
Structural polymorphism in protein cages and virus-like particles
Felicia
Lie
,
Taylor N.
Szyszka
and
Yu Heng
Lau
 *
*
School of Chemistry, The University of Sydney, Eastern Avenue, Sydney, NSW 2006, Australia. E-mail: yuheng.lau@sydney.edu.au
First published on 17th June 2023
Abstract
Protein cages and virus-like particles are often thought of as highly uniform structures that obey strict geometric rules for self-assembly. Yet, there is a growing number of examples where different architectures can emerge from the same native cage system through minor changes in experimental conditions or protein sequence. Access to diverse architectures can help tune the engineering of protein cages for biotechnology applications where shape and symmetry often affects function. In this review, we highlight the underappreciated diversity of polymorphic architectures that can be formed by protein cages and virus-like particles, categorising examples by their method of formation.
 Felicia Lie | Felicia Lie is a PhD candidate with Dr Yu Heng Lau in the School of Chemistry at the University of Sydney. She completed her undergraduate degree in Biological Chemistry at Nanyang Technological University in Singapore with a combined minor in Food Science from Wageningen University in the Netherlands. Her current research explores the design and evolution of encapsulin protein cages. |
 Taylor Szyszka | Dr Taylor Szyszka is a postdoctoral research associate in synthetic and chemical biology in the School of Chemistry at the University of Sydney. She received her PhD in Biochemistry from the University of Sydney, working with Prof. Joel Mackay. Her current work with Dr Yu Heng Lau is focused on building synthetic organelles for biomineralisation and nanoreactors for carbon fixation using encapsulin protein cages. |
 Yu Heng Lau | Dr Yu Heng Lau is a Senior Lecturer and research group leader in the School of Chemistry at the University of Sydney, Australia. Yu Heng obtained his PhD in Chemistry at the University of Cambridge with Prof. David Spring, then trained as a Sir Henry Wellcome Postdoctoral Fellow in Synthetic Biology at Harvard Medical School with Prof. Pamela Silver. His research program spans synthetic chemistry and synthetic biology, featuring two main themes: (1) developing cancer therapeutic leads that target protein-protein interactions at telomeres, and (2) exploring protein cages as synthetic organelles for hosting catalysis in engineered cells. |
1. Introduction
Protein-based compartments are ubiquitous in nature, from capsid shells that enclose viruses1 to metabolic organelles in bacteria,2–4 as well as cargo transport and storage vesicles in eukaryotes.5,6 Compartment-forming proteins serve as natural starting points for the construction of recombinant protein cages and virus-like particles that can be engineered to function as vaccines, nanoreactors, and targeted drug delivery vehicles. Across the diverse collection of natural and engineered cage-like protein architectures, a common underlying feature is the self-assembly of simple repeating protein subunits that results in a high degree of symmetry and uniformity (Fig. 1).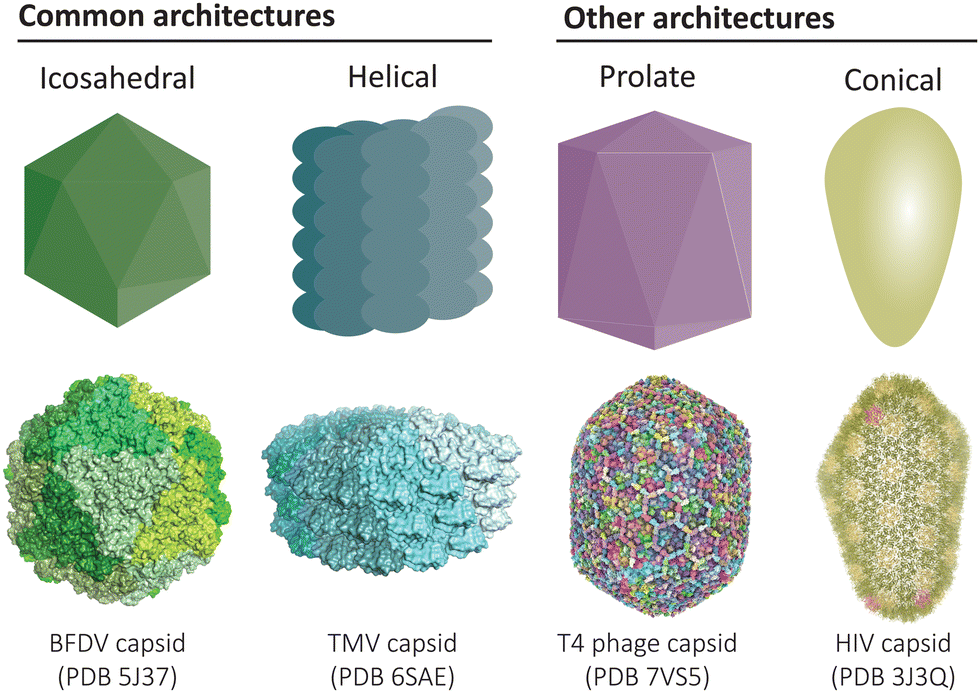 | ||
| Fig. 1 Common architectures found in protein cages and virus-like particles. Icosahedral and helical symmetries are most common,12,13 while there also exists elongated prolate structures and conical assemblies, amongst other less common architectures.14,15 | ||
The prevailing principles used to classify the symmetric structures of protein compartments originate from studies on viral capsids in the 1950s and 60s.7 Caspar and Klug's seminal theoretical framework from 1962 rationalises how multiple copies of a single repeating protein subunit can assemble to form architectures with helical and icosahedral symmetry, introducing the principle of quasi-equivalence to explain the latter case.8 These principles and associated nomenclature are still currently used to describe the structural organisation across all families of protein compartments. For example, the different sizes of icosahedral compartments are commonly classified according to their triangulation number (T = 1, 3, 4, 7 etc.), which corresponds to the number of protein subunits in the assembled structure.9 Exceptions to Caspar–Klug theory have more recently been accounted for using more generalisable classification systems.10,11
Despite the symmetry and uniformity of protein compartments, examples of structural polymorphism are increasingly being reported due to the improving resolution and accessibility of electron microscopy techniques (and other analytical methods such as light scattering and mass spectrometry that have been reviewed by Jacobson and co-workers).16 Polymorphism is an important feature that should not be overlooked, as the structure of a protein compartment is the key determinant of its function. Viruses and protein cages can be considered polymorphic if they can adopt different symmetries (e.g. helical and icosahedral), different sizes (e.g. different icosahedral T numbers), or a mixed collection of distinct but related architectural forms (e.g. irregular conical capsids). Polymorphism can be a native feature in some viruses,17 while other polymorphisms can arise from engineered changes in protein sequence or identity of enclosed cargo. Regardless of the source, structural polymorphism may be an inherent feature in some native cages and has important consequences in biotechnological applications such as antigen display.18,19
In this review, we will cover key examples of structural polymorphism, focusing on how structural diversity can emerge even in the simplest of protein compartments. Such compartments include virus-like particles (VLPs) with a single type of major capsid protein, and simple bacterial protein cages such as encapsulin nanocompartments20,21 and lumazine synthases22 that are encoded by only a single gene. We categorise the diverse ways in which different architectures can arise from the same native cage system.
For further reading on related topics beyond the scope of this review, we refer the reader to reports of polymorphic protein cages derived from more complex bacterial microcompartments that involve a greater number of unique shell proteins,23 as well as studies involving de novo designed cages where the aim is to generate uniform structures.24,25 We also note that disassembled states or partially assembled intermediates may also be considered as polymorphic states in some contexts, but we primarily focus on fully-assembled equilibrium states for this review.
1.1. Pleomorphism in native viruses
Before addressing how structural polymorphism can arise in protein cages and VLPs, we first note examples of natural polymorphism in live viruses. In this context, the existence of different structural states is sometimes referred to as pleomorphism.17 This phenomenon is seen in many enveloped viruses, presumably due to the lipid envelope serving as the protective outer layer, thus placing less importance on having a rigid closed protein capsid for protecting the enclosed genome.Structural pleomorphism can play an integral role in the viral life cycle, as in the case of the well-studied retrovirus HIV.26 The HIV capsid adopts a truncated spherical morphology in the immature state,27 before being converted to a conical structural in the final mature state.14 Even in the final mature structure, different forms are possible depending on the distribution of pentameric facets within the predominantly hexameric array.26,28
Pleomorphism may also affect the ability of a virus to enter its cellular host. Influenza viruses can naturally adopt a mix of infectious forms including both spherical and filamentous morphologies.29 It is thought that while the spherical form dominates in laboratory culture conditions, the filamentous form isolated from hosts may assist infection in vivo and help overcome host adaptations that inactivate viral glycoproteins.30,31
These natural examples of structural polymorphism indicate that the structural constraints on protein cages and virus-like particles are not always strict, and that a variety of architectures is often accessible from a simple set of capsid proteins, especially in the absence of strong selective pressure for structural uniformity.
2. Polymorphisms in recombinant protein cages and VLPs
Recombinant expression of protein cages and viral capsid proteins can lead to the formation of polymorphic assemblies. Structural polymorphism can be a consequence of omitting secondary components of the native assembly during expression (e.g. cargo or scaffolding proteins), or the in vitro buffer conditions used in experiments.2.1. Well-defined dimorphic states in recombinant wild-type cages
VLPs formed from the core antigen protein of hepatitis B virus (HBV) represent a well-studied example of capsid polymorphism (Fig. 2a). HBV capsids form icosahedral particles with either T = 3 or T = 4 symmetry. The dimorphic structure of HBV was initially determined by cryo-EM on samples produced in a recombinant E. coli expression system.32 Notably, the recombinant system faithfully reproduces the native capsid structure in this instance, as early studies reported observation of different sized spherical ‘Dane’ particles isolated from native hosts,33,34 which were subsequently confirmed to be dimorphic icosahedral particles,35,36 of which the more common T = 4 particle size is implied to be the active infectious particle. Dimorphism between T = 3 and T = 4 states are not unique to HBV particles, as the phenomenon has also been observed in recombinant capsids of bacteriophage MS2.37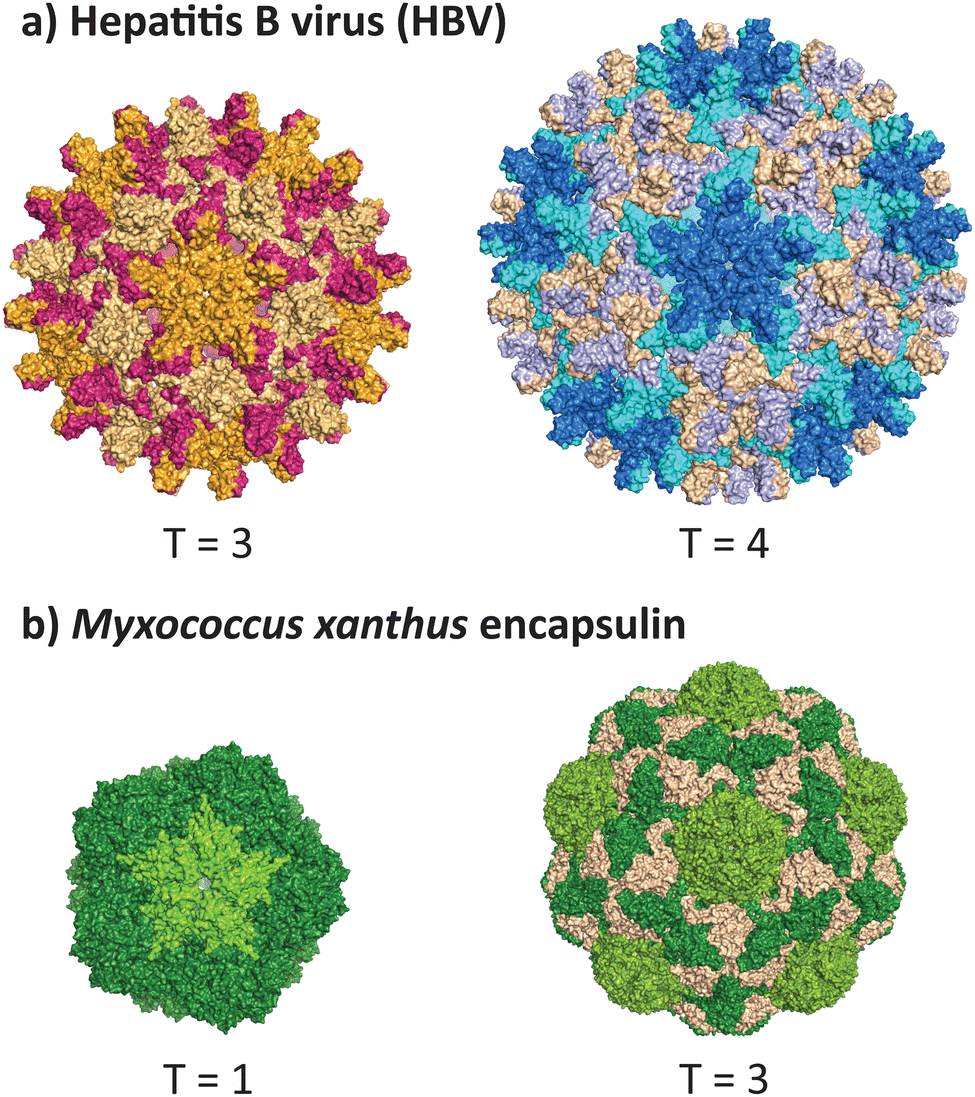 | ||
| Fig. 2 Dimorphism in recombinantly expressed wild-type VLPs and protein cages. (a) The hepatitis B virus can adopt the native T = 4 form (PDB 1QGT)40 and a smaller T = 3 form (PDB 6UI6).41 (b) The encapsulin protein cage from Myxococcus xanthus can adopt the native T = 3 form (PDB 7S20) and a smaller T = 1 form (PDB 7S21).39 | ||
A non-viral example of icosahedral dimorphism in wild-type sequences is the encapsulin nanocompartment from Myxococcus xanthus (Fig. 2b). The structure was reported to be a T = 3 icosahedral particle when isolated from the native bacterium,38 although recombinant expression of only the shell protein EncA resulted in a minor population of T = 1 particles, which were later found to occur only in the absence of the encapsulated cargo proteins EncB and EncC.39 This example demonstrates that recombinant expression does not always reflect the native state of a protein cage or VLP, and highlights the role of cargo in reinforcing a uniform morphology (see 2.3 and 2.4). When repurposing protein cages in engineering studies, the potential for multiple assembly states should be accounted for, as cage stability and other biomolecular properties may differ across various assembled states.
2.2. The role of scaffolding proteins in controlling polymorphism
While small icosahedral cages and VLPs are often able to assemble independently using only a single type of protein subunit, larger assemblies (T > 4) typically require assistance from additional scaffolding proteins to template the desired architecture. Theoretical studies on templating effects suggest that this phenomenon may indeed be a general property of large spherical crystals.42 Scaffolding proteins have been observed across a variety of dsDNA bacteriophages, as reviewed by Dokland.43 A well-studied example of a dsRNA system requiring scaffolding is the infectious bursal disease virus (IBDV) that natively forms a T = 13 assembly, where the mature capsid protein (VP2) assembles exclusively into a T = 1 structure when expressed alone without the scaffolding protein VP3.44,45An apparent exception to the rule is the HK97 bacteriophage (T = 7), where a separate scaffolding protein does not exist. Nevertheless, the delta domain of the major capsid protein (102 residues at the N-terminus) shares many similarities to the scaffolding protein of other dsDNA bacteriophages. Loss of the delta domain has been found to heavily reduce protein solubility, implying a crucial role for this domain in assisting with correct folding and assembly of the capsid protein.46
2.3. Polymorphisms scaffolded by nucleic acid encapsulation
Given the natural propensity of virus-derived capsid proteins to bind nucleic acids as part of their native function, the introduction of different DNA or RNA cargo into recombinant systems can serve as a scaffold to influence the resulting assembled morphology.The cowpea chlorotic mottle virus (CCMV) was found to assemble into different sized particles depending on the lengths of the ssRNA packaged.48 When the ssRNA length was approximately 3000 nucleotides, similar to that of wild type, 26 nm capsids resembling T = 3 particles were formed. Shorter ssRNA of less than 2000 nucleotides were packaged in particles with diameters resembling T = 2 or T = 3 assemblies, while longer ssRNA of greater than 4500 nucleotides were found in particles with sizes resembling T = 3 or T = 4 particles. Furthermore, for ssRNA of greater than 3200 nucleotides, paired particles could be observed sharing an RNA molecule through the pores of their capsids. These results suggest that the capsid protein favours specific curvatures, as the pairing of smaller particles that share RNA is preferred over the formation of larger particles. More recently, similarly polymorphism has been observed when packing ssDNA in CCMV, where the length affects the diameter of the spherical structures observed.49
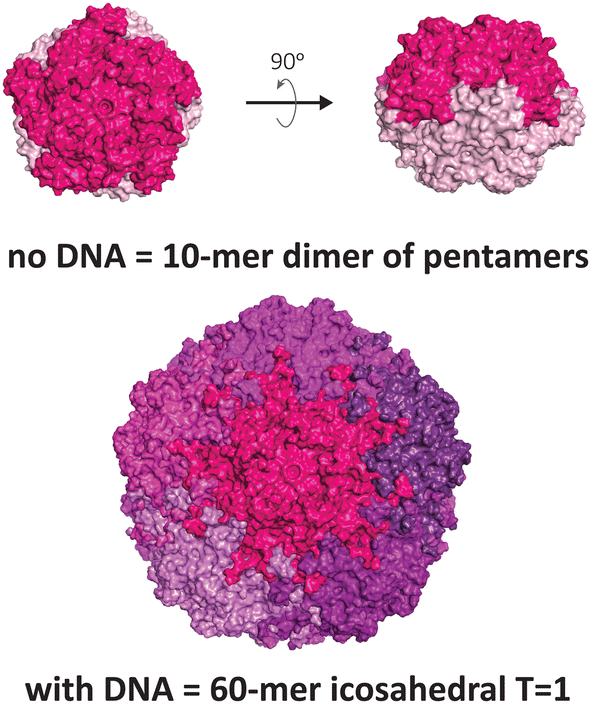
Another VLP that display unusual polymorphism under nucleic acid control is the capsid of the beak and feather disease virus (BFDV), a small circovirus with predominantly T = 1 icosahedral symmetry (Fig. 3). Crystallographic studies revealed the presence of an additional architecture consisting of ten protomers, involving an unusual direct dimeric interaction between two pentameric facets, mediated by the surfaces that normally face externally in the icosahedral form.12 Notably, introduction of ssDNA to the capsid protein strongly favours the icosahedral form. The observed dimer of pentamers is consistent with electron micrographs of related circoviruses in infected cells that show pleomorphic heterogeneity in particle sizes,50,51 thus it is postulated to be a functional complex that externally displays its DNA binding domain and nuclear localisation signal, enabling trafficking into the nucleus and subsequent packaging of the viral genome to produce the icosahedral form.
Artificially designed DNA origami structures such as sheets,52 tubes and rings53 can serve as templates for assembly of the CCMV capsid. The formation of origami-controlled architectures is driven by the electrostatic interactions of the negatively charged DNA backbone and positively charged residues in the capsid protein N-terminal region. Interestingly, multiple layers were observed to envelope the origami structure, depending on the molar ratio of CCMV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNA. Single particle reconstruction of a tubular six-helix bundle possessing single and double layers of capsid protein on DNA origami showed helical symmetry with capsid proteins arranged as hexamers. Caps of the tubes with a single protein layer were also modelled and found to be made up of six pentamers and one hexamer.
:DNA. Single particle reconstruction of a tubular six-helix bundle possessing single and double layers of capsid protein on DNA origami showed helical symmetry with capsid proteins arranged as hexamers. Caps of the tubes with a single protein layer were also modelled and found to be made up of six pentamers and one hexamer.
CCMV is also able to form tubes simply by encapsulating dsDNA with lengths of 100–1000 nucleotides.49 This phenomenon is suggested to arise from the stacking of dsDNA rather than coiling, thus creating a tube-like template. Indeed, the theoretical lengths of the tubes based on a fully stretched dsDNA scaffold deviated only slightly from experimental TEM measurements by around 10 nm. On the other hand, spherical structures were formed when ssDNA was encapsulated.
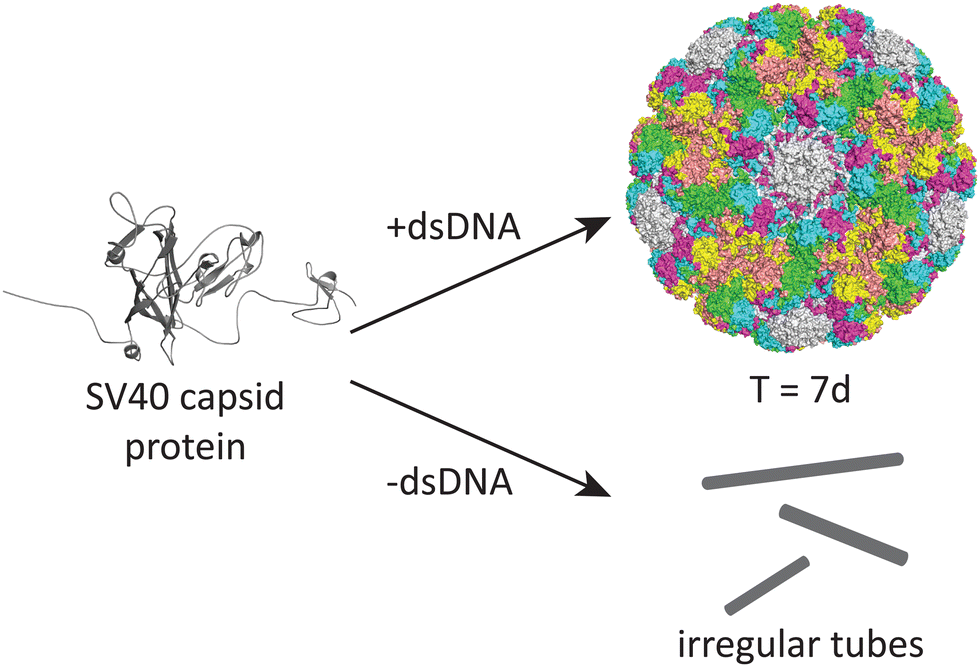
DNA may also play an important role in the assembly of the simian virus 40 (SV40), a 40 nm spherical particle which adopts a T = 7d assembly under appropriate assembly conditions (Fig. 4).54 In the absence of DNA, the native 40 nm spherical particle is not observed, while other structures including tubes and irregular particles can be obtained. The authors postulate that DNA may function as a scaffold that interacts with the VP1 pentamer while not excluding the possibility that DNA may induce conformational changes in VP1.
2.4. Polymorphisms scaffolded by artificial cargo
The scaffolding role of nucleic acids in controlling polymorphism can be mimicked using a range of synthetic artificial cargoes. The following examples show how artificial cargo can be used to recapitulate native-like control of capsid morphology, which may be useful in applications such as drug delivery and antigen display.The CCMV capsid has been found to encapsulate polystyrene sulfonate (PSS),56 a negatively-charged polymer that takes the place of the native RNA genome, while avoiding the complication of secondary or tertiary ssRNA structures arising from base pairing. Although different lengths of PSS were used, with molecular masses ranging from 0.4–3.4 MDa and hydrodynamic radii of 18–43 nm, only two discrete particle sizes were observed, corresponding to apparent T = 2 and T = 3 assemblies with 120 and 180 capsid proteins respectively.
CCMV has also been assembled around nano-emulsions formed from silicon oil (polydimethylsiloxane) in water and stabilised by sodium dodecyl sulfate (SDS).57 As the size of the emulsion droplets increased to be larger than native CCMV, regular capsomere arrangements were less prevalent, instead forming defective capsomeres with hexagonal web-like arrangements and scar like irregularities that suggest protein disorder at a sub-capsomere scale. Thus, size and curvature of the templating cargo influences the symmetry of the particle. Comparing this with the previous example of packaging PSS, it is postulated that the incompressibility of these emulsion droplets may explain the persistence of defects and overall differences in assembly behaviour.
Finally, size control of the closely related brome mosaic virus (BMV) capsid protein has also been achieved using gold nanoparticles, with different nanoparticle diameters templating particles resembling T = 1, pseudo T = 2 and T = 3 sizes.58
2.5. Polymorphisms controlled by buffer conditions
Aside from scaffolding effects, the buffer composition used in the study of protein cages and VLPs can affect their assembly states.Polyomaviridae are a family of viruses that have multiple polymorphic forms when the major capsid protein is expressed and assembled recombinantly. These viruses are assembled entirely from pentameric capsomeres, requiring non-equivalent interactions between pentameric units that break the Caspar–Klug quasi-equivalence principle. In recombinant systems, both the binding of calcium ions and the formation of disulphide bonds under non-reducing conditions are often needed to trigger the assembly of capsomeres into full VLPs.59,60
One of the earliest reports providing structural definition on the polymorphic nature of polyomaviruses was conducted on the polyomavirus major capsid protein VP1, exploring a range of assembly conditions including pH, calcium chloride and ammonium sulfate.61 Negatively stained electron micrographs indicated the formation of differently sized spherical particles and some filaments. The authors proposed a smaller T = 1 icosahedral assembly in addition to the regular T = 7d icosahedral assembly, as well as a possible octahedral 120-mer, although no studies since this publication from 1989 have confirmed this suggested structure. Indeed, similar buffer exploration studies on the closely related SV40 VP1 protein showed the formation of T = 1 icosahedral particles and filaments alongside the expected T = 7d, as well as intermediate-sized particles that were more heterogeneous and harder to conclusively deduce the structure of.62 Furthermore, studies on the VP1 protein of the BK virus, another member of the polyomaviridae, reported only the T = 1 and T = 7d forms in different buffer conditions.63
The capsids of cowpea chlorotic mottle virus (CCMV) and brome mosaic virus (BMV) are also established systems for studying capsid assembly featuring polymorphism. The initial reports of in vitro CCMV and BMV assembly by Bancroft, Heibert and co-workers in the late 1960s described heterogeneity in electron micrographs of their capsid assemblies under a variety of buffer conditions.64,65 Variations in pH and NaCl concentrations resulted in laminar and plate-like particles alongside the expected spherical particles, as well as tubular filaments. Later work in elucidating the T = 3 icosahedral structure of CCMV showed that a swollen capsid structure could be generated in the presence of EDTA to chelate Ca2+ which stabilises the native structure.66 In addition, assembly experiments at high protomer concentrations and acidic pH resulted in the formation of smaller particles as observed by negative stain EM, suggestive of pseudo T = 2 symmetry according to mass estimates from light scattering data.67 The various polymorphic forms have been analysed in terms of a phase diagram, showing the various assembled states as a function of ionic strength and pH.68
The lumazine synthase protein cage is a non-viral compartment that also forms different polymorphic states depending on the buffer conditions. Analysis of wild-type lumazine synthase from Bacillus subtilis (LSBS) by electron microscopy and small angle X-ray scattering (SAXS) showed the existence of three classes of particle sizes.69 The expected T = 1 icosahedral assembly was observed in phosphate buffer pH 8, whereas in Tris and borate buffers at various pH values, larger sized particles that potentially represent T = 3 and T = 4 structures were also observed, along with an array of deformed or incomplete assemblies. This work demonstrates how minor changes in buffer conditions can dramatically affect the polymorphology of a protein cage and highlights the general structural plasticity of lumazine synthases (also see Section 4).
Temperature is another parameter which can affect the morphology of VLPs. One well-documented example is the P22 bacteriophage capsid (T = 7 icosahedral symmetry), where structural changes associated with maturation can be recapitulated in vitro by heating the recombinantly produced capsid. The native P22 phage first assembles into a prohead that includes the coat protein and a scaffold protein. The scaffold protein is removed upon expansion of the prohead capsid into the mature capsid, being replaced by the viral genomic DNA (Fig. 5).70 This capsid expansion process was first triggered in vitro using SDS treatment,71 but is most commonly induced by heating to ∼65 °C.72 Further heating can result in a wiffle ball morphology where only the pentameric facets are removed.73 As such, the P22 capsid system demonstrates the versatility of capsid protomers to form not only different sizes but also different porosities.
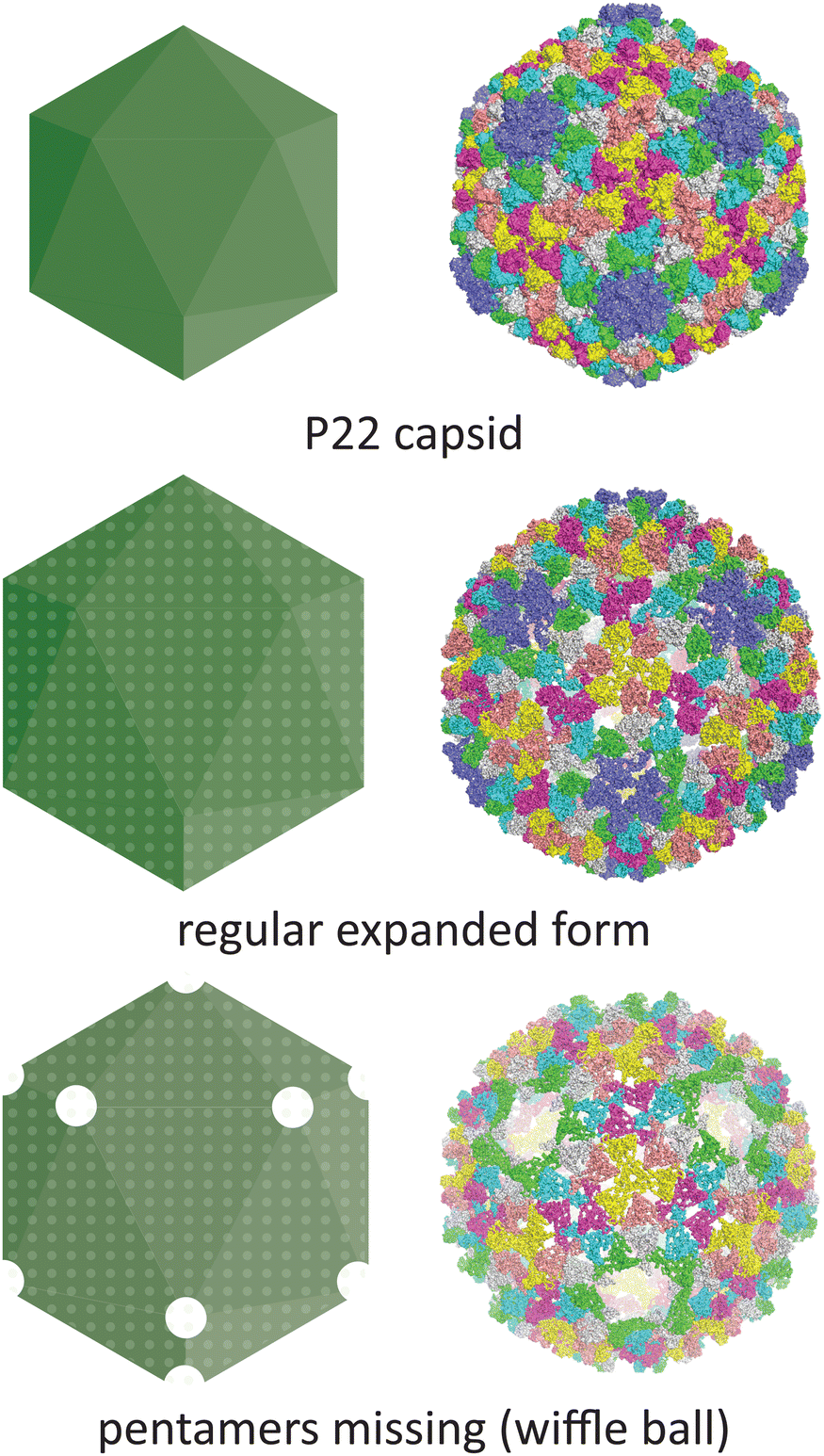 | ||
| Fig. 5 The conditions under which the P22 bacteriophage capsid (PDB 5UU5)74 is subjected to in vitro can lead to changes in morphology. Heating can lead to an expanded procapsid form (PDB 3IYI) and a porous wiffle ball structure with the pentamers missing from the assembly (PDB 3IYH).75 | ||
3. Polymorphisms arising from sequence modifications
This section discusses examples of polymorphic states arising from minor changes to the protein sequence, including the truncation of termini, point mutations, and loop insertions or deletions. The resulting change in inter-subunit interactions and protomer conformations can lead to a wide array of non-native structures with potential use in bionanotechnology applications.3.1. Icosahedral polymorphisms arising from terminal truncations
One of the most commonly studied sequence modifications in VLPs is the N-terminal truncation of picorna-like viruses. The largest of viral lineages according to Abrescia et al.,76 these viruses all share a single jelly-roll fold consisting of eight beta strands at the core of their capsid protein, leading to some broad similarities in their overall assembly characteristics.Across the large number of studies on positive-sense ssRNA viruses that commonly feature the jelly-roll fold, the N-terminus usually contains the RNA binding site and the residues that form the β-annulus at the quasi six-fold symmetric pore. Typically, N-terminal truncation leads to the formation of a mixture of assemblies where a decrease in the icosahedral T number is also observed (Table 1). This phenomenon arises as the pentamers generally remain intact while the hinge at the two-fold and three-fold symmetric axes is affected, leading to changes in curvature.
| Capsid | N-terminal residues deleted | Structural effects | Ref. |
|---|---|---|---|
| Brome mosaic virus (BMV) | 41–47 | Two distinct populations in vivo | Calhoun et al. (2007)80 |
| Brome mosaic virus (BMV) | 1–35 | T = 1 | Larson et al. (2005)81 |
| Flock house virus (FHV) | 1–31 | Heterogenous mixture of small bacilliform-like, irregular structures and wild-type T = 3 particles | Fan Dong et al. (1998)82 |
| Cowpea chlorotic mottle virus (CCMV) | 3–36 | Mixture of T = 3, 2 (60 subunit dimers), 1 (30 subunit dimers) | Tang et al. (2006)83 |
| Rabbit hemorrhagic disease virus (RHDV) | 1–29 | Primarily T = 1 with some T = 3 | Bárcena et al. (2004)84 |
| Tomato bushy stunt virus (TBSV) | 1–62 | Mixture of T = 1 and T = 3 | Hsu et al. (2006)85 |
| 1–72 | T = 1 only | ||
| Sesbania mosaic virus (SeMV) | 1–36 | Formation of a mixture of T = 1 and pseudo T = 2 | Lokesh et al. (2001)86 |
| 1–65 | T = 1 only | ||
| Southern bean mosaic virus (SBMV) | 1–61 | T = 1 | Erickson et al. (1985)87 |
| Grouper nervous necrosis (GNN) | 1–35 | T = 1 | Chen et al. (2015)88 |
| Hepatitis E virus (HEV) | 1–111 | T = 1 | Xing et al. (1999)89 |
Also part of the picorna-like lineage, the dsDNA human papillomavirus 16 (HPV16) forms a T = 1 icosahedral assembly, as opposed to its native T = 7 assembly, when 10 amino acids are removed from the N-terminus.77 Interestingly in the case of SV40 polyomavirus, another dsDNA picorna-like virus, the truncation of 34 residues from the C-terminus results in formation of aberrant assemblies and tubes.78 Further deletions resulted in the loss of assembly, with the protein remaining as pentamers.
A non-picorna-like virus that shows truncation-dependent polymorphism is the HBV capsid, which naturally assembles into T = 3 and T = 4 assemblies (see Section 2.1). When the C-terminus was shortened progressively up to 11 residues, the percentage of T = 4 assemblies decreased, while further truncation led to loss of capsid formation altogether.79
3.2. Other sequence modifications that affect size and morphology
Apart from terminal truncations, insertions and point mutations can also give rise to interesting polymorphic assemblies.In the capsid of rabbit haemorrhagic disease virus (RHDV), a 42 amino acid insertion of a feline calicivirus (FCV) derived epitope at the N terminus resulted in the formation of a mixture containing both wild-type T = 3 and larger T = 4 assemblies.90 However, not all N-terminal insertions resulted in polymorphic assemblies, as insertion of a chicken ovalbumin (OVA) epitope resulted in assemblies that were morphologically identical to the native T = 3 structure.91
Simple point mutations can also trigger shifts to alternative capsid morphologies. A S37P mutation in the MS2 bacteriophage capsid protein is sufficient to downsize the assembly from T = 3 to T = 1.92 Meanwhile, an A275T point mutation on the gp23 protein of the T4 bacteriophage capsid causes a change from the native prolate icosahedral assembly into a primarily isometric T = 13 icosahedral structure.93
In the assembly of VP1 from SV40, introduction of two point mutations have been found to convert the naturally polymorphic VLPs into a homogenous assembly.94 The mutant L102C/P300C forms an additional disulphide interaction between pentamers, thereby stabilising the overall assembly. The new interactions shift the assembly to a smaller particle of 24 nm diameter, which is likened to a T = 1 assembly that is clearly downsized from the native T = 7 size of 45 nm.
The HK97 bacteriophage capsid provides an unusual example of conditional polymorphic behaviour induced by a point mutation. An E219K mutation in the capsid protein was initially found to assemble into a complete icosahedral shell resembling the native prohead structure.95 When the particle was subsequently disassembled and reassembled in vitro through buffer changes, an all-hexameric wiffle ball porous morphology was observed where twelve pentamers were missing. The pores of the wiffle ball could be capped by the introduction of wild type pentameric facets. The formation of a spherical structure exclusively from hexamers is unusual as hexamers typically assemble into sheets or tubes. It is postulated that in this instance, the initial assembled state biased the conformation of the hexamers towards icosahedral assembly, which was retained during transient disassembly such that reassembly could occur even in the absence of pentamers.
Modifications that affect morphology have also been engineered into ferritins. By redesigning the surface interactions between subunits using Rosetta, along with optimisation of buffer conditions, the original 24-mer cage of the Thermotoga maritima ferritin (TmFtn) was converted into designer filaments, nanorods and nanoribbons.96
3.3. Mutants that convert icosahedral symmetric assemblies into tubular morphologies
While we previously discussed the role of nucleic acid cargo in forming tubular morphologies in Section 2.3, aberrant tubular structures have also been formed from icosahedral assemblies through the introduction of point mutations at key interaction sites or circular permutation.Mutants of the HK97 capsid affecting the key E153-R210 salt bridge between capsomeres have been reported to assemble into tubes or sheets.97 Another mutation at a salt bridge interaction which results in tubular structures is at the transient D231-K178 interaction between the G and E loops, where D231A and D231E results in aberrant assembled fragments, while K178D/L/R results in tubes of different widths.98
Tube formation can be triggered by circular permutation in the Aquifex aeolicus lumazine synthase (AaLS) protein cage. Connecting the N- and C-termini via a GTGGSGSS linker and creating new termini between residues 119 and 120 retains the overall monomer fold,99 but instead of a T = 1 icosahedrally-symmetric structure, a mixture of fragmented, spherical and tubular structures was observed. Interestingly, the spherical assemblies were dynamic, converting to tubular structures when incubated for a week at room temperature. On the other hand, the majority of the unassembled fragments remained disassembled.
3.4. Chimeric assemblies with variable ratios of different protein subunits
Heterogeneous structures can arise from assemblies that contain differing ratios of two or more structurally homologous subunits. Such chimeric assemblies can occur naturally, such as in mammalian ferritins assembled from two related subunit chains (H and L).100,101 This phenomenon constitutes a form of polymorphism, as the ratio of subunits in ferritin assemblies is variable across different cell types and in vivo. Indeed, chimeric ferritins can also be produced by assembling different sequence-engineered subunits in recombinant experiments in vitro.102Adeno-associated virus (AAV) capsids represent an important class of engineered VLPs used in gene delivery that can exhibit chimeric polymorphism. For example, co-expression of wild-type AAV capsid protein with an engineered version containing a protease cleavage site insertion led to chimeric mosaic capsids, with transduction efficiencies dependent on the ratio of subunits present.103 Computational tools have also been developed to help guide the formation of chimeric AAVs with appropriate subunit ratios for achieving efficient transduction.104
4. Polymorphic states with different symmetries
While the majority of polymorphic states reported in the literature involve conservative changes in icosahedral T number or a shift from icosahedra to tubes, there is a small but growing number of studies that show assembles that adopt radically different symmetries during protein engineering efforts (Fig. 6).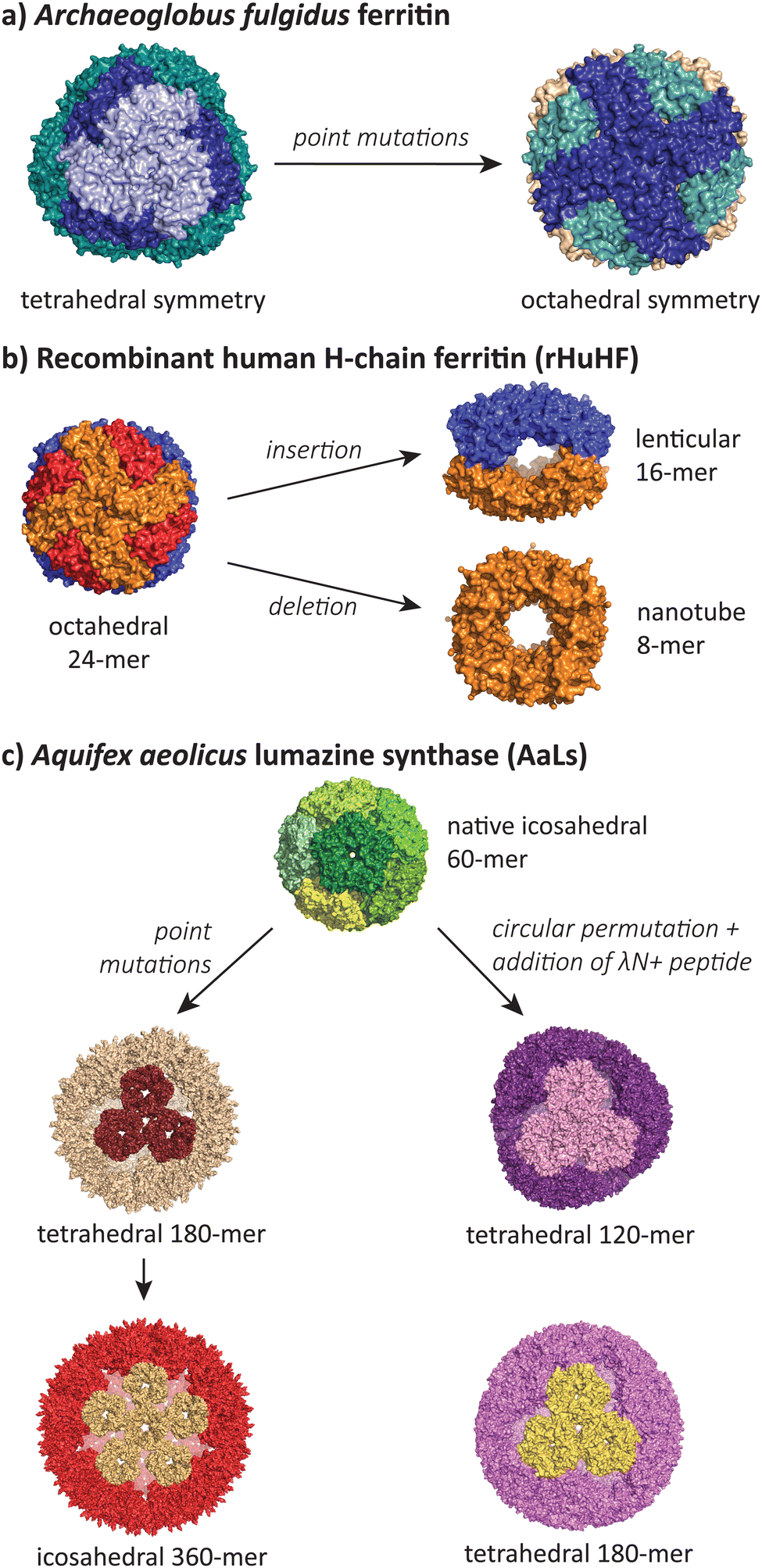 | ||
| Fig. 6 The most structurally divergent morphologies occur in protein cages, where sequence modifications can lead to completely different symmetries and subunit interaction patterns. (a) The Archaeoglobus fulgidus ferritin shifts from a tetrahedral symmetry (PDB 1SQ3) to an octahedral symmetry (PDB 3KX9) with two point mutations.105,106 (b) Human H-chain ferritin can be mutated from an octahedral assembly (PDB 2FHA)111 to a lenticular morphology (PDB 5GOU)107 and a tube-forming ring (PDB 5ZND).108 (c) The Aquifex aeolicus lumazine synthase protein cage can be converted from its native T = 1 icosahedral form (PDB 1HQK)112 to tetrahedral (PDB 5MQ3, 7A4F, 7A4G) and larger icosahedral forms (PDB 5MQ7).113,114 | ||
Lim and co-workers have reported a double mutant of the Archaeoglobus fulgidus ferritin that results in a switch from the unusual native porous tetrahedral symmetry into the more common closed octahedral symmetry found in all other ferritins (Fig. 6a).105,106 The two mutations responsible for the symmetry shift (K150A/R151A) both lie at the four-fold symmetric pore of the octahedral structure, indicating that while alanine can be accommodated at this pore, the original lysine and arginine residues provide charge repulsion and steric hindrance to favour the more open tetrahedral form.
Radical changes in the morphology of recombinant human H-chain ferritin (rHuHF) were observed when the sequence LNEQVKA was inserted at the C3–C4 interface (Fig. 6b).107 The cage morphology was observed to shift from a 24-mer rhombic dodecahedron with octahedral symmetry to a 16-mer with lenticular morphology and a lowered symmetry. Interestingly, the same effect could also be achieved by the insertion of different sequences with at least 6 residues (e.g. Ala6, Pro6, Leu6) at the same site, or when the site of insertion was shifted one residue either side of the original site. In a related study, the removal of 49 residues at the C-terminus resulted in the formation of an 8-mer in solution.108 When crystallised, a ring-like structure possessing D4 symmetry was observed to stack uniformly, resulting in the formation of tubes. Alignment of these tubes in a staggered manner led to the formation of a nanotube array with two kinds of pores. Finally, a deletion of residues 139–144 resulted in a spherical 48-mer while maintaining octahedral symmetry.109
Heddle and co-workers reported that a loop insertion into MS2 capsids results in a polymorphic collection of capsids that remain roughly spherical but break icosahedral symmetry.110 Insertion of either SpyTag variants or a random sequence into an external facing loop of MS2 led to a mix of T = 3, T = 4, D5, D3-A, and D3-B symmetries, as observed by cryo-EM. In all cases, the T = 3 form resembling wild-type symmetry was the dominant form. These findings suggest that these low abundance polymorphs have been missed in earlier studies due to limitations in resolving power that have since been overcome with technical advancements in microscopy.
The AaLS protein cage has also been a rich source of polymorphic assemblies arising from sequence engineering (Fig. 6c). When four amino acids projecting toward the lumen were mutated to glutamic acid (R83E, T86E, T120E, Q123E),115 the resulting assembly expanded in size from a T = 1 60-mer to a larger and more porous 180-mer, accompanied by a reduction in symmetry from icosahedral to tetrahedral.113 When this mutant AaLS was used as a template for directed evolution,116 seven more mutations with increased net negative charge led to further expansion into a 360-mer extended dodecahedron that reverted back to icosahedral symmetry.113 AaLS cages with tetrahedral symmetry114 were also observed after the addition of the cationic λN+ peptide to the N-terminus of circularly permuted AaLS.117 Notably, every polymorphic AaLS assembly still retains the same native-like pentamer as the basic assembly unit, only changing the interactions between pentamers to achieve the different assembled morphologies.
Overall, the examples in this section reveal that despite the ubiquity of icosahedral symmetry in spherical VLPs and protein cages, other symmetries are obtainable through minor sequence modifications. In the cases discussed here, symmetry changes can be achieved by breaking or altering key interactions at symmetry vertices. In cases where reduced symmetry occurs, such as icosahedral to tetrahedral, the resulting assemblies appear to be more porous due to non-ideal packing of the repeating subunits. Nevertheless, these non-canonical morphologies are still able to form stable assemblies that can be isolated and studied.
5. Conclusions and future perspectives
Given the diversity of polymorphic VLPs and protein cages discussed in this review, there is clearly a remarkable amount of plasticity in the way that the constraints of a closed capsid structure can be met. As simple VLPs and protein cages are inherently repetitive structures, small changes in the interactions between subunits can lead to large effects when propagated throughout an entire assembly.There is a perception that protein cages and VLPs form rigid, stable and uniform structures, perhaps propagated by the static nature of crystal and cryo-EM structures. Yet, several types of polymorphism we have discussed suggest that flexibility in subunit conformation and intra-subunit interactions are fundamental elements of such structures, going beyond Caspar–Klug quasi-equivalence. Of particular note is the prevalence of polymorphic mixtures, sometimes including different symmetries, where various assemblies can co-exist within the same experimental sample. These mixtures may represent kinetically trapped states or may alternatively indicate that the wild-type assembled structures are not unique thermodynamically favoured solutions. Regardless of which possibility is true, both are consistent with studies that implicate cargo as a critical component for templating structural homogeneity in the native context.
Technological improvements in electron microscopy have played a significant role in our understanding of polymorphism. Many of the early studies relied on the available scattering techniques, size-exclusion methods, and low-resolution electron microscopy, where the identity of the assembled states could not be definitively proven. As single particle cryo-EM reconstructions have become commonplace in recent years, many of these earlier claims are now being verified or corrected as molecular structures are elucidated. Nevertheless, there is still a literature bias towards studying the dominant morphological species, as asymmetric features and lower population species are harder to observe and more difficult to characterise.
As more examples of VLP and protein cage polymorphism are discovered, we will gain a greater understanding of the theoretical basis of their formation, and ultimately greater control over morphology in engineering contexts. The ability to programme specific polymorphisms is therefore set to play an increasingly important role in the future design of VLPs and protein cages for bio-nano technological applications, where precise control of symmetry and architecture are important factors that can determine efficacy of their applied functions.
Author contributions
All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. H. L. acknowledges funding from the Australian Research Council (DE19010062, DP230101045) and Westpac Scholars Trust (WRF2020).References
- W. H. Roos, I. L. Ivanovska, A. Evilevitch and G. J. L. Wuite, Cell. Mol. Life Sci., 2007, 64, 1484 CrossRef CAS PubMed
.
- J. A. Jones and T. W. Giessen, Biotechnol. Bioeng., 2021, 118, 491–505 CrossRef CAS PubMed
- C. A. Kerfeld, C. Aussignargues, J. Zarzycki, F. Cai and M. Sutter, Nat. Rev. Microbiol., 2018, 16, 277–290 CrossRef CAS PubMed
- H. B. McDowell and E. Hoiczyk, J. Bacteriol., 2022, 204, e00346 CrossRef CAS PubMed
- G. Jutz, P. Van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed
- E. D. Pastuzyn, C. E. Day, R. B. Kearns, M. Kyrke-Smith, A. V. Taibi, J. McCormick, N. Yoder, D. M. Belnap, S. Erlendsson, D. R. Morado, J. A. G. Briggs, C. Feschotte and J. D. Shepherd, Cell, 2018, 172, 275–288 CrossRef CAS PubMed
- F. H. C. Crick and J. D. Watson, Nature, 1956, 177, 473–475 CrossRef CAS PubMed
- D. L. Caspar and A. Klug, Cold Spring Harbor Symp. Quant. Biol., 1962, 27, 1–24 CrossRef CAS PubMed
-
B. V. V. Prasad and M. F. Schmid, in Viral Molecular Machines, ed. M. G. Rossmann and V. B. Rao, Springer, US, Boston, MA, 2012, pp. 17–47 Search PubMed
- R. Twarock and A. Luque, Nat. Commun., 2019, 10, 4414 CrossRef PubMed
- R. Schwartz, R. L. Garcea and B. Berger, Virology, 2000, 268, 461–470 CrossRef CAS PubMed
- S. Sarker, M. C. Terrón, Y. Khandokar, D. Aragão, J. M. Hardy, M. Radjainia, M. Jiménez-Zaragoza, P. J. de Pablo, F. Coulibaly, D. Luque, S. R. Raidal and J. K. Forwood, Nat. Commun., 2016, 7, 13014 CrossRef CAS PubMed
- F. Weis, M. Beckers, I. von der Hocht and C. Sachse, EMBO Rep., 2019, 20, e48451 CrossRef CAS PubMed
- G. Zhao, J. R. Perilla, E. L. Yufenyuy, X. Meng, B. Chen, J. Ning, J. Ahn, A. M. Gronenborn, K. Schulten, C. Aiken and P. Zhang, Nature, 2013, 497, 643–646 CrossRef CAS PubMed
- Q. Fang, W.-C. Tang, A. Fokine, M. Mahalingam, Q. Shao, M. G. Rossmann and V. B. Rao, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203272119 CrossRef CAS PubMed
- P. Kondylis, C. J. Schlicksup, A. Zlotnick and S. C. Jacobson, Anal. Chem., 2019, 91, 622–636 CrossRef CAS PubMed
-
M. Obr and F. K. M. Schur, Advances in Virus Research, Elsevier, 2019, vol. 105, pp. 117–159 Search PubMed
- T. G. W. Edwardson, M. D. Levasseur, S. Tetter, A. Steinauer, M. Hori and D. Hilvert, Chem. Rev., 2022, 122, 9145–9197 CrossRef CAS PubMed
- T. N. Szyszka, E. N. Jenner, N. Tasneem and Y. H. Lau, ChemSystemsChem, 2022, 4, e202100025 CrossRef CAS
- T. W. Giessen, Annu. Rev. Biochem., 2022, 91, 353–380 CrossRef PubMed
-
T. N. Szyszka, L. S. R. Adamson and Y. H. Lau, in Microbial Production of High-Value Products, ed. B. H. A. Rehm and D. Wibowo, Springer International Publishing, Cham, 2022, pp. 309–333 Search PubMed
- Y. Azuma, T. G. W. Edwardson and D. Hilvert, Chem. Soc. Rev., 2018, 47, 3543–3557 RSC
- M. Sutter, M. R. Melnicki, F. Schulz, T. Woyke and C. A. Kerfeld, Nat. Commun., 2021, 12, 3809 CrossRef CAS PubMed
- D. N. Woolfson, J. Mol. Biol., 2021, 433, 167160 CrossRef CAS PubMed
- Y. Hsia, J. B. Bale, S. Gonen, D. Shi, W. Sheffler, K. K. Fong, U. Nattermann, C. Xu, P.-S. Huang, R. Ravichandran, S. Yi, T. N. Davis, T. Gonen, N. P. King and D. Baker, Nature, 2016, 535, 136–139 CrossRef CAS PubMed
-
B. K. Ganser-Pornillos, M. Yeager and O. Pornillos, Viral Molecular Machines, Springer, Boston, MA, 2012, pp. 441–465 Search PubMed
- F. K. M. Schur, W. J. H. Hagen, M. Rumlová, T. Ruml, B. Müller, H.-G. Kräusslich and J. A. G. Briggs, Nature, 2015, 517, 505–508 CrossRef CAS PubMed
- S. Mattei, B. Glass, W. J. H. Hagen, H.-G. Kräusslich and J. A. G. Briggs, Science, 2016, 354, 1434–1437 CrossRef CAS PubMed
- J. S. Rossman and R. A. Lamb, Virology, 2011, 411, 229–236 CrossRef CAS PubMed
- J. Seladi-Schulman, J. Steel and A. C. Lowen, J. Virol., 2013, 87, 13343–13353 CrossRef CAS PubMed
- T. Li, Z. Li, E. E. Deans, E. Mittler, M. Liu, K. Chandran and T. Ivanovic, Nat. Microbiol., 2021, 6, 617–629 CrossRef CAS PubMed
- R. Crowther, Cell, 1994, 77, 943–950 CrossRef CAS PubMed
- D. S. Dane, C. H. Cameron and M. Briggs, Lancet, 1970, 295, 695–698 CrossRef PubMed
- L. M. Stannard and M. Hodgkiss, J. Gen. Virol., 1979, 45, 509–514 CrossRef CAS PubMed
- J. M. Kenney, C.-H. von Bonsdorff, M. Nassal and S. D. Fuller, Structure, 1995, 3, 1009–1019 CrossRef CAS PubMed
- K. A. Dryden, S. F. Wieland, C. Whitten-Bauer, J. L. Gerin, F. V. Chisari and M. Yeager, Mol. Cell, 2006, 22, 843–850 CrossRef CAS PubMed
- N. Martín Garrido, M. A. Crone, K. Ramlaul, P. A. Simpson, P. S. Freemont and C. H. S. Aylett, Mol. Microbiol., 2020, 113, 143–152 CrossRef PubMed
- C. A. McHugh, J. Fontana, D. Nemecek, N. Cheng, A. A. Aksyuk, J. B. Heymann, D. C. Winkler, A. S. Lam, J. S. Wall, A. C. Steven and E. Hoiczyk, EMBO J., 2014, 33, 1896–1911 CrossRef CAS PubMed
- E. Eren, B. Wang, D. C. Winkler, N. R. Watts, A. C. Steven and P. T. Wingfield, Structure, 2022, 30, 551–563 CrossRef CAS PubMed
- S. A. Wynne, R. A. Crowther and A. G. W. Leslie, Mol. Cell, 1999, 3, 771–780 CrossRef CAS PubMed
- W. Wu, N. R. Watts, N. Cheng, R. Huang, A. C. Steven and P. T. Wingfield, PLoS Comput. Biol., 2020, 16, e1007782 CrossRef CAS PubMed
- S. Li, P. Roy, A. Travesset and R. Zandi, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10971–10976 CrossRef CAS PubMed
- T. Dokland, Cell. Mol. Life Sci., 1999, 56, 580–603 CrossRef CAS PubMed
- J. R. Caston, J. L. Martinez-Torrecuadrada, A. Maraver, E. Lombardo, J. F. Rodriguez, J. I. Casal and J. L. Carrascosa, J. Virol., 2001, 75, 10815–10828 CrossRef CAS PubMed
- A. Ona, D. Luque, F. Abaitua, A. Maraver, J. R. Caston and J. F. Rodriguez, Virology, 2004, 322, 135–142 CrossRef CAS PubMed
- B. Oh, C. L. Moyer, R. W. Hendrix and R. L. Duda, Virology, 2014, 456–457, 171–178 CrossRef CAS PubMed
- M. A. Krol, N. H. Olson, J. Tate, J. E. Johnson, T. S. Baker and P. Ahlquist, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13650–13655 CrossRef CAS PubMed
- R. D. Cadena-Nava, M. Comas-Garcia, R. F. Garmann, A. L. N. Rao, C. M. Knobler and W. M. Gelbart, J. Virol., 2012, 86, 3318–3326 CrossRef CAS PubMed
- M. V. de Ruiter, R. M. van der Hee, A. J. M. Driessen, E. D. Keurhorst, M. Hamid and J. J. L. M. Cornelissen, J. Controlled Release, 2019, 307, 342–354 CrossRef CAS PubMed
- G. W. Stevenson, M. Kiupel, S. K. Mittal and C. L. Kanitz, Vet. Pathol., 1999, 36, 368–378 CrossRef CAS PubMed
- C. Rodriguez-Cariñg and J. Segalés, Vet. Pathol., 2009, 46, 729–735 CrossRef PubMed
- J. Mikkilä, A.-P. Eskelinen, E. H. Niemelä, V. Linko, M. J. Frilander, P. Törmä and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed
- I. Seitz, S. Saarinen, E.-P. Kumpula, D. McNeale, E. Anaya-Plaza, V. Lampinen, V. P. Hytönen, F. Sainsbury, J. J. L. M. Cornelissen, V. Linko, J. T. Huiskonen and M. A. Kostiainen, bioRxiv, 2022, preprint DOI:10.1101/2022.11.07.515152.
- H. Tsukamoto, M. Kawano, T. Inoue, T. Enomoto, R. Takahashi, N. Yokoyama, N. Yamamoto, T. Imai, K. Kataoka, Y. Yamaguchi and H. Handa, Genes Cells, 2007, 12, 1267–1279 CrossRef CAS PubMed
- T. Stehle, S. J. Gamblin, Y. Yan and S. C. Harrison, Structure, 1996, 4, 165–182 CrossRef CAS PubMed
- Y. Hu, R. Zandi, A. Anavitarte, C. M. Knobler and W. M. Gelbart, Biophys. J., 2008, 94, 1428–1436 CrossRef CAS PubMed
- C. B. Chang, C. M. Knobler, W. M. Gelbart and T. G. Mason, ACS Nano, 2008, 2, 281–286 CrossRef CAS PubMed
- J. Sun, C. DuFort, M.-C. Daniel, A. Murali, C. Chen, K. Gopinath, B. Stein, M. De, V. M. Rotello, A. Holzenburg, C. C. Kao and B. Dragnea, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1354–1359 CrossRef CAS PubMed
- M. W. O. Liew, Y. P. Chuan and A. P. J. Middelberg, Biochem. Eng. J., 2012, 67, 88–96 CrossRef CAS
- N. H. Dashti, R. S. Abidin and F. Sainsbury, ACS Nano, 2018, 12, 4615–4623 CrossRef CAS PubMed
- D. M. Salunke, D. L. Caspar and R. L. Garcea, Biophys. J., 1989, 56, 887–900 CrossRef CAS PubMed
- S. Kanesashi, K. Ishizu, M. Kawano, S. Han, S. Tomita, H. Watanabe, K. Kataoka and H. Handa, J. Gen. Virol., 2003, 84, 1899–1905 CrossRef CAS PubMed
- J. Nilsson, N. Miyazaki, L. Xing, B. Wu, L. Hammar, T. C. Li, N. Takeda, T. Miyamura and R. H. Cheng, J. Virol., 2005, 79, 5337–5345 CrossRef CAS PubMed
- J. B. Bancroft, C. E. Bracker and G. W. Wagner, Virology, 1969, 38, 324–335 CrossRef CAS PubMed
- E. Hiebert and J. B. Bancroft, Virology, 1969, 39, 296–311 CrossRef CAS PubMed
- J. A. Speir, S. Munshi, G. Wang, T. S. Baker and J. E. Johnson, Structure, 1995, 3, 63–78 CrossRef CAS PubMed
- A. Zlotnick, R. Aldrich, J. M. Johnson, P. Ceres and M. J. Young, Virology, 2000, 277, 450–456 CrossRef CAS PubMed
- L. Lavelle, M. Gingery, M. Phillips, W. M. Gelbart, C. M. Knobler, R. D. Cadena-Nava, J. R. Vega-Acosta, L. A. Pinedo-Torres and J. Ruiz-Garcia, J. Phys. Chem. B, 2009, 113, 3813–3819 CrossRef CAS PubMed
- X. Zhang, P. V. Konarev, M. V. Petoukhov, D. I. Svergun, L. Xing, R. H. Cheng, I. Haase, M. Fischer, A. Bacher, R. Ladenstein and W. Meining, J. Mol. Biol., 2006, 362, 753–770 CrossRef CAS PubMed
- R. Kant, A. Llauró, V. Rayaprolu, S. Qazi, P. J. De Pablo, T. Douglas and B. Bothner, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 1492–1504 CrossRef CAS PubMed
- W. Earnshaw, S. Casjens and S. C. Harrison, J. Mol. Biol., 1976, 104, 387–410 CrossRef CAS PubMed
- M. L. Galisteo and J. King, Biophys. J., 1993, 65, 227–235 CrossRef CAS PubMed
- C. M. Teschke, A. McGough and P. A. Thuman-Commike, Biophys. J., 2003, 84, 2585–2592 CrossRef CAS PubMed
- C. F. Hryc, D.-H. Chen, P. V. Afonine, J. Jakana, Z. Wang, C. Haase-Pettingell, W. Jiang, P. D. Adams, J. A. King, M. F. Schmid and W. Chiu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3103–3108 CrossRef CAS PubMed
- K. N. Parent, R. Khayat, L. H. Tu, M. M. Suhanovsky, J. R. Cortines, C. M. Teschke, J. E. Johnson and T. S. Baker, Structure, 2010, 18, 390–401 CrossRef CAS PubMed
- N. G. A. Abrescia, D. H. Bamford, J. M. Grimes and D. I. Stuart, Annu. Rev. Biochem., 2012, 81, 795–822 CrossRef CAS PubMed
- X. S. Chen, R. L. Garcea, I. Goldberg, G. Casini and S. C. Harrison, Mol. Cell, 2000, 5, 557–567 CrossRef CAS PubMed
- N. Yokoyama, M. Kawano, H. Tsukamoto, T. Enomoto, T. Inoue, R. Takahashi, A. Nakanishi, T. Imai, T. Wada and H. Handa, J. Biochem., 2007, 141, 279–286 CrossRef CAS PubMed
- A. Zlotnick, N. Cheng, J. F. Conway, F. P. Booy, A. C. Steven, S. J. Stahl and P. T. Wingfield, Biochemistry, 1996, 35, 7412–7421 CrossRef CAS PubMed
- S. L. Calhoun, J. A. Speir and A. L. N. Rao, Virology, 2007, 364, 407–421 CrossRef CAS PubMed
- S. B. Larson, R. W. Lucas and A. McPherson, J. Mol. Biol., 2005, 346, 815–831 CrossRef CAS PubMed
- X. F. Dong, P. Natarajan, M. Tihova, J. E. Johnson and A. Schneemann, J. Virol., 1998, 72, 6024–6033 CrossRef CAS PubMed
- J. Tang, J. M. Johnson, K. A. Dryden, M. J. Young, A. Zlotnick and J. E. Johnson, J. Struct. Biol., 2006, 154, 59–67 CrossRef CAS PubMed
- J. Bárcena, N. Verdaguer, R. Roca, M. Morales, I. Angulo, C. Risco, J. L. Carrascosa, J. M. Torres and J. R. Castón, Virology, 2004, 322, 118–134 CrossRef PubMed
- C. Hsu, P. Singh, W. Ochoa, D. J. Manayani, M. Manchester, A. Schneemann and V. S. Reddy, Virology, 2006, 349, 222–229 CrossRef CAS PubMed
- G. L. Lokesh, T. D. S. Gowri, P. S. Satheshkumar, M. R. N. Murthy and H. S. Savithri, Virology, 2002, 292, 211–223 CrossRef CAS PubMed
- J. W. Erickson, A. M. Silva, M. R. N. Murthy, I. Fita and M. G. Rossmann, Science, 1985, 229, 625–629 CrossRef CAS PubMed
- N.-C. Chen, M. Yoshimura, H.-H. Guan, T.-Y. Wang, Y. Misumi, C.-C. Lin, P. Chuankhayan, A. Nakagawa, S. I. Chan, T. Tsukihara, T.-Y. Chen and C.-J. Chen, PLoS Pathog., 2015, 11, e1005203 CrossRef PubMed
- L. Xing, K. Kato, T. Li, N. Takeda, T. Miyamura, L. Hammar and R. H. Cheng, Virology, 1999, 265, 35–45 CrossRef CAS PubMed
- D. Luque, J. M. González, J. Gómez-Blanco, R. Marabini, J. Chichón, I. Mena, I. Angulo, J. L. Carrascosa, N. Verdaguer, B. L. Trus, J. Bárcena and J. R. Castón, J. Virol., 2012, 86, 6470–6480 CrossRef CAS PubMed
- E. Crisci, H. Almanza, I. Mena, L. Córdoba, E. Gómez-Casado, J. R. Castón, L. Fraile, J. Bárcena and M. Montoya, Virology, 2009, 387, 303–312 CrossRef CAS PubMed
- M. A. Asensio, N. M. Morella, C. M. Jakobson, E. C. Hartman, J. E. Glasgow, B. Sankaran, P. H. Zwart and D. Tullman-Ercek, Nano Lett., 2016, 16, 5944–5950 CrossRef CAS PubMed
- Z. Chen, L. Sun, Z. Zhang, A. Fokine, V. Padilla-Sanchez, D. Hanein, W. Jiang, M. G. Rossmann and V. B. Rao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8184–E8193 CAS
- C. Xu, W. Zhu, H. Mao, W. Zhang, G. Yin, X. Zhang and F. Li, Small, 2020, 16, 2004484 CrossRef CAS PubMed
- Y. Li, J. F. Conway, N. Cheng, A. C. Steven, R. W. Hendrix and R. L. Duda, J. Mol. Biol., 2005, 348, 167–182 CrossRef CAS PubMed
- X. Zhang, Y. Liu, B. Zheng, J. Zang, C. Lv, T. Zhang, H. Wang and G. Zhao, Nat. Commun., 2021, 12, 4849 CrossRef CAS PubMed
- M. L. Hasek, J. B. Maurer, R. W. Hendrix and R. L. Duda, J. Mol. Biol., 2017, 429, 2474–2489 CrossRef CAS PubMed
- D. Tso, R. W. Hendrix and R. L. Duda, J. Mol. Biol., 2014, 426, 2112–2129 CrossRef CAS PubMed
- Y. Azuma, M. Herger and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 558–561 CrossRef CAS PubMed
- W. Wang, M. A. Knovich, L. G. Coffman, F. M. Torti and S. V. Torti, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 760–769 CrossRef CAS PubMed
- M. C. Sammarco, S. Ditch, A. Banerjee and E. Grabczyk, J. Biol. Chem., 2008, 283, 4578–4587 CrossRef CAS PubMed
- S. Kang, L. M. Oltrogge, C. C. Broomell, L. O. Liepold, P. E. Prevelige, M. Young and T. Douglas, J. Am. Chem. Soc., 2008, 130, 16527–16529 CrossRef CAS PubMed
- M. L. Ho, J. Judd, B. E. Kuypers, M. Yamagami, F. F. Wong and J. Suh, Cel. Mol. Bioeng., 2014, 7, 334–343 CrossRef CAS
- M. L. Ho, B. A. Adler, M. L. Torre, J. J. Silberg and J. Suh, ACS Synth. Biol., 2013, 2, 724–733 CrossRef CAS PubMed
- E. Johnson, D. Cascio, M. R. Sawaya, M. Gingery and I. Schröder, Structure, 2005, 13, 637–648 CrossRef CAS PubMed
- B. Sana, E. Johnson, P. Le Magueres, A. Criswell, D. Cascio and S. Lim, J. Biol. Chem., 2013, 288, 32663–32672 CrossRef CAS PubMed
- S. Zhang, J. Zang, W. Wang, H. Chen, X. Zhang, F. Wang, H. Wang and G. Zhao, Angew. Chem., Int. Ed., 2016, 55, 16064–16070 CrossRef CAS PubMed
- W. Wang, L. Wang, H. Chen, J. Zang, X. Zhao, G. Zhao and H. Wang, J. Am. Chem. Soc., 2018, 140, 14078–14081 CrossRef CAS PubMed
- S. Zhang, J. Zang, X. Zhang, H. Chen, B. Mikami and G. Zhao, ACS Nano, 2016, 10, 10382–10388 CrossRef CAS PubMed
- A. P. Biela, A. Naskalska, F. Fatehi, R. Twarock and J. G. Heddle, Commun. Mater., 2022, 3, 7 CrossRef PubMed
- P. D. Hempstead, S. J. Yewdall, A. R. Fernie, D. M. Lawson, P. J. Artymiuk, D. W. Rice, G. C. Ford and P. M. Harrison, J. Mol. Biol., 1997, 268, 424–448 CrossRef CAS PubMed
- X. Zhang, W. Meining, M. Fischer, A. Bacher and R. Ladenstein, J. Mol. Biol., 2001, 306, 1099–1114 CrossRef CAS PubMed
- E. Sasaki, D. Böhringer, M. van de Waterbeemd, M. Leibundgut, R. Zschoche, A. J. R. Heck, N. Ban and D. Hilvert, Nat. Commun., 2017, 8, 14663 CrossRef PubMed
- S. Tetter, N. Terasaka, A. Steinauer, R. J. Bingham, S. Clark, A. J. P. Scott, N. Patel, M. Leibundgut, E. Wroblewski, N. Ban, P. G. Stockley, R. Twarock and D. Hilvert, Science, 2021, 372, 1220–1224 CrossRef CAS PubMed
- F. P. Seebeck, K. J. Woycechowsky, W. Zhuang, J. P. Rabe and D. Hilvert, J. Am. Chem. Soc., 2006, 128, 4516–4517 CrossRef CAS PubMed
- B. Wörsdörfer, Z. Pianowski and D. Hilvert, J. Am. Chem. Soc., 2012, 134, 909–911 CrossRef PubMed
- N. Terasaka, Y. Azuma and D. Hilvert, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5432–5437 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |