DOI:
10.1039/D2SC07101K
(Perspective)
Chem. Sci., 2023,
14, 4205-4218
Redox catalysis via photoinduced electron transfer
Received
29th December 2022
, Accepted 7th March 2023
First published on 8th March 2023
Abstract
This perspective article highlights redox catalysis of organic and inorganic molecules via photoinduced electron transfer, which is well exploited for a number of important photoredox reactions including hydrogen evolution, water oxidation and a number of synthetic applications. Organic and inorganic photoredox catalysis is also combined with thermal transition metal redox catalysis to achieve overall photocatalytic redox reactions, which would otherwise not be possible by using photoredox catalysis or thermal redox catalysis alone. Both thermodynamic and kinetic data are discussed to understand the photoinduced electron-transfer processes of organic and inorganic photoredox catalysts in the light of the Marcus theory of electron transfer, providing a comprehensive and valuable guide for employing organic and inorganic redox catalysts via photoinduced electron transfer. The excited states of electron donors including radicals and anions act as super-reductants in the photoinduced electron-transfer reactions, whereas the excited states of electron acceptors including cations act as super-oxidants in the photoinduced electron-transfer reactions. Photoexcitation of simple electron donor–acceptor linked molecules with small reorganization energies of electron transfer results in formation of long-lived electron-transfer states, which can oxidize and reduce substrates to make various chemical transformations possible with use of transition metal redox catalysis. Finally molecular model systems of photosystems I and II are combined to achieve water splitting to evolve H2 and O2.
 Yong-Min Lee | Yong-Min Lee obtained his PhD in inorganic chemistry from Pusan National University in 1999 under the guidance of Professor Sung-Nak Choi. In 1999, he moved to the Center for Magnetic Resonance (CERM) at the University of Florence (Italy) as Postdoctoral fellow and Researcher under the supervision of Professor Claudio Luchinat and Professor Ivano Bertini. In 2006, he joined the Center for Dioxygen Chemistry at Ewha Womans University. Since 2009, he has been working at Ewha Womans University as a Special Appointment Professor. His research has been focusing on the roles of metal ions in biological systems. |
 Wonwoo Nam | Wonwoo Nam earned his PhD degree in Inorganic Chemistry from the University of California, Los Angeles (UCLA) under the supervision of Professor Joan S. Valentine in 1990. After working for one year as a postdoctoral fellow at UCLA, he was appointed an Assistant Professor at Hong Ik University in 1991. In 1994, he moved to Ewha Womans University, where he is currently a Distinguished Professor. His current research has been focusing on dioxygen activation, water oxidation, and important roles of metal ions in bioinorganic chemistry. |
 Shunichi Fukuzumi | Shunichi Fukuzumi received his bachelor's degree and PhD degree at Tokyo Institute of Technology in 1973 and 1978, respectively. After working as a postdoc at Indiana University in the USA (1978–1981), he became an Assistant Professor at Osaka University. He was promoted to a Full Professor in 1994 and a Distinguished Professor in 2013. His research has been focusing on artificial photosynthesis. He is currently a Professor of Ewha Womans University and a Professor Emeritus of Osaka University. |
1Introduction
Solar energy conversion in photosynthesis has been achieved by photoinduced multi-step charge-separation processes in the photosynthetic reaction centers (PRCs) in Photosystem II (PS-II), where electrons and protons are taken from water.1–4 These electrons and protons are used to generate a proton gradient and reduce NADP+ to NADPH via redox shuttle reactions in Photosystem I (PS-I) through the Z scheme, leading to the reduction of CO2 into carbohydrates.1,4 There have been extensive studies to mimic the charge-separation processes in PRCs using covalently and non-covalently linked organic electron donor–acceptor molecules.5–17 Photoexcitation of donor–acceptor linked molecules (D–A) results in electron transfer from the excited state of electron donor (D*) or acceptor (A*) to an electron acceptor (A) or donor (D) to produce the charge-separated state (D˙+–A˙−), which has both oxidizing ability due to D˙+ and the reducing ability due to A˙−.4 Photoexcitation of metal complexes composed of an electron donor metal (Mn+) and electron acceptor ligand (L) also results in generation of the metal-to-ligand charge-transfer (MLCT) state (M(n+1)+L˙−), which has both oxidizing ability due to M(n+1)+ and the reducing ability due to L˙−.18–20 In the presence of an electron donor substrate (SD) and an electron acceptor substrate (SA), electron transfer from SD to D˙+–A˙−, produced by photoexcitation of D–A [eqn (1)], results in formation of SD˙+ and D–A˙− [eqn (2)], which transfers an electron to SA to produce SA˙−, accompanied by regeneration of D–A [eqn (3)].21–23 A bond formation between SD˙+ and SA˙− may occur [eqn (4)] in competition with back electron transfer from SA˙− to SD˙+ [eqn (5)].21–23 In such a case, the overall reaction is the photocatalytic bond formation reaction between SD and SA with D–A, which acts as an organic photoredox catalyst [eqn (6)].21–23 D–A can be replaced by a metal complex (Mn+L) that affords the MLCT excited state upon photoexcitation.18–20| |  | (1) |
| | | SD + D˙+–A˙− → SD˙+ + D–A˙− | (2) |
| | | SA + D–A˙− → SA˙− + D–A | (3) |
| | 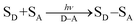 | (6) |
A substrate radical cation (SDH˙+), which is produced by electron transfer from SDH to D˙+–A˙− [eqn (7)], can react with a nucleophile (NH) to produce the adduct radical (SD–NH˙) and H+ [eqn (8)].21 On the other hand, SA˙−, which is produced by electron transfer from D–A˙− to SA [eqn (3)], is protonated to produce SAH˙ [eqn (9)].21 Then, hydrogen atom transfer from SD–NH to SAH˙ yields SD–N and SAH2 [eqn (10)].21 The overall reaction is the photocatalytic bond formation between SDH and NH with D–A, accompanied by the reduction of SA to SAH2 [eqn (11)].21 In this case as well, D–A can be replaced by Mn+L.21
| | | SDH + D˙+–A˙− → SDH˙+ + D–A˙− | (7) |
| | | SDH˙+ + NH → SD–NH˙ + H+ | (8) |
| | | SD–NH˙ + SAH˙ → SD–N + SAH2 | (10) |
| | 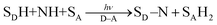 | (11) |
Both the photocatalytic bond formation between SD and SA [eqn (6)] and that between SDH and NH, accompanied by the reduction of SA to SAH2 [eqn (3), (9) and (10)] are started by electron transfer from substrates (SD and SDH) to D˙+–A˙− (or M(n+1)+L˙−) to produce SD˙+ and SDH˙+ [eqn (2) and (7)], respectively.19 Electron transfer from D–A˙− to SA should also occur to regenerate D–A [eqn (3)].21 Although there have been many studies on the photocatalytic reactions with D–A and Mn+L, the thermodynamics and kinetics of electron transfer between substrates and D˙+–A˙− (or M(n+1)+L˙−) have yet to be summarized by a systematic manner. In this review, we focus on the thermodynamics and kinetics of electron transfer between substrates (SD and SDH) and D˙+–A˙− (or M(n+1)+L˙−) [eqn (2), (3) and (7)], providing quantitative basis to predict the photocatalytic bond formation reactions [eqn (4) and (8)].
In the photocatalytic reactions, D–A acts as a photoredox catalyst [eqn (2) and (3)]. In these cases, D and A are not necessarily linked together. Photoinduced electron transfer from D to the excited state of A (A*) or photoinduced electron transfer from the excited state of D (D*) to A occurs to produce D˙+ and A˙−, which enable photocatalytic bond formation reactions [eqn (4) and (8)]. Once reactive species such as D˙+ and A˙− are produced, various redox reactions are made possible to occur via D˙+ and A˙−. This perspective focuses on photoinduced electron transfer reactions of D, A and D–A, which initiate various photoredox catalytic reactions. The rate constants of photoinduced electron transfer and back electron transfer are evaluated in light of the Marcus theory of electron transfer.24–26 Firstly electron transfer from D*, which are super-reductants, to A is discussed with the photoredox catalytic mechanisms. Secondly electron transfer from D to A*, which are super-oxidants, is discussed with the photoredox catalytic mechanisms. Finally photoredox catalytic reactions with PRC models (D–A) are discussed to show the combination of PS-I and PS-II models.
2 Super-reductants
2.1. Singlet excited states of NADH analogs
The singlet excited state of an NADH (dihydronicotinamide adenine dinucleotide) analog, 9,10-dihydro-10-methylacridine 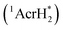 , has a largely negative one-electron reduction potential (−3.1 V vs. SCE) and an appreciable lifetime of 7.0 ns.27 Electron transfer from
, has a largely negative one-electron reduction potential (−3.1 V vs. SCE) and an appreciable lifetime of 7.0 ns.27 Electron transfer from  to various halogenated compounds (RX) occurs to produce dehalogenated compounds (RH) and the two-electron oxidation product, 10-methylacridinium ion (AcrH+), because the one-electron reduction potentials of RX are less negative (e.g., −2.78 V vs. SCE) than that of
to various halogenated compounds (RX) occurs to produce dehalogenated compounds (RH) and the two-electron oxidation product, 10-methylacridinium ion (AcrH+), because the one-electron reduction potentials of RX are less negative (e.g., −2.78 V vs. SCE) than that of  (−3.1 V vs. SCE).27 The rate constant of electron transfer from
(−3.1 V vs. SCE).27 The rate constant of electron transfer from  to PhCl was determined to be 6.0 × 108 M−1 s−1 at 298 K from the fluorescence quenching by PhCl.27 When AcrH2 was replaced by the didueterated compound (AcrD2), the rate constant of electron transfer from
to PhCl was determined to be 6.0 × 108 M−1 s−1 at 298 K from the fluorescence quenching by PhCl.27 When AcrH2 was replaced by the didueterated compound (AcrD2), the rate constant of electron transfer from 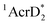 to PhCl was the same as that of
to PhCl was the same as that of  , showing no deuterium kinetic isotope effect.27 Electron transfer reduction of RX results in the C–X bond cleavage to produce R˙ and X−.28 The produced carbon-centered radical (R˙) abstracts a hydrogen atom from AcrH2˙+, which is produced by electron transfer from
, showing no deuterium kinetic isotope effect.27 Electron transfer reduction of RX results in the C–X bond cleavage to produce R˙ and X−.28 The produced carbon-centered radical (R˙) abstracts a hydrogen atom from AcrH2˙+, which is produced by electron transfer from  to RX, to form 10-methylacridinium ion (AcrH+) and RH [eqn (12)].27
to RX, to form 10-methylacridinium ion (AcrH+) and RH [eqn (12)].27| | 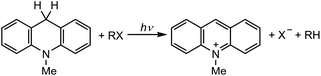 | (12) |
Because AcrH+ can be reduced by NaBH4 to regenerate AcrH2 [eqn (13)], AcrH2 acts as an organic photoredox catalyst for photocatalytic dehalogenation of RX by NaBH4.27
| | | NaBH4 + AcrH+ → AcrH2 + Na+ + BH3 | (13) |
Photoredox catalysis of AcrH2 was applied to trifluoromethylation of hydrazones with an electrophilic CF3-transfer reagent (CF3SO2Cl) as shown in Scheme 1.29 Upon photoexcitation of AcrH2 in the presence of CF3SO2Cl, electron transfer from 
 to CF3SO2Cl (Eredvs. SCE = −0.18 V) occurs rapidly to produce AcrH2˙+, CF3˙ and SO2Cl−, followed by radical addition of CF3˙ to the C
to CF3SO2Cl (Eredvs. SCE = −0.18 V) occurs rapidly to produce AcrH2˙+, CF3˙ and SO2Cl−, followed by radical addition of CF3˙ to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond to produce aminyl radical intermediate B in Scheme 1, which is stabilized by the lone pair of the adjacent nitrogen atom. B is oxidized by AcrH2˙+ to afford nitrenium intermediate C in Scheme 1, accompanied by regeneration of AcrH2. Finally, intermediate C undergoes deprotonation with a base to yield the trifluoromethylated product (Scheme 1).29
N bond to produce aminyl radical intermediate B in Scheme 1, which is stabilized by the lone pair of the adjacent nitrogen atom. B is oxidized by AcrH2˙+ to afford nitrenium intermediate C in Scheme 1, accompanied by regeneration of AcrH2. Finally, intermediate C undergoes deprotonation with a base to yield the trifluoromethylated product (Scheme 1).29
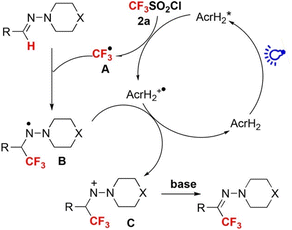 |
| | Scheme 1 Photocatalytic trifluoromethylation of hydrazones with an electrophilic CF3-transfer reagent (CF3SO2Cl) by AcrH2 photoredox catalysis. Reprinted from ref. 29 with permission form John Wiley and Sons (Copyright 2017). | |
The photoredox catalysis of AcrH2 was combined with palladium catalysis for arylation of arenes with aryldiazonium salts (Scheme 2).30 Photoirradiation of a methanol solution containing AcrH2, ArN2BF4, and Pd(OAc)2 with blue LED light for 12 h afforded C–H arylation product (86% yield).30 The reaction was initiated by electron transfer from  to ArN2BF4 to generate AcrH2˙+ and Ar˙, accompanied by N2 dissociation (Scheme 2).30 Ar˙ reacts with complex A in Scheme 2 to afford the palladium(III) complex B, which is then oxidized by AcrH2˙+via electron transfer from complex B to AcrH2˙+ to produce palladium(IV) complex C and regenerate the photocatalyst AcrH2 (Scheme 2).30 Finally, the reductive elimination of palladium(IV) complex C affords the formation of compound D, which is the arylated product, and regenerates the palladium(II) catalyst A.30 Thus, a dual catalytic system of palladium(II)/organic photoredox catalysts enables the arylation of arenes with ArN2+ over a wide substrate range under the mild conditions.30
to ArN2BF4 to generate AcrH2˙+ and Ar˙, accompanied by N2 dissociation (Scheme 2).30 Ar˙ reacts with complex A in Scheme 2 to afford the palladium(III) complex B, which is then oxidized by AcrH2˙+via electron transfer from complex B to AcrH2˙+ to produce palladium(IV) complex C and regenerate the photocatalyst AcrH2 (Scheme 2).30 Finally, the reductive elimination of palladium(IV) complex C affords the formation of compound D, which is the arylated product, and regenerates the palladium(II) catalyst A.30 Thus, a dual catalytic system of palladium(II)/organic photoredox catalysts enables the arylation of arenes with ArN2+ over a wide substrate range under the mild conditions.30
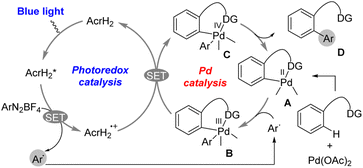 |
| | Scheme 2 Photocatalytic arylation of C–H bond by incorporating a palladium catalyst and AcrH2 photoredox catalyst. Reprinted from ref. 30 with permission form American Chemical Society (Copyright 2017). | |
The singlet excited state of 1-benzyl-1,4-dihydronicotinamide (BNAH), which is one of the NADH analogs, can also be used as a strong one-electron reductant for the photoinduced reduction of substrates, although the one-electron oxidation potential (−2.6 V vs. SCE)31 is less negative than that of  (−3.1 V vs. SCE).27 BNAH or BNA+ is used as a photoredox catalyst for hydrogenation of α,β-epoxy ketones and 1,2-diketones with HCO2H and Et3N to produce β-hydroxy and α-hydroxy ketones,32 because BNA+ can be converted to BNAH by a mixture of HCO2H/Et3N at room temperatures.33 The photocatalytic hydrogenation mechanism by BNAH is shown in Scheme 3. Firstly, electron transfer from 1BNAH* to α,β-epoxy ketone (compound 1 in Scheme 3) or benzil (i.e., diphenylethanedione; compound 3 in Scheme 3) occurs to produce BNAH˙+ and the radical anions of compounds 1 and 3 (species 5 and 6 in Scheme 3, respectively). Then, the carbon–oxygen bond cleavage occurs to convert species 5 (or species 6) into species 7 (or species 8), followed by proton transfer from BNAH˙+ to species 7 (or species 8) and an enol/keto tautomerization to produce species 9 (or species 10) and BNA˙ (Scheme 3). Because BNA˙ is a strong one-electron reductant,34 electron transfer from BNA˙ to species 9 (or species 10) to produce species 11 (or species 12), accompanied by regeneration of BNA+.33 The protonation of species 11 (or species 12) by water yields the final products (compounds 2 and 4 in Scheme 3).33
(−3.1 V vs. SCE).27 BNAH or BNA+ is used as a photoredox catalyst for hydrogenation of α,β-epoxy ketones and 1,2-diketones with HCO2H and Et3N to produce β-hydroxy and α-hydroxy ketones,32 because BNA+ can be converted to BNAH by a mixture of HCO2H/Et3N at room temperatures.33 The photocatalytic hydrogenation mechanism by BNAH is shown in Scheme 3. Firstly, electron transfer from 1BNAH* to α,β-epoxy ketone (compound 1 in Scheme 3) or benzil (i.e., diphenylethanedione; compound 3 in Scheme 3) occurs to produce BNAH˙+ and the radical anions of compounds 1 and 3 (species 5 and 6 in Scheme 3, respectively). Then, the carbon–oxygen bond cleavage occurs to convert species 5 (or species 6) into species 7 (or species 8), followed by proton transfer from BNAH˙+ to species 7 (or species 8) and an enol/keto tautomerization to produce species 9 (or species 10) and BNA˙ (Scheme 3). Because BNA˙ is a strong one-electron reductant,34 electron transfer from BNA˙ to species 9 (or species 10) to produce species 11 (or species 12), accompanied by regeneration of BNA+.33 The protonation of species 11 (or species 12) by water yields the final products (compounds 2 and 4 in Scheme 3).33
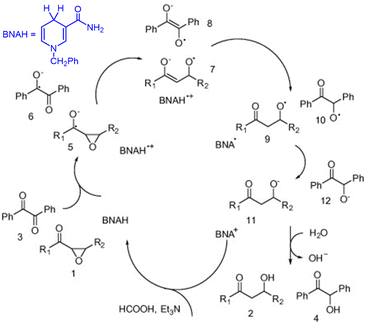 |
| | Scheme 3 Photocatalytic hydrogenation of α,β-epoxy ketones to β-hydroxy ketones with HCO2H and Et3N by BNAH photoredox catalysis. Reproduced from ref. 33 with permission form American Chemical Society (Copyright 2006). | |
2.2. Doublet excited states of radicals
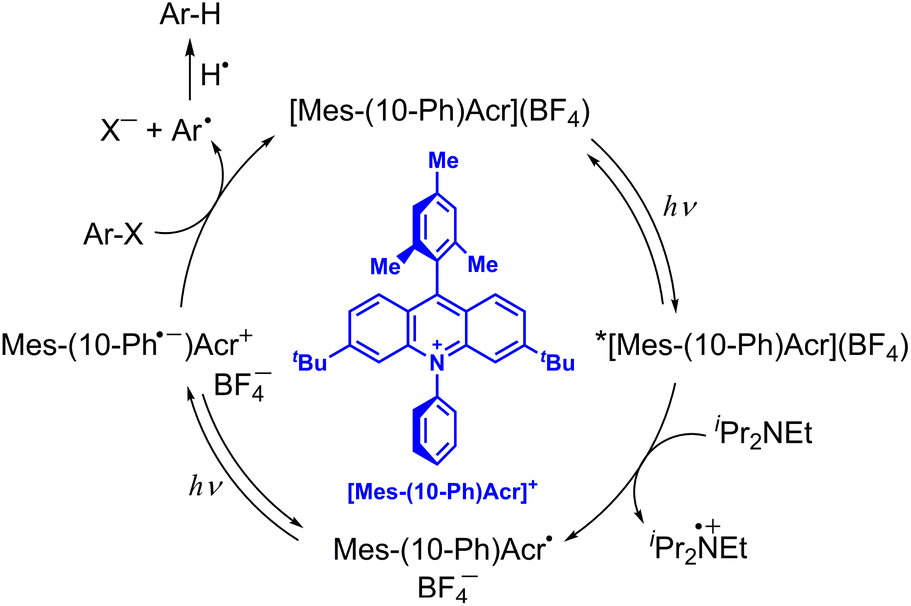
10-Methylacridinyl radical acts as a relatively strong reductant (Eredvs. SCE = −0.43 V),35 being unstable due to the fast dimerization reaction. However, 9-phenyl-10-methylacridinyl radical is stable due to the steric effect of 9-Ph moiety that prohibits the dimerization reaction.36 3,6-Di-tBu-9-mesityl-10-phenylacridinium tetra-fluoroborate, [Mes-(10-Ph)Acr](BF4), also provides the stable acridinyl radical (Mes-(10-Ph)Acr˙) upon the one-electron reduction of [Mes-(10-Ph)Acr](BF4).37 If the doublet excited state of the stable acridinyl radical ([Mes-(10-Ph)Acr˙]*) can be used as a reductant, the most negative Ered value was estimated to be −3.36 V vs. SCE.37 Such an extremely strong one-electron reductant has enabled to develop reductive dehalogenation of aryl halides even with electron-donating substituents to afford the desired dehalogenated products in good to excellent yields.37 As shown in Scheme 4, upon photoexcitation of [Mes-(10-Ph)Acr](BF4), electron transfer from diisopropylamine (iPr2NEt) to the Acr+ moiety of the electron-transfer state of [Mes-(10-Ph)Acr](BF4) occurs to produce iPr2NEt˙+ and the stable acridinyl radical (Mes-(10-Ph)Acr˙).37 The acridly radical is photoexcited to produce the charge-separated state in which the phenyl moiety becomes the phenyl radical anion (Mes-(10-Ph˙−)Acr+) that is capable to undergo dissociative electron transfer to Ar–X to produce aryl radical (Ar˙) and X−.37 Aryl radical abstracts hydrogen atom from iPr2NEt˙+ to produce Ar–H and regenerate the photocatalyst {[Mes-(10-Ph)Acr](BF4)}.37 When p-methoxychlorobenzene, which exhibits a largely negative half-peak potential (<−2.8 V vs. SCE), is employed as a substrate, the yield of p-methoxybenzene was 82%.37 This indicates that electron transfer from the excited state of the acridinyl radical ([Mes-(10-Ph)Acr˙]*) to p-methoxychlorobenzene may occur efficiently.37 However, the lifetime of [Mes-(10-Ph)Acr˙]* was much shorter than 100 ps. Fast electron transfer from the short-lived [Mes-(10-Ph)Acr˙]* to p-methoxychlorobenzene has yet to be confirmed by femtosecond laser transient absorption spectroscopy. The reductive detosylation of amines was also made possible by using the doublet excited state of Mes-(10-Ph)Acr˙.37
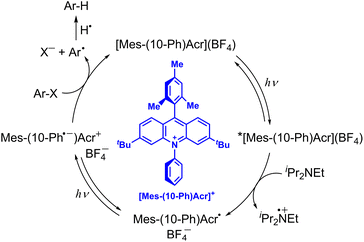 |
| | Scheme 4 Reductive dehydrogenation of aryl halide (Ar–X) with diisopropylamine by photoredox catalysis of [Mes-(10-Ph)Acr](BF4).37 | |
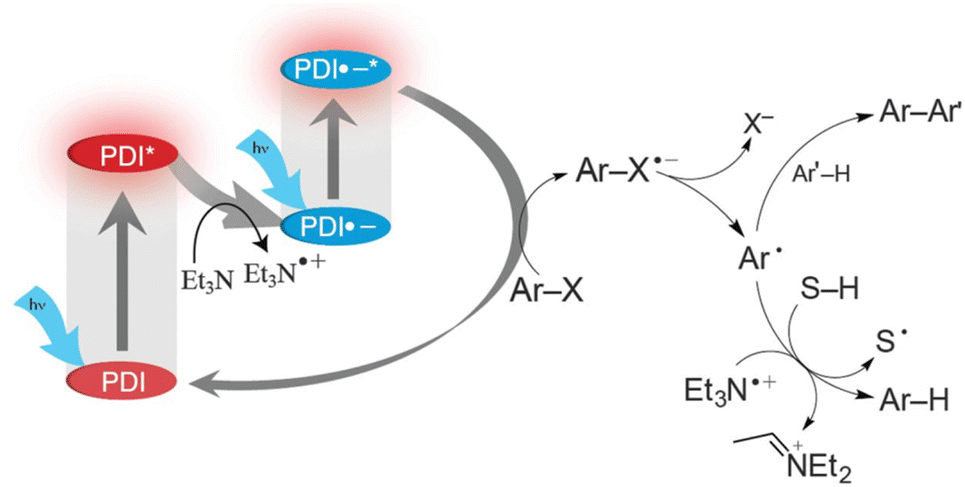
The doublet excited state of perylene diimide radical anion (PDI˙−) has also been used as a super-reductant that is capable of reducing aryl chlorides.38 The strong reducing excited state (*PDI˙−) was produced in situ by two subsequent visible light excitations starting from air-stable PDI, enabling to avoid the use of donor molecules, which are highly moisture- and air-sensitive. Only mixing of aryl chlorides (Ar–Cl compounds) with electron-withdrawing substituents, PDI and triethylamine (Et3N) under irradiation with visible light afforded the reduced products (i.e., Ar–H products) with good to excellent yields.38 The photocatalytic mechanism is shown in Scheme 5, where photoexcitation of a DMF solution containing PDI and Et3N under 455 nm blue light results in electron transfer from Et3N to PDI* to produce PDI˙− that is stable in the absence of dioxygen.38 PDI˙− is excited again by visible light to produce *PDI˙− and the dissociative electron transfer from *PDI˙− to aryl halides undergoes, resulting that aryl chlorides are reduced to aryl radical (Ar˙) species, which abstract a H-atom from Et3N˙+ to yield Ar–H products.38
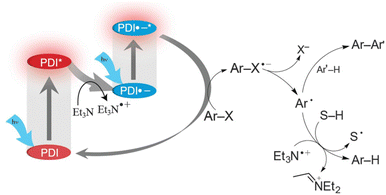 |
| | Scheme 5 Photocatalytic dehydrogenation of aryl halide (Ar–X) with triethylamine (Et3N) by consecutive photoinduced electron-transfer processes with perylene diimides (PDIs). Reprinted from ref. 38 with permission form the American Association for the Advancement of Science (Copyright 2014). | |
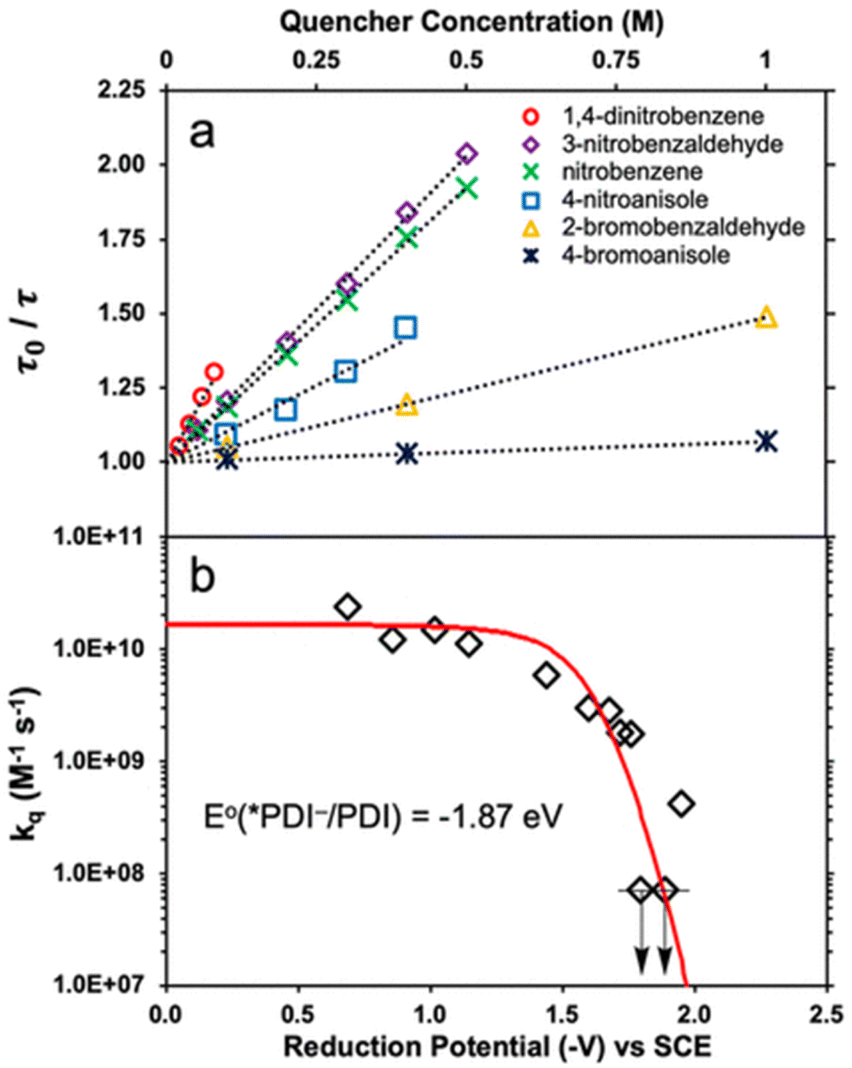
Transient absorption spectra of *PDI˙− were observed upon excitation (λex = 700 nm), exhibiting absorption bands at 460 and 600 nm due to 2(PDI˙−)*.39 The negative absorptions at 655–750 nm correspond to the ground state bleaching of PDI˙−.39 The excited state absorption at 460 nm due to *PDI˙− decayed, following first-order kinetics to give a lifetime (τ) of 160 ± 2 ps.39 The lifetime decreased with increasing concentrations of electron acceptors (Fig. 1a).39
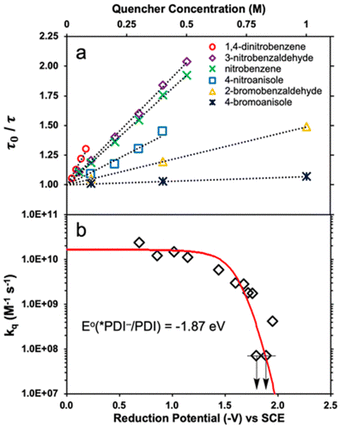 |
| | Fig. 1 (a) Stern–Volmer plots for lifetime quenching of *PDI˙−versus concentrations of electron acceptors. (b) Dependence of logarithm of kq of electron transfer from *PDI˙− to electron acceptors on the 1 e− reduction potentials (Eredvs. SCE) of electron acceptors. Red line represents the fit calculated using the Rehm–Weller equation. Reprinted from ref. 39 with permission form American Chemical Society (Copyright 2020). | |
The dependence of logarithm of the quenching constants (kq) of *PDI˙− by electron transfer to electron acceptors on the 1 e− reduction potentials of electron acceptors [E°(A/A˙−)] is shown in Fig. 1b, where the kq values of electron acceptors, which have E°(A/A˙−) values of more than −1.5 V vs. SCE, approached the diffusion limited value (∼2 × 1010 M−1 s−1).39 On the other hand, the kq values of electron acceptors, which have E°(A/A˙−) values of less than −1.5 V vs. SCE, exhibited the sharp drop in the region where E°(PDI/*PDI˙−) = E°(A/A˙−) (i.e., ΔGET = 0).39 The dependence of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kq on E°(A/A˙−) was well fitted with E°(PDI/*PDI˙−) value of −1.87 V vs. SCE and λ value of 0.84 eV using the Rehm and Weller empirical equation, which was reexamined by Farid, Dinnocenzo, Merkel, Young, Shukla and Guirado,40 that correlates kq and ΔGET with the reorganization energy for electron transfer [λ = 4 × ΔG‡(0)] (red line in Fig. 1b).39–41 Thus, *PDI˙− is a strong one-electron reductant that can reduce electron acceptors, which have the Ered values of more than −1.7 V. However, electron transfer from *PDI˙− to Ar–Cl was too slow to compete with the fast decay rate of 2(PDI˙−)*.39
kq on E°(A/A˙−) was well fitted with E°(PDI/*PDI˙−) value of −1.87 V vs. SCE and λ value of 0.84 eV using the Rehm and Weller empirical equation, which was reexamined by Farid, Dinnocenzo, Merkel, Young, Shukla and Guirado,40 that correlates kq and ΔGET with the reorganization energy for electron transfer [λ = 4 × ΔG‡(0)] (red line in Fig. 1b).39–41 Thus, *PDI˙− is a strong one-electron reductant that can reduce electron acceptors, which have the Ered values of more than −1.7 V. However, electron transfer from *PDI˙− to Ar–Cl was too slow to compete with the fast decay rate of 2(PDI˙−)*.39
The photoexcited state of 9,10-dicyanoanthracene radical anion (*DCA˙−) is also estimated to have an exceptionally negative 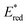 value of −3.2 V vs. SCE, which is comparable to some of the most oxidizable elemental metals such as Li (E vs. SCE = −3.3 V).42 In addition, *DCA˙− has a lifetime (τ) of 13.5 ns, which is long enough to undergo electron transfer reduction of a wide range of Ar–X compounds with Ered values as low as −2.94 V vs. SCE (Scheme 6).42 DCA˙− was produced using an H-type split cell with a porous carbon cathode and a Zn plate sacrificial anode by applying a constant cell voltage of 3.2 V.42 Photoirradiation of DCA˙− in the cathodic compartment with use of blue light enabled efficient coupling of 4-chlorobenzoate and B2pin3 (pin = pinacolato) as a radical acceptor to yield arylboronate (88%).42 This method has also been applied to the reductive borylation of 4-Cl-anisole, which has a very negative Ered value of −2.90 V vs. SCE.42 The mesolytic cleavage of a C–Br bond is significantly faster than that of a C–Cl bond, so that 1-Br-4-Cl-benzene was selectively converted to boronated–debrominated product.42
value of −3.2 V vs. SCE, which is comparable to some of the most oxidizable elemental metals such as Li (E vs. SCE = −3.3 V).42 In addition, *DCA˙− has a lifetime (τ) of 13.5 ns, which is long enough to undergo electron transfer reduction of a wide range of Ar–X compounds with Ered values as low as −2.94 V vs. SCE (Scheme 6).42 DCA˙− was produced using an H-type split cell with a porous carbon cathode and a Zn plate sacrificial anode by applying a constant cell voltage of 3.2 V.42 Photoirradiation of DCA˙− in the cathodic compartment with use of blue light enabled efficient coupling of 4-chlorobenzoate and B2pin3 (pin = pinacolato) as a radical acceptor to yield arylboronate (88%).42 This method has also been applied to the reductive borylation of 4-Cl-anisole, which has a very negative Ered value of −2.90 V vs. SCE.42 The mesolytic cleavage of a C–Br bond is significantly faster than that of a C–Cl bond, so that 1-Br-4-Cl-benzene was selectively converted to boronated–debrominated product.42
 |
| | Scheme 6 Reductive electrophotocatalysis with the photoexcited state of DCA˙− (*DCA˙−) for the reductive borylation of aryl chlorides and aryl bromides with bis(pinacolato)diboron (B2pin3). Reprinted from ref. 42 with permission form American Chemical Society (Copyright 2010). | |
2.3. Singlet excited states of organic anions
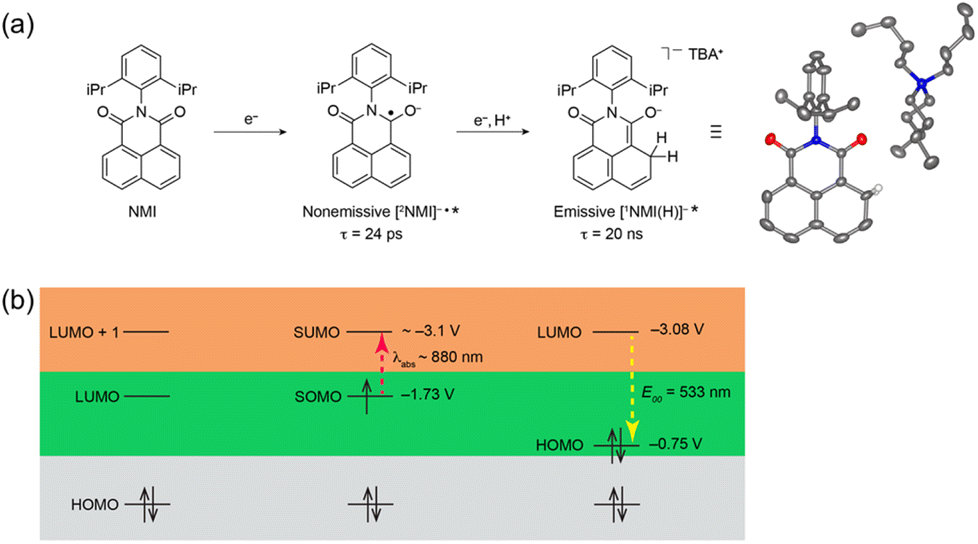
The electron-transfer reduction of naphthalene monoimide (NMI) gives the radical anion (NMI˙−), which has Eox value of −1.7 V vs. Fc/Fc+, in propylene carbonate (Fig. 2a).43 The femtosecond laser induced transient absorption measurements revealed that the doublet excited state of the radical anion ([2NMI˙−]*) has a lifetime (τ) of 24 ps, which was too short to undergo intermolecular photoredox reactions with substrates (Fig. 2a).43 In contrast, the singlet excited state of the two-electron reduced anion ([1NMI(H)−]*) species, which is called Meisenheimer complex, has the fluorescence lifetime of 20 ns.43 Together with a high excited state energy (−3.08 V vs. Fc/Fc+ in Fig. 2b), such a long fluorescent lifetime has enabled [1NMI(H)−]* to act as a super-reductant that can reduce Ar–X compounds to afford the products of C–C and C–P bond coupling via photoinduced electron transfer.43
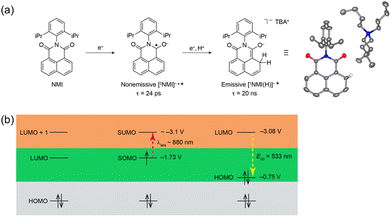 |
| | Fig. 2 (a) Chemical scheme of the formation of a singlet excited state ([1NMI(H)−]*) from naphthalene monoimide (NMI) via a doublet excited state ([2NMI˙−]*). (b) Energy level diagrams of NMI, [2NMI˙−]* and [1NMI(H)−]* with redox potentials vs. Fc/Fc+. Reprinted from ref. 43 with permission form American Chemical Society (Copyright 2021). | |
3 Super-oxidants
3.1. Excited states of quinones
The one-electron reduction potential of the triplet excited state 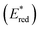 of 2,3-di-Cl-5,6-di-CN-p-benzoquinone (3DDQ*) is 3.18 V vs. SCE, which is highly positive enough to oxidize benzene by electron transfer to produce benzene radical cation, which can react with nucleophiles such as OH− to yield phenol (Scheme 7).44 Although electron transfer from benzene (Eoxvs. SCE = 2.48 V) to DDQ at the ground state (Eredvs. SCE = 0.51 V) is highly endergonic, electron transfer from benzene to 3DDQ* becomes exergonic to proceed.44 Thus, photoirradiation of benzene together with DDQ and H2O resulted in formation of phenol and DDQH2 [eqn (14)].44tButyl nitrite (TBN) was used as a recycling reagent to oxidize DDQH2 to regenerate DDQ under aerobic conditions.44 Phenol was not further oxidized although electron transfer from phenol to 3DDQ* occurred with the diffusion-limited rate constant. Back electron transfer from DDQ˙− to benzene radical cation may be quite exergonic, when this process is in the Marcus inverted region,24,45,46 where the back electron transfer is slowed down as compared to the case of phenol radical cation, which decayed by fast back electron transfer prior to the reaction with H2O. Thus, the back electron transfer controls the selectivity of the reactions via photoinduced electron transfer.
of 2,3-di-Cl-5,6-di-CN-p-benzoquinone (3DDQ*) is 3.18 V vs. SCE, which is highly positive enough to oxidize benzene by electron transfer to produce benzene radical cation, which can react with nucleophiles such as OH− to yield phenol (Scheme 7).44 Although electron transfer from benzene (Eoxvs. SCE = 2.48 V) to DDQ at the ground state (Eredvs. SCE = 0.51 V) is highly endergonic, electron transfer from benzene to 3DDQ* becomes exergonic to proceed.44 Thus, photoirradiation of benzene together with DDQ and H2O resulted in formation of phenol and DDQH2 [eqn (14)].44tButyl nitrite (TBN) was used as a recycling reagent to oxidize DDQH2 to regenerate DDQ under aerobic conditions.44 Phenol was not further oxidized although electron transfer from phenol to 3DDQ* occurred with the diffusion-limited rate constant. Back electron transfer from DDQ˙− to benzene radical cation may be quite exergonic, when this process is in the Marcus inverted region,24,45,46 where the back electron transfer is slowed down as compared to the case of phenol radical cation, which decayed by fast back electron transfer prior to the reaction with H2O. Thus, the back electron transfer controls the selectivity of the reactions via photoinduced electron transfer.| | 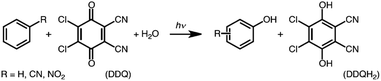 | (14) |
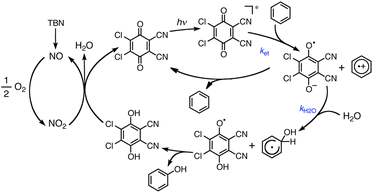 |
| | Scheme 7 A mechanism for photohydroxylation of benzene by dioxygen with DDQ used as a photoredox catalyst in the presence of TBN and H2O in MeCN under photoirradiation. Reprinted from ref. 44 with permission form American Chemical Society (Copyright 2013). | |
Since the singlet–triplet energy gap of Cl4Q is 0.64 eV, the singlet excited state of DDQ (1DDQ*) is estimated to have much stronger oxidizing ability than the triplet excited state of DDQ (3DDQ*).47 Thus, electron transfer from benzonitrile (PhCN) to 1DDQ* occurs to produce DDQ˙− and PhCN˙+ that is in equilibrium with the dimer radical cation [(PhCN)2˙+] (Scheme 8).47 The reaction of PhCN˙+ and H2O occurs to form the OH-adduct radical, which is oxidized by DDQH˙ to produce CN-substituted phenols and DDQH2 (Scheme 8).47 The ratio of ortho-, meta- and para-CN-phenols produced in the photohydroxylation of benzonitrile by 1DDQ*, was 44:18:38, respectively.47 Similarly, the photohydroxylation of nitrobenzene (PhNO2) also occurred via electron transfer from nitrobenzene to 1DDQ*.47 The ratio of ortho-, meta- and para-NO2-phenols produced in the photohydroxylation of nitrobenzene by 1DDQ*, was 45:40:15, respectively.47
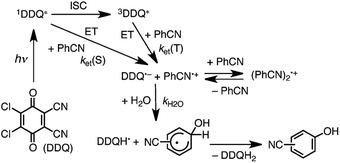 |
| | Scheme 8 Proposed mechanism for photohydroxylation of benzonitrile with DDQ and H2O in MeCN via1DDQ*. Reprinted from ref. 47 with permission form John Wiley and Sons (Copyright 2015). | |
The ratios of regioisomers were found to be determined by the electronic charges of radical cation of substrates, supported by the density functional theory.47 For benzonitrile radical cation, the negative charge at the ortho-position is smaller than that at the meta-position, while the para-position has a positive charge.47 As a result, OH− was mainly added to the two ortho-positions and a one para-position of benzonitrile radical cation, resulting in ortho- and para-CN-phenols, respectively.47 However, for nitrobenzene radical cation, since the para-position has the largest negative charge, other- and meta-NO2-phenols were mainly obtained as products in the photohydroxylation of nitrobenzene.47
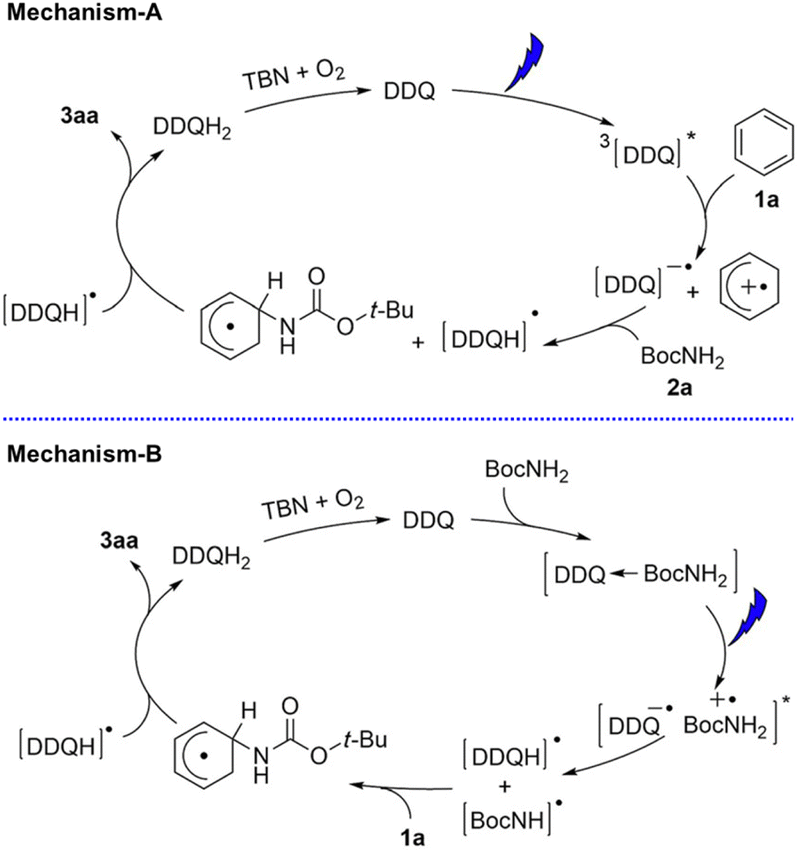
DDQ has also been used as an effective photoredox catalyst with tbutyl nitrite (TBN) for C–H amination of arenes by amines under visible light irradiation.48 Two photocatalytic mechanisms are shown in Scheme 9. Electron transfer from benzene to 3DDQ* occurs to produce benzene radical cation and DDQ˙− as the case of photohydroxylation of benzene (mechanism-A in Scheme 9).48 Benzene radical cation undergoes nucleophilic attack by the amine, followed by oxidation of the resulting species by DDQH˙ to yield the arene C–N substitution product.48 DDQ is regenerated by TBN.48 The radical cations generated from benzene and Cl-benzene react with various amine nucleophiles, whereas the radical cation of anisole is less reactive, yielding products with only strong nucleophiles such as pyrazole and other azoles.48
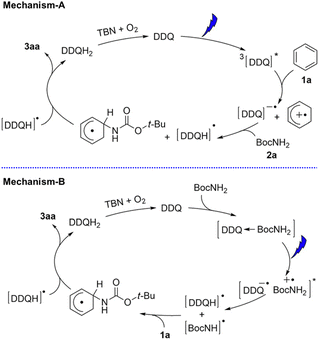 |
| | Scheme 9 Proposed mechanisms for photocatalytic C–H amination of different arenes with DDQ in the presence of TBN under photoirradiation. Reprinted from ref. 48 with permission form John Wiley and Sons (Copyright 2017). | |
Alternatively the charge-transfer complex formed between amines and DDQ in the ground state may be involved (mechanism-B in Scheme 9).48 Excitation by visible light results in hydrogen atom transfer from amines to DDQ via electron transfer followed by proton transfer.48 The generated amine radical attacks the arene and another H-atom abstraction yields the product and DDQH2.48 However, the photoexcitation of the charge-transfer band at 600 nm resulted in much less product as compared with the photoexcitation of DDQ.48 Thus, the proposed mechanism-A is more likely to be correct.
3.2. Excited states of radical dications
The higher one-electron reduction potential value than the triplet excited state of DDQ 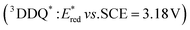 was reported for the excited state of a trisaminocyclopropenium radical dication
was reported for the excited state of a trisaminocyclopropenium radical dication 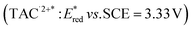 .49,50 TAC+ was electrochemically oxidized to form TAC˙2+, which was photoexcited with use of visible light to produce TAC˙2+* that is an excited-state species with sufficiently strong oxidizing power (3.33 V vs. SCE) to oxidize various substrates, including benzene and halogenated benzenes, via electron transfer, resulting in C–H/N–H coupling with azoles (Scheme 10).49,50 TAC˙2+* can also convert aryl olefins to the corresponding glycol monoesters with high chemo- and diastereoselectivity.51
.49,50 TAC+ was electrochemically oxidized to form TAC˙2+, which was photoexcited with use of visible light to produce TAC˙2+* that is an excited-state species with sufficiently strong oxidizing power (3.33 V vs. SCE) to oxidize various substrates, including benzene and halogenated benzenes, via electron transfer, resulting in C–H/N–H coupling with azoles (Scheme 10).49,50 TAC˙2+* can also convert aryl olefins to the corresponding glycol monoesters with high chemo- and diastereoselectivity.51
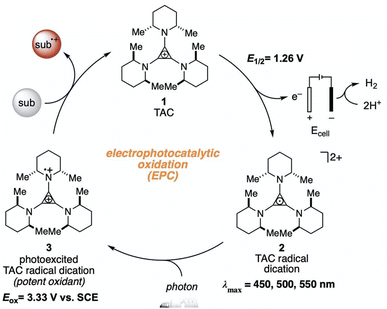 |
| | Scheme 10 Electrochemical generation of TAC˙2+ and photoinduced electron transfer oxidation of substrate by TAC˙2+*. Reprinted from ref. 49 with permission form the American Association for the Advancement of Science (Copyright 2021). | |
3.3. Excited states of Flavin-Sc3+ complexes
Metal (Mn+) ions acting as Lewis acids can bind to riboflavin-2′,3′,4′,5′-tetraacetate (Fl) to form the 1:1 and 1:2 complexes with Fl to metal ratios.52 The formation constants K1 and K2 of the 1:1 and 1:2 complexes of Fl with Sc3+ were determined to be 3.1 × 104 M−1 and 1.4 × 103 M−1, respectively.53 The fluorescence of 1(Fl-2Sc3+)* is quenched by electron transfer from electron donors to 1(Fl-2Sc3+)*.53 The quenching rate constants (kq) of electron transfer from electron donors to 1(Fl-2Sc3+)* increase as the 1 e− oxidation potentials (E0ox) of electron donors decrease, approaching a diffusion limited rate constant (Fig. 3).53 The 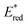 (vs. SCE) values of Fl-Mn+ complexes were in order of 1(Fl-2Sc3+)* (2.45 V) > 1(Fl-Yb3+)* (2.25 V) > 1(Fl-Mg2+)* (2.06 V) > 1Fl* (1.67 V), indicating that the order of
(vs. SCE) values of Fl-Mn+ complexes were in order of 1(Fl-2Sc3+)* (2.45 V) > 1(Fl-Yb3+)* (2.25 V) > 1(Fl-Mg2+)* (2.06 V) > 1Fl* (1.67 V), indicating that the order of 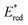 (vs. SCE) values of Fl-Mn+ complexes is in agreement of the order of the Lewis acidity of Mn+ bound to Fl.54 The
(vs. SCE) values of Fl-Mn+ complexes is in agreement of the order of the Lewis acidity of Mn+ bound to Fl.54 The 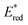 value of 1(Fl-2Sc3+)* is by 0.78 V more positive than that of 1Fl* (1.67 V) due to the strong binding of two Sc3+ ions to Fl˙−.53 Thus, 1(Fl-2Sc3+)* acts as a much stronger oxidant than 1Fl* as shown in Fig. 3.53 The combination of Fl with Sm3+ and iron complexes has also been reported to enable the visible-light driven aerobic C–H bond oxidation of alkyl benzenes.55,56
value of 1(Fl-2Sc3+)* is by 0.78 V more positive than that of 1Fl* (1.67 V) due to the strong binding of two Sc3+ ions to Fl˙−.53 Thus, 1(Fl-2Sc3+)* acts as a much stronger oxidant than 1Fl* as shown in Fig. 3.53 The combination of Fl with Sm3+ and iron complexes has also been reported to enable the visible-light driven aerobic C–H bond oxidation of alkyl benzenes.55,56
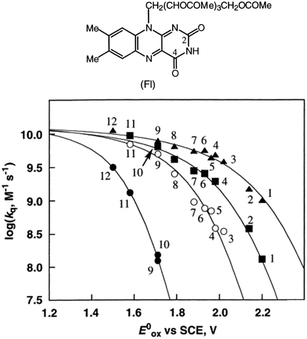 |
| | Fig. 3 Plots of logkq of electron transfer from electron donors [(1) PhCH3, (2) PhCH2CH3, (3) m-xylene, (4) o-xylene, (5) p-cymene, (6) p-xylene, (7) 1,2,3-tri-Me-benzene, (8) 1,2,4-tri-Me-benzene, (9) 1,2,3,4-tetra-Me-benzene, (10) 1,2,3,5-tetra-Me-benzene, (11) penta-Me-benzene and (12) m-di-MeO-benzene] to 1Fl* (10 μM) in the absence (●) and presence of Mn+ (1.0 × 10−2 M) [Mn+ = Sc3+ (▲), Yb3+ (■) and Mg2+ (○)] against 1 e− oxidation potentials (E0ox) of electron donors in MeCN at 298 K. Reprinted from ref. 53 with permission form American Chemical Society (Copyright 2001). | |
3.4. Excited states of MnIV(O)-(Sc3+)n complexes
As in the case of Fl-2Sc3+, two Sc3+ ions are bound to the oxo moiety of a MnIV-oxo complex bearing N-benzyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-diamino-ethane (BnTPEN) ligand to form [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2.57,58 Two HOTf molecules are also bound to a MnIV-oxo complex bearing N4Py ligand to form [(N4Py)MnIV(O)]2+-(HOTf)2.59 Upon Photoexcitation of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 in MeCN at 298 K, the long-lived photoexcited state with the lifetime of 6.4 μs was formed.60 When Sc(OTf)3 was replaced by Sc(NO3)3, the lifetime of the photoexcited state of [(BnTPEN)MnIV(O)]2+-Sc(NO3)3 increased to 7.1 μs.61 Such a long-lived excited state is assigned as the doublet 2E photoexcited state, which was formed by intersystem crossing from the 4E excited state, due to the spin forbidden decay to the quartet ground state as reported for Mn(IV) complexes.62,63
The transient absorption due to 2E excited state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 decayed via electron transfer from various electron donors to the 2E excited state to determine the rate constants of electron transfer (ket).60 Logarithm of ket increases with decreasing Eox values of electron donors to approach a diffusion-limited rate constant, as expressed by the Marcus equation of intermolecular electron transfer [eqn (15)],24,25
| | | (ket)−1 = (kdif)−1 + {Zexp[(−λ/4)(1 + ΔGet/λ)2/(kBT)]}−1 | (15) |
where
kdif is the diffusion rate constant,
Z is the collision frequency (10
11 M
−1 s
−1),
T is the absolute temperature,
kB is the Boltzmann constant and
λ is the reorganization energy of electron transfer.
60 The Gibbs energy change of electron transfer (Δ
Get) is given by
eqn (16),
| | 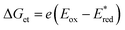 | (16) |
where
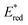
is the 1 e
− reduction potential of the excited
2E state of Mn
IV-oxo intermediate and
e is the elementary charge.
60 The best fit line using
eqn (15) in
Fig. 4 afforded that
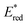
and the reorganization energy (
λ) values of {[(BnTPEN)Mn
IV(O)]
2+-(Sc(OTf)
3)
2}* are 2.1(1) V and 0.64(4) eV, respectively.
60 The reorganization energy of electron transfer from electron donors to {[(BnTPEN)Mn
IV(O)]
2+-(Sc(OTf)
3)
2}* (0.64(4) eV) is much smaller than that from electron donors to the ground state of [(BnTPEN)Mn
IV(O)]
2+-(Sc(OTf)
3)
2 (2.11 eV).
58 Such a large difference in the
λ values results from the small
λ value for the ligand-center electron transfer from electron donors to {[(BnTPEN)Mn
IV(O)]
2+-(Sc(OTf)
3)
2}*, which corresponds to the LMCT state, {[(BnTPEN)Mn
IV(O)]
2+-(Sc(OTf)
3)
2}*.
60 In contrast, electron transfer from electron donors to the ground state of [(BnTPEN)Mn
IV(O)]
2+-(Sc(OTf)
3)
2 occurs in the Mn
IV center with the large
λ value because of the significant change in the bond distance of Mn–O between the Mn
IV and Mn
III oxidation states.
58,59
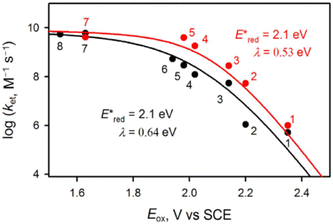 |
| | Fig. 4 Plots of logket of electron transfer from electron donors [(1) C6H6, (2) PhCH3, (3) PhCH2CH3, (4) m-xylene, (5) mesitylene, (6) PhCH2OH, (7) durene, and (8) naphthalene] to {[(BnTPEN)MnIV(O)]2+-Sc(NO3)3}* (red circles) and {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* (black circles) against Eox values of electron donors in TFE/MeCN (v/v 1:1) at 298 K. Reprinted from ref. 61 with permission form John Wiley and Sons (Copyright 2020). | |
The highly positive 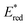 value of the 2E excited state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 and the small λ value enabled electron transfer from benzene (C6H6) to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* to generate C6H6˙+,60 which forms (C6H6)2˙+ in the presence of large excess C6H6.64 C6H6˙+ reacts with H2O to produce the OH adduct radical, which is oxidized by [(BnTPEN)MnIII(O)]+-(Sc(OTf)3)2 to generate PhOH with MnII species after removal of H+ (Scheme 11A).60 MnII species reacts rapidly with [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 to form [MnIII–O–MnIII]4+ species (Scheme 11B).60
value of the 2E excited state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 and the small λ value enabled electron transfer from benzene (C6H6) to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* to generate C6H6˙+,60 which forms (C6H6)2˙+ in the presence of large excess C6H6.64 C6H6˙+ reacts with H2O to produce the OH adduct radical, which is oxidized by [(BnTPEN)MnIII(O)]+-(Sc(OTf)3)2 to generate PhOH with MnII species after removal of H+ (Scheme 11A).60 MnII species reacts rapidly with [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 to form [MnIII–O–MnIII]4+ species (Scheme 11B).60
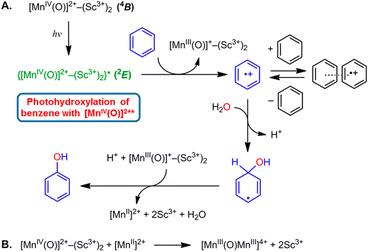 |
| | Scheme 11 (A) Proposed mechanism for the oxidation of C6H6 by {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}*.61 (B) The formation of [MnIII–O–MnIII]4+ species by reacting MnII with MnIV(O)-(Sc3+)2. Reprinted from ref. 60 with permission form American Chemical Society (Copyright 2018). | |
4 PRC models
4.1. 9-Mesityl-10-methylacridinium ion
A simple electron donor–acceptor-linked molecule with a small λ value of electron transfer and a high-lying triplet excited state is an ideal PRC model compound, which has highly oxidizing and reducing capability with a long lifetime of the electron-transfer state between the electron donor and acceptor.5 The acridinium ion (Acr+) is the best candidate as an electron acceptor moiety in such an electron donor–acceptor-linked molecule, because the λ value of electron self-exchange between Acr+ and its 1 e− reduced species is the smallest (0.3 eV) among redox-active organic compounds.5 Thus, a mesityl group (Mes) is directly linked as an electron-donor moiety at the 9-position of Acr+ to form 9-mesityl-10-methylacridinium ion (Acr+–Mes). According to the X-ray crystal structure of Acr+–Mes, the mesityl moiety is orthogonal to the acridinium moiety.65 In such a case, there is little orbital interaction between the Mes and Acr+ moieties. Photoexcitation of Acr+–Mes results in fast intramolecular electron transfer from the Mes moiety to the singlet excited state of the Acr+ moiety to produce the triplet electron-transfer (ET) state (Acr˙–Mes˙+) via intersystem crossing.65,66 Because the intramolecular back ET from Acr˙ unit to Mes˙+ unit is too slow to compete with intermolecular back ET, which obeyed second-order kinetics.65,66 In order to obtain the lifetime due to intramolecular back ET without contribution of the intermolecular back ET, Acr+–Mes was immobilized into tube-shaped nanosized mesoporous silica-alumina to prepare (Acr+–Mes@tAlMCM-41) by cation exchange.67 Photoirradiation of Acr+–Mes@tAlMCM-41 (λ > 390 nm) at 298 K results in formation of the triplet ET state [3(Acr˙–Mes˙+)*] via photoinduced ET from the Mes moiety to 1(Acr+)* moiety (Fig. 5A).67 The EPR spectrum of 3(Acr˙–Mes˙+)*@tAlMCM-41 was confirmed by overlapping the EPR signals of the Acr˙ and Mes˙+ moieties (Fig. 5B and C, respectively).67 The decay in the EPR signal intensity due to 3(Acr˙–Mes˙+)* followed first-order kinetics (Fig. 5D) to give the lifetime of 3 s for the intramolecular back ET in deaerated MeCN at 298 K.67 The lifetime of 3 s is the longest reported so far and is longer than the charge-separated state lifetime in the PRCs.67
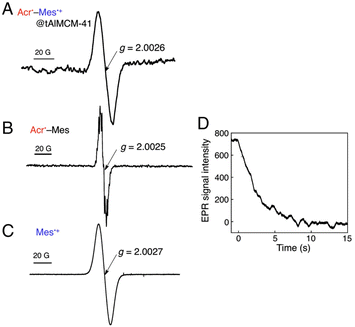 |
| | Fig. 5 (A) EPR spectrum of Acr˙–Mes˙+@tAlMCM-41 in MeCN at 298 K observed under photoirradiation (λ > 390 nm) for 2 min. (B) EPR spectrum of Acr˙–Mes generated by the photoinduced ET reduction of Acr+–Mes by BNAH (5.0 equiv.) in MeCN under photoirradiation. (C) EPR spectrum of mesitylene radial cation prepared by the photoinduced 1 e− oxidation of mesitylene with Hg(CF3COO)2 in CF3COOH at 298 K under photoirradiation. (D) Decay time course of the EPR signal intensity due to Acr˙–Mes˙+@tAlMCM-41 in MeCN at 298 K. Reprinted with permission from ref. 67. Copyright 2012, National Academy of Sciences. | |
4.2. PS-II model
The first functional model of PS-II was reported for the photocatalytic reduction of p-benzoquinone derivatives [X–Q: DDQ, p-benzoquinone (Q), Cl4Q, 2,5-di-Me-p-benzoquinone (PXQ) and duroquinone (DQ)] as plastoquinone analogs with the concomitant 4e−/4H+ oxidation of water to evolve O2 [eqn (17)] in the presence of [FeII(N4Py)]2+ (N4Py = 1,1-di(pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine)68 acting as a water oxidation catalyst (WOC).69| | 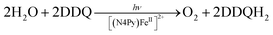 | (17) |
The photocatalytic mechanism of the oxidation of H2O by DDQ with [(N4Py)FeII]2+ is shown in Scheme 12.69 Electron transfer from iron(II) complex to 3DDQ* with H2O occurs to form FeIII(OH) and DDQH˙ species (Scheme 12, pathway a). Subsequent electron transfer from FeIII(OH) to 3DDQ* occurs to form FeIV(O) and DDQH˙ species (Scheme 12, pathway b).70–72 Finally electron transfer from FeIV(O) to 3DDQ* also occurs to generate FeV(O) intermediate (Scheme 12, pathway c).69 From the slope of plot of the decay rate constant of 3DDQ* against concentration of iron(IV)-oxo, the second-order rate constant of electron transfer from iron(IV)-oxo to 3DDQ* was determined to be 9.4 × 109 M−1 s−1.69 Iron(V)-oxo reacts rapidly with H2O to produce FeIII(OOH) species and H+ (Scheme 12, pathway d).69 On the other hand, DDQH˙ disproportionates to produce DDQ and DDQH2.69 FeIII(OOH) is oxidized thermally by DDQ via H-atom transfer to produce iron(III)-superoxo species, FeIII(O2˙−) (Scheme 12, pathway e), accompanied by release of O2 and regeneration of iron(II) complex (Scheme 12, pathway f).69 Meanwhile [(N4Py)FeIII(OOH)]2+ can be generated independently from the reaction of [(N4Py)FeII]2+ and H2O2,72 and the production of O2 was confirmed through a thermal reaction between [(N4Py)FeIII(OOH)]2+ and DDQ, indicating that [(N4Py)FeIII(OOH)]2+ is thermally oxidized by DDQ.69
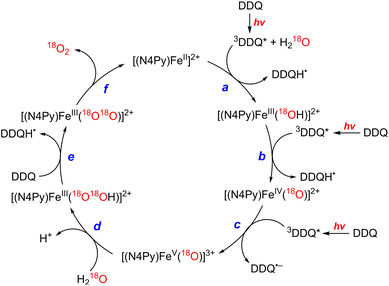 |
| | Scheme 12 A mechanism of the photocatalytic 4e−/4H+ oxidation of H2O by DDQ as an oxidant and [FeII(N4Py)]2+ as a WOC. Reprinted from ref. 69 with permission form American Chemical Society (Copyright 2019). | |
The rate constants of electron transfer from [(N4Py)FeIV(O)]2+ to other 3X–Q* derivatives were also determined to be close to the diffusion limited value.69 Thus, X–Q can oxidize H2O to O2 with Fe(II) complex under photoirradiation in the PS-II model reactions.69
4.3. PS-I model
Because of the extremely long lifetime of Acr˙–Mes˙+ with the strong oxidizing and reducing capability, Acr+–Mes has acted an efficient organic photoredox catalyst in various organic transformations, which have been reviewed well.73–82 One notable example is the combination of a PRC model (Acr+–Mes) and an H2 evolution catalyst [CoIII(dmgH)2pyCl (py = pyridine and dmgH− = N,N′-Dihydroxy-2,3-butanediimine monoanion)],83–86 which mimics the photocatalytic function of PS-I (Scheme 13).87 Solar energy is harvested by the Acr+ moiety of Acr+–Mes to generate 3(Acr˙–Mes˙+).87 Electron transfer from QH2 to the Mes˙+ unit of 3(Acr˙–Mes˙+) occurs with the diffusion-limited value to form Acr˙–Mes and X–QH2˙+. Then, electron transfer from Acr˙–Mes to CoIII complex (i.e., CoIII(dmgH)2pyCl) occurs to generate CoII species, accompanied by reproduction of Acr+–Mes.87 QH2˙+ is deprotonated rapidly to produce semiquinone radical (QH˙).87 Then proton-coupled electron transfer (PCET) from QH˙ to CoII species occurs to form Q and the CoIII–H− species that reacts with H+ to evolve H2, accompanied by reproduction of an H2 evolution catalyst (CoIII(dmgH)2pyCl).87 The deuterium kinetic isotope effect (KIE = 2.0) was observed for the H2 evolution rate by use of D2O instead of H2O.88,89 This indicates that PCET from QH˙ (QD˙) to CoII species may be the rate-determining step for the photocatalytic H2 evolution in the PS-I model reaction.87
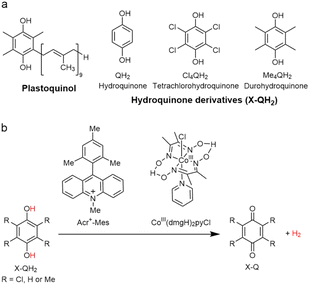 |
| | Scheme 13 (a) Plastoquinol and its analogs.84 (b) Photocatalytic hydrogen evolution using the PS-I model system consisting of hydroquinone derivatives, 9-mesityl-10-methylacridinium ion as a PRC model and CoIII(dmgH)2pyCl. Reprinted from ref. 87 with permission form American Chemical Society (Copyright 2020). | |
4.4. Combination of PS-I and PS-II molecular models
A PS-II molecular model (Scheme 12) has been combined with a PS-I molecular model (Scheme 14) to mimic the photocatalytic function of photosynthesis as shown in Fig. 6a, where the two cells for PS-I and PS-II molecular models were connected in a H2O-TFE phase with use of a glass membrane.90 Only hydroquinone (QH2) derivatives can pass through the glass membrane, but other catalysts cannot.90 Photoirradiation of a H2O–TFE/toluene solution containing [(N4Py)FeII]2+ and quinone in the left cell as well as a H2O–TFE/toluene solution containing Acr+–Mes and CoIII(dmgH)2pyCl in the right cell (Fig. 6a) resulted in evolution of O2 and H2 from the left and right cells, respectively (Fig. 6b and c), with 100% yield in both cases on the basis of the initial concentration of quinone (Q) in accordance with the stoichiometry of mimicry of photosynthesis [eqn (18)].90| |  | (18) |
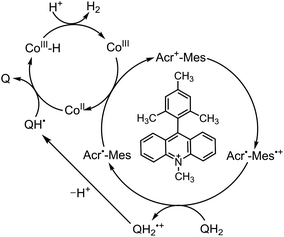 |
| | Scheme 14 Proposed PS-I model mechanism of photocatalytic H2 evolution from hydroquinone (QH2) derivatives with Acr+–Mes and CoIII complex. Reprinted from ref. 87 with permission form American Chemical Society (Copyright 2020). | |
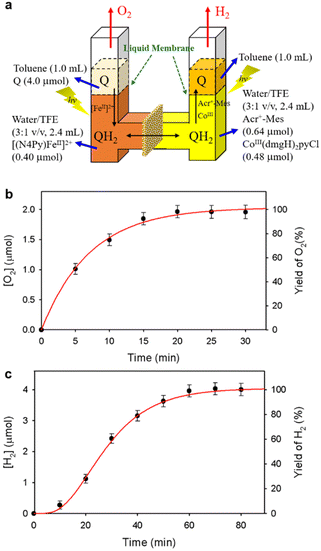 |
| | Fig. 6 (a) A photocatalytic system used an H-type cell for the photocatalytic production of H2 and O2 from H2O. (b) Time profile of the production of O2 in the photocatalytic H2O oxidation. (c) Time profile of the production of H2 in the photocatalytic H2O reduction. Reprinted from ref. 90 with permission form American Chemical Society (Copyright 2022). | |
When the toluene phases of the left and right cells are connected with a glass filter (Fig. 7a), photoirradiation of both parts resulted in formation of much larger amounts of H2 (Fig. 7b) as compared with the initial concentration of quinone.90 The turnover number for the H2 evolution were 160 ± 21 on the basis of the initial concentration of quinone.90 Thus, quinone acts as a photocatalyst for photocatalytic H2O splitting as shown in Scheme 15.90 This is the first case to achieve photocatalytic H2O splitting to produce H2 and O2 by combining the molecular models of PS-I and PS-II in homogeneous catalytic system (e.g., in solution).
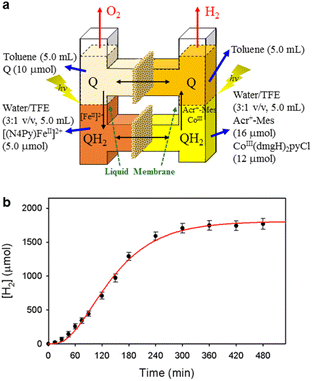 |
| | Fig. 7 (a) A photochemical cell used for photocatalytic production of H2 and O2 from H2O. (b) Time profile of the production of H2 in the photocatalytic H2O splitting under photoirradiation. Reprinted from ref. 90 with permission form American Chemical Society (Copyright 2022). | |
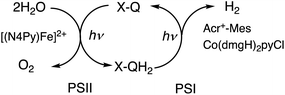 |
| | Scheme 15 A scheme for the photocatalytic production of H2 and O2 from H2O by the combination of the molecular models of PS-I and PS-II with use of X–Q/X–QH2 as plastoquinone/plastoquinol analogue in photosynthesis. Reprinted from ref. 90 with permission form American Chemical Society (Copyright 2022). | |
5 Conclusions
The excited states of organic electron donors become much stronger electron donors as compared with the ground states, undergoing various reduction reactions, which would otherwise be impossible to occur at the ground states. For example, the singlet excited state of an NADH (dihydronicotinamide adenine dinucleotide) analog, 9,10-dihydro-10-methylacridine 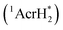 has a largely negative one-electron reduction potential
has a largely negative one-electron reduction potential 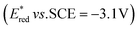 and a lifetime of 7.0 ns to enable to reduce various alkyl halides. The more negative
and a lifetime of 7.0 ns to enable to reduce various alkyl halides. The more negative 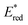 value (−3.36 V) is evaluated for the doublet excited state of stable acridinyl radical (Mes–Acr˙), which acts as a super-reductant. The excited states of radical anions and anions also act as super-reductants that enabled to reduce substrates, which are usually difficult to be reduced by the ground state reductants. By the same token, the excited states of organic electron acceptors such as DDQ and TAC˙2+ become much stronger electron acceptors as compared with the ground states, undergoing various oxidation reactions, which would otherwise be impossible to occur at the ground states. The binding of Sc3+ ions to flavin and Mn(IV)-oxo complexes enhanced the oxidizing ability. The excited state further enhanced the oxidizing ability, acting as super-oxidants.
value (−3.36 V) is evaluated for the doublet excited state of stable acridinyl radical (Mes–Acr˙), which acts as a super-reductant. The excited states of radical anions and anions also act as super-reductants that enabled to reduce substrates, which are usually difficult to be reduced by the ground state reductants. By the same token, the excited states of organic electron acceptors such as DDQ and TAC˙2+ become much stronger electron acceptors as compared with the ground states, undergoing various oxidation reactions, which would otherwise be impossible to occur at the ground states. The binding of Sc3+ ions to flavin and Mn(IV)-oxo complexes enhanced the oxidizing ability. The excited state further enhanced the oxidizing ability, acting as super-oxidants.
A simple electron donor–acceptor-linked molecule with a small reorganization energy of electron transfer such as Acr+–Mes acts as an ideal photosynthetic reaction center (PRC) model compound, which has high oxidizing and reducing capability with a long lifetime of the ET state. The PCR model compound was used to construct a PS-I model system in which H2 was evolved from plastoquinol analogues with a cobalt complex as an H2 evolution catalyst.
The PS-I model was combined with a PS-II model with use of the excited state of plastoquinone analogues and water oxidation catalyst to achieve the photofunction of photosynthesis. Because H2 can reduce NAD+ to NADH by redox catalysis of metal complexes,91,92 NADH can be produced from water by adding to NAD+ reduction catalysts to the molecular photocatalytic system in Scheme 15. By the same token, CO2 can be reduced by water by adding CO2 reduction catalysts93–95 to the molecular photocatalytic system in Scheme 15. Thus, redox catalysis via photoinduced electron transfer discussed in this perspective may further be expanded to construct more useful photocatalytic systems for solar fuel production and green redox reactions. The development of stronger super reductants and super oxidants together with PRC models with the stronger reducing and oxidizing power would further expand the scope of redox catalysis via photoinduced electron transfer.
Author contributions
All the authors contributed to the discussion, editing and revision of this work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the contributions of their collaborators and coworkers cited in the listed references, JSPS (No. 16H02268 to S. F.), NRF of Korea (NRF-2021R1A3B1076539 to W. N and NRF-2020R1I1A1A01074630 to Y.-M. L.).
Notes and references
- R. Croce and H. van Amerongen, Science, 2020, 369, eaay2058 CrossRef CAS PubMed.
- J.-R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- M. M. Najafpour, I. Zaharieva, Z. Zand, S. M. Hosseini, M. Kouzmanova, M. Hołyńska, I. Tranca, A. W. Larkum, J.-R. Shen and S. I. Allakhverdiev, Coord. Chem. Rev., 2020, 409, 213183 CrossRef CAS.
- M. M. Najafpour and S. I. Allakhverdiev, J. Photochem. Photobiol., B, 2015, 152, 173–424 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609–620 RSC.
- M. J. Llansola-Portoles, D. Gust, T. A. Moore and A. L. Moore, C. R. Chim., 2017, 20, 296–313 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 2, 4575–4587 RSC.
- O. Ito and F. D'Souza, Molecules, 2012, 17, 5816–5835 CrossRef CAS PubMed.
- S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283–2297 RSC.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427–1439 RSC.
- C. B. KC and F. D'Souza, Coord. Chem. Rev., 2016, 322, 104–141 CrossRef CAS.
- M. E. El-Khouly, E. El-Mohsnawy and S. Fukuzumi, J. Photochem. Photobiol. C.: Photochem. Rev., 2017, 31, 36–83 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Biochem. Soc. Trans., 2018, 46, 1279–1288 CrossRef CAS PubMed.
- A. Zieleniewska, F. Lodermeyer, A. Roth and D. M. Guldi, Chem. Soc. Rev., 2018, 47, 702–714 RSC.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Tetrahedron, 2020, 76, 131024 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Coord. Chem. Rev., 2020, 408, 213184 CrossRef CAS.
-
S. Fukuzumi, Electron Transfer: Mechanisms and Applications, Wiley-VCH, Weinheim, 2020 Search PubMed.
- S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC.
- S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- N. Ishikawa and S. Fukuzumi, J. Am. Chem. Soc., 1990, 112, 8864–8870 CrossRef.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat, F. M'Halla and J. M. Savéant, J. Am. Chem. Soc., 1980, 102, 3806–3813 CrossRef CAS.
- B. Huang, X.-S. Bu, J. Xu, J.-J. Dai, Y.-S. Feng and H.-J. Xu, Asian J. Org. Chem., 2018, 7, 137–140 CrossRef CAS.
- J. Jiang, W.-M. Zhang, J.-J. Dai, J. Xu and H.-J. Xu, J. Org. Chem., 2017, 82, 3622–3630 CrossRef CAS PubMed.
- S. Fukuzumi, K. Hironaka and T. Tanaka, J. Am. Chem. Soc., 1983, 105, 4722–4727 CrossRef CAS.
- Q. Huang, J.-W. Wu and H.-J. Xu, Tetrahedron Lett., 2013, 54, 3877–3881 CrossRef CAS.
- H.-J. Xu, Y.-C. Liu, Y. Fu and Y.-D. Wu, Org. Lett., 2006, 8, 3449–3451 CrossRef CAS PubMed.
- S. Fukuzumi, S. Koumitsu, K. Hironaka and T. Tanaka, J. Am. Chem. Soc., 1987, 109, 305–316 CrossRef CAS.
- S. Fukuzumi, Y. Tokuda, T. Kitano, T. Okamoto and J. Otera, J. Am. Chem. Soc., 1993, 115, 8960–8968 CrossRef CAS.
- S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459–8467 CrossRef CAS PubMed.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 586, 76–80 CrossRef PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- C. J. Zeman, S. Kim, F. Zhang and K. S. Schanze, J. Am. Chem. Soc., 2020, 142, 2204–2207 CrossRef CAS PubMed.
- S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2010, 142, 2087–2092 CrossRef PubMed.
- A. J. Rieth, M. I. Gonzalez, B. Kudisch, M. Nava and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 14352–14359 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, H. Imahori and D. M. Guldi, Chem. – Eur. J., 2003, 9, 1585–1593 CrossRef CAS PubMed.
- M. Murakami, K. Ohkubo and S. Fukuzumi, Chem. – Eur. J., 2010, 16, 7820–7832 CrossRef CAS PubMed.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chem. – Eur. J., 2015, 21, 2855–2861 CrossRef CAS PubMed.
- S. Das, P. Natarajan and B. König, Chem. – Eur. J., 2017, 23, 18161–18165 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS PubMed.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 7247–7252 CrossRef CAS PubMed.
- S. Fukuzumi, S. Kuroda and T. Tanaka, J. Am. Chem. Soc., 1985, 107, 3020–3027 CrossRef CAS.
- S. Fukuzumi, K. Yasui, T. Suenobu, K. Ohkubo, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 10501–10510 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, Chem. – Eur. J., 2000, 6, 4532–4535 CrossRef CAS PubMed.
- M. S. S. V. Mouli, S. Katyal and A. K. Mishra, Synlett., 2022 DOI:10.1055/a-1928-3417.
- B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef PubMed.
- J. Chen, Y.-M. Lee, K. M. Davis, X. Wu, M. S. Seo, K.-B. Cho, H. Yoon, Y. J. Park, S. Fukuzumi, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013, 135, 6388–6391 CrossRef CAS PubMed.
- H. Yoon, Y.-M. Lee, X. Wu, K.-B. Cho, R. Sarangi, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 9186–9194 CrossRef CAS PubMed.
- J. Chen, H. Yoon, Y.-M. Lee, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Chem. Sci., 2015, 6, 3624–3632 RSC.
- N. Sharma, J. Jung, K. Ohkubo, Y.-M. Lee, M. E. El-Khouly, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2018, 140, 8405–8409 CrossRef CAS PubMed.
- N. Sharma, Y.-M. Lee, W. Nam and S. Fukuzumi, Isr. J. Chem., 2020, 60, 1049–1056 CrossRef CAS.
- R. W. Kitzmann, J. Moll and K. Heinze, Photochem. Photobiol. Sci., 2022, 21, 1309–1331 CrossRef PubMed.
- V. Baslon, P. J. Harris, C. Reber, E. H. Colmer, A. T. Jackson, P. A. Forshaw, M. J. Smith, R. Adam Kinney and J. Teser, Can. J. Chem., 2017, 95, 547–552 CrossRef CAS.
- P. B. Merkel, P. Luo, J. P. Dinnocenzo and S. Farid, J. Org. Chem., 2009, 74, 5163–5173 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Okubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- T. Tsudaka, H. Kotani, K. Ohkubo, T. Nakagawa, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, Chem. – Eur. J., 2017, 23, 1306–1317 CrossRef CAS PubMed.
- S. Fukuzumi, K. Doi, A. Itoh, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15572–15577 CrossRef CAS PubMed.
- J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kin, E. Münck, W. Nam and L. Que Jr., J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS PubMed.
- Y. H. Hong, J. Jung, T. Nakagawa, N. Sharma, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2019, 141, 6748–6754 CrossRef CAS PubMed.
- H. Kotani, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kojima, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2017, 333, 44–56 CrossRef CAS.
- S. Hong, Y.-M. Lee, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2009, 131, 13910–13911 CrossRef CAS PubMed.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- X. Zhang, K. P. Pakesh, L. Ravindar and H.-L. Quin, Green Chem., 2018, 20, 4790–4833 RSC.
- A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
- A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 2–26 CrossRef PubMed.
- T. Bortolato, S. Cuadros, G. Simionato and L. Dell'Amico, Chem. Commun., 2022, 58, 1263–1283 RSC.
- M. V. Bobo, J. J. Kuchta III and A. K. Vannucci, Org. Biomol. Chem., 2021, 19, 4816–4834 RSC.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54–73 CrossRef CAS.
- D. Dolui, S. Khandelwal, P. Majumder and A. Dutta, Chem. Commun., 2020, 56, 8166–8181 RSC.
- G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2020, 59, 14838–14846 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Bull. Korean Chem. Soc., 2021, 42, 1558–1568 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Bull. Korean Chem. Soc., 2020, 41, 1217–1232 CrossRef CAS.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367–374 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, J. Inorg. Biochem., 2019, 199, 110777 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee, H. S. Ahn and W. Nam, Chem. Sci., 2018, 9, 6017–6034 RSC.
- L. Chen, G. Chen, C.-F. Leung, C. Cometto, M. Robert and T.-C. Lau, Chem. Soc. Rev., 2020, 49, 7271–7283 RSC.
- T. Kojima, ChemPhotoChem, 2021, 5, 512–520 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Wonwoo
Nam
*ab,
Wonwoo
Nam



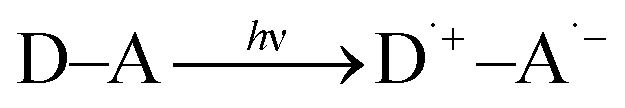

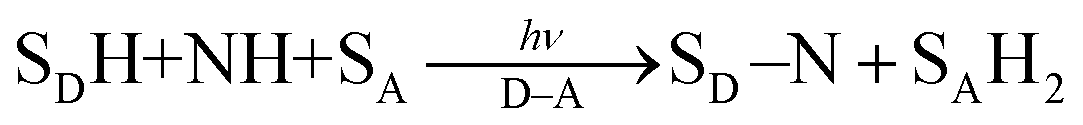
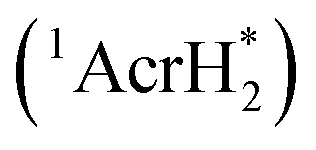 , has a largely negative one-electron reduction potential (−3.1 V vs. SCE) and an appreciable lifetime of 7.0 ns.27 Electron transfer from
, has a largely negative one-electron reduction potential (−3.1 V vs. SCE) and an appreciable lifetime of 7.0 ns.27 Electron transfer from 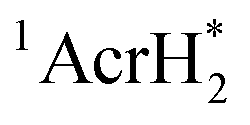 to various halogenated compounds (RX) occurs to produce dehalogenated compounds (RH) and the two-electron oxidation product, 10-methylacridinium ion (AcrH+), because the one-electron reduction potentials of RX are less negative (e.g., −2.78 V vs. SCE) than that of
to various halogenated compounds (RX) occurs to produce dehalogenated compounds (RH) and the two-electron oxidation product, 10-methylacridinium ion (AcrH+), because the one-electron reduction potentials of RX are less negative (e.g., −2.78 V vs. SCE) than that of 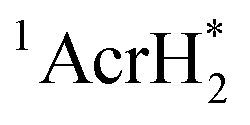 (−3.1 V vs. SCE).27 The rate constant of electron transfer from
(−3.1 V vs. SCE).27 The rate constant of electron transfer from  to PhCl was determined to be 6.0 × 108 M−1 s−1 at 298 K from the fluorescence quenching by PhCl.27 When AcrH2 was replaced by the didueterated compound (AcrD2), the rate constant of electron transfer from
to PhCl was determined to be 6.0 × 108 M−1 s−1 at 298 K from the fluorescence quenching by PhCl.27 When AcrH2 was replaced by the didueterated compound (AcrD2), the rate constant of electron transfer from 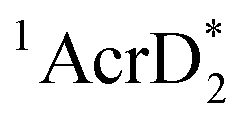 to PhCl was the same as that of
to PhCl was the same as that of 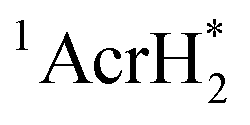 , showing no deuterium kinetic isotope effect.27 Electron transfer reduction of RX results in the C–X bond cleavage to produce R˙ and X−.28 The produced carbon-centered radical (R˙) abstracts a hydrogen atom from AcrH2˙+, which is produced by electron transfer from
, showing no deuterium kinetic isotope effect.27 Electron transfer reduction of RX results in the C–X bond cleavage to produce R˙ and X−.28 The produced carbon-centered radical (R˙) abstracts a hydrogen atom from AcrH2˙+, which is produced by electron transfer from  to RX, to form 10-methylacridinium ion (AcrH+) and RH [eqn (12)].27
to RX, to form 10-methylacridinium ion (AcrH+) and RH [eqn (12)].27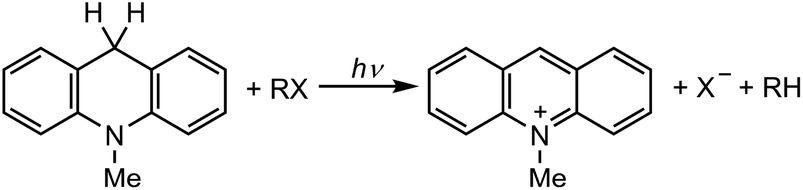
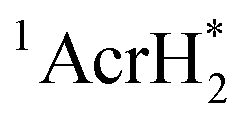
 to CF3SO2Cl (Eredvs. SCE = −0.18 V) occurs rapidly to produce AcrH2˙+, CF3˙ and SO2Cl−, followed by radical addition of CF3˙ to the C
to CF3SO2Cl (Eredvs. SCE = −0.18 V) occurs rapidly to produce AcrH2˙+, CF3˙ and SO2Cl−, followed by radical addition of CF3˙ to the C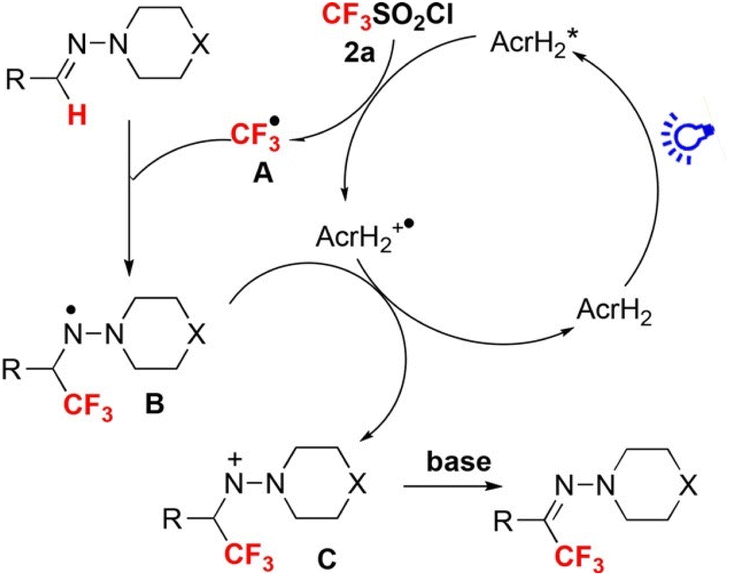
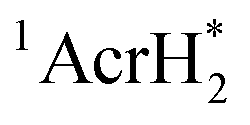 to ArN2BF4 to generate AcrH2˙+ and Ar˙, accompanied by N2 dissociation (Scheme 2).30 Ar˙ reacts with complex A in Scheme 2 to afford the palladium(III) complex B, which is then oxidized by AcrH2˙+via electron transfer from complex B to AcrH2˙+ to produce palladium(IV) complex C and regenerate the photocatalyst AcrH2 (Scheme 2).30 Finally, the reductive elimination of palladium(IV) complex C affords the formation of compound D, which is the arylated product, and regenerates the palladium(II) catalyst A.30 Thus, a dual catalytic system of palladium(II)/organic photoredox catalysts enables the arylation of arenes with ArN2+ over a wide substrate range under the mild conditions.30
to ArN2BF4 to generate AcrH2˙+ and Ar˙, accompanied by N2 dissociation (Scheme 2).30 Ar˙ reacts with complex A in Scheme 2 to afford the palladium(III) complex B, which is then oxidized by AcrH2˙+via electron transfer from complex B to AcrH2˙+ to produce palladium(IV) complex C and regenerate the photocatalyst AcrH2 (Scheme 2).30 Finally, the reductive elimination of palladium(IV) complex C affords the formation of compound D, which is the arylated product, and regenerates the palladium(II) catalyst A.30 Thus, a dual catalytic system of palladium(II)/organic photoredox catalysts enables the arylation of arenes with ArN2+ over a wide substrate range under the mild conditions.30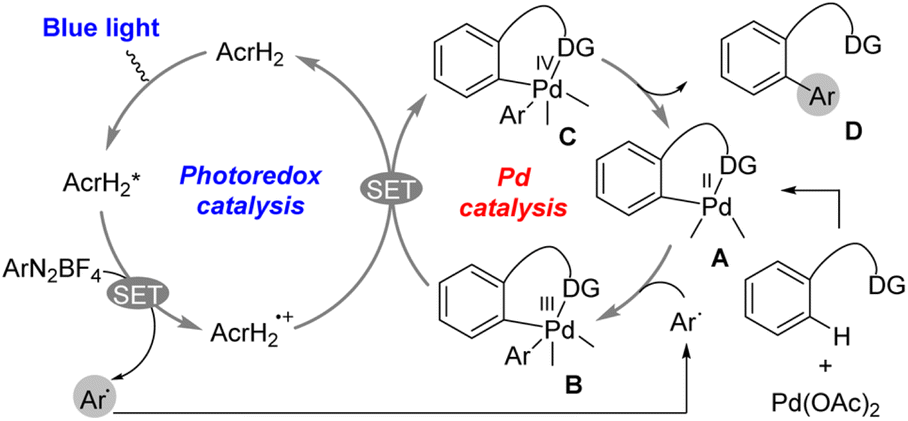
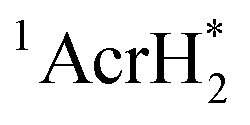 (−3.1 V vs. SCE).27 BNAH or BNA+ is used as a photoredox catalyst for hydrogenation of α,β-epoxy ketones and 1,2-diketones with HCO2H and Et3N to produce β-hydroxy and α-hydroxy ketones,32 because BNA+ can be converted to BNAH by a mixture of HCO2H/Et3N at room temperatures.33 The photocatalytic hydrogenation mechanism by BNAH is shown in Scheme 3. Firstly, electron transfer from 1BNAH* to α,β-epoxy ketone (compound 1 in Scheme 3) or benzil (i.e., diphenylethanedione; compound 3 in Scheme 3) occurs to produce BNAH˙+ and the radical anions of compounds 1 and 3 (species 5 and 6 in Scheme 3, respectively). Then, the carbon–oxygen bond cleavage occurs to convert species 5 (or species 6) into species 7 (or species 8), followed by proton transfer from BNAH˙+ to species 7 (or species 8) and an enol/keto tautomerization to produce species 9 (or species 10) and BNA˙ (Scheme 3). Because BNA˙ is a strong one-electron reductant,34 electron transfer from BNA˙ to species 9 (or species 10) to produce species 11 (or species 12), accompanied by regeneration of BNA+.33 The protonation of species 11 (or species 12) by water yields the final products (compounds 2 and 4 in Scheme 3).33
(−3.1 V vs. SCE).27 BNAH or BNA+ is used as a photoredox catalyst for hydrogenation of α,β-epoxy ketones and 1,2-diketones with HCO2H and Et3N to produce β-hydroxy and α-hydroxy ketones,32 because BNA+ can be converted to BNAH by a mixture of HCO2H/Et3N at room temperatures.33 The photocatalytic hydrogenation mechanism by BNAH is shown in Scheme 3. Firstly, electron transfer from 1BNAH* to α,β-epoxy ketone (compound 1 in Scheme 3) or benzil (i.e., diphenylethanedione; compound 3 in Scheme 3) occurs to produce BNAH˙+ and the radical anions of compounds 1 and 3 (species 5 and 6 in Scheme 3, respectively). Then, the carbon–oxygen bond cleavage occurs to convert species 5 (or species 6) into species 7 (or species 8), followed by proton transfer from BNAH˙+ to species 7 (or species 8) and an enol/keto tautomerization to produce species 9 (or species 10) and BNA˙ (Scheme 3). Because BNA˙ is a strong one-electron reductant,34 electron transfer from BNA˙ to species 9 (or species 10) to produce species 11 (or species 12), accompanied by regeneration of BNA+.33 The protonation of species 11 (or species 12) by water yields the final products (compounds 2 and 4 in Scheme 3).33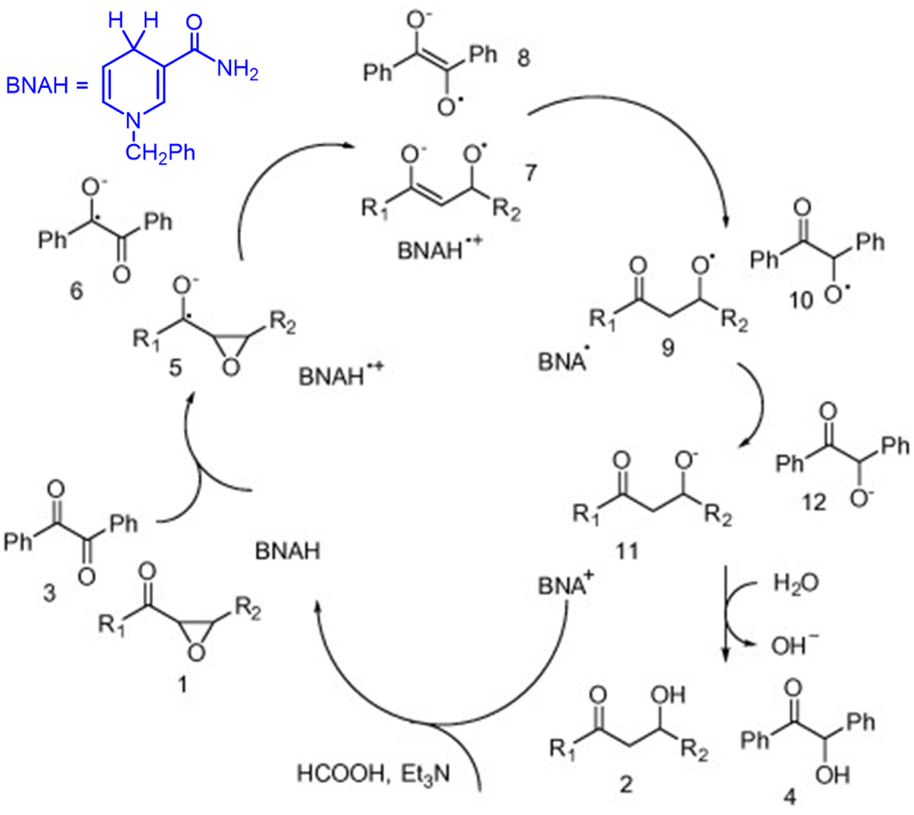
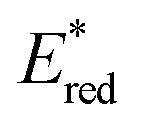 value of −3.2 V vs. SCE, which is comparable to some of the most oxidizable elemental metals such as Li (E vs. SCE = −3.3 V).42 In addition, *DCA˙− has a lifetime (τ) of 13.5 ns, which is long enough to undergo electron transfer reduction of a wide range of Ar–X compounds with Ered values as low as −2.94 V vs. SCE (Scheme 6).42 DCA˙− was produced using an H-type split cell with a porous carbon cathode and a Zn plate sacrificial anode by applying a constant cell voltage of 3.2 V.42 Photoirradiation of DCA˙− in the cathodic compartment with use of blue light enabled efficient coupling of 4-chlorobenzoate and B2pin3 (pin = pinacolato) as a radical acceptor to yield arylboronate (88%).42 This method has also been applied to the reductive borylation of 4-Cl-anisole, which has a very negative Ered value of −2.90 V vs. SCE.42 The mesolytic cleavage of a C–Br bond is significantly faster than that of a C–Cl bond, so that 1-Br-4-Cl-benzene was selectively converted to boronated–debrominated product.42
value of −3.2 V vs. SCE, which is comparable to some of the most oxidizable elemental metals such as Li (E vs. SCE = −3.3 V).42 In addition, *DCA˙− has a lifetime (τ) of 13.5 ns, which is long enough to undergo electron transfer reduction of a wide range of Ar–X compounds with Ered values as low as −2.94 V vs. SCE (Scheme 6).42 DCA˙− was produced using an H-type split cell with a porous carbon cathode and a Zn plate sacrificial anode by applying a constant cell voltage of 3.2 V.42 Photoirradiation of DCA˙− in the cathodic compartment with use of blue light enabled efficient coupling of 4-chlorobenzoate and B2pin3 (pin = pinacolato) as a radical acceptor to yield arylboronate (88%).42 This method has also been applied to the reductive borylation of 4-Cl-anisole, which has a very negative Ered value of −2.90 V vs. SCE.42 The mesolytic cleavage of a C–Br bond is significantly faster than that of a C–Cl bond, so that 1-Br-4-Cl-benzene was selectively converted to boronated–debrominated product.42
 of 2,3-di-Cl-5,6-di-CN-p-benzoquinone (3DDQ*) is 3.18 V vs. SCE, which is highly positive enough to oxidize benzene by electron transfer to produce benzene radical cation, which can react with nucleophiles such as OH− to yield phenol (Scheme 7).44 Although electron transfer from benzene (Eoxvs. SCE = 2.48 V) to DDQ at the ground state (Eredvs. SCE = 0.51 V) is highly endergonic, electron transfer from benzene to 3DDQ* becomes exergonic to proceed.44 Thus, photoirradiation of benzene together with DDQ and H2O resulted in formation of phenol and DDQH2 [eqn (14)].44tButyl nitrite (TBN) was used as a recycling reagent to oxidize DDQH2 to regenerate DDQ under aerobic conditions.44 Phenol was not further oxidized although electron transfer from phenol to 3DDQ* occurred with the diffusion-limited rate constant. Back electron transfer from DDQ˙− to benzene radical cation may be quite exergonic, when this process is in the Marcus inverted region,24,45,46 where the back electron transfer is slowed down as compared to the case of phenol radical cation, which decayed by fast back electron transfer prior to the reaction with H2O. Thus, the back electron transfer controls the selectivity of the reactions via photoinduced electron transfer.
of 2,3-di-Cl-5,6-di-CN-p-benzoquinone (3DDQ*) is 3.18 V vs. SCE, which is highly positive enough to oxidize benzene by electron transfer to produce benzene radical cation, which can react with nucleophiles such as OH− to yield phenol (Scheme 7).44 Although electron transfer from benzene (Eoxvs. SCE = 2.48 V) to DDQ at the ground state (Eredvs. SCE = 0.51 V) is highly endergonic, electron transfer from benzene to 3DDQ* becomes exergonic to proceed.44 Thus, photoirradiation of benzene together with DDQ and H2O resulted in formation of phenol and DDQH2 [eqn (14)].44tButyl nitrite (TBN) was used as a recycling reagent to oxidize DDQH2 to regenerate DDQ under aerobic conditions.44 Phenol was not further oxidized although electron transfer from phenol to 3DDQ* occurred with the diffusion-limited rate constant. Back electron transfer from DDQ˙− to benzene radical cation may be quite exergonic, when this process is in the Marcus inverted region,24,45,46 where the back electron transfer is slowed down as compared to the case of phenol radical cation, which decayed by fast back electron transfer prior to the reaction with H2O. Thus, the back electron transfer controls the selectivity of the reactions via photoinduced electron transfer.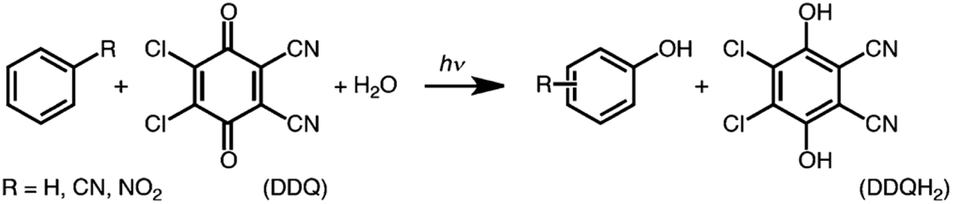


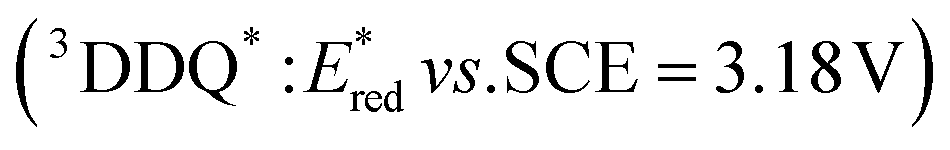 was reported for the excited state of a trisaminocyclopropenium radical dication
was reported for the excited state of a trisaminocyclopropenium radical dication 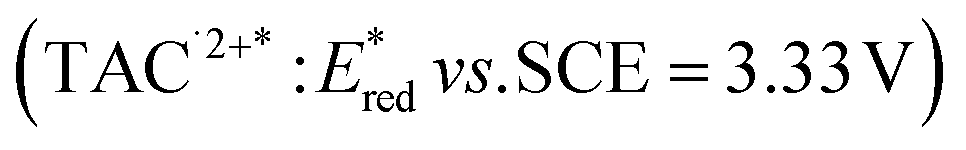 .49,50 TAC+ was electrochemically oxidized to form TAC˙2+, which was photoexcited with use of visible light to produce TAC˙2+* that is an excited-state species with sufficiently strong oxidizing power (3.33 V vs. SCE) to oxidize various substrates, including benzene and halogenated benzenes, via electron transfer, resulting in C–H/N–H coupling with azoles (Scheme 10).49,50 TAC˙2+* can also convert aryl olefins to the corresponding glycol monoesters with high chemo- and diastereoselectivity.51
.49,50 TAC+ was electrochemically oxidized to form TAC˙2+, which was photoexcited with use of visible light to produce TAC˙2+* that is an excited-state species with sufficiently strong oxidizing power (3.33 V vs. SCE) to oxidize various substrates, including benzene and halogenated benzenes, via electron transfer, resulting in C–H/N–H coupling with azoles (Scheme 10).49,50 TAC˙2+* can also convert aryl olefins to the corresponding glycol monoesters with high chemo- and diastereoselectivity.51
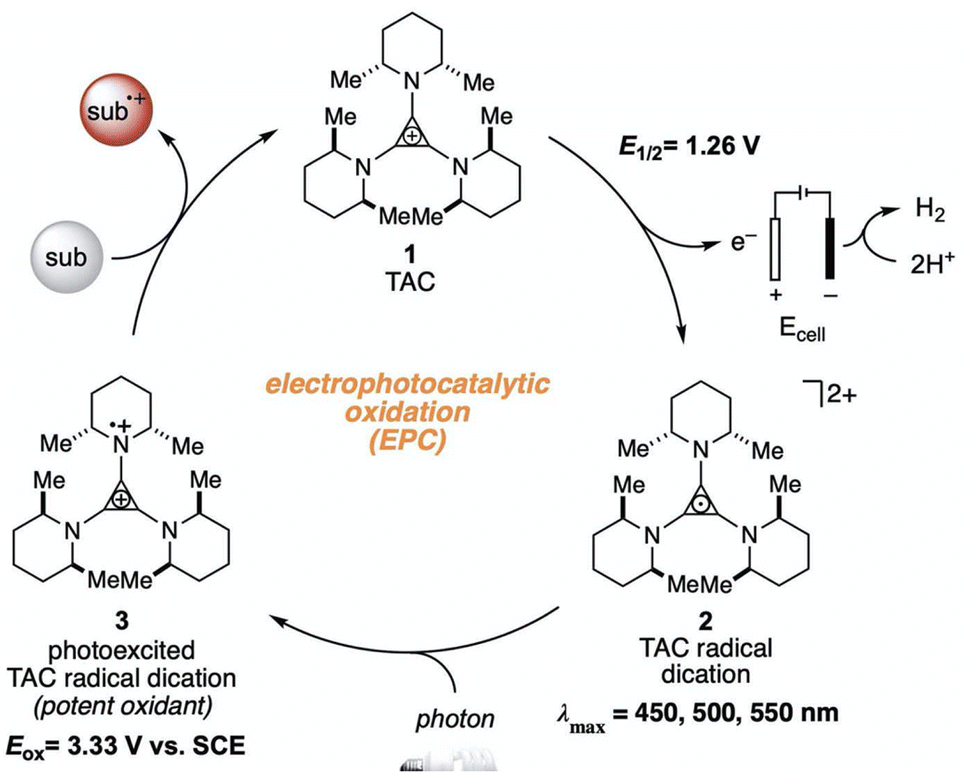
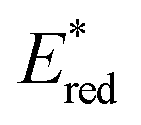 (vs. SCE) values of Fl-Mn+ complexes were in order of 1(Fl-2Sc3+)* (2.45 V) > 1(Fl-Yb3+)* (2.25 V) > 1(Fl-Mg2+)* (2.06 V) > 1Fl* (1.67 V), indicating that the order of
(vs. SCE) values of Fl-Mn+ complexes were in order of 1(Fl-2Sc3+)* (2.45 V) > 1(Fl-Yb3+)* (2.25 V) > 1(Fl-Mg2+)* (2.06 V) > 1Fl* (1.67 V), indicating that the order of 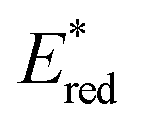 (vs. SCE) values of Fl-Mn+ complexes is in agreement of the order of the Lewis acidity of Mn+ bound to Fl.54 The
(vs. SCE) values of Fl-Mn+ complexes is in agreement of the order of the Lewis acidity of Mn+ bound to Fl.54 The  value of 1(Fl-2Sc3+)* is by 0.78 V more positive than that of 1Fl* (1.67 V) due to the strong binding of two Sc3+ ions to Fl˙−.53 Thus, 1(Fl-2Sc3+)* acts as a much stronger oxidant than 1Fl* as shown in Fig. 3.53 The combination of Fl with Sm3+ and iron complexes has also been reported to enable the visible-light driven aerobic C–H bond oxidation of alkyl benzenes.55,56
value of 1(Fl-2Sc3+)* is by 0.78 V more positive than that of 1Fl* (1.67 V) due to the strong binding of two Sc3+ ions to Fl˙−.53 Thus, 1(Fl-2Sc3+)* acts as a much stronger oxidant than 1Fl* as shown in Fig. 3.53 The combination of Fl with Sm3+ and iron complexes has also been reported to enable the visible-light driven aerobic C–H bond oxidation of alkyl benzenes.55,56
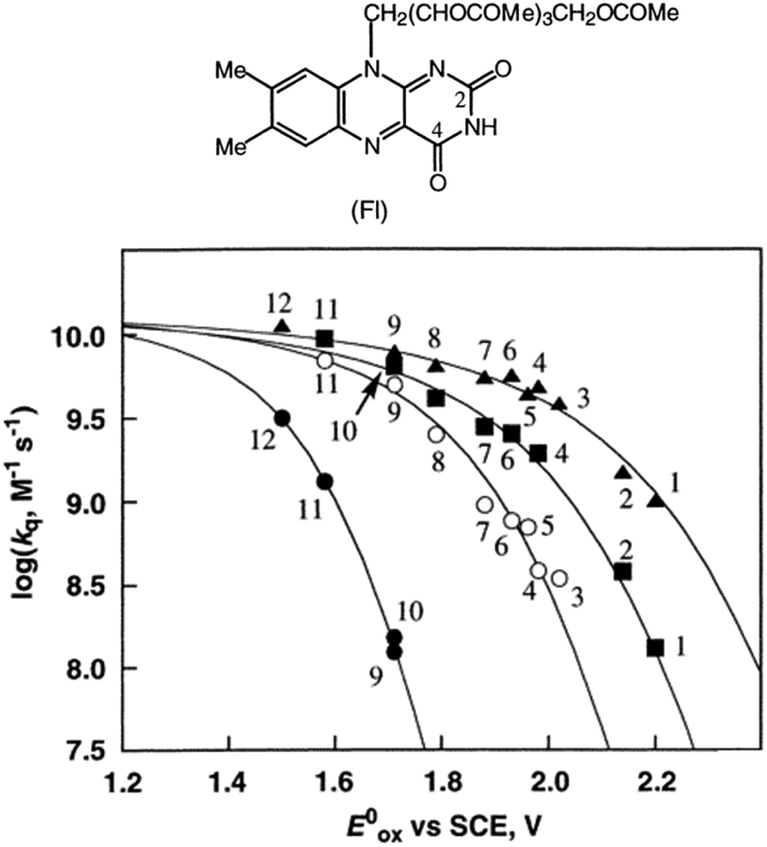

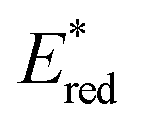 is the 1 e− reduction potential of the excited 2E state of MnIV-oxo intermediate and e is the elementary charge.60 The best fit line using eqn (15) in Fig. 4 afforded that
is the 1 e− reduction potential of the excited 2E state of MnIV-oxo intermediate and e is the elementary charge.60 The best fit line using eqn (15) in Fig. 4 afforded that  and the reorganization energy (λ) values of {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* are 2.1(1) V and 0.64(4) eV, respectively.60 The reorganization energy of electron transfer from electron donors to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* (0.64(4) eV) is much smaller than that from electron donors to the ground state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 (2.11 eV).58 Such a large difference in the λ values results from the small λ value for the ligand-center electron transfer from electron donors to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}*, which corresponds to the LMCT state, {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}*.60 In contrast, electron transfer from electron donors to the ground state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 occurs in the MnIV center with the large λ value because of the significant change in the bond distance of Mn–O between the MnIV and MnIII oxidation states.58,59
and the reorganization energy (λ) values of {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* are 2.1(1) V and 0.64(4) eV, respectively.60 The reorganization energy of electron transfer from electron donors to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* (0.64(4) eV) is much smaller than that from electron donors to the ground state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 (2.11 eV).58 Such a large difference in the λ values results from the small λ value for the ligand-center electron transfer from electron donors to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}*, which corresponds to the LMCT state, {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}*.60 In contrast, electron transfer from electron donors to the ground state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 occurs in the MnIV center with the large λ value because of the significant change in the bond distance of Mn–O between the MnIV and MnIII oxidation states.58,59
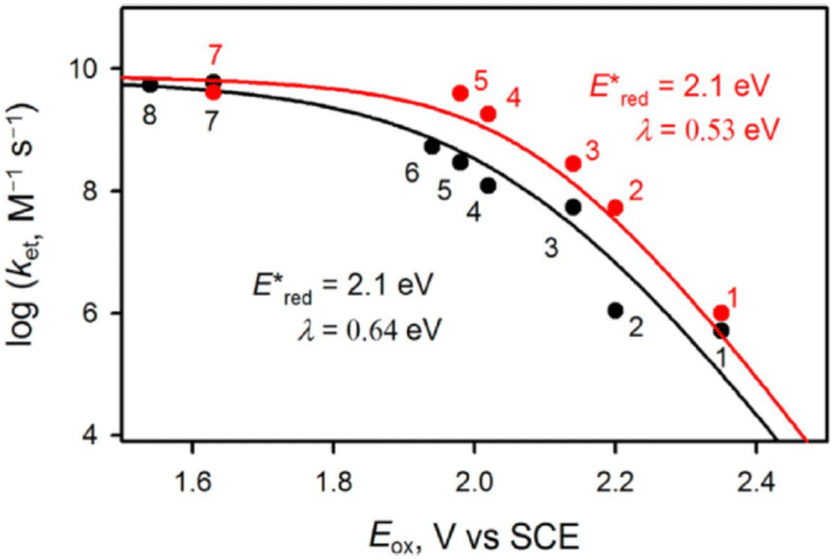
 value of the 2E excited state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 and the small λ value enabled electron transfer from benzene (C6H6) to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* to generate C6H6˙+,60 which forms (C6H6)2˙+ in the presence of large excess C6H6.64 C6H6˙+ reacts with H2O to produce the OH adduct radical, which is oxidized by [(BnTPEN)MnIII(O)]+-(Sc(OTf)3)2 to generate PhOH with MnII species after removal of H+ (Scheme 11A).60 MnII species reacts rapidly with [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 to form [MnIII–O–MnIII]4+ species (Scheme 11B).60
value of the 2E excited state of [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 and the small λ value enabled electron transfer from benzene (C6H6) to {[(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2}* to generate C6H6˙+,60 which forms (C6H6)2˙+ in the presence of large excess C6H6.64 C6H6˙+ reacts with H2O to produce the OH adduct radical, which is oxidized by [(BnTPEN)MnIII(O)]+-(Sc(OTf)3)2 to generate PhOH with MnII species after removal of H+ (Scheme 11A).60 MnII species reacts rapidly with [(BnTPEN)MnIV(O)]2+-(Sc(OTf)3)2 to form [MnIII–O–MnIII]4+ species (Scheme 11B).60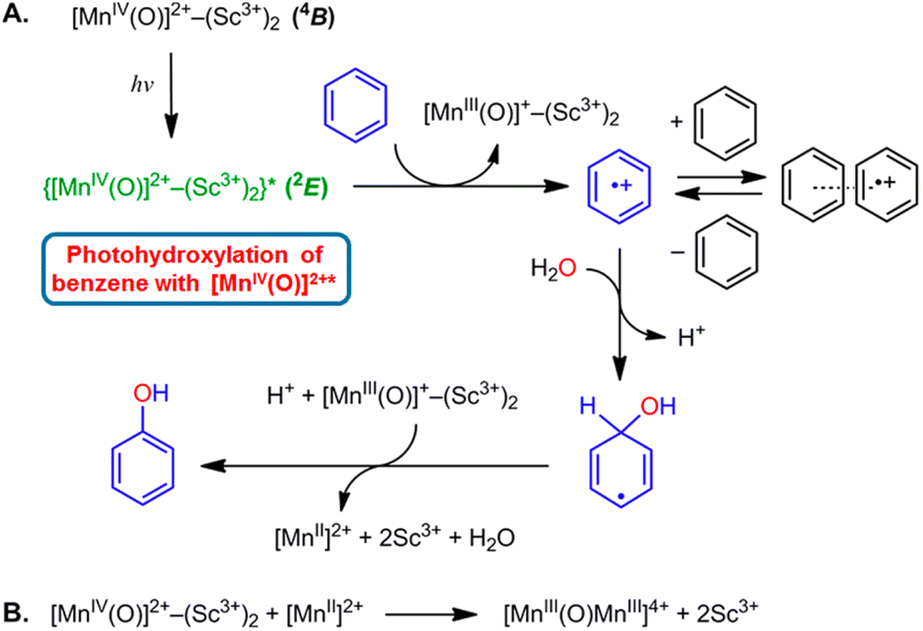

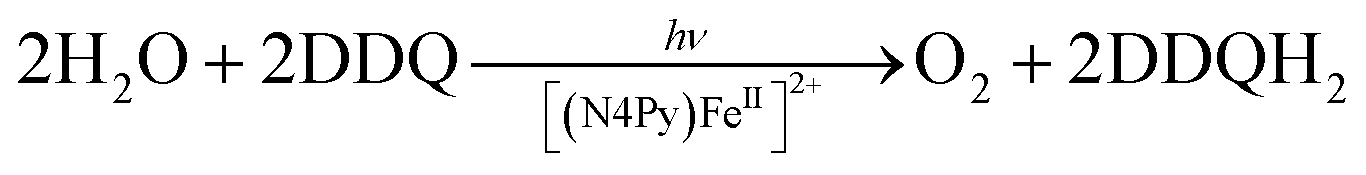
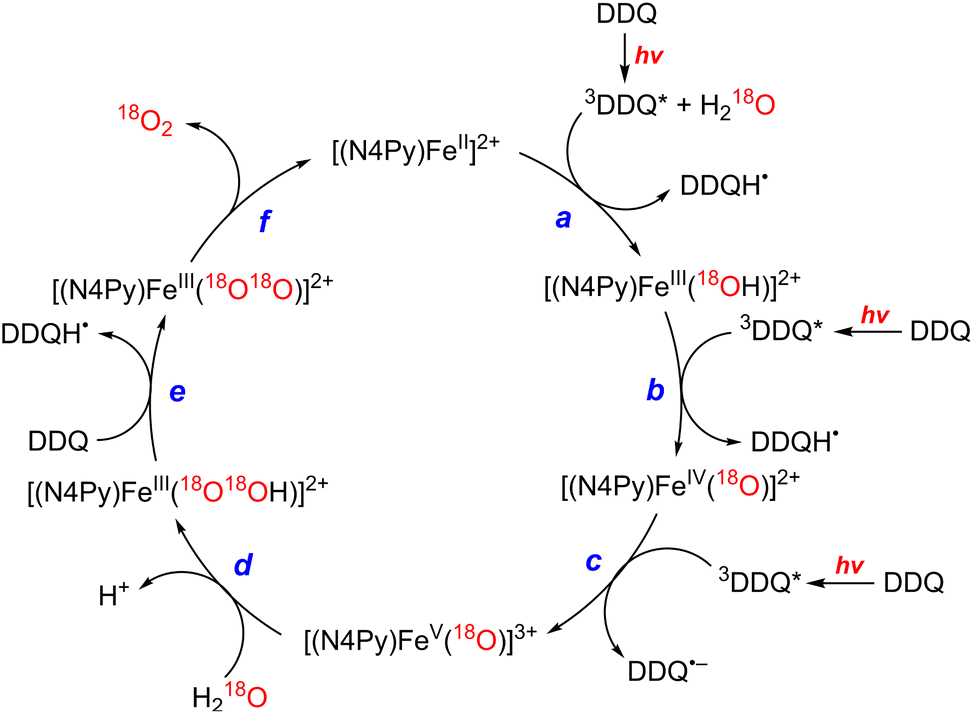
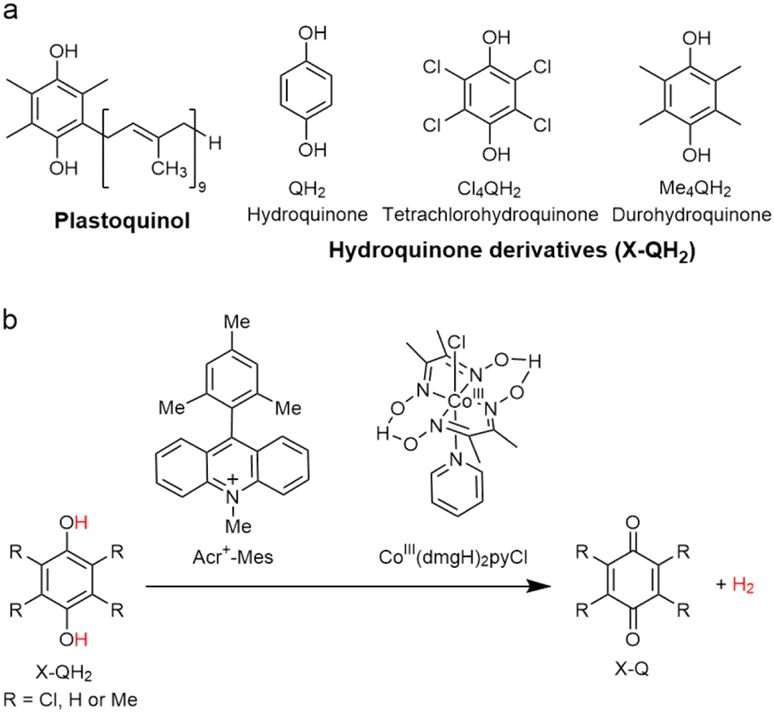
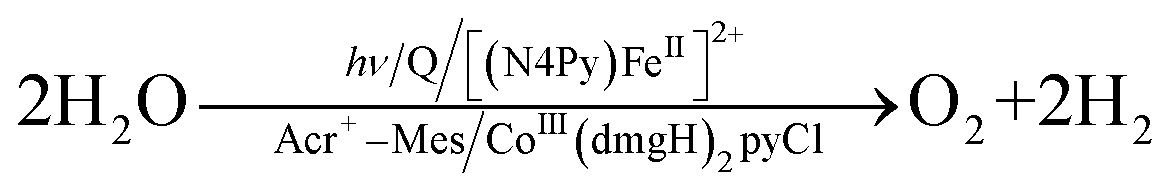
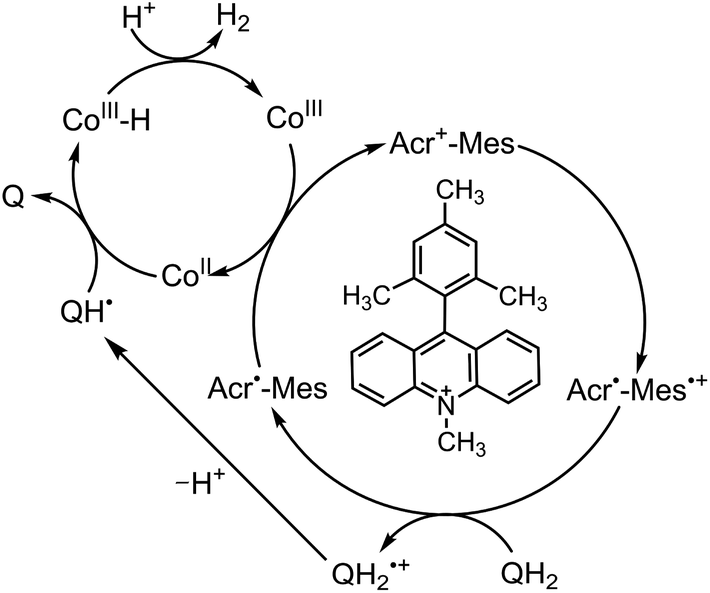
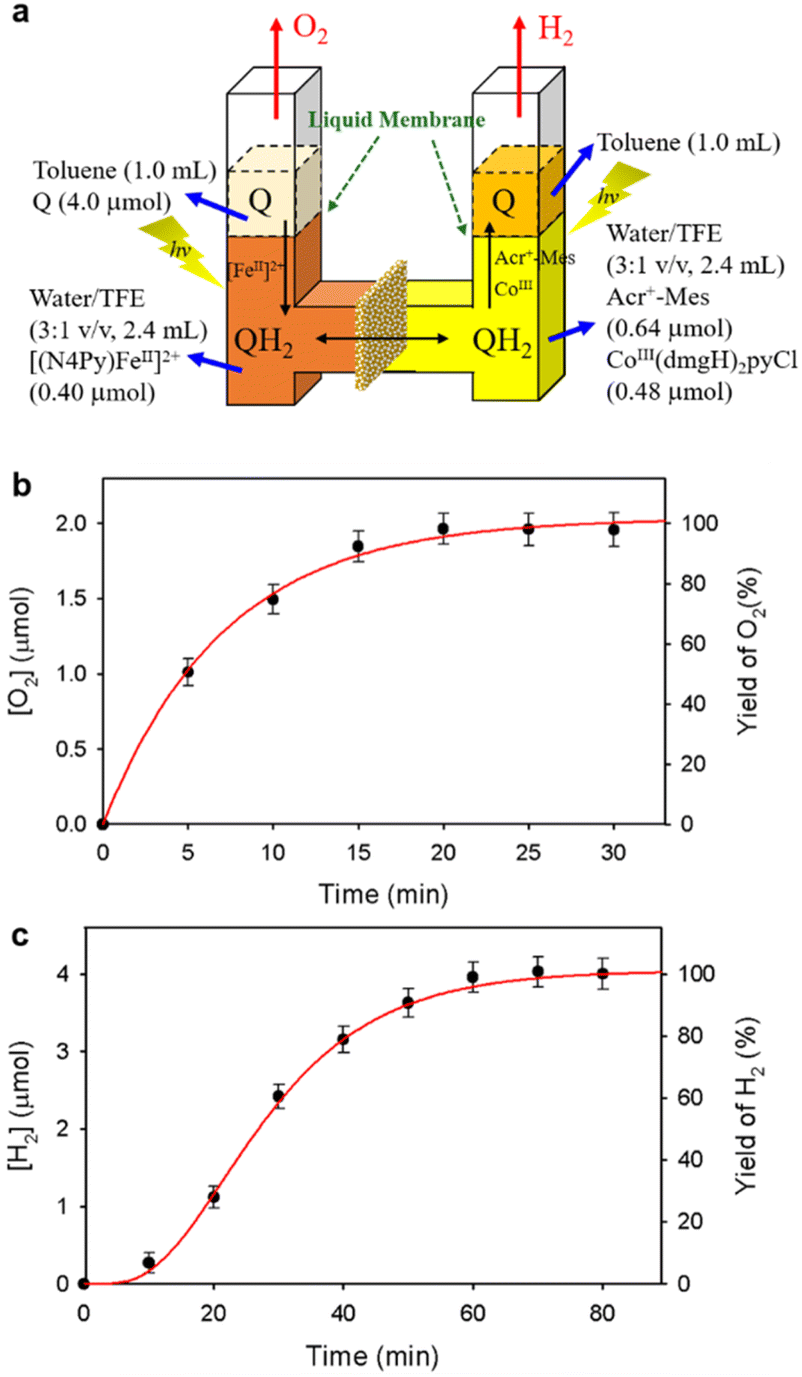
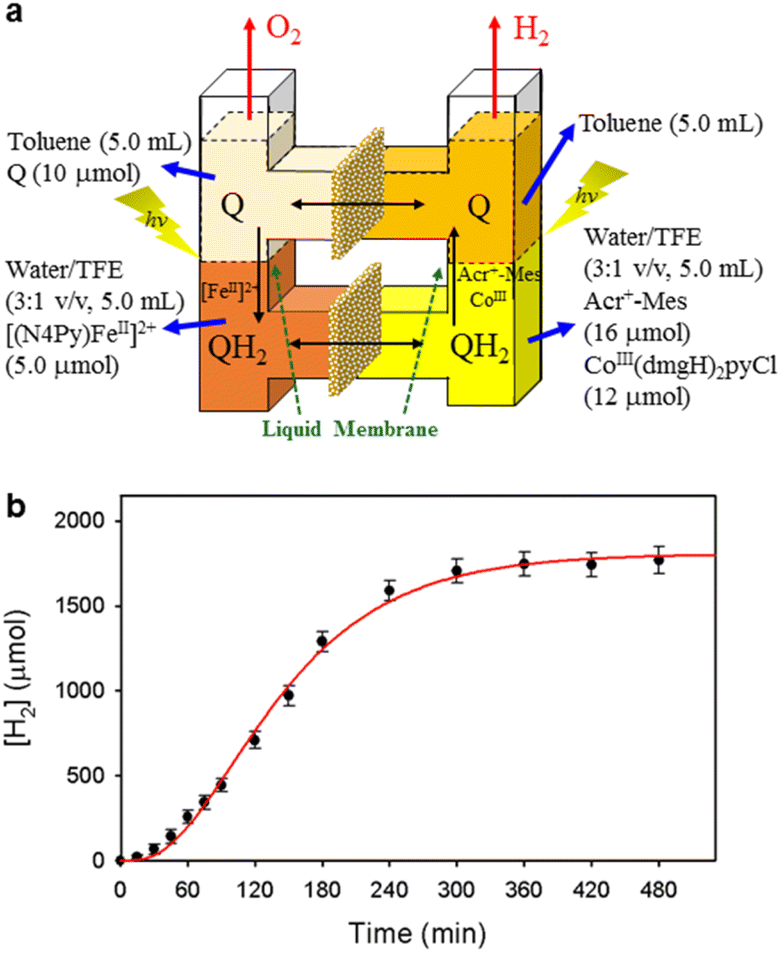
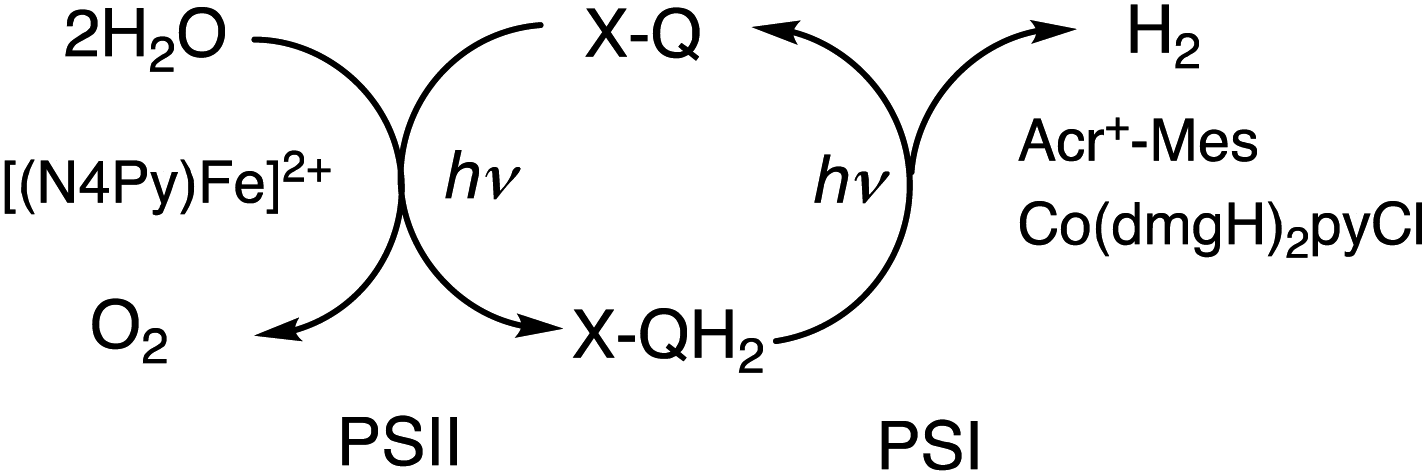
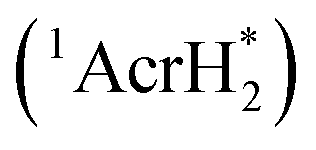 has a largely negative one-electron reduction potential
has a largely negative one-electron reduction potential  and a lifetime of 7.0 ns to enable to reduce various alkyl halides. The more negative
and a lifetime of 7.0 ns to enable to reduce various alkyl halides. The more negative 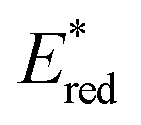 value (−3.36 V) is evaluated for the doublet excited state of stable acridinyl radical (Mes–Acr˙), which acts as a super-reductant. The excited states of radical anions and anions also act as super-reductants that enabled to reduce substrates, which are usually difficult to be reduced by the ground state reductants. By the same token, the excited states of organic electron acceptors such as DDQ and TAC˙2+ become much stronger electron acceptors as compared with the ground states, undergoing various oxidation reactions, which would otherwise be impossible to occur at the ground states. The binding of Sc3+ ions to flavin and Mn(IV)-oxo complexes enhanced the oxidizing ability. The excited state further enhanced the oxidizing ability, acting as super-oxidants.
value (−3.36 V) is evaluated for the doublet excited state of stable acridinyl radical (Mes–Acr˙), which acts as a super-reductant. The excited states of radical anions and anions also act as super-reductants that enabled to reduce substrates, which are usually difficult to be reduced by the ground state reductants. By the same token, the excited states of organic electron acceptors such as DDQ and TAC˙2+ become much stronger electron acceptors as compared with the ground states, undergoing various oxidation reactions, which would otherwise be impossible to occur at the ground states. The binding of Sc3+ ions to flavin and Mn(IV)-oxo complexes enhanced the oxidizing ability. The excited state further enhanced the oxidizing ability, acting as super-oxidants.