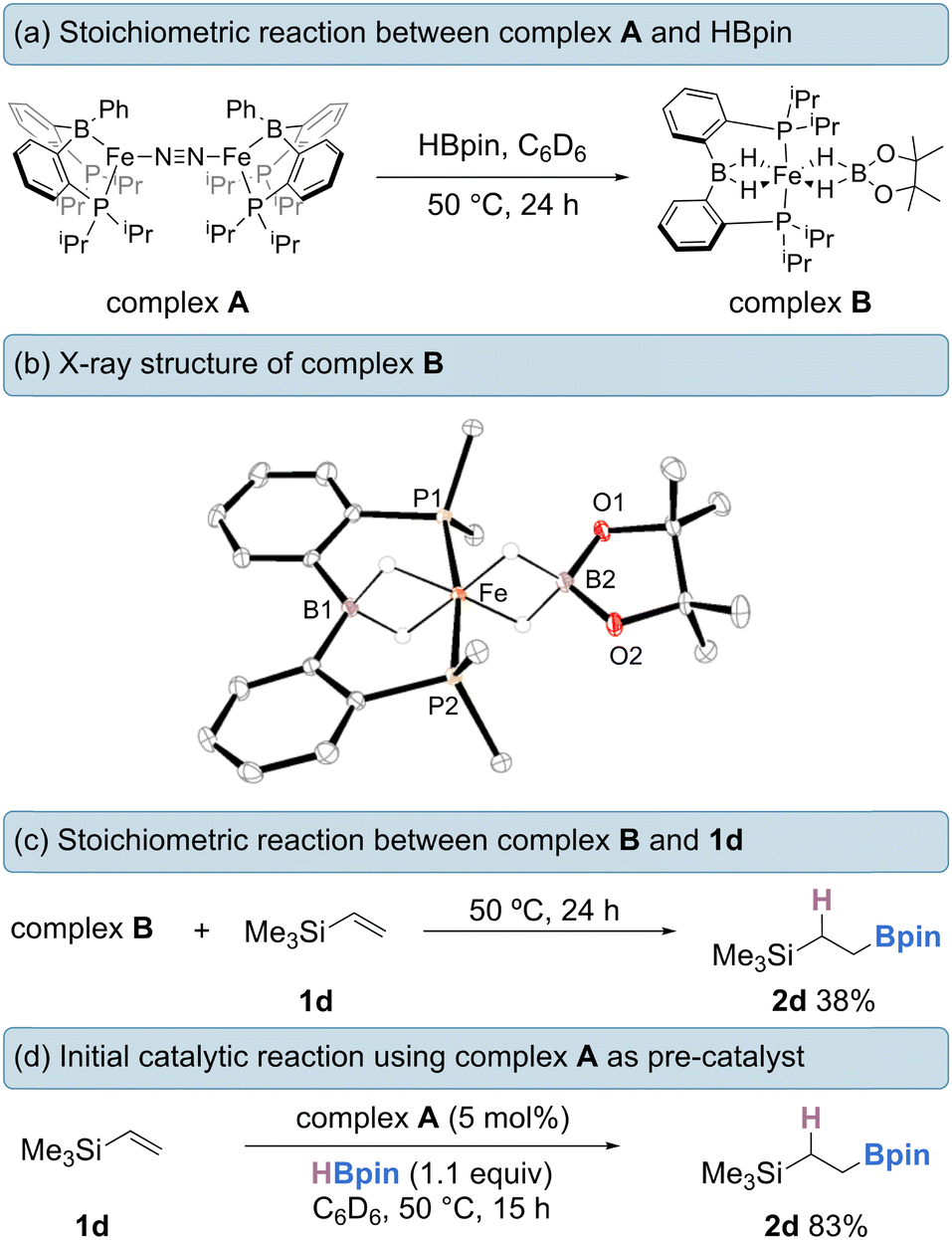 | ||
| Scheme 2 (a) Stoichiometric reaction of complex A with HBpin. (b) solid state structure of complex B with selected atomic labelling; methyl groups and hydrogen atoms not bonded to Fe omitted for clarity, ellipsoids drawn at the 50% probability level. (c) stoichiometric reaction of B with olefin and (d) initial catalytic reaction. | ||
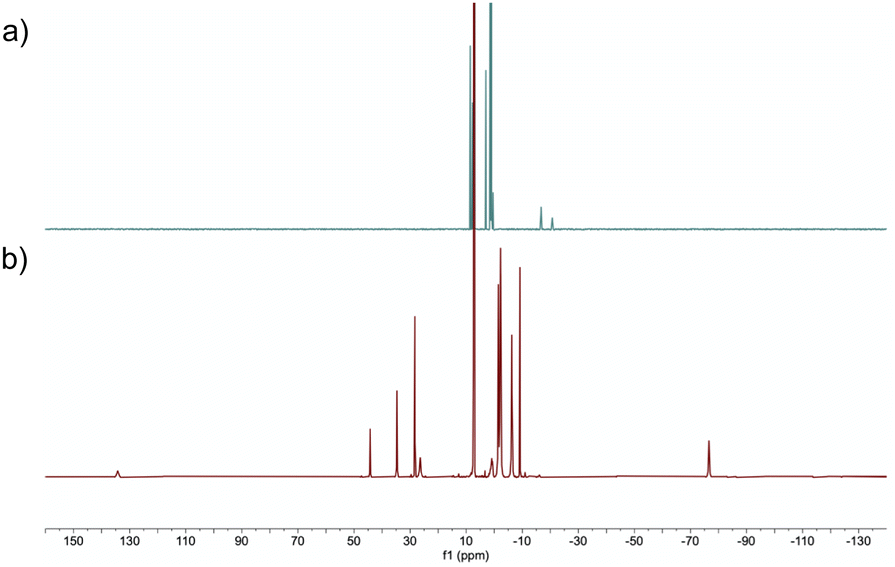 | ||
| Fig. 1 1H NMR spectra of (a) complex B and (b) complex A. | ||
The solid-state structure of complex B was confirmed by single-crystal X-ray diffraction from crystals obtained by slow-evaporation from diethyl ether (Scheme 2b). Complex B adopts a distorted octahedral geometry with a P–Fe–P bond angle of 170.32°. The Fe–B1 distance of 2.082(2) Å and Fe–B2 distance of 2.037(2) Å lie within the reported range for Fe(II) hydridoborates.15,17–20 The B1–H distances in complex B are both 1.30 Å, whereas the B2–H bonds are slightly longer at 1.35 Å and 1.36 Å. Both sets of values are comparable with reported hydridoborate species of η3-H2Bpin and η3-H2BR2 fragments.15a,16
Complex B could presumably form via initial oxidative addition of HBpin by complex A, followed by reductive elimination of PhBpin (observed by NMR spectroscopy) to generate an iron(0) species. Analogous transformations have been reported with rhodium complexes bearing the same ligand framework.21 Oxidative addition of another equivalent of HBpin to the new Fe(0) species, followed by metathesis of the iron-boryl bond with HBpin, furnishes an iron(II)-hydride species. Finally, HBpin is captured and bonded to the Fe–H unit in the η3-H2Bpin mode, leading to complex B, an 18-valence electron complex. Further investigations into the formation of complex B are currently ongoing within our laboratory.
The stoichiometric treatment of complex B with vinyltrimethylsilane (1d) led to the formation of the desired alkylboronate ester 2d in 38% yield (Scheme 2c). When employed as a pre-catalyst (5 mol%), complex B gave none of the desired product. Using complex A as the catalyst (5 mol%), the intermolecular hydroboration of 1d with 1.1 equivalents of HBpin proceeded smoothly, giving the anti-Markovnikov hydroboration product 2d in 83% yield, suggesting that complex A could be a suitable pre-catalyst for olefin hydroboration (Scheme 2d).
To examine the feasibility of A as a potential pre-catalyst for hydroboration, vinyltrimethylsilane (1d) was selected as the model substrate with HBpin as the terminal reductant. After systematic evaluation of various reaction parameters (see ESI† for full optimization), the desired hydroboration product 2d was obtained in 98% yield (Table 1, entry 1), with 5 mol% complex A as the pre-catalyst, 1.1 equivalents of HBpin at 50 °C under neat conditions (Table 1, entry 1). With reduced catalyst loading the reaction took place, albeit with much lower yield (entry 2). In the absence of the iron catalyst, no reaction was observed (entry 3). Solvent effects were also explored. The solvent-free reaction gave superior yields, however non-polar solvents such as benzene or methylcyclohexane lead to a good yield of 2d whereas reactions in Lewis basic solvents such as diethyl ether led to lower yields (entries 4–6). Performing the reaction for shorter durations or lower temperatures lowered the product yield (entries 7 and 8). Finally, the reaction also proceeded when higher equivalents of HBpin were used, with little effect on the yield (entry 9).
|
|
||
|---|---|---|
| Entry | Deviation from standanrd conditions | Yieldb (%) |
| a Conditions: 1d (0.205 mmol), HBpin (0.225 mmol), complex A (5 mol%) at 50 °C for 15 h. b isolated yields. | ||
| 1 | None | 98 |
| 2 | 2 mol% catalyst | 19 |
| 3 | Without complex A | 0 |
| 4 | Benzene as solvent | 83 |
| 5 | Methylcyclohexane as solvent | 81 |
| 6 | Diethyl ether as solvent | 68 |
| 7 | 6 hours reaction time | 45 |
| 8 | Room temperature | 9 |
| 9 | 1.5 equivalents of HBpin | 95 |
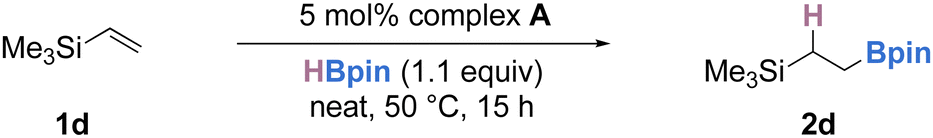
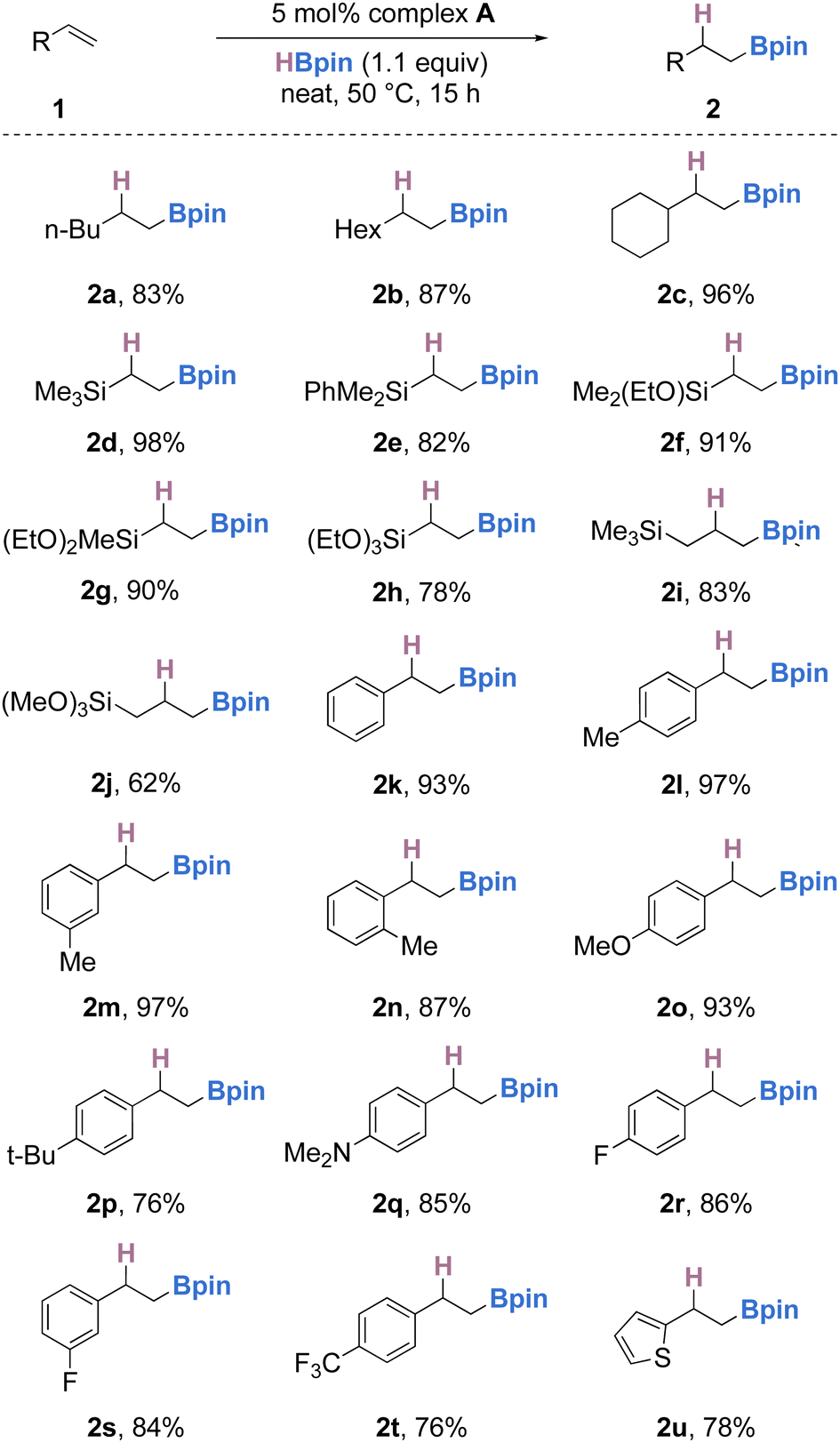
With optimized conditions in hand (5 mol% of A, 1.0 equiv. of olefin, 1.1 equiv. of pinacolborane at 50 °C under neat conditions for 15 hours), we then turned our attention to the scope and limitations of this transformation (Scheme 3). Firstly, simple aliphatic terminal olefins such as 1-hexene, 1-octene and vinyl-cyclohexane were selectively hydroborated, furnishing the anti-Markovnikov alkylboronate esters (2a–c) in 83–96% yield. Simple vinyl silanes bearing either alkyl, aryl or alkoxy substituents (or a combination thereof) also underwent smooth hydroboration to the anti-Markovnikov products in good to excellent yields (2d–j). Styrene and a range of functionalized styrenes bearing electron donating groups such as methyl, methoxy or dimethylamino were successfully hydroborated to the corresponding alkylboronates in 76–97% yield (2k–q). Electron-deficient styrenes bearing fluorides or trifluoromethyl substituents were also well-tolerated (2r–t); however, when other halide (chloride or bromide) substituted styrenes were used, an intractable mixture of products was formed. Simple olefins bearing heterocycles, such as thiophene, could also be hydroborated in 78% yield (2u). From these results, it is evident that electronic effects of the substituents on the aryl ring have a slight effect, with more electron rich olefins undergoing hydroboration in slightly higher yields (2ovs. 2kvs. 2t). The effect of steric hindrance had little effect on the outcome of the reaction, although ortho-substituted styrenes delivered the olefin in slightly diminished yields when compared to the meta/para substituted styrenes (2lvs. 2mvs. 2n). Olefins bearing an epoxide on the lateral chain or 1,1-disubstituted olefins either gave an inseparable mixture or showed no reactivity with pinacolborane under optimized conditions.
 | ||
| Scheme 3 Scope of olefin hydroboration. | ||
Hydroboration of p-acetoxystyrene (1v) with 1 equivalent of HBpin led to a mixture of products containing the desired hydroboration product, but ester reduction was also observed. However, performing the reaction with 3 equivalents of HBpin led to the clean formation of 2v in 60% yield (Scheme 4).
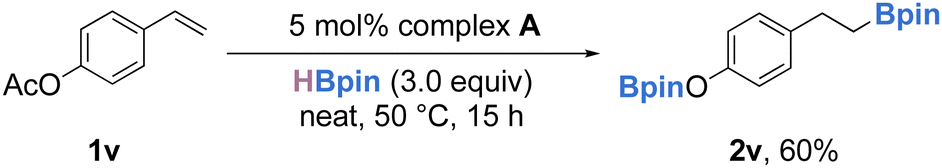 | ||
| Scheme 4 Hydroboration of p-acetoxystyrene. | ||
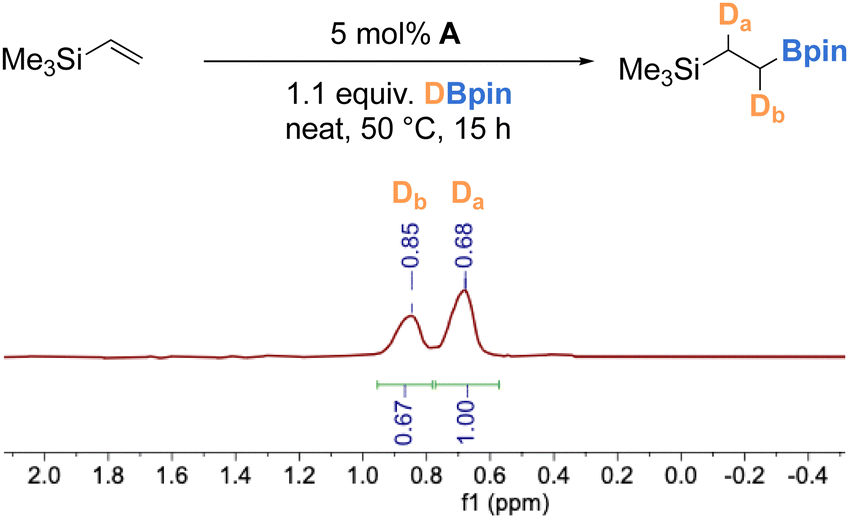
To gain mechanistic insight into the catalytic hydroboration reaction using complex A, a deuterium labelling experiment was performed with DBpin. Using our optimized conditions for catalytic hydroboration of vinyltrimethylsilane, deuterium was incorporated into the desired alkylboronate ester product with a roughly 0.67 (Db, δ = 0.85 ppm) to 1 (Da, δ = 0.68 ppm) distribution of deuterium at the terminal and internal positions (Scheme 5). This observation is in accord with the presence of a borane-derived iron hydride/deuteride intermediate over the duration of the catalytic reaction, and indicates that the olefin insertion step is reversible and proceeds without selectivity.
 | ||
| Scheme 5 Deuterium labelling study to gain mechanistic insight into the catalytic reaction. | ||
In conclusion, complex A reacts with excess HBpin to generate [(η3-H2iPrDPB)Fe(η3-H2Bpin)] (complex B) which has been characterised by multinuclear NMR, IR spectroscopy and single crystal X-ray diffraction. Complex A was also found to be a competent pre-catalyst for the hydroboration of a range of olefins leading to the anti-Markovnikov alkylboronates in good yields. We posit that the cooperative nature of the system herein is crucial for reactivity of the DPB-iron complexes, and this is something that we are exploring in our laboratory.
We thank the University of Manchester (EPSRC DTG) for a studentship (LAG) and the UK EPSRC for funding (DW, EP/W023172/1). We also thank Ralph Adams (NMR spectroscopy), Martin Jennings (elemental analysis) and Gareth Smith (mass spectrometry) for analytical services. We thank the referees for the suggestions provided for improving this manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Hydrofunctionalization, ed. V. P. Ananikov and M. Tanaka, Springer, Berlin, 2011 Search PubMed; (b) J. Seyden-Penne, Re-ductions by Alumino and Borohydrides in Organic Synthesis, Wiley-VCH, New York, 2nd edn, 1997 Search PubMed; (c) Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC.
- C. M. Vogels and S. A. Westcott, Curr. Org. Chem., 2005, 9, 687–699 CrossRef CAS.
- (a) Catalysis without Precious Metals, ed. Bullock, R. M., Wiley-VCH, Weinheim, 2010 Search PubMed; (b) Non-Noble Metal Catalysis: Molecular Approaches and Reactions, ed. R. J. Klein-Gebbink, and M.-E. Moret, Wiley-VCH, Weinheim, 2019 Search PubMed.
- S. R. Tamang and M. Findlater, Molecules, 2019, 24, 3194 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS.
- J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- M. L. Shegavi and S. K. Bose, Catal. Sci. Technol., 2019, 9, 3307–3336 RSC.
- (a) L. Zhang, D. Peng, X. Leng and Z. Huang, Angew. Chem., 2013, 125, 3764–3768 CrossRef; (b) M. D. Greenhalgh and S. P. Thomas, Chem. Commun., 2013, 49, 11230–11232 RSC; (c) R. Gilbert-Wilson, W.-Y. Chu and T. B. Rauchfuss, Inorg. Chem., 2015, 54, 5596–5603 CrossRef CAS PubMed; (d) K.-N. T. Tseng, J. W. Kampf and N. K. Szymczak, ACS Catal., 2015, 5, 411–415 CrossRef CAS; (e) M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem. – Eur. J., 2016, 22, 11605–11608 CrossRef CAS PubMed; (f) T. Ogawa, A. J. Ruddy, O. L. Sydora, M. Stradiotto and L. Turculet, Organometallics, 2017, 36, 417–423 CrossRef CAS; (g) X. Chen, Z. Cheng and Z. Lu, Org. Lett., 2017, 19, 969–971 CrossRef CAS PubMed; (h) A. J. MacNair, C. R. Millet, G. S. Nichol, A. Ironmonger and S. P. Thomas, ACS Catal., 2016, 6, 7217–7221 CrossRef CAS; (i) J. Chen, T. Xi and Z. Lu, Org. Lett., 2014, 16, 6452–6455 CrossRef CAS PubMed.
- J. V. Obligacíon and P. J. Chirik, Org. Lett., 2013, 15, 2680–2683 CrossRef PubMed.
- J. Zheng, J.-B. Sortais and C. Darcel, ChemCatChem, 2014, 6, 763–766 CrossRef CAS.
- (a) W. Hill Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef PubMed; (b) S. N. MacMillan, W. Hill Harman and J. C. Peters, Chem. Sci., 2014, 5, 590–597 RSC; (c) M. A. Nesbit, D. L. M. Suess and J. C. Peters, Organometallics, 2015, 34, 4741–4752 CrossRef CAS; (d) H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 10, 3053–3062 CrossRef PubMed; (e) H. Fong and J. C. Peters, Inorg. Chem., 2015, 11, 5124–5134 CrossRef PubMed; (f) D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 4938–4941 CrossRef CAS PubMed; (g) D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12580–12583 CrossRef CAS PubMed.
- (a) R. Sun, W. H. Deng, B. Yu, Y. Lu, X. Zhai, R. Z. Liao, C.-H. Tung and W. Wang, Organometallics, 2022, 41, 2504–2512 CrossRef CAS; (b) Q. Liang, H. A. G. Mayerstein and D. Song, Organometallics, 2023, 42(9), 816–824 CrossRef CAS.
- T.-Z. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed.
- M. P. Mehn, S. D. Brown, T. K. Paine, W. W. Brennessel, C. J. Cramer, J. C. Peters and J. L. Que, Dalton Trans., 2006, 1347–1351 RSC.
- C. A. Ghilardi, P. Innocenti, S. Midollini and A. Orlandini, J. Chem. Soc., Dalton Trans., 1985, 605–609 RSC.
- R. Langer, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 7196–7209 CrossRef CAS PubMed.
- Y. Ohki, T. Hatanaka and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 17174–17186 CrossRef CAS PubMed.
- W.-C. Shih, W. Gu, M. C. MacInnis, D. E. Herbert and O. V. Ozerov, Organometallics, 2017, 36, 1718–1726 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and spectral characterisation. CCDC 2260204. See DOI: https://doi.org/10.1039/d3cc00494e |
| This journal is © The Royal Society of Chemistry 2023 |