
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular tunnelling junctions with robust high rectification based on assembly effects†
Max
Roemer‡
 a,
Xiaoping
Chen‡
bc,
Yuan
Li
bh,
Lejia
Wang
b,
Xiaojiang
Yu
d,
Pierre-André
Cazade
e,
Cameron
Nickle
f,
Romena
Akter
f,
Enrique
Del Barco
f,
Damien
Thompson
*e and
Christian A.
Nijhuis
*bg
a,
Xiaoping
Chen‡
bc,
Yuan
Li
bh,
Lejia
Wang
b,
Xiaojiang
Yu
d,
Pierre-André
Cazade
e,
Cameron
Nickle
f,
Romena
Akter
f,
Enrique
Del Barco
f,
Damien
Thompson
*e and
Christian A.
Nijhuis
*bg
aThe University of Sydney, School of Chemistry, Sydney, NSW 2109, Australia
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: c.a.nijhuis@utwente.nl
cCollege of Chemistry, Chemical Engineering and Environment, Fujian Provincial Key Laboratory of Modern Analytical Science and Separation Technology, Minnan Normal University, Zhangzhou, 363000, China
dSingapore Synchrotron Light Source, National University of Singapore, 5 Research Link, Singapore 117603, Singapore
eDepartment of Physics, Bernal Institute, 34 University of Limerick, Limerick V94 T9PX, Ireland. E-mail: damien.thompson@ul.ie
fUniversity of Central Florida, Physics Department, Orlando, FL 32816, USA
gUniversity of Twente, Faculty of Science and Technology (TNW), Hybrid Materials for Opto-Electronics (HMOE), 7500 AE, Enschede, The Netherlands
hKey Laboratory of Organic Optoelectronics, Department of Chemistry, Tsinghua University, Beijing, 100084, P.R. China
First published on 11th September 2024
Abstract
The performance of large-area molecular diodes can in rare cases approach the lower limit of commercial semiconductor devices but predictive structure–property design remains difficult as the rectification ratio (R) achieved by self-assembled monolayer (SAM) based diodes depends on several intertwined parameters. This paper describes a systematic approach to achieve high rectification in bisferrocenyl-based molecular diodes, HSCnFc–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Fc (n = 9–15) immobilised on metal surfaces (Ag, Au and Pt). Experiments supported by molecular dynamics simulations show that the molecular length and bottom electrode influence the SAM packing, which affects the breakdown voltage (VBD), the associated maximum R (Rmax), and the bias at which the Rmax is achieved (Vsat,R). From the electrical characterisation of the most stable Pt–SCnFc–CC–Fc//GaOx/EGaIn junctions, we found that VBD, Vsat,R, and Rmax all scale linearly with the spacer length of Cn, and that Rmax for all the SAMs consistently exceeds the “Landauer limit” of 103. Our data shows that the robust switching of M–SCnFc–CC–Fc//GaOx/EGaIn junctions is the result of the combined optimisation of parameters involving the molecular structure, the type of metal substrate, and the applied operating conditions (bias window), to create stable and high-performance junctions.
C–Fc (n = 9–15) immobilised on metal surfaces (Ag, Au and Pt). Experiments supported by molecular dynamics simulations show that the molecular length and bottom electrode influence the SAM packing, which affects the breakdown voltage (VBD), the associated maximum R (Rmax), and the bias at which the Rmax is achieved (Vsat,R). From the electrical characterisation of the most stable Pt–SCnFc–CC–Fc//GaOx/EGaIn junctions, we found that VBD, Vsat,R, and Rmax all scale linearly with the spacer length of Cn, and that Rmax for all the SAMs consistently exceeds the “Landauer limit” of 103. Our data shows that the robust switching of M–SCnFc–CC–Fc//GaOx/EGaIn junctions is the result of the combined optimisation of parameters involving the molecular structure, the type of metal substrate, and the applied operating conditions (bias window), to create stable and high-performance junctions.
Introduction
Molecular diodes are attractive building blocks for nanoscale electronic circuits,1–5 and their development has been a focal point since the landmark paper by Aviram and Ratner proposing the first molecular diode in 1974.6 The performance of a diode is usually expressed in terms of its rectification ratio (R), which defines how much electrical current density (J, in A cm−2) traverses the junction in the ON vs. OFF states. R is simply the ratio of |Jon|/|Joff|, i.e., R = |J(−V)|/|(J(V))|, where V is the applied voltage in V, and R is the ratio of absolute J at forward bias to that at reverse bias. Ideal diodes would allow all current to pass through the diode in the ON state, but not in the OFF state. However, this ideal behaviour is not reached as a small amount of current (leakage current) always flows also in the OFF state. In molecular diodes, this leakage current is dictated by many factors, resulting in smaller values for R than ideal cases. Macroscopic semiconductor-based diodes operate at R of ≥105, while molecular diodes have been generally limited to orders of magnitude smaller values of R because of the molecular resistance in the ON state and large leakage currents (due to defects or molecular disorder) in the OFF state.7–16 Recently, exceptionally well-performing molecular diodes have been reported with R of several thousand based on rigid biferrocenes with interlocked cyclopentadienyl rings in SAM based junctions or nonadiyne in single-molecule junctions,17,18 and even of several tens of thousands in switchable junctions, which operate with a different mechanism via directional counterion migration in SAMs of viologens.19Our previous studies showed that the performance of SAM-based diodes depends on the choice of the bottom electrode,20 electrode–molecule anchoring group,21 molecular structure,12,22 and tilt angle of the active group,9 which all impact the supramolecular packing of the SAM and its ability to effectively transport current in the ON state while blocking leakage currents in the OFF state.23 Even seemingly subtle changes in the molecular structure can have profound macroscopic implications as nicely demonstrated by well-known odd–even effects.24,25 SAMs of molecules on Ag with odd numbers of methylene units (nc = odd) pack better than the ones with even numbers (nc = even), which results in odd–even effects for surface coverages, electrical behaviours and the SAM packing properties predicted from molecular dynamics (MD) simulations. For example, the odd–even effects in packing energies of SAMs derived from n-alkanethiolates (where an odd or even number of CH2 moieties determines the orientation of the functional terminal group) lead to odd–even effects in exchange kinetics during SAM formation,26 surface dipole and work-function,27 dielectric constant28 and optical properties.29 Such odd–even effects also result in current oscillations in molecular junctions22,30–32 and impact molecular diode performance.30
The mechanism of rectification of molecular diodes derived from S(CH2)11Fc (Fc is ferrocenyl),10,22,33–41 or derivatives such as S(CH2)11Fc2,11 is well understood. At positive bias, the highest occupied molecular orbital (HOMO) does not participate in charge transport resulting in low conductivity, while at negative bias, the HOMO falls in the conduction window leading to high conductivity. We showed recently that molecular junctions of Pt–SC15Fc–CC–Fc//GaOx/EGaIn (EGaIn stands for eutectic alloy of gallium and indium) can rectify with R = 6.3 × 105 by an electrostatic mechanism that maximises the number of molecules contributing to charge transport in only the forward direction of applied bias when the diodes are ON,42 allowing them to bypass the Landauer limit for single-level dominated charge transport.42–44 In these diodes, the SAM structure changes with applied electric field (or applied bias) due to the electrostatic forces between the SAM and the top EGaIn electrode. This effect led to an increase in the number of conducting molecules with increasing applied bias for only one bias polarity, boosting the diode performance. This example highlights that molecular diode performance can be bias dependent.
To rationally design molecular diodes that are stable and operate with high R for potential future applications in nanoscale electronics, it is important to understand the strengths and weaknesses of such systems by systematically taking both electronic and (supra)molecular structure of the SAMs into consideration. Here, we go beyond that first SC15Fc–CC–Fc diode demonstration. Being aware that very long molecules become highly resistive, and very short molecules give disordered SAMs (resulting in large leakage currents at reverse bias when the diodes should block the current), we identified an optimal range of molecular lengths to obtain well-performing molecular diodes by systematically exploring the alkyl chain length-dependence of the SCnFc–CC–Fc junctions with nc = 9–15. Furthermore, the choice of the bottom electrode, either Ag, Au or Pt, results in changes in energy level alignments, differentiating electronic effects from molecular effects in the M–SCnFc–CC–Fc//GaOx/EGaIn junctions, but also affects the stability of the junctions. We identified Pt as the bottom electrode of choice and we determined the breakdown voltage (VBD) and subsequently measured those junctions at the maximum possible bias range. We show that R depends on the applied bias range and that the maximal possible bias range depends on the length of the alkyl chains and type of electrode material. The larger VBD allows us to increase the operating bias window, leading to an improved value of R. This work shows that the design of optimally performing junctions is complicated by several intertwined factors that all need to be optimised, illustrating that it is still challenging to rationally design molecular junctions.
Results and discussion
The molecular tunnelling junctions
The junctions consist of thiolate–metal contacts with the bottom electrode and non-covalent interactions with the EGaIn top electrode (Fig. 1). The variations of the alkyl chain length and the type of metal substrate (Fig. 1b) allow us to probe odd–even effects of the SAM, the corresponding electrical characteristics, the evolution in the trend of charge transport rates, and the corresponding rectification ratios, as well as characterising the stability of the junctions as measured by the breakdown voltage.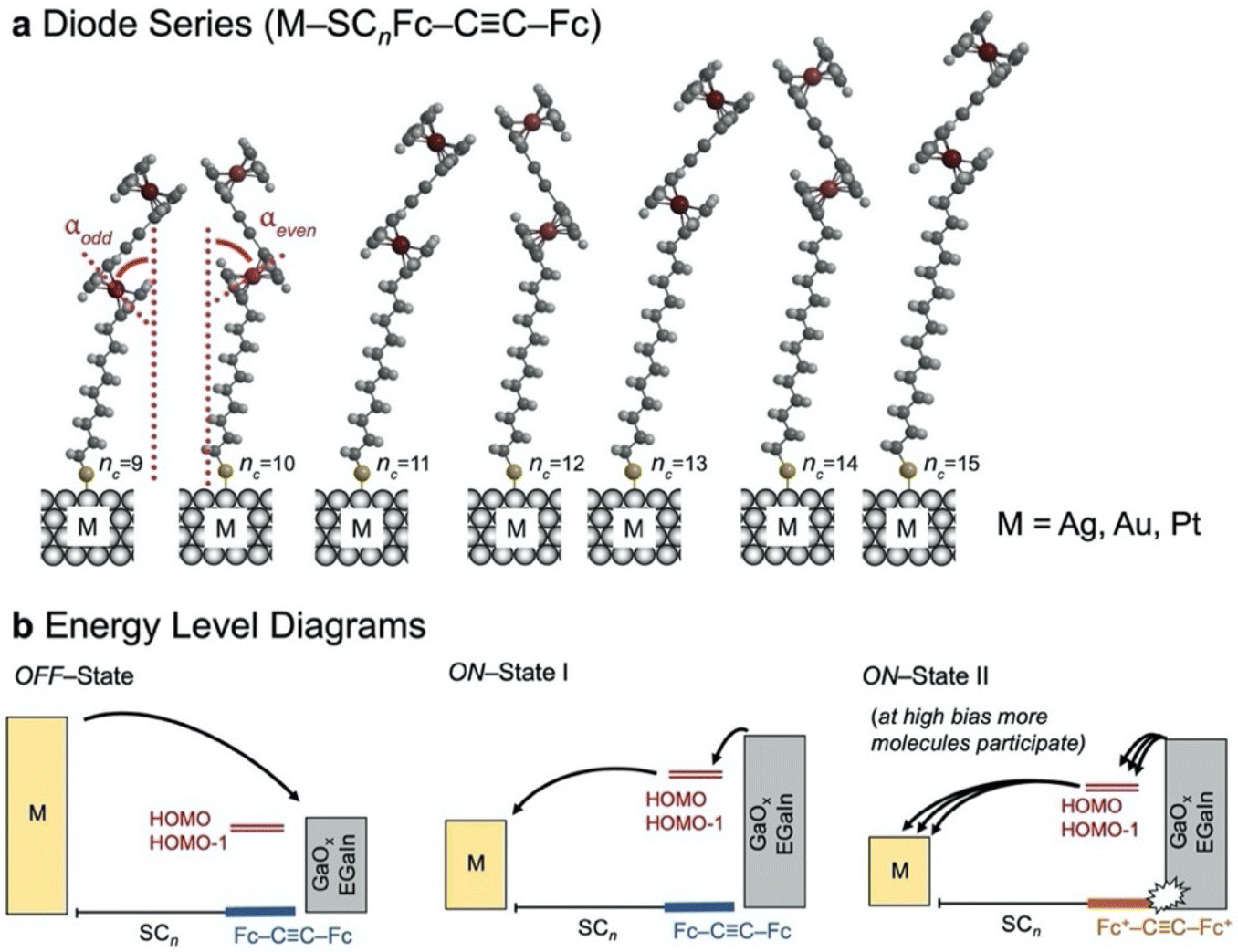 | ||
| Fig. 1 (a) Representation of the series of HSCnFc–CC–Fc (nc = 9–15) immobilised on a noble metal surface (M). The odd and even numbered diodes differ in orientation and tilt angle (αodd and αeven) of the Fc–CC–Fc head group. (b) Energy level diagrams for the diode in the OFF-state, the ON-state I and the ON-state II at which the current flow is enhanced due to Coulomb interactions between the oxidised positively charged Fc+–CC–Fc+ and the negatively charged EGaIn-electrode increasing the number of molecules involved in charge transport as indicated by the multiple arrows. The Fc–CC–Fc head group provides two near-degenerate levels HOMO and HOMO−1 for charge transport. | ||
The HSCnFc–CC–Fc precursors were synthesised and characterised (see ESI†). The melting points of the HSCnFc–CC–Fc molecules show an odd–even effect with a similar trend as observed for the corresponding monoferrocenyl thiols (HSCnFc),45i.e., the odd numbered derivatives melt at lower temperature than the even numbered ones highlighting an odd–even effect in the packing structure. Such odd–even effects are well known for n-alkanes, in the solid and the liquid states.46,47 Since the non-templated assembly already shows an odd–even effect, naturally odd–even effects are also expected in the monolayer packing structure. Fig. 1 illustrates the different SCnFc–CC–Fc SAMs with nc = 9–15 on metallic substrates M (M = Au, Ag, or Pt). The terminal Fc–CC–Fc moiety follows an odd–even effect in the tilt angle of the head group (α) with respect to the surface normal. This odd–even effect originates from the number of methylene units in the alkyl chain (nc) and the fixed M–C–S angle, as illustrated in Fig. 1a.24 However, in SAMs with large head groups, the SAM-packing structure may be driven by head group – head group interactions.18,42,48
Surface characterisations of the SAMs
We prepared stable SAMs on Ag, Au, and Pt, and characterised them using cyclic voltammetry (CV), X-ray photoelectron spectroscopy (XPS), ultra-violet photoelectron spectroscopy (UPS), and near-edge X-ray absorption fine spectroscopy (NEXAFS). We have previously characterised M–SC15Fc–CC–Fc by these techniques.42 Here, we provide XPS, UPS, and NEXAFS datasets for two further derivatives, M–SC14Fc–CC–Fc and M–SC13Fc–CC–Fc (see ESI† for details), and CV data for all SAMs on Au and Pt. In addition, all SAMs were characterised with MD simulations.
Fig. 2a and b shows the measured surface coverages (ΓCV, in nmol cm−2) of SCnFc–CC–Fc SAMs on Au and Pt derived from CV (Fig. S2 and S3 show the CV curves and Tables S1 and S2† the corresponding data). We have previously characterised M–SC15Fc–CC–Fc (M = Au, Pt) by CV, and the data of this derivative was taken from our previous work.42 The ΓCV values show distinct odd–even effects on both Au and Pt surfaces, with odd numbers of nc giving higher ΓCV, indicating a more ordered and tightly packed structured SAM than the nc = even derivatives.
 | ||
| Fig. 2 Surface coverage (ΓCV) of SCnFc–CC–Fc SAMs on Au (a) and Pt (b) derived from CV. Au–SCnFc–CC–Fc and Pt–SCnFc–CC–Fc were used as working electrodes, saturated Ag/AgCl as reference electrode, and Pt as counter electrode. A 1.0 M HClO4 aqueous solution was used as supporting electrolyte and the scan rate was 1.00 V s−1. The error bars represent the standard deviation from measurements of three samples for each kind of SAM. The red dotted lines serve as guides for the eye only. Results from the molecular dynamics calculations for the Pt–SCnFc–CC–Fc SAMs. (c) SAM heights (dSAM,MD) and surface coverages (ΓMD) on Pt. (d) Computed damping of the head group motion with increasing nc, (e) Computed odd–even effects in packing energy differences (ΔE) for the full molecule and projected on to the different parts of the molecules, i.e., the alkyl chain, the head group, and alkyl-head cross interactions. (f) Computed packing energy components (E) for the different components of the molecules of the Pt–SCnFc–CC–Fc series. | ||
On the other hand, previous work showed that for SAMs of HSCnFc derivatives on Au this effect is largely reversed with respect to Pt (driven by differences in the M–S–C bond angles of 104° for Au and close to 180° for Pt)32 as most odd-numbered members of the series exhibited lower ΓCV.22,49 For the current Fc–CC–Fc systems, the odd–even effect on Au and Pt electrodes was alike, i.e., odd numbered SAMs on both metals gave higher values of ΓCV than the even numbered SAMs. Furthermore, we did not detect back-bending in the CV curves up to nc = 15 (see ESI, Fig. S2 and S3†), which is in contrast to the series of HSCnFc showing substantial back-bending for the longer derivatives (nc ≥ 14), resulting in additional peaks in the CV curves.22,49 Instead, for the current series, we observed a gradual increase in the ΓCV as a function of nc. The observed differences are likely due to packing effects related to the large head group. The data suggests that the packing of the SCnFc–CC–Fc SAMs is driven by the large Fc–CC–Fc group, overruling the effects of the M–S–C bond angles that directed the packing and properties of the SCnFc series.22,49 Our observations are supported by MD calculations of the SAM heights (dSAM,MD, in nm) and surface coverages (ΓMD, in nmol cm−2) on Pt (Fig. 2c). As expected for well-packed SAMs, the dSAM,MD increases with nc as the molecule becomes longer. The SAM structural order, as quantified by the damping of root-mean-square fluctuation (RMSF) values of non-hydrogen atoms, improves with length and shows a clear odd–even effect (Fig. 2d). The improved structuring in odd-numbered SAMs on Pt is reflected also in the computed odd–even packing energy differences (ΔE, in kcal mol−1), with large components coming from the head groups (Fig. 2e). The MD calculations show that the odd–even effect in order and ΔE is reflected in the tilt angles α of the head groups (Fig. S10 for SAMs on Pt and Fig. S11† for the SAMs on Au).
We note that the experimental odd–even effect in ΓCV cannot be observed in the modelling of ΓMD, which is due to the protocol of the modelling procedure. The SAM model was created by allowing the molecules to move freely on the surface until they form packed monolayers and this equilibrated physisorbed configuration is then fixed to the surface by switching on the metal–thiolate bonds. Although this method works well to capture different types of odd–even effects in the chemisorbed SAMs (such as oscillations in tilt angles and molecular packing energies), it does not capture the more subtle odd–even effects in surface coverages, which would require a more gradual “strengthening” of the metal–thiolate bonds in the model as the SAM forms. The computed packing energy contributions (E) of different components of the molecules (Fig. 2f), show that the combination of head group and alkyl group stabilisation promotes creation of tightly-packed, upright SAMs for all nc = 9–15 with gradual improvement in stability for longer chains. The simulations predict that at higher nc = 12–15, the alkyl chain is long enough and its packing strong enough to replace alkyl–head group cross-interactions as the primary driving force assisting head group packing in stabilising the SAMs.
Electrical characterisation of the SAMs
We measured J(V) curves and determined R across the M–SCnFc–CC–Fc//GaOx/EGaIn junctions following the procedure reported previously.22,42 From statistically large numbers of J(V) curves, we determined the Gaussian log-average values of J, 〈log10|J|〉G, at each measured V to construct the 〈log10|J|〉Gvs. V curves. Details of data recording and analysis are given in the ESI,† and the J(V) curves and corresponding histograms are shown in Fig. S14–S16.† We have previously collected J(V) curves of M–SC15Fc–CC–Fc (M = Ag, Au, Pt), and the data of this derivative was taken from our previous work.42
For the three SAM series on Ag, Au and Pt (Fig. 3a–c), the 〈log10|J|〉G decreased with increasing nc, which is as expected as the tunnelling barrier width increases with increasing nc. We observe a clear odd–even oscillation on Pt (Fig. 3c) as the odd-numbered derivatives produce smaller values of J than the even numbered ones. For junctions on Ag (Fig. 3a), this odd–even effect is weakly present but absent for junctions on Au (Fig. 3b).
 | ||
| Fig. 3 Measured electrical properties 〈log10|J|〉G (a–c) and 〈log10R〉G (d–f) vs. nc for M–SCnFc–CC–Fc//GaOx/EGaIn junctions at low bias of ±1.0 V. The respective metals are indicated in each panel. The error bars represent the σlog,G from the Gaussian fit of log10|J| and log10R. | ||
All the junctions rectify electrical current at ±1.0 V and we observe a gradual increase of R with increasing nc, i.e., from nc = 9 to 15 on all three metal surfaces (Fig. 3d–f). For instance, the R for SAMs on Ag increased by approximately an order of magnitude from 〈log10R〉G of 2.0 ± 0.5 to 3.0 ± 0.3. This increase in R is likely due to the increasing surface coverage at increasing nc along with the improvement in molecular packing, and so reduced leakage currents.22,23 As established above by the ΓCV and the MD simulations, the longer chain derivatives pack better as the alkyl–alkyl interactions become more pronounced with increasing number of nc, which translates into better diode performance (Fig. 2). A strong odd–even effect was evident for 〈log10R|〉G on Ag (Fig. 3d), with the odd-numbered derivatives operating at consistently higher 〈log10R|〉G. These effects were less pronounced on Pt (Fig. 3f†) and partially reversed on Au (Fig. 3e). The reason could be that the surface morphologies of template-stripped Ag, Au, and Pt differ from each other.42,50,51 This observation agrees with our earlier work where we also showed that the odd–even effect on Au is smaller than on Ag, likely due to the fact that the tilt angle of the molecular backbone on Ag (and Pt) is smaller than on Au.32 In light of the surface characterisation described above, the trend of R on Au indicates a transition around nc = 12 which can be explained by a transition from an alkyl chain packing driven SAM to a head group packing dominated SAM (Fig. 2 and S11†). Probably for this series, the tilt angle of the molecular backbone (the alkyl chain) of the SAM of 30° (as opposed to reported tilts of 0–10° for SAMs on Ag and Pt)48 explains why such a clear transition was not visible for the other two types of SAMs.
Electrical stability of the junctions
Above we have shown that the monolayers on Pt are well-organised and densely-packed and that SAMs with nc = odd performing better than those with nc = even. We have established previously that the SAMs on Pt substrates are more stable and can withstand larger applied bias than those with Ag and Au electrodes.42 For these reasons, we focus on junctions on Pt with nc = odd to study how the value of R depends on applied voltage. To do so, we need to establish the maximum bias range we can apply for each value of nc = odd before a junction electrically shorts.
Fig. 4 shows the VBD (the applied maximum bias at which the junctions start to short) and the associated electric fields (EBD, EBD = VBD/dSAM,MD). We have previously determined the break-down voltage of Pt–SC15Fc–CC–Fc, and the data of this derivative was taken from our previous work.42 The general finding is that the VBD increases linearly with increasing nc at both positive and negative bias directions, while the electric fields across the junctions remain constant, with EBD of 1.31 ± 0.08 and −1.03 ± 0.02 GV m−1 at positive and negative voltages, respectively. This further confirms that increasing the nc improves the stability of the Pt–SCnFc–CC–Fc//GaOx/EGaIn junctions in terms of VBD. This finding is in agreement with the breakdown behaviour of n-alkylthiolate based EGaIn junctions,52 and silicon based molecular wires of varying lengths in single-molecule junctions.53
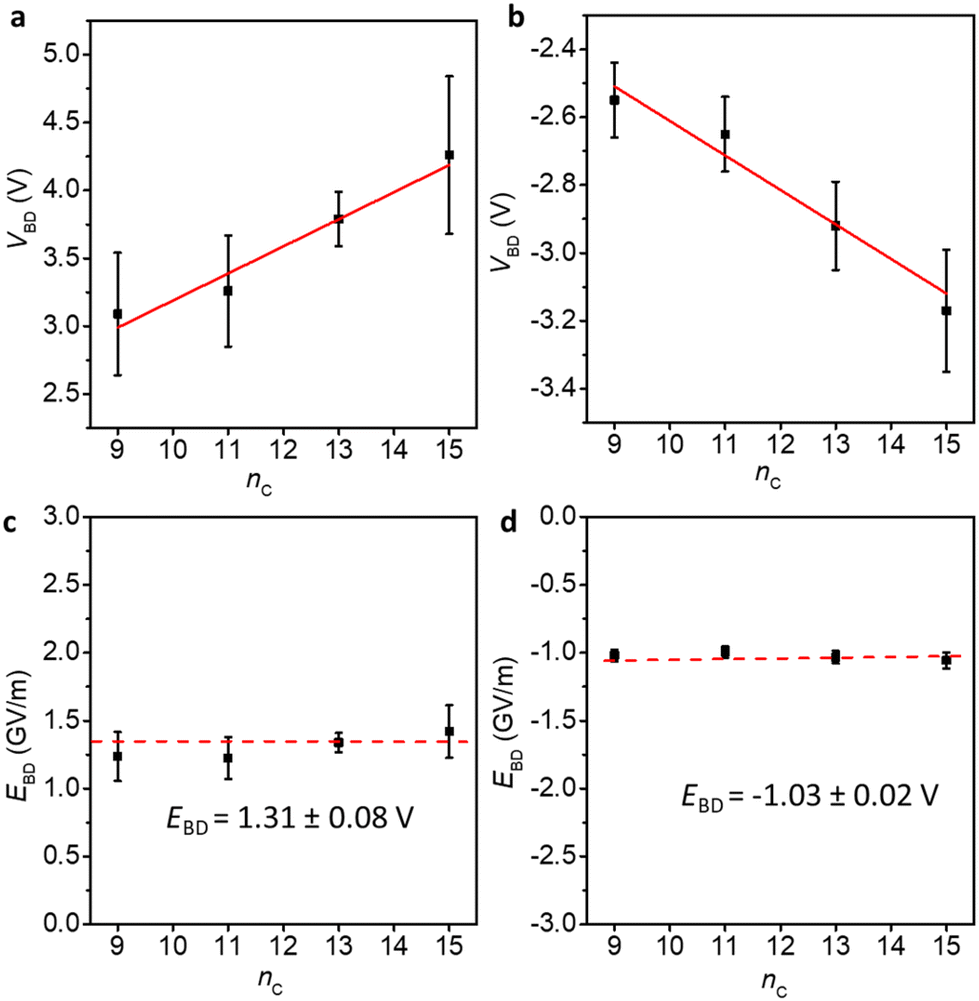 | ||
| Fig. 4 Breakdown voltages VBD (a and b) and breakdown fields EBD (c and d) for junctions of Pt–SCnFc–CC–Fc//GaOx/EGaIn (nc = 9, 11, 13, 15) at positive and negative bias, respectively. VBD represents the breakdown voltage, EBD represents the breakdown field. The error bars in VBD represent the standard deviation (σ) from the Gaussian fits in Fig. S17.† The VBD of nc = 15 is taken from our previous report.42 The error bars in EBD are calculated from the error bars of VBD (EBD = VBD/dSAM,MD). | ||
High bias operation
Through determination of the breakdown voltages, we have established the maximum safe bias windows in which the junctions are stable and do not short. Now we apply this knowledge to measure junctions of those derivatives at the maximum bias range. Fig. 5 shows the 〈log10|J|〉Gvs. applied bias V for Pt–SCnFc–CC–Fc//GaOx/EGaIn junctions (nc = 9, 11, 13, and 15) at high bias and the corresponding values of R vs. V which are also summarised in Table 1.
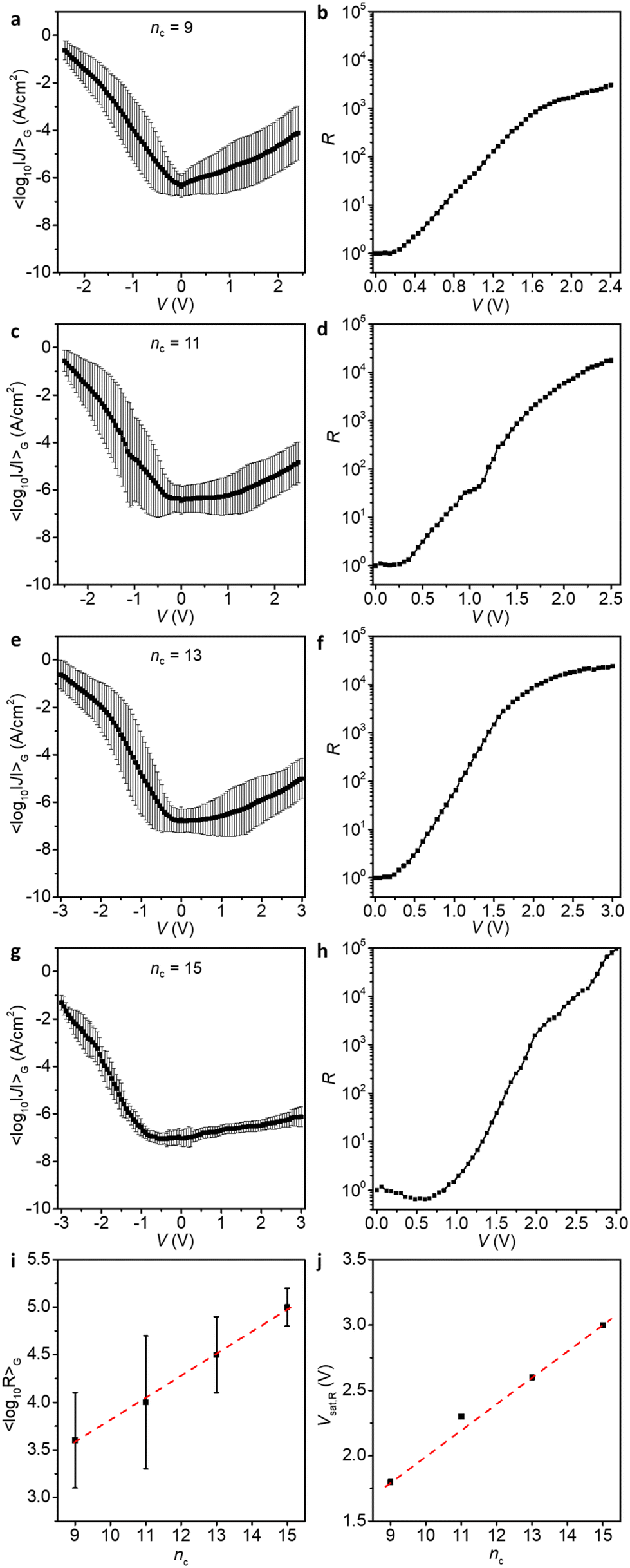 | ||
| Fig. 5 Curves of 〈log10|J|〉Gvs. applied bias V (a, c, e and g) and R against applied bias (b, d, f and h) of Pt–SCnFc–CC–Fc//GaOx/EGaIn (nc = 9, 11, 13 and 15) at bias of ±2.4, ±2.5, ±3.0, and ±3.0 V, respectively. Error bar represents the σlog,G at each bias. (i) 〈log10R〉G for junctions of Pt–SCnFc–CC–Fc//GaOx/EGaIn (nc = 9, 11, 13 and 15) at the highest applied bias (as in panels a, c, e and d). (j) The plot of Vsat,Rvs. nc. The error bars in panel i are σlog,G and the dashed red lines in i and j are guides to the eye. | ||
C–Fc//GaOx/EGaIn (nc = 9, 11, 13, and 15) at high bias
| n c | 9 | 11 | 13 | 15 |
|---|---|---|---|---|
| Bias [V] | ±2.4 | ±2.5 | ±3.0 | ±3.0 |
| R | 4.0 × 103 | 1.0 × 104 | 3.1 × 104 | 1.0 × 105 |
As shown in Fig. 5i, 〈log10R〉G for junctions at the highest applied bias (〈log10R〉G,max) increases linearly with increasing nc, which indicates that the longer alkyl chain ensures a high VBD, meaning that a high forward voltage can be applied without suffering from an increase in leakage current at large reverse bias. Interestingly, the voltage at which the R saturates (Vsat,R) also shows a linearly positive dependence on the number of nc (Fig. 5j).
The onset voltage at which R begins to evolve (Vonset,R), may be used as a semi-quantitative guide to determine when HOMO and HOMO−1 begin entering the conduction window, which is approximately 0.3 V for all junctions (with the exception for junctions with nc = 15 which are too resistive and the current is dominated by capacitive current near 0 V, and therefore we cannot determine the onset voltage reliably).42 In principle, the Vsat,R represents the bias at which the HOMO and HOMO−1 have fully entered the conduction window, and therefore R no longer increases with increasing bias.42 Calculations by Zhang and co-workers indicated that other frontier orbitals, up to HOMO−5, can potentially enter the bias window and contribute to the conduction in (theoretical) single-molecule junctions of the SCnFc–CC–Fc.54 It would be interesting to investigate in more detail the potential role of deeper orbitals in our large-area junctions.
Interestingly, a clear transition can be seen around 1.8 to 2.0 V, nc = 9, 11 and 13, but the curves still continue to increase at higher voltages. The behaviour is different for junctions with nc = 15 which show a continuous increase in the current all the way to the maximum applied voltage of 3.0 V. This behaviour can be explained as follows. We have reported before42 that at forward bias, when the HOMO and HOMO−1 fall in the conduction window (Fig. 1b, ON-State I), the bisferrocene is oxidised resulting in electrostatic attraction between the ferrocenium cations and the negatively charged top-electrode (Fig. 1b, ON-State II). This electrostatic attraction is voltage driven, and for nc = 15 clearly the largest voltages of up to 3 V are accessible explaining the superior performance in terms of the value of R of the diode with nc = 15. We did not investigate molecular diodes with nc > 15 as already at nc = 15 the currents measured in the low voltage ranges were below the detection limit of our system.
Fitting employing a single-level tunnelling model
We successfully modelled the behaviour of the junctions employing a theoretical model for the Landauer formulism developed previously.42Eqn (1) describes a modified version of the Landauer single-level model that accounts for the multiple junctions present in a SAM junction.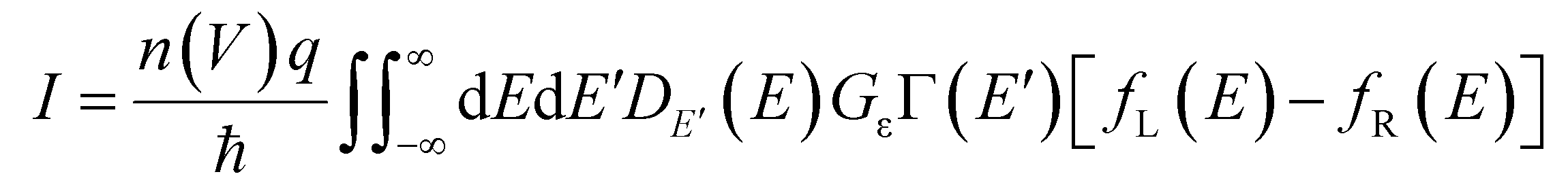 | (1) |
Further, a function was used to describe the number of molecules contributing to conduction as a function of the bias voltage. The individual components of eqn (1) have been explained in detail in our previous work,42 and are further outlined in the ESI (pages S62–S63†). Fitting was conducted by implementing several constraints and the low and high bias regimes were treated slightly differently to account for the differences in the onset voltage (see pages S64–S68† for details). Overlays of the fits of the J(V) curves and the experimental results, for the whole series of diodes, are shown in Fig. S18.† Overall, a strong agreement between the theoretical calculations and the experimental data was evident. The data shows as well that the asymmetry across the junctions increased with the molecular length, and that the molecule–electrode coupling varied as a function of molecular length. This further confirms that the molecular potentiometer rule applies in the current series, by demonstrating that an asymmetric position of the molecular orbitals involved in the charge transport in the junction results in a high rectification ratio. In addition, this agrees with theoretical models developed for other rectifiers such as an alkyl cobaltocene,55 and aryl systems with two alkylthiol anchoring groups of varying lengths.56
Conclusions
In summary, we show that the evolution of R for SAM based molecular diodes depends on the molecular length, the bias range, and the type of metal used for the bottom electrode. We optimised these parameters to achieve consistently high R values of 103–105, above the Landauer limit and approaching the lower limit of CMOS performance. The different molecular lengths and metals affect the supramolecular structure of the SAM, and therefore impact the junction stability and leakage current. Increasing the lengths of the molecule from nc = 9 to 15 resulted in a ∼1.3 fold increase of the VBD, therefore allowing to operate these stable molecular diodes at a high operating bias of V = 3 V. Remarkably, the tight surface packing ensured low leakage currents at reverse bias despite the high driving voltages. Subtle changes in the packing structure driven by odd–even effects as well as the important role of the bottom electrode material affect diode performance in accordance with previous findings,9–12,18,22,57 with Pt electrodes supporting densely packed, stable SAMs. Our findings give new insights into why diodes that “look good on paper” often showed disappointing performance, with our dataset showing that systematic improvement requires simultaneous tuning of both applied bias and (supra)molecular effects induced by supramolecular and molecule–substrate interactions. We therefore believe that our findings will inform future studies aiming to improve molecular electronic devices.Author contributions
M.R. synthesised the compounds. X.C and L.Y. characterised the compounds electrically. L.W. and M.R. characterised the SAMs by CV. X.Y. characterised the SAMs by XPS, UPS and NEXAFS. P.A.C. and D.T. performed the MD calculations. C.N., R.A. and E.D.B. performed the fitting. C.A.N. conceptualised and led the study. M.R., X.C. and C.A.N. wrote the manuscript, and all authors commented on the manuscript and discussed the data.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Minister of Education (MOE) for supporting this research under award no. MOE2015-T2-1-050. Prime Minister's Office, Singapore under its Medium sized centre program is also acknowledged for supporting this research. The authors would also like to acknowledge the Singapore Synchrotron Light Source (SSLS) for providing the facility necessary for spectroscopic measurements. We thank Dr Li Jiang for providing the Au, Ag and Pt substrates. X. C. thanks the support under the Natural Science Foundation of Fujian Province, China (Grant No. 2022J05173). D. T. thanks Science Foundation Ireland (SFI) for financial support under Grant Number 12/RC/2275_P2 (SSPC), and for provision of computing resources at the SFI/Higher Education Authority Irish Centre for High-End Computing (ICHEC).References
- J. C. Cuevas and E. Scheer, Molecular Electronics, An Introduction to Theory and Experiment, World Scientific Publishing, 2010 Search PubMed.
- Y. Han and C. A. Nijhuis, Chem. – Asian J., 2020, 15, 3752–3770 CrossRef CAS PubMed.
- N. Xin, J. Guan, C. Zhou, X. Chen, C. Gu, Y. Li, M. A. Ratner, A. Nitzan, J. F. Stoddart and X. Guo, Nat. Rev. Phys., 2019, 1, 211–230 CrossRef.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- M. Naher, M. Roemer, G. A. Koutsantonis and P. J. Low, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 38–80 Search PubMed.
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- B. Capozzi, J. Xia, O. Adak, E. J. Dell, Z. F. Liu, J. C. Taylor, J. B. Neaton, L. M. Campos and L. Venkataraman, Nat. Nanotechnol., 2015, 10, 522–527 CrossRef CAS PubMed.
- M. Elbing, R. Ochs, M. Koentopp, M. Fischer, C. von Hanisch, F. Weigend, F. Evers, H. B. Weber and M. Mayor, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8815–8820 CrossRef CAS PubMed.
- P. Song, S. Guerin, S. J. R. Tan, H. V. Annadata, X. Yu, M. Scully, Y. M. Han, M. Roemer, K. P. Loh, D. Thompson and C. A. Nijhuis, Adv. Mater., 2018, 30, 1706322 CrossRef PubMed.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 17814–17827 CrossRef CAS PubMed.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2010, 132, 18386–18401 CrossRef CAS PubMed.
- L. Yuan, N. Nerngchamnong, L. Cao, H. Hamoudi, E. del Barco, M. Roemer, R. K. Sriramula, D. Thompson and C. A. Nijhuis, Nat. Commun., 2015, 6, 6324 CrossRef CAS PubMed.
- G. M. Morales, P. Jiang, S. Yuan, Y. Lee, A. Sanchez, W. You and L. Yu, J. Am. Chem. Soc., 2005, 127, 10456–10457 CrossRef CAS PubMed.
- I. Díez-Pérez, J. Hihath, Y. Lee, L. Yu, L. Adamska, M. A. Kozhushner, I. I. Oleynik and N. Tao, Nat. Chem., 2009, 1, 635–641 CrossRef PubMed.
- A. R. Garrigues, L. Yuan, L. Wang, E. R. Mucciolo, D. Thompon, E. del Barco and C. A. Nijhuis, Sci. Rep., 2016, 6, 26517 CrossRef CAS PubMed.
- M. L. Perrin, E. Galan, R. Eelkema, F. Grozema, J. M. Thijssen and H. S. J. van der Zant, J. Phys. Chem. C, 2015, 119, 5697–5702 CrossRef CAS.
- A. C. Aragonès, N. Darwish, S. Ciampi, F. Sanz, J. J. Gooding and I. Díez-Pérez, Nat. Commun., 2017, 8, 15056 CrossRef PubMed.
- L. Yuan, R. Breuer, L. Jiang, M. Schmittel and C. A. Nijhuis, Nano Lett., 2015, 15, 5506–5512 CrossRef CAS PubMed.
- Y. Han, C. Nickle, Z. Zhang, H. P. A. G. Astier, T. J. Duffin, D. Qi, Z. Wang, E. del Barco, D. Thompson and C. A. Nijhuis, Nat. Mater., 2020, 19, 843–848 CrossRef CAS PubMed.
- L. Yuan, L. Jiang, B. Zhang and C. A. Nijhuis, Angew. Chem., Int. Ed., 2014, 53, 3377–3381 CrossRef CAS PubMed.
- L. Wang, L. Yuan, L. Jiang, X. Yu, L. Cao and C. A. Nijhuis, J. Phys. Chem. C, 2019, 123, 19759–19767 CrossRef CAS.
- N. Nerngchamnong, L. Yuan, D.-C. Qi, J. Li, D. Thompson and C. A. Nijhuis, Nat. Nanotechnol., 2013, 8, 113–118 CrossRef CAS PubMed.
- D. Thompson and C. A. Nijhuis, Acc. Chem. Res., 2016, 49, 2061–2069 CrossRef CAS PubMed.
- F. Tao and S. L. Bernasek, Chem. Rev., 2007, 107, 1408–1453 CrossRef CAS PubMed.
- P. Cyganik, A. Terfort and M. Zharnikov, Nano Res., 2023, 17, 4231–4243 CrossRef.
- T. Felgenhauer, H. T. Rong and M. Buck, J. Electroanal. Chem., 2003, 550–551, 309–319 CrossRef CAS.
- D. M. Alloway, M. Hofmann, D. L. Smith, N. E. Gruhn, A. L. Graham, R. Colorado, V. H. Wysocki, T. R. Lee, P. A. Lee and N. R. Armstrong, J. Phys. Chem. B, 2003, 107, 11690–11699 CrossRef CAS.
- F. Ben Amara, E. R. Dionne, S. Kassir, C. Pellerin and A. Badia, J. Am. Chem. Soc., 2020, 142, 13051–13061 CrossRef CAS PubMed.
- Y. Feng, E. R. Dionne, V. Toader, G. Beaudoin and A. Badia, J. Phys. Chem. C, 2017, 121, 24626–24640 CrossRef CAS.
- M. Baghbanzadeh, F. C. Simeone, C. M. Bowers, K.-C. Liao, M. Thuo, M. Baghbanzadeh, M. S. Miller, T. B. Carmichael and G. M. Whitesides, J. Am. Chem. Soc., 2014, 136, 16919–16925 CrossRef CAS PubMed.
- M. M. Thuo, W. F. Reus, C. A. Nijhuis, J. R. Barber, C. Kim, M. D. Schulz and G. M. Whitesides, J. Am. Chem. Soc., 2011, 133, 2962–2975 CrossRef CAS PubMed.
- L. Yuan, D. Thompson, L. Cao, N. Nerngchangnong and C. A. Nijhuis, J. Phys. Chem. C, 2015, 119, 17910–17919 CrossRef CAS.
- R. Arielly, M. Vadai, D. Kardash, G. Noy and Y. Selzer, J. Am. Chem. Soc., 2014, 136, 2674–2680 CrossRef CAS PubMed.
- B. Cui, Y. Xu, G. Ji, H. Wang, W. Zhao, Y. Zhai, D. Li and D. Liu, Org. Electron., 2014, 15, 484–490 CrossRef CAS.
- D. Duche, U. Planchoke, F.-X. Dang, J. L. Rouzo, M. Bescond, J.-J. Simon, T. S. Balaban and L. Escoubas, J. Appl. Phys., 2017, 121, 115503 CrossRef.
- H. Jeong, Y. Jang, D. Kim, W.-T. Hwang, J.-W. Kim and T. Lee, J. Phys. Chem. C, 2016, 120, 3564–3572 CrossRef CAS.
- E. D. Mentovich, N. Rosenberg-Shraga, I. Kalifa, M. Gozin, V. Mujica, T. Hansen and S. Richter, J. Phys. Chem. C, 2013, 117, 8468–8474 CrossRef CAS.
- L. Müller-Meskamp, S. Karthäuser, H. J. W. Zandvliet, M. Homberger, U. Simon and R. Waser, Small, 2009, 5, 496–502 CrossRef PubMed.
- S. Seo, E. Hwang, Y. Cho, J. Lee and H. Lee, Angew. Chem., Int. Ed., 2017, 56, 12122–12126 CrossRef CAS PubMed.
- J. Trasobares, D. Vuillaume, D. Théron and N. Clément, Nat. Commun., 2016, 7, 12850 CrossRef CAS PubMed.
- S. Wang, M.-Z. Wei, G.-C. Hu, C.-K. Wang and G.-P. Zhang, Org. Electron., 2017, 49, 76–84 CrossRef CAS.
- X. Chen, M. Roemer, L. Yuan, W. Du, D. Thompson, E. del Barco and C. A. Nijhuis, Nat. Nanotechnol., 2017, 12, 797–803 CrossRef CAS PubMed.
- D. Thompson, E. d. Barco and C. A. Nijhuis, Appl. Phys. Lett., 2020, 117, 030502 CrossRef CAS.
- N. Clement and A. Fujiwara, Nat. Nanotechnol., 2017, 12, 725–726 CrossRef CAS PubMed.
- N. Nerngchamnong, L. Yuan, D. C. Qi, J. Li, D. Thompson and C. A. Nijhuis, Nat. Nanotechnol., 2013, 8, 113–118 CrossRef CAS PubMed.
- K. Yang, Z. Cai, A. Jaiswal, M. Tyagi, J. S. Moore and Y. Zhang, Angew. Chem., Int. Ed., 2016, 55, 14090–14095 CrossRef CAS PubMed.
- R. Boese, H.-C. Weiss and D. Bläser, Angew. Chem., Int. Ed., 1999, 38, 988–992 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed.
- N. Nerngchamnong, D. Thompson, L. Cao, L. Yuan, L. Jiang, M. Roemer and C. A. Nijhuis, J. Phys. Chem. C, 2015, 119, 21978–21991 CrossRef CAS.
- L. B. Newcomb, I. D. Tevis, M. B. Atkinson, S. M. Gathiaka, R. E. Luna and M. Thuo, Langmuir, 2014, 30, 11985–11992 CrossRef CAS PubMed.
- E. A. Weiss, G. K. Kaufman, J. K. Kriebel, Z. Li, R. Schalek and G. M. Whitesides, Langmuir, 2007, 23, 9686–9694 CrossRef CAS PubMed.
- L. Yuan, L. Jiang and C. A. Nijhuis, Adv. Funct. Mater., 2018, 28, 1801710 CrossRef.
- H. Li, T. A. Su, V. Zhang, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, J. Am. Chem. Soc., 2015, 137, 5028–5033 CrossRef CAS PubMed.
- J.-M. Zhao, L.-Y. Chen, Y.-J. Li, N.-P. Shi, Y.-Z. Sun, H. Huang and G.-P. Zhang, Phys. E, 2021, 130, 114691 CrossRef CAS.
- R. Liu, S. H. Ke, W. Yang and H. U. Baranger, J. Chem. Phys., 2006, 124, 024718 CrossRef PubMed.
- P. E. Kornilovitch, A. M. Bratkovsky and R. Stanley Williams, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 165436 CrossRef.
- C. A. Nijhuis, W. F. Reus, J. R. Barber, M. D. Dickey and G. M. Whitesides, Nano Lett., 2010, 10, 3611–3619 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical data of the molecules, details of the SAM formation and characterisation data (CV, XPS, UPS, NEXAFS), details of the MD calculations, electrical characterisation data and details of the fitting. See DOI: https://doi.org/10.1039/d4nr01514b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |