
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast sensitivity-controlled and specific detection of extracellular vesicles using optical force with antibody-modified microparticles in a microflow system†
Kana
Fujiwara‡
abc,
Yumiko
Takagi‡
ab,
Mamoru
Tamura
 bd,
Mika
Omura
be,
Kenta
Morimoto
be,
Ikuhiko
Nakase
*be,
Shiho
Tokonami
*bc and
Takuya
Iida
*ab
bd,
Mika
Omura
be,
Kenta
Morimoto
be,
Ikuhiko
Nakase
*be,
Shiho
Tokonami
*bc and
Takuya
Iida
*ab
aDepartment of Physics, Graduate School of Science, Osaka Metropolitan University, 1-2, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8570, Japan. E-mail: t-iida@omu.ac.jp
bResearch Institute for Light-induced Acceleration System (RILACS), Osaka Metropolitan University, 1-2, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8570, Japan. E-mail: tokonami@omu.ac.jp; i-nakase@omu.ac.jp
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka Metropolitan University, 1-2, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8570, Japan
dDepartment of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, 1-3, Machikaneyama-cho, Toyonaka, Osaka 560-8531, Japan
eDepartment of Biological Chemistry, Graduate School of Science, Osaka Metropolitan University, 1-2, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8570, Japan
First published on 12th July 2023
Abstract
Extracellular vesicles (EVs), including nanoscale exosomes and ectosomes, hold promise as biomarkers that provide information about the cell of origin through their cargo of nucleic acids and proteins, both on their surface and within. Here, we develop a detection method of EVs based on light-induced acceleration of specific binding between their surface and antibody-modified microparticles, using a controlled microflow with three-dimensional analysis by confocal microscopy. Our method successfully detected 103–104 nanoscale EVs in liquid samples as small as a 500 nanoliters within 5 minutes, with the ability to distinguish multiple membrane proteins. Remarkably, we achieved the specific detection of EVs secreted from living cancer cell lines with high linearity, without the need for a time-consuming ultracentrifugation process that can take several hours. Furthermore, the detection range can be controlled by adjusting the action range of optical force using a defocused laser, consistent with the theoretical calculations. These findings demonstrate an ultrafast, sensitive, and quantitative approach for measuring biological nanoparticles, enabling innovative analyses of cell-to-cell communication and early diagnosis of various diseases, including cancer.
New conceptsIn this study, we introduce an unconventional method for rapid, sensitive, and specific detection of biological nanoparticles containing information about the original cell. Our approach allows for control of the detection range under manipulating the action range and strength of the optical force in conjunction with three-dimensional analysis using confocal microscopy. This method significantly improves the sensitivity, the precision, and the detection time in comparison with conventional methods, such as ELISA, while maintaining the specificity of the antibodies used. Notably, we demonstrate successful detection of EVs with target membrane proteins secreted from living cells, without the need for ultracentrifugation processes. Our developed method leverages the effects of optical and microfluidic pressures to accelerate molecular recognition, enabling selective trapping of nanoscale biological samples. This approach overcomes the bottlenecks of previous research on detection efficiency and operation time associated with the complexities of conventional methods, and holds promise for the next generation of liquid biopsy in cancer marker detection and other biological applications. |
Biological cells, including human cells, comprise various vesicle structures such as endosomes, lysosomes, and peroxisomes and autophagosomes. Nanoscale extracellular vesicles (EVs) such as exosomes and ectosomes are secreted outside the cell for intercellular communication.1–6 Nanoscale EVs including exosomes, with a diameter of 50–150 nm, have gained increasing attention not only as carriers of intercellular communication but also as biomarkers for various diseases and capsules for drug delivery, as they contain microRNAs, mRNAs, and proteins from their cells of origin.7–9 Therefore, the rapid and sensitive detection of nanoscale EVs from trace samples would be particularly valuable in the early diagnosis of intractable diseases, for example, cancer and Alzheimer's disease.10,11 Nanoscale EVs are characterized by membrane proteins with four transmembrane domains called tetraspanins,12 which are typically detected using assays involving selective binding of antibodies to these proteins. For example, conventional protein detection methods such as enzyme-linked immunosorbent assay (ELISA)13,14 rely on molecular recognition such as an antigen–antibody reaction and quantitatively evaluate the target molecules through a chromogenic reaction. Also, western blotting can be used to detect specific proteins in protein mixtures by combining electrophoresis with antigen–antibody reactions.15,16 Furthermore, a surface plasmon resonance (SPR) sensor can also detect target proteins based on the fact that the SPR wavelength of a metal is altered depending on the refractive index of the medium in contact with the surface.17,18 However, these methods require complex pre-treatment and operation processes with several hours required for detection. As an alternative, ExoCounter19 was designed to specifically and quantitatively detect exosomes; however, this method is based on ELISA, which requires a large sample volume and several hours for detection. Moreover, particle counting methods have the advantage of quantifying EVs but lack specificity in distinguishing these vesicles from other particles in clinical samples.20
Although these conventional methods for the detection and interaction analysis of nanoscale EVs and proteins have a proven track record in research applications, clinical practices require high sensitivity in addition to the advantages of simplicity and speed. For example, the SARS-CoV-2 virus, which is responsible for the COVID-19 pandemic, is a biological nanoparticle with a similar size to nanoscale EVs.21 Rapid and accurate detection of infected individuals can help prevent disease clusters and the spread of virus infection. In addition, biopsies in cancer screening, which involve tissue excision from organs and are associated with risks of human error, patient discomfort, and time-consuming procedures, can be replaced with liquid biopsy using body fluids such as blood, saliva, and urine to detect disease-related proteins. Liquid biopsy is gaining increasing attention as a non-invasive alternative for diagnosis.22–24 This process is simpler, quicker, and less invasive as it requires only the collection of body fluids for testing. Simplicity, speed of pre-treatment, and high sensitivity in measurement are important attributes of liquid biopsies.
Pre-treatment such as separation by ultracentrifugation is necessary for detecting nanoscale EVs, representing a significant bottleneck in research and development. If nanoscale EVs can be easily guided and concentrated at a targeted location for the observation, these limitations of current detection methods may be overcome. Research on optical manipulation at the nanoscale and microscale has made great progress in recent years. Optical tweezers have been developed to manipulate and trap microscopic materials without destruction and contact, using gradient forces proportional to the intensity gradient of electromagnetic fields generated by a focused laser. These tweezers can capture cells, microbes, and biomolecules;25–27 control the motion of semi-conductor quantum dots and metallic nanoparticles;28,29 and are now being used across the fields of physics, chemistry, and biology. There are also successful examples of the assembly of nanoparticles and bacteria using thermo-hydrodynamic effects (i.e., light-induced convection) and bubbles generated in high-temperature conditions as aggregation sites, based on the photothermal effect of optically responsive materials (substrates and particles).30,31 Trace DNA detection through the formation of sub-millimetre-order aggregates has also been proposed, which uses the synergistic effect of light-induced force and photothermal convection to accelerate the double-strand formation between target DNA and single-stranded DNA modified on the surface of gold nanoparticles.32
Furthermore, the analysis of proteins using light-induced convection and bubbles due to the marked photothermal effect in dense systems of metal nanoparticles has been attempted.33 Additionally, this light–matter interaction was further enhanced in a narrow space in a microfluidic channel using pressure-driven flow, which enabled the detection of femtogram-level proteins with photothermal probe particles34 and accelerated the antigen–antibody reaction of attogram-level proteins with non-thermal probe particles.35 Thus, detection of proteins with high specificity would be expected using antibody-modified microparticles without light absorption to avoid thermal damage while using the same narrow space. However, it is unclear whether the same method can be applied to the highly sensitive and rapid detection of biological nanoparticles expressing proteins on their surface, such as EV. Moreover, there have been limited discussions on the effects of the intensity and action range of the optical force on the detection efficiency.
Therefore, the aim of this study was to develop a method that can effectively detect nanoscale EVs derived from several types of cancer cells by distinguishing the surface membrane-expressed proteins from others. Toward this end, we used the human colorectal cancer cell line HCT116 and the human lung cancer cell line A549 as models, with surface expression of CD9 and CD63 as the targets for optical condensation detection in a narrow space under the optical system, as demonstrated in Fig. 1a and b. Probe microparticles (MPs) modified with antibodies (anti-CD9 and anti-CD63 antibodies) that could specifically bind to these proteins were introduced into the microfluid channels. We then attempted to control the action range and intensity of the optical force, and further explored the mechanism of light-induced assembly and the possibility of micro-detection under the assistance of pressure-driven flow.
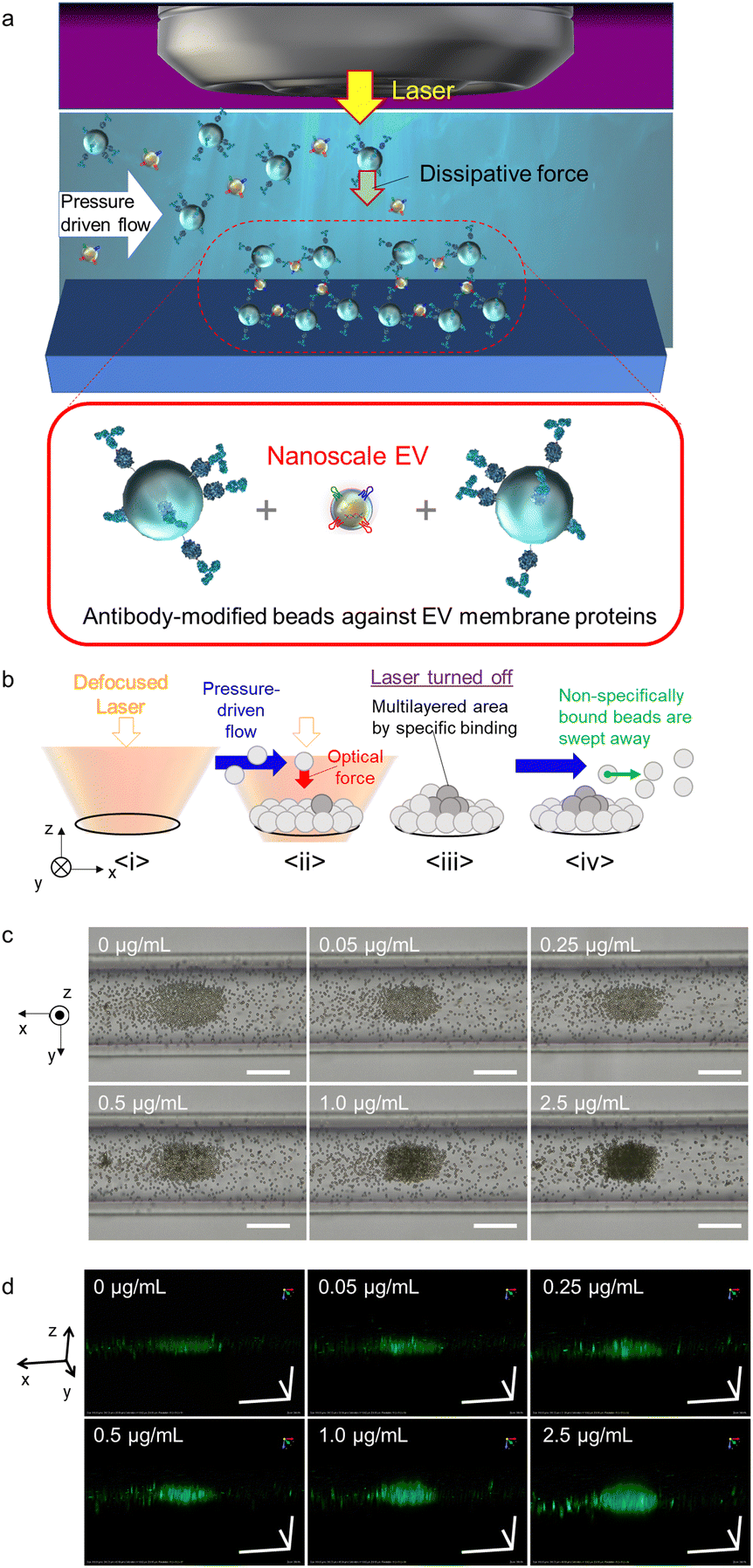 | ||
| Fig. 1 (a) Schematic diagram of the selective light-induced assembly of antibody-modified probe microparticles (MPs) mediated by nanoscale EVs in a microchannel. (b) Mechanism of the formation of trapping sites and multi-layered structures via target EVs between antibody-modified probe MPs. (c) Transmission optical microscope images of light-induced assembly with and without different concentrations of HCT116 cell-derived standard EVs. The pressure-driven flow is in the +x-direction, where the direction appears to be inverted in the optical microscope image. (d) Confocal three-dimensional images of light-induced assembly with and without different concentrations of HCT116 standard EVs. Each scale bar is 50 μm in all panels of (c) and (d). | ||
A combination of nanoscale EVs derived from HCT116 colorectal cancer cells was introduced into a microfluidic channel with a flow rate of 0.1 μL min−1 along with anti-CD63 antibody-modified probe MPs that selectively binds to surface proteins on nanoscale EVs. Green fluorochrome-doped polymer beads with a diameter of 2 μm were used as probe MPs to generate a strong dissipative force through Mie scattering at a wavelength of 1064 nm (Fig. 1c and d). The nanoscale EVs and probes were introduced into the channel at a constant rate using pressure-driven flow via a syringe pump. Optical condensation was achieved by applying a dissipative force to the probe MPs through irradiation with a continuous-wave (CW) laser of 1064 nm wavelength and 265 mW power, focused by a 40× objective lens with a numerical aperture of 0.6 from above under pressure-driven flow. The position of the laser focal point was set at 65 μm below the bottom of the channel for light-induced assembly (refer to Fig. S1 in the ESI† for details of the optical system used for optical condensation). The laser irradiation was set for 5 minutes to trap the nanoscale EVs and probe MPs at the solid–liquid interface at the bottom of the channel. The structures formed at the bottom of the channel were then observed 30 seconds after irradiation using two-dimensional (2D) transmission imaging under halogen-lamp irradiation and three-dimensional (3D) confocal imaging at an excitation wavelength of 488 nm (refer to Experimental). The higher the concentration of EV standard, the larger the size of the dark area with low transmittance in the 2D transmission image (Fig. 1c), and the larger the stereo image of green fluorescence in the 3D confocal image (Fig. 1d). The damage due to the photothermal effect was negligible because both the probe MPs and the nanoscale EVs have almost no optical absorption in the infrared wavelength range.
The assembly area (the area of the first layer of assembled probe MPs indicating the trapping site modified with antibodies) and the multi-layered area (black region obtained from the optical transmission image) as shown in Fig. 1b and c, and the ratio of the multi-layered area to the assembly area was plotted on the vertical axis as a function of the concentration of HCT116 EV standard (representing the concentration of the membrane protein) (Fig. 2a–c). The size of the 3D structure was then plotted as a function of the concentration of EV standard in the xz-cross-section area of the 3D confocal image (see Fig. S2 in the ESI†), showing the same concentration dependence, as demonstrated in Fig. 2b and c. This indicates that the aggregation formed by the optical condensation can be represented as 3D accumulated structures. A negative control experiment of light-induced assembly was performed by introducing calnexin as a target protein along with anti-CD63 antibody-modified probe MPs to confirm that these structures were formed by the antigen-antibody reaction. Calnexin is a molecular chaperone in the endoplasmic reticulum, which is involved in the folding and assembly of newly synthesized proteins, thus serving as a negative control for HCT116-derived nanoscale EVs.36 As a result, even at high concentrations of calnexin, a limited multi-layer structure was formed with no concentration dependence (Fig. S3 and S4 in the ESI†). These results indicated that light-induced acceleration of specific binding of the anti-CD63 antibodies on the surface of the probe MPs to the CD63 on the surface of HCT116-derived nanoscale EVs led to a multi-layered 3D structure of microparticles (Fig. 1c, d and Fig. 2). This also indicates that quantitative detection by optical condensation of nanoscale EVs can be achieved within only 5 min. The concentration of nanoscale EVs in 1 mg mL−1 of stock solution was 3 × 1011 EVs per mL. Furthermore, 0.05 μg mL−1 corresponds to 1.5 × 107 EVs per mL, which means 3.75 × 103 nanoscale EVs were contained in 500 nL after mixing probe MPs with the target sample liquid.
 | ||
| Fig. 2 Calibration curve of HCT116 standard nanoscale EVs for light-induced acceleration in a microchannel. (a) Assembly area, (b) multi-layered area, and (c) ratio of the multi-layered area at each concentration of EV standard. Inset: Low concentration range. Error bars are standard deviations (n = 3). | ||
CD9, a membrane protein, is also expressed on the surface of nanoscale EVs derived from HCT116 cells. Similar experiments conducted using anti-CD9 antibody-modified probe MPs revealed that the ratio of the multi-layered area was higher compared to targeting CD63, as depicted in Fig. 3a. The relationship between the detected amount in optical condensation detection and the absorbance measurement in ELISA (Fig. 3c) showed a significant correlation, indicating that high linearity and low standard deviation can be achieved even at concentrations that are one to two orders of magnitude lower than the limit of detection (1.25 μg mL−1). In the case of A549 cells, CD63 exhibited a higher signal compared to CD9 in the detection of nanoscale EVs. As seen in Fig. 3b, the ratio of the multi-layered area was larger, suggesting that the expression level of CD63 is higher than that of CD9, which is consistent with the trend observed in ELISA (Fig. 3d).
 | ||
| Fig. 3 (a and b) Ratio of the multi-layered area of anti-CD63 antibody-modified probe microparticles (MPs) and anti-CD9 antibody-modified probe MPs at each exosome concentration (CD63/CD63 means that target EV was sandwiched by Anti-CD63 antibody-modified probe MPs, and CD9/CD9 means that target EV was sandwiched by Anti-CD9 antibody-modified probe MPs.). The detection targets are HCT116- and A549-derived exosomes, respectively. (c and d) Measurement results of CD63/CD63 sandwich ELISA (anti-CD63 antibodies are used for capture and detection) and CD9/CD9 sandwich ELISA (anti-CD9 antibodies are used for capture and detection). The detection targets are HCT116- and A549-derived nanoscale EVs, respectively. Error bars present standard deviations (n = 3). | ||
In contrast to ELISA, which requires 100 μL of sample and 5–6 h for detection, including incubation and washing processes, the optical condensation detection method developed in this study enables rapid quantitative evaluation after only 5 min of laser irradiation, with higher sensitivity by one to two orders of magnitude. Only approximately 500 nL of liquid passes through the cross-section of the microchannel in 5 min, which is a very small sample volume. Additionally, the laser spot can be adjusted to the sub-millimetre area to collect many probe particles and target nanoscale EVs. Furthermore, the automatic washing process by pressure-driven flow eliminates the complexity of the operation and reduces the detection time. These features of our developed microflow-type optical condensation overcome conventional methods from the viewpoints of detection time, sample volume, and limit of detection, as shown in Table 1.
| Method | Detection time | Label | Sample volume | Limit of detection (μg mL−1) |
|---|---|---|---|---|
| This work | 5 minutes | Unnecessary | 500 nL | 0.05 |
| ELISA | 5–6 hours | Necessary | 100 μL | 1–2 |
| SPR sensor | 3–4 hours | Unnecessary | 100 μL | 1 |
We further aimed to increase the sensitivity of microflow-type optical condensation detection by controlling the optical force (the dissipative force due to photon momentum transfer) through modulation of the laser focal point (Fig. 5 is the simulation result corresponding to the experiment in Fig. 4). The ratio of the multi-layered area to the first layer, as an indicator of the aggregate size, was evaluated for each dispersed target concentration. Although a positive correlation was determined between the target concentration and the percentage of the multi-layered area, the effect of the range and intensity of optical force on the light-induced assembly of probe particles was further investigated by changing the focal position of the laser. The size of the laser spot can be controlled by shifting the laser focal point by 45, 65, and 94 μm in the vertical direction below the channel bottom. The more the focal point is shifted downward, the wider the range of light-induced assembly that is observed (Fig. S5 and S6 in the ESI†). The distance from the bottom of the channel to the focusing position may be defined as the “focusing distance” (FD). The slope was maximal at FD = 45 μm, where the spot size was the smallest, and with relatively small error bars at a linearity of 1.0 μg mL−1 or less (Fig. S7 in the ESI†). This is considered to be due to the increased laser power (intensity) per unit area and the enhanced dissipative force as a result of the smaller spot size. It is worth noting that the lower limit of detection of the conventional ELISA method is 1.25 μg mL−1, whereas using our method, the sensitivity increased by more than one order of magnitude and a calibration curve with good linearity (p < 0.001, R2 = 0.998) was obtained at FD = 65 μm. Furthermore, it is significant to note that such high-sensitivity detection was achieved after only 5 min of laser irradiation. However, a significant decrease was observed in the slope of the ratio of the multi-layered area with FD = 45 μm in the region between 1.0 and 2.5 μg mL−1 compared to the low-concentration region with a target concentration of 1.0 μg mL−1 or less. Such a decrease may be due to the saturation of the trapping sites formed in the first layer since the area of the trapping site gets smaller for the smaller FD. It is expected that the detection range may be controlled by modulating the spot diameter. These results indicate the possibility of stable multi-layer formation and control of the detection range of the target by increasing the dissipative force with increasing laser intensity.
The effect of spot diameter on aggregate formation was investigated through theoretical analysis (Fig. 5) by solving the equation of motion based on the Euler method considering the discrete element model with optical force, interparticle contact force, drag force, gravitational force, and random force described in ref. 35. During the process of light-induced assembly, probe particles dispersed randomly were trapped by a dissipative force exerted by the large laser spot causing them to accumulate into a monolayer due to interaction with the channel wall (Fig. 5a, b and c; cohesive energy density, 0 J m−3). If the target objects were located near the beam waist, they would bind to antibodies on the surface of the probe particles, leading to cross-linking of the beads, and formation of a multi-layered structure, with the first layer serving as the trapping site. The size of the spot diameter at the bottom of the channel depending on FD, had a significant impact on the light-induced assembly process (Fig. 5d–f; cohesive energy density, 100 J m−3). A smaller FD resulted in a narrower spot diameter at the bottom of the channel and a higher laser intensity at the laser spot. Since the dissipative force is proportional to the scattering cross-section of particles and the laser intensity, and the area of the light-induced aggregates was dependent on the focal position. Additionally, the smaller the FD, the larger the multi-layered structure obtained along the optical axis. Although the maximum slope occurred at FD = 45 μm in Fig. 4c, the ratio of the multi-layered area was larger at FD = 65 μm at very low concentrations. This could be attributed to the fact that the spot diameter at FD = 45 μm was too small for the channel width, resulting in reduced accumulation efficiency as targets passing outside the laser spot could not be trapped. On the other hand, at FD = 94 μm, the weaker dissipative force of the laser beam resulted in unstable aggregates that were almost completely swept away in the latter half of the simulation (Movie S6, ESI†), as observed in Fig. 4 and Fig. S7 (ESI†). These results suggest that the detection range and sensitivity can be controlled via the optical force by appropriately setting the association between the spot diameter and channel width.
 | ||
| Fig. 4 (a) Schematic diagram of the relationship between the focusing distance (FD) and the laser spot diameter at the bottom of the microchannel. (b and c) The ratio of the multi-layered area at each FD. Error bars present the standard deviations (n = 3). The assembly area and multi-layered area at each FD are presented in Fig. S5 of the ESI.† | ||
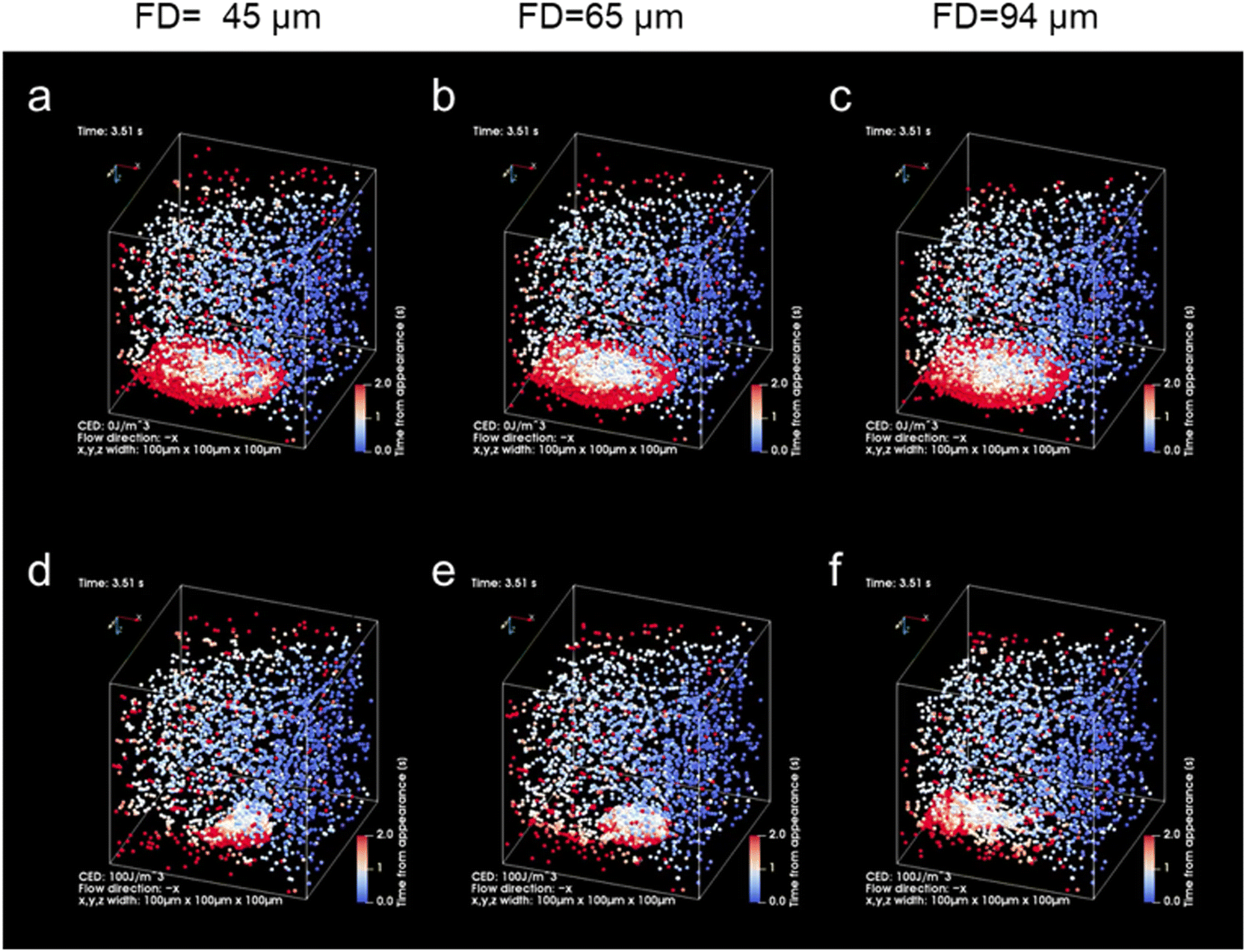 | ||
| Fig. 5 Theoretical results of light-induced assembly of microparticles at each focusing distance (FD; 45 μm, 65 μm, 94 μm) by optical force in a microchannel (volume flow rate: 0.05 μL min−1, laser power: 265 mW). The laser was irradiated during the initial 3 s and was then turned off subsequently. The colour of the particles indicates the time elapsed since the particle appeared from the inlet (blue: 0 s, white: 1 s, red: 2 s). (a–c) Cohesive energy density, 0 J m−3 (see also Movies S1, S3 and S5 in the ESI†). (d–f) Cohesive energy density, 100 J m−3 (see also Movies S2, S4 and S6 in the ESI†). | ||
Next, after the procedure in Fig. S8 (ESI†), we performed the specific detection of the nanoscale EVs from the supernatant of our cultivated HCT116 cell line with high linearity after the ultracentrifuge (Fig. 6a), where these EVs may contain 6.68 × 104 nanoscale EVs expressing membrane protein CD63 on their surfaces from the corresponding ratio of the multi-layered area (broken red lines: 0.89 μg mL−1 in the inset of Fig. 2c and 11.4 μg mL−1 in Fig. 6a). Remarkably, in Fig. 6b after the simple process in Fig. 6c, we also confirmed positive correlation in the detection curve after the light-induced detection of proteins by anti-CD63 antibody-modified MPs from the supernatant of HCT116 cells even without ultracentrifugation. Although the protein concentration was slightly over estimated in comparison with Fig. 6a, the high linearity in the region lower than 40 μg mL−1 indicates the selective detection of nanoscale EVs expressing CD63 on their surfaces. In fact, nanoparticles of 100 nm in diameter were observed by scanning electron microscope (SEM) and the nanopore analytical system, as shown in Fig. S9 (ESI†). These results strongly support that we can omit the ultracentrifuge process requiring several hours in the detection process of nanoscale EVs by using our developed microflow-type optical condensation method.
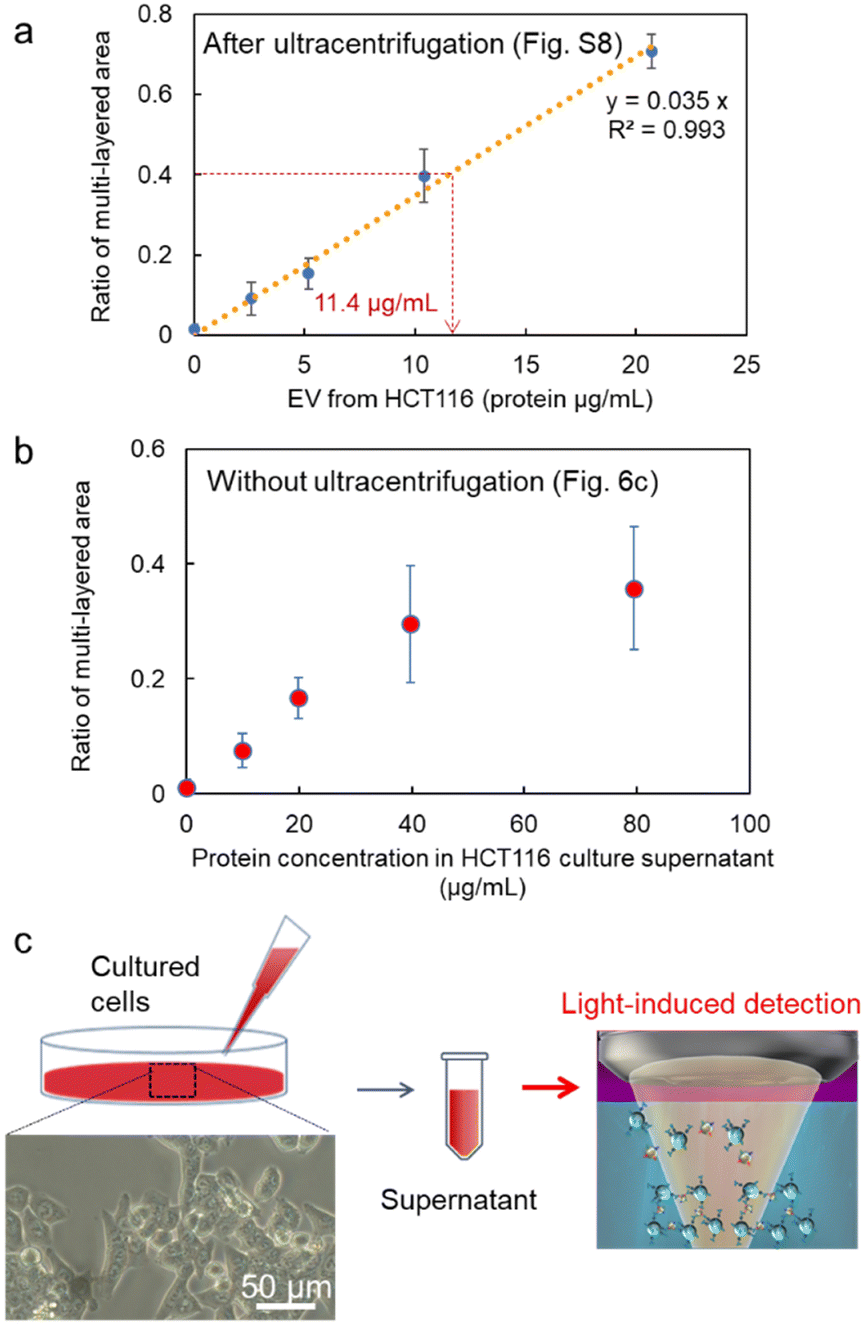 | ||
| Fig. 6 Detection curves of EVs from the diluted cell supernatant of the cultured HCT116 cell line, (a) after the ultracentrifugation process in Fig. S8 (ESI†), and (b) without the ultracentrifugation as shown in (c). Error bars are standard deviations (n = 3). The inset of (c) is the optical microscope image of HCT 116 cells (40×). | ||
Conclusions
This study presents an unconventional approach for controlling the detection range of nanoscale extracellular vesicles (EVs) by adjusting the action range and intensity of optical force. The phenomenon of the light-induced assembly of probe particles and nanoscale EVs using a focused laser beam in a microfluidic channel was investigated, revealing that laser irradiation for only 5 minutes could detect 103–104 nanoscale EVs. This method significantly increases the sensitivity of detection by over one order of magnitude and reduces the detection time by nearly two orders of magnitude compared to those of the conventional methods like ELISA. The results also indicated that the expression levels of membrane proteins CD63 and CD9 were different in nanoscale EVs derived from colorectal cancer cells (HCT116) and lung cancer cells (A549), even at concentrations below the lower detection limit of ELISA. Furthermore, this microflow-type optical condensation approach has the advantage of analysing biomolecules that cannot be amplified by polymerase chain reaction (PCR), whereas DNA and RNA can be amplified with PCR. The elucidation of the mechanism in this study represents a significant achievement, demonstrating that this method can be applied to the selective and rapid detection of EVs, including nanoscale EVs, without the need for the time consuming ultracentrifugation process. These findings provide a guiding principle for high-throughput detection of not only nanoscale EVs but also a variety of biological nanoparticles that serve as biomarkers, leading to the establishment of a foundation for early disease diagnosis.Experimental
Microflow-type optical condensation system for light-induced acceleration of antigen–antibody binding
The optical system used for the experiment in the light-induced acceleration of HCT116-derived nanoscale EVs and anti-CD63 probe MPs is presented in Fig. S1 of the ESI.† An upright optical microscope (ECLIPSE Ni-E; Nikon, Tokyo, Japan) was used. The sample liquid (30 μL; mixture of 15 μL dispersion liquid of HCT116-derived nanoscale EVs and 15 μL dispersion liquid of anti-CD63 probe MPs) was introduced into a glass syringe (micro-syringe 1725 TLL Lure Lock 81120, 250 μL, Hamilton, USA), and the flow rate was controlled using a syringe pump (78-0210J, KD Scientific). The direction of pressure-driven flow (+x-direction) appears to be inverted in the optical image due to the optical system of our microscope (both the x- and y-directions are inverted). Thereafter, a near-infrared CW laser with a wavelength of 1064 nm (FLS-1064-2000F, Sigma Koki, Japan) was introduced from a backport adapter (LMS-M1064-20001S/LN, Sigma Koki, Japan) and focused using an objective (CFI S Plan Fluor ELWD 40XC, 0.6 NA) on the bottom of the microchannel (width 100 μm × height 100 μm; MiniLuer 0144 COC, ASICON, Japan).The laser spot diameter was 2.6 μm at the focal point, and the laser power was determined with a laser power meter (UP17O-H5 and TUNER, Gentec Electro-Optics, Canada), and the spot position was controlled to the defocused condition to generate optical pressure over a wide region. Optical transmission images of the assembled structure were recorded using a CCD camera (DS-Filc-L3, Nikon, Japan) under irradiation with white light from a halogen lamp after 5 min of laser irradiation. The cross-sectional areas of the assembled structure and multi-layered structure (black region) were analysed using commercial software (NIS Elements, Nikon, Japan).
Preparation of nanoscale EVs and antibody-modified probe MPs
HCT116-derived nanoscale EVs (HansaBioMed OU, HBM-HCT) and A549-derived nanoscale EVs (HansaBioMed OU, HBM-A549) were used as the standard sample of biomarkers to be detected, and anti-CD63 antibody (Cosmo Bio Inc., SHI-EXO-M02-B) and anti-CD9 antibody (Cosmo Bio Inc., SHI-EXO-M01-B) were biotinylated and used as the probe molecules. The stock solutions (1 mg mL−1) of each exosome dispersion were diluted to the desired concentration for the experiment with 10 mM phosphate buffer (pH 6.7) (FUJIFILM, 164-27135) as a solvent.Each antibody was modified with streptavidin–Fluoresbrite YG-coated microspheres (Polysciences, 24159), which were with 2-μm diameter. The stock solutions (1 mg mL−1) of the biotinylated antibodies were diluted to 20 μg mL−1 with the same phosphate buffer as that used for the exosome dilution. First, the microspheres were diluted to 5.30 × 108 particles per mL with phosphate buffer and washed five times via centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 5 min). After the last wash, the microspheres were resuspended in phosphate buffer at 1.06 × 109 particles per mL. The prepared solutions of the streptavidin-conjugated microspheres and biotinylated antibodies were mixed in equal amounts and incubated for 1 h at 42 °C. The mixture was then removed from the incubator and washed thrice. The prepared antibody-modified probe MPs were left to stand overnight at 4 °C.
000 × g, 5 min). After the last wash, the microspheres were resuspended in phosphate buffer at 1.06 × 109 particles per mL. The prepared solutions of the streptavidin-conjugated microspheres and biotinylated antibodies were mixed in equal amounts and incubated for 1 h at 42 °C. The mixture was then removed from the incubator and washed thrice. The prepared antibody-modified probe MPs were left to stand overnight at 4 °C.
Negative control
A calnexin standard [Human Calnexin ELISA Kit (Colorimetric), Novus Biologicals, NBP2-75816] was used as a negative control target, which was diluted with 10 mM phosphate buffer (pH 6.7). The measured concentration was set near the lower detection limit of ELISA (0.63 ng mL−1).Confocal optical microscopy
A confocal laser microscope (Nikon C2Si, Nikon, Japan) was used to observe the multilayer structures of the probe particles. An excitation laser (wavelength 488 nm) was introduced above the sample. A pinhole was placed in front of the detector to obtain 3D images by capturing several layers of confocal images in the optical axis direction.ELISA
The ELISA kits used in this study were CD63-Capture Human Exosome ELISA Kit (FUJIFILM, 290-83601) and CD9-Capture Human Exosome ELISA Kit (FUJIFILM, 290-83701). Notably, 10 mM phosphate buffer (pH 6.7) was used instead of the sample reaction buffer and antibody reaction buffer from the kits to enable effective comparison with the optical condensation system. In addition, the biotinylated antibodies (anti-CD9 and anti-CD63 antibodies) used in this system were diluted to 250 ng mL−1 rather than the antibodies included in these kits. All other procedures were performed according to the manufacturer's protocol.Cell cultures
Human colorectal-derived HCT116 cells (CCL-247, ATCC) were purchased from ATCC. The cells were cultured in 100 mm dishes using McCoy's 5A Modified Medium (Gibco, Life Technologies Corporation) containing 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Life Technologies Corporation) and incubated at 37 °C under 5% CO2 in a humidified incubator.Isolation of EVs
HCT116 cells (3 × 106 cells) were seeded on 100 mm dishes in McCoy's medium containing 10% FBS for 24 h. After washing with serum-free McCoy's (3 mL × 5), the cells were cultured in McCoy's medium containing 10% EV-free FBS (Exosome-Depleted FBS, System Biosciences LLC) (10 mL) for 24 h. The secreted EVs were isolated by centrifuging the collected cell culture medium (300 g) for 10 min at 4 °C, and the supernatant (2000 g) for 10 min at 4 °C and followed by another centrifugation (10000 g) for 30 min at 4 °C to remove the cell debris. The supernatant was then centrifuged (150000 g) for 80 min at 4 °C (Himac CP65β, Hitachi, Tokyo, Japan) in duplicate, and the EV pellet was collected in phosphate-buffered saline (PBS). The concentrations of isolated EVs were determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc).
Author contributions
T. I., S. T. and I. N. initiated the research and contributed to the study design. K. F. Y. T., T. I., and S. T. performed the experiment of light-induced assembly of extracellular vesicles (EVs). K. F. and Y. T. performed a comparative experiment with ELISA. K. F., M. O., K. M., and I. N. performed extraction of EVs from the cultivated cell line and nanopore measurement. M. T. and T. I. performed the theoretical calculations. K. F., Y. T., T. I. and S. T. prepared the figures and the manuscript. The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Professors Takaaki Satoh and Ayumu Taguchi for constructive discussions. This study was supported by the JST-Mirai Program (No. JPMJMI18GA, No. JPMJMI21G1), Grant-in-Aid for Scientific Research (A) (No. JP17H00856, No. JP21H04964), JST FOREST Program (No. JPMJFR201O), Grant-in-Aid for Scientific Research (B) (No. JP18H03522), Scientific Research on Innovative Areas (No. JP16H06507), Grant-in-Aid for Early-Career Scientists (No. JP20K15196) from Japan Society for the Promotion of Science KAKENHI, and the Key Project Grant Program of Osaka Prefecture University. The authors thank Editage (http://www.editage.com) for English language editing.Notes and references
- X. Jin, Y. Zhang, A. Alharbi, A. Hanbashi, A. Alhoshani and J. Parrington, Trends Pharmacol. Sci., 2020, 41, 582–594 CrossRef CAS PubMed
.
- A. S. Abu Lila and T. Ishida, Biol. Pharm. Bull., 2017, 40, 1–10 CrossRef CAS PubMed
- C. Lässer, V. Seyed Alikhani, K. Ekström, M. Eldh, P. T. Paredes, A. Bossios, M. Sjöstrand, S. Gabrielsson, J. Lötvall and H. Valadi, J. Transl. Med., 2011, 9, 9 CrossRef PubMed
- T. Takenaka, S. Nakai, M. Katayama, M. Hirano, N. Ueno, K. Noguchi, T. Takatani-Nakase, I. Fujii, S. S. Kobayashi and I. Nakase, Int. J. Pharm., 2019, 572, 118762 CrossRef CAS PubMed
- M. Mathieu, N. Névo, M. Jouve, J. I. Valenzuela, M. Maurin, F. J. Verweij, R. Palmulli, D. Lankar, F. Dingli, D. Loew, E. Rubinstein, G. Boncompain, F. Perez and C. Théry, Nat. Commun., 2021, 12, 4389 CrossRef CAS PubMed
- K. W. Witwer and C. Théry, J. Extracell. Vesicles, 2019, 8, 1 Search PubMed
- S. E. L. Andaloussi, L. Mäger, X. O. Breakefield and M. J. Wood, Nat. Rev. Drug Discovery, 2013, 12, 347–357 CrossRef PubMed
- I. K. Herrmann, M. J. A. Wood and G. Fuhrmann, Nat. Nanotechnol., 2021, 16, 748–759 CrossRef CAS PubMed
- A. Hoshino, A. Hoshino, H. S. Kim, L. Bojmar, K. E. Gyan, M. Cioffi, J. Hernandez, C. P. Zambirinis, G. Rodrigues, H. Molina, S. Heissel, M. T. Mark, L. Steiner, A. Benito-Martin, S. Lucotti, A. Di Giannatale, K. Offer, M. Nakajima, C. Williams, L. Nogués, F. A. Pelissier Vatter, A. Hashimoto, A. E. Davies, D. Freitas, C. M. Kenific, Y. Ararso, W. Buehring, P. Lauritzen, Y. Ogitani, K. Sugiura, N. Takahashi, M. Alečković, K. A. Bailey, J. S. Jolissant, H. Wang, A. Harris, L. M. Schaeffer, G. García-Santos, Z. Posner, V. P. Balachandran, Y. Khakoo, G. P. Raju, A. Scherz, I. Sagi, R. Scherz-Shouval, Y. Yarden, M. Oren, M. Malladi, M. Petriccione, K. C. De Braganca, M. Donzelli, C. Fischer, S. Vitolano, G. P. Wright, L. Ganshaw, M. Marrano, A. Ahmed, J. DeStefano, E. Danzer, M. H. A. Roehrl, N. J. Lacayo, T. C. Vincent, M. R. Weiser, M. S. Brady, P. A. Meyers, L. H. Wexler, S. R. Ambati, A. J. Chou, E. K. Slotkin, S. Modak, S. S. Roberts, E. M. Basu, D. Diolaiti, B. A. Krantz, F. Cardoso, A. L. Simpson, M. Berger, C. M. Rudin, D. M. Simeone, M. Jain, C. M. Ghajar, S. K. Batra, B. Z. Stanger, J. Bui, K. A. Brown, V. K. Rajasekhar, J. H. Healey, M. de Sousa, K. Kramer, S. Sheth, J. Baisch, V. Pascual, T. E. Heaton, M. P. La Quaglia, D. J. Pisapia, R. Schwartz, H. Zhang, Y. Liu, A. Shukla, L. Blavier, Y. A. DeClerck, M. LaBarge, M. J. Bissell, T. C. Caffrey, P. M. Grandgenett, M. A. Hollingsworth, J. Bromberg, B. Costa-Silva, H. Peinado, Y. Kang, B. A. Garcia, E. M. O'Reilly, D. Kelsen, T. M. Trippett, D. R. Jones, I. R. Matei, W. R. Jarnagin and D. Lyden, Cell, 2020, 182, 1044–1061 CrossRef CAS PubMed
- A. Möller and R. J. Lobb, Nat. Rev. Cancer, 2020, 20, 697–709 CrossRef PubMed
- C. Z. J. Lim, Y. Zhang, Y. Chen, H. Zhao, M. C. Stephenson, N. R. Y. Ho, Y. Chen, J. Chung, A. Reilhac, T. P. Loh, C. L. H. Chen and H. Shao, Nat. Commun., 2019, 10, 1144 CrossRef PubMed
- Z. Andreu and M. Yáñez-Mó, Front. Immunol., 2014, 5, 442 Search PubMed
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871–874 CrossRef CAS PubMed
- M. P. Bui, S. Ahmed and A. Abbas, Nano Lett., 2015, 15, 6239–6246 CrossRef CAS PubMed
- W. N. Burnette, Anal. Biochem., 1981, 112, 195–203 CrossRef CAS PubMed
- I. Nakase, K. Noguchi, I. Fujii and S. Futaki, Sci. Rep., 2016, 6, 34937 CrossRef CAS PubMed
- B. Liedberg, C. Nylander and I. Lunström, Sens. Actuators, 1983, 4, 299 CrossRef CAS
- A. Vázquez-Guardado, F. Mehta, B. Jimenez, A. Biswas, K. Ray, A. Baksh, S. Lee, N. Saraf, S. Seal and D. Chanda, Nano Lett., 2021, 21, 7505–7511 CrossRef PubMed
- Y. Kabe, M. Suematsu, S. Sakamoto, M. Hirai, I. Koike, T. Hishiki, A. Matsuda, Y. Hasegawa, K. Tsujita, M. Ono, N. Minegishi, A. Hozowa, Y. Murakami, M. Kubo, M. Itonaga and H. Handa, Clin. Chem., 2018, 64, 1463–1473 CrossRef CAS PubMed
- E. H. Koritzinsky, J. M. Street, R. A. Star and P. S. T. Yuen, J. Cell. Physiol., 2017, 232, 1587–1590 CrossRef CAS PubMed
- J. Zhang, T. Xiao, Y. Cai, C. L. Lavine, H. Peng, H. Zhu, K. Anand, P. Tong, A. Gautam, M. L. Mayer, R. M. Walsh Jr., S. Rits-Volloch, D. R. Wesemann, W. Yang, M. S. Seamn, J. Lu and B. Chen, Science, 2021, 374, 1353–1360 CrossRef CAS PubMed
- E. Crowley, F. Di Nicolantonio, F. Loupakis and A. Bardelli, Nat. Rev. Clin. Oncol., 2013, 10, 472–484 CrossRef CAS PubMed
- K. A. Walker, J. Chen, J. Zhang, M. Fornage, Y. Yang, L. Zhou, M. E. Grams, A. Tin, N. Daya, R. C. Hoogeveen, A. Wu, K. J. Sullivan, P. Ganz, S. L. Zeger, E. F. Gudmundsson, V. Emilsson, L. J. Launer, L. L. Jennings, V. Gudnason, N. Chatterjee, R. F. Gottesman, T. H. Mosley, E. Boerwinkle, C. M. Ballantyne and J. Coresh, Nat. Aging, 2021, 1, 473–489 CrossRef PubMed
- S. B. Nimse, M. D. Sonawane, K. S. Song and T. Kim, Analyst, 2016, 141, 740–755 RSC
- A. Ashkin, J. M. Dziedzic, J. E. Bjorkholm and S. Chu, Opt. Lett., 1986, 11, 288 CrossRef CAS PubMed
- A. Ashkin, J. M. Dziedzic and T. Yamane, Nature, 1987, 330, 769–771 CrossRef CAS PubMed
- A. Ashkin, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4853–4860 CrossRef CAS PubMed
- T. Iida and H. Ishihara, Phys. Rev. Lett., 2003, 90, 057403 CrossRef PubMed
- T. Iida, J. Phys. Chem. Lett., 2012, 3, 332–336 CrossRef CAS PubMed
- S. Fujii, K. Kanaizuka, S. Toyabe, K. Kobayashi, E. Muneyuki and M. Haga, Langmuir, 2011, 27, 8605–8610 CrossRef CAS PubMed
- Y. Yamamoto, E. Shimizu, Y. Nishimura, T. Iida and S. Tokonami, Opt. Mater. Express, 2016, 6, 1280–1285 CrossRef CAS
- T. Iida, Y. Nishimura, M. Tamura, K. Nishidia, S. Ito and S. Tokonami, Sci. Rep., 2016, 6, 37768 CrossRef CAS PubMed
- Y. Nishimura, K. Nishida, Y. Yamamoto, S. Ito, S. Tokonami and T. Iida, J. Phys. Chem. C, 2014, 118, 18799–18804 CrossRef CAS
- M. Ueda, Y. Nishimura, M. Tamura, S. Ito, S. Tokonami and T. Iida, APL Photonics, 2019, 4, 010802 CrossRef
- T. Iida, S. Hamatani, Y. Takagi, K. Fujiwara, M. Tamura and S. Tokonami, Commun. Biol., 2022, 5, 1053 CrossRef CAS PubMed
- I. Nakase, N. Ueno, M. Matsuzawa, K. Noguchi, M. Hirano, M. Omura, T. Takenaka, A. Sugiyama, N. B. Kobayashi, T. Hashimoto, T. Takatani-Nakase, E. Yuba, I. Fujii, S. Futaki and T. Yoshida, FEBS Open Bio, 2021, 11, 753–767 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 and Movies S1–S6. See DOI: https://doi.org/10.1039/d2nh00576j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |