
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvesting in entropy: the strategy of cucurbit[n]urils to accelerate the intramolecular Diels–Alder cycloaddition reaction of tertiary furfuryl amines†
Karen de la Vega-Hernández a,
Marcos G. Suero*ab and
Pablo Ballester*ab
a,
Marcos G. Suero*ab and
Pablo Ballester*ab
aInstitute of Chemical Research of Catalonia (ICIQ-CERCA), The Barcelona Institute of Science and Technology (BIST), Avgda. Països Catalans 16, 43007, Tarragona, Spain. E-mail: mgsuero@iciq.es; pballester@iciq.es
bICREA, Passeig Lluís Companys, 23, 08018 Barcelona, Spain
First published on 3rd May 2024
Abstract
Cucurbit[n]urils, renowned for their host–guest chemistry, are becoming versatile biomimetic receptors. Herein, we report that cucurbit[7]uril (CB[7]) accelerates the intramolecular Diels–Alder (IMDA) reaction for previously elusive and unreactive tertiary N-methyl-N-(homo)allyl-2-furfurylamines by up to 4 orders of magnitude under mild conditions. Using 1H NMR titrations and ITC experiments, we characterize the dissimilar thermodynamic and kinetic properties of the complexes. We also determine the activation parameters (ΔG≠, ΔH≠ and ΔS≠) leading to the transition state of the IMDA reactions, both in the bulk and included in CB[7], to shed light on the key role of the receptor on the acceleration observed. CB[7] acts as an “entropy trap” utilizing guest binding to primarily pay the entropy penalty for reorganizing the substrate in a high-energy reactive conformation that resembles the geometry of the highly ordered transition state required for the IMDA reaction. This study underscores the potential of cucurbit[n]urils as artificial active sites, emulating specific aspects of enzymatic catalysis.
Introduction
Reminiscent of a hula-hoop at the nanoscale, cucurbit[n]urils (CB[n]s) have arisen as versatile rigid ring-shaped receptors. Their cavities contain two open-portals and are sculpted to surround most of the surface of the included guests.1–3 CB[n]s are composed of glycoluril units linked together by methylene bridges, where n represents the number of recurrent units in the ring (Fig. 1, top). Accordingly, the different members of the cucurbit[n]uril family display dissimilar portal sizes and cavity volumes. Their binding selectivity is mainly governed by the size and shape complementarity principle.4 Owing to the hydrophobic nature of the CB[n]s' cavity and their water solubility, they are excellent hosts for the binding/inclusion of organic molecules of reduced polarity. The binding process is mainly driven by the hydrophobic effect. In addition, the carbonyl groups defining the rims of the portals confer CB[n]s with cation binding properties (charge–dipole interaction). Notably, the fast-paced growth of the host–guest chemistry of CB[n]s has been echoed with several applications in the fields of catalysis,1,5,6 sensing,7,8 and drug delivery.9,10 | ||
| Fig. 1 Molecular structures of cucurbit[n]urils (top). Line-drawing structures of tertiary N-methyl-N-(homo)allyl-2-furfurylamines FA-1 and FA-2, along with their Diels–Alder cycloadducts P-1 and P-2. Schematic representation of the binding equilibrium of the amines with CB[7], the subsequent irreversible intramolecular Diels–Alder (IMDA) reaction occurring in the inclusion complexes, and the binding equilibrium with the cycloadducts (bottom). | ||
Researchers working in the field of supramolecular catalysis are actively exploring the use of synthetic receptors as molecular vessels able to mimic certain aspects of enzymatic catalysis.1,11–13 These aspects include: molecular recognition, catalytic efficiency, catalytic environment (providing confined spaces), substrate specificity, regio- and stereo-control of the reaction compared to the bulk. In this regard, CB[n]s have not been left behind, and have been used as molecular vessels in photochemical reactions,14–16 oxidation reactions,17 solvolysis reactions,18 azide–alkyne click chemistry,19,20 1,3-dipolar cycloaddition,21 and Diels–Alder (DA) reactions.22,23 Recently, Assaf, Nau and co-workers reported that the inclusion of the intermolecular Diels–Alder dimerization of cyclopentadiene in CB[7] produced an acceleration factor, kincluded/kbulk, of 4 × 105 M.22 This magnitude is very close to the 106 M factor (ΔΔS≠ = 28.3 e.u.) theoretically predicted by Page and Jencks regarding an intramolecular analog.24 In the same vein, a few years before, Scherman and co-workers investigated the acceleration levels experienced by inclusion in CB[7] of the intramolecular Diels–Alder (IMDA) reaction of secondary N-allyl-2-furfurylamines.23 They reported a maximum acceleration factor, determined as kincluded/kbulk, of ∼103 at 37 °C. This value corresponds to a difference in entropies of activation for the IMDA reactions occurring in the bulk and included in CB[7] of c.a. 13.7 e.u. They suggested that the binding of the secondary N-allyl-2-furfurylamines in the cavity of CB[7] was driven not only by the hydrophobic effect caused by the inclusion of the non-polar residues but also by the establishment of multiple cation–dipole interactions between the protonated secondary amine group and the seven carbonyl functions defining the rims of the container's portals.
Diels–Alder reactions are enormously useful in organic synthesis as ring-forming reactions.25–28 The archetypal DA reaction is a [4π + 2π] cycloaddition in which a diene (the 4π component) combines with a dienophile (the 2π component) to produce a 6-membered ring. The DA reaction is governed by orbital symmetry considerations: it proceeds through a suprafacial interaction of both components (4π and 2π), involving a highly ordered transition state,29 which is reflected in large and negative activation entropies (i.e. – 33 e.u.).24 In the intramolecular version of DA reactions, both the diene and the dienophile are part of the same molecule. This characteristic increases the effective concentration of the reactive sites, causing a reduction of the activation entropy, and, in some cases, produces a favorable preorganization for the reaction. Preorganization and lower activation entropy are related because the preorganized ground-state conformation of the substrate has an entropy close to that of the transition state.30,31 However, having both reactive sites in the same molecule does not guarantee the fulfillment of the optimum geometrical requirements (preorganization) for the transition state (TS) of the IMDA reaction, or that the more populated conformers of the molecule locate the diene and the dienophile groups in close spatial proximity. These limitations seem to be present in the case of secondary N-allyl-2-furfurylamines and tertiary N-methyl-N-allyl-2-furfurylamines for which the IMDA is not significantly favored. An effective strategy to accelerate these disfavored IMDA reactions in the bulk relied on installing steric buttresses on the N atom.32–34 The idea at work has to do with restricting the conformational freedom of the molecules and their reactive groups. Nevertheless, this strategy requires the synthesis of elaborated substrates, and the subsequent removal -when possible- of the bulky protecting groups. In short, the efficient synthesis of cycloadducts deriving from the IMDA reaction of tertiary N-methyl-N-(homo)allyl-2-furfurylamines is yet to be achieved.
Herein, we report the acceleration of the IMDA reaction of unactivated tertiary N-methyl-N-allyl-2-furfurylamine (FA-1) and N-methyl-N-(homo)allyl-2-furfurylamine (FA-2) by inclusion in CB[7] (Fig. 1, bottom). We investigated the binding properties of CB[7] with the two tertiary amines by means of 1H NMR spectroscopic titrations and ITC experiments. We also conducted kinetic studies of the IMDA reactions of FA-1 and FA-2, both in the presence and absence of CB[7]. Furthermore, we evaluated the effect of the use of CB[7] as molecular vessel in the reactions' kinetic parameters (ΔG≠, ΔH≠ and ΔS≠) and compared them with those of the reaction in the bulk. Finally, we tested the use of CB[8] and a series of cyclodextrins (α-, β- and γ-CDs) as mediators for the studied IMDA reaction. CB[7] was unique in preorganizing the included FA-1 and FA-2 tertiary amines in a reactive conformation capable of accelerating their IMDA reactions.
Results and discussion
The interaction of FA-1 with CB[7] in D2O solution was probed using 1H NMR spectroscopy (Fig. 2, see also ESI†). The 1H NMR spectrum of a mixture of FA-1 and CB[7] revealed that the proton signals of the furan and alkene groups of FA-1 moved significantly upfield (Δδ (Ha) = −0.83 ppm; Δδ (Hg) = −1.37 ppm), compared to those of free FA-1 in solution (Fig. 2a and b). This result indicated that the furan (diene) and the alkene (dienophile) groups were included in the cavity of CB[7], where they experienced the shielding effect caused by the seven glycoluril units. In the presence of more than 1.0 equiv. of FA-1, we detected two separate sets of signals for the protons of the amine. The new set of signals corresponded to that of FA-1 free in solution (Fig. 2c). Taken together, the obtained results indicated the quantitative formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex, FA-1⊂CB[7], in the equimolar mixture of binding partners, for which we estimated an association constant value (Ka) larger than 104 M−1. The binding process displayed slow exchange dynamics on the 1H NMR chemical-shift timescale.
:1 inclusion complex, FA-1⊂CB[7], in the equimolar mixture of binding partners, for which we estimated an association constant value (Ka) larger than 104 M−1. The binding process displayed slow exchange dynamics on the 1H NMR chemical-shift timescale.
 | ||
| Fig. 2 Selected region of the 1H NMR spectra (400 MHz, D2O, 298 K) of: (a) FA-1 (0.005 M); (b) FA-1⊂CB[7] (1.05:1, 0.005 M); (c) FA-1⊂CB[7] (2:1, 0.005 M); (d) P-1⊂CB[7] (1:1, 0.005 M), acquired after 16 h of evolution of spectrum b; and (e) P-1 (0.005 M). FA-1 in red and P-1 in blue. Primed labels correspond to proton signals of bound species. *Residual solvent peak. | ||
We monitored the intramolecular Diels–Alder reaction of FA-1 included in CB[7] using kinetic 1H NMR experiments. After 16 h, we observed the complete conversion of the FA-1⊂CB[7] complex into the corresponding P-1⊂CB[7] counterpart (Fig. 2d). The diagnostic signals of the P-1⊂CB[7] complex were identified by comparison with those of the 1H NMR spectrum of a solution containing an equimolar mixture of the DA cycloadduct P-1 and CB[7] (Fig. S13†).‡
Next, we determined accurate binding constants and other thermodynamic parameters of the binding processes of FA-1 and P-1 with CB[7] using isothermal titration calorimetry experiments in D2O solution (ITC, Table 1, Fig. S20–S22†).35,§ It is worth mentioning here that the accurate determination of the thermodynamic parameters for the FA-1⊂CB[7] complex was possible because its background IMDA reaction (uncatalyzed) and that in the presence of CB[7] (catalyzed) were negligible under the used titration conditions. The binding constant values (Ka) of CB[7] for FA-1 and P-1 were in the same order of magnitude (105 M−1). Remarkably, CB[7] showed some selectivity in the binding of FA-1 suggesting that the IMDA reaction catalyzed by substoichiometric amounts of CB[7] might experience catalytic turnover. The large enthalpic contribution to the binding of FA-1 and P-1 by CB[7] (Table 1) supported that the processes were driven by the non-classical hydrophobic effect. In the specific case of CB[n]s binding, this has been associated to the release of high-energy water molecules included in their hydrophobic cavities.36 Not surprisingly, the formation of the two inclusion complexes with CB[7] were associated with a significant entropic cost mainly deriving from the association of two molecules in 1:1 complexes (loss of at least three degrees of freedom).
| Complex | K a [M−1] | ΔG [kcal mol−1] | ΔHb [kcal mol−1] | TΔS [kcal mol−1] | k off [s−1] | k on [M−1 s−1] |
|---|---|---|---|---|---|---|
| a Error values in Ka and ΔH are reported as standard deviations and propagated to ΔG, ΔS, and kon. b Determined by ITC. c Determined from 1H–1H EXSY experiments. d Calculated from Ka = kon/koff. | ||||||
| FA-1⊂CB[7] | (8.1 ± 0.6) × 105 | −8.1 ± 0.7 | −14.8 ± 0.5 | −6.7 ± 0.5 | 1.1 | (8.9 ± 0.6) × 105 |
| P-1⊂CB[7] | (6.1 ± 0.3) × 105 | −7.9 ± 0.6 | −14.1 ± 0.5 | −6.2 ± 0.4 | 1.2 | (7.3 ± 0.3) × 105 |
We also kinetically characterized the formed 1:1 complexes, FA-1⊂CB[7] and P-1⊂CB[7], by means of 2D-EXSY experiments. We used separate solutions of the complexes containing an excess of the guest (2.0 equiv.) with respect to CB[7]. Using the integral values of the diagonal and cross-peaks, due to chemical exchange, of selected proton signals of the guests, we determined the dissociation rate constants of the two complexes, koff, which were approximately 1 s−1 (Table 1).37 In turn, the rate constants for the formation of the complexes (kon) were calculated to be of the order of 105 M−1 s−1 indicating that the binding processes were not controlled by diffusion.
We used kinetic 1H NMR spectroscopy at 298 K to monitor the IMDA reaction of FA-1 yielding the cycloadduct P-1. We performed the reaction in three separate NMR tubes containing exclusively FA-1 (5 mM), an equimolar mixture of FA-1 (5 mM) and CB[7], and FA-1 (10 mM) with a substoichiometric amount of CB[7] (0.1 equiv.). The distribution of the species as a function of time was determined using integral values of selected proton signals of all detectable species. We fit the experimental concentration data vs. time to suitable theoretical models in order to obtain the rate constant values of the IMDA reactions of FA-1 in the bulk and included in CB[7]. For the case of the reaction in the bulk, we used a simple theoretical model for a unimolecular irreversible reaction obtaining kbulk as (1.1 ± 0.1) × 10−7 s−1. For the IMDA reaction of FA-1 chaperoned by CB[7], we used a more elaborated theoretical kinetic model considering: (a) the reversible formation of the FA-1⊂CB[7] complex (eqn (1)), (b) its irreversible transformation into the P-1⊂CB[7] counterpart (included IMDA reaction) (eqn (2)), and (c) the reversible dissociation of the latter into their binding partners (eqn (3)) (Fig. 1). Although the rate of the transformation of FA-1 into P-1 occurring in the bulk (background reaction) is almost negligible compared to that of the included reaction, we also considered the former in the elaborated model (eqn (4)).
 | (1) |
 | (2) |
 | (3) |
 | (4) |
We fit the kinetic experimental data to the theoretical models using COPASI kinetic modelling software (Fig. 3). For the elaborated model, we considered the rate constant of the included IMDA reaction, kincluded, as the only variable, and fixed the kons and koffs values for the FA-1⊂CB[7] and P-1⊂CB[7] complexes to those determined in the previous kinetic and thermodynamic characterization study. The value of kbulk was also fixed to that determined in the initial kinetic experiment. The fit of the experimental kinetic data to the elaborated theoretical kinetic model for the chaperoned CB[7] reaction was acceptable for both experimental conditions (i.e. 1:1 and 1:0.1 FA-1:CB[7]). Despite we observed small deviations between experimental and theoretical curves, we used the fits to calculate an average value of kincluded of (3.5 ± 1.8) × 10−5 s−1. The ratio kincluded/kbulk was used to quantify the ∼300 fold acceleration experienced by the inclusion of the IMDA reaction of FA-1 in CB[7].¶ This acceleration value can be translated into a difference in activation entropy for the reactions in the bulk and included in CB[7] of 11.3 e.u. (ΔΔS≠ = Rln(kincluded/kbulk)) and provides an entropy change of 2.8 e.u. per internal rotation (4 C–C single bonds) accompanying the double cyclization.
 | ||
| Fig. 3 Kinetic profiles of the concentration of the species (M) vs. time (h) for the IMDA reaction in D2O at 298 K of: (a) FA-1 with 1.0 equiv. of CB[7]; and (b) FA-1 with 0.1 equiv. of CB[7]. Dashed lines represent the fit of the kinetic data to the elaborated model. Concentrations were calculated as the average of two different experiments and the error bars correspond to the standard deviations. | ||
The experimental kinetic data for the reaction of FA-1 in the presence of substoichiometric amounts of CB[7] evidenced the existence of at least 4 turnovers of the container in the first 180 h of the reaction.
The interesting results obtained in the acceleration of the IMDA reaction of FA-1 by inclusion in CB[7], prompted us to investigate its use for promoting the IMDA reaction of FA-2. FA-2 being a tertiary homoallylic-furfuryl amine required the freezing of one additional C–C single bond rotation in the TS of the IMDA reaction and upon double ring closure. On the one hand, this explains the observed diminution in rate constant for the IMDA reaction of FA-2 in the bulk (kbulk = <10−8 s−1) compared to FA-1. According to Page and Jencks, the loss of one additional internal rotation in the formation of a 6-membered ring, as is the case of P-2, increases 0.8 kcal mol−1 the free energy of activation.24 Considering this, we predicted that the entropic rate acceleration factor for including the IMDA reaction of FA-2 into CB[7] at 298 K (800 cal/298 K = 2.8 e.u.) might be at least 4 fold larger than the one measured for the inclusion of FA-1. Alternatively, we may assume that the binding of FA-2 into the container pays for the cost of freezing an additional rotation. This corresponds to our estimate of 2.8 e.u. at 298 K, that in turn should produce an acceleration entropic factor for the included IMDA reaction of FA-2, kincluded/kbulk, 4 times larger than the one measured for FA-1, that is 4 × 330 ∼ 103 or an overall difference of entropic activation of 5 × 2.8 = 14 e.u. for the IMDA reaction of FA-2 included in CB[7]. In our reasoning, we hypothesized that upon inclusion in CB[7], FA-1 requires the freezing of four rotations that are assumed to be comparable with FA-2. In any case, as pointed out by Page and Jencks, owing to the specific geometric and electronic requirements of the TS, it is very difficult to derive the magnitudes of “rotamer distribution” to rate acceleration factors. Factor values of 230 have also been suggested for freezing each internal rotation based on comparison of rates of ring closures of different sized rings systems in acyl transfer reactions.24
We started by evaluating the interaction of FA-2 with CB[7] using 1H NMR titration experiments. The addition of 1.0 equiv. of FA-2 to a mM D2O solution of CB[7] produced upfield shifts in the proton signals compared to the free amine. When 2.0 equiv. of FA-2 were added, the proton signals of the amine broadened and moved downfield towards the chemical shifts of the free counterpart. These observations were indicative of the inclusion of FA-2 in the hydrophobic cavity of CB[7] and the existence of a binding process showing intermediate chemical exchange dynamics on the chemical-shift timescale (Fig. 4b–c). We used HypNMR2008 to mathematically analyze the chemical shift changes experienced by FA-2 and CB[7]. The fit of the titration data to a 1:1 theoretical model was good, supporting the assignment of a 1:1 stoichiometry to the formed complex, FA-2⊂CB[7], and returning a binding constant value of (5.5 ± 0.5) × 103 M−1. We were surprised by the fact that the increase in a methylene unit (15 Å3) in FA-2 compared to FA-1 provoked a difference of two orders of magnitude in the binding constants of their complexes with CB[7]. In fact, molecular modelling studies showed a nice fit for both FA-1 and FA-2 in the cavity of CB[7] and did not anticipate the experimental result (Fig. 5). We surmise that the addition of one methylene unit in FA-2 compared to FA-1 had a significant effect in increasing the number of possible rotamers, a.k.a. rotamer distribution, for the former compared to the latter. Some of the rotamers of FA-2 may bind weaker than others or even don't bind at all, owing to dissimilar complementarity in size and shape with the cavity of CB[7]. This particularity is not taken into account in the data fitting used for the determination of the binding constant for the FA-2⊂CB[7] complex. For this reason, we refer to the determined Ka (FA-2⊂CB[7]) value as an apparent binding constant that should not be directly compared to the Ka (FA-1⊂CB[7]) value owing to the different rotamer distribution of the two guests. In the equimolar mixture of FA-2 and CB[7] at 5 mM concentration, the FA-2⊂CB[7] complex is present in 80% yield.
 | ||
| Fig. 4 Selected region of the 1H NMR spectra (400 MHz, D2O, 298 K) of: (a) FA-2 (0.005 M); (b) FA-2⊂CB[7] (1:1, 0.005 M); (c) FA-2⊂CB[7] (2:1, 0.005 M); (d) P-2⊂CB[7] (1:1, 0.005 M), acquired after 10 h of evolution of spectrum b; and (e) mixture of free FA-2 and P-2⊂CB[7] (1:1), acquired after 10 h of evolution of spectrum c. FA-2 in red and P-2 in blue. Primed labels correspond to proton signals of bound species. *Residual solvent peak. | ||
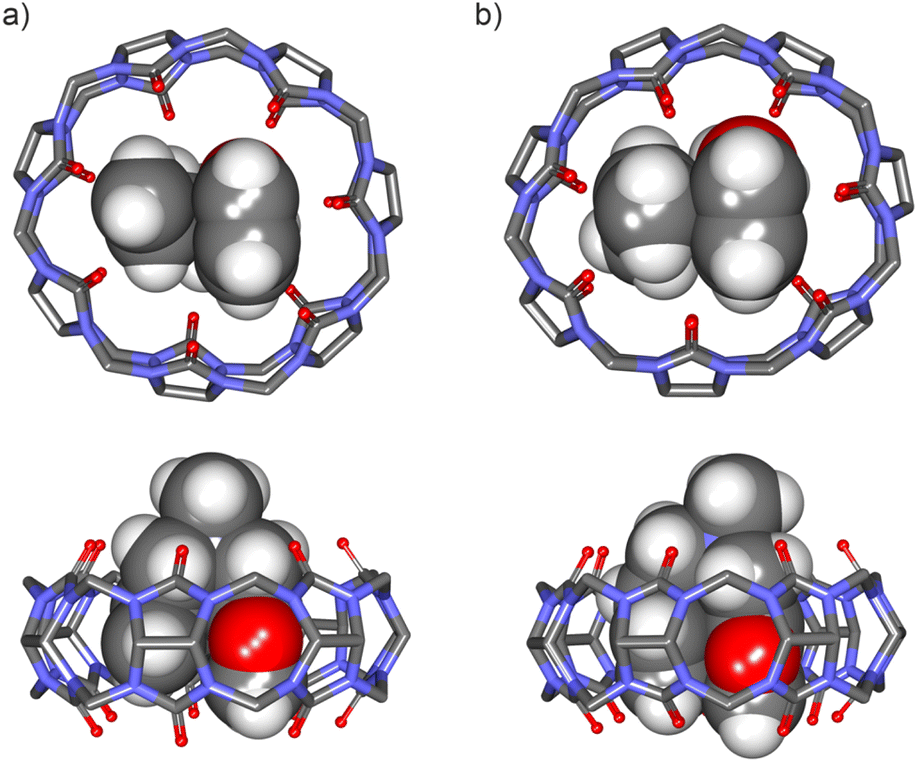 | ||
| Fig. 5 Energy-minimized structures at the PM6 level of theory of 1:1 inclusion complexes: (a) FA-1⊂CB[7] (bottom view and side view), and (b) FA-2⊂CB[7] (bottom view and side view). CB[7] is shown in line-stick representation with non-polar hydrogen atoms omitted for clarity. Bound guests are depicted as CPK models. See Fig. 1 for the line-drawing structures of the compounds. | ||
In the absence of CB[7], a 5 mM D2O solution of FA-2 produced the IMDA adduct P-2 in just 7%, after heating the solution at 45 °C for 34 days (at rt we did not observe the formation of P-2). Pleasingly, at the same concentration and after standing at 25 °C for 10 h, the 1H NMR spectrum of an equimolar mixture of CB[7] and FA-2 showed the exclusive observation of the proton signals of the inclusion complex P-2⊂CB[7] (Fig. 4d). We used kinetic 1H NMR to monitor the IMDA reactions of FA-2 in the absence and presence of CB[7]. To calculate/estimate the corresponding kincluded rate constant, we fitted the changes in concentration vs. time using COPASI and the elaborated kinetic model. On the other hand, the value of kbulk was determined using initial rates.||
We used the ratio of the determined rate constants, kbulk < 10−8 s−1 and kincluded = 1.2 × 10−4 s−1, to calculate/estimate an acceleration factor larger than 104 for the inclusion of the IMDA reaction of FA-2 in CB[7].** This translates into a reduction in entropy of activation of more than 18 e.u. for the IMDA reaction of FA-2 included in CB[7] compared to the bulk. Our prediction was that the reduction in entropy of activation for the IMDA reaction of FA-2 upon inclusion in CB[7] would be 14 e.u. Our simple prediction cannot account for the experimental results. Most likely, the inclusion of FA-2 in CB[7] is capable of modifying and/or stabilizing the geometric and electronic requirements of the reaction TS, as well as providing other non-considered entropic advantages.
We also experimentally undertook the study of the IMDA reaction of FA-2 at 25 °C in the presence of substoichiometric amounts of CB[7]. In such conditions, the IMDA reaction of FA-2 progressed until the substoichiometric amount of CB[7] was consumed in the formation of the P-2⊂CB[7] complex. This result evidenced the existence of an undesired product inhibition process in this reaction. For the reaction performed with 0.5 equiv. of CB[7], the final 1H NMR spectrum showed the presence of a 50:50 mixture of free FA-2 and P-2⊂CB[7] complex. This outcome indicated that the binding constant of the P-2⊂CB[7] complex should be more than two orders of magnitude larger than that of the FA-2⊂CB[7] complex (Fig. S23†). We estimated an association constant value (Ka) for the P-2⊂CB[7] complex of the order of 106 M−1 (Fig. 4e) explaining the observed product inhibition and lack of turnover.†† Remarkably, the binding constant of P-2⊂CB[7] seemed to be larger than that of P-1⊂CB[7], while those of the complexes of the starting materials (FA-1 and FA-2) followed a reverse ordering. We could displace P-2 from the cavity of CB[7] by adding incremental amounts of 2-adamantanone (ADA) (Ka (adamantane guests⊂CB[7]) > 1010 M−1).38 During the guest exchange experiment we observed separate signals for the protons of free and bound P-2 evidencing that the binding process featured slow chemical exchange dynamics on the 1H NMR chemical-shift timescale, and that the P-2⊂CB[7] complex was also kinetically stable (koff ∼ 1 s−1).
In order to gain some insight on the activation parameters leading to the TS of the IMDA reactions of FA-1 and FA-2, both in the bulk and included in CB[7], we determined their rate constants at different temperatures and performed the corresponding Eyring plots (Fig. S32 and S46†). The plots showed good linearity allowing the determination of the activation reaction parameters, ΔH≠ and ΔS≠ (Table 2).
| Guest | Host | ΔH≠ [kcal mol−1] | ΔΔH≠ [kcal mol−1] | ΔS≠ [cal mol−1 K−1] | ΔΔS≠ [cal mol−1 K−1] | TΔS≠b [kcal mol−1] | ΔG≠b [kcal mol−1] | Rate accelerationc (kincluded/kbulk) | ΔΔS≠ = Rln(kincluded/kbulk) [cal mol−1 K−1] |
|---|---|---|---|---|---|---|---|---|---|
| a FA-1⊂CB[7] (1:1, 0.005 M), FA-2⊂CB[7] (1:1, 0.005 M), FA-1 (0.005 M), and FA-2 (0.005 M) in D2O. Error values in ΔH≠ and ΔS≠ are reported as standard deviations and propagated to TΔS≠ and ΔG≠. b 298 K. c An acceleration factor of 1000 was determined in ref. 23 at 310 K for the IMDA of a secondary furfuryl amine included in CB[7]. |
|||||||||
| FA-1 | CB[7] | 23.7 ± 0.1 | 1.4 | 2.3 ± 0.4 | 17.5 | 0.7 ± 0.1 | 23.0 ± 0.2 | 330 | 11 |
| FA-1 | — | 22.3 ± 0.5 | −15.2 ± 1.6 | −4.5 ± 0.5 | 26.9 ± 0.2 | ||||
| FA-2 | CB[7] | 14.1 ± 0.8 | 1.3 | −29.3 ± 2.7 | 23.3 | −8.7 ± 0.8 | 22.8 ± 1.1 | >11000 |
>18 |
| FA-2 | — | 12.8 ± 0.4 | −52.6 ± 1.4 | −15.7 ± 0.4 | 28.4 ± 0.6 | ||||
The comparison of the obtained parameters' values was useful in rationalizing the acceleration produced by the inclusion of the reaction in the molecular vessel. For the reactions in the bulk, the entropic terms were more negative than for those included in CB[7]. Using the determined reaction activation entropies, we calculated differences of 17.5 e.u. and 23.3 e.u. for the IMDA reactions of FA-1 and FA-2, respectively, when included in CB[7] compared to the bulk. The experimentally determined ΔS≠ values are larger than those derived from the acceleration factors at 298 K. Notably, the magnitude of the enthalpic activation term remained almost constant independently of the medium of the reaction. Taken together, these results indicated that the acceleration observed for the inclusion of the IMDA reactions of the tertiary amines in CB[7] was predominantly caused by entropic effects. We propose that the cavity of CB[7] acted as an artificial active site. The tight confinement of the substrates forced them to adopt a high-energy reactive conformation. The energy of binding to CB[7] partially paid for the entropic cost of the substrate to adopt a conformation that is close to that of the transition state (preorganization). This hypothetical mechanism for the acceleration of reactions by inclusion in synthetic receptors and enzymes has been termed by Jencks as the “Circe effect”.39
We also investigated the temperature dependence of the acceleration factor provided by inclusion of the IMDA reactions in the cavity of CB[7]. We observed a subtle increase in its magnitude that we quantified as 0.02–0.03 e.u. K−1 (see Table S13† for a cumulative table with all relevant rate constants from CB[n]-promoted IMDA reactions and control experiments at different temperatures, as well as the acceleration factors). At this point, we are not able to provide a sound explanation to this observation. We simply propose that the temperature effect on TS is more significant in confined species.
We performed a control experiment to verify the pivotal role of the CB[7] cavity in accelerating the included IMDA reactions. Thus, the addition of 1.0 equiv. of FA-1 or FA-2, into a solution containing the preformed and thermodynamically more stable ADA⊂CB[7] complex did not induce any detectable acceleration of the reaction (Fig. S28 and S39†).
Next, we assayed the larger cavity offered by CB[8] as reaction vessel. The binding processes of both FA-1 and FA-2 with CB[8] displayed fast exchange dynamics on the 1H NMR chemical-shift timescale (Fig. S18 and S19†). We estimated apparent binding constants for both complexes, FA-1⊂CB[8] and FA-2⊂CB[8], in the range of 103 M−1. Regarding the acceleration factors of the reactions, CB[8] accelerated the IMDA reaction of FA-1 41 times compared to the reaction in the bulk solution (see ESI†), whereas FA-2 did not result in any observable conversion after 2 weeks in the presence of CB[8], neither at 25 °C nor at 40 °C. The chemical shift changes experienced by the protons of FA-1 and FA-2 guests in the 1H NMR spectroscopic titration experiments supported the inclusion of their reactive functions in the cavity of CB[8]. We used the volumes of the substrates (164 Å3 for FA-1, and 179 Å3 for FA-2) and the reported cavity volume of CB[7] (242 Å3)3 to calculate packing coefficient (PC) values40 for the complexes FA-1⊂CB[7] and FA-2⊂CB[7], which were found to be 68% and 74%, respectively. In contrast, for CB[8] (367 Å3),3 the PC values of the inclusion complexes decreased to 45% and 49%, respectively. Based on this, we hypothesized that the IMDA reactions that took place inside the cavity of CB[7] happened in a tight-packing regime.22 On the other hand, due to the larger cavity of CB[8] and the loose-packing nature of the complexes, the included reactants were conformationally less organized by the container, which explains the observed reduction in the acceleration factors.
Lastly, a series of cyclodextrins (α-CD, β-CD and γ-CD) were evaluated as molecular vessels for the inclusion of the IMDA reaction of FA-1. CDs have a polar hydrophilic outer shell – which makes them water-soluble –, and a hydrophobic interior.18 They are good hosts for suitable hydrophobic molecules and feature similar cavity sizes than CB[n]s. Moreover, it was described that β-CD increased the yields of the IMDA cyclization of unsubstituted alkenylfuran.41 Unfortunately, using 1H NMR titration experiments, we observed that the CD series did not show significant binding affinity for FA-1 (Ka < 10 M−1). Moreover, after 60 h, we were not able to detect the diagnostic signals of the P-1 cycloadduct in any of the mixtures of the CD hosts and FA-1 (Fig. S52–S59†).
Conclusions
In summary, we demonstrated that CB[7] accelerates, under mild conditions, the IMDA reactions of previously elusive and unreactive tertiary N-methyl-N-(homo)allyl-2-furfurylamines with acceleration factors of up to 4 orders of magnitude. The substrates' conformational selection occurring on complex formation (FA-1⊂CB[7] and FA-2⊂CB[7]) entailed a suitable spatial approximation and placement of the reactive ends for the reaction to occur inside the cavity. The assessment of the activation parameters unveiled the crucial role of CB[7] in accelerating the IMDA reactions: inclusion in CB[7] paid for the entropy penalty of reorganizing the substrates in a conformation more closely resembling the TS geometry. This study further highlighted the potential of cucurbit[n]urils to mimic certain facets of enzymatic catalysis.Data availability
We have no experimental data for depositing. All relevant experimental data is included in the ESI.†Author contributions
Conceptualization, K. V. H.; data curation, K. V. H.; formal analysis, K. V. H. and P. B.; funding acquisition, K. V. H., M. G. S. and P. B.; methodology, K. V. H.; supervision, M. G. S. and P. B.; writing – original draft, K. V. H.; writing – review & editing, K. V. H., M. G. S. and P. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 801474, the Ministerio de Ciencia, Innovación y Universidades/Agencia Estatal de Investigación (MICIU/AEI/10.13039/501100011033; PID2020-114020GB-100), the Severo Ochoa Excellence Accreditation 2020-2023 (CEX2019-000925-S) and AGAUR (2021SGR00851). We also thank CERCA Programme/Generalitat de Catalunya and ICIQ Foundation for financial support.Notes and references
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Cucurbit[n]urils for Supramolecular Catalysis, Chem.–Eur. J., 2020, 26, 15446–15460 CrossRef CAS PubMed
.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Cucurbituril-Based Molecular Recognition, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed
- K. I. Assaf and W. M. Nau, Cucurbiturils: from synthesis to high-affinity binding and catalysis, Chem. Soc. Rev., 2015, 44, 394–418 RSC
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, Cucurbituril chemistry: a tale of supramolecular success, RSC Adv., 2012, 2, 1213–1247 RSC
- B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, From Containers to Catalysts: Supramolecular Catalysis within Cucurbiturils, Chem.–Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed
- L. Zheng, S. Sonzini, M. Ambarwati, E. Rosta, O. A. Scherman and A. Herrmann, Turning Cucurbit[8]uril into a Supramolecular Nanoreactor for Asymmetric Catalysis, Angew. Chem., Int. Ed., 2015, 54, 13007–13011 CrossRef CAS PubMed
- G. Ghale and W. M. Nau, Dynamically Analyte-Responsive Macrocyclic Host–Fluorophore Systems, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed
- F. Biedermann, D. Hathazi and W. M. Nau, Associative chemosensing by fluorescent macrocycle–dye complexes – a versatile enzyme assay platform beyond indicator displacement, Chem. Commun., 2015, 51, 4977–4980 RSC
- S. Walker, R. Oun, F. J. McInnes and N. J. Wheate, The Potential of Cucurbit[n]urils in Drug Delivery, Isr. J. Chem., 2011, 51, 616–624 CrossRef CAS
- N. Basílio and U. Pischel, Drug Delivery by Controlling a Supramolecular Host–Guest Assembly with a Reversible Photoswitch, Chem.–Eur. J., 2016, 22, 15208–15211 CrossRef
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Supramolecular catalysis. Part 2: artificial enzyme mimics, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC
- Q. Zhang, L. Catti, V. R. I. Kaila and K. Tiefenbacher, To catalyze or not to catalyze: elucidation of the subtle differences between the hexameric capsules of pyrogallolarene and resorcinarene, Chem. Sci., 2017, 8, 1653–1657 RSC
- A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, DNA-based asymmetric catalysis, Chem. Soc. Rev., 2010, 39, 2083–2092 RSC
- S. Y. Jon, Y. H. Ko, S. H. Park, H.-J. Kim and K. Kim, A facile, stereoselective [2 + 2] photoreaction mediated by cucurbit[8]uril, Chem. Commun., 2001, 1938–1939, 10.1039/B105153A
- R. Wang, L. Yuan and D. H. Macartney, Cucurbit[7]uril Mediates the Stereoselective [4+4] Photodimerization of 2-Aminopyridine Hydrochloride in Aqueous Solution, J. Org. Chem., 2006, 71, 1237–1239 CrossRef CAS PubMed
- Y. Kang, X. Tang, H. Yu, Z. Cai, Z. Huang, D. Wang, J.-F. Xu and X. Zhang, Supramolecular catalyst functions in catalytic amount: cucurbit[8]uril accelerates the photodimerization of Brooker's merocyanine, Chem. Sci., 2017, 8, 8357–8361 RSC
- Y.-H. Wang, H. Cong, F.-F. Zhao, S.-F. Xue, Z. Tao, Q.-J. Zhu and G. Wei, Selective catalysis for the oxidation of alcohols to aldehydes in the presence of cucurbit[8]uril, Catal. Commun., 2011, 12, 1127–1130 CrossRef CAS
- N. Basilio, L. García-Río, J. A. Moreira and M. Pessêgo, Supramolecular Catalysis by Cucurbit[7]uril and Cyclodextrins: Similarity and Differences, J. Org. Chem., 2010, 75, 848–855 CrossRef CAS PubMed
- J. A. Finbloom, K. Han, C. C. Slack, A. L. Furst and M. B. Francis, Cucurbit[6]uril-Promoted Click Chemistry for Protein Modification, J. Am. Chem. Soc., 2017, 139, 9691–9697 CrossRef CAS PubMed
- X. Feng, F. Zhao, R. Qian, M. Guo, J. Yang, R. Yang and D. Meng, Supramolecular Catalyst Functions in Catalytic Amount: Cucurbit[7]uril Accelerates Click Reaction in Water, ChemistrySelect, 2021, 6, 10739–10745 CrossRef CAS
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, Cycloaddition induced by cucurbituril. A case of Pauling principle catalysis, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS
- F. N. Tehrani, K. I. Assaf, R. Hein, C. M. E. Jensen, T. C. Nugent and W. M. Nau, Supramolecular Catalysis of a Catalysis-Resistant Diels–Alder Reaction: Almost Theoretical Acceleration of Cyclopentadiene Dimerization inside Cucurbit[7]uril, ACS Catal., 2022, 12, 2261–2269 CrossRef CAS
- A. Palma, M. Artelsmair, G. Wu, X. Lu, S. J. Barrow, N. Uddin, E. Rosta, E. Masson and O. A. Scherman, Cucurbit[7]uril as a Supramolecular Artificial Enzyme for Diels–Alder Reactions, Angew. Chem., Int. Ed., 2017, 56, 15688–15692 CrossRef CAS PubMed
- M. I. Page and W. P. Jencks, Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect, Proc. Natl. Acad. Sci. U.S.A., 1971, 68, 1678–1683 CrossRef CAS PubMed
- M. Chaudhary, S. Shaik, M. Magan, S. Hudda, M. Gupta, G. Singh and P. Wadhwa, A Brief Review on Recent Developments in Diels-Alder Reactions, Curr. Org. Synth., 2024, 21, 1–22 CrossRef
- C. L. Bouchez, M. Rusch and M.-H. Larraufie, Diels-Alder Cycloaddition in Medicinal Chemistry, Curr. Org. Chem., 2016, 20, 2358–2378 CrossRef
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, The Diels–Alder Reaction in Total Synthesis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed
- K.-i. Takao, R. Munakata and K.-i. Tadano, Recent Advances in Natural Product Synthesis by Using Intramolecular Diels−Alder Reactions, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed
- M. V. Basilevskii, A. G. Shamov and V. A. Tikhomirov, Transition state of the Diels-Alder reaction, J. Am. Chem. Soc., 1977, 99, 1369–1372 CrossRef CAS
- S. Horiuchi, T. Murase and M. Fujita, Diels–Alder Reactions of Inert Aromatic Compounds within a Self-Assembled Coordination Cage, Chem.–Asian J., 2011, 6, 1839–1847 CrossRef CAS PubMed
- J. Åqvist, M. Kazemi, G. V. Isaksen and B. O. Brandsdal, Entropy and Enzyme Catalysis, Acc. Chem. Res., 2017, 50, 199–207 CrossRef PubMed
- N. Choony, A. Dadabhoy and P. G. Sammes, The trityl group as a removable steric buttress in cycloaddition reactions, Chem. Commun., 1997, 513–514, 10.1039/A700313G
- N. Choony, A. Dadabhoy and P. G. Sammes, On the use of removable steric buttresses in cycloaddition reactions, J. Chem. Soc., Perkin Trans. 1, 1998, 2017–2022, 10.1039/A802415D
- D. Hallooman, D. Cudian, M. Ríos-Gutiérrez, L. Rhyman, I. A. Alswaidan, M. I. Elzagheid, L. R. Domingo and P. Ramasami, Understanding the Intramolecular Diels-Alder Reactions of N-Substituted N-Allyl-Furfurylamines: An MEDT Study, ChemistrySelect, 2017, 2, 9736–9743 CrossRef CAS
- M. V. Rekharsky and Y. Inoue, Solvent and Guest Isotope Effects on Complexation Thermodynamics of α-, β-, and 6-Amino-6-deoxy-β-cyclodextrins, J. Am. Chem. Soc., 2002, 124, 12361–12371 CrossRef CAS PubMed
- W. M. Nau, M. Florea and K. I. Assaf, Deep Inside Cucurbiturils: Physical Properties and Volumes of their Inner Cavity Determine the Hydrophobic Driving Force for Host–Guest Complexation, Isr. J. Chem., 2011, 51, 559–577 CrossRef CAS
- J. C. Cobas and M. Martín-Pastor, EXSYCalc Version 1.0 Search PubMed
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, New Ultrahigh Affinity Host−Guest Complexes of Cucurbit[7]uril with Bicyclo[2.2.2]octane and Adamantane Guests: Thermodynamic Analysis and Evaluation of M2 Affinity Calculations, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed
- W. P. Jencks, in Advances in Enzymology - and Related Areas of Molecular Biology, ed. A. Meister, Wiley, 1975, vol. 43, ch. 4, pp. 219–410 Search PubMed
- S. Mecozzi and J. Rebek, Julius, The 55 % Solution: A Formula for Molecular Recognition in the Liquid State, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS
- D. D. Sternbach and D. M. Rossana, Cyclodextrin catalysis in the intramolecular Diels-Alder reaction with the furan diene, J. Am. Chem. Soc., 1982, 104, 5853–5854 CrossRef CAS
- P. Brun, J. Zylber, G. Pepe and J.-P. Reboul, Intramolecular versus intermolecular Diels-Alder reaction in the cyclisation reaction of furfuryl amines with maleic anhydride, Heterocycl. Commun., 1994, 1, 13–16 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, binding studies, theoretical kinetic models, kinetic data, and energy-minimized structures of the inclusion complexes. See DOI: https://doi.org/10.1039/d4sc01816h |
| ‡ Cycloadducts P-1 and P-2 were synthesized using CB[7] as reaction vessel. The displacement of the bound products to the bulk solution was achieved by addition of a competitive guest (2-adamantanone). P-1 was isolated and characterized by a full set of high-resolution spectra (Fig. S5–S7). The relative configuration was deduced from 1H–1H NOESY and GOESY experiments (Fig. S8–S9). The determined exo-configuration is in agreement with previous reports in the literature for the DA cycloadducts obtained from secondary (see ref. 26) and tertiary furfuryl amines (for details, see: P. Brun, J. Zylber, G. Pepe and J.-P. Reboul, Heterocycl. Commun. 1994, 1, 13–16).42 |
| § We decided to use D2O as solvent to take into account any isotopic effect of the solvent in the thermodynamic parameters of binding. This will become relevant in the mathematical analysis of the kinetic data. |
| ¶ We obtained an analogous value for the acceleration of the included IMDA of FA-1 using initial rates in the quantification of the rate constants for the reaction in the bulk and in the presence of CB[7] at 298 K. |
| || The slow kinetics of the IMDA reaction of FA-2 in the bulk forced us to use the initial rate method in the determination of the reaction rate constant. In contrast, the kinetics of the reaction chaperoned by CB[7] were too fast to be analyzed using the same method. At high temperatures, the reaction evolved in more than 10% extent just after two consecutive experimental measurements (<10 min). |
| ** We could not experimentally determine the kinetic stability of the FA-2⊂CB[7] complex. We estimated it to be similar to that of FA-1⊂CB[7] approx. 1 s−1. For this reason, we refer to the value of kincluded returned from the fit of the kinetic data as calculated/estimated. |
| †† The addition of a less polar cosolvent (10% CD3OD, Fig. S45) as a non-destructive strategy to achieve catalytic turnover was not successful in our case producing analogous results to those obtained in pure water. |
| This journal is © The Royal Society of Chemistry 2024 |