
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect-engineered graphitic carbon nitride on carbon cloth supports for the photoelectrocatalytic degradation of organophosphate pesticides†
Giacomo
Marchiori
a,
Roberta
Seraglia
b,
Gian Andrea
Rizzi
ab,
Chiara
Maccato
 *ab,
Mattia
Benedet
ab,
Emanuela
Callone
c,
Sandra
Dirè
c,
Alberto
Gasparotto
ab and
Davide
Barreca
b
*ab,
Mattia
Benedet
ab,
Emanuela
Callone
c,
Sandra
Dirè
c,
Alberto
Gasparotto
ab and
Davide
Barreca
b
aDepartment of Chemical Sciences – Padova University and INSTM, Via Marzolo, 1 – 35131 Padova, Italy. E-mail: chiara.maccato@unipd.it
bCNR-ICMATE and INSTM – Department of Chemical Sciences – Padova University, Via Marzolo 1, and Corso Stati Uniti 4 – 35127, Padova, Italy
c“Klaus Müller” Magnetic Resonance Laboratory, Department of Industrial Engineering – Trento University, Via Sommarive, 9 – 38123 Trento, Italy
First published on 1st October 2024
Abstract
The effective degradation of persistent aqueous pollutants, such as fenitrothion (FNT), a widely used organophosphate pesticide, represents a major urgency for the protection of human health and the environment. In this regard, this study is focused on the fabrication of green photoelectrocatalysts based on graphitic carbon nitride (gCN), capable of generating hydrogen peroxide (H2O2) to trigger electro-Fenton processes for FNT degradation. In particular, electrophoretic deposition of gCN onto carbon cloth (CC) substrates was performed starting from gCN powders designed via thermal condensation of urea mixed with acetylacetone (AcAc). The resulting defect engineering promoted an improved gCN light harvesting capability and an enhanced separation of photogenerated charge carriers. The obtained supported materials featured an attractive electrochemical reactivity and operational stability, opening the door to their possible real-world end-use. The present work illustrates, as a proof-of-concept, the potential of gCN-based photoelectrocatalysts in water treatment technologies, offering a sustainable solution in a greener perspective to mitigate the environmental impact of hazardous pollutants.
1. Introduction
The presence in the environment and in water of organophosphate compounds, widely used in agriculture as pesticides, has raised a significant concern due to their harmful effects on animal and human health even at trace levels.1,2 These issues, along with their persistence against the most common treatments, have stimulated extensive efforts aimed at the development of methods enabling their efficient monitoring and removal.3,4 To this aim, an attractive solution is provided by advanced oxidation processes (AOPs),5 which are based on the generation of reactive oxygen species (ROS, among which ˙OH) promoting the oxidative decontamination of polluted aqueous solutions. Among the most successful AOPs, electro-Fenton (EF) routes offer various concurrent advantages for the degradation of numerous organic contaminants, encompassing fertilizers, pesticides, dyes, surfactants, and drugs active ingredients.6In view of real-world applications of EF processes, the development of low-cost, abundant, active and metal-free catalysts is highly required,7 since systems based on precious metals have a high environmental impact and also suffer from low selectivity and durability.8 In this regard, an appealing alternative is offered by 2D materials and, in particular, by graphitic carbon nitride (gCN; EG ≈ 2.7 eV). The latter, a low-cost semiconductor comprising naturally abundant elements, is a rising star in the field of photo(electro)catalysis thanks to the non-toxicity, the capability of absorbing Vis radiation, and the broad variability of morphological, structural, and electronic properties.9 In fact, the chemico-physical characteristics of gCN-based materials are directly dependent on the adopted synthetic routes, usually involving thermal condensation of N-containing organic precursors (such as urea, melamine, cyanamide and thiourea).9 Various studies have so far been focused on gCN photo(electro)catalysts for water and air purification, CO2 reduction, H2O2 production, and water splitting,10–14 but, to our knowledge, the use of such systems for the degradation of FNT, an aromatic organophosphate pesticide,15 has never been reported to date.
In the present work, gCN was used to develop photocathodes for the efficient generation of H2O2 to foster EF processes for FNT degradation. The target systems were synthesized starting from urea alone and from urea mixed with AcAc. The functionalization with AcAc was aimed at graphitic carbon nitride defect engineering,16,17 in order to favourably boost the resulting photoelectrocatalytic activity. The corresponding powdered materials were used as precursors for the preparation of supported photoelectrocatalysts via electrophoretic deposition (EPD) (Scheme 1), an amenable route utilized in our research group to fabricate a variety of gCN-based electrocatalysts for the oxygen evolution reaction (OER).18–20 The target systems were grown on flexible CC substrates, ensuring an excellent electrical conductivity and good corrosion resistance.21 To assess the influence of AcAc introduction on the system properties, both chemico-physical characterization and functional (photo)electrochemical tests were carried out on both gCN containing AcAc and bare gCN obtained by urea as such. The monitoring of FNT degradation during EF experiments was performed by high performance liquid chromatography-mass spectrometry (HPLC-MS), analysing the working solutions as a function of time during degradation experiments. In the following, particular emphasis is given to the interrelations between material structure, composition, morphology, optical properties, and functional performances.
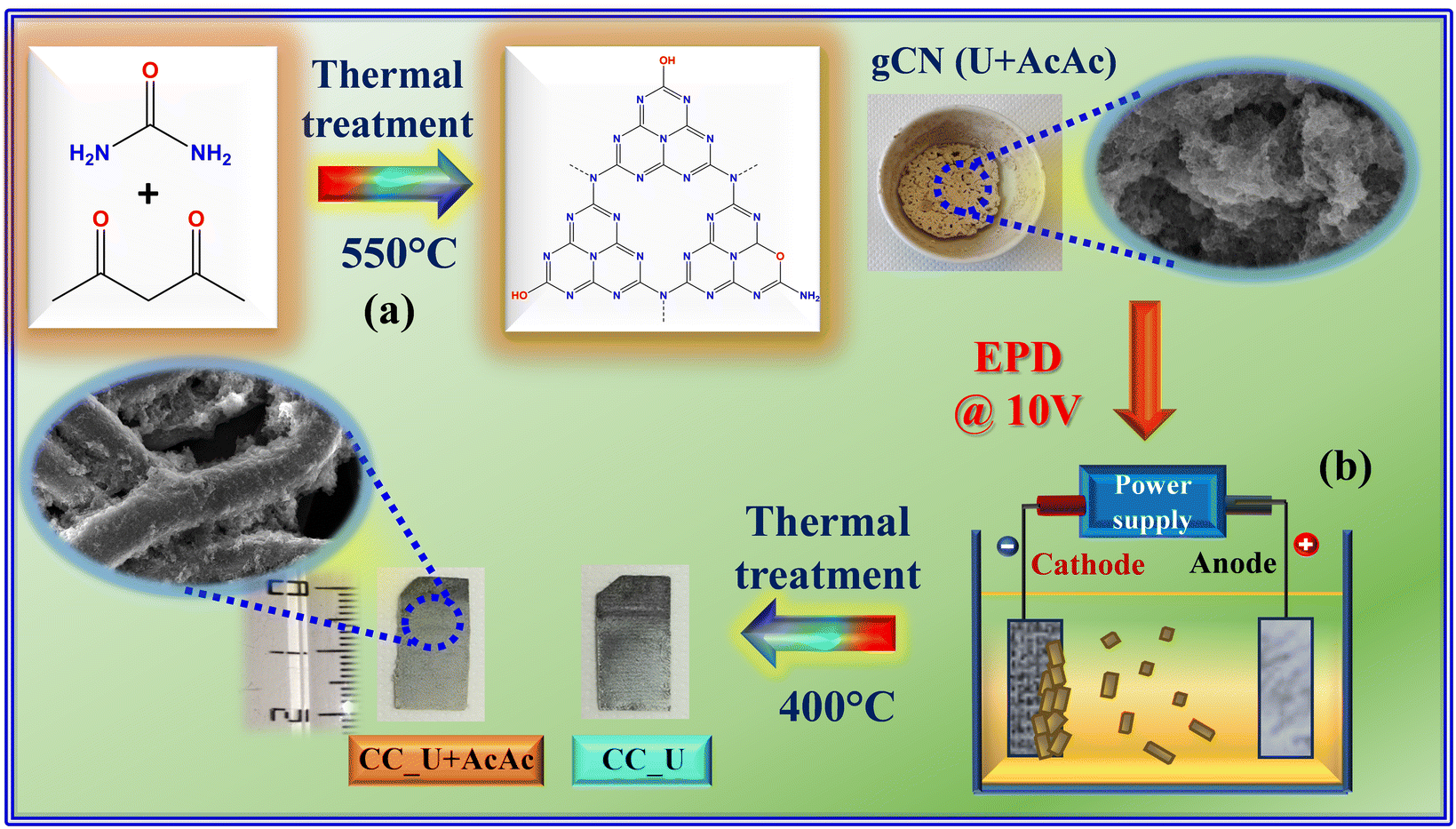 | ||
| Scheme 1 Sketch of the procedure adopted in the present work for (a) the synthesis of gCN powders and (b) the subsequent preparation of gCN-containing electrode materials. U = urea; AcAc = acetylacetone; CC = carbon cloth; EPD = electrophoretic deposition. The suspensions for EPD were prepared by dispersing 40 mg of finely grinded carbon nitride powders into a beaker containing 50 mL of acetone and 4 mg of freshly dissolved I2 (Sigma-Aldrich, ≥99.8%), followed by sonication for 30 min. For more details on the experimental procedures, see ESI,† §S1. | ||
2. Experimental
2.1 Synthesis
gCN powders were synthesized following previous work,16,17 and performing a subsequent optimization of the thermal treatment conditions. Deposition onto CC substrates was carried out via EPD using previously reported operational parameters18,22 (see also ESI,† §S1). Henceforth, powders obtained from urea alone and from urea and acetylacetone will be referred to as gCN (U) and gCN (U + AcAc), whereas the corresponding CC-supported specimens will be denoted as CC_U and CC_U + AcAc, respectively.2.2 Characterization
X-ray diffraction (XRD) measurements were collected using a Bruker AXS D8 Advance Plus diffractometer, equipped with a Cu Kα X-ray source (λ = 1.54 Å). The average crystal size was estimated through the Scherrer equation. Analyses were performed at the PanLab facility (Department of Chemical Sciences, Padova University) founded by MIUR Dipartimento di Eccellenza grant “NExuS”. Fourier transform-infrared (FT-IR) diffuse reflectance spectra were recorded by diluting gCN within dried KBr powders, using a Jasco FT/IR-4100 instrument (resolution = 4 cm−1). UV-Vis diffuse reflectance and photoluminescence (PL) spectra were acquired on an FLS1000 (Edinburgh Instruments) spectrophotometer (excitation wavelength = 330 nm; spectral bandwidth = 1 nm). Band gap (EG) values were estimated using the Tauc equation – plots of [F(R)hν]nvs. hν, where F(R) is the Kubelka–Munk function, hν is the photon energy, and R is the measured reflectance, assuming indirect and allowed transitions (n = 1/2).2313C and 15N solid state nuclear magnetic resonance (NMR) analyses were carried out with a Bruker Avance 400WB spectrometer with a cross polarization/magic angle spinning (CPMAS) double-band 4 mm probe (ESI†, §S.2).X-ray photoelectron spectroscopy (XPS) analysis was performed using an ESCALAB™ QXi spectrometer funded by “Sviluppo delle infrastrutture e programma biennale degli interventi del Consiglio Nazionale delle Ricerche (2019)”, at a working pressure of ≈5 × 10−9 mbar, using a monochromatized Al Kα X-ray source (hv = 1486.6 eV). Binding energy (BE) values were corrected for charging by assigning a value of 284.8 eV to the adventitious C1s component. Atomic percentages (at%) were calculated by peak area integration, using ThermoFisher sensitivity factors. Peak fitting was carried out by XPSPEAK (version 4.1) software,24 using Gaussian–Lorentzian sum functions. Field emission scanning electron microscopy (FE-SEM) analyses were carried out using a Zeiss SUPRA 40VP instrument, at primary beam accelerating voltages between 5 and 20 kV. Photoelectrochemical and electro-Fenton degradation tests were conducted by employing the prepared supported materials as working electrodes in 0.1 M Na2SO4 aqueous solution, in a three-electrode configuration (see also Fig. S1†). A HPLC-MS apparatus, equipped with an electrospray ionization (ESI) ion source, was employed to monitor FNT degradation. Further data on characterization and functional tests are reported in the ESI† (§S2).
3. Results and discussion
In this work, attention was first dedicated to the FT-IR characterization of the starting powders, revealing the formation of carbon nitride-based materials containing an appreciable amount of –NHx (x = 1, 2) and –OH groups (see also Fig. S2, Table S1 and ESI,† page S8–S9). Accordingly, the powder XRD patterns (Fig. 1a) were dominated by a peak at 2θ = 27.5° attributed to the interplanar (002) stacking of gCN sheets, whereas the low-intensity signal at 2θ = 13.1° was ascribed to the periodic arrangement of tri-s-triazine units in (100) crystallographic planes.25–28 According to the Scherrer equation, both samples featured an average crystallite size of ≈5 nm. The lower intensity of both signals in the gCN (U + AcAc) sample was attributed to AcAc functionalization, resulting in a higher content of defects responsible, in turn, for a decreased crystallinity.16,17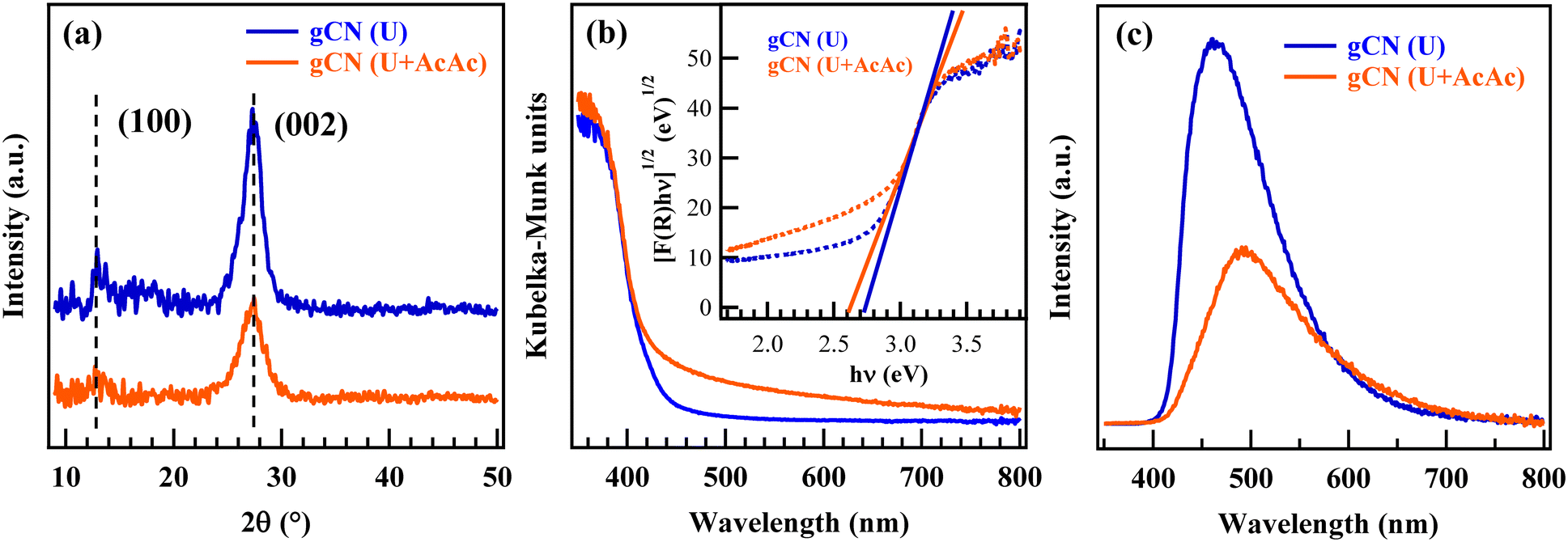 | ||
| Fig. 1 Physico-chemical characterization of gCN (U) and gCN (U + AcAc) powders: (a) XRD patterns; (b) UV-Vis spectra and (inset) Tauc plots; (c) PL spectra. | ||
Optical absorption spectra (Fig. 1b) were dominated by a strong absorption for λ < 440 nm, associated with gCN interband electronic transitions.25,27,28 Different from gCN (U), gCN (U + AcAc) also displayed a moderate, but well detectable, absorption tail at λ > 440 nm, which was ascribed to the presence of inter-band states due to AcAc functionalization, associated with defect formation (see also XPS data below).16,17 The extrapolated EG values (Fig. 1b, inset) were 2.74 and 2.62 eV for gCN (U) and gCN (U + AcAc), respectively, confirming a broadened optical absorption range for the latter system,16,17 a favourable issue for an improved Vis-light exploitation. PL spectra (Fig. 1c) displayed a strong fluorescence emission, centred at ≈470 and 500 nm for gCN (U) and gCN (U + AcAc), respectively, due to the radiative relaxation of excited electrons from the conduction to the valence band.25–28 The red shift of the emission maximum could be related to the lower gCN (U + AcAc) band gap, as previously discussed. The lowered PL intensity in the case of gCN (U + AcAc) proved that AcAc functionalization was effective in suppressing charge carriers' recombination, a beneficial feature in view of the target end use (see also below).
Material composition was preliminarily investigated by nuclear magnetic resonance (NMR) (ESI,† §S3.2 and Fig. S3), which supported the formation of tris-s-triazine units in gCN.2913C CPMAS spectra of both samples presented two resonances at 165 and 157 ppm, attributable respectively to C–NHx and [N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N] moieties in a ≈1
N] moieties in a ≈1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, as revealed by quantitative 13C MAS experiments. It is worth mentioning that C–NHx resonance could be satisfactorily fitted with two components, whose ratio changed between the two samples, suggesting different amounts of C–NH2 and C–NH–C defects (Table S2, ESI†). The 15N CPMAS spectra presented four resonances centred at ≈195, 157, 136 and 117 ppm due to C–NC, N–(C)3, –NH– and –NH2 groups,29 respectively, with small line shape differences, in agreement with 13C results and other spectroscopic data.
:1 ratio, as revealed by quantitative 13C MAS experiments. It is worth mentioning that C–NHx resonance could be satisfactorily fitted with two components, whose ratio changed between the two samples, suggesting different amounts of C–NH2 and C–NH–C defects (Table S2, ESI†). The 15N CPMAS spectra presented four resonances centred at ≈195, 157, 136 and 117 ppm due to C–NC, N–(C)3, –NH– and –NH2 groups,29 respectively, with small line shape differences, in agreement with 13C results and other spectroscopic data.
Important complementary information was provided by XPS. Wide scan spectra (Fig. 2a) highlighted the presence of only C, N, and O. C1s signals (Fig. 2b and S4a†) were decomposed into four components: C0, attributed to both adventitious carbon and the CC substrate;30,31C1, due to both terminal C–NHx (x = 1, 2) moieties20,22,23,32 and C–O groups;17,31C2, the most intense band, assigned to C atoms in N–CN moieties;19,20,33C3, related to π electronic excitations.34,35 For the AcAc-functionalized specimen, C1 contribution turned out to be higher (Table S3†), demonstrating that AcAc functionalization resulted in a higher content of defective sites, since these amino-groups should not be present in a fully condensed gCN structure. This conclusion was in line with the analysis of N1s photopeaks, which resulted from the contribution of four bands (Fig. 2c and S4b†): N0, the main one, due to bi-coordinated N centres (CN–C);17,19,20,22N1, corresponding to N–(C)3 nitrogen atoms in gCN;17,19,32,33N2, related to uncondensed –NHx groups;19,20N3, due to π electronic excitations.36,37 The higher relative weight of N2 in CC_U + AcAc with respect to CC_U (Table S4†) indicated a higher content of defects resulting from the presence of –NHx, which can act as capturing sites, minimizing charge carrier recombination and enhancing the system photoactivity,23 as confirmed by PL analyses.17,34,38 Further details are reported in the ESI† (§S4; Fig. S5–S7, Tables S5 and S6). Overall, XPS results highlighted that CC_U + AcAc possessed a higher content of defects, proving that AcAc functionalization is a viable tool to achieve carbon nitride defect engineering.
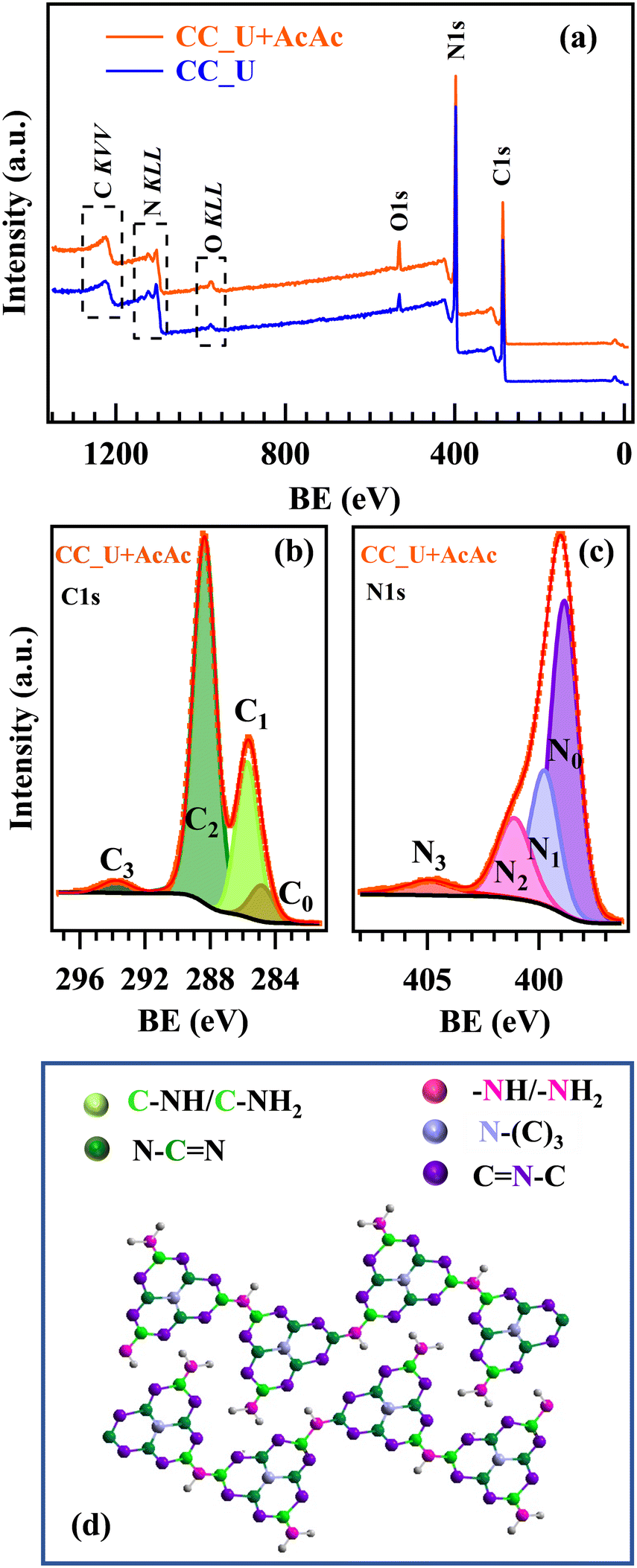 | ||
| Fig. 2 (a) XPS survey of CC_U and CC_U + AcAc samples. (b) C1s and (c) N1s photoelectron peaks for CC_U + AcAc. (d) Sketch of the gCN structure:18,23 colour codes identifying non-equivalent C and N atoms are the same used for the different C1s and N1s fitting components. | ||
FE-SEM analyses evidenced that bare CC (Fig. 3a) was characterized by the presence of interwoven fibres (average diameter ≈10 μm), whose assembly resulted in a network with an open morphology. For both CC_U and CC_U + AcAc (Fig. 3b and c), gCN deposition, yielding aggregates with a sheet-like morphology, resulted in an increase of the overall active area. The latter feature favourably affects catalytic activity, thanks to: i) a higher density of surface active sites; ii) an enhanced injection of photogenerated charge carries into the electrolyte, suppressing detrimental recombination.18,25–28 A detailed micrograph inspection revealed that, for CC_U (Fig. 3b), gCN particles were aggregated in confined areas, leaving various uncovered CC regions. Conversely, a more homogeneous carbon nitride dispersion was observed for CC_U + AcAc (Fig. 3c), for which gCN aggregates ‘welded’ in some cases the spaces between contiguous fibres, penetrating into the cloth meshes. Such a difference can be rationalized basing on the above results. In fact, the powder attachment to the CC substrate likely occurs through the establishment of bonds involving terminal –NHx and –OH groups, a process promoted by the annealing treatment.39 In fact, the larger amount of uncondensed amino groups, observed for gCN (U + AcAc), favoured a better deposit adhesion to the underlying CC. This result underscores another important advantage brought about by defect engineering resulting from AcAc functionalization.
 | ||
| Fig. 3 Representative FE-SEM micrographs for: (a) bare carbon cloth; (b) CC_U sample; (c) CC_U + AcAc sample. | ||
Overall, the characterization results reported so far suggest that carbon nitride flakes in gCN (U + AcAc) are internally less condensed (and, hence, more amino-rich) than those in gCN (U). As discussed below, this result might explain the better catalytic activity of the electrode material prepared from gCN (U + AcAc) powders towards the O2 → H2O2 reduction reaction. In fact, although the exact nature and behaviour of the various gCN defect types is still a matter of debate, –NHx groups have been reported to promote charge separation (acting as trapping agents for holes) and interfacial electron transfer. In addition, their Lewis basicity is also beneficial to the photoreduction reaction, positively impacting material functional performances.40,41
Prior to FNT degradation tests, photoelectrochemical characterization was carried out by employing gCN materials as working electrodes (Fig. 4a). Cyclic voltammetry (CV, Fig. S8†) showed two significant reduction peaks at more positive potential values than H2 evolution: the first one, attributed to the 2e− oxygen reduction reaction (ORR) yielding the formation of H2O2 from O2; the second one, associated with the H2O2 → H2O reduction and the direct 4e− ORR resulting in the formation of H2O from O2 (see ESI†, page S16).42 The reduction peaks were also evident in linear sweep voltammetry (LSV), showing that, under illumination, the signal for the 2e− ORR was shifted towards a more positive bias (Fig. 4b and c). The corresponding shift values (≈30 and ≈50 mV for CC_U and CC_U + AcAc) demonstrated that AcAc functionalization yielded an improvement in the system behaviour. Chronoamperometry (CA) measurements under the same conditions used for EF tests (Fig. 4d) did not reveal any significant current density decrease vs. time, a result highlighting the appreciable electrocatalyst stability, an important prerequisite for eventual real-world applications.
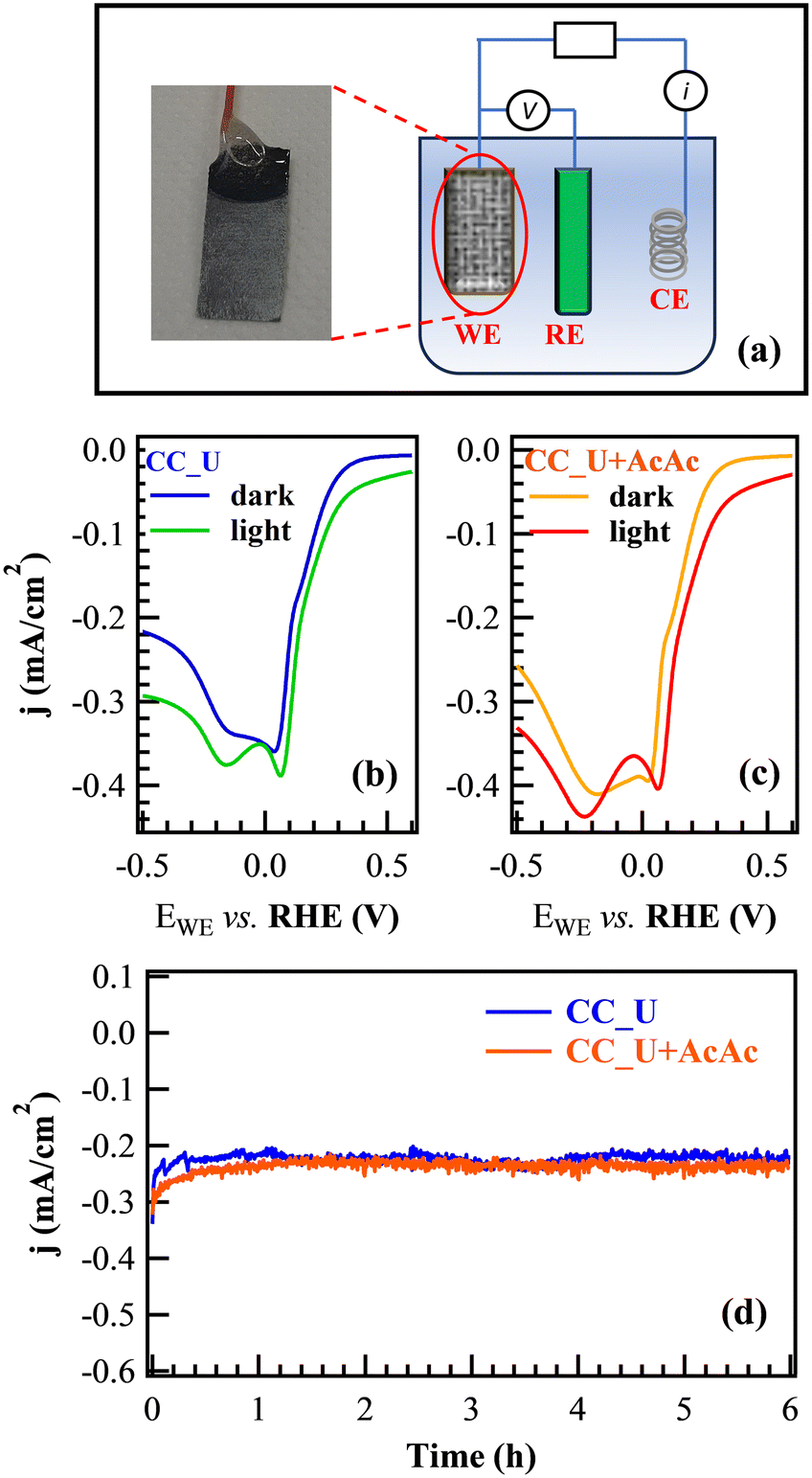 | ||
| Fig. 4 (a) Diagram of the cell used for electrochemical tests (WE = working electrode; RE = reference electrode; CE = counter-electrode). (b and c) LSV in the dark and under illumination for CC_U and CC_U + AcAc. (d) CA curves recorded at a bias of +0.06 V vs. the reversible hydrogen electrode (RHE). | ||
The attention was subsequently dedicated to EF experiments. As sketched in Fig. 5a, O2 is reduced through a 2e− mechanism, leading to the formation of H2O2 “activated” by the Fenton cycle6,43 generating, in turn, HO2˙ and especially ˙OH, capable of boosting FNT degradation:
| Fe2+ + H2O2 + H+ → Fe3+ + H2O + ˙OH (E° = 1.65 V) | (1) |
| Fe3+ + H2O2 → Fe2+ + HO2˙ + H+ (E° = 2.80 V) | (2) |
| Fe3+ + HO2˙ → Fe2+ + O2 + H+ | (3) |
| Fe3+ + e− → Fe2+ | (4) |
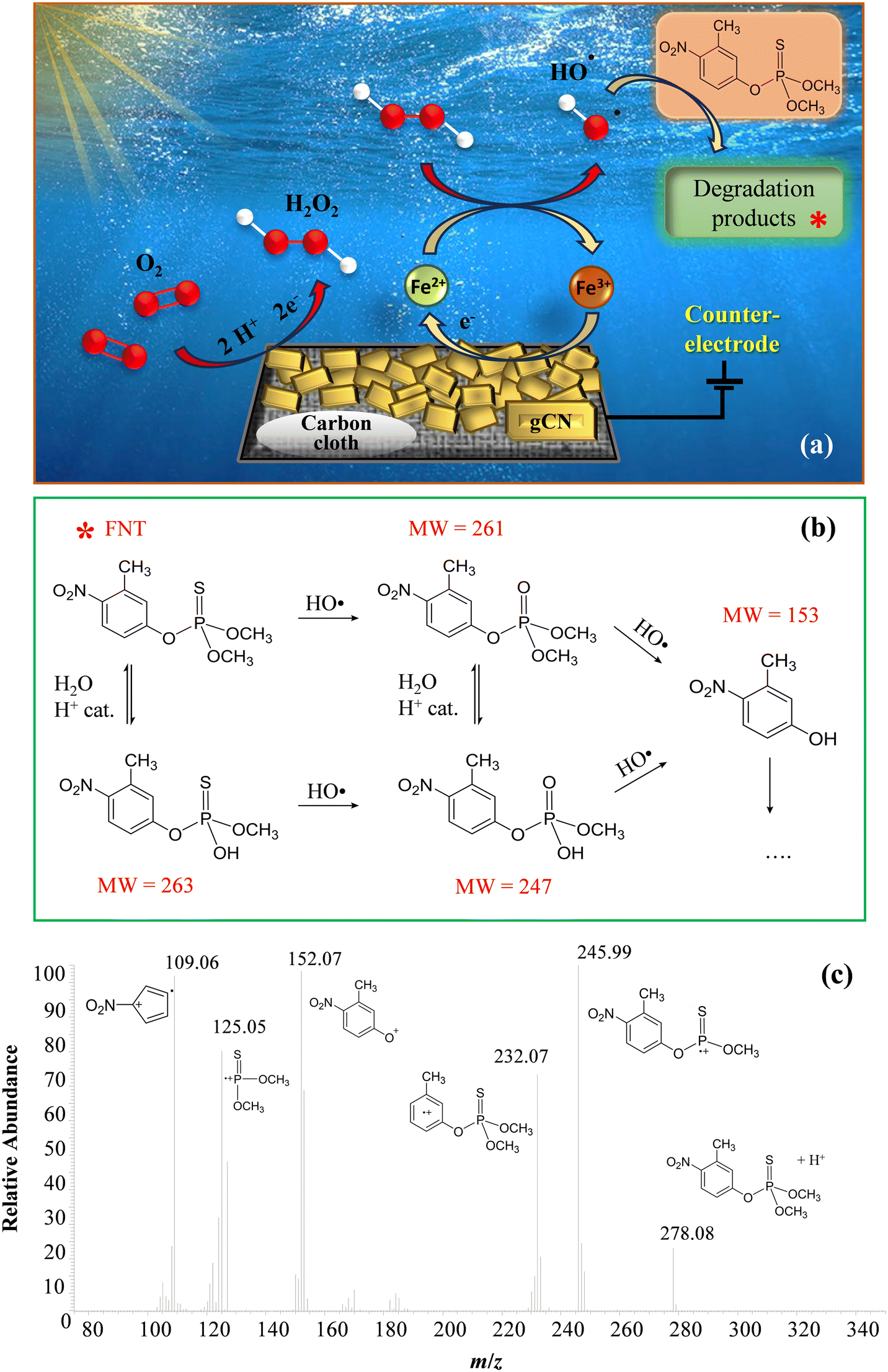 | ||
| Fig. 5 (a) Sketch of the reactions involved in the electro-Fenton process for FNT degradation. (b) Possible FNT degradation pathways and labelling of the species found in solution under the adopted conditions (MW = molecular weight). (c) Representative ESI-MS2 fragmentation spectrum for FNT. | ||
Mass spectrometry measurements, in accordance with the literature,44,45 allowed the identification of four different species, besides FNT, present in the working solution even before the real beginning of EF experiments (Fig. 5b). The occurrence of these compounds was associated with the acidification of the solution itself, which could have led to their formation from the pristine FNT through partial oxidation and hydrolysis processes. Fig. 5b presents the possible FNT degradation pathways taking place under the presently adopted conditions. After the start of EF tests, the newly generated ROS promoted the degradation of the indicated organic species, likely following similar reaction pathways. It is worth highlighting that, even if 3-methyl-4-nitrophenol (MW = 153) was the lowest molecular weight product among the identified ones, subsequent oxidative processes could produce even lighter organic species, undetectable via HPLC-MS measurements, due to their low concentrations and/or very short lifetimes in solution.
The relative concentration vs. time trends of the identified species are shown in Fig. 6a and b. Unexpectedly, FNT was never detected by HPLC-MS analysis, not even in the solution portions corresponding to time “0”, at variance with the blank degradation test (Fig. S9†). This result was ascribed to the initial adsorption of FNT molecules onto the photoelectrocatalyst surface, removing an appreciable amount of pesticide from the working solution. In fact, adsorbed FNT can likely be partially degraded directly onto the electrodes, yielding thus the other species found in the liquid phase.
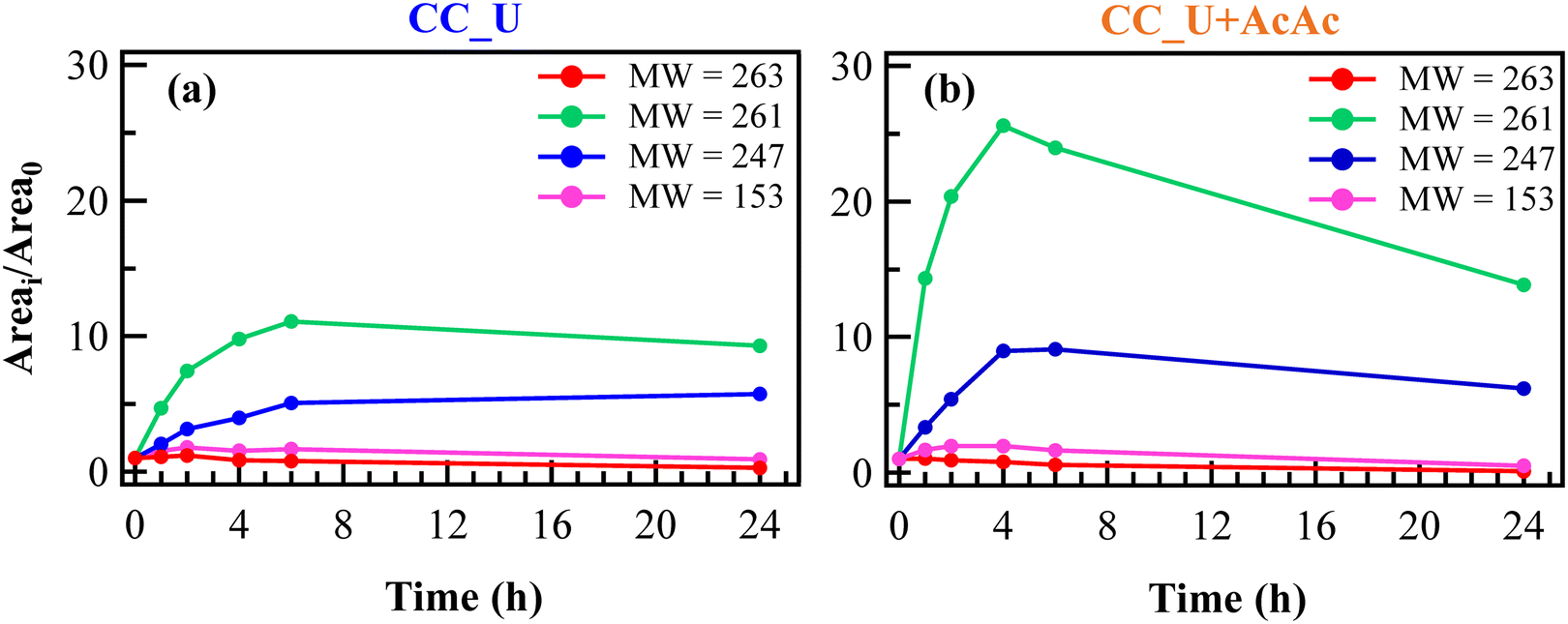 | ||
| Fig. 6 Evolution of FNT degradation products during electro-Fenton experiments, using CC_U (a) and CC_U + AcAc (b) as working electrodes. For all species, the Areai/Area0 ratio was calculated as the ratio between the chromatographic peak areas at the i-th time and at time = 0 h. | ||
The trends of FNT-derived species in EF tests were significantly different compared to the corresponding ones in the blank degradation experiment (see Fig. S9†), during which the relative concentrations remained almost constant for 24 h. Notably, in EF tests the relative concentrations of species with MW = 247 and 261 (see Fig. 5b and 6a and b) underwent an increase during the first hours, more significant in the case of CC_U + AcAc. For the same system, a subsequent concentration decrease for these species was observed till the end of the test, demonstrating once again the beneficial role of defect engineering through AcAc functionalization. For comparison, results pertaining to FNT degradation on bare CC are reported in Fig. S10.†
In order to confirm the generation of H2O2 and oxidizing radical species via EF, further experiments were performed in solutions of coumarin (1-benzopyran-2-one), used thanks to its ability to act as a scavenger for ˙OH radicals.46 In the presence of the latter, coumarin can be converted into 7-hydroxycoumarin (7-OHC), featuring a very intense fluorescence emission at λ ≈ 460 nm, well distinct from the coumarin emission maximum at λ ≈ 380 nm (Fig. 7a). The direct monitoring of 7-OHC formation provides a qualitative indication on the presence of ˙OH,46 otherwise elusive due to their high reactivity and short lifetimes in solution (≈10−9 s). Coumarin tests on both CC_U and CC_U + AcAc (Fig. 7b and c) resulted in the appearance of a strong emission signal at 460 nm at 3 and 6 h, absent for the mother solution and the samples at time “0”. As stated above, this signal demonstrates the effective generation of ˙OH radicals in solution. It is also worth noting that, during EF tests conducted on FNT and coumarin, the formation of a well-adherent deposit on the working electrodes (Fig. 8a) took place (see also the pertaining characterization in the ESI,† Fig. S11–S16 and Tables S7–S10). Its dark orange colour suggested the formation of insoluble Fe(III) compounds, whose occurrence was indeed confirmed by XPS analyses (see also ESI,† §S5.3). At variance with fresh samples (Fig. 2a), survey spectra (Fig. S11†) showed the presence of Na, Fe, S and P. Whereas the occurrence of Na and S (Fig. S12†) could be traced back to the used Na2SO4 electrolyte, P signals (Fig. S13†) arose from FNT decomposition products. The Fe2p signal shape and positions [Fig. 8c; BE(Fe2p3/2) = 711.7 eV] were in line with literature data for FeOOH,47 whose formation could result from the continuous H+ consumption during EF processes (2e− and 4e− ORR, ESI†, page S16), causing a pH increase in proximity of the electrode surface, promoting thus its precipitation48 (Fig. 8b). This phenomenon explains why the formation of the observed deposits occurred only on the working electrode's external region during EF tests. However, S2p and P2p BEs were also consistent with the presence of Fe(III) phosphate and sulphate30 (ESI,† pages S19 and S20), besides FeOOH. C1s signals (ESI,† Fig. S14) pointed out the presence of C–OH, C–O–C, esters, and carboxylic acids,31,47 resulting from the partial oxidation of exposed CC supports and the adsorption of FNT degradation products. Nevertheless, N1s signals (Fig. 8d and e) did not undergo dramatic variations in comparison to the pristine materials (Fig. 2c and S4b†), evidencing that the formation of iron-containing deposits did not induce significant alterations of the underlying gCN.
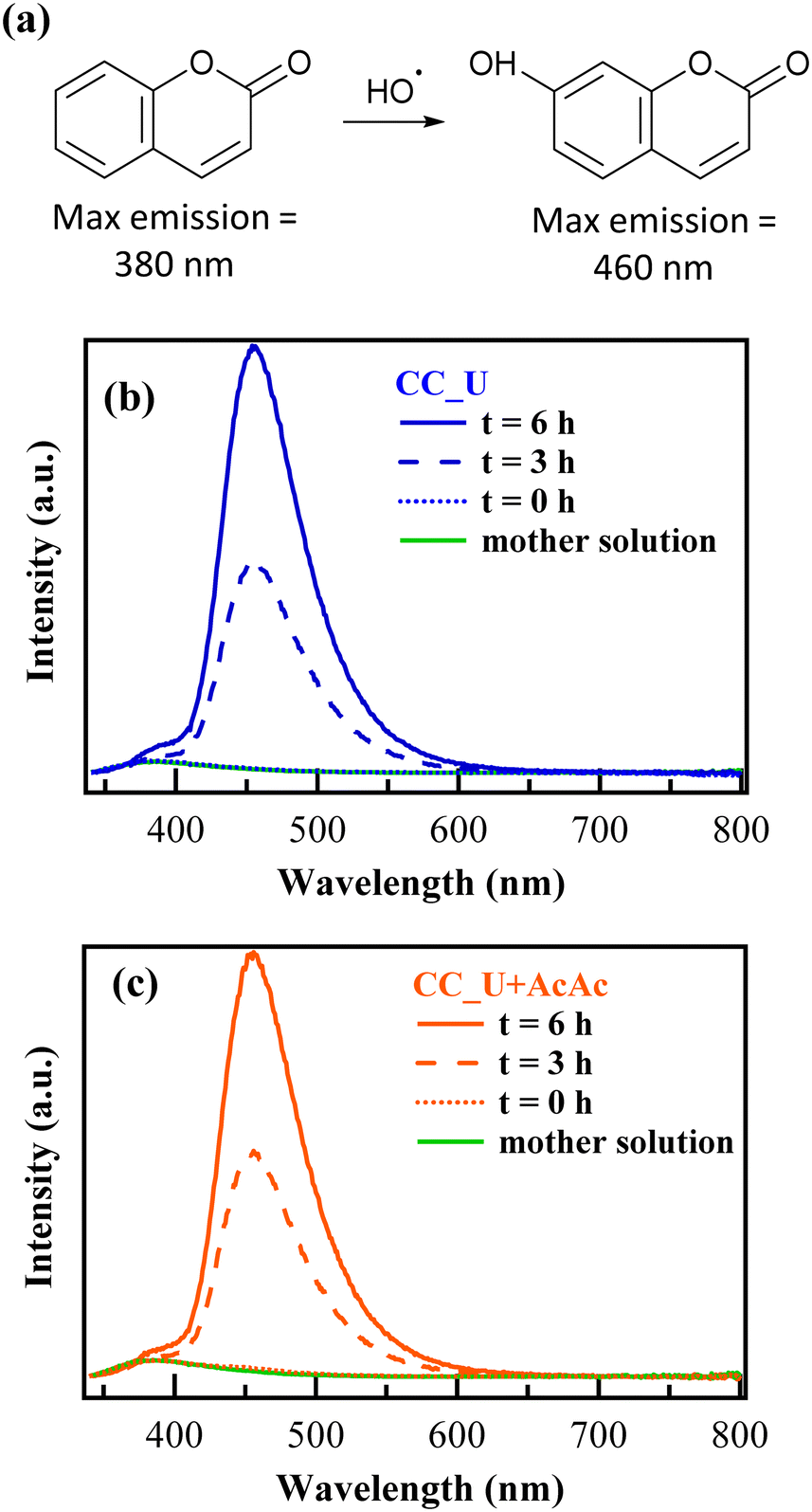 | ||
| Fig. 7 (a) Oxidation of coumarin to 7-hydroxycoumarin. (b and c) Emission spectra of 1 mM coumarin solutions as a function of time using respectively CC_U and CC_U + AcAc as photocathodes in electro-Fenton experiments. | ||
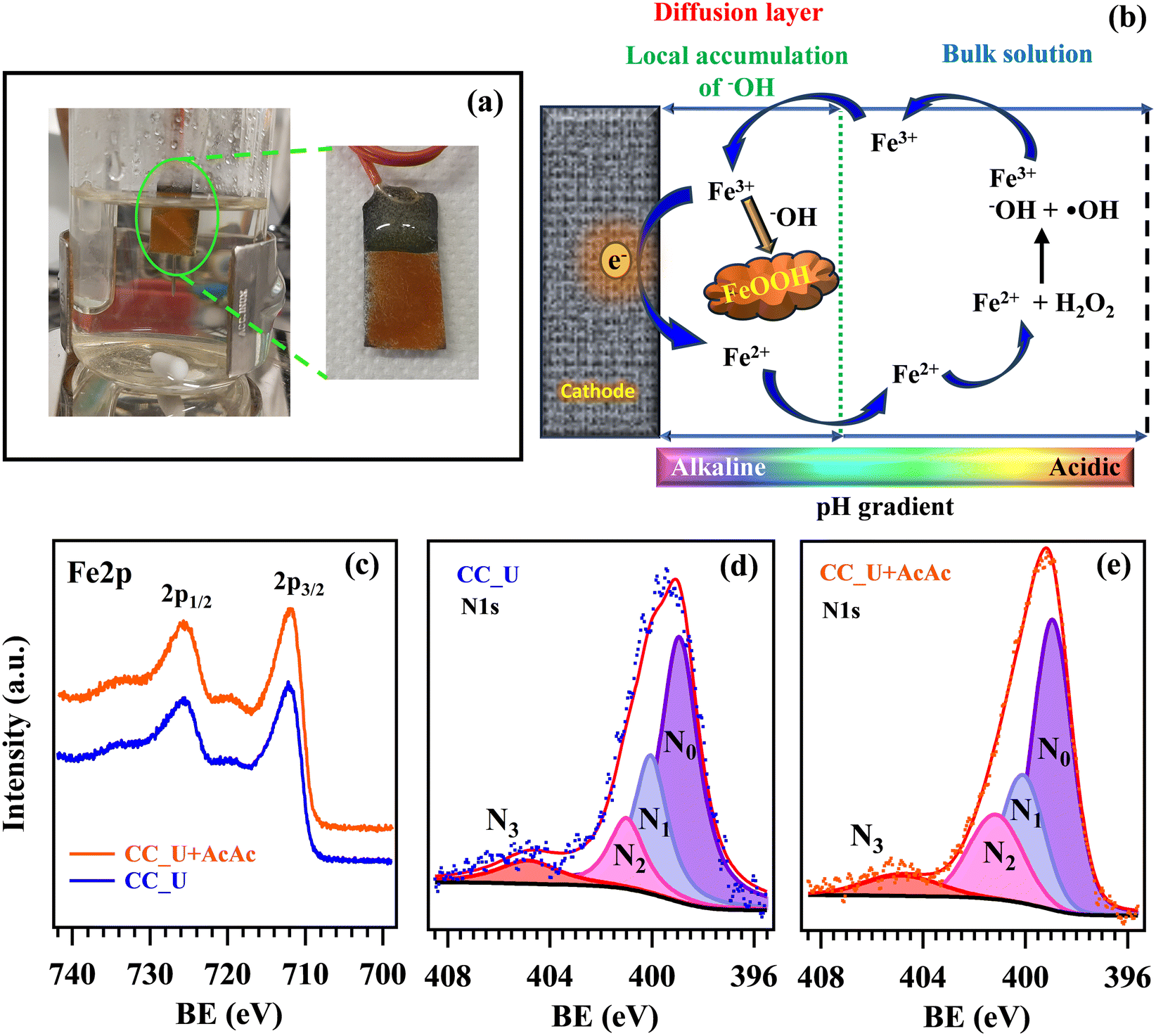 | ||
| Fig. 8 (a) Image of the CC_U + AcAc sample after the test for 24 h as a working electrode in EF processes for FNT degradation. (b) Sketch of the route yielding insoluble Fe(III) deposits onto the electrode's surface. XPS signals after EF tests for Fe2p (c) and N1s (d and e). | ||
4. Conclusions
In summary, this work was dedicated to the fabrication and defect engineering of graphitic carbon nitride photoelectrocatalysts for FNT decomposition. The target systems were immobilized on flexible carbon cloths via EPD, starting from carbon nitride powders obtained from urea as such or mixed with AcAc. A thorough multi-technique investigation enabled to elucidate the interplay between processing conditions and the resulting system's physico-chemical and functional properties. In a nutshell, AcAc functionalization promoted gCN adhesion to the substrate, resulting in a more homogeneous distribution of carbon nitride aggregates. The obtained results demonstrated the successful development of gCN supported systems, whose reactivity towards FNT degradation was mainly improved through defect engineering thanks to a suppressed electron–hole recombination.The performed tests evidenced a remarkable electrochemical stability of the present electrocatalysts, an important starting point for practical applications. Material characterization after degradation tests highlighted the formation of insoluble Fe-containing species on the electrode surfaces, which, however, did not result in an appreciable performance decrease and in significant alterations of the underlying carbon nitride. The obtained results, which, to the best of our knowledge, have no literature precedents, open challenging perspectives for the implementation of the electro-Fenton technique and the subsequent scale-up of the target technology in view of real-world water purification activated by natural sunlight. In addition, more detailed studies on the formation mechanisms and the potential toxicological effects of FNT-derived products represent important issues for future research developments beyond the present study.
Data availability
All the data can be found within the manuscript and ESI† files.Author contributions
Giacomo Marchiori: investigation, data curation, formal analysis, writing – original draft. Roberta Seraglia: investigation, data curation, writing – original draft. Gian Andrea Rizzi: methodology, conceptualization, writing – review and editing. Chiara Maccato: methodology, conceptualization, supervision, resources, investigation, writing – original draft. Mattia Benedet: investigation, data-curation, formal analysis. Emanuela Callone and Sandra Dirè: investigation, data curation, writing – reviewing and editing. Alberto Gasparotto: investigation, data curation, writing – reviewing and editing. Davide Barreca: supervision, resources, investigation, data curation, formal analysis, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Council of Research (Progetti di Ricerca @CNR-avviso 2020-ASSIST), Padova University (P-DiSC#04BIRD2020-UNIPD EUREKA, P-DiSC#02BIRD2023-UNIPD RIGENERA, DOR 2021–2024), INSTM Consortium (INSTM21PDGASPAROTTO-NANOMAT, INSTM21PDBARMAC-ATENA), and PRIN 2022474YE8 SCI-TROPHY project, financed by the European Union - Next Generation EU - Bando PRIN 2022 – M4.C2.1.1.References
- L. G. Sultatos, J. Toxicol. Environ. Health, 1994, 43, 271–289 CrossRef CAS.
- T. O. Ajiboye, P. O. Oladoye, C. A. Olanrewaju and G. O. Akinsola, Environ. Nanotechnol., Monit. Manage., 2022, 17, 100655 CAS.
- I. A. Saleh, N. Zouari and M. A. Al-Ghouti, Environ. Technol. Innovation, 2020, 19, 101026 CrossRef.
- H. Mali, C. Shah, B. H. Raghunandan, A. S. Prajapati, D. H. Patel, U. Trivedi and R. B. Subramanian, J. Environ. Sci., 2023, 127, 234–250 CrossRef CAS.
- Y. Deng and R. Zhao, Curr. Pollut. Rep., 2015, 1, 167–176 CrossRef CAS.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS.
- C. Hu, Y. Xiao, Y. Zou and L. Dai, Electrochem. Energy Rev., 2018, 1, 84–112 CrossRef CAS.
- X. Lyu, W.-N. Zhang, G. Li, B.-W. Shi, Y.-N. Zhang, H. Chen, S.-C. Li and X. Wang, ACS Appl. Nano Mater., 2020, 3, 8586–8591 CrossRef.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- L. Chen and J. Song, Adv. Funct. Mater., 2017, 27, 1702695 CrossRef.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2013, 2, 353–358 CrossRef.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- W. Niu and Y. Yang, ACS Energy Lett., 2018, 3, 2796–2815 CrossRef CAS.
- B. Kalinic, L. Girardi, P. Ragonese, A. Faramawy, G. Mattei, M. Frasconi, R. Baretta, S. Bogialli, M. Roverso, G. A. Rizzi and C. Maurizio, Appl. Surf. Sci., 2022, 596, 153552 CrossRef CAS.
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, X. Li, B. Li, W. Dong and Y. Zhou, Appl. Catal., B, 2020, 272, 118970 CrossRef CAS.
- J. Yang, Z. Ji and S. Zhang, ACS Appl. Energy Mater., 2023, 6, 3401–3412 CrossRef CAS.
- M. Benedet, G. A. Rizzi, A. Gasparotto, O. I. Lebedev, L. Girardi, C. Maccato and D. Barreca, Chem. Eng. J., 2022, 448, 137645 CrossRef CAS.
- M. Benedet, A. Gallo, C. Maccato, G. A. Rizzi, D. Barreca, O. I. Lebedev, E. Modin, R. McGlynn, D. Mariotti and A. Gasparotto, ACS Appl. Mater. Interfaces, 2023, 15, 47368–47380 CrossRef CAS PubMed.
- M. Benedet, G. A. Rizzi, O. I. Lebedev, V. Roddatis, C. Sada, J.-L. Wree, A. Devi, C. Maccato, A. Gasparotto and D. Barreca, J. Mater. Chem. A, 2023, 11, 21595–21609 RSC.
- E. Scattolin, M. Benedet, G. A. Rizzi, A. Gasparotto, O. I. Lebedev, D. Barreca and C. Maccato, ChemSusChem, 2024, e202400948 CrossRef CAS.
- S. Benedoue, M. Benedet, A. Gasparotto, N. Gauquelin, A. Orekhov, J. Verbeeck, R. Seraglia, G. Pagot, G. A. Rizzi, V. Balzano, L. Gavioli, V. Di Noto, D. Barreca and C. Maccato, Nanomaterials, 2023, 13, 1035 CrossRef CAS.
- M. Benedet, G. A. Rizzi, A. Gasparotto, N. Gauquelin, A. Orekhov, J. Verbeeck, C. Maccato and D. Barreca, Appl. Surf. Sci., 2023, 618, 156652 CrossRef CAS.
- https://xpspeak.software.informer.com/4.1/ .
- W. Zhang, Q. Zhang, F. Dong and Z. Zhao, Int. J. Photoenergy, 2013, 2013, 1–9 Search PubMed.
- Y. Zheng, Z. Zhang and C. Li, J. Photochem. Photobiol., A, 2017, 332, 32–44 CrossRef.
- V. Devthade, D. Kulhari and S. S. Umare, Mater. Today: Proc., 2018, 5, 9203–9210 CAS.
- M. Ismael, Y. Wu, D. H. Taffa, P. Bottke and M. Wark, New J. Chem., 2019, 43, 6909–6920 RSC.
- Y. Hu, Y. Shim, J. Oh, S. Park, S. Park and Y. Ishii, Chem. Mater., 2017, 29, 5080–5089 CrossRef CAS.
- https://srdata.nist.gov/xps, accessed October, 2023.
- I. Bertóti, M. Mohai and K. László, Carbon, 2015, 84, 185–196 CrossRef.
- M. Benedet, A. Gasparotto, G. A. Rizzi, D. Barreca and C. Maccato, Surf. Sci. Spectra, 2022, 29, 024001 CrossRef CAS.
- M. Benedet, G. A. Rizzi, D. Barreca, A. Gasparotto and C. Maccato, Surf. Sci. Spectra, 2023, 30, 014004 CrossRef CAS.
- P. Qiu, C. Xu, H. Chen, F. Jiang, X. Wang, R. Lu and X. Zhang, Appl. Catal., B, 2017, 206, 319–327 CrossRef CAS.
- P. Qiu, H. Chen and F. Jiang, RSC Adv., 2014, 4, 39969–39977 RSC.
- T. Ma, J. Bai and C. Li, Vacuum, 2017, 145, 47–54 CrossRef CAS.
- Y. Fu, C. a. Liu, C. Zhu, H. Wang, Y. Dou, W. Shi, M. Shao, H. Huang, Y. Liu and Z. Kang, Inorg. Chem. Front., 2018, 5, 1646–1652 RSC.
- X. Wang, J. Meng, X. Zhang, Y. Liu, M. Ren, Y. Yang and Y. Guo, Adv. Funct. Mater., 2021, 31, 2010763 CrossRef CAS.
- L. Cheng, H. Zhang, X. Li, J. Fan and Q. Xiang, Small, 2021, 17, 2005231 CrossRef CAS PubMed.
- X. Zhan, Y. Zhao, Y. Sun, C. Lei, H. Wang and H. Shi, Chemosphere, 2022, 307, 136087 CrossRef CAS.
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS.
- J.-y. Chen, N. Li and L. Zhao, J. Power Sources, 2014, 254, 316–322 CrossRef CAS.
- E. Brillas and S. Garcia-Segura, Sep. Purif. Technol., 2020, 237, 116337 CrossRef CAS.
- M. Kerzhentsev, C. Guillard, J.-M. Herrmann and P. Pichat, Catal. Today, 1996, 27, 215–220 CrossRef CAS.
- J.-M. Herrmann, Catal. Today, 1999, 53, 115–129 CrossRef CAS.
- V. Leandri, J. M. Gardner and M. Jonsson, J. Phys. Chem. C, 2019, 123, 6667–6674 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin Elmer Corporation, Eden Prairie, MN, USA, 1992 Search PubMed.
- F. Deng, H. Olvera-Vargas, M. Zhou, S. Qiu, I. Sires and E. Brillas, Chem. Rev., 2023, 123, 4635–4662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00259h |
| This journal is © The Royal Society of Chemistry 2025 |