
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Donor-free 9-aluminafluorenes: molecular structures and reactivity†
Paula L.
Lückert
,
Jannik
Gilmer
,
Alexander
Virovets
 ,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
Institut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt, Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
First published on 2nd December 2024
Abstract
Aluminum-doped polycyclic aromatic hydrocarbons (PAHs) are underexplored despite the broad applications of boron-containing PAHs in areas such as catalysis and optoelectronics. We disclose the donor-free, sterically unprotected 9-methyl-9-aluminafluorene (Me-AlFlu; 2), synthesized by heating a 9,9-dimethyl-9-stannafluorene and AlMe3 in hexanes. The compound is a dimer, (2)2, with trans-positioned AlMe substituents in the solid state. In solution, (2)2 shows a dynamic cis/trans-interconversion rather than a monomer-dimer equilibrium (Tol-d8, RT). Lewis bases L cleave (2)2 into monomeric adducts 2·L (L = OEt2, thf, pyridine). Lewis acidic AlBr3 transforms (2)2 into a 2,2′-(Br2Al)2-1,1′-biphenyl (3), crystallographically characterized as dimeric (3)2. (3)2 is a synthetic equivalent for the elusive free Br-AlFlu: Treatment with donor molecules furnishes Br-AlFlu·L adducts (L = OEt2, pyridine); the three-coordinate, monomeric aluminafluorene Mes*-AlFlu was prepared from (3)2, Mes*Li, and a 2,2′-dilithio-1,1′-biphenyl in quantitative yield (Mes* = 2,4,6-(tBu)3C6H2).
Introduction
Doping organic π-electron systems with other p-block elements is an effective strategy to impart new chemical and physical properties to these species.1 Specifically, the combination of a polycyclic aromatic hydrocarbon (PAH) such as fluorene with boron as a dopant to generate 9-borafluorenes (BFlus) can have a particularly pronounced effect,2,3 as a conjugation barrier (i.e., the CH2 fragment in the carbonaceous species) is removed and a vacant B(pz) orbital is introduced instead, which can now: (i) mediate electron delocalization and bring about novel optoelectronic properties,4 (ii) facilitate reduction,5,6 and (iii) act as a Lewis acid to promote bond-activation reactions7 or the expansion of the five-membered central borole ring.8Compared to the extensive research on BFlus, their heavier homologues, the 9-aluminafluorenes (AlFlus),9,10 are far less well explored. This is unfortunate, because AlFlus are expected to exhibit a lower degree of aryl-heteroatom double-bond character than BFlus,11 leading to a greater propensity to form structurally intriguing aggregates through Al⋯π(Ar) complexes or Al–C–Al′ two-electron–three-center (2e3c) bonds. Relative to open-chain arylaluminum compounds, AlFlus should possess a structurally enforced enhanced Lewis acidity due to their small endohedral C–Al–C angle. This angle (108° in a regular five-membered ring) deviates more from the ideal 120° angle of three-coordinate AlR3 species than from the corresponding angles of perfectly tetrahedral (109.5°) adducts. By the same token, the behavior of Al-based Lewis acids is more diverse than that of their B-based counterparts, as Al sites, unlike B centers, can readily accommodate coordination numbers larger than four.
In 1962, Eisch et al. reported the formation of Ph-AlFlu through the metalative cyclization of o-biphenylyl(diphenyl)aluminum at 200 °C. Their claim was mainly based on the analysis of hydrolysis and iodinolysis products.12,13 The topic lay dormant until 2015, when Chujo and Tanaka used salt-metathesis protocols to synthesize AlFlus carrying Al-bonded phenyl rings with one or two chelating (dimethylamino)methyl substituents at their ortho positions (Fig. 1). Their research focused on the emission properties of the obtained four- and five-coordinate AlFlus.14,15 More recently, Braunschweig et al. disclosed the synthesis of various aluminafluorenes R-AlFlu [R = 1,2,4-(tBu)3C5H2 (92%; Fig. 1), Ph2(tBu)Si (44%), 2-C4H3S (79%), tBu (23%; Fig. 1), Br (53%)]. The compounds were again prepared from 2,2′-dilithio-1,1′-biphenyl by salt-metathesis reactions and isolated and structurally characterized as their ether adducts – with the exception of the η5-cyclopentadienide derivative, which is monomeric in the solid state, and the tBu derivative, which crystallizes as a dimer.16
 | ||
| Fig. 1 Known mono- and dimeric 9-aluminafluorenes featuring 2,6-bis[(dimethylamino)methyl]phenyl (BDMAPh), 1,2,4-(tBu)3C5H2 (Cp3t), and tert-butyl (tBu) substituents. | ||
One aim of our study outlined herein was to develop straightforward, high-yield synthesis protocols for base-free R-AlFlus featuring (i) the small substituent R = Me to minimize steric shielding of the Al center, and (ii) the reactive substituent R = Br for late-stage derivatization. Particular emphasis was placed on the molecular structure of Me-AlFlu in non-donor solvents and in the solid state, as well as on the synthesis of the first base-free, three-coordinate, monomeric aluminafluorene, Mes*-AlFlu (Mes* = 2,4,6-(tBu)3C6H2). All our AlFlus were equipped with tBu groups in their 2,7-positions to enhance solubility in non-polar solvents and to facilitate NMR-spectroscopic analysis.
Results and discussion
Syntheses
The base-free Me-AlFlu (2) was synthesized by heating the 9,9-dimethyl-9-stannafluorene 1 with 1 equiv. of AlMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 in either hexanes or C6H6/toluene (Scheme 1). The only by-product formed is the volatile and relatively inert SnMe4.18,19 An advantage of using hexanes as the solvent is that the dimer (2)2 precipitates in pure form already upon cooling the reaction mixture to room temperature (yield: 74%); when C6H6/toluene is employed, the yield of (2)2 is higher (91%), but some further workup is required. In the presence of the donor molecules Et2O, THF, or pyridine, (2)2 is cleanly split into its constituting monomers to furnish the monoadducts 2·OEt2, 2·thf, or 2·py (Scheme 1).
17 in either hexanes or C6H6/toluene (Scheme 1). The only by-product formed is the volatile and relatively inert SnMe4.18,19 An advantage of using hexanes as the solvent is that the dimer (2)2 precipitates in pure form already upon cooling the reaction mixture to room temperature (yield: 74%); when C6H6/toluene is employed, the yield of (2)2 is higher (91%), but some further workup is required. In the presence of the donor molecules Et2O, THF, or pyridine, (2)2 is cleanly split into its constituting monomers to furnish the monoadducts 2·OEt2, 2·thf, or 2·py (Scheme 1).
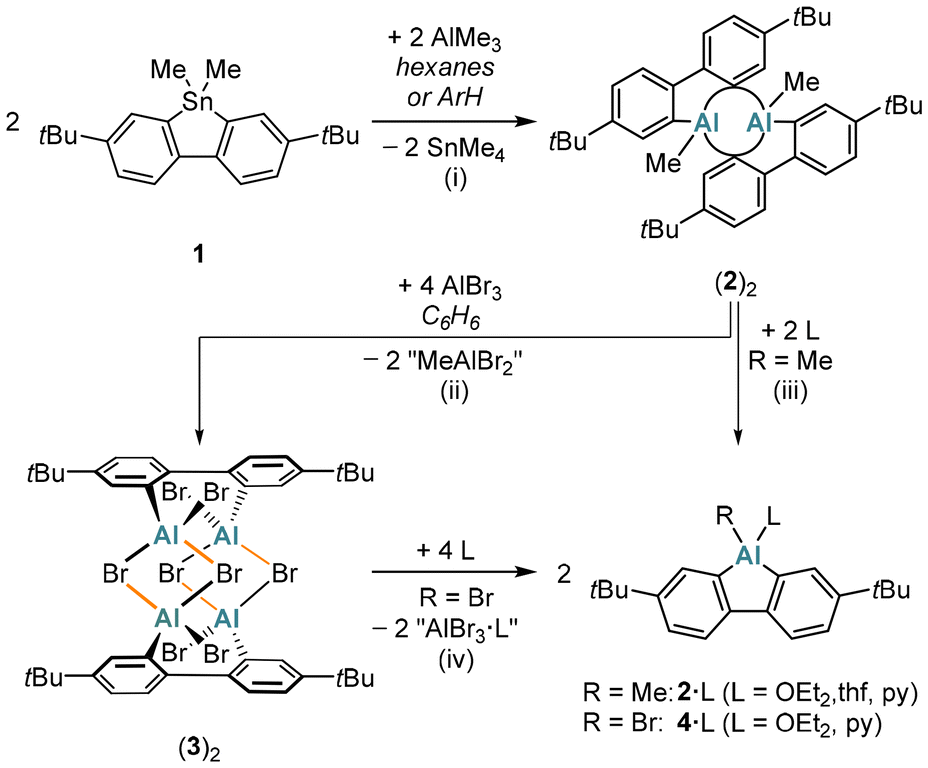 | ||
| Scheme 1 Synthesis of donor-free (2)2 through Sn/Al exchange between the 9-stannafluorene 1 and AlMe3 (ArH: C6H6/toluene). The addition of AlBr3 to (2)2 furnishes (3)2. Lewis bases (L: Et2O, THF, or pyridine), cleave (2)2 or (3)2 into the monomeric adducts 2·L or 4·L. (i) Hexanes, 140 °C, 3 d (74% yield) or C6H6/toluene, 120 °C, 3 d (91% yield); sealed glass ampoule. (ii) C6H6, room temperature, 1 d (95% yield). (iii) 2·OEt2: in Et2O, room temperature; 2·thf: C6D6, room temperature; 2·py: C6H6, room temperature (quantitative conversions). (iv) 4·OEt2: C6H6, room temperature (quantitative conversion); 4·py: C6D6, room temperature (not isolated). Note: in (3)2, four bonds were arbitrarily chosen as formally intermolecular (highlighted in orange) to facilitate the distinction between the monomers M and M′. | ||
Treatment of (2)2 with 4 equiv. of AlBr317 in C6H6 results not only in quantitative AlMe/AlBr exchange but also in the incorporation of two AlBr3 molecules to afford dimeric 2,2′-(Br2Al)2-1,1′-biphenyl [(3)2, 95%; Scheme 1]. Upon addition of Et2O to (3)2 in C6H6, the donor adduct of Br-AlFlu, 4·OEt2, precipitates quantitatively as a colorless solid. In terms of yield, our overall synthesis cascade to 4·OEt2 improves upon the published protocol16 by about 40 percentage points. Although pyridine can also reconstitute the AlFlu scaffold from (3)2, it proved challenging to separate the target product 4·py from by-products such as [AlBr2(py)4][X] ([5][X]; X = Br−, AlBr4−; Fig. S44 and S45†).
A particularly notable application of (3)2 as a synthetic equivalent of donor-free Br-AlFlu is the preparation of Mes*-AlFlu (6): sequential addition of Mes*Li (4 equiv.) and 2,2′-dilithio-4,4′-di-tert-butyl-1,1′-biphenyl (2 equiv.) to (3)2 in C6H6 gave 6 in 97% yield (Scheme 2).
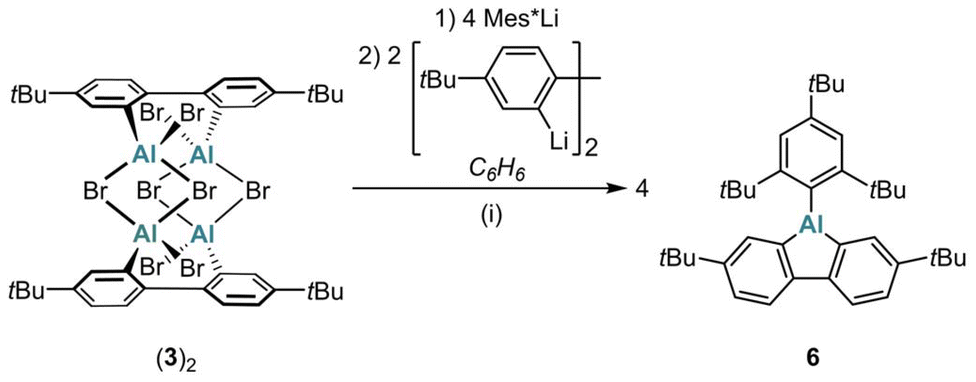 | ||
| Scheme 2 Synthesis of Mes*-AlFlu (6) using (3)2 as a synthetic equivalent of the elusive Br-AlFlu. (i) C6H6, room temperature, 1 d (97% yield). | ||
Solid-state structures
In the solid state, Me-AlFlu forms centrosymmetric dimers, with the Al-bonded Me substituents adopting a trans-configuration (trans-(2)2; Fig. 2).20 The individual monomers, M and M′, are linked by two Al⋯C interactions, resulting in two Al(1)⋯Al(1)′ bridging aryl rings (Arb) and two terminal rings (Art), with bridging [C(11)] and terminal [C(21)] ipso-C atoms. The position of Arb is asymmetric between Al(1) and Al(1)′, as indicated by the differing angles Al(1)–C(11)⋯C(14) = 153.13(17)° and Al(1)′–C(11)⋯C(14) = 128.38(16)°. The fact that the Al(1)′–C(11)–C(14) angle is significantly closer to 90° than the Al(1)–C(11)–C(14) angle can still be viewed as a remnant of the initial intermolecular Al⋯π(Ar) complex when the two heterofluorene units first encountered each other. Correspondingly, the ‘intermonomer’ Al(1)′–C(11) bond (2.148(3) Å) is longer by 0.055 Å than the ‘intramonomer’ Al(1)–C(11) bond (2.093(3) Å; cf. Al(1)–C(21) = 1.971(3) Å). The range of C–C bond lengths in Arb (1.382(5)–1.427(5) Å) is close to that in Art (1.389(5)–1.409(5) Å), indicating that the bridging mode does not lead to a systematic bond-length alternation. However, the two C–C bonds involving the bridging C(11) atom are slightly longer than the other four (1.409(4) and 1.427(5) Å vs. 1.382(5)–1.401(5) Å). Finally, we note that trans-(2)2 has very similar structural parameters to Braunschweig's (tBu-AlFlu)2,16 while the comparable 9-borafluorene dimer (H-BFlu)2 shows one B–(μ-H)–B two-electron–three-center bond and one B⋯B′-bridging aryl ring (the three other rings remain terminally bonded).21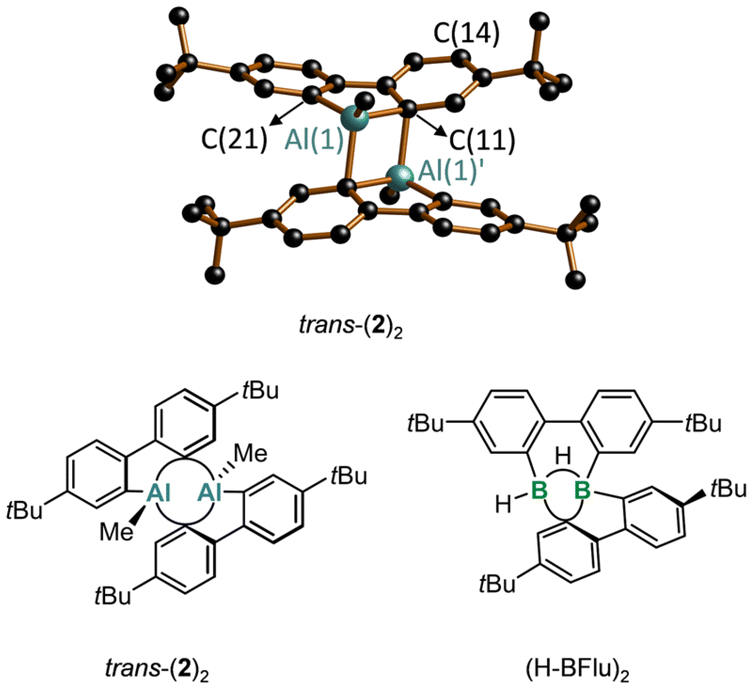 | ||
| Fig. 2 Top: molecular structure of trans-(2)2 in the solid state; H atoms omitted for clarity (C: black, Al: turquoise). Bottom: structural formulae of trans-(2)2 and of the comparable 9-borafluorene dimer (H-BFlu)2. | ||
X-ray crystallography reveals that the compound (3)2 no longer contains the 9-aluminafluorene motif but instead forms a centrosymmetric 2,2′-(Br2Al)2-1,1′-biphenyl dimer (Fig. 3). The two Br2Al substituents in each monomer adopt an approximate s-trans configuration with a torsion angle Al(1)–C(11)–C(21)–Al(2) of 129.55(19)° [Al(1)–C(11) = 1.945(5) Å, Al(2)–C(21) = 1.956(4) Å]. Four Br atoms occupy bridging positions between Al centers of different monomers, assembling the cage-like structure of (3)2. The underlying structural feature, a four-membered R(Br)Al–(μ-Br)2–Al(Br)R ring, is common not only for aluminum tribromide (R = Br) but also for numerous dibromo(organo)alanes.22
 | ||
| Fig. 3 Left: molecular structure of (3)2 in the solid state, viewed from two different perspectives; tBu-groups in the 2,7-positions of the biphenyl backbones and H atoms omitted for clarity. Right: molecular structure of 6 in the solid state; H atoms omitted for clarity (C: black, Al: turquoise, Br: brown). | ||
The 2,4,6-(tBu)3C6H2-substituted Mes*-AlFlu (6) exists as a monomeric species with a three-coordinate Al center in the crystal lattice (Fig. 3). The sum of C–Al–C angles is 360°, confirming a trigonal-planar ligand environment, although the endocyclic C(11)–Al(1)–C(21) bond angle is nearly rectangular (91.79(6)°). All three Al–C bonds are of equal length, regardless of whether they are endo- or exocyclic, or whether the respective ipso-C(pz) orbital is positioned parallel or orthogonal to the vacant Al(pz) orbital [Al(1)–C(11)/C(21)/C(31) = 1.9611(14)/1.9516(14)/1.9606(13) Å]. Within the five-membered AlC4 core, the length of the central C(12)–C(22) bond (1.5024(19) Å) approaches that of a typical C–C single bond (1.54 Å),23 while the benzannulated bonds are significantly shorter [C(11)–C(12)/C(21)–C(22) = 1.4208(18)/1.4134(18) Å]. The other ten C–C bonds within the biphenyl fragment fall within a narrow range of 1.389(2)–1.4041(19) Å, closely matching the corresponding bonds in C6H6 (1.39 Å).24 Taken together, this analysis of bond lengths suggests that the AlFlu moiety of 6 preserves two largely unperturbed Clar sextets within its two C6H3 fragments, with no indication of a delocalized (antiaromatic) π-system, nor any significant Al(1)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(11)/C(21) double-bond character in the AlC4 heterocycle.
C(11)/C(21) double-bond character in the AlC4 heterocycle.
The donor adducts 2·OEt2, 2·py, and 4·py were subjected to X-ray analysis to confirm that (2)2 and (3)2 can indeed serve as precursors of Me-AlFlu and Br-AlFlu, respectively (Fig. S40, S41, S43†). Furthermore, compared to donor-free 6, the C–C bond lengths within the C6H3–C6H3 units of 2·py and 4·py were found to differ by no more than 3σ (and much less for most bonds).25 This observation again suggests that the vacant Al(pz) orbital exerts no significant electron-withdrawing mesomeric effect on the π-electron system.
NMR analysis
At room temperature, 2 gives severely broadened 1H NMR signals, providing limited diagnostic value (Tol-d8; Fig. 4 and S7†). At 70 °C, two sharp resonances are detectable in the aliphatic region of the spectrum (integral ratio 3H:18H); the aromatic region contains one broad feature and two doublets with coupling constants of about 8.2 Hz (Fig. 4 and S6†). At −30 °C, the 1H NMR spectrum of 2 is characterized by two well-resolved sets of signals attributable to two different but closely similar components (Fig. 4 and S8†); the same is true for the 13C{1H} NMR spectrum (Fig. S9†). The proton-integral values of the two sets indicate a minor-to-major component ratio of approximately 0.12:1 (Fig. S8†). Focusing on the major component, the 1H NMR spectrum reveals one singlet at −0.67 ppm (6H), and two additional singlets at 1.47 and 1.28 ppm (2 × 18H), assignable to two equivalent AlMe substituents and two pairs of non-equivalent tBu groups, respectively. In the aromatic region, four doublets (4 × 2H; 2 × 3JH,H = 8.2 Hz, 2 × 4JH,H = 2.2 Hz) and two doublets of doublets (2 × 2H) are observed, indicative of two pairs of non-equivalent C6H3 fragments. In principle, these NMR features would align with both the molecular structure of the cis- and trans-(2)2 dimer (as observed in the solid state). Vice versa, the minor signal set likely arises from trans- or cis-(2)2. At low temperatures, both isomers are present in an (essentially) static mixture, while some dynamic rearrangement equilibrium is established at higher temperatures. This preliminary conclusion raises two questions: (i) Does cis- or trans-(2)2 dominate at low temperatures? (ii) Is the dynamic equilibrium at high temperatures due to monomer/dimer association/dissociation, or is it the result of a rapidly interconverting cis/trans dimeric form of (2)2?
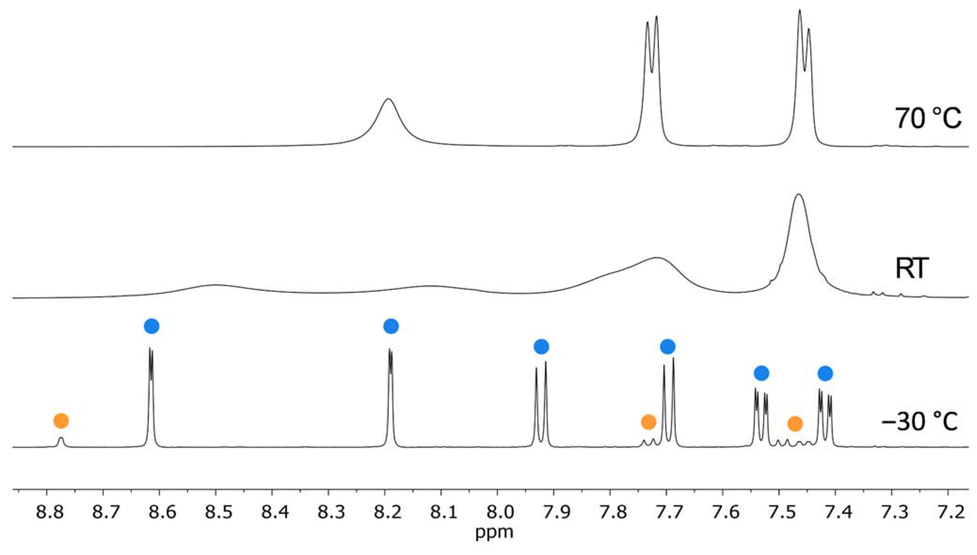 | ||
Fig. 4 Aromatic regions of 1H NMR spectra of (2)2 in Tol-d8 (500.2 MHz). Top: 70 °C. Middle: room temperature. Bottom: −30 °C.  : trans-(2)2. : trans-(2)2.  : cis-(2)2. : cis-(2)2. | ||
To address question (i), quantum-chemical calculations predict that the crystallographically characterized trans-(2)2 is 1.6 kcal mol−1 more favorable in energy than cis-(2)2 (Scheme S1†; experimental value, determined at −30 °C from the proton-integral values of the minor/major component: ΔG° = 0.7 kcal mol−1). Furthermore, the relative proportion of the minor component increases with solvent polarity, consistent with the existing dipole moment of cis-(2)2 (1H NMR spectroscopic control; Table S1 and Fig. S1, S2†). Finally, the computed 13C chemical shift values for cis/trans-(2)2 align more closely with the assumption that the major component is trans-(2)2 rather than vice versa (Tables S9–S11†). It is therefore safe to assume that the major component in an equilibrating cis/trans-(2)2 mixture is the trans isomer.
Regarding question (ii), we note that the computed energy required for cleaving trans-(2)2 into its constituting monomers is 19.4 kcal mol−1 (in CH2Cl2). In contrast, the computed energy barrier of the cis/trans interconversion of (2)2 is only ΔG‡ = 14.8 kcal mol−1, which agrees well with the value of ≈14.5 kcal mol−1 experimentally determined from the coalescence temperature (Tc) in conjunction with the maximum peak separation (Δν) in the slow-exchange limit (CD2Cl2; see the ESI† for full details). The observed NMR features are therefore more convincingly attributed to a dynamic cis/trans equilibrium rather than to a monomer/dimer association/dissociation equilibrium.
In the temperature range of −30 to 70 °C, (3)2 exhibited only extremely broadened signals in the 1H and 13C{1H} NMR spectra, providing no structural information.
The 1H and 13C{1H} NMR spectra of all adducts formed between our R-AlFlus and Lewis bases are in accord with the proposed molecular structures, as is the case for ligand-free 6 (see the ESI† for the fully assigned spectra). In addition to aiding in structure elucidation, 13C{1H} NMR spectroscopy is also a valuable tool for mapping the π-charge density distribution in conjugated systems, as the shielding of a specific C(sp2) atom depends linearly on the corresponding π-electron density at that position.26 Given this background, we compared the 13C chemical shift values of the C atoms constituting the C6H3–C6H3 fragment of 6 with those of the equivalent atoms in the corresponding fragments of the adducts 2·OEt2, 2·thf, 2·py, 4·OEt2, and 4·py. Except for the Al-bonded ipso-C atoms, whose shift differences varied from δ(6)–δ(adduct) = 3.3 to −4.7 ppm without a systematic trend, the Δδ(13C) values for all other structurally analogous C atoms were less than ±1.8 ppm. In other words, we found no evidence of an overall 13C-deshielding effect or π-electron depletion in 6 that could be attributed to a mesomerically electron-withdrawing Al(sp2) center.
13C{1H} NMR spectroscopy on 2·py and 4·py provides a method to evaluate the relative Lewis acidities of free, monomeric Me-AlFlu and Br-AlFlu: in pyridine complexes of main-group elements, stronger acids induce increased shielding of the C-2,6 and deshielding of the C-3,4,5 nuclei of the ligand.27 For 2·py/4·py, our observations consistently indicate that Me-AlFlu is the stronger acid, comparable in this respect to BPh3.28 X-ray crystallography, however, offers a contrasting view: 4·py exhibits a shorter Al–N bond and a more pyramidalized Al center, implying higher Lewis acidity for Br-AlFlu.28 Given the small differences in the key NMR and structural parameters between 2·py and 4·py, these conflicting observations highlight the limitation of relying on a single method to determine Lewis acidity, emphasizing the need for complementary approaches.
Conclusions
We synthesized the donor-free 9-aluminafluorene Me-AlFlu (2), which was characterized as its dimer (2)2 through X-ray crystallography and VT NMR spectroscopy (Tol-d8). The key to this success was the highly selective reaction between the 9,9-dimethyl-9-stannafluorene 1 and AlMe3,17,18 which proceeds in non-donor solvents and releases volatile SnMe4 as the sole by-product. Unlike the bulky tert-butyl group in tBu-AlFlu,16 the sterically less demanding methyl substituent in Me-AlFlu allows relatively unhindered access to the electrophilic Al center, as demonstrated by the straightforward formation of various base adducts 2·L (L = OEt2, thf, py). In terms of an umpolung of the Al center, exploring the reduction of 2 on a preparative scale could be promising (a non-benzannulated alumole has previously been reduced to its corresponding dianion).10 The resulting product, [2]2−, could potentially serve as an Al-centered nucleophile – analogous to the [H-BFlu]2− dianion, which is a valuable B-centered nucleophile.3,6,29Treatment of (2)2 with AlBr3 furnishes the 2,2′-(Br2Al)2-1,1′-biphenyl (3)2. Although this ring-opened product no longer retains the AlFlu motif, it rearranges back to afford Br-AlFlu adducts, such as 4·L (L = OEt2, py), in the presence of Lewis bases. The use of Mes*Li, which provides the extremely bulky, negatively charged Lewis base [Mes*]−, grants unprecedented access to three-coordinate, monomeric aluminafluorenes, specifically Mes*-AlFlu (6), via LiBr elimination. This reaction proves the utility of (3)2 as a synthetic equivalent for the still-elusive free Br-AlFlu. A comparison of characteristic structural and NMR features of 6 with those of 2·L/4·L reveals that the three-coordinate Al center exerts only a negligible π-electron withdrawing effect and does not mediate significant π-electron delocalization.
Author contributions
P. L. L. performed the experimental studies and characterized all new compounds. P. L. L. and J. G. performed the quantum-chemical calculations. A. V. performed the X-ray crystal structure analyses of all compounds. H.-W. L. and M. W. supervised the project. The manuscript was written by P. L. L. and M. W. and edited by all co-authors.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Gabriele Sentis for performing all VT NMR measurements. We thank the center for scientific computing (CSC) Frankfurt and the University of Kent for providing HPC resources that contributed to the computational investigation of this work.References
- (a) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds, Chem. Rev., 2022, 122, 565–788 CrossRef CAS; (b) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications, Chem. Rev., 2017, 117, 3479–3716 CrossRef; (c) L. Ji, S. Griesbeck and T. B. Marder, Recent developments in and perspectives on three-coordinate boron materials: a bright future, Chem. Sci., 2017, 8, 846–863 RSC.
- (a) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, 9-Borafluorenes: Synthesis, Properties, and Reactivity, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed; (b) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Doping Polycyclic Aromatics with Boron for Superior Performance in Materials Science and Catalysis, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- H. Budy, J. Gilmer, T. Trageser and M. Wagner, Anionic Organoboranes: Delicate Flowers Worth Caring for, Eur. J. Inorg. Chem., 2020, 4148–4162 CrossRef CAS.
- (a) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, Dibenzoborole-Containing π-Electron Systems: Remarkable Fluorescence Change Based on the “On/Off” Control of the pπ−π* Conjugation, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS; (b) A. Wakamiya, K. Mishima, K. Ekawa and S. Yamaguchi, Kinetically stabilized dibenzoborole as an electron-accepting building unit, Chem. Commun., 2008, 579–581 RSC; (c) A. Iida and S. Yamaguchi, Thiophene-Fused Ladder Boroles with High Antiaromaticity, J. Am. Chem. Soc., 2011, 133, 6952–6955 CrossRef CAS PubMed; (d) A. Iida, A. Sekioka and S. Yamaguchi, Heteroarene-fused boroles: what governs the antiaromaticity and Lewis acidity of the borole skeleton?, Chem. Sci., 2012, 3, 1461–1466 RSC.
- (a) A. Hübner, M. Bolte, H.-W. Lerner and M. Wagner, Extensive Structural Rearrangements upon Reduction of 9H-9-Borafluorene, Angew. Chem., Int. Ed., 2014, 53, 10408–10411 CrossRef; (b) A. Hübner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, A Preorganized Ditopic Borane as Highly Efficient One- or Two-Electron Trap, J. Am. Chem. Soc., 2015, 137, 3705–3714 CrossRef; (c) K. E. Wentz, A. Molino, S. L. Weisflog, A. Kaur, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard, Stabilization of the Elusive 9-Carbene-9-Borafluorene Monoanion, Angew. Chem., Int. Ed., 2021, 60, 13065–13072 CrossRef CAS PubMed; (d) H. Budy, T. Kaese, M. Bolte, H.-W. Lerner and M. Wagner, A Chemiluminescent Tetraaryl Diborane(4) Tetraanion, Angew. Chem., Int. Ed., 2021, 60, 19397–19405 CrossRef CAS.
- J. Gilmer, H. Budy, T. Kaese, M. Bolte, H.-W. Lerner and M. Wagner, The 9H-9-Borafluorene Dianion: A Surrogate for Elusive Diarylboryl Anion Nucleophiles, Angew. Chem., Int. Ed., 2020, 59, 5621–5625 CrossRef CAS.
- (a) P. A. Chase, W. E. Piers and B. O. Patrick, New Fluorinated 9-Borafluorene Lewis Acids, J. Am. Chem. Soc., 2000, 122, 12911–12912 CrossRef CAS; (b) A. Hübner, A. M. Diehl, M. Bolte, H.-W. Lerner and M. Wagner, High-Temperature Reactivity of the Strongly Electrophilic Pristine 9H-9-Borafluorene, Organometallics, 2013, 32, 6827–6833 CrossRef; (c) J. Gilmer, T. Trageser, L. Čaić, A. Virovets, M. Bolte, H.-W. Lerner, F. Fantuzzi and M. Wagner, Catalyst-free diboration and silaboration of alkenes and alkynes using bis(9-heterofluorenyl)s, Chem. Sci., 2023, 14, 4589–4596 RSC; (d) J. Gilmer, M. Bolte, A. Virovets, H.-W. Lerner, F. Fantuzzi and M. Wagner, A Hydride-Substituted Homoleptic Silylborate: How Similar is it to its Diborane(6)-Dianion Isostere?, Chem. – Eur. J., 2023, 29, e202203119 CrossRef CAS PubMed.
- (a) A. Hübner, Z.-W. Qu, U. Englert, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Main-Chain Boron-Containing Oligophenylenes via Ring-Opening Polymerization of 9-H-9-Borafluorene, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef; (b) Y. Shoji, N. Tanaka, S. Muranaka, N. Shigeno, H. Sugiyama, K. Takenouchi, F. Hajjaj and T. Fukushima, Boron-mediated sequential alkyne insertion and C–C coupling reactions affording extended π-conjugated molecules, Nat. Commun., 2016, 7, 12704 CrossRef; (c) K. R. Bluer, L. E. Laperriere, A. Pujol, S. Yruegas, V. A. K. Adiraju and C. D. Martin, Coordination and Ring Expansion of 1,2-Dipolar Molecules with 9-Phenyl-9-borafluorene, Organometallics, 2018, 37, 2917–2927 CrossRef CAS; (d) Y. Murata, K. Matsunagi, J. Kashida, Y. Shoji, C. Özen, S. Maeda and T. Fukushima, Observation of Borane–Olefin Proximity Interaction Governing the Structure and Reactivity of Boron-Containing Macrocycles, Angew. Chem., Int. Ed., 2021, 60, 14630–14635 CrossRef CAS; (e) T. Bischof, X. Guo, I. Krummenacher, L. Beßler, Z. Lin, M. Finze and H. Braunschweig, Alkene insertion reactivity of a o-carboranyl-substituted 9-borafluorene, Chem. Sci., 2022, 13, 7492–7497 RSC.
- For selected publications on alumoles, the non-benzannulated congeners of AlFlus, see: (a) H. Hoberg and R. Krause-Göing, Darstellung und Eigenschaften von Pentaphenylaluminacyclopentadien, J. Organomet. Chem., 1977, 127, C29–C31 CrossRef CAS; (b) T. Wasano, T. Agou, T. Sasamori and N. Tokitoh, Synthesis, structure and reactivity of a 1-bromoalumole, Chem. Commun., 2014, 50, 8148–8150 RSC; (c) T. Agou, T. Wasano, T. Sasamori and N. Tokitoh, Syntheses and Structures of Stable 1-Aminoalumole Derivatives, Organometallics, 2014, 33, 6963–6966 CrossRef CAS; (d) Y. Zhang, J. Wei, W.-X. Zhang and Z. Xi, Lithium Aluminate Complexes and Alumoles from 1,4-Dilithio-1,3-Butadienes and AlEt2Cl, Inorg. Chem., 2015, 54, 10695–10700 CrossRef CAS PubMed; (e) N. Tokitoh, T. Agou, T. Wasano and T. Sasamori, Synthesis and properties of stable alumoles, Phosphorus, Sulfur, Silicon Relat. Elem., 2016, 191, 584–587 CrossRef CAS; (f) V. Y. Lee, H. Sugasawa, O. A. Gapurenko, R. M. Minyaev, V. I. Minkin, H. Gornitzka and A. Sekiguchi, 1-Chloroalumole, Organometallics, 2022, 41, 467–471 CrossRef CAS.
- T. Agou, T. Wasano, P. Jin, S. Nagase and N. Tokitoh, Syntheses and Structures of an “Alumole” and Its Dianion, Angew. Chem., Int. Ed., 2013, 52, 10031–10034 CrossRef CAS.
- (a) T. D. Coyle, S. L. Stafford and F. G. A. Stone, Chemical and Spectroscopic Evidence for the Occurrence of π-Character in Carbon–Boron Bonds, J. Chem. Soc., 1961, 3103–3108 RSC; (b) A. Foord, B. Beagley, W. Reade and I. A. Steer, A gas-phase electron-diffraction study of trivinylborane, J. Mol. Struct., 1975, 24, 131–137 CrossRef CAS; (c) Y. Yamamoto and I. Moritani, Carbon-13 Nuclear Magnetic Resonance Studies of Organoboranes. The Relative Importance of Mesomeric B–C π-Bonding Forms in Alkenyl- and Alkynylboranes, J. Org. Chem., 1975, 40, 3434–3437 CrossRef CAS.
- J. J. Eisch and W. C. Kaska, The Novel Synthesis of Aluminoles by the Metalative Cyclization of Unsaturated Organoaluminum Compounds, J. Am. Chem. Soc., 1962, 84, 1501–1502 CrossRef CAS.
- J. J. Eisch and W. C. Kaska, The Synthesis of Aluminoles via the Addition and Cyclization Reactions of Arylaluminum Compounds, J. Am. Chem. Soc., 1966, 88, 2976–2983 CrossRef CAS.
