
Stabilization of Cu+ sites by amorphous Al2O3 to enhance electrochemical CO2 reduction to C2+ products†
Hailian
Cheng
ab,
Shuaiqiang
Jia
*ab,
Jiapeng
Jiao
ab,
Xiao
Chen
ab,
Ting
Deng
ab,
Cheng
Xue
ab,
Mengke
Dong
ab,
Jianrong
Zeng
d,
Chunjun
Chen
ab,
Haihong
Wu
 *ab,
Mingyuan
He
ab and
Buxing
Han
*abc
*ab,
Mingyuan
He
ab and
Buxing
Han
*abc
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, State Key Laboratory of Petroleum Molecular & Process Engineering, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai, 200062, China. E-mail: sqjia@chem.ecnu.edu.cn; hhwu@chem.ecnu.edu.cn; hanbx@iccas.ac.cn
bInstitute of Eco-Chongming, 20 Cuiniao Road, Chenjia Town, Chongming District, Shanghai, 202162, China
cBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China
dShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, 201204, China
First published on 15th January 2024
Abstract
Catalytic conversion of CO2 to produce fuels and chemicals is of great importance, and the electrocatalytic CO2 reduction reaction (eCO2RR) is considered one of the most attractive pathways. Multi-carbon (C2+) products are more desirable in many cases. To date, Cu-based catalysts, especially Cu+ sites, have been found to be the most efficient for the production of C2+ products. However, the retention of Cu+ sites at high cathodic potentials in the eCO2RR remains a great challenge. In this study, we designed and synthesized CuAl-oxide-derived (CuxAly-OD, x and y are the mass percentages of Cu and Al) catalysts for the eCO2RR to C2+ products. During the eCO2RR process, the predominant Cu species changed to Cu+, and the resulting electrocatalyst showed a high faradaic efficiency (FE) of 81.6% for C2+ products in alkaline aqueous solutions. Experimental studies showed that the presence of a stable amorphous Al2O3 phase stabilized the Cu+ sites by oxidation state control, leading to high selectivity and activity for the production of C2+ products. This work provides a strategy for improving the stability of Cu+ in the catalyst to enhance the performance of the eCO2RR.
Introduction
The catalytic conversion of CO2 to produce fuels and chemicals is of great importance, as it ameliorates both environmental concerns and the energy crisis.1–4 The electrocatalytic CO2 reduction reaction (eCO2RR), in particular, is considered one of the most attractive pathways for carbon cycle rebalancing due to its mild operating conditions, ease of operation, product adjustment, and potential for synergy with renewable energy sources.5–8 To date, Cu-based catalysts have been found to be the most efficient for the production of multi-carbon (C2+) hydrocarbons and oxygenated products.9–11 However, Cu-based catalysts still have certain limitations, including poor stability,12 poor selectivity for specific products, and competition from the hydrogen evolution reaction (HER).13To address these challenges, various strategies have been employed to improve the selectivity, activity, and stability of Cu-based catalysts for the eCO2RR, including surface treatment of Cu electrodes,14–16 alloying,17–20 doping of other metal elements to modulate the electronic structure21–23 and formation of coordination compounds.24–29 It is noteworthy that in order to stabilize the active sites of Cu catalysts, researchers have attempted to use metal–organic frameworks (MOFs) with specially designed ligands to achieve the stabilization of the Cu2+ site,16,30 but unfortunately, the Cu-MOF catalysts reported so far did not continuously stabilize Cu2+ during electrolysis but were electrochemically reduced to Cu0.31 At the same time, it has also been reported that Cu2+ is incorporated into oxide matrices to form Cu–M–O solid solutions in order to stabilize Cu2+ during the eCO2RR process.32 However, most of the target products obtained by the above strategy of stabilizing Cu2+ active sites were methane or other monocarbon products. Therefore, the development of catalysts with stabilized Cu+ sites for enhancing the dimerization of the reaction intermediate *CO and hence the selectivity of higher value-added C2+ products remains challenging.
Here, we prepared a catalyst capable of stabilizing the Cu+ site in the eCO2RR process by introducing the metal Al to form a CuxAly-OD nanosheet catalyst. Remarkably, using the Cu100Al7.88-OD catalyst as an electrode in a flow cell with a 1 M KOH electrolyte, we obtained a total current density of 692.6 mA cm−2 with a faradaic efficiency (FE) of 81.6% for C2+ products, which was twice as high as that of FEC2+ over a pure CuO-OD catalyst. In situ Raman spectroscopy measurements and X-ray absorption spectroscopy (XAS) experiments showed that the broadened orbital in interfacial Al2O3 offers a lower orbital for extra electrons than Cu+, which can effectively retain nearby Cu+, and the highly active Cu+ sites significantly enhance C–C coupling, improving the selectivity and activity of the eCO2RR to C2+ products.
Results and discussion
The synthesis of CuxAly-OD electrodes involves a two-step process (Fig. 1a). First, the CuxAly-oxide (named CuxAly-Ox) catalysts were prepared by the co-precipitation of CuCl2·2H2O and AlCl3·6H2O salts (the details of the synthesis method are provided in the ESI†), and then transferred to a hydrothermal reactor and placed in a 100 °C oven for 18 h. Further CuxAly-OD electrodes were obtained by the redox reaction cyclic voltammetry (CV) technique. The mass percentages of Cu and Al in the as-synthesized CuxAly-Ox catalysts were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) (Table S1†). The Cu100Al7.88-Ox catalyst was used as a representative example. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images showed that Cu100Al7.88-Ox exhibited a two-dimensional nanosheet morphology along with a rough edge structure (Fig. 1b and c). According to the ESI Fig. S1,† with the increase of Al doping, the morphology of the catalyst changed from a two-dimensional nanosheet structure to a flower-like structure. The high-resolution transmission electron microscopy (HRTEM) image (Fig. 1d) showed that the lattice fringes in the Cu100Al7.88-Ox catalyst were 0.250 nm and 0.274 nm, corresponding to the distance of the (111) and (110) planes of CuO, respectively.33,34 It is noteworthy that no lattice fringes belonging to Al were found in HRTEM images, which may be because Al2O3 in the catalyst was an amorphous physical phase (Fig. S2 and S3†). Meanwhile, from the energy-dispersive X-ray spectroscopy (EDS) elemental mapping images (Fig. 1e), Cu, Al, and O were uniformly distributed in the Cu100Al7.88-Ox catalyst. | ||
| Fig. 1 Preparation and characterization of catalysts. (a) Schematic illustration of the process to prepare CuxAly-Ox catalysts. (b) SEM, (c) TEM, (d) HRTEM, and (e) EDS elemental mapping images of the Cu100Al7.88-Ox catalyst. | ||
The electronic and chemical structures of the as-synthesized catalysts were characterized by different methods. The X-ray diffraction (XRD) patterns of the CuxAly catalysts before (CuxAly-Ox) and after CV activation (CuxAly-OD) are shown in Fig. 2a and b. It can be seen that before CV activation, the Cu phase of all catalysts was only CuO (JCPDS#48-1548). However, after CV activation the pure CuO catalyst changed into Cu0, whereas the CuxAly-OD catalyst was Cu2O (JCPDS#05-0667). It is noteworthy that we did not find the diffraction peaks of Al in the XRD pattern, probably due to the insufficient crystallinity of Al2O3 in the catalysts prepared by this method, which existed as an amorphous phase.35 X-ray photoelectron spectroscopy (XPS) experiments were used to gain insight into the electronic interaction between Cu and Al as well as the surface chemical states of each element in Cu100Al7.88-Ox and Cu100Al7.88-OD catalysts. Fig. 2c shows that the valence state of Cu in both CuO and Cu100Al7.88-Ox catalysts is Cu2+ accompanied by strong satellite peaks. After CV activation (Fig. 2d), the CuO electrode was easily reduced to Cu0 in the catalyst without Al doping, while CuxAly-OD mostly retained Cu+. The XPS spectra of Al 2p showed that Al was present in the physical phase of Al2O3 both before and after the activation of the Cu100Al7.88-Ox catalyst (Fig. S4†). Combined with the XRD and XPS results, it can be observed that the CuO catalyst was reduced to metallic Cu0, which proved that Cu2+ without oxidation state control protection can be easily reduced, further illustrating the importance of amorphous Al2O3 in stabilizing the Cu+ site.
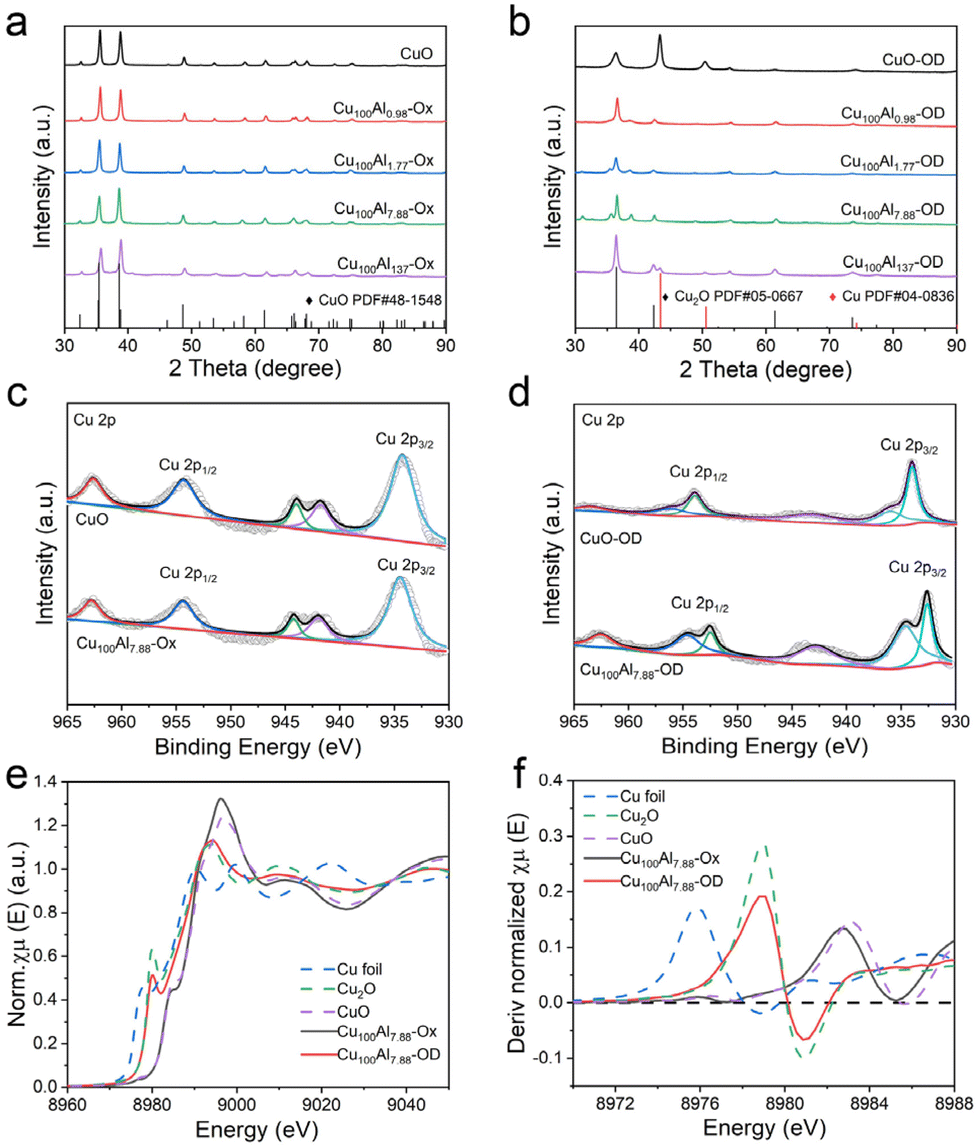 | ||
| Fig. 2 XRD patterns of CuO and CuxAly-Ox catalysts (a) before and (b) after CV activation in a CO2-saturated 1 M KOH electrolyte. Cu 2p XPS spectra of CuO and Cu100Al7.88-Ox catalysts (c) before CV activation and (d) after CV activation in a CO2-saturated 1 M KOH electrolyte. (e) Normalized Cu K-edge XANES spectra and (f) derivative normalized spectra of different catalysts. For comparison, reference spectra from a Cu foil, Cu2O, and CuO are also shown. | ||
To further investigate the chemical state and electronic structure of Cu in the Cu100Al7.88-Ox and Cu100Al7.88-OD catalysts, synchrotron X-ray absorption spectroscopy (XAS) was performed on the catalysts prepared in this work, and the spectra of Cu foil, Cu2O and CuO as references were obtained. As shown in Fig. 2e, normalized X-ray absorption near edge structure spectra (XANES) of the Cu K-edge showed that the Cu species in Cu100Al7.88-Ox exhibited an oxidized state (+2), the same as the CuO reference sample, whereas the Cu species in Cu100Al7.88-OD exhibited the same +1 valence state as the Cu2O reference sample. Moreover, this was further confirmed by the corresponding derivative normalized spectra (Fig. 2f) and the k3-weighted Cu K edge Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) spectra (Fig. S5†), which indicated that the presence of amorphous Al2O3 could stabilize Cu+ to prevent its reduction to Cu0. This is consistent with the results of the XPS and XRD analyses.
The eCO2RR catalytic performances of CuxAly-OD catalysts were tested in a flow cell. 1 M KOH was used as the electrolyte, and the catalysts were loaded onto a gas diffusion layer (GDL). Gaseous products were probed and quantified during the eCO2RR using gas chromatography (GC) and liquid products were examined after the completion of the electrochemical reaction via1H nuclear magnetic resonance (NMR). The linear sweep voltammetry (LSV) curves were measured in CO2- and Ar-saturated 1 M KOH electrolytes, the Cu100Al7.88-OD electrode exhibited a higher current density in the CO2-saturated electrolyte compared to the results in the Ar-saturated electrolyte, indicating the occurrence of an eCO2RR on the Cu100Al7.88-OD electrode (Fig. 3a). We tested all the CuxAly-OD and CuO catalysts, including CuO-OD, Cu100Al0.98-OD, Cu100Al1.77-OD, Cu100Al7.88-OD, and Cu100Al137-OD, and compared their eCO2RR performances. As shown in Fig. 3b, gaseous products (H2, CO, CH4, and C2H4) and liquid products (HCOOH, CH3COOH, CH3CH2OH (EtOH), and CH3CH2CH2OH (PrOH)) were detected in the present catalytic system. According to the product distribution, the decreasing FE of H2 and increasing FE of carbon products become obvious after Al doping into Cu catalysts. At a potential of −1.68 V vs. RHE, the FE of C2+ products over the Cu100Al7.88-OD electrode could reach 81.6% with a current density as high as 692.6 mA cm−2, in which the partial current density of C2+ products could reach 565.2 mA cm−2 (Fig. S6†). The catalyst maintained a high C2+ yield as the applied potential increased. In contrast, pure CuO catalysts showed a maximum FEC2+ of only 41.8% compared to a potential of −1.68 V vs. RHE, and produced more H2 and C1 products, respectively. Fig. 3c shows the FEC2+ and jtotal over different CuxAly-OD electrodes at −1.68 V vs. RHE, and it was found that the amount of Al doping was crucial for the selectivity of the C2+ product. A comparison of the electrocatalytic performances of the catalysts with different Cu/Al mass ratios revealed that the C2+ product exhibited good FE and partial current density at the Cu100Al7.88-OD electrode (Fig. S7–S10†). In addition to catalytic activity, stability is also one of the important criteria for evaluating catalyst performance. Subsequently, we conducted a stability test in a flow cell (Fig. 3d), in which the current density of the catalyst and FEC2+ remained unchanged after 8 h of electrolysis at −1.68 V vs. RHE, indicating that Cu100Al7.88-OD had a good stability for the eCO2RR.
 | ||
| Fig. 3 Electrochemical performances of the CuxAly-OD catalyst. (a) LSV curves for Cu100Al7.88-OD in CO2-saturated and Ar-saturated 1 M KOH electrolytes. (b) FE of the products over Cu100Al7.88-OD at different applied potentials. (c) FEC2+ and total current densities over CuxAly-OD at −1.68 V vs. RHE. (d) Electrochemical stability test of the Cu100Al7.88-OD electrode at −1.68 V vs. RHE in a flow cell. (e) Comparison of double-layer capacitance in a 1 M KOH electrolyte with CuxAly-OD. (f) Nyquist plots of CuxAly-OD in the 1 M KOH electrolyte. Data were obtained at ambient temperature and pressure with a CO2 stream of 20 sccm. | ||
To explore the reasons for the excellent performance of the Cu100Al7.88-OD electrode, electrochemically active surface area (ECSA) and electrochemical impedance spectra (EIS) of CuxAly-OD electrodes were analyzed. The ECSA of the catalyst was then estimated through the electrochemical double-layer capacitance (Cdl) measurements. The CV curves with various scan rates in the non-faradaic region were recorded. As shown in Fig. 3e, the linear slopes show that the Cdl value for the Cu100Al7.88-OD electrode (18.65 mF cm−2) is larger than those of the other catalysts, which demonstrates that doping of a suitable amount of Al can generate more active sites than the pure Cu catalyst. The EIS was also carried out to probe the effect of the metal ratios of the catalysts on the charge transport kinetics at the open circuit potential (OCP). It showed that the charge transfer resistance of the Cu100Al7.88-OD electrode was lower than that of other catalysts, indicating a favorable kinetics toward the eCO2RR (Fig. 3f).36
To gain insight into characterizing both the structural evolution of catalyst surfaces and reaction intermediates at electrode/electrolyte interfaces during electrochemical reactions, in situ Raman spectroscopy was used to monitor the change of interfacial species and highly active sites of the Cu100Al7.88-OD electrode under different electrolysis times and potentials (Fig. 4). As shown in Fig. 4a, after prolonged electrolysis, the Cu100Al7.88-OD electrode showed a Raman peak at 620 cm−1, which was attributed to Cu2O.37 This absorption peak corresponds to the peak at this location under OCP conditions and remains stable for more than 3000 s of continuous electrolysis, indicating that the Cu+ site is stable during the eCO2RR. The in situ Raman results demonstrate that the broadened orbital in interfacial amorphous Al2O3 offers a lower orbital for extra electrons than Cu+, which can effectively retain nearby Cu+.38,39 It is worth noting that the catalyst was not exposed to ambient air during the Raman test, so the Cu+ site was generated after CV activation and remained stable during the eCO2RR process. The retention of the band at around 530 cm−1 for Cu-OH can be explained by the increase of the local pH near the catalyst surface at high reaction potentials (−1.68 V vs. RHE).40 Interestingly, the Raman peaks at 1330 cm−1 belong to the CO2− in-stretching vibrations, which confirms the first intermediate during the eCO2RR over the Cu100Al7.88-OD electrode.41 The peaks around 1880 cm−1 are assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching vibrations. Further investigation of the mechanism of the C2+ products from the Cu100Al7.88-OD electrode is very interesting. In particular, the peaks located in the range of 1800–2000 cm−1 are associated with the bridge site of *CO absorption (*CObridge), which can favor *CO–CO coupling.42
O stretching vibrations. Further investigation of the mechanism of the C2+ products from the Cu100Al7.88-OD electrode is very interesting. In particular, the peaks located in the range of 1800–2000 cm−1 are associated with the bridge site of *CO absorption (*CObridge), which can favor *CO–CO coupling.42
 | ||
| Fig. 4 In situ Raman spectra of (a) the Cu100Al7.88-OD electrode at −1.68 V vs. RHE with different reaction times during the eCO2RR in a 0.5 M KOH electrolyte and (b) the Cu100Al7.88-OD electrode at various potentials during the eCO2RR in a 0.5 M KOH electrolyte. All potentials are given vs. RHE. | ||
The in situ Raman spectra of the Cu100Al7.88-OD electrode at various potentials are shown in Fig. 4b; a Raman peak at 1012 cm−1 (representing a small amount of bicarbonate) and a Raman peak at 1068 cm−1 (representing carbonate) were detected in the Cu100Al7.88-OD electrode. According to previous studies, the density of localized proton donors near the catalyst surface can be estimated from the ratio of HCO3−/CO32−.43 As the applied potential decreases, the HCO3−/CO32− ratio increases, indicating a decrease in proton donors near the Cu100Al7.88-OD electrode surface. These alkaline conditions promote the formation of C2+ products, which is consistent with our electrochemical results (Fig. 3c).44,45
Conclusions
In summary, we have proposed a method to stabilize the Cu+ oxidation state with a suitable amount of Al2O3, and the catalyst can promote CO2 electroreduction to C2+ products with high catalytic activity, selectivity, and stability. Combined in situ spectroscopic and electrochemical measurements indicate that Cu+ stabilized by the amorphous Al2O3 enhances the adsorption stability of the *CO intermediate, which in turn promotes its further dimerization (C–C coupling) and thus significantly improves the selectivity for C2+ products. This discovery effectively solves the stability problem of Cu-based catalysts for the eCO2RR and provides an effective way to design low-cost, energy-efficient catalysts.Author contributions
H. L. C., S. Q. J., H. H. W. and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. H. L. C. and S. Q. J. performed most of the experiments and analyzed the experimental data. J. P. J., X. C., T. D., C. X. M. K. D. and C. J. C. provided help in materials synthesis and characterization. H. L. C., S. Q. J., X. C. and J. R. Z. conducted XAS measurements and analyzed the results. H. H. W., M. Y. H. and B. X. H. co-supervised the whole project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Key R&D Program of China (2023YFA1507901, 2020YFA0710201), the National Natural Science Foundation of China (22003070, 22293015, 22121002), the China Postdoctoral Science Foundation (2023M731096), and the Research Funds of Happiness Flower ECNU (2020ST2203). We thank the staff of beamline BL13SSW at Shanghai Synchrotron Radiation Facility for the support for the experiments of in situ XAS measurements.References
- R. E. Sharwood, Science, 2022, 378, 137–138 CrossRef CAS PubMed.
- Z. Zhang, T. N. Borhani and A. G. Olabi, Energy, 2020, 205, 118057 CrossRef CAS.
- M. E. Chu, C. J. Chen, Y. H. Wu, X. P. Yan, S. Q. Jia, R. T. Feng, H. H. Wu, M. Y. He and B. X. Han, Green Energy Environ., 2022, 7, 792–798 CrossRef CAS.
- S. Jia, Q. Zhu, M. Chu, S. Han, R. Feng, J. Zhai, W. Xia, M. He, H. Wu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 10977 CrossRef CAS PubMed.
- G. Zhang, S. D. Straub, L. Shen, Y. Hermans, P. Schmatz, A. M. Reichert, J. P. Hofmann, I. Katsounaros and B. J. M. Etzold, Angew. Chem., Int. Ed., 2020, 59, 18095–18102 CrossRef CAS PubMed.
- X. X. Chang, M. He, Q. Lu and B. J. Xu, Sci. China: Chem., 2023, 66, 96–106 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- D. Yang, X. Liu, Y. H. Dai, Y. Zhu and Y. H. Yang, Chem. J. Chin. Univ., 2022, 43, 20220198 Search PubMed.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- T. R. Wei, S. S. Zhang, Q. Liu, Y. Qiu, J. Luo and X. J. Liu, Acta Phys.-Chim. Sin., 2023, 39, 2207026 Search PubMed.
- L. Xu, J. Feng, L. Wu, X. Song, X. Tan, L. Zhang, X. Ma, S. Jia, J. Du, A. Chen, X. Sun and B. Han, Green Chem., 2023, 25, 1326–1331 RSC.
- H. Zhang, C. He, S. Han, Z. Du, L. Wang, Q. Yun, W. Cao, B. Zhang, Y. Tian and Q. Lu, Chin. Chem. Lett., 2022, 33, 3641–3649 CrossRef CAS.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard III and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- O. Christensen, S. Zhao, Z. Sun, A. Bagger, J. V. Lauritsen, S. U. Pedersen, K. Daasbjerg and J. Rossmeisl, ACS Catal., 2022, 12, 15737–15749 CrossRef CAS.
- Z. Xin, Z. Yuan, J. Liu, X. Wang, K. Shen, Y. Chen and Y.-Q. Lan, Chin. Chem. Lett., 2023, 34, 107458 CrossRef CAS.
- S. J. Kang, J. H. Won, H. Choi, W. Sim, M. K. Kim, S. Sultan, Y. Kwon and H. M. Jeong, J. Energy Chem., 2022, 66, 68–73 CrossRef CAS.
- C. Choi, J. Cai, C. Lee, H. M. Lee, M. Xu and Y. Huang, Nano Res., 2021, 14, 3497–3501 CrossRef CAS.
- Y. Xu, C. Li, Y. Xiao, C. Wu, Y. Li, Y. Li, J. Han, Q. Liu and J. He, ACS Appl. Mater. Interfaces, 2022, 14, 11567–11574 CrossRef CAS PubMed.
- X. Zhang, Y. Zhou, H. Zhang, H. Li, K. Liu, H. Li, H. Pan, J. Hu, J. Fu, S. Chen and M. Liu, J. Energy Chem., 2021, 63, 625–632 CrossRef CAS.
- Y. Ji and G. Zheng, Sci. China: Chem., 2021, 64, 1111–1112 CrossRef CAS.
- Z. Zhao, X. Li, J. Wang, X. Lv and H. B. Wu, J. CO2 Util., 2021, 54, 101741 CrossRef CAS.
- J. Chen, X. Wei, R. Cai, J. Ren, M. Ju, X. Lu, X. Long and S. Yang, ACS Mater. Lett., 2022, 4, 497–504 CrossRef CAS.
- Z. Zhang, H. Tian, L. Bian, S. Liu, Y. Liu and Z. Wang, J. Energy Chem., 2023, 83, 90–97 CrossRef CAS.
- J. Wang, H. Tan, Y. Zhu, H. Chu and H. Chen, Angew. Chem., Int. Ed., 2021, 60, 17254–17267 CrossRef CAS PubMed.
- T. C. Chou, C. C. Chang, H. L. Yu, W. Y. Yu, C. L. Dong, J. J. Velasco-Vélez, C. H. Chuang, L. C. Chen, J. F. Lee, J. M. Chen and H. L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- Z. Wu, F. Gao and M. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- H. Xiao, W. A. Goddard 3rd, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou, P. Zapol, J. Cabana, L. A. Curtiss and A. Salehi-Khojin, Adv. Mater., 2021, 33, 2004393 CrossRef CAS PubMed.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- J. Jang, K. Lee, H. Shin, H. S. Lee, B.-H. Lee, J. Jeong, J. Kim, W. Hwang, S. Park, M. S. Bootharaju, S. Back, J. Shim, J. H. Kim, T. Hyeon and Y. Sung, J. Mater. Chem. A, 2023, 11, 19066–19073 RSC.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS PubMed.
- T. N. Nguyen, Z. Chen, A. S. Zeraati, H. S. Shiran, S. M. Sadaf, M. G. Kibria, E. H. Sargent and C. T. Dinh, J. Am. Chem. Soc., 2022, 144, 13254–13265 CrossRef CAS PubMed.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustainable Chem., 2022, 34, 100589 CrossRef CAS.
- B. Jia, L. Li, C. Xue, J. Kang, L. Liu, T. Guo, Z. Wang, Q. Huang and S. Guo, Adv. Mater., 2023, 35, 2305587 CrossRef CAS PubMed.
- S. Sultan, H. Lee, S. Park, M. M. Kim, A. Yoon, H. Choi, T.-H. Kong, Y.-J. Koe, H.-S. Oh, Z. Lee, H. Kim, W. Kim and Y. Kwon, Energy Environ. Sci., 2022, 15, 2397–2409 RSC.
- G. Kastlunger, L. Wang, N. Govindarajan, H. H. Heenen, S. Ringe, T. Jaramillo, C. Hahn and K. Chan, ACS Catal., 2022, 12, 4344–4357 CrossRef CAS.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- M. Moradzaman and G. Mul, ChemElectroChem, 2021, 8, 1478–1485 CrossRef CAS.
- C. Liu, X. Zhang, J. Huang, M. Guan, M. Xu and Z. Gu, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS.
- P. Li, J. Bi, J. Liu, Y. Wang, X. Kang, X. Sun, J. Zhang, Z. Liu, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04492k |
| This journal is © The Royal Society of Chemistry 2024 |