
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimeric tetrabromo-p-quinodimethanes: synthesis and structural/electronic properties†
Diego J.
Vicent‡
 a,
Manuel
Pérez-Escribano‡
b,
Abel
Cárdenas-Valdivia
c,
Ana
Barragán
d,
Joaquín
Calbo
b,
José I.
Urgel
d,
David
Écija
*d,
José
Santos
*a,
Juan
Casado
*c,
Enrique
Ortí
*b and
Nazario
Martín
*ad
a,
Manuel
Pérez-Escribano‡
b,
Abel
Cárdenas-Valdivia
c,
Ana
Barragán
d,
Joaquín
Calbo
b,
José I.
Urgel
d,
David
Écija
*d,
José
Santos
*a,
Juan
Casado
*c,
Enrique
Ortí
*b and
Nazario
Martín
*ad
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: jsantosb@pdi.ucm.es; nazmar@quim.ucm.es
bInstituto de Ciencia Molecular, Universidad de Valencia, 46980 Paterna, Spain. E-mail: enrique.orti@uv.es
cDepartamento de Química Física, Universidad de Málaga, 229071 Malaga, Spain. E-mail: casado@uma.es
dIMDEA-Nanociencia, C/ Faraday, 9, Campus de Cantoblanco, 28049 Madrid, Spain. E-mail: david.ecija@imdea.org
First published on 24th August 2023
Abstract
Despite their great potential as molecular building blocks for organic synthesis, tetrabromo-p-quinodimethanes (TBQs) are a relatively unknown family of compounds. Herein, we showcase a series of five derivatives incorporating two tetrabromo-anthraquinodimethane (TBAQ) units linked by π-conjugated spacers of different nature and length. The resulting dimers TBQ1–5 are fully characterised by means of thorough spectroscopic measurements and theoretical calculations. Interestingly, owing to the steric hindrance imposed by the four bulky bromine atoms, the TBAQ fragments adopt a characteristically warped geometry, somehow resemblant of a butterfly, and the novel dimers show a complex NMR pattern with signal splittings. To ascertain whether dynamic processes regarding fluxional inversion of the butterfly configurations are involved, first-principles calculations assessing the interconversion energy barriers are performed. Three possible stereoisomers are predicted involving two diastereomers, thus accounting for the observed NMR spectra. The rotational freedom of the TBAQ units around the π-conjugated linker influences the structural and electronic properties of TBQ1–5 and modulates the electronic communication between the terminal TBAQ moieties. The role of the linker on the electronic properties is investigated by Raman and UV-vis spectroscopies, theoretical calculations and UV-vis measurements at low temperature. TBQ1–5 are of interest as less-explored structural building precursors for a variety of scientific areas. Finally, the sublimation, self-assembly and reactivity on Au(111) of TBQ3 is assessed.
Introduction
Bottom-up synthetic approaches for preparing complex organic structures can be highly time and resources consuming. Thus, organic chemists tend to make use of pre-made molecular fragments, modularly assembling them into more complex structures. These so-called molecular building blocks are usually endowed with different reactive functional groups supported onto a scaffold of low-to-medium complexity. The use of appropriate building blocks, therefore, allows speeding up the synthetic tasks as well as decreasing the synthetic costs.Molecules bearing gem-dibromoolefine functionalities have been employed as intermediates in the well-known Ramirez–Corey–Fuchs homologation of carbonyls to alkynes.1,2 They are readily attained from either ketones or aldehydes by reacting with carbon tetrabromide and triphenylphosphine. Upon lithiation, gem-dibromoolefines undergo Fritsch–Buttenberg–Wiechell (FBW) rearrangement to yield the corresponding alkyne.3,4 Besides their use in FBW rearrangements, this functionality has found use in the synthesis of [4]radialene.5 Nonetheless, it is as substrates for palladium cross-coupling reactions where gem-dibromoolefines have found a broader use, allowing Suzuki,6 Stille7 and Sonogashira couplings.8 When the gem-dibromoolefines are stemming from p-quinones, the resulting tetrabromo-p-quinodimethanes (TBQs) may undergo dehalogenative homocoupling on coinage metal surfaces, namely Au(111) and Ag(111), forming one-dimensional (1D) acene and periacene polymers that may exhibit appealing non-trivial topological electronic properties, depending upon the acene nature and length of the polymer.9–13
As building blocks bearing four reactive bromine atoms, TBQs are excellent platforms for the incorporation of up to four substituents. This high degree of functionalisation is ideal for some organic electronics applications, as in the case of hole transporting materials (HTMs) for perovskite solar cells (PSCs), where the presence of several electroactive arylamine groups strongly enhances the charge transport. For instance, when triarylamine functionalities are inserted into an anthraquinone-based TBQ (TBAQ), the resulting HTM exhibits high photovoltaic performance and stability in large area PSCs.14 The steric hindrance of the anthracene peri hydrogens allows conceiving molecules with switchable geometry, which makes TBQs ideal platforms for synthetising materials with aggregation-induced emission (AIE) and photochromism properties.15–17 If the core of the TBQ is a pentacene, then the substitution with phenyl derivatives yields a structure that is prone to undergo a cyclodehydrogenation reaction to provide hexabenzocoronenes, which have potential as semiconducting materials for field-effect transistor applications.18 Another example of their potential in materials science is found in molecules with self-assembly capabilities.19
Regarding their chemical structure, TBQ building blocks can be considered as precursors for the synthesis of π-extended analogues of Thiele's and Chichibabin's hydrocarbons. Owing to their quinoid character, they have a strong tendency to stabilise open-shell chemical structures when suitably substituted.20,21 For example, the deposition of a bipentacene TBQ on a Au(111) surface allowed the synthesis of peripentacene and the unequivocal characterization of its open-shell character.22
The versatility of the TBQ functionality as building block for preparing a wide variety of molecules of high interest in materials science and organic electronics prompted us to design, synthesise and expand the spectrum of TBQ derivatives by preparing a series of anthracene-based TBQ dimers (Scheme 1). The newly synthesised TBQ1–5 molecules are endowed with eight bromine atoms located on the two anthra-p-quinodimethane units which are, in turn, covalently connected by different spacers that modulate the conjugation and/or electronic communication between them. The structural, electronic, optical and vibrational experimental properties of the TBAQ building block and of the TBQ dimers were jointly studied with first-principles calculations and spectroscopic techniques to deepen into the understanding of their intramolecular characteristics. Furthermore, the sublimation, self-assembly and reactivity of a TBQ archetype (TBQ3) was explored on Au(111). Altogether, our findings could eventually seed supramolecular and material properties based on these high-bromine functional molecules.
