
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-enhanced dehydrogenation of formic acid on Pd-based hybrid plasmonic nanostructures
Jiannan
Zhu
a,
Jiawei
Dai
a,
You
Xu
a,
Xiaoling
Liu
a,
Zhengyun
Wang
a,
Hongfang
Liu
 a and
Guangfang
Li
*ab
a and
Guangfang
Li
*ab
aKey Laboratory of Material Chemistry for Energy Conversion and Storage (Ministry of Education), Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, PR China. E-mail: guangfl@hust.edu.cn
bShenzhen Huazhong University of Science and Technology Research Institute, Shenzhen, 518000, PR China
First published on 9th November 2023
Abstract
Coupling visible light with Pd-based hybrid plasmonic nanostructures has effectively enhanced formic acid (FA) dehydrogenation at room temperature. Unlike conventional heating to achieve higher product yield, the plasmonic effect supplies a unique surface environment through the local electromagnetic field and hot charge carriers, avoiding unfavorable energy consumption and attenuated selectivity. In this minireview, we summarized the latest advances in plasmon-enhanced FA dehydrogenation, including geometry/size-dependent dehydrogenation activities, and further catalytic enhancement by coupling local surface plasmon resonance (LSPR) with Fermi level engineering or alloying effect. Furthermore, some representative cases were taken to interpret the mechanisms of hot charge carriers and the local electromagnetic field on molecular adsorption/activation. Finally, a summary of current limitations and future directions was outlined from the perspectives of mechanism and materials design for the field of plasmon-enhanced FA decomposition.
Introduction
The global environmental problems and energy crises caused by the exhaustion of fossil fuels have forced us to search for feasible alternatives. As green and efficient energy, hydrogen (H2) is the most promising alternative to traditional fossil fuels because of its high calorific value and non-toxic emission.1–3 However, the storage and transportation of hydrogen is the biggest challenge for large-scale hydrogen energy utilisation. Organic liquid hydrogen carriers provide an effective guide for hydrogen storage and transportation. Formic acid (FA, HCOOH) is considered as a promising hydrogen carrier owing to its nontoxicity, wide availability from biomass raw materials, and considerable hydrogen capacity (4.4 wt%).4–8 H2 release from FA is thermodynamically feasible, while the kinetics is extremely slow. Pd is viewed as the most efficient heterogeneous catalyst for FA dehydrogenation (HCOOH → H2 + CO2).7,9,10 However, its catalytic performance is still not satisfying.During the past decades, much effort has been devoted to utilizing light energy to boost the catalytic dehydrogenation of Pd catalysts.11–15 In particular, when coupled with plasmonic noble metals such as Au or Ag, the Pd-based plasmonic hybrid mechanism demonstrated a greatly improved catalytic selectivity, activity, and toxicity resistance for catalytic FA decomposition under light illumination in a mild environment.11,14,16 This hybrid plasmonic system simultaneously acts as a light absorber and a catalytically active site. Such an enhancement mechanism originates from the plasmonic effect.
Upon illumination, plasmonic nanoparticles (i.e., Au, Ag, Cu, Al) support localized surface plasmon resonance (LSPR)-collective oscillations of electrons at the surface when the natural frequency of the electron coincides with the incident wavelength.20–32 A key LSPR feature is the large extinction associated with the surface oscillating electromagnetic field (Fig. 1a),17 which is closely related to the size, composition and geometry of plasmonic particles.21,25,33–37 As shown in Fig. 1b, a decay of electron collective oscillations generates radiative scattering of photons or non-thermal energetic carriers (hot electrons and hot holes) at the femtosecond scale, which then redistributed to form thermal hot carriers through electron–electron scattering.18 Finally, the electron–hole pair relaxation occurs via electron–phonon and phonon–phonon scattering, and the energy dissipates into the environment over a longer time horizon, resulting in localized heating effects. For pure Au and Ag nanoparticles, their extinction ranges concentrate in the visible region and are widely used as platforms for light absorption (Fig. 1c).19 The FA dehydrogenation rate can be significantly enhanced through the LSPR-induced electromagnetic field and hot charge carriers by rationally combining a plasmonic metal with Pd. Examples of plasmon-enhanced FA dehydrogenation are summarized in Table 1. Nevertheless, the mechanism of LSPR effect on FA dehydrogenation is still undefined. It is significant to reveal the fundamentals of the electromagnetic field and hot charge carriers on molecular adsorption/activation to deepen the understanding of this growing research field.
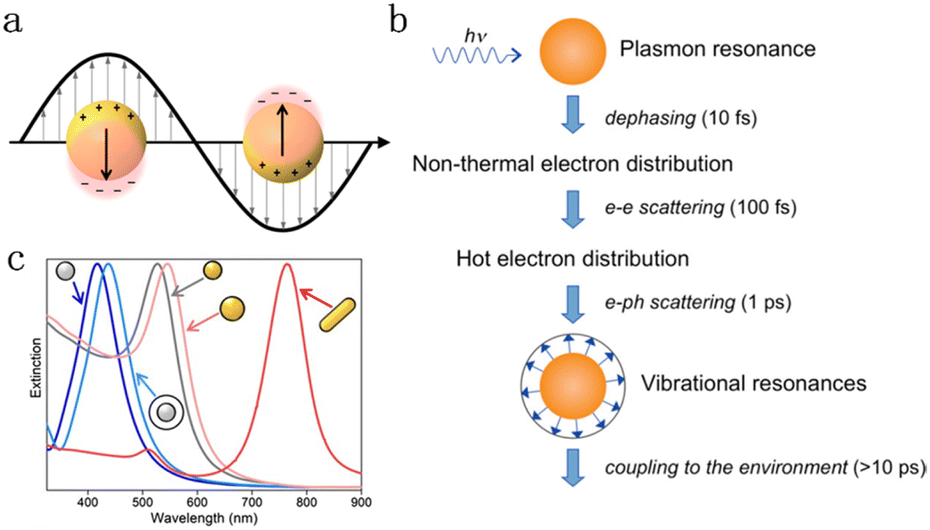 | ||
| Fig. 1 (a) Surface enhanced electromagnetic field caused by collective electron oscillations.17 Reproduced with permission from ref. Copyright 2018 American Chemical Society. (b) Decay sequence and approximate time scale of metal nanoparticles after light excitation.18 Reproduced with permission from ref. Copyright 2011 American Chemical Society. (c) Extinction spectra of Ag (gray) and Au (yellow) nanoparticles with different sizes, shapes, and dielectric environments (with/without a white silica shell).19 Reproduced with permission from ref. Copyright 2023 Wiley-VCH. | ||
| No. | Catalysts | H2 evolution rate [mmol g−1 h−1] | Light intensity [mW cm−2] | Temperature [°C] | Reference | |
|---|---|---|---|---|---|---|
| Dark | Light | |||||
| 1 | Pd-covered Au NRs | 0.4 | 5.3 | 100 | 5 | 16 |
| 2 | 1.8 | 6.7 | 25 | |||
| 3 | 7.5 | 22 | 40 | |||
| 4 | Pd-tipped Au NRs | 0.6 | 8.7 | 5 | ||
| 5 | 2.7 | 10.5 | 25 | |||
| 6 | 10 | 32.5 | 40 | |||
| 7 | Pd-covered Ag@Au HNPs | ∼0 | 3 | 500 | 15 | 14 |
| 8 | 3 | 7.5 | 25 | |||
| 9 | 12 | 15 | 50 | |||
| 10 | Pd-dotted Ag@Au HNPs | 2 | 29 | 15 | ||
| 11 | 10 | 35 | 25 | |||
| 12 | 25 | 105 | 50 | |||
| 13 | Au@Pd shell | 0.5 | 1 | 125 | 40 | 38 |
| 14 | Au@AuPd shell | 1 | 5 | |||
| 15 | Au–Pd satellites | 35 | 90 | |||
| 16 | Au–AuPd core–satellites | 70 | 191 | |||
| 17 | Au1Pd3/GO | 8.2 | 16.4 | 100 | 25 | 13 |
| 18 | Au1Pd2/GO | 11.7 | 22.2 | |||
| 19 | Au1Pd1/GO | 7 | 16.6 | |||
| 20 | Au2Pd1/GO | 3.7 | 9.4 | |||
| 21 | Au3Pd1/GO | 1.2 | 3.5 | |||
| 22 | Pd/GO | 0.9 | 1.6 | |||
| 23 | Au@Pd/UiO-66(Zr85Ti15) | 1517 | 1875 | 320 | 30 | 39 |
| 24 | PdAg@g-C3N4 | 6.1 | 9.8 | — | 25 | 40 |
| 25 | — | 27.6 | 50 | |||
| 26 | AgPd/CN-3% | 2.1 | 5.9 | — | 30 | 41 |
| 27 | NiAuPd | 29 | 69 | 121 | 5 | 12 |
| 28 | 183 | 341 | 25 | |||
| 29 | 441 | 641 | 45 | |||
| 30 | Au1@Pd | 15.9 | 58.5 | 121 | 25 | 11 |
| 31 | Au2@Pd | 14.6 | 42.1 | |||
| 32 | Au3@Pd | 8.4 | 14.7 | |||
| 33 | Au4@Pd | 5.3 | 12.9 | |||
In this review, we introduce the recent progress in plasmon-enhanced FA dehydrogenation, summarize the influence of the size/geometry on plasmon catalysis, and discuss the impact of electronic structure from the perspective of Fermi-level engineering and alloying. Then, we summarize the mechanisms of hot charge carriers and local field work in FA dehydrogenation. Some representative cases are covered to discuss the plasmonic energy dispersion in space and the unique advantages of LSPR in reactant adsorption/activation, which may inspire exploring and understanding the plasmon-enhanced FA dehydrogenation. Finally, we present an outlook and propose some suggestions applicable, but not limited to, obtaining higher FA dehydrogenation activity via hybrid plasmonic metal nanostructures.
Plasmon-enhanced activity
Au and Ag are the most popular plasmonic metals for plasmonic catalysis, and their LSPR properties are heavily dependent upon their morphology, size and composition.42–45 For plasmon-enhanced FA dehydrogenation, the core–shell heterostructure is the most commonly reported nanostructure, and the typical fabrication is the reduction of a Pd-containing salt precursor on previously synthesized colloid plasmonic seeds, i.e., the seed-mediated method.46–50 Generally, pre-synthesized plasmonic nanoparticles of different shapes/sizes are selected as cores, which can coarsely determine the overall LSPR properties. Then, by adjusting the relevant thermodynamic and kinetic factors, the growth mode of the shell metal can be precisely controlled. Therefore, geometric control engineering can be well achieved in terms of the core and shell in principle. This section focuses on the effects of geometry-determined optical properties on FA decomposition performance.Size effect
The extinction spectra of Au nanostructures show a strong dependence on their size when exceeding the quasi-static limit.51–53 For spherical Au nanoparticles with a size larger than 5 nm, strong plasmon resonances occur in the visible region (about 520–600 nm). When the diameter is less than 5 nm, the LSPR is dramatically suppressed, which significantly blue shifts and eventually disappears once the particle size < 2 nm. The particle size dramatically affects the optical properties of plasmonic nanostructures, such as absorption and scattering efficiencies, and the intensity of near-field enhancements, all of which make great contributions to catalytic performance in plasmonic catalysis. A spherical metal with a diameter smaller than the incident wavelength can be approximated as a dipole. The following formula (1) can express the corresponding particle polarizability α under excitation,54–56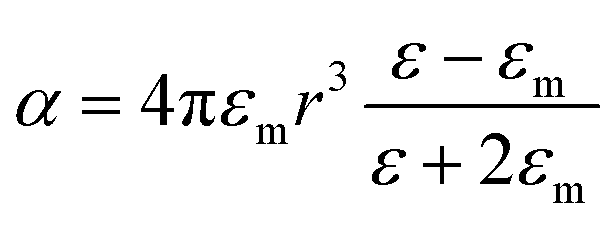 | (1) |
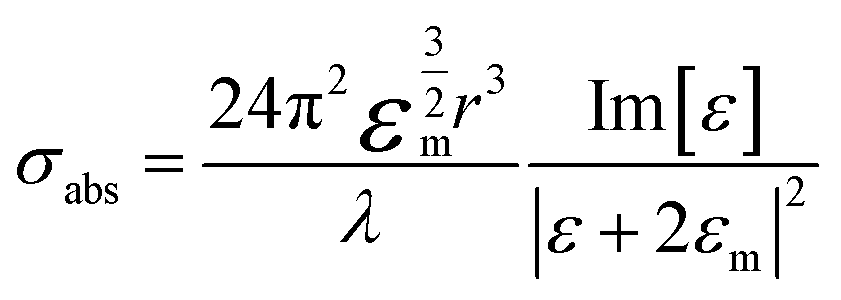 | (2) |
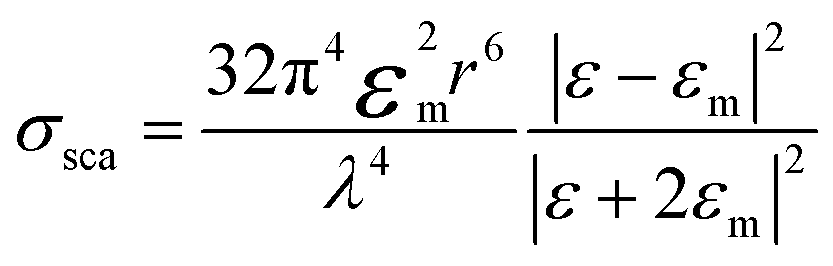 | (3) |
| σext = σabs + σsca | (4) |
It can be found that the absorption is proportional to r3, while the scattering is proportional to r6. When r is much smaller than the incident wavelength, extinction is essentially dominated by absorption. The scattering ratio in extinction increases significantly with the increase of the size.
The influence of the size effect may be different in different reaction systems, since different rate-determining steps are involved, and the size-dependent near and far field properties may not contribute constantly in different plasmon-enhanced processes. For example, Christopher et al. studied the plasmon-enhanced O2 dissociation on Ag nanostructures with various sizes and found that the quantum efficiency was consistent with the near-field enhancement rather than the hot carrier density in different photon wavelength tests (Fig. 2a), which clearly proved that O2 dissociation was related to the strong LSPR local electromagnetic field.57 However, as reported by Li and co-workers in 2017, Ag nanoparticles with intermediate sizes (25 and 50 nm), which balanced the light absorption and local electromagnetic field, exhibited the best photocatalytic rates (Fig. 2b).58 In our previous work, we synthesized different-sized Au@Pd nanospheres with narrow size distributions to clarify the role of size effect in FA dehydrogenation over Pd-based hybrid plasmonic catalysts.11 The Au core size varied from 56 to 121 nm to ensure visible light range absorption, while the thickness of the shell comprising Pd nanodots was kept consistent. The optical properties of Au@Pd core–satellite nanospheres were dominated by the Au core, including the LSPR position, absorption, and scattering. With the increase of the Au diameter, the absorption ratio of the hybrid plasmonic catalyst decreases, while the far-field scattering and near-field intensity increase instead. The smallest Au@Pd nanospheres, exhibiting the highest absorption/extinction ratio, demonstrated the best catalytic performance (Fig. 2c and d). Interestingly, large, relatively high scattering nanospheres demonstrated a higher-than-expected catalytic enhancement factor, approaching the same value as the smallest ones (Fig. 2e) by moderately raising the reaction temperature or shortening the interparticle distance by increasing the catalyst concentration, which was ascribed to the reduced interparticle spacing that promotes the secondary absorption of scattered photons (multiple scattering effects) and drastic near-field enhancement between the neighbouring particle region.59–62 Therefore, in the case of FA dehydrogenation on Pd-based hybrid plasmon metal, both hot charge carriers and the near-field were involved in accelerating dehydrogenation.
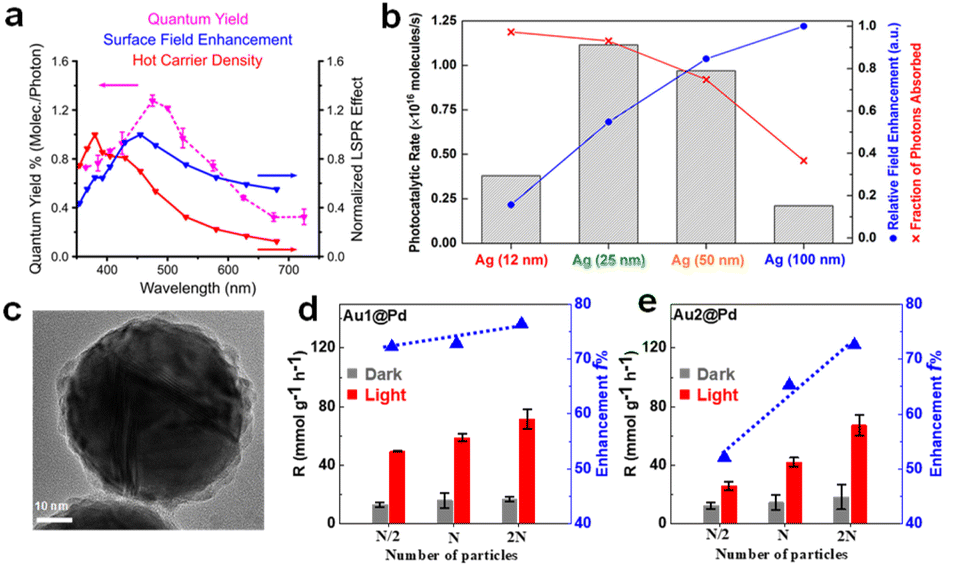 | ||
| Fig. 2 (a) Correlation between the wavelength dependent O2 dissociation quantum yield and LSPR effects including field enhancement and carrier density.57 Reproduced with permission from ref. Copyright 2019 American Chemical Society. (b) The photocatalytic rates, field enhancements, and fractions of photons absorbed in the catalyst bed for four Ag@SiO2/Pt heterostructures with different Ag NP sizes.58 Reproduced with permission from ref. Copyright 2017 American Chemical Society. (c) TEM image of an individual Au1@Pd NP. The correlation of H2 evolution rate and enhancement f with the particle concentration, (d) Au1@Pd and (e) Au2@Pd samples, respectively. N represents the original particle numbers.11 Reproduced with permission from ref. Copyright 2022 American Chemical Society. | ||
Morphology effect
Unlike Au nanospheres, Au nanorods (NRs) exhibit more special plasmon resonances due to their anisotropic structure.37,46,63,64 The electron oscillates along the short axis and resonates with the light in the 510–530 nm wavelength range, named transverse surface plasmon resonance absorption (TSPR); meanwhile, another vital LSPR along the long axis is attributed to the longitudinal LSPR, which typically distributes in the visible to near-infrared region. Considering its strong oscillation, the longitudinal LSPR exhibits a more significant impact on optical properties and sequential catalytic activity. For example, Zheng synthesized two kinds of anisotropic Au–Pd nanorods, Pd-covered Au NRs with a thin Pd layer completely covering the Au NRs (Fig. 3a) and Pd-tipped Au NRs with Pd nanocrystals selectively deposited at the Au NR tips (Fig. 3b).16 Such structural difference results in different photochemical conversion efficiencies; Pd-tipped Au-NRs exhibit better plasmon-enhanced activity for FA dehydrogenation, while a comparable light-driven enhancement factor was observed for both samples. Single-particle photoluminescence (PL) spectroscopy was applied to reveal the plasmon-induced pathway, especially the interfacial interaction between Au and Pd. The results in Fig. 3c and d show that the quenching efficiency of longitudinal LSPR reached 87–95%, while TSPR exhibited a similar intensity with pure Au NRs for both samples. According to the accepted PL mechanism, the quenching results from the effective charge separation, which avoids the recombination of the hot charge carriers; the luminescence mainly originates from the LSPR radiative decay of Au NRs, and PL damping indicates that the radiation tunnelling becomes suppressed and more plasmon energy disperses through the electron transfer from Au to Pd. The finite-difference time-domain (FDTD) method was also carried out to investigate the effect of the plasmon resonance energy transfer (PRET) process to further confirm the interaction of Pd with the Au NR, which showed that the enhancement of electric field intensity around the tips was much stronger than that around the sides, revealing that the plasmon energy can be effectively utilized by locating the active metal at the sites with the intense field.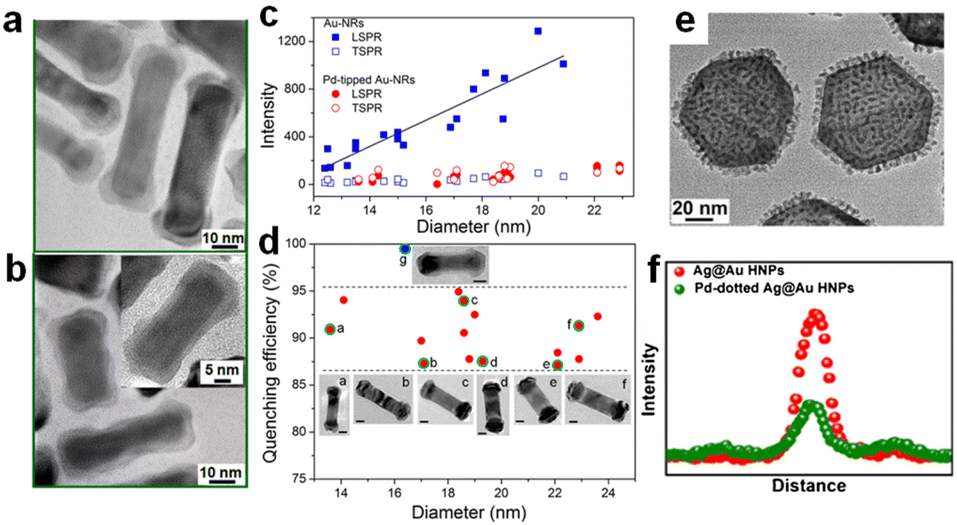 | ||
| Fig. 3 The TEM images of (a) Pd-tipped Au NRs and (b) Pd-covered Au NRs. (c) The relationship between PL intensities and diameter in different plasmon modes of single Au NRs and Pd-tipped Au NRs. (d) The PL quenching efficiency of single Pd-tipped (red dots) and Pd-covered (blue dots) Au NRs with different diameters.16 Reproduced with permission from ref. Copyright 2015 American Chemical Society. (e) The TEM image of Pd-dotted Ag@Au. (f) The PL intensity of individual Ag@Au and Pd-dotted Ag@Au.14 Reproduced with permission from ref. Copyright 2020 Elsevier. | ||
Such a selective deposition of active species at the field-enhanced sites has also been applied in other plasmonic hybrid catalysts. For instance, Xiao et al. prepared Pt end-deposited bimetallic nanostructures (ePt-Au NBPs) over Au nanobipyramids (Au NBPs), which have a similar LSPR property to Au NRs.65 Compared to the all-deposited aPt-Au NBPs, the maintained strong field at the sharp tips can promote the generation and migration of the carriers and then boost the spontaneous reconstruction of the Pt surface to produce more active sites.
This design strategy has also been validated in theoretical studies. Linic et al. studied the absorption/scattering of Au@Pt NRs through the finite element method (FEM).66 The absorption intensity of longitudinal LSPR becomes much higher than that of pure Au NRs, while no significant changes appear in the TSPR region, which is attributed to the ratio of the dielectric function imaginary part for Pt to Au being much higher at low energy than in high plasmon mode, resulting in the plasmon energy being preferentially dissipated in the Pt shell at the longitudinal LSPR position rather than TSPR.
By switching nanorods to nanoplates, Huang et al. reported that Pd-dotted Ag@Au nanoplates exhibited an improved catalytic activity normalized to Pd mass under identical reaction conditions.14 They selectively deposited Pd nanodots on the surface of Ag@Au nanoplates and obtained a two-shell geometry (Fig. 3e), including continuous Pd coverage (Pd-covered Ag@Au) and discrete Pd decoration (Pd-dotted Ag@Au), in which Pd-dotted Ag@Au manifested a higher FA dehydrogenation rate and plasmon enhancement factor. As shown in Fig. 3f, single-particle PL intensity experiments revealed significant quenching in Pd-dotted Ag@Au compared to Ag@Au, verifying a transfer of hot electrons from Ag@Au to Pd dots and leading to surface charge heterogeneity. Such structure–activity difference results from the combined effect of the optical absorption and the local field. Compared to a fully covered Pd shell, discrete Pd dots do not completely block the absorption of photons by Ag@Au. The FDTD further confirmed that the uplifted Pd dots exhibit a steep gradient field. However, unlike the selective deposition mentioned above, such Pd dots did not intentionally couple with the field sites but affected the electromagnetic spatial distribution through their non-plasmonic geometry and thus enhanced chemisorption and bond activation.
Coupling LSPR with Fermi level engineering
In conventional thermal FA dehydrogenation, a mature strategy is to promote surface charge heterogeneity (increase the electron density on the Pd surface) to obtain a higher dehydrogenation rate.67,68 For example, Tsang and co-workers prepared a series of core–shell nanostructures in which the shell is a thin Pd layer, while the cores are composed of various metals (M) with different work functions.7 A simple linear relationship between the HCOOH decompose rates, and the work function of M fcc (111) can be obtained, indicating that the electron redistribution forced by Fermi level equilibrium plays a vital role in promoting FA dehydrogenation. Moreover, as shown in Fig. 4a, Jiang et al. utilized the difference in work functions between Co, Au, and Pd to prepare an efficient CoAuPd/C catalyst, which effectively regulated the electron density of Pd sites and exhibited the best FA dehydrogenation activity at room temperature.69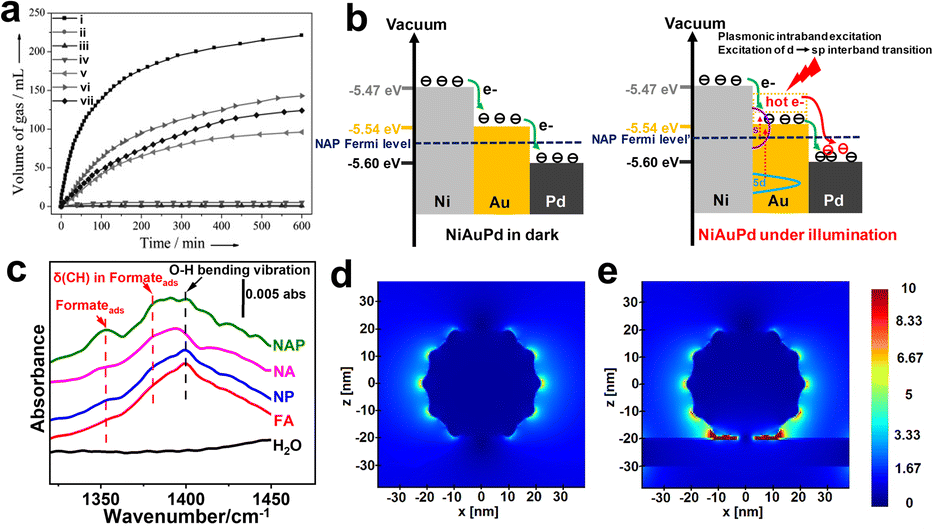 | ||
| Fig. 4 (a) Real-time gas volume production diagram in the presence of (i) Co0.30Au0.35Pd0.35/C, (ii) Co/C, (iii) Au/C, (iv) Co0.30Au0.70/C, (v) Pd/C, (vi) Co0.30Pd0.70/C, and (vii) Au0.50Pd0.50/C at room temperature.69 Reproduced with permission from ref. Copyright 2013 Wiley-VCH. (b) Schematic diagram illustrates the charge transfer within NiAuPd under darkness (left) or under radiation (right). (c) ATR-FTIR spectra of HCOOH and formate-related intermediate co-adsorption over various catalysts. The distribution of the electromagnetic field of (d) AuPd, and (e) trimetallic NiAuPd in space.12 Reproduced with permission from ref. Copyright 2022 Wiley-VCH. | ||
Inspired by this, we reported a trimetallic NiAuPd heterogeneous catalyst, which synergistically combines Fermi level engineering and plasmon effect of promoting the surface charge heterogeneity, resulting in an enhanced intermediate chemisorption and ultimately promoted plasmonic catalysis (Fig. 4b).12 Under darkness, electrons transfer from Ni (high Fermi level) to Au and Pd due to the work function differences, and Au acts as an electron-mediated channel to promote charge migration. Such intrinsic electronic structure enables NiAuPd to exhibit a significant O–H cleavage and formate-related intermediate chemisorption (Fig. 4c). Under irradiation, compared with the AuPd or NiO–AuPd, a higher Fermi level in NiAuPd allows hot electrons to upshift into a more energetic state after absorbing a photon. Furthermore, the FDTD in Fig. 4d and e demonstrates that the narrow gaps between Au–Pd and Ni plate create a fierce field; such hot spots can boost C–H activation through a direct energy flow.
Combination of alloying and support
Different from the core/shell heterostructure discussed above, Pd-based alloys have also been well studied in FA dehydrogenation due to the alloying effect, which can delicately modify the electronic structure of the catalytically active metal via d-orbital overlap or metal surface segregation.70–74 Various alloyed nanocatalysts have demonstrated improved catalytic behaviors via strain/ligand effect and/or ensemble effect in traditional thermal catalysis.75–78 During the past decades, Pd-based plasmon alloys have also been explored to combine plasmonic enhancement with intrinsic alloy effect, while most reports involve semiconductor supports, because the photon/plasmon response can lead to dramatic changes in the electronic structure and thus the overall reactivity of the complex catalyst through the electronic interaction between the plasmonic metal and semiconductor resulting from the Mott–Schottky effect. It has been well demonstrated that the charge kinetics of photocatalytic reactions could be able to accelerate photocatalytic FA decomposition by integration of plasmonic-based alloys (AgPd and AuPd) and semiconductors to make Mott–Schottky photocatalysts. Carbon nitride (g-C3N4), one of the most important semiconductors,79–81 has been widely used as a semiconductor support in hydrogen generation due to its efficient handover of photogenerated electrons to plasmonic hybrid nanoparticles. For example, a monodisperse alloy of AgPd on a graphite carbon nitride semiconductor (AgPd/CN) was used for photocatalytic evolution of hydrogen from formic acid.41,82 Stucky et al. prepared the AgPd alloy shown in Fig. 5a–d on the surface of CN.41 The electron transfer from CN and AgPd leads to an electron-enrichment of Pd and thus affords more catalytic activity and stability for the H2 evolution under visible light. In another example, small (3 nm) AuPd nanoparticles were loaded on carbon nitride nanospheres (AuPd/CNS) for the design of Mott–Schottky catalysts, Liu et al. found that Mott–Schottky, alloying and plasmonic effects can be used in conjunction with each other to efficiently accelerate the electron transfer from photoresponsive super small carbon nitride nanospheres to plasmonic alloys (Fig. 5e), which eventually leads to a remarkable photocatalytic activity for the photocatalytic hydrogen evolution from formic acid.15 | ||
| Fig. 5 The (a) TEM and (b) HR-TEM images of AgPd/CN. (c) The qualitative line scan of an individual AgPd nanoparticle over CN. (d) The XRD patterns of different samples prepared by the same method.41 Reproduced with permission from ref. Copyright 2017 Royal Society of Chemistry. (e) The mechanism diagram of photocatalytic hydrogen evolution from formic acid for AuPd/CNS with/without irradiation.15 Reproduced with permission from ref. Copyright 2019 Elsevier. (f) Possible mechanism for plasmon-enhanced H2 production by Au@Pd/UiO-66.39 Reproduced with permission from ref. Copyright 2017 American Chemical Society. | ||
In addition to building the Mott–Schottky effect, some other materials such as metal organic frameworks (MOFs),39 carbon-based materials,13,83 and wide-bandgap oxides (e.g., ZrO2)84,85 are often employed. For example, Yamashita et al. loaded Au@Pd nanoparticles on the surface of an amine-modified MOF (UiO-66(Zr100−xTix)) for boosting FA dehydrogenation under light.39 More importantly, the amine functionality in the MOF and the electron-rich state of Pd upon plasmon excitation synergistically promote FA decomposition. As shown in Fig. 5f, weakly alkaline –NH2 acts as a proton scavenger to positively promote O–H dissociation and form –+HNH2. After photon absorption, the electron-rich state on the Pd surface helps the chemisorbed formate intermediates break C–H. Thus, the weakly basic synergistic combination of amine groups and LSPR effects accounts for its high catalytic activity.
Mechanism insight
Strong LSPRs enable plasmonic metals to convert incident photons to hot carriers, strong electromagnetic fields, or thermal heat for directing and improving chemical reactions. By combining plasmonically active metals with traditionally catalytic metals, plasmonic hybrid nanostructures could simultaneously promote light conversion and molecular solid adsorption. Some efforts have been devoted to the plasmon-induced contribution, while the mechanism is more complex. This section will discuss plasmon enhancement from the perspective of hot carriers and local electromagnetic fields.Hot charge carriers
In plasmonic catalysis, a hot charge carrier is widely regarded as one of the main mechanisms in promoting bond activation.20,86,87 For a simple hybrid system composed of a plasmonic metal and an adsorbed molecule, there are two recognized charge transfer pathways, indirect and direct charge transfer.20,22,88,89 In an indirect path, hot carriers are generated within the metal through Landau damping, and then injected into neighbouring chemisorbed molecules before fundamentally thermalizing into a low-energy carrier or recombination. However, in a direct charge transfer, plasmonic nanoparticles and adsorbed molecules are not treated as two individual electronic states, but a new hybrid electronic state formed at the metal–molecule interface, which can participate in LSPR excitation, energy decay, and distribution, and this process is also known as chemical interface damping (CID).90,91 More importantly, this direct route is believed to be more efficient because it is momentum conserved without involving energy loss induced by electron–electron and electron–phonon scattering within metal body.Experiments and theoretical simulations have also verified that direct charge transfer can take place in solid-state hybrid plasmonic systems. For example, Linic's group deposited a thin Pt layer (1 nm) on the surface of Ag nanocubes (75 nm edge length).92 It is found that absorption is the main decay route for Ag–Pt, and the ratio of absorption is larger for a Pt shell (Ag–Pt) compared to the Ag shell (pure) for identical Ag nanocubes, while the radiative decay occupied a larger proportion in extinction for pure Ag nanocubes. That is, almost all the electromagnetic field energy of Ag is dissipated through the Pt shell (forming e−–h+ pair), which is attributed to the fact that the imaginary part of Pt is larger than that of Ag in the visible range, and the plasmonic local field further enhances this absorption channel.
After summarizing the current views on the spatial dispersion of LSPR energy in the metal–metal system, we still need to know the role of hot charge carriers in promoting FA dehydrogenation. Generally speaking, upon light irradiation, the adsorbed state could be excited by LSPR, and the hot carrier injection (or direct excitation within the molecule/interfacial metal-adsorbent) makes the complex reach a charged (or excited) state, thus obtaining nuclear kinetic energy and chemical bond activation. Specifically, Zheng et al. studied the PL spectra of the single particles of Au–Pd impregnated with FA solution (Fig. 6a), and the results showed that the PL intensity was significantly quenched, indicating that the hot electrons were injected into the FA molecule.93 When the surface of Au–Pd was covered with Al2O3 to isolate the charge transfer channel, the PL quenching was not observed, which further confirms that the hot electron is an essential factor in enhancing FA dehydrogenation. In addition, density functional theory (DFT) calculations revealed that adding an electron on the surface of Pd atoms can effectively reduce the Gibbs free energy of the adsorbed state of FA molecules and intermediates (Fig. 6b), illustrating that optimizing the interfacial electronic properties can facilitate the dehydrogenation.94 In addition to hot electrons, hot holes are also believed to promote FA dehydrogenation. As shown in Fig. 6c, Moskovits et al. proposed that hot electrons/holes boost the neutralization rate cooperatively to reduce the activation energy, in which holes are injected into negatively charged formates to accelerate C–H fracture, while hot electrons neutralize –H+ to accelerate H2 desorption.95
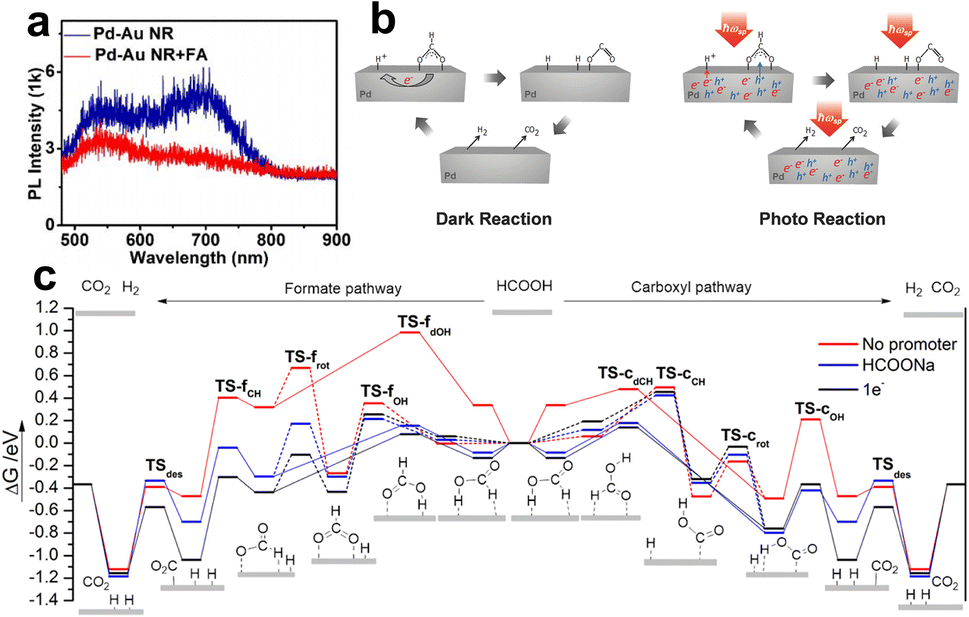 | ||
| Fig. 6 (a) The PL intensities of individual Pd-tipped Au NRs and the signal after immersing in FA aqueous (1 M).93 Reproduced with permission from ref. Copyright 2021 American Chemical Society. (b) Gibbs free energy reaction profiles (eV) for FA dehydrogenation over Pd (111) via the formate (left) and carboxyl (right) pathway. The slab surface is modified by various methods: no promoter, HCOONa (HCOO− and Na+), and 1e−.94 Reproduced with permission from ref. Copyright 2017 American Chemical Society. (c) Illustrations of the mechanism in plasmon accelerating FA decomposition.95 Reproduced with permission from ref. Copyright 2016 Wiley-VCH. | ||
Local electromagnetic field
For surface-chemisorbed molecules, the local electromagnetic field can significantly modify their bond length, configuration, etc, which can flatten the potential energy surface (PES) and reduce the activation energy barrier, although the molecules cannot be excited to the excited state within the femtosecond scale lifetime range.96 Beyond that, the selective effect of local electromagnetic fields on adsorption configurations has been discovered in recent years. Compared with nonpolar or weak-polar reactants, the plasmon force can more easily influence molecules with large dipole moments, thus changing the diffusion and adsorption behavior. For instance, Zhu and co-workers observed a photo-switching product selectivity in the alkyne–hydroamination reaction, where the product selectivity depends on the ratio of surface chemisorbed reactants.84 Alkynes and anilines exhibit different chemisorption types with/without irradiation. In the dark, numerous alkynes are adsorbed on the surface sites and produce Diyne. Upon illumination, the alkynes are released from the surface, while the light-polarizable aniline is preferentially adsorbed and driven by plasmon forces to obtain Imine. Considering the large dipole moment of HCOOH and some additives (e.g., HCOONa), such plasmon force can also optimize the interfacial adsorption configuration in FA dehydrogenation.The local field-assisted FA activation was judged by the Au–Pd core–shell and core–satellite nanostructures. Emiliano and collaborators prepared the Au@Pd shell and Au@AuPd shell, and assembled the Au–Pd satellite and Au–AuPd satellite to study FA dehydrogenation under darkness and light.38 The results in Fig. 7a and b show that the geometric structure can profoundly influence the dehydrogenation activity. Among these, the Au–AuPd satellite has the highest dehydrogenation rate and enhancement factor due to the gap between the reactor and the plasmon antenna, allowing the electromagnetic field to climb up to almost 30 times larger than the surface of the Au@Pd core–shell (Fig. 7c). Meanwhile, the higher dielectric function imaginary enables the e−–h+ to almost form in Pd after the plasmon decay (Fig. 7d).
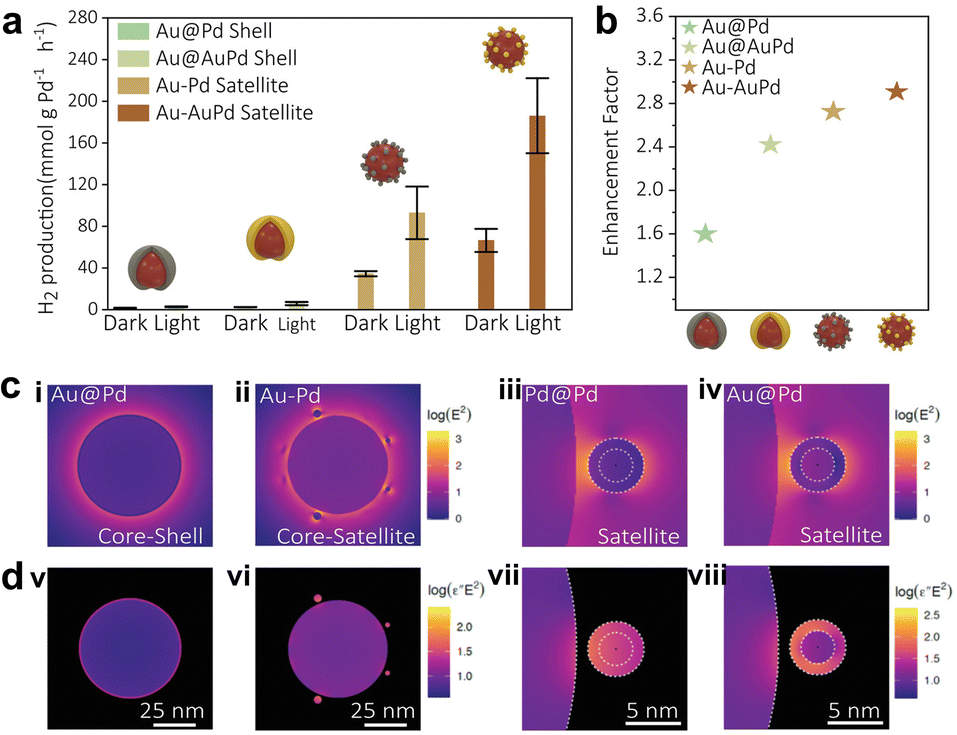 | ||
| Fig. 7 (a) The H2 production of various bimetallic nanocatalysts under dark conditions and upon illumination, and (b) their corresponding enhancement factors (the ratio between light and dark performances). (c) Electric field distribution for various bimetallic nanocatalysts, (i) Au@Pd core–shell, (ii) Au–Pd core–satellite, (iii) a Pd satellite and (iv) Au@Pd satellite placed 1 nm away from an antenna, and (d) corresponding absorption maps of (v) Au@Pd core–shell, (vi) Au–Pd core–satellite, (vii) a Pd satellite and (viii) Au@Pd satellite.38 Open Access ref. from 2022 Wiley-VCH. | ||
Beyond FA dehydrogenation, hot spots located at interparticle gaps were extensively studied in the antenna–reactor geometry. In this typical system, no obvious interfacial hybrid state was created between the antenna and reactor, which are usually physically separated or isolated by insulators (such as SiO2).87,97–101 According to Sytwu's work, an antenna–reactor system where Au–Pd is physically mixed, the β phase Pd nanocubes exhibit a faster transition rate to the α phase under illumination.102,103 And the Pd closer to the plasmon antenna always transitions faster than those far away from the antenna. Hot spots contribute to the nucleation at Pd nanocube edges rather than the corners (low-coordination sites), implying that the electromagnetic field assists in creating new sites, even though these sites are energetically unfavorable.
As a powerful tool, DFT calculation can well investigate the influence of electromagnetic fields on reaction thermodynamics, intermediates, hybrid electronic states, and so on.86,104–106 The hybridized orbitals at the interface are redistributed under the action of electromagnetic fields, which may contribute to the direct dissipation of plasmon energy in these metal-molecular states. Such momentum conservation excitation avoids the metallic electron–electron and electron–phonon relaxation, resulting in some selective activation scenarios, and some unexpected phenomena. In a recent study, the projected density of states (PDOS) was used to analyze interfacial hybridization states when CuPd was employed as an adsorption site for CO2.105 When an electric field was introduced to simulate the LSPR collective electron oscillation, the hybridized O1 2P and O2 2P shifted away from the Fermi level and became more overlapped, further stabilizing the interfacial adsorption configuration. More impressive, new quasi-isolated states are generated near the Fermi level, contributing to the oscillation of more electronic states and the extension of hot carrier lifetimes.
Perspective and outlook
Although great progress has been made during the past decades in the field of light-enhancing FA dehydrogenation, to move forward, further improvement complemented by both theoretical and experimental advances is essential to establish a more quantitative understanding of hybrid plasmonic metal nanostructures. This section focuses on major limitations and further development for plasmon-enhanced FA dehydrogenation.(1) Mechanism investigation. Compared with thermal catalysis, the physicochemical understanding of plasmon-involved chemical reactions is still far from clear, such as plasmon-induced configuration transformation and specific contributions of hot carriers and electromagnetic fields. In addition, some additives (e.g., HCOONa) are widely employed to boost H2 evolution in traditional thermal catalysis, while the role of additives in plasmonic catalysis still needs to be determined. Advanced characterization and theoretical simulations provide promising approaches to explore the underlying mechanism in plasmon-enhanced FA dehydrogenation. Advanced techniques with fine temporal, spatial, and energy resolution, such as tip-enhanced Raman scattering (TERS),107–110 surface-enhanced Raman scattering (SERS),111–113 and synchrotron infrared nanospectroscopy (SINS),114,115etc., can be used to obtain molecular vibrations, intermediate conversion, and energy dissipation at a picosecond level or in a specific nanoscale region, which is helpful to determine multiple effects. Theoretical simulation provides another convenience for obtaining high-quality information, including chemisorbed structure, hybrid electronic density of states, intermediates, and so on, which can deepen the understanding of plasmon energy conversion and guide the rational design of catalyst systems.86
(2) Plasmonic catalyst design principles. The ability of light absorption is essential for plasmonic catalysis. Within the mean free path, the optical absorption is positively correlated with the square of field strength and the dielectric function imaginary part but inversely correlated with the metal size. Therefore, high field intensity, large imaginary parts, and small size are required for strong optical absorption. However, there is an intrinsic dilemma between absorption and the local field.116 Typically, a small imaginary part can bring an intense field but minimize plasmon decay to form energetic hot carriers; meanwhile, nanoparticles with large size have a strong electromagnetic field, but most LSPR energy decays through photon radiation. Fortunately, absorption-local fields can be balanced through geometric construction (e.g., protruding tips), coupling with high imaginary part metals (Pd, Pt), assembling interparticle hotspots, and tuning the particle size.
Except the LSPR properties, the critical role of catalytic electronic states has been gradually unraveled, which largely determines the adsorption/desorption of molecules on the catalyst surface.87,117 Considering that the surface plasmon energy distribution is non-homogeneous, local fields or hot carriers can greatly influence interfacial states, such as molecular polarities, electronic states, adsorption, etc., in the regions with significant LSPR features. However, over the sites with poor plasmon energy, there are some blank spots where LSPR does not effectively affect the sites, and a thermal-driven process may not be negligible. Therefore, from the perspective of obtaining higher catalytic efficiency, optimizing the intrinsic ground electronic states may be one of the vital strategies. In addition to Fermi level engineering and metallic alloying we mentioned above, there exist some other approaches such as geometry or faceting control of activity metals, and intermetallic tuning (i.e., ligand, ensemble, and strain effect) which have been successfully established for modifying electronic states. For example, it has been demonstrated that Pd–Au interface sites exhibit high activity and selectivity for FA decomposition by optimization of the arrangement of Pd and Au atoms through an ensemble effect.118 The Pd–PdO interface could decompose FA at a high rate with overcoming CO toxicity due to a special electronic state.119 Therefore, to further improve catalytic performances, much attention should be paid to rationally designing efficient catalysts by considering both solar energy utilization and intrinsic electronic properties.
In addition, cheaper and more abundant Cu and Al elements can be developed as substitutes for precious metals, but the potential effects of easily oxidized surfaces on optical properties and catalytic rates must be recognized.
(3) Integrating local heat. The local surface plasmon decays in a non-radiative way, finally causing local heat. For FA dehydrogenation, a thermodynamically favorable process, heating should facilitate a higher yield. Most reported plasmon-involved systems apply a water bath to eliminate the heating effect induced by irradiation. From the point of view of energy utilization efficiency, this is wasteful because most energy is lost through heat transfer. However, attention should be paid to the quantitative distinction between thermal and non-thermal effects. Although the decoupling of these effects is greatly limited by the extended timescales and highly confined space, some studies and reviews have focused on this key question.59,97,120,121 The contribution of hot carriers, local field, and local heat was distinguished through accurate temperature detection, optimized catalytic system design, or the application of in situ spectroscopy. In addition, excessive heating may also lead to poor product selectivity, generating carbon monoxide during HCOOH decomposition.11,14,122,123 Therefore, the local thermal effect induced by LSPR is a double-edged sword. How to well balance photothermal and selectivity is one of the critical issues that must be addressed to make full use of plasmon energy.
With increasing attention and ongoing efforts on plasmonic catalysis, we look forward to more exciting discoveries, such as further improvements in selectivity/activity in this area, and predict that plasmonic catalysts probably will become the next generation of photocatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1102600), National Natural Science Foundation of China (22005109), Guangdong Basic and Applied Basic Research Foundation (2020A1515110860), and the Innovative Foundation of Huazhong University of Science and Technology (5003013070). We are grateful for all funding to support this work.Notes and references
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Li, X. Wei, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 19550–19571 CrossRef CAS PubMed.
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- S. Niaz, T. Manzoor and A. H. Pandith, Renewable Sustainable Energy Rev., 2015, 50, 457–469 CrossRef CAS.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664 RSC.
- Z. Li and Q. Xu, Acc. Chem. Res., 2017, 50, 1449–1458 CrossRef CAS PubMed.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- A. Gemenetzi, Y. Deligiannakis and M. Louloudi, ACS Catal., 2023, 13, 9905–9917 CrossRef CAS.
- X. Liu, D. Wang and Y. Li, Nano Today, 2012, 7, 448–466 CrossRef CAS.
- M. Karatok, K. Duanmu, C. R. O'Connor, J. A. Boscoboinik, P. Sautet, R. J. Madix and C. M. Friend, Chem. Sci., 2020, 11, 6492–6499 RSC.
- J. Zhu, J. Huang, J. Dai, R. Chen, X. Fu, Z. Wang, H. Liu and G. Li, ACS Appl. Energy Mater., 2022, 5, 10013–10022 CrossRef CAS.
- J. Zhu, J. Huang, J. Dai, L. Jiang, Y. Xu, R. Chen, L. Li, X. Fu, Z. Wang, H. Liu and G. Li, ChemSusChem, 2023, 16, e202202069 CrossRef CAS PubMed.
- P. Liu, X. Gu, H. Zhang, J. Cheng, J. Song and H. Su, Appl. Catal., B, 2017, 204, 497–504 CrossRef CAS.
- F. Tong, Z. Lou, X. Liang, F. Ma, W. Chen, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2020, 277, 119226 CrossRef CAS.
- S. B. Zhang, M. Li, J. K. Zhao, H. Wang, X. L. Zhu, J. Y. Han and X. Liu, Appl. Catal., B, 2019, 252, 24–32 CrossRef CAS.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2015, 137, 948–957 CrossRef CAS PubMed.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- G. V. Hartland, Chem. Rev., 2011, 111, 3858–3887 CrossRef CAS PubMed.
- C. Brissaud, L. V. Besteiro, J. Y. Piquemal and M. Comesana-Hermo, Sol. RRL, 2023, 7, 2300195 CrossRef CAS.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- S. Linic, P. Christopher, H. Xin and A. Marimuthu, Acc. Chem. Res., 2013, 46, 1890–1899 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- M. W. Knight, N. S. King, L. Liu, H. O. Everitt, P. Nordlander and N. J. Halas, ACS Nano, 2014, 8, 834–840 CrossRef CAS PubMed.
- H. Jing and H. Wang, Chem. Mater., 2015, 27, 2172–2180 CrossRef CAS.
- H. Jing, Q. Zhang, N. Large, C. Yu, D. A. Blom, P. Nordlander and H. Wang, Nano Lett., 2014, 14, 3674–3682 CrossRef CAS PubMed.
- Y. Xin, K. Yu, L. Zhang, Y. Yang, H. Yuan, H. Li, L. Wang and J. Zeng, Adv. Mater., 2021, 33, 2008145 CrossRef CAS PubMed.
- X. Sun, J. Yang, L. Sun, G. Yang, C. Liu, Y. Tao, Q. Cheng, C. Wang, H. Xu and Q. Zhang, ACS Nano, 2022, 16, 19174–19186 CrossRef CAS PubMed.
- M. Ivanchenko and H. Jing, Chem. Mater., 2023, 35, 4598–4620 CrossRef CAS.
- J. Gargiulo, M. Herran, I. L. Violi, A. Sousa-Castillo, L. P. Martinez, S. Ezendam, M. Barella, H. Giesler, R. Grzeschik, S. Schluecker, S. A. Maier, F. D. Stefani and E. Cortes, Nat. Commun., 2023, 14, 3813 CrossRef CAS PubMed.
- H. Cheng, K. Fuku, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2015, 3, 5244–5258 RSC.
- P. Zhang, T. Wang and J. Gong, Adv. Mater., 2015, 27, 5328–5342 CrossRef CAS PubMed.
- M. Wang, M. Ye, J. Iocozzia, C. Lin and Z. Lin, Adv. Sci., 2016, 3, 1600024 CrossRef PubMed.
- J. H. Park, Y. W. Cho and T. H. Kim, Biosens., 2022, 12, 180 CrossRef CAS PubMed.
- M. Fang, X. Tan, Z. Liu, B. Hu and X. Wang, Research, 2021, 2021, 9794329 CrossRef CAS PubMed.
- M. Sun, Z. Wang and H. Wang, Chem. Mater., 2022, 34, 1965–1975 CrossRef CAS.
- G. G. Li, M. Sun, E. Villarreal, S. Pandey, S. R. Phillpot and H. Wang, Langmuir, 2018, 34, 4340–4350 CrossRef CAS PubMed.
- Q. Zhang, L. Han, H. Jing, D. A. Blom, Y. Lin, H. L. Xing and H. Wang, ACS Nano, 2016, 10, 2960–2974 CrossRef CAS PubMed.
- M. Herran, A. Sousa Castillo, C. Fan, S. Lee, W. Xie, M. Doeblinger, B. Auguie and E. Cortes, Adv. Funct. Mater., 2022, 32, 2203418 CrossRef CAS.
- M. Wen, K. Mori, Y. Kuwahara and H. Yamashita, ACS Energy Lett., 2017, 2, 1–7 CrossRef CAS.
- H. Liu, X. Y. Liu, W. W. Yang, M. Q. Shen, S. Geng, C. Yu, B. Shen and Y. S. Yu, J. Mater. Chem. A, 2019, 7, 2022–2026 RSC.
- L. P. Xiao, Y. S. Jun, B. H. Wu, D. Y. Liu, T. T. Chuong, F. A. Jie and G. D. Stucky, J. Mater. Chem. A, 2017, 5, 6382–6387 RSC.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- Z. Yin, Y. Wang, C. Song, L. Zheng, N. Ma, X. Liu, S. Li, L. Lin, M. Li, Y. Xu, W. Li, G. Hu, Z. Fang and D. Ma, J. Am. Chem. Soc., 2018, 140, 864–867 CrossRef CAS PubMed.
- R. Liu, Z. K. Zhou, Y. C. Yu, T. Zhang, H. Wang, G. Liu, Y. Wei, H. Chen and X. H. Wang, Phys. Rev. Lett., 2017, 118, 237401 CrossRef PubMed.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- Y. Chen, Z. Fan, Z. Zhang, W. Niu, C. Li, N. Yang, B. Chen and H. Zhang, Chem. Rev., 2018, 118, 6409–6455 CrossRef CAS PubMed.
- Y. Xia, K. D. Gilroy, H. C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- M. Ivanchenko, A. L. Carroll, A. B. Brothers and H. Jing, Nanoscale Adv., 2022, 5, 88–95 RSC.
- Q. Zhang, N. Large, P. Nordlander and H. Wang, J. Phys. Chem. Lett., 2014, 5, 370–374 CrossRef CAS PubMed.
- S. Kim, S. Lee and S. Yoon, ACS Appl. Mater. Interfaces, 2022, 14, 4163–4169 CrossRef CAS PubMed.
- L. T. M. Huynh, S. Kim and S. Yoon, ACS Photonics, 2022, 9, 3260–3267 CrossRef CAS.
- L. Wang, M. Hasanzadeh Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Marago and M. A. Iati, J. Phys.: Condens.Matter, 2017, 29, 203002 CrossRef PubMed.
- B. Seemala, A. J. Therrien, M. Lou, K. Li, J. P. Finzel, J. Qi, P. Nordlander and P. Christopher, ACS Energy Lett., 2019, 4, 1803–1809 CrossRef CAS.
- K. Li, N. J. Hogan, M. J. Kale, N. J. Halas, P. Nordlander and P. Christopher, Nano Lett., 2017, 17, 3710–3717 CrossRef CAS PubMed.
- R. C. Elias and S. Linic, J. Am. Chem. Soc., 2022, 144, 19990–19998 CrossRef CAS PubMed.
- X. Jiang, J. Huang, Z. Bi, W. Ni, G. Gurzadyan, Y. Zhu and Z. Zhang, Adv. Mater., 2022, 34, 2109330 CrossRef CAS PubMed.
- G. W. P. Adhyaksa, S. W. Baek, G. I. Lee, D. K. Lee, J. Y. Lee and J. K. Kang, ChemSusChem, 2014, 7, 2461–2468 CrossRef CAS PubMed.
- S. M. Meyer, J. Pettine, D. J. Nesbitt and C. J. Murphy, J. Phys. Chem. C, 2021, 125, 16268–16278 CrossRef CAS.
- H. Jing and H. Wang, CrystEngComm, 2014, 16, 9469–9477 RSC.
- Q. Zhang, Y. Zhou, E. Villarreal, Y. Lin, S. Zou and H. Wang, Nano Lett., 2015, 15, 4161–4169 CrossRef CAS PubMed.
- M. Chen, Z. Ye, L. Wei, J. Yuan and L. Xiao, J. Am. Chem. Soc., 2022, 144, 12842–12849 CrossRef CAS PubMed.
- S. Chavez, U. Aslam and S. Linic, ACS Energy Lett., 2018, 3, 1590–1596 CrossRef CAS.
- K. Jiang, K. Xu, S. Zou and W. B. Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed.
- X. Qin, H. Li, S. Xie, K. Li, T.-W. Jiang, X.-Y. Ma, K. Jiang, Q. Zhang, O. Terasaki, Z. Wu and W. B. Cai, ACS Catal., 2020, 10, 3921–3932 CrossRef CAS.
- Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Haevecker, A. Knop Gericke and R. Schloegl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef CAS PubMed.
- H. Xin, A. Vojvodic, J. Voss, J. K. Norskov and F. Abild-Pedersen, Phys. Rev. B, 2014, 89, 115114 CrossRef.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- J. Li, H. M. Yin, X. B. Li, E. Okunishi, Y. L. Shen, J. He, Z. K. Tang, W. X. Wang, E. Yucelen, C. Li, Y. Gong, L. Gu, S. Miao, L.-M. Liu, J. Luo and Y. Ding, Nat. Energy, 2017, 2, 17111 CrossRef CAS.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg, H. Zong, D. Su, J. T. Miller, T. Mueller and C. Wang, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS PubMed.
- Y. Feng, G. Wang, Y. Chang, Y. Cheng, B. Sun, L. Wang, C. Chen and H. Zhang, Nano Lett., 2019, 19, 4478–4489 CrossRef CAS PubMed.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 CrossRef CAS PubMed.
- L. Y. Zhang, C. X. Guo, H. Cao, S. Wang, Y. Ouyang, B. Xu, P. Guo and C. M. Li, Chem. Eng. J., 2022, 431, 133237 CrossRef CAS.
- J. N. Zhu, X. Q. Zhu, F. F. Cheng, P. Li, F. Wang, Y. W. Xiao and W. W. Xiong, Appl. Catal., B, 2019, 256, 117830 CrossRef CAS.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G. J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- B. Gholipour, A. Zonouzi, M. Shokouhimehr and S. Rostamnia, Sci. Rep., 2022, 12, 13583 CrossRef CAS PubMed.
- C. Xu, X. Wang and J. Zhu, J. Phys. Chem. C, 2008, 112, 19841–19845 CrossRef CAS.
- E. Peiris, S. Sarina, E. R. Waclawik, G. A. Ayoko, P. Han, J. Jia and H. Y. Zhu, Angew. Chem., Int. Ed., 2019, 58, 12032–12036 CrossRef CAS PubMed.
- S. Sarina, S. Bai, Y. Huang, C. Chen, J. Jia, E. Jaatinen, G. A. Ayoko, Z. Bao and H. Zhu, Green Chem., 2014, 16, 331–341 RSC.
- Y. Dong, C. Hu, H. Xiong, R. Long and Y. Xiong, ACS Catal., 2023, 13, 6730–6743 CrossRef CAS.
- W. Jiang, B. Q. L. Low, R. Long, J. Low, H. Loh, K. Y. Tang, C. H. T. Chai, H. Zhu, H. Zhu, Z. Li, X. J. Loh, Y. Xiong and E. Ye, ACS Nano, 2023, 17, 4193–4229 CrossRef CAS PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- S. Linic, S. Chavez and R. Elias, Nat. Mater., 2021, 20, 916–924 CrossRef CAS PubMed.
- S. A. Lee and S. Link, Acc. Chem. Res., 2021, 54, 1950–1960 CrossRef CAS PubMed.
- T. G. Habteyes, S. Dhuey, E. Wood, D. Gargas, S. Cabrini, P. J. Schuck, A. P. Alivisatos and S. R. Leone, ACS Nano, 2012, 6, 5702–5709 CrossRef CAS PubMed.
- U. Aslam, S. Chavez and S. Linic, Nat. Nanotechnol., 2017, 12, 1000–1005 CrossRef CAS PubMed.
- F. Tong, X. Liang, F. Ma, X. Bao, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, B. Huang and Z. Zheng, ACS Catal., 2021, 11, 3801–3809 CrossRef CAS.
- P. Wang, S. N. Steinmann, G. Fu, C. Michel and P. Sautet, ACS Catal., 2017, 7, 1955–1959 CrossRef CAS.
- B. Wu, J. Lee, S. Mubeen, Y. S. Jun, G. D. Stucky and M. Moskovits, Adv. Opt. Mater., 2016, 4, 1041–1046 CrossRef CAS.
- Y. Zhang, L. Yan, M. Guan, D. Chen, Z. Xu, H. Guo, S. Hu, S. Zhang, X. Liu, Z. Guo, S. Li and S. Meng, Adv. Sci., 2022, 9, 2102978 CrossRef CAS PubMed.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef PubMed.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- D. F. Swearer, H. Zhao, L. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain, E. Ringe, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, 8916–8920 CrossRef CAS PubMed.
- Z. Zhu, R. Tang, C. Li, X. An and L. He, Adv. Sci., 2023, 2302568, DOI:10.1002/advs.202302568.
- R. Ninakanti, F. Dingenen, R. Borah, H. Peeters and S. W. Verbruggen, Top. Curr. Chem., 2022, 380, 40 CrossRef CAS PubMed.
- M. Vadai, D. K. Angell, F. Hayee, K. Sytwu and J. A. Dionne, Nat. Commun., 2018, 9, 4658 CrossRef PubMed.
- K. Sytwu, M. Vadai, F. Hayee, D. K. Angell, A. Dai, J. Dixon and J. A. Dionne, Science, 2021, 371, 280–283 CrossRef CAS PubMed.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- C. Hu, X. Chen, J. Low, Y. Yang, H. Li, D. Wu, S. Chen, J. Jin, H. Li, H. Ju, C. Wang, Z. Lu, R. Long, L. Song and Y. Xiong, Nat. Commun., 2023, 14, 221 CrossRef CAS PubMed.
- F. X. Tong, X. Z. Liang, M. Liu, Z. Y. Wang, Y. Y. Liu, P. Wang, H. F. Cheng, Y. Dai, Z. K. Zheng and B. B. Huang, ACS Catal., 2022, 12, 3558–3565 CrossRef CAS.
- E. Cortes, R. Grzeschik, S. A. Maier and S. Schluecker, Nat. Rev. Chem, 2022, 6, 259–274 CrossRef PubMed.
- L. Langelueddecke, P. Singh and V. Deckert, Appl. Spectrosc., 2015, 69, 1357–1371 CrossRef CAS PubMed.
- T. Touzalin, A. L. Dauphin, S. Joiret, I. T. Lucas and E. Maisonhaute, Phys. Chem. Chem. Phys., 2016, 18, 15510–15513 RSC.
- C. Gao, W. Lin, J. Wang, R. Wang and J. Wang, Plasmonics, 2018, 13, 1343–1358 CrossRef CAS.
- J. F. Li, Y. J. Zhang, S. Y. Ding, R. Panneerselvam and Z. Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS PubMed.
- A. B. Zrimsek, N. Chiang, M. Mattei, S. Zaleski, M. O. McAnally, C. T. Chapman, A. I. Henry, G. C. Schatz and R. P. Van Duyne, Chem. Rev., 2017, 117, 7583–7613 CrossRef CAS PubMed.
- S. Y. Ding, E. M. You, Z. Q. Tian and M. Moskovits, Chem. Soc. Rev., 2017, 46, 4042–4076 RSC.
- I. D. Barcelos, A. R. Cadore, A. B. Alencar, F. C. B. Maia, E. Mania, R. F. Oliveira, C. C. B. Bufon, A. Malachias, R. O. Freitas, R. L. Moreira and H. Chacham, ACS Photonics, 2018, 5, 1912–1918 CrossRef CAS.
- H. A. Bechtel, S. C. Johnson, O. Khatib, E. A. Muller and M. B. Raschke, Surf. Sci. Rep., 2020, 75, 100493 CrossRef CAS.
- P. Christopher and M. Moskovits, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, 2017, vol. 68, pp. 379–398 Search PubMed.
- K. Sytwu, M. Vadai and J. A. Dionne, Adv. Phys.: X, 2019, 4, 1619480 CAS.
- W. Y. Yu, G. M. Mullen, D. W. Flaherty and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 11070–11078 CrossRef CAS PubMed.
- Q. Lv, Q. L. Meng, W. W. Liu, N. Sun, K. Jiang, L. P. Ma, Z. Q. Peng, W. B. Cai, C. P. Liu, J. J. Ge, L. M. Liu and W. Xing, J. Phys. Chem. C, 2018, 122, 2081–2088 CrossRef CAS.
- K. Chen and H. Wang, Nano Lett., 2023, 23, 2870–2876 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhou, X. Fu, E. Villarreal, L. Sun, S. Zou and H. Wang, J. Phys. Chem. C, 2019, 123, 26695–26704 CrossRef CAS.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- I. Dutta, S. Chatterjee, H. Cheng, R. K. Parsapur, Z. Liu, Z. Li, E. Ye, H. Kawanami, J. S. C. Low, Z. Lai, X. J. Loh and K. W. Huang, Adv. Energy Mater., 2022, 12, 2103799 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |