
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe ACE2 receptor accelerates but is not biochemically required for SARS-CoV-2 membrane fusion†
Marcos
Cervantes‡
a,
Tobin
Hess‡
a,
Giorgio G.
Morbioli§‡
 a,
Anjali
Sengar
a and
Peter M.
Kasson
*ab
a,
Anjali
Sengar
a and
Peter M.
Kasson
*ab
aDepartments of Molecular Physiology and Biomedical Engineering, University of Virginia, Charlottesville, VA 22908, USA. E-mail: kassonlab@gmail.com
bScience for Life Laboratory and Department of Molecular and Cellular Biology, Uppsala University, Uppsala, SE 75123, USA
First published on 5th June 2023
Abstract
The SARS-CoV-2 coronavirus infects human cells via the ACE2 receptor. Structural evidence suggests that ACE2 may not just serve as an attachment factor but also conformationally activate the SARS-CoV-2 spike protein for membrane fusion. Here, we test that hypothesis directly, using DNA-lipid tethering as a synthetic attachment factor in place of ACE2. We find that SARS-CoV-2 pseudovirus and virus-like particles are capable of membrane fusion without ACE2 if activated with an appropriate protease. Thus, ACE2 is not biochemically required for SARS-CoV-2 membrane fusion. However, addition of soluble ACE2 speeds up the fusion reaction. On a per-spike level, ACE2 appears to promote activation for fusion and then subsequent inactivation if an appropriate protease is not present. Kinetic analysis suggests at least two rate-limiting steps for SARS-CoV-2 membrane fusion, one of which is ACE2 dependent and one of which is not. Since ACE2 serves as a high-affinity attachment factor on human cells, the possibility to replace it with other factors implies a flatter fitness landscape for host adaptation by SARS-CoV-2 and future related coronaviruses.
Introduction
SARS-CoV-2 has caused a global pandemic since its emergence in humans, with over 600 million confirmed infections and 6.5 million deaths as of October 2022.1 Viral entry and infection are mediated by the SARS-CoV-2 spike protein, which binds to ACE2 receptors on the cell surface,2,3 is activated via proteolytic cleavage,4 and then drives membrane fusion between the viral envelope and a cellular membrane. Since the spike protein performs multiple roles, dissecting the functional requirements of each can be challenging. Separating out these requirements, however, will yield an understanding not only of how SARS-CoV-2 currently infects cells but a better ability to predict future evolution of this and similar viruses.The SARS-CoV-2 spike protein binds to ACE2 via its receptor-binding domain (RBD). Structural studies have identified “down” and “up” conformations of the RBD in the spike trimer, with “up” conformations capable of binding ACE2. Single-molecule FRET experiments have also shown that the RBD occupies a conformational equilibrium that is modulated by ACE2. Further computational studies have postulated a broader range of “down” and “up” conformations,5,6 and additional structures of SARS-CoV-2 spike in complex with ACE2 have suggested that ACE2 binding can destabilize the trimer.7 This destabilization is believed important to activation for membrane fusion, which requires proteolysis at the S2′ site on the spike protein.7–9 Such proteolysis can be performed by a number of enzymes, most notably TMPRSS2 on the cell surface, but also endosomal cathepsins and extracellular proteases.10–13
These observations naturally yield a model where ACE2 binding primes SARS-CoV-2 spike for proteolysis, release of the fusion peptides, and ultimately membrane fusion and entry. Such receptor-activated fusion has been observed in many strains of HIV, where receptor/co-receptor ligation is critical for conformational activation of the envelope protein.14–17 In contrast, receptor binding by influenza hemagglutinin appears primarily to localize the virus near the plasma membrane, since replacement of physiological receptors with synthetic tethers anchored in the viral membrane can functionally reconstitute fusion with identical kinetics.18 Here, we ask where SARS-CoV-2 falls upon this continuum in the requirement of receptor binding for conformational activation of the spike protein.
To accomplish this, we use DNA-lipid tethers that can irreversibly insert into membranes and permit programmable self-assembly.19 Complementary strands of DNA conjugated to lipids inserted in viral particles and synthetic liposomes can thus target the particle to the liposome in the absence of physiological receptors. The triggers for fusion can then be chemically reconstituted and tested precisely. Fusion can be detected using single-virus optical microscopy, where fluorescent dyes in the virus or the target membrane report on viral state changes occurring through the fusion process.20,21 This approach of DNA-lipid tethering and single-virus fusion has previously been used to characterize entry by multiple different viral families, including orthomyxoviruses18 and flaviviruses.22
In the orientation used, where the lipids are at the opposite ends of the DNA double helix after hybridization, fusion is not observed prior to triggering; this has been extensively controlled using both synthetic liposomes and influenza virus,18,23 and is also observed here with SARS-CoV-2. Separating receptor binding from membrane fusion permits examination of the biochemical requirements for viral entry, which may be distinct from the most common pathways a given virus takes for infection. There has been extensive work on the cellular pathways for SARS-CoV-2 entry.12,24–28 As the evolution of SARS-CoV-2 has shown, however, the most common entry pathways of a virus can change even over the space of a few years – the Wuhan strain primarily entered via TMPRSS2-mediated fusion, whereas the BA.1 strain primarily entered via cathepsin-mediated fusion.26,27 Understanding the biochemical requirements for entry thus provides critical knowledge to help predict future evolutionary pathways of SARS-CoV-2 and their respective vulnerability to therapeutic countermeasures.
Results
We measured SARS-CoV-2 spike-mediated binding and fusion to both synthetic and cellular membranes, using synthetic liposomes to ensure the absence of endogenous ACE2 receptor and employing Vero cell plasma membrane vesicles as a control. Experiments were performed using three SARS-CoV-2 spikes: Wuhan, D614G/N501Y and B.1.1.529 (omicron) and using two viral particle types: pseudoviruses on an HIV core and virus-like particles created by co-expressing S, E, M, and N proteins. These were chosen to permit testing of SARS-CoV-2 entry requirements under BSL-2 conditions. In all cases, viral particles and target membranes were separately incubated with complementary DNA strands conjugated to DPPE lipids. Target membranes were then immobilized in a microfluidic flow cell, and viral particles were allowed to bind. After washing away unbound particles, soluble proteases were added to trigger fusion. In prior work, trypsin has been shown to activate SARS-CoV-2 S for fusion in the presence of ACE2 receptor, with resulting fusion kinetics indistinguishable from S protein activated by either TMPRSS2 or cathepsin B or L.29 We thus use trypsin as the major protease in these DNA-tethering experiments, also presenting comparisons to TMPRSS2. Individual fusion events were monitored as a function of time since protease addition (Fig. 1).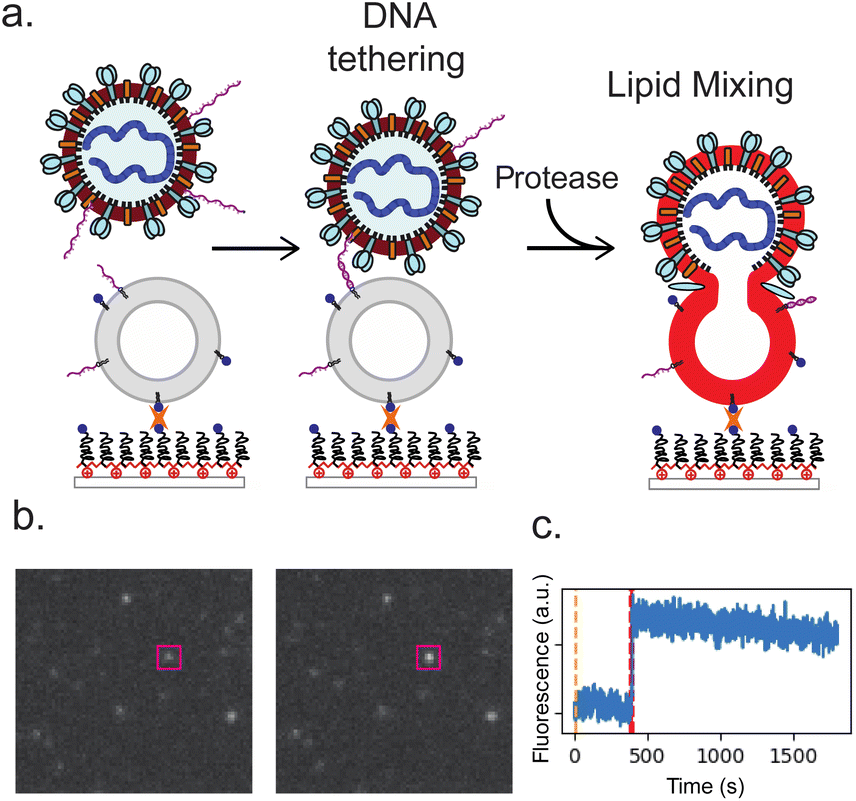 | ||
| Fig. 1 DNA-tethering and fusion of SARS-CoV-2 pseudoviruses and virus-like particles (VLP). The experiment design is schematized in panel (a), where DNA-functionalized viral particles are added to a microfluidic flow cell and allowed to bind to protein-free liposomes functionalized with complementary DNA. After unbound particles are washed away, fusion is initiated by addition of a soluble protease and monitored via lipid mixing, detected as fluorescence dequenching of Texas Red dye in the VLP or pseudoviral envelope. Representative images of a 10.1 × 9.6 μm sub-micrograph before and after lipid mixing are shown in panel (b) with a fusing particle outlined in magenta. The corresponding fluorescence intensity trace is plotted in panel (c). | ||
We demonstrate fusion of viral particles to target membranes in three scenarios, where the physiological receptors and proteases supporting entry are progressively replaced with exogenous factors to probe their biochemical requirements. First, particles can fuse to Calu-3 plasma membrane vesicles where both ACE2 receptor and TMPRSS2 protease are displayed. Second, they can fuse to vero plasma membrane vesicles where ACE2 receptor is present, if exogenous protease has been added to activate the spike protein. Third, and the main subject of this work, they can fuse to protein-free synthetic liposomes if the particles are tethered via DNA-lipids and exogenous protease is added to activate the spike protein.
Fusion events occurred only when viral particles were tethered to target membranes and only in the presence of protease. Fusion was primarily monitored via lipid mixing between the viral particle and the target membrane, detected via dequenching of Texas Red dye in the viral envelope and the resulting fluorescence enhancement. Lipid dye dequenching appears to be a good probe for the role of receptor binding and proteolytic cleavage in viral membrane fusion and entry: in prior work we have demonstrated that virus-like particles activated by exogenous protease can bind ACE2 and productively enter cells in a manner that correlates with lipid mixing, even when endosomal acidification is inhibited.29 DNA-mediated viral binding was specific; when DNA was omitted, the number of adsorbed particles dropped >25-fold (Fig. S1†). Similarly, fusion events were not observed in the absence of protease addition: for three separate experiments, no fusion was observed over 206 viral particles tracked. This is consistent with prior work we performed on DNA-tethered influenza virus where fusion required low pH, the endogenous trigger for influenza fusion.18
Fig. 2 compares viral fusion kinetics for the three viral particle types used: Wuhan spike pseudotyped on an HIV core, omicron (B.1.1.529) spike pseudotyped on an HIV core, and Wuhan D614G/N501Y virus-like particles. In this experiment, viral particles were tethered to synthetic liposomes using DNA-lipids, and fusion was initiated by addition of soluble protease. To facilitate comparison of kinetics, cumulative distribution functions are plotted where 100% represents all fusing viral particles. In general, a left-shift in the single-event fusion statistics represents an increase in fusion kinetics, while the shape of the curve contains information on the underlying kinetic mechanism, as we will discuss later. Fusion efficiencies are compared in Table S1.† Both pseudoviruses tested yielded identical fusion rates, while the VLPs tested yielded rates that were slightly yet significantly faster (p ≤ 0.001, Kolmogorov–Smirnov test with Bonferroni correction for VLPs against all other samples shown in Fig. 2 and 3 except omicron with 200 μg per mL trypsin; that sample yielded p = 0.04). This difference in fusion rate could result from differences in spike protein density on the virus-like particles versus the HIV core, although such differences were not detected via immunofluorescence (Fig. S2†), but it could also be due to the presence of E and M in the virus-like particles, the percentage of active spike on the VLP surface, or the D614G/N501Y mutations. In particular, the D614G mutation has been previously characterized as favoring the “up” conformation of the spike receptor-binding domain.30–32
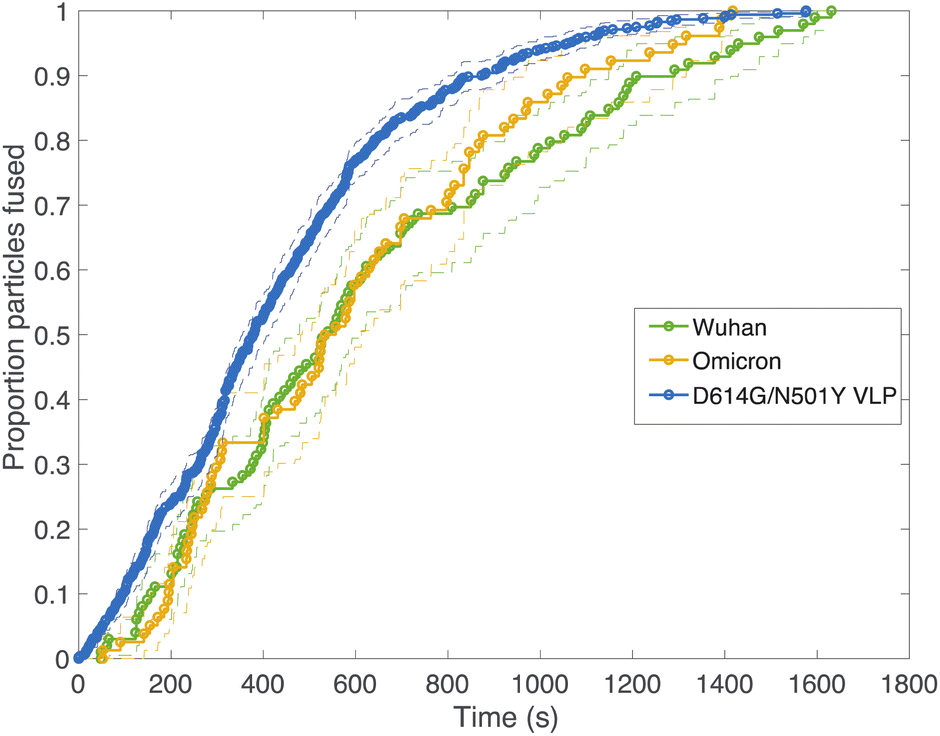 | ||
| Fig. 2 Cumulative distribution functions for fusion by different DNA-tethered SARS-CoV-2 spike particles. Lipid mixing is used as a surrogate for viral membrane fusion, and fusion kinetics are compared between HIV-based pseudoviruses displaying Wuhan or omicron spike versus virus-like particles displaying D614G N501Y spike. Bootstrapped 90% confidence intervals are plotted in dashed lines. The D614G/N501Y virus-like particles fused significantly faster than any of the Wuhan or omicron samples except Wuhan at 1000 μg per mL trypsin (p-values via 2-sample Kolmogorov–Smirnov test of 0.001 for omicron at 200 μg mL−1, 0.04 for Wuhan at 200 μg mL−1, 7 × 10−4 for omicron at 500 μg mL−1, 7 × 10−4 for Wuhan at 500 μg mL−1, 0.002 for omicron at 1000 μg mL−1 and 0.11 for Wuhan at 1000 μg per mL trypsin). | ||
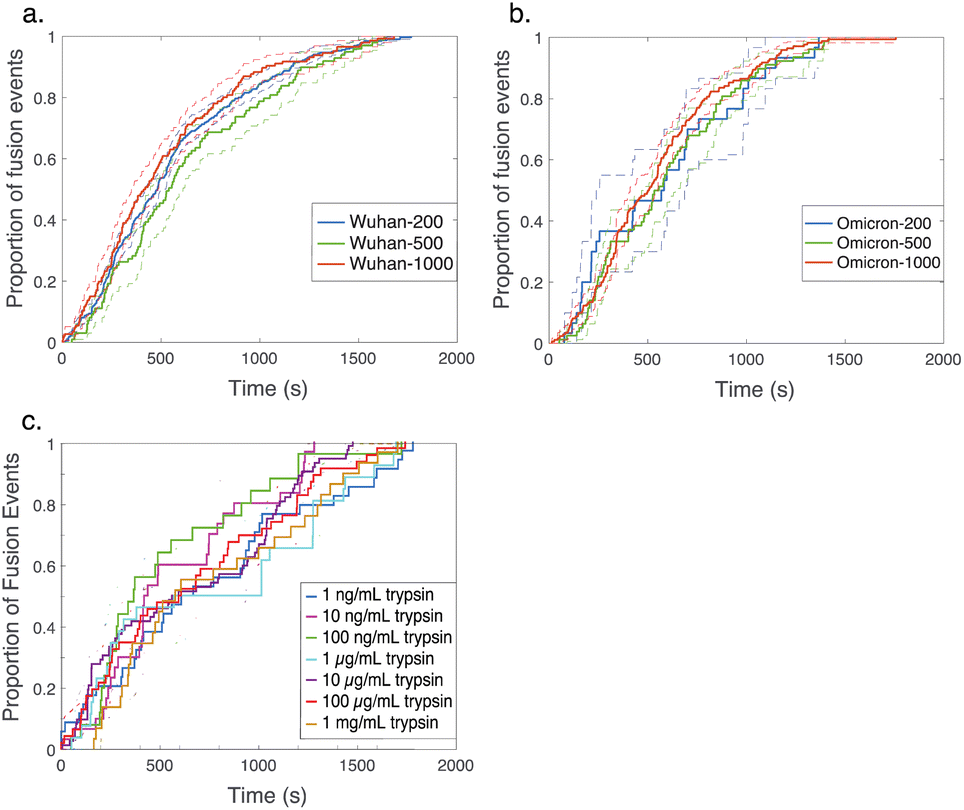 | ||
| Fig. 3 Cumulative distribution functions for fusion by DNA-tethered SARS-CoV-2 spike particles at different trypsin concentrations. Wuhan-pseudotyped particles are plotted in (a) at 200, 500, and 1000 μg per mL trypsin, and omicron-pseudotyped particles are plotted in (b) over the same concentration range. Bootstrapped 90% confidence intervals are plotted in dashed lines. Wuhan and omicron-pseudotyped particles fused at statistically indistinguishable rates at each trypsin concentration tested (p-values of 0.49 at 200 μg mL−1, 0.84 at 500 μg mL−1, and 0.09 at 1000 μg mL−1, via 2-sample Kolmogorov–Smirnov test) and similarly across trypsin concentrations for each of omicron and Wuhan pseudovirus samples. Panel (c) shows the trypsin concentration extended over a million-fold range with no significant change in kinetics observed for Wuhan pseudovirus (all pairwise cumulative distribution functions non-significant via 2-sample Kolmogorov–Smirnov tests). Confidence intervals are omitted from this panel for visual clarity but are shown in Fig. S3.† | ||
In order to probe the rate-limiting factors for SARS-CoV-2 fusion, we first varied the protease used for activation. In both the Wuhan and omicron backgrounds, fusion kinetics were insensitive to trypsin concentration over a range from 200 to 1000 μg mL−1 (Fig. 3), and Wuhan was further tested and insensitive to trypsin concentration over a range from 1 ng mL−1 to 1000 μg mL−1 (Fig. 3c). TMPRSS2 did not efficiently activate omicron spikes for fusion, consistent with prior reports,26,27 but addition of 40 μg mL−1 soluble TMPRSS2 to D614G/N501Y virus-like particles yielded kinetics indistinguishable from 200 μg per mL trypsin (Fig. S4†). These two results suggest that proteolytic cleavage is likely not the rate-limiting step for fusion of DNA-tethered viral particles, since changing protease concentration would be expected to alter enzyme–substrate complex formation and changing protease identity alters kcat. TMPRSS2 and trypsin both cleave to form S2′ fragments over the concentration and time ranges tested here. We have previously reported SDS-PAGE results with the Wuhan spike displayed on HIV pseudovirus particles demonstrating these fragments.29 These gels also show that both enzymes can further cleave spike protein to inactivate the virus if incubated for longer times.
Since multiple spectroscopic and structural studies have suggested ACE2 engagement may also alter SARS-CoV-2 S protein conformation,31,33–36 we used soluble ACE2 to test whether the protein can play an activating role separate from its attachment role. When soluble ACE2 was added to the flow cell simultaneous to protease (after viral particle binding), we observed a significant speedup in lipid mixing kinetics with both the Wuhan and the omicron spike proteins (Fig. 4). This suggests that the spike protein conformations required for fusion are accessible in the absence of ACE2 yet are promoted by ACE2 and indeed can be promoted by ACE2 addition in trans. We also compared fusion driven by DNA tethering, trypsin activation, and soluble ACE2 to binding and fusion mediated by physiological ACE2 and TMPRSS2 displayed on the surface of Calu-3 plasma membrane vesicles. In this case, Calu-3 plasma membrane vesicles were used instead of synthetic target liposomes, and physiological receptors were used instead of DNA-lipids for viral attachment. These results (Fig. 4c) show that the time between particle binding and fusion is faster than the time between protease introduction and fusion. Since Calu-3 plasma membrane vesicles present ACE2 and TMPRSS2 on the membrane surface (Fig. S5†) as opposed to in solution, this result suggests that either the difference between two-dimensional and three-dimensional reaction kinetics or possibly co-localization of ACE2 and TMPRSS2 on the plasma membrane surface may play a role in driving faster fusion kinetics.
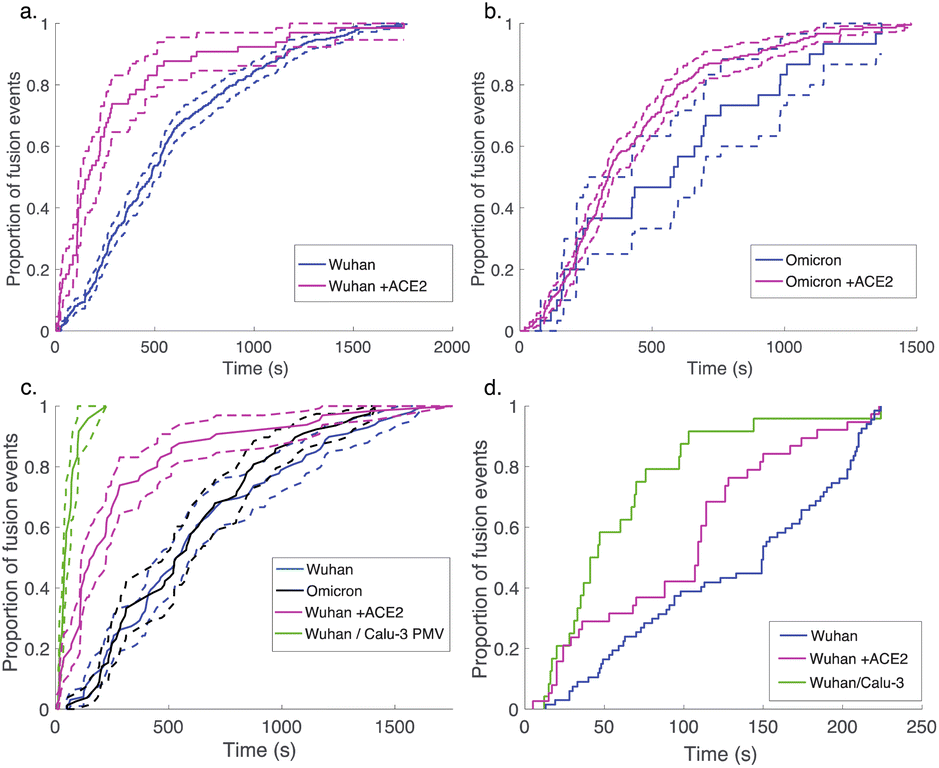 | ||
| Fig. 4 Addition of ACE2 speeds fusion of DNA-tethered pseudovirions. Wuhan-pseudotyped particles are plotted in (a) with and without 40 μg mL−1 soluble human ACE2, and omicron-pseudotyped particles are plotted in (b). Rates of lipid mixing by DNA-tethered Wuhan pseudovirions with and without ACE2 are compared to rates of lipid mixing by Wuhan pseudovirions to Calu-3 plasma membrane vesicles (PMV) containing both ACE2 and TMPRSS2 in (c). Bootstrapped 90% confidence intervals are plotted in dashed lines. Because the longest waiting time measured for the plasma membrane vesicles was 224 s, cumulative distribution functions for the subset of DNA-tethered Wuhan pseudoviruses fusing in ≤224 s were replotted in (d) to account for any potential sampling bias. When compared by either method, fusion to PMV was significantly faster (p < 0.001, 2-sample Kolmogorov–Smirnov test). | ||
We also tested adding ACE2 to the flow cell after DNA-binding but 15 minutes prior to protease addition. In this case, we observed 8-fold fewer fusion events (Fig. S5†), suggesting that ACE2 drives SARS-CoV-2 spike protein towards a conformational state that is fusion-enhanced but may also lead to inactivation if proteolytic cleavage does not occur. This is likely analogous to the well-known effect of pH on influenza hemagglutinin, where exposure to low pH activates hemagglutinin for fusion but also leads to inactivation over time if a target membrane is not present.37
To better understand the process of ACE2-mediated activation and inactivation, we performed a timecourse experiment, where ACE2 was added to DNA-tethered viral particles several minutes before protease. The results show that on a ∼5 minute scale the fusion kinetics once again resemble tethered particles without ACE2 (Fig. 5). This suggests that the rate for ACE2-mediated inactivation of spike (kinact, Fig. 5a) is fast relative to ACE2 dissociation from inactive spike (koff,2) and/or binding of spike by ACE2 (kon). Dissociation from active spike (koff,1) is likely not rate-limiting because addition of a high concentration of ACE2 also results in kinetics resembling the absence of ACE2. Prior surface plasmon resonance experiments on ACE2:full-length spike interactions yield koff estimates between 4 × 10−3 s−1 and 4 × 10−2 s−1.35,38 These prior measurements give a general guide but are not precisely comparable, since they were performed using full-length spike but not viral particles and report a composite of dissociation from active spike (koff,1) and from inactive spike (koff,2).
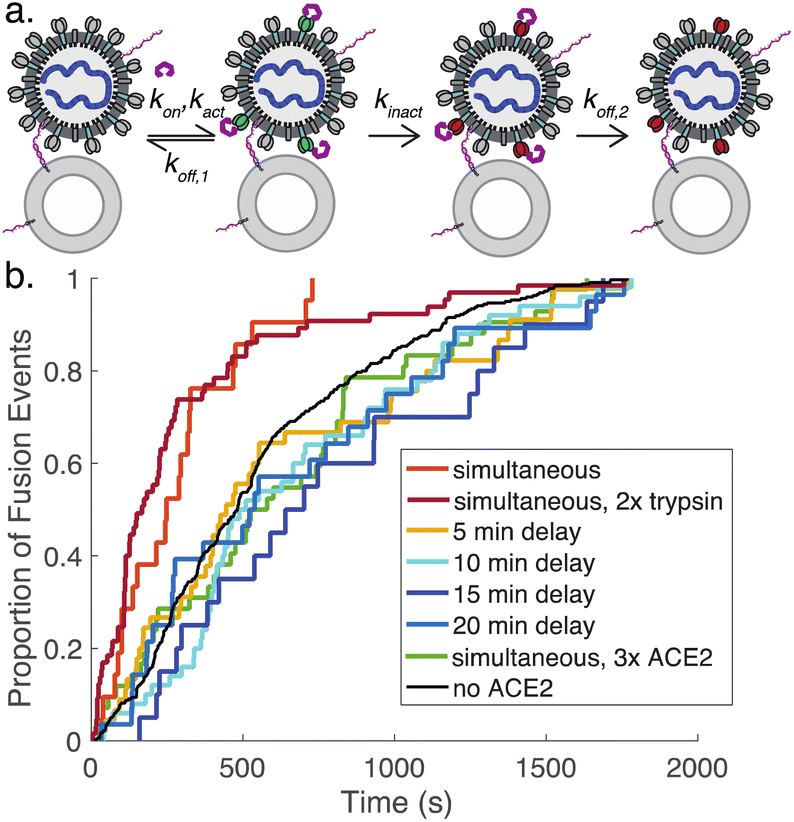 | ||
| Fig. 5 Prolonged exposure to ACE2 likely inactivates spike proteins. DNA-tethered pseudovirions were exposed to ACE2 either simultaneous to protease addition or a designated interval prior. Simultaneous exposure speeds membrane fusion, but prior exposure yields fusion kinetics indistinguishable from DNA-tethered pseudovirions exposed to protease without ACE2. This leads to a schema (panel a) where ACE2 binds to, activates, and ultimately inactivates individual spike proteins. Based on the data available, inactivation is fast relative to ACE2 dissociation and/or association. Panel (b) plots cumulative distribution curves for fusion when 40 μg mL−1 ACE2 was added either simultaneous to or the designated interval before addition of 100 μg per mL trypsin. For comparison, fusion kinetics are also plotted for 200 μg per mL trypsin and 100 μg per mL trypsin with 120 μg mL−1 ACE2. The only distributions that were significantly different were simultaneous addition of 40 μg mL−1 ACE2 with either 100 μg mL−1 or 200 μg per mL trypsin (p < 0.005 via Kolmogorov–Smirnov test). | ||
Comparative analysis of the effect of ACE2 on fusion mediated by the Wuhan versus the omicron spike proteins shows a markedly greater enhancement for Wuhan versus omicron. To further examine the source of this enhancement, gamma function fits were calculated for fusion by Wuhan and omicron pseudoviruses in the absence and presence of ACE2, fitting time-courses both independently and using a set of global parameters. The fraction of particles undergoing lipid mixing f is given by
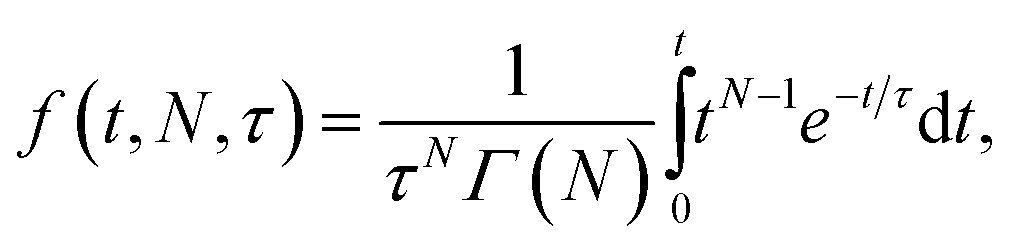
 . This functional form describes the idealized kinetics if fusion results from N events each with waiting time τ,20,29 and the best global fits to the observed data were obtained for constant τ and variable N (Fig. 6). This distribution is the simplest analytical function used to model fusion resulting from N independent processes with the same waiting time or N sequential processes with the same waiting time;20 more complex models exist to model more heterogeneous pathways. The addition of ACE2 decreased N from 2.4 to 1.8 for omicron and from 2.5 to 1.0 for Wuhan, so the number of kinetically evident, rate-limiting steps in membrane fusion was reduced by the presence of ACE2. Based on this, we conclude the most likely explanation for ACE2's action is that ACE2 promotes a conformational change in the SARS-CoV-2 spike protein required for fusion. This change is one of ≥2 rate-limiting steps for fusion. The Wuhan spike is activated by soluble ACE2 such that the ACE2-related conformational change is no longer rate-limiting. These results support the idea that the conformational change promoted by ACE2 is energetically accessible in the absence of ACE2 receptor but occurs more slowly if receptor is not present. For the omicron spike, however, ACE2 somewhat reduces the activation free energy, but this conformational change still contributes to the rate-limiting step. This model is consistent with other data suggesting that the omicron spike is less conformationally labile than the Wuhan spike.38 These results also suggest that the rate-limiting steps for SARS-CoV-2 fusion involve a stepwise process rather than a concerted mechanism.
. This functional form describes the idealized kinetics if fusion results from N events each with waiting time τ,20,29 and the best global fits to the observed data were obtained for constant τ and variable N (Fig. 6). This distribution is the simplest analytical function used to model fusion resulting from N independent processes with the same waiting time or N sequential processes with the same waiting time;20 more complex models exist to model more heterogeneous pathways. The addition of ACE2 decreased N from 2.4 to 1.8 for omicron and from 2.5 to 1.0 for Wuhan, so the number of kinetically evident, rate-limiting steps in membrane fusion was reduced by the presence of ACE2. Based on this, we conclude the most likely explanation for ACE2's action is that ACE2 promotes a conformational change in the SARS-CoV-2 spike protein required for fusion. This change is one of ≥2 rate-limiting steps for fusion. The Wuhan spike is activated by soluble ACE2 such that the ACE2-related conformational change is no longer rate-limiting. These results support the idea that the conformational change promoted by ACE2 is energetically accessible in the absence of ACE2 receptor but occurs more slowly if receptor is not present. For the omicron spike, however, ACE2 somewhat reduces the activation free energy, but this conformational change still contributes to the rate-limiting step. This model is consistent with other data suggesting that the omicron spike is less conformationally labile than the Wuhan spike.38 These results also suggest that the rate-limiting steps for SARS-CoV-2 fusion involve a stepwise process rather than a concerted mechanism.
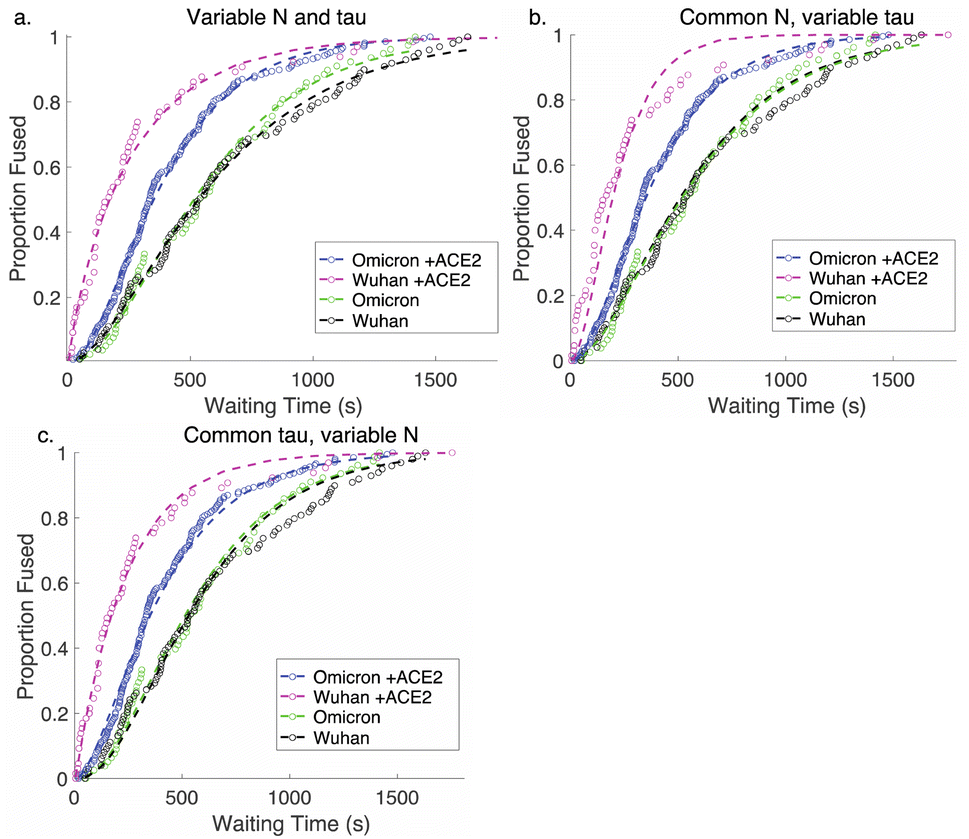 | ||
| Fig. 6 Gamma-function fits to fusion kinetics. Gamma-function fits were calculated for lipid-mixing CDFS of omicron and Wuhan pseudoviruses with and without ACE2. These fits were calculated either (a) varying N and tau independently, (b) fixing N globally across the data sets and varying tau, or (c) fixing tau and varying N. Fit parameters are listed in Table S2.† | ||
Discussion
Using DNA-lipid tethers, we have shown that the ACE2 receptor is not biochemically required for SARS-CoV-2 entry: if another factor can successfully bind the virus to a target membrane, protease activation can successfully trigger membrane fusion by the SARS-CoV-2 spike protein. However, when soluble ACE2 is added in trans, it substantially speeds fusion. Together, these results yield a model where the protease-activatable conformations of SARS-CoV-2 are conformationally accessible in the absence of ACE2 receptor but are substantially promoted by the presence of ACE2. This is the functional equivalent of prior single-molecule FRET studies showing that RBD motions of the spike protein are accessible in the absence of ACE2 but promoted by its presence.39 Our data do not prove that the RBD motions correspond to the fusion-activatable conformations, but it is reasonable to hypothesize that they at least lead to such.Kinetic analysis of both Wuhan and omicron variant spike proteins in the absence and presence of ACE2 suggest that ACE2 activation drives one of the rate-limiting steps for fusion; in the presence of ACE2, Wuhan displays only one kinetically evident rate-limiting step, while omicron is less potently activated by ACE2 and retains >1 rate-limiting step. This is consistent with the more “closed” conformational equilibria of omicron and decreased ACE2 responsiveness previously reported.38 It is satisfying that in analogy to many other viral spike proteins, ACE2 activation in the absence of the factors required for full fusion can lead to spike inactivation. This is also consistent with structural data on S1 dissociation by liganded SARS-CoV-2 spike protein.7
These results complement recent single-molecule FRET studies showing that the SARS-CoV-2 spike protein exists on the surface of lentiviral particles in a dynamic equilibrium between “closed” and “open” states, and that soluble ACE2 promotes open conformations.39,40 The precise relationship between the spike conformational equilibria and the pathway towards fusion remains incompletely determined, but the spectroscopic, structural, and now fusion kinetics data suggest that ACE2 promotes conformational changes in the spike protein that are on-pathway for fusion. The single-molecule FRET data imply, and our data demonstrate, that the fusion-active conformations are energetically accessible in the absence of ACE2, but that the addition of ACE2 removes what is otherwise a rate-limiting free energy barrier on the fusion pathway.
The ability of SARS-CoV-2 spike to mediate VLP and HIV pseudovirus fusion to both simple synthetic liposomes and plasma membrane vesicles also provides an interesting counterpoint to observations of VSV pseudovirus trafficking in cells, where endocytosis appeared required for entry as well as mildly acidic pH.28 It has long been observed that cell–cell fusion pathways mediated by SARS-CoV-2 spike as well as other viral fusion proteins can differ biochemically and mechanistically from virus-cell fusion.41,42 Here, we show in a clean biochemical assay that relatively simple liposomes can support VLP fusion at neutral pH in the presence of an appropriate protease. The most likely entry pathway in cells may impose additional requirements, whether due to membrane residence time, increased efficiency of fusion in endocytic compartments, or other factors. This is why both observationally defining entry pathways in cells and biochemically defining requirements for fusion are critically important for understanding SARS-CoV-2 entry.
Experimental
Full experimental details are given in the ESI.† Briefly, HIV pseudoviruses were produced using previously published protocols43 and plasmids that were gifts of Jesse Bloom. Omicron spike pseudotyping was performed using the plasmid pTwist-SARS-CoV-2 Δ18 B.1.1.529, a gift from Alejandro Balazs.44 Virus-like particles were produced using plasmids encoding the M, N, E, and S proteins from SARS-CoV-2 in addition to a luciferase RNA carrying the SARS-CoV-2 PS9 sequence using previously published protocols.45 Plasmids were a gift from Jennifer Doudna. Viral particles were labeled with Texas Red-DHPE at a quenching concentration. Display of spike protein was verified via single-particle immunofluorescence (Fig. S2†). All handling of pseudoviral and virus-like particles was performed under BSL-2 conditions using institutionally approved protocols.Target liposomes were composed of 68.75 mol% POPC, 20% DOPE, 10% cholesterol, 1% biotin-DPPE, 0.25% Oregon Green-DHPE and extruded at 100 nm. Plasma membrane vesicles used for comparison were produced as previously reported,29 and display of TMPRSS2 and ACE2 molecules was verified via single-particle immunostaining (Fig. S4†). DNA functionalization of both viral particles and target liposomes was performed by adding DNA-lipids to either particles or liposomes at a concentration of 0.03 mol% lipid for liposomes and 0.2 μM for viral particles. The DNA sequences consist of a 24-mer for liposomes and a complementary 24-mer with a 24-mer poly-T spacer for viral particles; full sequences are given in the ESI.† These particular sequences have been extensively validated for tethering influenza viral particles in the past.18 Soluble ACE2 consisted of dimeric human ACE2, residues 1–740.
Biotinylated liposomes were bound to a PEGylated glass coverslip inside a microfluidic flow cell using PLL-PEG-biotin and neutravidin as previously described.46 DNA-functionalized viral particles were allowed to bind for 1–1.5 hours in the dark at room temperature, and then unbound virus was washed away, the flow cell chamber was brought to 37 °C, and the fusion reaction was initiated by addition of soluble protease at pH 7.4. Video micrographs were acquired via epifluorescence microscopy using a 100×, 1.49 NA oil immersion objective and an Andor Zyla 4.2 sCMOS camera. Micrographs were recorded at 1 s intervals using a 150 ms exposure time. Fluorescence time series data were analyzed using previously reported protocols18,29 with Matlab code available from https://github.com/kassonlab/micrograph-spot-analysis. The number of fusion events compiled into each cumulative distribution function (CDF) is given in Table S1.†
Conclusions
The SARS-CoV-2 virus can utilize a variety of pathways for cell entry, depending both on cell type and viral variant. It is thus critical to determine the biochemical requirements for entry in order to ascertain both the mechanisms of viral fusion and the potential for future evolutionary plasticity. Here, we do so using DNA tethers as attachment factors in place of ACE2. When so attached, both pseudoviruses and virus-like particles can fuse to synthetic liposomes, triggered by addition of a protease at neutral pH. ACE2 is thus a high-affinity attachment factor but does not play a required role in conformational activation of the spike protein. By adding soluble ACE2 in trans, however, we demonstrate that ACE2 does promote fusion, speeding a required step in conformational activation so that it ceases to become rate-limiting in the Wuhan variant. Thus, a mechanistic picture of SARS-CoV-2 activation emerges where ACE2 lies intermediate between influenza receptors which do not conformationally activate the viral glycoprotein and HIV receptors/co-receptors, which are typically required. This intermediate role permits SARS-CoV-2 to efficiently leverage ACE2 but likely imparts a greater degree of evolutionary plasticity, since it has the potential to infect host cells using secondary attachment factors.Data availability
Analyzed data are deposited on Zenodo at doi: 10.5281/zenodo.7853006.Author contributions
M. C. and T. H. performed experiments, analyzed data, contributed to the initial draft, and contributed to editing. G. G. M. conceptualized the study, performed experiments, analyzed data, and contributed to editing. A. S. performed experiments, analyzed data, and contributed to editing. P. M. K. conceptualized the study, analyzed data, contributed to the initial draft, and contributed to editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jesse Bloom, Jennifer Doudna, and Alejandro Balazs for the kind gift of plasmids. CR3022 antibody for viral immunostaining (NR-52392) was obtained through BEI Resources, NIAID. We also thank W. Mothes and R. Rawle for helpful discussions. This work was supported by grants from the Commonwealth Health Research Board (207-01-18), UVA Global Infectious Diseases Institute, and Knut and Alice Wallenberg Foundation (KAW2020.0209) to P. M. K.Notes and references
- W. H. Organization, WHO Coronavirus dashboard, https://covid19.who.int/, accessed October 2022 Search PubMed.
- P. Zhou, X. L. Yang, X. G. Wang, B. Hu, L. Zhang, W. Zhang, H. R. Si, Y. Zhu, B. Li, C. L. Huang, H. D. Chen, J. Chen, Y. Luo, H. Guo, R. D. Jiang, M. Q. Liu, Y. Chen, X. R. Shen, X. Wang, X. S. Zheng, K. Zhao, Q. J. Chen, F. Deng, L. L. Liu, B. Yan, F. X. Zhan, Y. Y. Wang, G. F. Xiao and Z. L. Shi, Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- M. Letko, A. Marzi and V. Munster, Nat. Microbiol., 2020, 5, 562–569 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Kruger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Muller, C. Drosten and S. Pohlmann, Cell, 2020, 181, 271–280 CrossRef CAS PubMed.
- T. Sztain, S.-H. Ahn, A. T. Bogetti, L. Casalino, J. A. Goldsmith, R. S. McCool, F. L. Kearns, J. A. McCammon, J. S. McLellan, L. T. Chong and R. E. Amaro, Nat. Chem., 2021, 1–6 Search PubMed.
- M. I. Zimmerman, J. R. Porter, M. D. Ward, S. Singh, N. Vithani, A. Meller, U. L. Mallimadugula, C. E. Kuhn, J. H. Borowsky, R. P. Wiewiora, M. F. D. Hurley, A. M. Harbison, C. A. Fogarty, J. E. Coffland, E. Fadda, V. A. Voelz, J. D. Chodera and G. R. Bowman, Nat. Chem., 2021, 13, 651–659 CrossRef CAS PubMed.
- D. J. Benton, A. G. Wrobel, P. Xu, C. Roustan, S. R. Martin, P. B. Rosenthal, J. J. Skehel and S. J. Gamblin, Nature, 2020, 588, 327–330 CrossRef CAS PubMed.
- R. N. Kirchdoerfer, N. Wang, J. Pallesen, D. Wrapp, H. L. Turner, C. A. Cottrell, K. S. Corbett, B. S. Graham, J. S. McLellan and A. B. Ward, Sci. Rep., 2018, 8, 15701 CrossRef PubMed.
- J. A. Jaimes, J. K. Millet and G. R. Whittaker, iScience, 2020, 101212 CrossRef CAS PubMed.
- D. Bestle, M. R. Heindl, H. Limburg, O. Pilgram, H. Moulton, D. A. Stein, K. Hardes, M. Eickmann, O. Dolnik and C. Rohde, Life Sci. Alliance, 2020, 3, e202000786 CrossRef PubMed.
- J. Koch, Z. M. Uckeley, P. Doldan, M. Stanifer, S. Boulant and P.-Y. Lozach, bioRxiv, 2020, preprint, bioRxiv:2020.2012.2022.423906, DOI:10.1101/2020.12.22.423906.
- J. Koch, Z. M. Uckeley, P. Doldan, M. Stanifer, S. Boulant and P.-Y. Lozach, EMBO J., 2021, 40, e107821 CrossRef CAS PubMed.
- G. Papa, D. L. Mallery, A. Albecka, L. G. Welch, J. Cattin-Ortolá, J. Luptak, D. Paul, H. T. McMahon, I. G. Goodfellow, A. Carter, S. Munro and L. C. James, PLoS Pathog., 2021, 17, e1009246 CrossRef CAS PubMed.
- C. Sood, M. Marin, C. S. Mason and G. B. Melikyan, PLoS One, 2016, 11, e0148944 CrossRef PubMed.
- X. Ma, M. Lu, J. Gorman, D. S. Terry, X. Hong, Z. Zhou, H. Zhao, R. B. Altman, J. Arthos, S. C. Blanchard, P. D. Kwong, J. B. Munro and W. Mothes, Elife, 2018, 7, e34271 CrossRef PubMed.
- M. M. Shaik, H. Peng, J. Lu, S. Rits-Volloch, C. Xu, M. Liao and B. Chen, Nature, 2019, 565, 318–323 CrossRef CAS PubMed.
- H. Wang, C. O. Barnes, Z. Yang, M. C. Nussenzweig and P. J. Bjorkman, Cell Host Microbe, 2018, 24, 579–592 CrossRef CAS PubMed.
- R. J. Rawle, S. G. Boxer and P. M. Kasson, Biophys. J., 2016, 111, 123–131 CrossRef CAS PubMed.
- C. Yoshina-Ishii, G. P. Miller, M. L. Kraft, E. T. Kool and S. G. Boxer, J. Am. Chem. Soc., 2005, 127, 1356–1357 CrossRef CAS PubMed.
- D. Floyd, J. R. Ragains, J. J. Skehel, S. C. Harrison and A. M. van Oijen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15382–15387 CrossRef CAS PubMed.
- L. Wessels, M. W. Elting, D. Scimeca and K. Weninger, Biophys. J., 2007, 93, 526–538 CrossRef CAS PubMed.
- R. J. Rawle, E. R. Webster, M. Jelen, P. M. Kasson and S. G. Boxer, ACS Cent. Sci., 2018, 4(11), 1503–1510 CrossRef CAS PubMed.
- Y. H. Chan, B. van Lengerich and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979–984 CrossRef CAS PubMed.
- M. Laporte, V. Raeymaekers, R. Van Berwaer, J. Vandeput, I. Marchand-Casas, H.-J. Thibaut, D. Van Looveren, K. Martens, M. Hoffmann, P. Maes, S. Pöhlmann, L. Naesens and A. Stevaert, PLoS Pathog., 2021, 17, e1009500 CrossRef CAS PubMed.
- M. Dittmar, J. S. Lee, K. Whig, E. Segrist, M. Li, B. Kamalia, L. Castellana, K. Ayyanathan, F. L. Cardenas-Diaz, E. E. Morrisey, R. Truitt, W. Yang, K. Jurado, K. Samby, H. Ramage, D. C. Schultz and S. Cherry, Cell Rep., 2021, 35, 108959 CrossRef CAS PubMed.
- B. Meng, A. Abdullahi, I. A. T. M. Ferreira, N. Goonawardane, A. Saito, I. Kimura, D. Yamasoba, P. P. Gerber, S. Fatihi, S. Rathore, S. K. Zepeda, G. Papa, S. A. Kemp, T. Ikeda, M. Toyoda, T. S. Tan, J. Kuramochi, S. Mitsunaga, T. Ueno, K. Shirakawa, A. Takaori-Kondo, T. Brevini, D. L. Mallery, O. J. Charles, S. Baker, G. Dougan, C. Hess, N. Kingston, P. J. Lehner, P. A. Lyons, N. J. Matheson, W. H. Owehand, C. Saunders, C. Summers, J. E. D. Thaventhiran, M. Toshner, M. P. Weekes, P. Maxwell, A. Shaw, A. Bucke, J. Calder, L. Canna, J. Domingo, A. Elmer, S. Fuller, J. Harris, S. Hewitt, J. Kennet, S. Jose, J. Kourampa, A. Meadows, C. O’Brien, J. Price, C. Publico, R. Rastall, C. Ribeiro, J. Rowlands, V. Ruffolo, H. Tordesillas, B. Bullman, B. J. Dunmore, S. Fawke, S. Gräf, J. Hodgson, C. Huang, K. Hunter, E. Jones, E. Legchenko, C. Matara, J. Martin, F. Mescia, C. O’Donnell, L. Pointon, J. Shih, R. Sutcliffe, T. Tilly, C. Treacy, Z. Tong, J. Wood, M. Wylot, A. Betancourt, G. Bower, C. Cossetti, A. De Sa, M. Epping, S. Fawke, N. Gleadall, R. Grenfell, A. Hinch, S. Jackson, I. Jarvis, B. Krishna, F. Nice, O. Omarjee, M. Perera, M. Potts, N. Richoz, V. Romashova, L. Stefanucci, M. Strezlecki, L. Turner, E. M. D. D. De Bie, K. Bunclark, M. Josipovic, M. Mackay, J. Allison, H. Butcher, D. Caputo, D. Clapham-Riley, E. Dewhurst, A. Furlong, B. Graves, J. Gray, T. Ivers, E. Le Gresley, R. Linger, S. Meloy, F. Muldoon, N. Ovington, S. Papadia, I. Phelan, H. Stark, K. E. Stirrups, P. Townsend, N. Walker, J. Webster, I. Scholtes, S. Hein, R. King, E. P. Butlertanaka, Y. L. Tanaka, T. Ikeda, J. Ito, K. Uriu, Y. Kosugi, M. Suganami, A. Oide, M. Yokoyama, M. Chiba, C. Motozono, H. Nasser, R. Shimizu, Y. Yuan, K. Kitazato, H. Hasebe, S. Nakagawa, J. Wu, M. Takahashi, T. Fukuhara, K. Shimizu, K. Tsushima, H. Kubo, Y. Kazuma, R. Nomura, Y. Horisawa, K. Nagata, Y. Kawai, Y. Yanagida, Y. Tashiro, K. Tokunaga, S. Ozono, R. Kawabata, N. Morizako, K. Sadamasu, H. Asakura, M. Nagashima, K. Yoshimura, P. Cárdenas, E. Muñoz, V. Barragan, S. Márquez, B. Prado-Vivar, M. Becerra-Wong, M. Caravajal, G. Trueba, P. Rojas-Silva, M. Grunauer, B. Gutierrez, J. J. Guadalupe, J. C. Fernández-Cadena, D. Andrade-Molina, M. Baldeon, A. Pinos, J. E. Bowen, A. Joshi, A. C. Walls, L. Jackson, D. Martin, K. G. C. Smith, J. Bradley, J. A. G. Briggs, J. Choi, E. Madissoon, K. Meyer, P. Mlcochova, L. Ceron-Gutierrez, R. Doffinger, S. A. Teichmann, A. J. Fisher, M. S. Pizzuto, A. de Marco, D. Corti, M. Hosmillo, J. H. Lee, L. C. James, L. Thukral, D. Veesler, A. Sigal, F. Sampaziotis, I. G. Goodfellow, N. J. Matheson, K. Sato and R. K. Gupta, Nature, 2022, 603(7902), 706–714 CrossRef CAS PubMed.
- B. J. Willett, J. Grove, O. A. MacLean, C. Wilkie, N. Logan, G. D. Lorenzo, W. Furnon, S. Scott, M. Manali, A. Szemiel, S. Ashraf, E. Vink, W. T. Harvey, C. Davis, R. Orton, J. Hughes, P. Holland, V. Silva, D. Pascall, K. Puxty, A. da Silva Filipe, G. Yebra, S. Shaaban, M. T. G. Holden, R. M. Pinto, R. Gunson, K. Templeton, P. R. Murcia, A. H. Patel, The COVID-19 Genomics UK (COG-UK) Consortium, J. Haughney, D. L. Robertson, M. Palmarini, S. Ray and E. C. Thomson, medRxiv, 2022, preprint, medRxiv:2022.2001.2003.21268111, DOI:10.1101/2022.01.03.21268111.
- A. J. B. Kreutzberger, A. Sanyal, A. Saminathan, L.-M. Bloyet, S. Stumpf, Z. Liu, R. Ojha, M. T. Patjas, A. Geneid, G. Scanavachi, C. A. Doyle, E. Somerville, R. B. D. C. Correia, G. Di Caprio, S. Toppila-Salmi, A. Mäkitie, V. Kiessling, O. Vapalahti, S. P. J. Whelan, G. Balistreri and T. Kirchhausen, Proc. Natl. Acad. Sci. U. S. A, 2022, 119, e2209514119 CrossRef CAS PubMed.
- A. Sengar, M. Cervantes, S. T. Bondalapati, T. Hess and P. M. Kasson, J. Virol., 2023, 97(5), e0199222 CrossRef PubMed.
- Y. Cai, J. Zhang, T. Xiao, H. Peng, S. M. Sterling, R. M. Walsh, S. Rawson, S. Rits-Volloch and B. Chen, Science, 2020, 369, 1586 CrossRef CAS PubMed.
- T. Zhou, Y. Tsybovsky, J. Gorman, M. Rapp, G. Cerutti, G.-Y. Chuang, P. S. Katsamba, J. M. Sampson, A. Schön, J. Bimela, J. C. Boyington, A. Nazzari, A. S. Olia, W. Shi, M. Sastry, T. Stephens, J. Stuckey, I. T. Teng, P. Wang, S. Wang, B. Zhang, R. A. Friesner, D. D. Ho, J. R. Mascola, L. Shapiro and P. D. Kwong, Cell Host Microbe, 2020, 28, 867–879 CrossRef CAS PubMed.
- S. M. C. Gobeil, K. Janowska, S. McDowell, K. Mansouri, R. Parks, K. Manne, V. Stalls, M. F. Kopp, R. Henderson, R. J. Edwards, B. F. Haynes and P. Acharya, Cell Rep., 2021, 34, 108630 CrossRef CAS PubMed.
- J. Shang, G. Ye, K. Shi, Y. Wan, C. Luo, H. Aihara, Q. Geng, A. Auerbach and F. Li, Nature, 2020, 58(7807), 221–224 CrossRef PubMed.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Science, 2020, 367, 1444–1448 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292 CrossRef CAS PubMed.
- T. Stegmann, F. P. Booy and J. Wilschut, J. Biol. Chem., 1987, 262, 17744–17749 CrossRef CAS PubMed.
- J. Zhang, Y. Cai, C. L. Lavine, H. Peng, H. Zhu, K. Anand, P. Tong, A. Gautam, M. L. Mayer, S. Rits-Volloch, S. Wang, P. Sliz, D. R. Wesemann, W. Yang, M. S. Seaman, J. Lu, T. Xiao and B. Chen, Cell Rep., 2022, 39, 110729 CrossRef CAS PubMed.
- M. Lu, P. D. Uchil, W. Li, D. Zheng, D. S. Terry, J. Gorman, W. Shi, B. Zhang, T. Zhou, S. Ding, R. Gasser, J. Prévost, G. Beaudoin-Bussières, S. P. Anand, A. Laumaea, J. Grover, L. Liu, D. D. Ho, J. R. Mascola, A. Finzi, P. D. Kwong, J. S. Blanchard and W. Mothes, Cell Host & Microbe, 2020, 28, 880–891 CrossRef CAS PubMed.
- Z. Yang, Y. Han, S. Ding, W. Shi, T. Zhou, A. Finzi, P. D. Kwong, W. Mothes and M. Lu, mBio, 2022, e0322721, DOI:10.1128/mbio.03227-21.
- K. E. Zawada, K. Okamoto and P. M. Kasson, J. Mol. Biol., 2018, 430, 594–601 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber and S. Pöhlmann, Mol. Cell, 2020, 78, 779–784 CrossRef CAS PubMed.
- K. H. D. Crawford, R. Eguia, A. S. Dingens, A. N. Loes, K. D. Malone, C. R. Wolf, H. Y. Chu, M. A. Tortorici, D. Veesler, M. Murphy, D. Pettie, N. P. King, A. B. Balazs and J. D. Bloom, Viruses, 2020, 12(5), 513 CrossRef CAS PubMed.
- W. F. Garcia-Beltran, K. J. St. Denis, A. Hoelzemer, E. C. Lam, A. D. Nitido, M. L. Sheehan, C. Berrios, O. Ofoman, C. C. Chang, B. M. Hauser, J. Feldman, A. L. Roederer, D. J. Gregory, M. C. Poznansky, A. G. Schmidt, A. J. Iafrate, V. Naranbhai and A. B. Balazs, Cell, 2022, 185, 457–466 CrossRef CAS PubMed.
- A. M. Syed, T. Y. Taha, T. Tabata, I. P. Chen, A. Ciling, M. M. Khalid, B. Sreekumar, P.-Y. Chen, J. M. Hayashi, K. M. Soczek, M. Ott and J. Doudna, Science, 2021, 374, 1626–1632 CrossRef CAS PubMed.
- A. M. Villamil Giraldo and P. Kasson, J. Phys. Chem. Lett., 2020, 11, 7190–7196 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06967a |
| ‡ These authors contributed equally. |
| § Present address: Department of Chemistry, Laboratory for Living Devices, Tufts University, Medford, MA 02155, United States. |
| This journal is © The Royal Society of Chemistry 2023 |