 | ||
| Scheme 1 Synthetic route to the dimeric tetrabromo-p-quinodimethanes TBQ1–5. | ||
Results and discussion
Synthesis and structural properties
The synthesis of TBQ-based building blocks can be achieved starting from relatively unexpensive products (Scheme 1). Reaction of 4,4′-oxydiphthalic anhydride (1) with AlCl3 in benzene, followed by treatment of the resulting product with polyphosphoric acid (PPA) provides anthraquinone dimer 2 in good yield.23 To obtain TBQ1, standard Ramirez olefination conditions employing CBr4 and PPh3 were applied,1 although the yields were low to moderate. On the other hand, to synthesise TBQ2–5 the key starting material was 2-bromoanthraquinone (3), which may be purchased from commercial retailers or readily attained from bromobenzene and phthalic anhydride. Product TBQ2 is obtained starting from 3 in two steps. First step involves the one-pot two-fold Sonogashira cross-coupling of trimethylsilylacetylene with two molecules of 3, enabled by the use of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as base, with moderate yield. When the resulting acetylene-bridged anthraquinone dimer (4) is exposed to standard Ramirez olefination conditions, inseparable complex reaction mixtures were obtained. The study of the crudes by mass spectroscopy revealed peaks with isotopic profiles compatible with molecules carrying various bromine atoms. Following a previous study by M. Lauten and coworkers,24 where phosphites demonstrated enhanced reactivity towards ketones compared to phosphines, tri-iso-propylphosphite [P(OiPr)3] was employed. Use of 2 and 4 equivalents of CBr4 and P(OiPr)3, respectively, per carbonyl equivalent, led to partial olefination products of 4, although no complex mixtures were observed. Full conversion of the starting material to TBQ2 was finally achieved by adding three extra loads of CBr4 and P(OiPr)3.To synthesise TBQ5 from precursor 6, featuring a butadiyne bridge, 2-ethynylanthraquinone was quantitatively homocoupled via Hay coupling. For preparing TBQ4 from precursor 7, the starting material 3 was coupled with 1,4-diethynylbenzene by Sonogashira reaction. Finally, precursor 8 used to obtain TBQ3 was prepared by Suzuki reaction of 3 with 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene. Final products TBQ3–5 were obtained following the same procedure as for TBQ2 (Scheme 1). The FTIR spectra of TBQ1–5 evidence the lack of the characteristic carbonyl band of the precursing quinones, along with the arising of an intense sharp band at ca. 760 cm−1 ascribed to the out-of-plane bending of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CBr2 moieties. Featuring eight bromine atoms per molecule, TBQ1–5 show a very characteristic isotopic profile (see Fig. S13–S19†).
CBr2 moieties. Featuring eight bromine atoms per molecule, TBQ1–5 show a very characteristic isotopic profile (see Fig. S13–S19†).
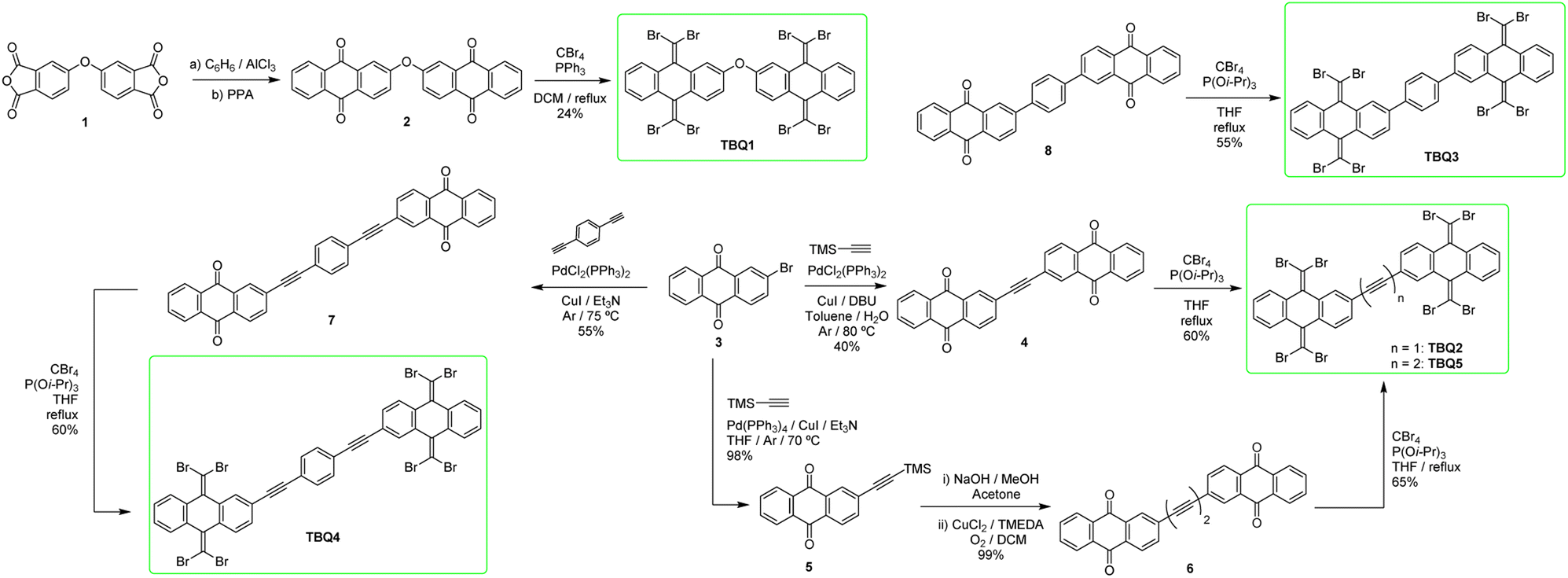
Prior to theoretically characterizing the high-bromine TBQ1–5 dimers, the structure of the TBAQ building block was inspected and compared with that computed for the widely studied cyano-based tetracyanoanthra-p-quinodimethane (TCAQ) analogue. Geometry optimizations were performed within the density functional theory (DFT) framework at the BMK/6-31G(d,p) level of theory in gas phase (see the Computational methods section in the ESI† for full computational details and methodology benchmark). In the case of TCAQ, the fully planar D2h conformation is a high-order saddle point with four imaginary vibrational modes (see Section S6 in the ESI†).25–27 When considering TBAQ, the D2h conformation shows up to five imaginary vibrational frequencies (Section S7†). Starting from this planar structure, the search for the global minimum-energy conformer was performed by distorting the molecular geometry along the imaginary vibrational modes with symmetry constraints (Fig. 1). Three conformations, two of them equivalent to each other and possessing a C2h point group of symmetry (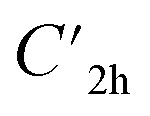 and
and 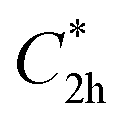 ), and the D2 conformation, are transition states and a high-order saddle point, respectively, that further present one or two imaginary frequencies. These three structures are calculated ca. 20 kcal mol−1 more stable than the planar D2h molecular geometry. From the D2 conformation, a local minimum-energy, chair-like structure is predicted with no imaginary frequency and C2h symmetry, which is computed 31.3 kcal mol−1 more stable than the D2h conformer (Fig. 1). Finally, the global minimum-energy structure, displaying a saddle- or butterfly-like disposition, belongs to the C2v point group of symmetry, and is calculated 53.8 kcal mol−1 lower in energy than the planar D2h structure. Noticeably, the butterfly inversion through D2h is highly unfavorable for TBAQ (53.8 kcal mol−1) compared to TCAQ (22.3 kcal mol−1). However, as the intermediate transition states (
), and the D2 conformation, are transition states and a high-order saddle point, respectively, that further present one or two imaginary frequencies. These three structures are calculated ca. 20 kcal mol−1 more stable than the planar D2h molecular geometry. From the D2 conformation, a local minimum-energy, chair-like structure is predicted with no imaginary frequency and C2h symmetry, which is computed 31.3 kcal mol−1 more stable than the D2h conformer (Fig. 1). Finally, the global minimum-energy structure, displaying a saddle- or butterfly-like disposition, belongs to the C2v point group of symmetry, and is calculated 53.8 kcal mol−1 lower in energy than the planar D2h structure. Noticeably, the butterfly inversion through D2h is highly unfavorable for TBAQ (53.8 kcal mol−1) compared to TCAQ (22.3 kcal mol−1). However, as the intermediate transition states (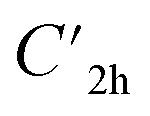 ,
, 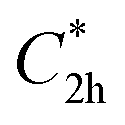 and D2) connect with the C2v global minimum, the butterfly shape of TBAQ can be inverted with an activation barrier of 33.3 kcal mol−1, to be compared with a value of 18.4 kcal mol−1 for TCAQ. Nonetheless, the butterfly interconversion process is still significantly impeded at room temperature.
and D2) connect with the C2v global minimum, the butterfly shape of TBAQ can be inverted with an activation barrier of 33.3 kcal mol−1, to be compared with a value of 18.4 kcal mol−1 for TCAQ. Nonetheless, the butterfly interconversion process is still significantly impeded at room temperature.
 | ||
| Fig. 1 Relative electronic energy (a) and molecular structure (b) of the different conformations localized at the BMK/6-31G(d,p) level on the potential energy surface of TBAQ. In (a), the number of imaginary frequencies of each structure is indicated within parentheses, and the arrows indicate the evolution of the structure upon deformation along the imaginary modes. In (b), side (left) and top (right) views are displayed. Color coding: H in white, C in gray and Br in yellow. | ||
To gain insight into the structural changes driving the relative energy difference between the C2v global minimum-energy structure and the planar D2h transition state for TBAQ in comparison to TCAQ, we investigated the two main structural deformations that follow the interconversion from D2h to C2v; i.e., the planar-to-butterfly change and the widening of the Br–C–Br and NC–C–CN bond angles (see Section S8† for details). Theoretical calculations at the BMK/6-31G(d,p) level indicate that the change from planar to butterfly shape, in going from D2h to C2v, leads to a net stabilization of −37.4 and −10.7 kcal mol−1 for TBAQ and TCAQ, respectively. The larger stabilization observed for TBAQ is consistent with the presence of the bulkier Br atoms, compared to the cyano groups of TCAQ, as the electron localization function evidences (Fig. S25†).28 In line with the previous reasoning, in TBAQ the broadening of the Br–C–Br angle from 98° in D2h to 124° in C2v provides a larger stabilization (−16.1 kcal mol−1) than the relaxation of the NC–C–CN angle of TCAQ (from 97° in D2h to 111° in C2v with a stabilization of −11.1 kcal mol−1), thus supporting the larger steric interactions between Br atoms.
The 1H and 13C magnetic resonances of the TBQ1–5 dimers reveal interesting structural aspects of these molecular systems (Fig. S3–S12†). Considering the substitution pattern of the phenyl rings through which the anthracenes are linked by (Scheme 1), the 1H-NMR spectra should show three sets of signals integrating for one proton each: the lone peri proton (position C1) showing as a doublet, coupled with a small 4J coupling constant to the proton in meta (position C3); the latter appearing as a double doublet due to the coupling with the proton in ortho to it (position C4); this last proton showing itself as a doublet. Instead, some of these signals split in some of the TBQs, i.e., H–C1 and H–C3 of TBQ1 (Fig. S3†) and TBQ2 (Fig. 2). In the case of these dimers bearing shorter linkers, the proximity of both TBAQ subunits seems to create different magnetic environments for the above-mentioned protons. As larger spacers are introduced, e.g. in TBQ5, the splitting effect tends to vanish, as both TBAQs stop magnetically affecting each other (see Fig. 2). The splitting effect is also observed in the case of the 13C-NMR signals, mainly concerning the signal of the carbon nuclei involved in the alkyne fragment and the ones close to it (C1, C2 and C3) (see Fig. S4, S6 and S9†).
 | ||
| Fig. 2 Comparison of the 1H-NMR spectra of TBQ2 and TBQ5 showing the splitting of the peri hydrogen signal of the former, consistent with the presence of two isomeric species. | ||
In order to assess the possibility of having rotamers with different butterfly-to-butterfly relative orientations (same vs. opposite orientation with respect to the molecular plane) that may explain the NMR signal splitting described above, the energy barrier for single-bond TBAQ rotation was evaluated for TBQ1–5 dimers at the BMK/6-31G(d,p) level including solvent conditions (CH2Cl2) (see Section S9 of the ESI† for further details). Fig. 3 displays the rotational barrier profile calculated for TBQ2 as a representative example (see Fig. S26–S29† for the other dimers). Theoretical calculations indicate that the butterfly rotation along dihedral angle θ is accessible at room temperature, as torsion barriers in the range of 0.31–3.47 kcal mol−1 are predicted for the highly containing bromine dimers (Table S2†). The largest energy barriers are predicted for TBQ1 and TBQ3 due to the evident H⋯H steric hindrance upon rotation. Analysis of the Br⋯H–C1 distance (Fig. S30†) confirms a significant change in length upon butterfly rotation (Table S3†). This distance is significantly shorter for TBQ1 and TBQ2, achieving minimum values of 5.12 and 6.17 Å, respectively, which could explain the 1H-NMR signal splitting recorded experimentally for the H–C1 peri proton in these derivatives bearing shorter linkers (Fig. 2).
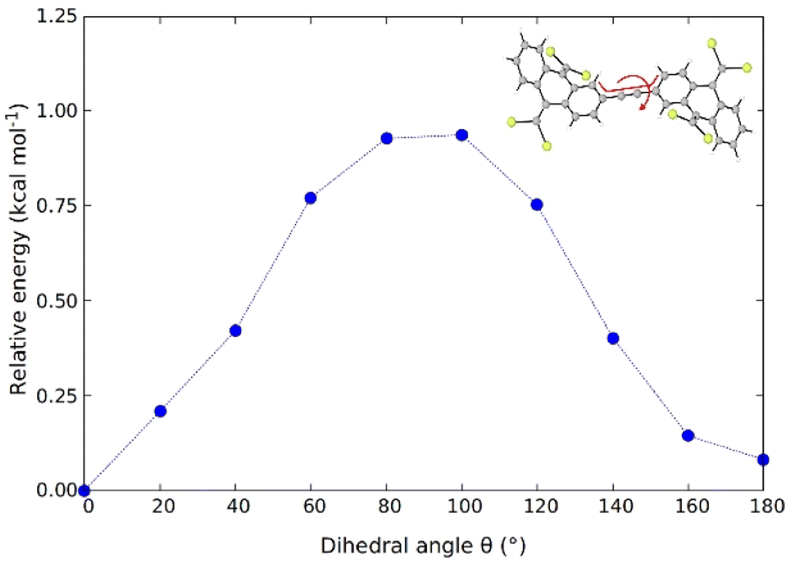 | ||
| Fig. 3 Rotational energy barrier profile calculated for TBQ2 at the BMK/6-31(d,p)+PCM(CH2Cl2) level along the torsion of the dihedral angle θ (in red) defining the relative orientation of the TBAQ moieties. | ||
A detailed analysis of the possible configurational isomers of TBQ2 (Fig. S1†) reveals the presence of three possible isomers, i.e., a pair of enantiomers and a meso form. Attempts to isolate the diastereomers by chromatographic means were to no avail due to the limited solubility of TBQ2 in the eluting mixture and the negligible chromatographic separation between diastereomers. However, 1H-NMR spectra of the collected fractions show diminished intensity for one of the doublets constituting the split signal of the H–C1 peri proton (see Fig. S12†), as would be expected from a fraction enriched in one of the diastereomers.
Vibrational spectroscopic properties
The solid-state Raman spectra of the TBQ dimers together with that recorded for TBAQ are shown in Fig. 4a. The BMK/6-31G(d,p) Raman spectrum simulated for TBQ2 compares very well with the experimental spectrum in the whole spectral region, as may be observed in Fig. S63,† allowing to make a vibrational assignment of the experimental bands in terms of the main components of the vibrational normal modes (Fig. S64 and S65†). The experimental bands of TBQ2 at 1601 and 1593 cm−1 can be correlated with the theoretical bands at 1593 and 1579 cm−1, respectively. The two modes are composed by the CC stretching modes [ν(CC)] of the anthraquinone core with CC displacements featuring the breathing mode of the six-member rings (i.e., vibrational normal modes 1 and 3 of benzene, see Fig. 4b) and coupled with the outermost methylene CC stretches of the CCBr2 moieties, or ν(CC)mode1-3 + ν(CCBr2). On the other hand, the experimental bands of TBQ2 at 1582 and 1561 cm−1 correspond to those theoretically predicted at 1569 and 1553 cm−1, respectively, and can be described as CC stretching modes carrying out the 8a mode of benzene in which the bonds of the central six-member ring, which are parallel to the external methylene double bonds, simultaneously stretch, or ν(CC)mode8 + ν(CCBr2). The BMK/6-31G(d,p) theoretical Raman spectra calculated for TBAQ at the C2v, C2h and D2h geometries in Fig. 1 (Fig. S66†) reveals that the ν(CC) + ν(CCBr2) bands experience a progressive increment of their wavenumbers upon sequential C2v → C2h → D2h planarization of the central anthracene core.
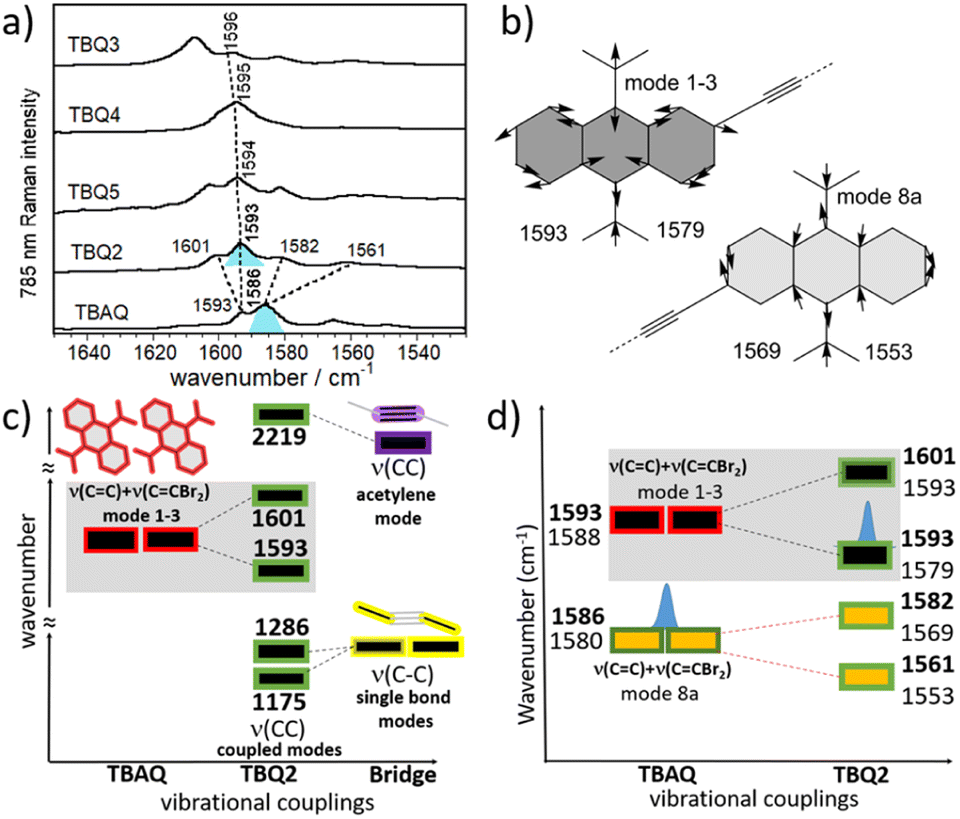 | ||
| Fig. 4 (a) Raman spectra of the compounds in solid state at 298 K, employing a 785 nm laser (blue-shaded bands are those mainly discussed in the text). (b) BMK/6-31G(d,p) theoretical vibrational ν(CC) modes (wavenumbers in cm−1) of TBQ2 showing the main CC displacements and related to the modes 1, 3 and 8a of benzene. (c) Wavenumber evolution of the ν(CC) vibrational modes (mode 1–3 of benzene) from the TBAQ units (in red) upon vibrational coupling with the central ν(CC) acetylene spacer (in purple) and with the ν(CC) of the single connecting bonds (in yellow). Vibrational splittings are qualitative. (d) Vibrational splitting of the 1–3 and 8a modes simultaneously from TBAQ to TBQ2. Experimental values are shown in bold (blue-shaded bands are those equally denoted in the experimental spectra). | ||
The experimental vibrational Raman intensity patterns of TBQ2 and TBAQ in the 1600 cm−1 region are apparently very similar, the wavenumbers of the corresponding bands showing an overall upshift in passing from TBAQ to TBQ2 (Fig. 4a). For instance, the strongest Raman band at 1586 cm−1 in TBAQ shifts to 1593 cm−1 in TBQ2 (blue-shaded in Fig. 4a). This strong band keeps slightly upshifting in the dimers, up to by +3 cm−1 on TBQ2 → TBQ3. There are two possible reasons behind these wavenumber upshift upon dimerization: (i) the planarization of the anthracene core, as observed for TBAQ along C2v → C2h → D2h symmetry transformations (Fig. S66†), and (ii) the role of the linker in the dynamics and electronic structure of the TBAQ units upon formation of TBQ2. Comparison of the optimized minimum-energy geometries calculated for the TBAQ molecule and the TBAQ subunits within the dimers indicates a negligible planarization of the butterfly moiety in TBQ1–5, with root-mean-square deviations of the atomic positions in the dimers with respect to TBAQ smaller than 0.01 Å and a folding of the anthracene skeleton differing in only 0.2° (Table S4†). On the other hand, the vibrational couplings suggested in point (ii) are qualitatively presented in Fig. 4c and d (and Fig. S64 and S65†), where the evolution of the ν(CC) modes upon assembly and subsequent vibrational mixing of two TBAQ monomers with a central acetylene (and with two CC single bonds) is shown. The 1–3 and 8a benzenoid-like modes of TBAQ are calculated at 1588 and 1580 cm−1, respectively, in good correlation with the Raman peaks observed at 1593 and 1586 cm−1. Interestingly, in the theoretical Raman spectra of TBAQ and TBQ2, the most active Raman bands correspond to modes of different vibrational nature, the 8a mode in TBAQ at 1580 cm−1 and the 1–3 mode in TBQ2 at 1579 cm−1. This change in the vibrational structure of the strongest Raman bands reveals the effect of the dimerization in the vibrational spectra, which modifies the electronic polarizability tensor in such a way that its largest variation between TBAQ and TBQ2 occurs in different vibrational modes. Elongation of the bridge between the two TBAQ units in compounds TBQ3–5 slightly modifies the vibrational mixing and similar frequencies are detected in the Raman spectra. Conversely, vibrational bands of the same nature are found to downshift upon dimerization, what is in accordance with the common finding that the Raman wavenumbers always decrease with the increment of π-conjugation in oligomeric series.29–31
Fig. 5 displays the normalized infrared and Raman spectra of TBAQ and TBQ2 in the spectral regions where the strongest bands are measured, i.e., around 1600 cm−1 for the ν(CC) modes in the Raman spectrum and at 750 cm−1 for the ν(CBr) modes in the infrared spectrum (theoretically compared in Fig. S67 and S68†). In general, for the two molecules under comparison and concerning their vibrational bands in common (i.e., those of TBAQ), it can be seen that each strong Raman band around 1600 cm−1 has a weak infrared analogue feature. Conversely, in the region around 750 cm−1, each intense infrared band has a corresponding weak Raman feature. This reveals that all these vibrations display non-vanishing infrared and Raman intensities, which is in agreement with a C2v molecular symmetry in TBAQ and a local C2v conformation for the TBAQ units in TBQ2 as well. Nonetheless, it is observed in Fig. 5 that every Raman (infrared) active mode of TBQ2 is much less active in the infrared (Raman) (dotted lines correlate vibrational modes in the Raman and infrared spectra) than the corresponding signals in TBAQ. This observation might be indicative of a symmetry alteration upon formation of the dimer, whose molecular structure evolves closer to a form with an inversion centre around the central acetylene bridge.
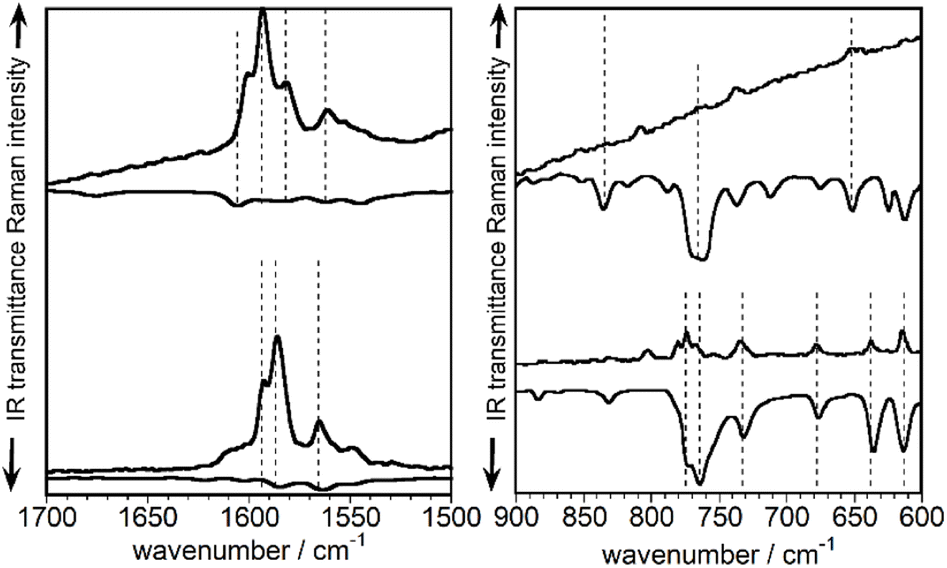 | ||
| Fig. 5 Comparison between the 785 nm solid-state laser Raman (pointing up) and the infrared (pointing down) spectra of TBAQ (below) and TBQ2 (above) at 298 K, normalized to the strongest Raman and infrared bands. | ||
Electronic structure and optical properties
The electronic structure of the high-bromine dimers TBQ1–5 was studied at the DFT-BMK/6-31G(d,p) level in dichloromethane. Theoretical calculations indicate that the highest-occupied molecular orbital (HOMO) is mainly delocalized over the bridge and the inner benzene ring of the anthraquinone moieties (see Fig. S31†). Similarly, the lowest-unoccupied molecular orbital (LUMO) is centred over the bridge and extends to the TBAQ moieties, especially for the less conjugated TBQ1 and TBQ3 derivatives. Increasing the π-conjugation of the bridge leads to a systematic destabilization of the HOMO level from −6.81 eV in TBQ1 to −6.43 eV in TBQ4, whereas the LUMO lowers in energy from −1.34 eV in TBQ1 to −1.82 eV in TBQ5 (Table S5†). As a result, the HOMO–LUMO energy gap varies in a range of values from 5.47 eV for TBQ1 to 4.69 eV for TBQ4, suggesting a strong shift in the optical properties along the family of TBQ dimers (see below).The large destabilization of the LUMO of TBAQ (−1.18 eV) and TBQ dimers (from −1.34 to −1.82 eV, Table S5†) compared to TCAQ (−3.49 eV) remarks that the reduction of the TBAQ moiety is significantly hindered compared to TCAQ. As a consequence, TBQ1–5 show no reduction activity in the cyclic voltammetry experiments performed within the solvent scan window. It is noteworthy, that exocyclic bromovinylenes are prone to decomposition via debromination, thus limiting the experimental characterization of their redox properties.
The experimental optical characterisation of TBQ1–5 is summarized in Fig. 6. All TBQ dimers present absorption features in the UV region between 250 and 370 nm. The lowest energy absorption of TBQ1 (300 nm) and TBQ3 (314 nm) is blue shifted compared to the other TBQs, due to the restricted conjugation imposed by their oxygen and phenyl linkers. When an alkyne linker is introduced in TBQ2, extension of the conjugation effect is evidenced by a red shift of the absorption (349 nm). Increasing the length of the π-conjugated bridge leads to lower-energy absorptions in TBQ4 and TBQ5 (368 and 365 nm, respectively). Interestingly, the dimers with alkyne spacers (TBQ2 and TBQ5) show similar absorption profiles featuring four bands, the most energetic two lying at ca. 263 and 315 nm, and the lower-energy ones peaking at 325 and 349 nm for TBQ2 and 339 and 365 nm for TBQ5. All TBQ derivatives exhibit weak emission features in non-deaerated solutions, with small Stokes shifts of about 10 nm for TBQ2, TBQ4 and TBQ5, and 30 and 55 nm for TBQ1 and TBQ3, respectively.
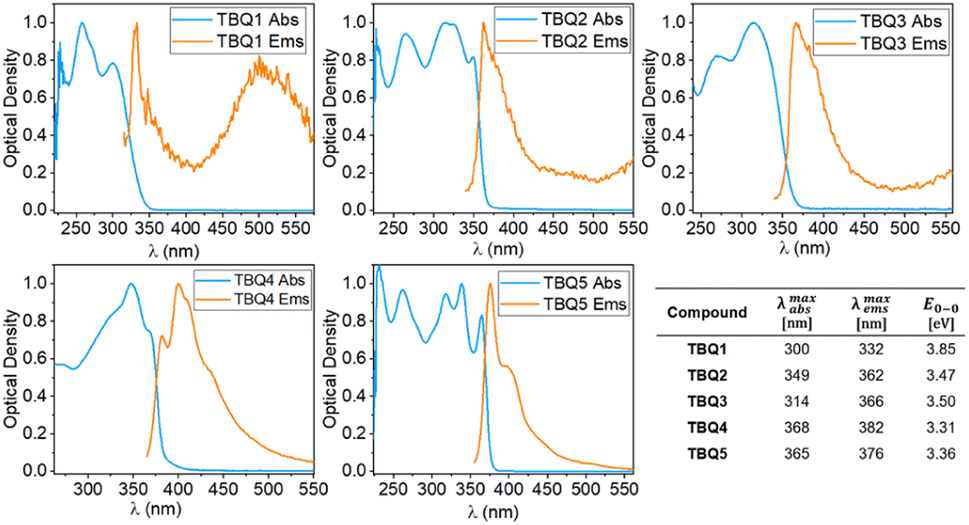 | ||
| Fig. 6 Optical properties of TBQ1–5 in CH2Cl2 solution at 25 °C. The blue plot corresponding to the normalised absorption and the orange to the normalised emission. | ||
Theoretical calculations were performed by means of the time-dependent DFT (TD-DFT) approach to unveil the nature of the low-lying absorption bands for TBQ1–5. The lowest-energy absorption band experimentally recorded at 300–365 nm results from the electronic transition to the first singlet excited state S1 (Fig. S33–S37†), except for TBQ1, for which the transition to the S2 state is predicted near S1 and also contributes to this band with an oscillator strength (f) of 0.551 (Fig. S33†). The S0 → S1 transition mostly originates in the HOMO → LUMO monoexcitation (Table S6†), and, according to the topology of these orbitals, is described by a local excitation mainly centred in the linker and the inner anthraquinone halves. The electronic nature of the S1 excitation is confirmed by the natural transition orbitals involved in the transition (Fig. S38–S42†),32 which suggest a small charge-transfer from the linker to the TBAQ moieties, especially for the less-conjugated TBQ1 and TBQ3 derivatives.
The effect of the internal rotation of the TBAQ units on the first singlet excited states was disclosed to better understand the complex low-lying absorption pattern with several peaks recorded for TBQ2 and TBQ5 (Section S13†). TD-DFT calculations indicate that the torsion of the TBAQ moiety along θ significantly shifts the S1 excitation energy, e.g., from 335 nm at θ = 0° to 300 nm at θ = 100° for TBQ2 (Fig. S43†). The torsion also impacts on the position and intensity of higher-energy states as predicted for the excited states S2 and S4 of TBQ5 at intermediate rotation angles (θ = 80–100°, Fig. S45†). These results highlight that the position of the absorption peaks recorded experimentally and the shape of the absorption spectra, especially in TBQ2 and TBQ5, might come from the large rotational freedom of these TBQ dimers.
The study of the evolution of the absorption features of TBQ2 as a function of the temperature on cooling (Fig. 7) reveals progressive spectral changes that mainly involve: (i) the red-shift of the absorption features, which is characteristic of π-conjugated chromophores, and (ii) a hyperchromic shift of the lowest-energy bands of the spectrum (i.e., the band at 349 nm red shifts to 355 nm and becomes the strongest of the spectrum at 80 K). In addition, the resolution of new vibronic bands is observed, such as the 333/339 nm pair. This temperature-dependent behaviour is typically found in flexible π-conjugated compounds, in which the blockade of the torsional vibrational motions produces a greater population of more planar conformers in detriment of the more distorted ones.33 These conformationally frozen structures give way to the vibronic resolution and consequently the 0–0 vibronic bands become the most active bands of the spectrum.
 | ||
| Fig. 7 Variable-temperature UV-vis electronic absorption spectra of TBQ2 in 2-methyltetrahydrofurane upon cooling from 298 K to 80 K. The dotted grey line corresponds to the spectrum recorded at room temperature after cooling. | ||
The variable-temperature UV-vis experimental data are nicely supported by the rotational energy barriers obtained from quantum-chemical calculations. The predicted maximal energy barriers of ca. 1 kcal mol−1 for TBQ2 are assimilable to the amount of thermal energy removed on cooling from 298 to 80 K (i.e., 0.43 kcal mol−1). These results highlight that at room temperature the molecule has free rotation, with a broad set of thermally populated rotational conformers, whereas the rotation barrier becomes harder to overcome as the temperature diminishes. Therefore, only the most stable planar conformers are present at low temperature, and give rise to the vibronically-resolved UV-vis spectrum at 80 K.
Self-assembly and reactivity on Au(111)
Finally, we have explored the capabilities of TBQs to steer molecular architectures on surfaces. To this aim, we deposited a submonolayer coverage of TBQ3 on a pristine Au(111) surface kept at room temperature. As illustrated in Fig. 8a, after such deposition, a disordered molecular assembly is found, where characteristic bright protrusions coming in pairs are detected coexisting with irregular darker features. By a proper comparison with previous findings from our group, it is feasible to rationalize such molecular assembly.9TBQ3 can be interpreted as two TBAQ units connected by a phenyl bridge. The deposition of TBAQ on Au(111) leads to a regular self-assembly in which the molecular species are intact, displaying a butterfly conformation and exhibiting two distinct adsorption geometries: either the phenyl moieties pointing up (observed in STM as two elongated bright protrusions) or down.9 Similar results are visualized for the assembly of TBQ3 on Au(111), though some of them appear in pairs. Thus, we rationalize that upon sublimation over 80% of TBQ3 molecules cleave by the phenyl bridge, therefore TBAQ fragments coexist with pristine TBQ3 on the surface. Importantly, adsorbed pristine TBQ3 species (10–20%) are visualized as two consecutive pairs of two elongated protrusions, as illustrated in Fig. 8b.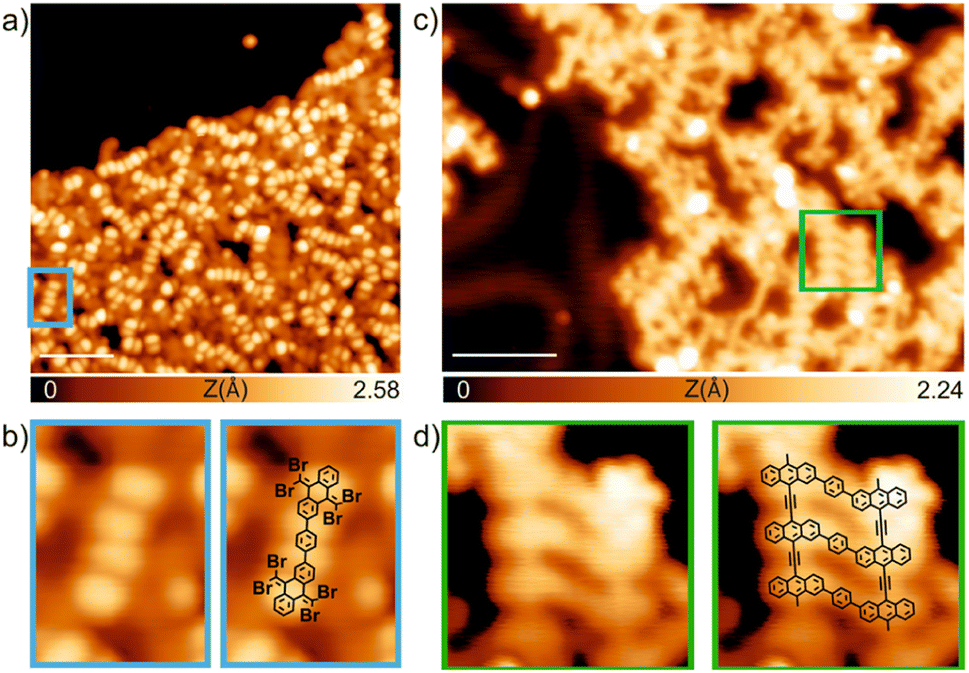 | ||
| Fig. 8 On-surface deposition and reactivity of TBQ3 species on Au(111). (a) Overview STM image of the sublimation of a submonolayer coverage of TBQ3 on a pristine Au(111) surface kept at room temperature. Vb = 1 V, It = 100 pA, scale bar: 4.0 nm. (b) Left: Zoomed-in image of the blue rectangle indicated in panel (a) showing an intact molecule with two consecutive pairs of elongated bright protrusions. Right: Superposition of the chemical model. (c) Long-range STM image after the annealing of (a) at 150 °C. Vb = 0.5 V, It = 50 pA, scale bar: 4 nm. (d) Left: Zoomed-in image of the green highlighted area in panel (c). Right: Superposition of the chemical model. | ||
Next, such molecular assembly was annealed to steer debromination and homocoupling reactions (Fig. 8c). We observed the formation of molecular wires, many of them corresponding to polymers arising from the homocoupling of debrominated TBAQ fragments. Notably, the surviving TBQ3 species, from the sublimation and adsorption at room temperature, have been able to react upon annealing, giving rise to segments of conjugated ladder polymers, as illustrated in Fig. 8d, though featuring frequent side reactivity due to concomitant intra- and inter-molecular reactions.
Conclusion
In summary, five novel dimeric molecules (TBQ1–5) based on the tetrabromoanthra-p-quinodimethane (TBAQ) motif have been synthesized. Owing to their high bromine content nature, these systems are excellent platforms for preparing more complex, highly substituted derivatives, which makes them very appealing and versatile building blocks. Theoretical DFT calculations reveal that the experimentally observed NMR signal splitting neither arise from slow rotation of the TBAQ moieties along the separating linker, nor from the fluxional inversion of the butterfly configuration of the TBAQ core. Featuring activation barriers in the 0.3–3.5 kcal mol−1 range for bond rotation, TBQ1–5 dimers can freely rotate at room temperature. On the other side, the high energy barriers predicted for the fluxional inversion of the butterfly configuration makes it unaffordable at low-to-moderate temperatures. Considering the characteristic butterfly geometry of the TBAQ subunits, when two such units are chemically bound, three possible stereoisomers involving a pair of enantiomers and a meso form are produced, thus explaining the observed NMR splitting. Interestingly, the Raman shifts to higher frequencies of the vibrational modes of TBQ1–5 when increasing the length of the bridges linking the TBAQ moieties, point to an enhanced π-electron delocalization of the butterfly configuration of the TBAQs, which is further evidenced in the electronic and optical properties. The modulation of such π-conjugation, even in small amounts, is a key aspect in the design of molecular building blocks and precursors given the enhancement or multiplicative effects these might have in the electronic structure of the final targeted products.The preliminary on-surface chemistry study demonstrates the capabilities of TBQ species to be sublimated, adsorbed and homocoupled on surfaces, though still further research efforts are necessary to improve the sublimation and to promote the formation of regular polymers.
The results here reported unveil the structural, spectroscopic and electronic properties of these less-known but useful TBAQ-based building blocks and pave the way to a better understanding for the variety of further potential derivatives of interest in different scientific areas. Work is currently in progress in this regard.
Data availability
The BMK/6-31G(d,p)(PCM)-optimized structures calculated for TBAQ, TCAQ and TBQ1-5 are available at the ZENODO repository (https://doi.org/10.5281/zenodo.8038098). Computational results and structures can also be retrieved from the authors upon request.Author contributions
N. M. and J. S. conceived the project. D. J. V., M. P.-E., A. C. V., and A. B. performed the investigation. J. S., M. P.-E., A. B., J. I. U., D. E., and J. C. analysed the results. J. S., M. P.-E, and J. C. wrote the manuscript. All authors reviewed and edited the manuscript. N. M., E. O. and J. C. supervised the project.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the Spanish MCIN/AEI (projects PID2019-108532GB-I00, PID2020-114653RB-I00, PID2020-119748GA-I00, PID2021-127127NB-I00, PID2021-128569NB-I00, and TED2021-131255B-C44 funded by MCIN/AEI/10.13039/501100011033, Centro de Excelencia Severo Ochoa SEV-2016-0686, Unidad de Excelencia María de Maeztu CEX2019-000919-M and AEI/FEDER funds), the Comunidad de Madrid (project QUIMTRONIC-CM (Y2018/NMT-4783)), the Generalitat Valenciana (projects PROMETEO/2020/077 and MFA/2022/017), the Junta de Andalucía (PROYEXCEL-0328) and ERC-2020-SyG (Project: 951224) “TOMATTO” are acknowledged. This study was also supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) (MAD2D-CM)-MRR project. M. P.-E. thanks grant PRE2021-097082 funded by MCIN/AEI and “ESF Investing in your future”.Notes and references
- N. B. Desai, N. McKelvie and F. Ramirez, J. Am. Chem. Soc., 1962, 84, 1745–1747 CrossRef.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef.
- S. Eisler, A. D. Slepkov, E. Elliott, T. Luu, R. McDonald, F. A. Hegmann and R. R. Tykwinski, J. Am. Chem. Soc., 2005, 127, 2666–2676 CrossRef CAS PubMed.
- W. A. Chalifoux and R. R. Tykwinski, Nat. Chem., 2010, 2, 967–971 CrossRef CAS PubMed.
- M. Iyoda, S. Tanaka, H. Otani, M. Nose and M. Oda, J. Am. Chem. Soc., 1988, 110, 8494–8500 CrossRef CAS.
- C.-Q. Chen, T. Chen, J.-X. Liu, G.-F. Zhang, C. Li, W.-L. Gong, Z.-J. Xiong, N.-H. Xie, B. Z. Tang and M.-Q. Zhu, Macromolecules, 2015, 48, 7823–7835 CrossRef.
- M. Shimizu, K. Nishimura, M. Mineyama and H. Fuji, Org. Process Res. Dev., 2019, 23, 1740–1745 CrossRef CAS.
- P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2004, 126, 3108–3112 CrossRef CAS PubMed.
- A. Sánchez-Grande, B. de la Torre, J. Santos, B. Cirera, K. Lauwaet, T. Chutora, S. Edalatmanesh, P. Mutombo, J. Rosen, R. Zboril, R. Miranda, J. Björk, P. Jelínek, N. Martín and D. Écija, Angew. Chem., Int. Ed., 2019, 58, 6559–6563 CrossRef PubMed.
- B. Cirera, A. Sánchez-Grande, B. de la Torre, J. Santos, S. Edalatmanesh, E. Rodríguez-Sánchez, K. Lauwaet, B. Mallada-Faes, R. Zbořil, R. Miranda, O. Gröning, P. Jelínek, N. Martín and D. Écija, Nat. Nanotechnol., 2020, 15, 437–443 CrossRef CAS PubMed.
- A. Sánchez-Grande, J. I. Urgel, A. Cahlík, J. Santos, S. Edalatmanesh, E. Rodríguez-Sánchez, K. Lauwaet, P. Mutombo, D. Nachtigallov, R. Nieman, H. Lischka, B. de la Torre, R. Miranda, O. Gröning, N. Martín, P. Jelínek and D. Écija, Angew. Chem., Int. Ed., 2020, 59, 17747–17752 CrossRef.
- H. González-Herrero, J. I. Mendieta-Moreno, S. Edalatmanesh, J. Santos, N. Martín, D. Écija, B. de la Torre and P. Jelinek, Adv. Mater., 2021, 33, 2104495 CrossRef PubMed.
- A. Sánchez-Grande, J. I. Urgel, I. García-Benito, J. Santos, K. Biswas, K. Lauwaet, J. M. Gallego, J. Rosen, R. Miranda, J. Björk, N. Martín and D. Écija, Adv. Sci., 2022, 9, 2200407 CrossRef PubMed.
- G. Wu, Y. Zhang, R. Kaneko, Y. Kojima, A. Islam, K. Sugawa, J. Otsuki and S. Liu, J. Power Sources, 2020, 454, 227938 CrossRef CAS.
- Z. He, L. Shan, J. Mei, H. Wang, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, X. Gu, Q. Miao and B. Z. Tang, Chem. Sci., 2015, 6, 3538–3543 RSC.
- Y. Yin, H. Hu, Z. Chen, H. Liu, C. Fan and S. Pu, Dyes Pigm., 2021, 184, 108828 CrossRef CAS.
- J. Li, L. Gao, T. Lu, Z. Feng, D. Jiang, C. Du, K. Wang, P. Lu and B. Zou, Adv. Opt. Mater., 2021, 9, 2100813 CrossRef CAS.
- S. Kumar, S. Pola, C.-W. Huang, M. M. Islam, S. Venkateswarlu and Y.-T. Tao, J. Org. Chem., 2019, 84, 8562–8570 CrossRef CAS PubMed.
- Y. Dorca, C. Naranjo, P. Delgado-Martínez, R. Gómez and L. Sánchez, Chem. Commun., 2019, 55, 6070–6073 RSC.
- Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed.
- T. Nishiuchi, S. Aibara, H. Sato and T. Kubo, J. Am. Chem. Soc., 2022, 144, 7479–7488 CrossRef CAS PubMed.
- A. Sánchez-Grande, J. I. Urgel, L. Veis, S. Edalatmanesh, J. Santos, K. Lauwaet, P. Mutombo, J. M. Gallego, J. Brabec, P. Beran, D. Nachtigallová, R. Miranda, N. Martín, P. Jelínek and D. Écija, J. Phys. Chem. Lett., 2021, 12, 330–336 CrossRef PubMed.
- N. Martín, I. Pérez, L. Sánchez and C. Seoane, J. Org. Chem., 1997, 62, 870–877 CrossRef.
- Y.-Q. Fang, O. Lifchits and M. Lautens, Synlett, 2008, 3, 413–417 Search PubMed.
- U. Schubert, S. Hünig and A. Aumüller, Liebigs Ann. Chem., 1985, 6, 1216–1222 CrossRef.
- E. Ortí, R. Viruela and P. M. Viruela, J. Mater. Chem., 1995, 5, 1697 RSC.
- E. Ortí, R. Viruela and P. M. Viruela, J. Phys. Chem., 1996, 100, 6138–6146 CrossRef.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed., 1997, 36, 1808–1832 CrossRef CAS.
- J. Casado, K. Takimiya, T. Otsubo, F. J. Ramírez, J. J. Quirante, R. P. Ortiz, S. R. González, M. M. Oliva and J. T. L. Navarrete, J. Am. Chem. Soc., 2008, 130, 14028–14029 CrossRef CAS PubMed.
- V. Hernández, J. Casado, F. J. Ramírez, G. Zotti, S. Hotta and J. T. L. Navarrete, J. Chem. Phys., 1996, 104, 9271–9282 CrossRef.
- C. Castiglioni, M. Gussoni, J. T. Lopez Navarrete and G. Zerbi, Solid State Commun., 1988, 65, 625–630 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- D. Wasserberg, S. C. J. Meskers, R. A. J. Janssen, E. Mena-Osteritz and P. Bäuerle, J. Am. Chem. Soc., 2006, 128, 17007–17017 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01615c |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2023 |