- T. Matsumoto, H. Takamine, K. Tanaka and Y. Chujo, Synthesis and Characterization of Heterofluorenes with Five-coordinated Group 13 Elements, Chem. Lett., 2015, 44, 1658–1660 CrossRef CAS.
- T. Matsumoto, K. Tanaka, K. Tanaka and Y. Chujo, Synthesis and characterization of heterofluorenes containing four-coordinated group 13 elements: theoretical and experimental analyses and comparison of structures, optical properties and electronic states, Dalton Trans., 2015, 44, 8697–8707 RSC.
- R. Drescher, L. Wüst, C. Mihm, I. Krummenacher, A. Hofmann, J. Goettel and H. Braunschweig, Synthesis, structure and insertion reactivity of Lewis acidic 9-aluminafluorenes, Dalton Trans., 2021, 50, 10400–10404 RSC.
- We are aware that the compounds AlMe3 and AlBr3 are monomeric neither in solution nor in the solid state. However, for simplicity, the monomeric forms were used in calculating the quantities employed.
- P. L. Lückert, J. Gilmer, A. Virovets, H.-W. Lerner and M. Wagner, Donor-free 9,10-dihydro-9,10-dialuminaanthracenes, Chem. Sci. 10.1039/D4SC06940D.
- SnMe4 can in principle be recovered and used to recycle the Me2SnCl2 required to synthesize 1.
- Two polymorphs of (Me-AlFlu)2 have been structurally characterized by X-ray diffraction: The denser α-form crystallizes in the triclinic space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one unique molecule at a general position and a second molecule at an inversion center (Z = 3). The β-form, which is discussed in the main text, crystallizes in the monoclinic space group P21/n with one crystallographically unique molecule at an inversion center. The key structural parameters of all the molecules are essentially identical (Fig. S39†); full details are available in the ESI.†.
with one unique molecule at a general position and a second molecule at an inversion center (Z = 3). The β-form, which is discussed in the main text, crystallizes in the monoclinic space group P21/n with one crystallographically unique molecule at an inversion center. The key structural parameters of all the molecules are essentially identical (Fig. S39†); full details are available in the ESI.†. - A. Hübner, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Confirmation of an Early Postulate: B–C–B Two-Electron–Three-Center Bonding in Organo(hydro)boranes, Angew. Chem., Int. Ed., 2012, 51, 12514–12518 CrossRef.
- C. Elschenbroich, Organometallics, WILEY-VCH, Weinheim, Germany, 2006 Search PubMed.
- M. A. Fox and J. K. Whitesell, Organic Chemistry, Jones and Bartlett, Sudbury, Massachusetts, USA, 2003, p. 77 Search PubMed.
- E. D. Glendening, R. Faust, A. Streitwieser, K. P. C. Vollhardt and F. Weinhold, The Role of Delocalization in Benzene, J. Am. Chem. Soc., 1993, 115, 10952–10957 CrossRef CAS.
- The molecular structure of 2·OEt2 was excluded from this comparison due to its significantly larger error margins.
- (a) M. Karplus and J. A. Pople, Theory of Carbon NMR Chemical Shifts in Conjugated Molecules, J. Chem. Phys., 1963, 38, 2803–2807 CrossRef CAS; (b) R. H. Levin and J. D. Roberts, Nuclear magnetic resonance spectroscopy. Ring-current effects upon carbon-13 chemical shifts, Tetrahedron Lett., 1973, 24, 135–138 CrossRef; (c) H. Günther, H. Schmickler, H. Königshofen, K. Recker and E. Vogel, Does a “Ring Current Effect” Exist for 13C-Nuclear Magnetic Resonance? A Study of Bridged Annulenes, Angew. Chem., Int. Ed., 1973, 12, 243–245 CrossRef; (d) D. G. Farnum, Charge Density-NMR Chemical Shift Correlations in Organic Ions, Adv. Phys. Org. Chem., 1975, 11, 123–175 CrossRef CAS.
- (a) A. Schnurr, M. Bolte, H.-W. Lerner and M. Wagner, Cyclic Phosphonium Bis(fluoroaryl)boranes – Trends in Lewis Acidities and Application in Diels–Alder Catalysis, Eur. J. Inorg. Chem., 2012, 112–120 CrossRef CAS; (b) M. Henkelmann, A. Omlor, M. Bolte, V. Schünemann, H.-W. Lerner, J. Noga, P. Hrobárik and M. Wagner, A free boratriptycene-type Lewis superacid, Chem. Sci., 2022, 13, 1608–1617 RSC.
- 13C{1H} NMR data (the numbering scheme used here differs from that used in the ESI†): 147.3 (C-2,6), 125.0 (C-3,5), 139.9 (C-4) for 2·py; 149.1 (C-2,6), 124.3 (C-3,5), 137.8 (C-4) for 4·py; 148.3 (C-2,6), 125.6 (C-3,5), 141.0 (C-4) for BPh3·py. X-ray analysis: Al–N = 1.9969(16)/2.0160(17) Å, N–Al–COG = 105.44/112.06° for two crystallographically independent molecules of 2·py. Al–N = 1.964(2) Å, N–Al–COG = 122.26° for 4·py. COG: centroid of the AlC4 heterocycle.
- H. Budy, S. E. Prey, C. D. Buch, M. Bolte, H.-W. Lerner and M. Wagner, Nucleophilic borylation of fluorobenzenes with reduced arylboranes, Chem. Commun., 2022, 58, 254–257 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, X-ray crystallographic data and computational details. CCDC 2394332–2394341. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03148b |
| This journal is © The Royal Society of Chemistry 2025 |