
Breaking the deep-red light absorption barrier of iridium(III)-based photosensitizers
Gloria
Vigueras
 *a,
Gilles
Gasser
a and
José
Ruiz
*b
*a,
Gilles
Gasser
a and
José
Ruiz
*b
aChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: gloria.vigueras@um.es
bDepartamento de Química Inorgánica, Universidad de Murcia, Biomedical Research Institute of Murcia (IMIB-Arrixaca), E-30100 Murcia, Spain. E-mail: jruiz@um.es
First published on 30th December 2024
Abstract
Activating photosensitizers with long-wavelength excitation is an important parameter for effective photodynamic therapy due to the minimal toxicity of this light, its superior tissue penetration, and excellent spatial resolution. Unfortunately, most Ir(III) complexes suffer from limited absorption within the phototherapeutic window, rendering them ineffective against deep-seated and/or large tumors, which poses a significant barrier to their clinical application. To address this issue, several efforts have been recently made to shift the absorption of Ir(III) photosensitizers to the deep-red/near-infrared region by using different strategies: functionalization with organic fluorophores, including porphyrinoid compounds, and ligand design via π-extension and donor–acceptor interactions. In this Frontier, we highlight such new developments and the ongoing challenges in this field.
1. Introduction
Photodynamic therapy (PDT) is a clinically-approved modality for cancer and infection treatment that relies on the interplay between light, a photosensitizer (PS), and molecular oxygen. When exposed to light of a suitable wavelength, PSs can undergo various photochemical processes. The PS is first excited to a nanosecond-lived singlet state (S1) that rapidly transitions to a more stable triplet state via intersystem crossing (ISC). The triplet state (T1) PS has a sufficient lifetime to generate reactive oxygen species (ROS) through electron transfer mechanisms (type-I PDT), and/or singlet oxygen (1O2) through energy transfer to ground-state triplet oxygen (3O2) (type-II PDT). These ROS inflict damage on tumor cells by inducing various cell death mechanisms.1 Recently, an alternative type-III process has been suggested where the PS can damage biological substrates without the participation of oxygen.2,3Light irradiation is crucial for PDT, with its effectiveness being determined by the photosensitizer's absorption spectrum and the tissue's optical properties.4,5 Shorter wavelengths, like ultraviolet (UV) or blue light, are absorbed more readily by biological tissues, limiting their penetration depth. In contrast, deep-red and near-infrared (NIR) light, within the ‘tissue transparency window’ (600–900 nm), can penetrate much deeper, reaching depths of several millimetres (Fig. 1)6 and reducing the risk of acute photo-damage.7 This deeper penetration is crucial for treating tumors that are located beneath the skin surface.
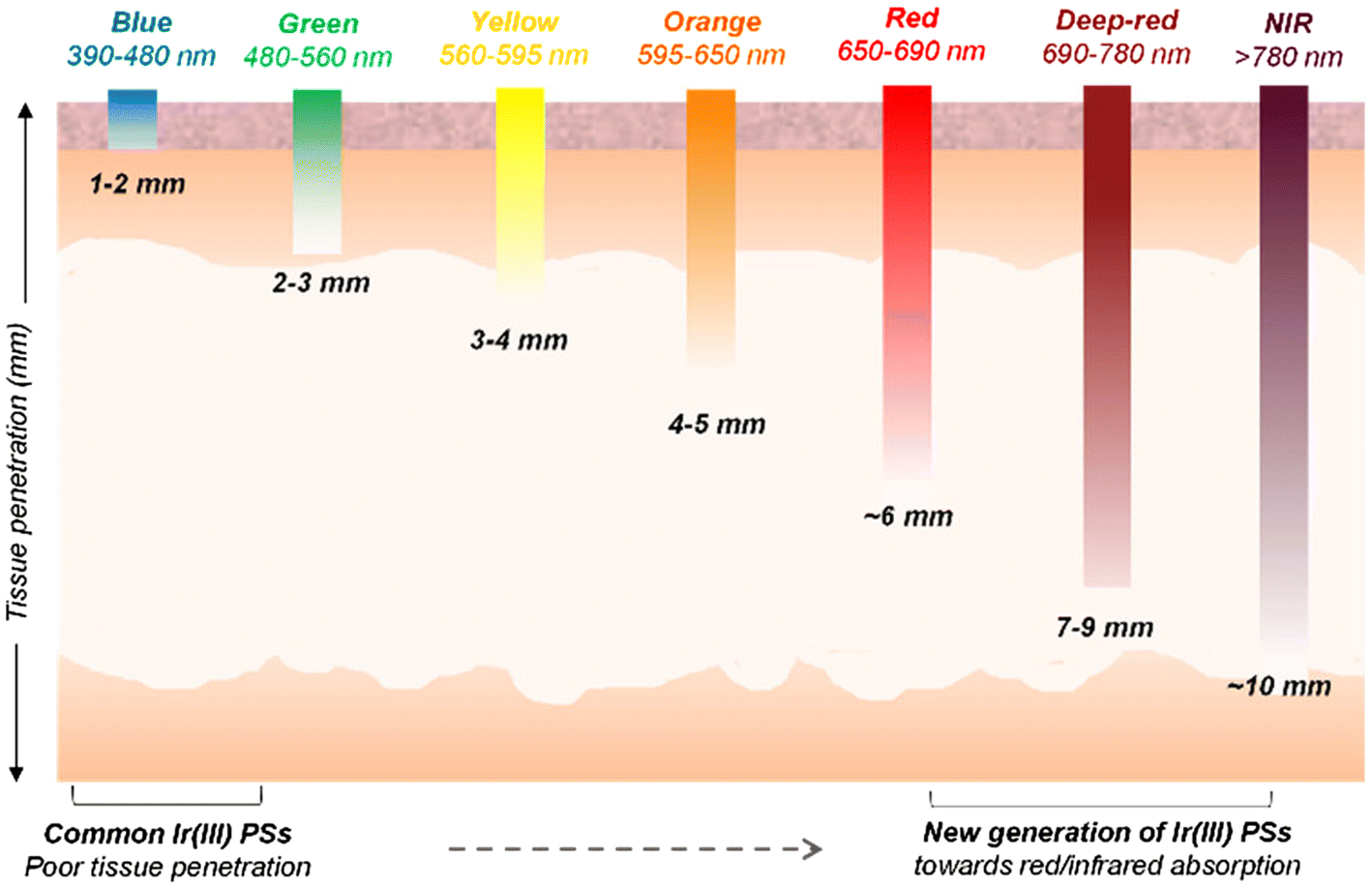 | ||
| Fig. 1 Wavelengths of light exhibiting different tissue penetration depths. Longer wavelengths, especially those in the deep-red/near-infrared region of the electromagnetic spectrum, have the ability to permeate deeper into tissues than high-energy light, which often has limited tissue penetration. Adapted from ref. 6. | ||
Numerous PSs have been employed in clinical or preclinical PDT,8 including porphyrin, chlorin, and phthalocyanine derivatives,9,10 all of which share a tetrapyrrole macrocycle structure. This characteristic structure is known for its efficient light absorption and singlet oxygen generation. Additionally, PSs such as methylene blue, Rose Bengal, and hypericin have found application in clinical settings or trials.11,12 Other photoactive agents, including boron-dipyrromethene (BODIPY) and cyanine dyes, fullerenes, semiconductors, and aggregation-induced emission fluorogens,13–15 also exhibit photodynamic properties suitable for oncological applications. Beyond this type of PSs, transition metal complexes have emerged as a promising and productive area of study for PDT. Among these, most screened compounds are octahedral polypyridine Ru(II) complexes,16,17 which have demonstrated unique photophysical and photochemical properties. Notably, TLD-1433, a ruthenium-based photosensitizer, is the first of its kind to enter human clinical PDT trials and is currently under evaluation in a phase II trial for treating non-muscle-invasive bladder cancer via intravesical infusion (NCT03945162).
Cyclometalated iridium(III) complexes with the architecture [Ir(C^N)2(N^N)]+ (C^N = cyclometalated ligand, N^N = diimine ligand) are widely studied as luminescent materials18 and PSs due to their exceptional photostability, their extended triplet excited state lifetimes and high quantum yields of both emission and ROS production.19 These compounds can be readily tuned by chemical variations of the coordinated and auxiliary ligands. It is worth remembering that within the auxiliary ligands, we can highlight two types: (a) ancillary ligands, which generally do not contribute directly to the electronic transitions responsible for photophysical properties, and play a supportive role by stabilizing the complex (e.g., acac ligands), and (b) chromophoric ligands, which are π-conjugated ligands (usually diimine ligands) that actively participate in light absorption and emission processes, often responsible for metal-to-ligand charge transfer (MLCT) or intraligand charge transfer (ILCT) transitions.20 In this Frontier, we will focus on the modification of chromophoric ligands, via functionalization with fluorophores or via π-extension and donor–acceptor interactions.
Bis-cyclometalated Ir(III) complexes are known to have spatially separated frontier orbitals the make it possible to independently tune the N^N centered lowest unoccupied molecular orbital (LUMO) and C^N centered highest occupied molecular orbital (HOMO) levels.21 Thus, these compounds show efficient tuning of the emission color and wide Stokes’ shifts that allow easily distinguishing emission overexcitation and eliminating self-quenching processes.22–26 Furthermore, Ir(III) compounds can be easily synthesized and purified and have great air and moisture stability.27 However, a significant drawback of iridium-based PSs is their low molar absorption coefficients at high wavelengths above 500 nm.28–30 This is primarily attributed to two factors: (i) the ligands commonly used to construct these complexes, such as phenylpyridine and bipyridine, typically absorb light in the ultraviolet region; and (ii) the spin-forbidden transition directly populating triplet excited states, which reduces its absorption intensity despite falling within the visible light range.31,32 While it is true that lowering the LUMO level of the ligands can extend absorption into the deep-red/NIR region, this strategy alone often falls short for PDT due to the challenges in tissue penetration and phototherapeutic effectiveness. Although deep-red/NIR absorption improves tissue penetration, the accompanying decrease in absorption cross-section (molar absorptivity) at these wavelengths often necessitates higher light intensities, which can cause collateral damage to healthy tissues and reduce the therapeutic index. This happens for example in Photofrin®, which has a low molar absorption coefficient (ε630 ≈ 3 × 103 M−1 cm−1).33
In recent years, significant efforts have been made to activate Ir(III)-based PSs with deep-red/NIR light. Strategies such as the attachment of upconversion nanoparticles (UCNPs) have been presented as an alternative to achieve NIR-triggered generation of ROS.34 However, the efficiency of UCNPs is usually very low, which is an important bottleneck limiting clinical applications.35,36 On the other hand, the use of NIR two-photon (2P) absorption often proves inefficient. This approach requires a 2P excitation laser source with a minimal irradiation volume, limiting its effectiveness for treating large areas.37–39
Herein, we highlight two different strategies that focus on obtaining deep-red/NIR absorbing iridium(III)-based PSs constructed through (1) functionalization with organic fluorophores (e.g. BODIPYs, rhodamines, porphyrinoids, etc.), and (2) ligand design via π-extension and donor–acceptor interactions. While both approaches involve extending conjugation in the complex, we categorize them based on whether the extended conjugation arises from a separate fluorophore (section 2) or is intrinsic to the ligand design (section 3). We will also discuss the significant challenges that hinder the clinical applications of this new class of iridium-based PSs.
2. Functionalization with organic fluorophores
This section focuses on systems where fluorophores (e.g., organic dyes or porphyrins) are chemically attached to the metal complex. The aim here is to harness the intrinsic photophysical properties of the fluorophores (in this case their absorption in deep-red/NIR region) by integrating them in the chromophoric ligand. The photophysical mechanisms in these systems are typically dominated by the intrinsic behavior of the fluorophore.40 While the metal center can influence these properties, the electronic transitions (e.g., π–π* transitions) remain primarily fluorophore-based. The metal's role often includes modifying the electronic structure, facilitating intersystem crossing, or stabilizing the fluorophore.41Among organic fluorophores with red to NIR absorption, BODIPY,40,42–47 coumarin,48,49 cyanine,50 rhodamine,51–53 and xanthene,54 have been introduced into iridium complexes for achieving long-wavelength luminescent π–π* transitions and the long-lived 3MLCT states of the metal complex thanks to the strong spin–orbit coupling induced by the heavy iridium ion. As a result, high ROS production and low energy long-wavelength excitation have been achieved. As shown in Fig. 2 no significant wavelength red-shift was observed upon functionalization compared to the free fluorescent compounds, suggesting minimal alteration to the molecular orbitals of the ligands during the complexation process.40
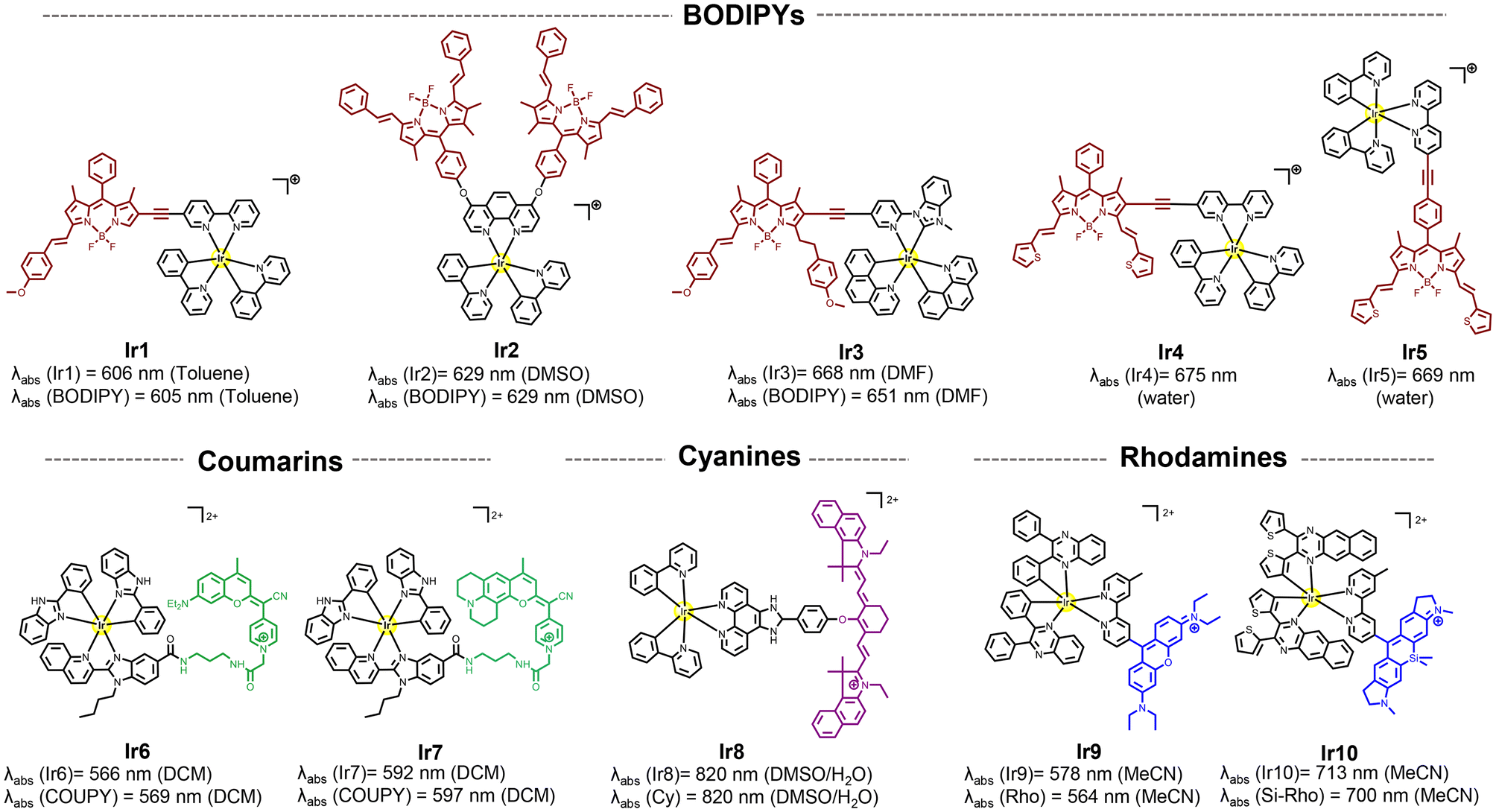 | ||
| Fig. 2 Selected structures of Ir(III) complexes functionalized to different organic fluorophores and their reported absorption wavelengths. | ||
In the BODIPY-containing conjugates Ir1–Ir3 (Fig. 1), methoxy-styryl units were used due to their electron-donating ability and π-conjugation nature, which produced a bathochromically shifted absorption into the red/deep-red region in the final compounds.45 The direct conjugation of the Ir complex and BODIPY through a –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– bond in Ir4 facilitated the ISC process to produce ROS with a singlet oxygen quantum yield (ΦΔ) of 0.95 (Table 1). In contrast, free rotation of the phenyl group in Ir5 disturbed the electron transfer process,46 which decreased its ΦΔ to 0.07 and its toxicity under red light irradiation (Table 1).
C– bond in Ir4 facilitated the ISC process to produce ROS with a singlet oxygen quantum yield (ΦΔ) of 0.95 (Table 1). In contrast, free rotation of the phenyl group in Ir5 disturbed the electron transfer process,46 which decreased its ΦΔ to 0.07 and its toxicity under red light irradiation (Table 1).
| λ abs/nm (solvent) | ε 104/M−1 cm−1 | Φ Δ (λirrad/nm) | IC50 lighta/μM (λirrad/nm) | PIh or cell viability dark | Cancer cell line | Ref. | |
|---|---|---|---|---|---|---|---|
a Normoxia.
b Micelles-Ir.
c Free Ir complex.
d ROS quantum yield.
e ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ir16-NPs.
f Free Ir18.
g Ir18-NPs.
h PI = defined as IC50 dark/IC50 light. Ir16-NPs.
f Free Ir18.
g Ir18-NPs.
h PI = defined as IC50 dark/IC50 light.
|
|||||||
| Ir1 | 606 (toluene) | 11.40 | 0.53 (611) | 6.18 (635) | 1.3 | LCC cells | 40 |
| Ir2 | 629 (DMSO) | 19.50 | 0.06 (632) | <40% cell viability at 12 μM (632) | — | HeLa cells | 43 |
| Ir3 | 668 (DMF) | 8.50 | 0.31 (660) | 2.01b | >6b | 4T1 cells | 45 |
| 6.59c (660) | >1.8c | ||||||
| Ir4 | 675 (water) | — | 0.95 (630) | 0.7 ± 0.3 (630) | >57 | A549 cells | 46 |
| Ir5 | 669 (water) | — | 0.07 (630) | 9.6 ± 0.9 (630) | >4 | A549 cells | 46 |
| Ir6 | 566 (DCM) | 4.40 | 0.34 (532) | 0.70 ± 0.06 (520) | >357.1 | A2780cis cells | 48 and 49 |
| Ir7 | 592 (DCM) | 2.60 | 0.16 (532) | 1.04 ± 0.02 (520) | >240.4 | A2780cis cells | 49 |
| Ir8 | 820 (DMSO/H2O) | — | 0.04 (808) | 27% cell viability at 50 μM (808) | >80% cell viability at 50 μM | 4T1 cells | 50 |
| Ir9 | 578 (MeCN) | 6.80 | 0.73 (514) | <30% cell viability at 2 μM (white) | >85% cell viability at 2 μM | MCF-7 cells | 53 |
| Ir10 | 713 (MeCN) | 8.90 | 0.69 (808) | <40% cell viability at 4 μM (808) | >85% cell viability at 4 μM | 4T1 cells | 52 |
| TPP-NPs | 650 (water) | 0.34 | 0.54 (635) | 0.22 (635) | >95% cell viability | HeLa cells | 61 |
| Ir11-NPs | 650 (water) | 0.38 | 0.72 (635) | 0.145 (635) | >95% cell viability | HeLa cells | 61 |
| Ir12-NPs | 650 (water) | 0.41 | 0.89 (635) | 0.057 (635) | >95% cell viability | HeLa cells | 61 |
| Ir13-NPs | 650 (water) | 0.40 | — | — | — | — | 62 |
| Ir14-NPs | 650 (water) | 0.68 | — | 0.47 (white) | >95% cell viability | HeLa cells | 62 |
| ZnPc-NCs | 678 (water) | 7.10 | 0.54 (620) | 0.72 ± 0.02 (630) | >138.9 | HeLa cells | 41 |
| Ir15-NCs | 679 (water) | 6.80 | 0.58 (620) | 1.2 ± 0.2 (630) | >83.3 | HeLa cells | 41 |
| Ir16 | 762 (MeOH) | 0.27 | 0.55 (808) | 3.65 ± 0.16e (808) | >95% cell viability | A549 cells | 64 |
| Ir17 | 508 (MeOH) | 1.60 | 0.34 (472) | 4.11 ± 0.14 (633) | 23 | A375 cells | 67 |
| Ir18 | 814 (MeOH:H2O) |
0.90 | 0.15 (808) | 14.4f | >95% cell viability | A549 cells | 65 |
| 7.1g (808) | |||||||
| Ir19 | 680 (DCM) | 9.00 | 0.37d (680) | 7.00 μg mL−1 (680) | — | HeLa cells | 73 |
The photophysical properties of far-red/NIR-emitting coumarins, nicknamed COUPYs, are highly influenced by the structural modifications carried out within the coumarin scaffold.49 In particular, the incorporation of strong electron-donating groups (EDGs) at position 7 of the coumarin skeleton through the fusion of the julolidine system in Ir7 was found to induce a red-shift in the absorption of the conjugate compared to Ir648 (see Fig. 1). Another important aspect is that the conjugation of the COUPY fluorophore to the iridium complex increased by a factor of 10 the 1O2 quantum yield of all the resulting conjugates compared with the free coumarins.49 In addition, all Ir(III)-COUPY conjugates were able to promote superoxide (O2˙−) generation in PBS through the type-I PDT mechanism, which could overcome the limitation of traditional type-II PDT agents under low oxygen environments.
Ir-cyanine conjugate Ir8 showed an intense absorption band in the NIR region (820 nm, Fig. 1) owing to the coordination of a phenanthrimidazole ligand containing a symmetric heptamethine cyanine fluorophore.50 The heptamethine chain contains a six-membered carbocyclic ring to increase the rigidity of the dye molecule, which is known to improve photostability and reduce aggregation in solution.55
A strategy for improving 1O2 generation ability of the rhodamine-containing cyclometalated iridium(III) system was achieved by simply replacing the cyclometalating ligand from 2-phenylpyridine to 2,3-diphenylquinoxaline in Ir9 (Fig. 1). In addition, the replacement of the oxygen bridge atom by silicon into the rhodamine moiety produced a substantial red-shift in the absorption spectrum of the parent compound.56 This shift results from interactions between the σ* orbital of the silicon atom and the π* orbitals of nearby carbon atoms, which provide the unique σ*–π* conjugation. Therefore, Ir10 exhibited strong deep-red absorption at 713 nm and a high singlet oxygen quantum yield (ΦΔ = 0.69; Table 1). Remarkably, even when a tumor was located 8.4 mm beneath a simulated epidermis, Ir10 achieved significant tumor ablation of 4-fold compared to the control (PBS).52
On the other hand, as commented in the introduction, the majority of clinically approved PSs are based on a tetrapyrrolic structural core.57 These porphyrinoid compounds, which include porphyrins, chlorins, bacteriochlorins58 and phthalocyanines (Pc),59 are well-known for their strong absorption in the red and NIR regions. However, such PSs suffer from poor photostability and slow body clearance, causing photosensitivity.17,60 In addition, these compounds show an intrinsic aggregation and they usually present poor solubility in aqueous media.61
To address these drawbacks and the issue of short absorption in iridium complexes, a promising strategy is to combine both types of molecules, creating a synergistic effect. This approach aims to achieve long-wavelength excitation and the formation of long-lived 3MLCT states, thereby enhancing ROS generation and increasing photostability for improved therapeutic efficacy.
Time-dependent density-functional theory (TD-DFT) calculations demonstrated that the iridium–porphyrin compounds Ir11 and Ir12 (Fig. 3) were able to combine the respective advantages of porphyrinoid-based compounds and transition metal complexes:61 (a) the conjugates retained the long-wavelength excitation and NIR emission of porphyrin itself; (b) they possessed highly effective ISC to obtain a considerably more long-lived triplet photoexcited state. Interestingly, the small energy gap separation between HOMO and LUMO and high spin–orbit coupling constant increased with the number of peripheral Ir centers incorporated in the tetraphenylporphyrin (TPP) unit, thereby increasing the molar absorption coefficient at long wavelengths (635 nm) and 1O2 production (ΦΔ = 0.89 for Ir12, Table 1). However, their synthesis and purification processes were complicated and led to low overall yields. To try to solve these problems, the N^O ligand was replaced with an N^N ligand in the iridium complex to obtain mononuclear and tetranuclear cationic iridium-TPP conjugates (Ir13 and Ir14, Fig. 3).62 The new cationic molecules had a high-yielding synthesis from readily available starting materials and white light photosensitization was efficiently achieved (Table 1).
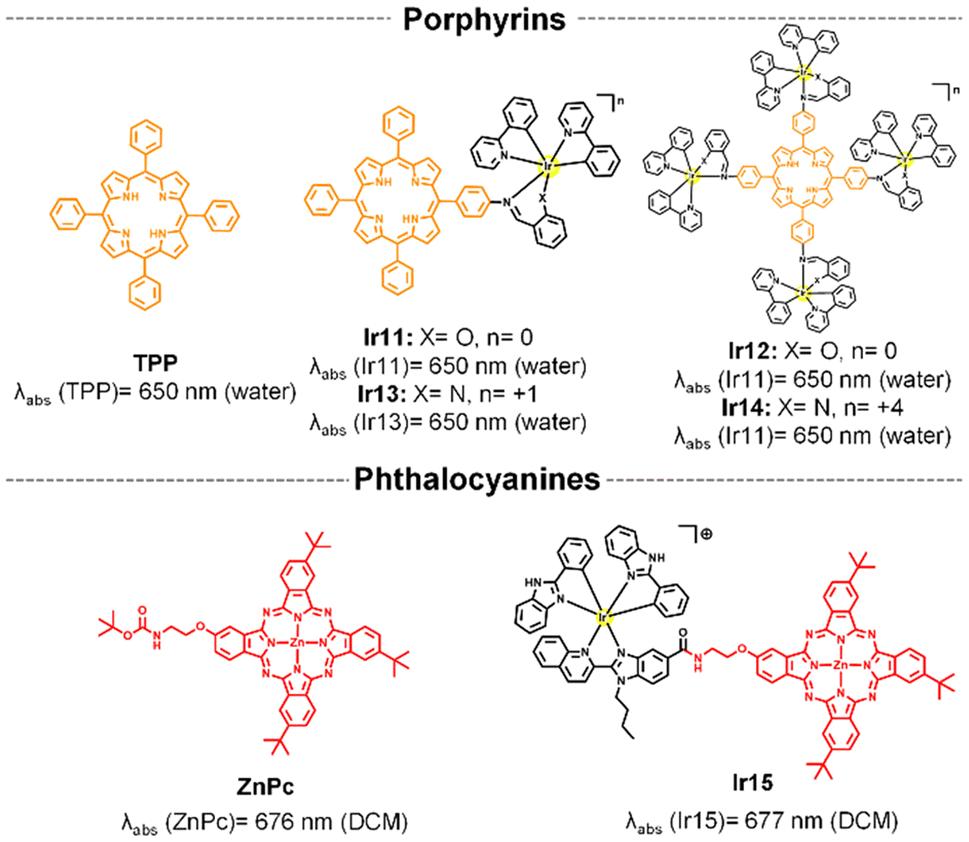 | ||
| Fig. 3 Structure of porphyrinoid compounds and their Ir-based conjugates. | ||
Although porphyrins have been used to achieve absorption in the red-light region, this absorption is of relatively low intensity.63 In contrast, Pcs show strong light absorption in the deep-red to NIR spectrum and they have minimal absorption in the visible range, reducing potential skin damage. In this context, the Ir(III)-phthalocyanine conjugate (Ir15, Fig. 3)41 showed great long-wavelength absorption at 677 nm and a higher photostability than free ZnPc. In order to increase water solubility and cell membrane permeability, Ir15 conjugate and parent zinc phthalocyanine (ZnPc) were encapsulated in amphoteric redox-responsive polyurethane–polyurea hybrid nanocapsules. The nanoencapsulated Ir15 achieved high photocytotoxicity under 630 nm light irradiation (Table 1), owing to dual type I and type II ROS photogeneration.
3. Ligand design via π-extension and donor–acceptor interactions
This section emphasizes the design of extended π-conjugated systems or donor–acceptor frameworks within the ligand itself to modulate electronic and optical properties of the final complex. The strategy is rooted in tuning the electronic distribution within the ligand scaffold rather than relying on a separate fluorophore entity. The photophysical mechanisms in these systems often involve charge-transfer transitions, such as MLCT or ligand-to-ligand charge transfer (LLCT). These mechanisms are intrinsic to the metal–ligand interaction and are highly tunable based on the nature of the π-system or donor–acceptor interactions within the ligand.64,65 Such designs allow for the creation of entirely new chromophores with unique optical properties, expanding beyond the scope of traditional fluorophore-functionalized complexes.One potential way to red-shift the MLCT absorption of Ir(III) complexes is to elongate the ligand π system to lower the π* orbital.64,66 In this sense, Ir16 was designed to include a rigid and planar N^N ligand with an extended π-conjugation system (Fig. 4). Ir16 showed strong absorption in the deep-red with a maximum at 762 nm that tailed over 950 nm. Remarkably, the compound exhibited a high 1O2 quantum yield of 0.56 (Table 1), which is the highest value among the 808 nm excited PSs reported so far. On the other hand, Ir1767 (Fig. 4) incorporated π-conjugated oligothiophenes into its ligand framework—a key structural feature also found in the Ru(II) photosensitizer TLD1433.68 This modification effectively red-shifts the ground-state absorption spectrum and extends the T1 excited-state lifetime. Increasing the number of thienyl units from 0 to 4 resulted in a progressive bathochromic shift to the orange spectral regions. Notably, under red-light excitation (633 nm), Ir17 demonstrated the most potent PDT effects with a phototherapeutic index (PI) of 23 (Table 1).
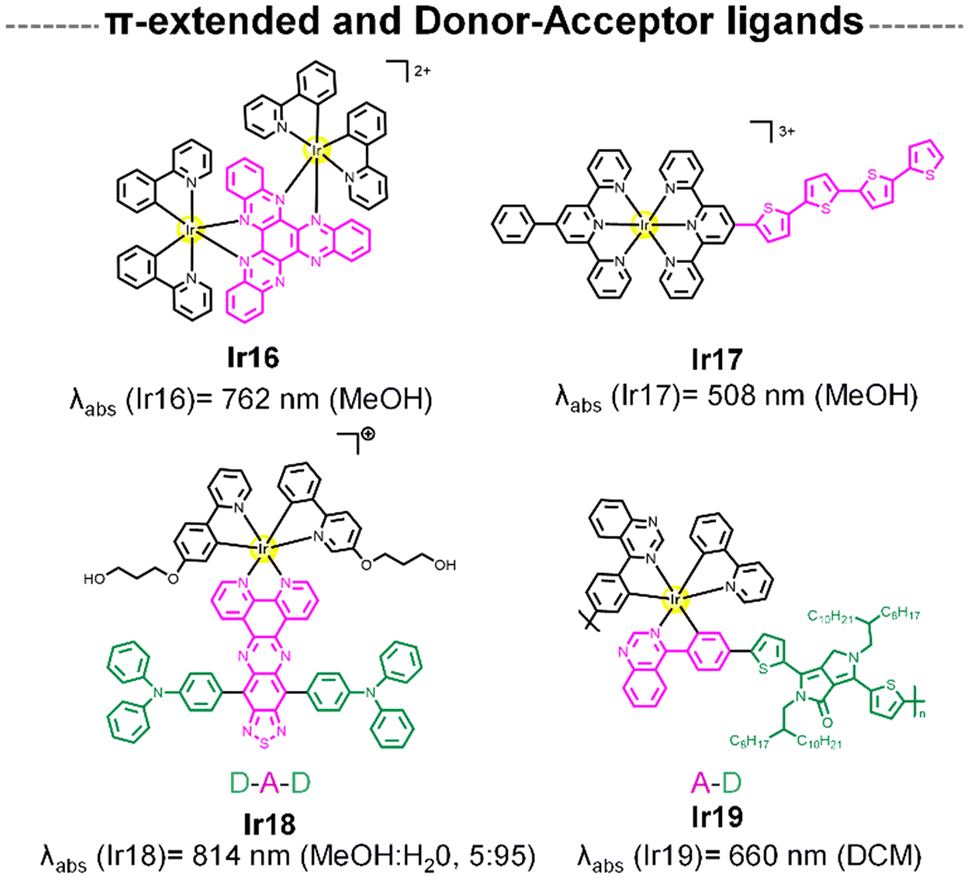 | ||
| Fig. 4 Structure of Ir complexes with of π-extended and donor–acceptor-type N^N ligand and its absorption wavelengths. | ||
Organic dyes with donor–acceptor (D–A) type electronic structures, also named push–pull molecules, are a well-known class of NIR-absorbing compounds due to their small HOMO–LUMO energy gap (the HOMO of the donor and the LUMO of the acceptor are at comparable energy levels; Fig. 5A). In these compounds the electron-donating and electron-withdrawing moieties are alternatively arranged along the conjugated structure, reducing the bandgap between the singlet–triplet state (ΔEST), which is also favourable to generate 1O2 (Fig. 5B).69,70 In addition, the significant intramolecular charge transfer (ICT) of the D–A–D molecules could provide a non-radiative deactivation pathway from the singlet excited state for heat generation.71 Some examples in the literature have demonstrated that the use of this type of push–pull structures as ligands in iridium complexes can shift their absorption to long wavelengths.65,72 In Ir18, a D–A–D type N^N ligand was designed by using triphenylamine (TPA) and [1,2,5]thiadiazolo-[3,4-i]dipyrido[a,c]phenazine (TDP) as the electron donor and acceptor respectively (Fig. 4).65Ir18 exhibited a deep-red absorption with an absorption maximum at 716 nm, which the authors assigned to the ICT transition. Interestingly, the maximum absorption peak of the complex was gradually red-shifted to 814 nm with the increase of the water fraction up to 95%, probably attributed to the formation of J-type aggregates of Ir18 in aqueous solution. Remarkably, the Ir complex showed no ROS production in the monomer state but had significant ROS generation in the aggregation state. Recently, Ir19 (Fig. 4) which contains a donor–acceptor conjugated polymer, has been reported to show a strong absorption in the red region (ε680 ≈ 9 × 104 M−1 cm−1). The metallopolymer was able to generate O2˙− intracellularly upon 680 nm laser irradiation, resulting in sufficient phototoxicity to induce cell apoptosis.73
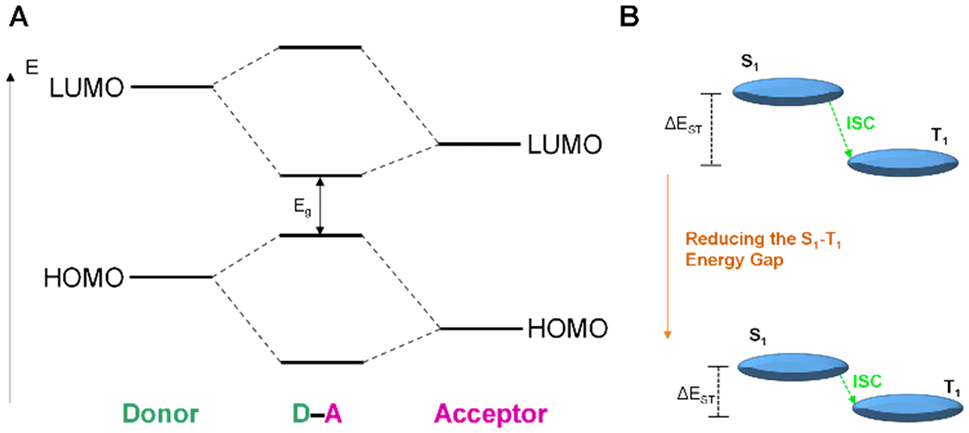 | ||
| Fig. 5 (A) Schematic diagram presenting the HOMO–LUMO energy levels of donor–acceptor molecules and the molecular interaction between them. (B) A physical model representing the S1–T1 ISC method and the proposed tactic in donor–acceptor molecules. Adapted from ref. 69. | ||
4. Conclusions and outlook
This Frontier gives an overview of recent literature on novel deep-red/NIR absorbing iridium(III) photosensitizers.Due to the low autofluorescence, scattering, and absorption of biological tissue in the deep-red/NIR region, compounds that can be excited with this type of light offer deeper penetration depth and superior signal-to-noise ratios for biomedical applications. While iridium(III) complexes exhibit many desirable properties such as high intersystem crossing, photobleaching resistance, large Stokes’ shifts, and high emission quantum yield, their traditional lack of deep-red/NIR absorption has restricted their potential. This limitation requires the use of high-energy, short-wavelength laser excitation, leading to poor tissue penetration and potential phototoxicity. To address these challenges, researchers have explored different approaches for designing deep-red-absorbing Ir-based PSs, drawing inspiration from organic small molecule chromophores. Two key strategies were envisioned: (1) functionalization with organic fluorophores including porphyrinoid compounds, and (2) ligand design via π-extension and donor–acceptor interactions. While both approaches involve extending conjugation in the complex, we categorize them based on whether the extended conjugation arises from a separate fluorophore (1) or is intrinsic to the ligand design (2), each leveraging unique mechanisms to achieve desired optical properties. The first approach utilizes fluorophores, such as organic dyes or porphyrins, chemically integrated into the metal complex through the modification of the N^N ligand. This strategy capitalizes on the inherent photophysical characteristics of the fluorophore, particularly their absorption in the deep-red or NIR region, with the metal center playing a supportive role by influencing electronic structure or stabilizing the fluorophore. In these complexes, an increase in the molar extinction coefficient (ranging from 0.4 to 19 × 104 M−1 cm−1) was observed compared with the free organic compound, which offers an additional advantage to this type of PSs. In addition, the choice of the linker seems to be crucial for the interaction between metal centre and the organic dye in order to facilitate the ISC of the fluorophore by heavy atom effect. On the other hand, the presence of the Ir(III) complex in the compounds can enhance the production of other types of ROS, such as superoxide (O2˙−) via the type-I PDT pathway. This could potentially circumvent the drawbacks of conventional type-II PDT agents in oxygen-deficient settings. It is noteworthy that many of these compounds also show NIR emission, which could be an advantage for the combination of phototherapy with fluorescence-guided surgery (FGS).
In contrast, the second approach focuses on designing extended π-conjugated systems or donor–acceptor frameworks directly within the ligand scaffold. Unlike fluorophore-based systems, these complexes often result in novel chromophores with unique and tunable optical properties, showcasing the versatility of ligand design in expanding the scope of photophysical applications. π-Conjugated systems effectively lower the π* orbital energy, leading to absorption in the deep-red or NIR regions. These designs often enhance PDT performance by increasing singlet oxygen quantum yields and improving phototoxicity under red-light excitation. In addition, D–A ligand structures also show promise, as they create strong ICT transitions and reduce singlet–triplet energy gaps. These features support red-shifted absorption and, in some cases, aggregation-induced ROS generation, broadening their applicability in aqueous environments.
However, the synthesis and purification of these types of compounds are sometimes complicated, and the yields are usually low. Moreover, poor solubility in aqueous media and the intrinsic aggregation of some porphyrinoid-based compounds limit clinical application. Therefore, nanoparticles (NPs), including polymeric nanocarriers, liposomes, micelles, and other biocompatible materials, are usually required to enhance the solubility and biocompatibility of these PSs. However, the choice of nanoparticle type must align with the target application, as factors such as biocompatibility and interaction with cellular environments depend on the physicochemical properties of the nanoparticle system.
Despite these challenges, the development of iridium(III) complexes with deep-red/NIR absorption represents a unique opportunity to improve the efficacy of PDT in the treatment of deep-seated or large tumors. In the near future, it will be crucial to optimize synthetic routes for higher yield and scalability, explore new ligand designs that further fine-tune NIR absorption properties, and conduct rigorous in vivo testing in preclinical models. These studies will provide valuable insights into pharmacokinetics, biodistribution, and overall efficacy of iridium(III)-based PSs in complex biological systems. Additionally, evaluating the long-term toxicity and immunogenicity of these compounds will be key to translating them into clinical practice.
We hope this Frontier will stimulate additional research to fully unlock the enormous potential that still exists for this special class of photoactive metal complexes.
Abbreviations
| 2P | Two-photon |
| acac | Acetylacetonate |
| BODIPY | Boron-dipyrromethene |
| COUPY | Nickname far-red emitting coumarin |
| EDG | Electron donating group |
| FGS | Fluorescence-guided surgery |
| HOMO | Highest occupied molecular orbital |
| ICT | Intramolecular charge transfer |
| ILCT | Intraligand charge transfer |
| ISC | Intersystem crossing |
| LLCT | Ligand-to-ligand charge transfer |
| LUMO | Lowest unoccupied molecular orbital |
| MLCT | Metal-to-ligand charge transfer |
| NCs | Nanocapsules |
| NIR | Near-infrared |
| NPs | Nanoparticles |
| PDT | Photodynamic therapy |
| PBS | Phosphate buffered saline |
| Pc | Phthalocyanine |
| PI | Phototherapeutic index |
| PS | Photosensitizer |
| ROS | Reactive oxygen species |
| S1 | Singlet excited state |
| T1 | Triplet excited state |
| TPA | Triphenylamine |
| TD-DFT | Time-dependent density-functional theory |
| TDP | [1,2,5]Thiadiazolo-[3,4-i]dipyrido[a,c]phenazine |
| TPP | Tetraphenylporphyrin |
| UV | Ultraviolet |
| UNCPs | Upconversion nanoparticles |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. V. thanks the European Union's Horizon 2020 research under the Marie Skłodowska-Curie Postdoctoral Fellow (101106108). J. R. was supported by the Spanish Ministerio de Ciencia e Innovación – Agencia Estatal de Investigación (MCI/AEI/10.13039/501100011033) and FEDER funds (project PID2021-122850NB-I00).References
- Y. Zhang, B.-T. Doan and G. Gasser, Chem. Rev., 2023, 123, 10135–10155 CrossRef CAS PubMed
.
- Y. Tang, Y. Li, B. Li, W. Song, G. Qi, J. Tian, W. Huang, Q. Fan and B. Liu, Nat. Commun., 2024, 15, 1–13 Search PubMed
- L. C.-C. Lee and K. K.-W. Lo, Small Methods, 2024, 2400563 CrossRef CAS PubMed
- S. Mallidi, S. Anbil, A.-L. Bulin, G. Obaid, M. Ichikawa and T. Hasan, Theranostics, 2016, 6, 2458–2487 CrossRef CAS PubMed
- M. M. Kim and A. Darafsheh, Photochem. Photobiol., 2020, 96, 280–294 CrossRef CAS PubMed
- Y. Xu, C. V. Chau, J. Lee, A. C. Sedgwick, L. Yu, M. Li, X. Peng, J. S. Kim and J. L. Sessler, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2314620121 CrossRef CAS PubMed
- V. V. Barun, A. P. Ivanov, A. V. Volotovskaya and V. S. Ulashchik, J. Appl. Spectrosc., 2007, 74, 430–439 CrossRef CAS
- G. Obaid, J. P. Celli, M. Broekgaarden, A.-L. Bulin, P. Uusimaa, B. Pogue, T. Hasan and H.-C. Huang, Nat. Rev. Bioeng., 2024, 2, 752–769 CrossRef CAS
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC
- M. J. Garland, C. M. Cassidy, D. Woolfson and R. F. Donnelly, Future Med. Chem., 2009, 1, 667–691 CrossRef CAS PubMed
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC
- X. Li, J. Kim, J. Yoon and X. Chen, Adv. Mater., 2017, 29, 1606857 CrossRef PubMed
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404–1414 CrossRef CAS PubMed
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed
-
E. Longhi and L. De Cola, in Iridium(III) in Optoelectronic and Photonics Applications, John Wiley & Sons, Ltd, 2017, pp. 205–274 Search PubMed
- Z. Fan, Y. Rong, T. Sadhukhan, S. Liang, W. Li, Z. Yuan, Z. Zhu, S. Guo, S. Ji, J. Wang, R. Kushwaha, S. Banerjee, K. Raghavachari and H. Huang, Angew. Chem., Int. Ed., 2022, 61, e202202098 CrossRef CAS PubMed
-
J. C. Deaton and F. N. Castellano, in Iridium(III) in Optoelectronic and Photonics Applications, 2017, pp. 1–69 Search PubMed
- C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155–177 CrossRef CAS
- A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34–76 CrossRef CAS
- E. C.-L. Mak, Z. Chen, L. C.-C. Lee, P. K.-K. Leung, A. M.-H. Yip, J. Shum, S.-M. Yiu, V. W.-W. Yam and K. K.-W. Lo, J. Am. Chem. Soc., 2024, 146, 25589–25599 CrossRef CAS PubMed
- K. M. Kuznetsov, I. S. Kritchenkov, J. R. Shakirova, V. V. Gurzhiy, V. V. Pavlovskiy, V. V. Porsev, V. V. Sokolov and S. P. Tunik, Eur. J. Inorg. Chem., 2021, 2021, 2163–2170 CrossRef CAS
- R. Tu, J. Liu, W. Chen, F. Fu and M.-J. Li, Dalton Trans., 2023, 52, 13137–13145 RSC
- Z.-Y. Pan, B.-F. Liang, Y.-S. Zhi, D.-H. Yao, C.-Y. Li, H.-Q. Wu and L. He, Dalton Trans., 2023, 52, 1291–1300 RSC
- D.-L. Ma, D. S.-H. Chan and C.-H. Leung, Acc. Chem. Res., 2014, 47, 3614–3631 CrossRef CAS PubMed
- G. Vigueras, L. Markova, V. Novohradsky, A. Marco, N. Cutillas, H. Kostrhunova, J. Kasparkova, J. Ruiz and V. Brabec, Inorg. Chem. Front., 2021, 8, 4696–4711 RSC
- C. Jones, M. Martinez-Alonso, H. Gagg, L. Kirby, J. A. Weinstein and H. E. Bryant, J. Med. Chem., 2024, 67, 16157–16164 CrossRef CAS PubMed
- E. Zafon, I. Echevarría, S. Barrabés, B. R. Manzano, F. A. Jalón, A. M. Rodríguez, A. Massaguer and G. Espino, Dalton Trans., 2022, 51, 111–128 RSC
- F. Monti, A. Baschieri, L. Sambri and N. Armaroli, Acc. Chem. Res., 2021, 54, 1492–1505 CrossRef CAS PubMed
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC
- L. G. Arnaut and M. M. Pereira, Chem. Commun., 2023, 59, 9457–9468 RSC
- S. Liu, J. Han, Y. Chang, W. Wang, R. Wang, Z. Wang, G. Li, D. Zhu and M. R. Bryce, Chem. Commun., 2022, 58, 10056–10059 RSC
- B. del Rosal and D. Jaque, Methods Appl. Fluoresc., 2019, 7, 022001 CrossRef CAS PubMed
- J. Zhao, X. Zhang, L. Fang, C. Gao, C. Xu and S. Gou, Small, 2020, 16, 2000363 CrossRef CAS PubMed
- J. Karges, H. Chao and G. Gasser, JBIC, J. Biol. Inorg. Chem., 2020, 25, 1035–1050 CrossRef CAS PubMed
- L. Wang, J. Karges, F. Wei, L. Xie, Z. Chen, G. Gasser, L. Ji and H. Chao, Chem. Sci., 2023, 14, 1461–1471 RSC
- D. Chen, T. Shao, H. Zhao, F. Chen, Z. Fang, Y. Tian and X. Tian, Dyes Pigm., 2023, 215, 111271 CrossRef CAS
- P. Majumdar, X. Yuan, S. Li, B. Le Guennic, J. Ma, C. Zhang, D. Jacquemin and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC
- J. Bonelli, E. Ortega-Forte, G. Vigueras, J. Follana-Berná, P. Ashoo, D. Abad-Montero, N. Isidro, M. López-Corrales, A. Hernández, J. Ortiz, E. Izquierdo-García, M. Bosch, J. Rocas, Á. Sastre-Santos, J. Ruiz and V. Marchán, ACS Appl. Mater. Interfaces, 2024, 16, 38916–38930 CrossRef CAS PubMed
- E. Palao, R. Sola-Llano, A. Tabero, H. Manzano, A. R. Agarrabeitia, A. Villanueva, I. López-Arbeloa, V. Martínez-Martínez and M. J. Ortiz, Chem. – Eur. J., 2017, 23, 10139–10147 CrossRef CAS PubMed
- N. E. Aksakal, E. Tanrıverdi Eçik, H. H. Kazan, G. Yenilmez Çiftçi and F. Yuksel, Photochem. Photobiol. Sci., 2019, 18, 2012–2022 CrossRef CAS PubMed
- G. Liang, N. Montesdeoca, D. Tang, B. Wang, H. Xiao, J. Karges and K. Shang, Biomaterials, 2024, 309, 122618 CrossRef CAS PubMed
- B. Liu, J. Jiao, W. Xu, M. Zhang, P. Cui, Z. Guo, Y. Deng, H. Chen and W. Sun, Adv. Mater., 2021, 33, 2100795 CrossRef CAS PubMed
- N. Xu, G.-D. Zhang, Z.-Y. Xue, M.-M. Wang, Y. Su, H. Fang, Z.-H. Yu, H.-K. Liu, H. Lu and Z. Su, Chem. Eng. J., 2024, 497, 155022 CrossRef CAS
- B. Liu, S. Monro, M. A. Jabed, C. G. Cameron, K. L. Colón, W. Xu, S. Kilina, S. A. McFarland and W. Sun, Photochem. Photobiol. Sci., 2019, 18, 2381–2396 CrossRef CAS PubMed
- V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Angew. Chem., Int. Ed., 2019, 58, 6311–6315 CrossRef CAS PubMed
- A. Rovira, E. Ortega-Forte, C. Hally, M. Jordà-Redondo, D. Abad-Montero, G. Vigueras, J. I. Martínez, M. Bosch, S. Nonell, J. Ruiz and V. Marchán, J. Med. Chem., 2023, 66, 7849–7867 CrossRef CAS PubMed
- Q. Yang, H. Jin, Y. Gao, J. Lin, H. Yang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 15417–15425 CrossRef CAS PubMed
- C. Liu, Q. Ding, Y. Liu, Z. Wang, Y. Xu, Q. Lu, X. Chen, J. Liu, Y. Sun, R. Li, Y. Yang, Y. Sun, S. Li, P. Wang and J. S. Kim, Inorg. Chem., 2024, 63, 13059–13067 CrossRef CAS PubMed
- J. Liu, X. Yang, S. Wu, P. Gong, F. Pan, P. Zhang, C.-S. Lee, C. Liu and K. M.-C. Wong, J. Mater. Chem. B, 2024, 12, 3710–3718 RSC
- L. Zhou, F. Wei, J. Xiang, H. Li, C. Li, P. Zhang, C. Liu, P. Gong, L. Cai and K. M.-C. Wong, Chem. Sci., 2020, 11, 12212–12220 RSC
- Y. Wu, J. Wu and W.-Y. Wong, Biomater. Sci., 2021, 9, 4843–4853 RSC
- A. Gandioso, E. Izquierdo-García, P. Mesdom, P. Arnoux, N. Demeubayeva, P. Burckel, B. Saubaméa, M. Bosch, C. Frochot, V. Marchán and G. Gasser, Chem. – Eur. J., 2023, 29, e202301742 CrossRef CAS PubMed
- M. Fu, Y. Xiao, X. Qian, D. Zhao and Y. Xu, Chem. Commun., 2008, 1780–1782 RSC
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed
- Q. Zhang, J. He, W. Yu, Y. Li, Z. Liu, B. Zhou and Y. Liu, RSC Med. Chem., 2020, 11, 427–437 RSC
- D. Li, S. Cai, P. Wang, H. Cheng, B. Cheng, Y. Zhang and G. Liu, Adv. Healthcare Mater., 2023, 12, 2300263 CrossRef CAS PubMed
- D. Van Straten, V. Mashayekhi, H. S. De Bruijn, S. Oliveira and D. J. Robinson, Cancers, 2017, 9, 19 CrossRef PubMed
- L. Zhang, Y. Geng, L. Li, X. Tong, S. Liu, X. Liu, Z. Su, Z. Xie, D. Zhu and M. R. Bryce, Chem. Sci., 2021, 12, 5918–5925 RSC
- Z. Wang, L. Li, W. Wang, R. Wang, G. Li, H. Bian, D. Zhu and M. R. Bryce, Dalton Trans., 2023, 52, 1595–1601 RSC
- R. D. Arasasingham and T. C. Bruice, Inorg. Chem., 1990, 29, 1422–1427 CrossRef CAS
- J. Zhao, Y. Gao, R. Huang, C. Chi, Y. Sun, G. Xu, X.-H. Xia and S. Gou, J. Am. Chem. Soc., 2023, 145, 11633–11642 CrossRef CAS PubMed
- J. Zhao, K. Yan, G. Xu, X. Liu, Q. Zhao, C. Xu and S. Gou, Adv. Funct. Mater., 2021, 31, 2008325 CrossRef CAS
- T. J. Whittemore, T. A. White and C. Turro, J. Am. Chem. Soc., 2018, 140, 229–234 CrossRef CAS PubMed
- X. Sun, H. D. Cole, G. Shi, V. Oas, A. Talgatov, C. G. Cameron, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2024, 63, 21323–21335 CrossRef CAS PubMed
- J. A. Roque III, H. D. Cole, P. C. Barrett, L. M. Lifshits, R. O. Hodges, S. Kim, G. Deep, A. Francés-Monerris, M. E. Alberto, C. G. Cameron and S. A. McFarland, J. Am. Chem. Soc., 2022, 144, 8317–8336 CrossRef CAS PubMed
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. Mei, L. A. Padilha, T. R. Ensley, H. Hu, S. Webster, D. J. Hagan, E. W. Van Stryland, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805–3817 CrossRef CAS
- Z. Y. Jin, H. Fatima, Y. Zhang, Z. Shao and X. J. Chen, Adv. Ther., 2022, 5, 2100176 CrossRef CAS
- S. Gao, G. Wei, S. Zhang, B. Zheng, J. Xu, G. Chen, M. Li, S. Song, W. Fu, Z. Xiao and W. Lu, Nat. Commun., 2019, 10, 2206 CrossRef PubMed
- J. Kasparkova, A. Hernández-García, H. Kostrhunova, M. Goicuría, V. Novohradsky, D. Bautista, L. Markova, M. D. Santana, V. Brabec and J. Ruiz, J. Med. Chem., 2024, 67, 691–708 CrossRef CAS PubMed
- Z. Zhang, Z. Wei, J. Guo, J. Lyu, B. Wang, G. Wang, C. Wang, L. Zhou, Z. Yuan, G. Xing, C. Wu and X. Zhang, Nat. Commun., 2024, 15, 170 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |