
Green synthesis of poly ε-caprolactone using a metal-free catalyst via non-covalent interactions†
Shweta
Sagar
 a,
Priyanku
Nath
a,
Shiva Lall
Sunar
a,
Aranya
Ray
a,
Mridula
Choudhary
a,
Alok
Sarkar
*b,
Saurabh K.
Singh
*a and
Tarun K.
Panda
*a
a,
Priyanku
Nath
a,
Shiva Lall
Sunar
a,
Aranya
Ray
a,
Mridula
Choudhary
a,
Alok
Sarkar
*b,
Saurabh K.
Singh
*a and
Tarun K.
Panda
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi – 502 284, Sangareddy, Telangana, India. E-mail: tpanda@chy.iith.ac.in; sksingh@chy.iith.ac.in
bMomentive Performance Materials Pvt. Ltd, Survey No. 09, Hosur Road, Electronic City (west), Bangalore-560100, India. E-mail: alok.sarkar@momentive.com
First published on 19th November 2024
Abstract
The ring-opening polymerization (ROP) of ε-caprolactone (CL) catalyzed by a metal-free initiator N,N′-dibutyl-N,N,N′,N′-tetramethylethane1,2-diammonium bromide [nBuMe2NCH2CH2NnBuMe2]Br2 (DBTMEDA)Br2 has been investigated. The catalyst (DBTMEDA)Br2 promotes polymerization under mild conditions without any external initiator. Polymerization was demonstrated in a controlled and living manner, producing PCLs with a precisely controlled molecular weight of up to 50 kDa with narrow polydispersity. Density Functional Theory (DFT) calculations indicated the involvement of a C–H⋯O type non-covalent interaction between DBTMEDA cations and the carbonyl group of ε-CL in the monomer activation step. Remarkably, DBTMEDA can also be easily recovered and reused for up to six consecutive cycles without an appreciable decrease in catalytic activity.
Introduction
Green chemistry aims to increase the usage of monomers from natural resources and implement safer, more efficient, and eco-friendly chemical processes. Non-covalent interactions, even though very weak in nature, play a dominant role in pharmaceutical design, supramolecular chemistry, molecular biology, sensing applications, material and crystal engineering, and a host of other fields in the chemical sciences.1–6 Immense efforts have been put forth by theoretical as well experimental scientists to quantitatively establish the role of non-covalent interactions, which include hydrogen bonding, electrostatic effects, π–π, cation–π, and hydrophobic interactions, and van der Waals forces.7–12 Notable contributions of these forces to rate accelerations and stereoselectivities have also been exemplified in several enzymatic catalyses.13,14 In a similar direction, Zhao and coworkers have leveraged the strong hydrogen-bonding ability of a commercially available fluorinated alcohol to perform efficient ROP of α-amino acid N-carboxyanhydrides with no cocatalyst.15Poly(ε-caprolactone) (PCL) is a high-demand synthetic polyester with attractive features such as excellent biodegradability, biocompatibility, good thermal stability, and ease of manufacture.16 Since the first synthesis of PCL by the Carothers group in the early 1930s, various PCL polymers have been commercialized and applied in many fields today, ranging from the biomedical and pharmaceutical fields to the agricultural field. Recently, with the advancement of tissue engineering, there has been a strong surge in demand for PCL, with the total estimated volume expected to reach 909.81 billion USD by 2029.17 Compared to the traditional polycondensation methods, metal complex-induced ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) is more effective and provides better controllability for the production.18
Numerous well-defined discrete metal complexes have been developed for the ROP of ε-CL.19–26 However, the presence of traces of metal residues in commercial products is one of the challenges in using these polymers in biomedical, drug delivery, tissue engineering, and micro-electronics applications.27–30 Both organocatalysts and enzymes have been intensively investigated for the ROP of cyclic esters as alternative metal-free catalysts.31–35 Enzyme catalysts demonstrate high selectivity and promising catalytic activity under mild reaction conditions.36 However, enzyme-catalyzed industrial processes still face the challenge of improving the reactivity of enzymes in non-aqueous media. To overcome these problems, difunctional systems combining an H-bond donor compound with an organic base (H-bond acceptor) have been designed.37–41 In this approach, the ester moiety of the cyclic monomer is activated by the H-bond donor. Concomitantly, the base accepts an H-bond from an externally added alcohol to initiate the polymerization. Besides the desired H-bond association with the monomer and initiating alcohol, the H-bond donor and acceptor compounds can interact, which may represent a limitation of a difunctional H-bond donor catalyst. Such interactions lowering the amount of the active catalyst in solution or even completely quenching the catalytic activity have been reported.42,43
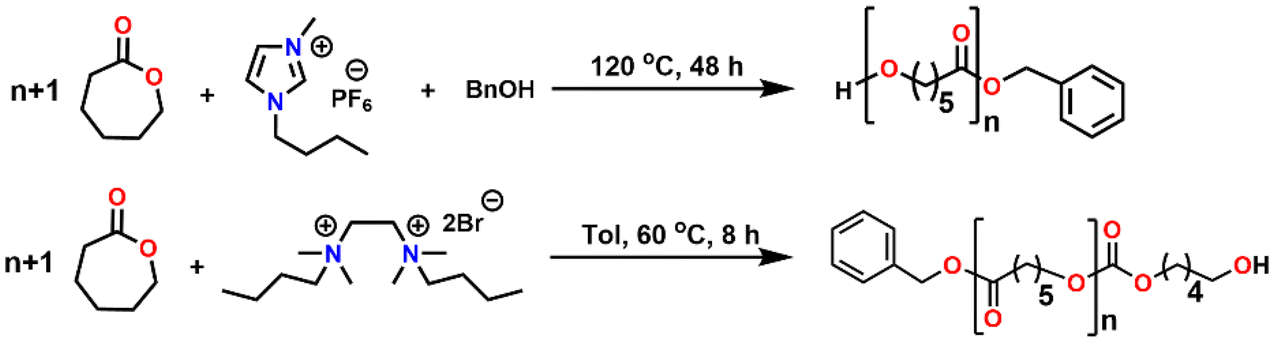
Ionic liquids (ILs) as organocatalysts are one of the hot topics of chemistry in catalysis research. ILs are liquid compounds of cations and anions that display ionic-covalent crystalline structures.44 Significant features of ILs that make them suitable as catalysts are their flexibility of adjusting acidity/basicity by simple cation/anion exchange reactions.45–48 Despite the widespread utility of ILs in various inter-disciplinary areas of fields, their application in ROP of ε-CL remained limited to only specific aromatic imidazolium-salt containing alkyl chains such as 1-butyl-3-methylimidazolium hexafluorophosphate [Bmim][PF6], 1-methyl-3-methyl-imidazolium hexafluorophosphate [Memim][PF6] and bis(N-(N′-butylimidazolium)alkane) (Fig. 1).49–51 Also, these ILs are of high cost and environmentally toxic and have high purity requirements.52–54 High temperature, slow reaction, and requirement of an external initiator remain the critical disadvantages of these organocatalysts compared to conventional metal catalysts.
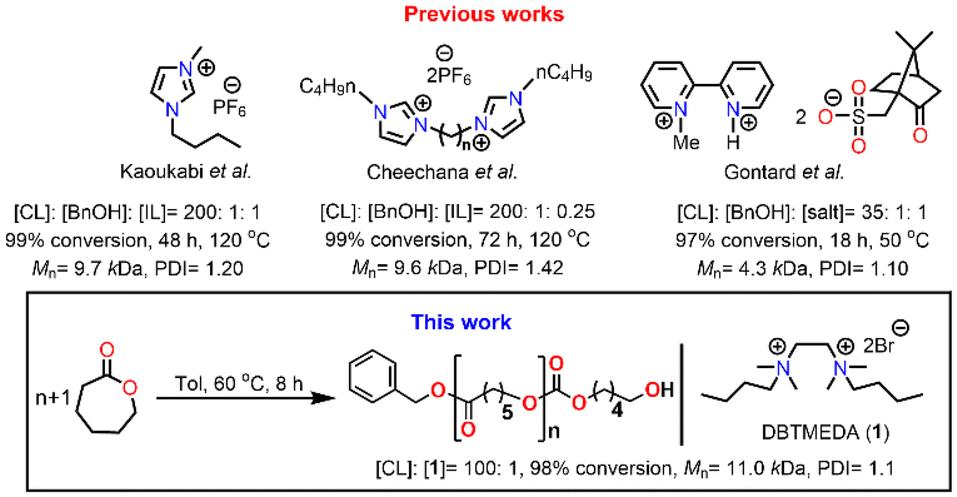 | ||
| Fig. 1 Previously reported organocatalysts and the current work for ROP of ε-CL. | ||
On the other hand, aliphatic quaternary-ammonium-salts containing alkyl chains exhibit stronger ionic interactions because of their localized cationic charge on the ammonium group and enhanced van der Waals interactions among the alkyl chains.55 Therefore, we anticipated that they might enable faster ROP by their stronger ionic interaction with the monomer than their aromatic imidazolium counterparts. As catalysts for ROP, aliphatic quaternary-ammonium-salts can present numerous other advantages, including their green character, simple one-step synthesis from readily available commercial materials, and ease of handling. Furthermore, their characterization and solution properties have been well established,55–57 but to the best of our knowledge, their application as catalysts for the ROP is unknown. Herein, we describe our study of an aliphatic dicationic quaternary ammonium salt, DBTMEDA (1), catalyzing the ROP of ε-CL (Fig. 1). Syntheses of PCL with precise and predictable control over the molecular weight were achieved with >95% conversion within 8 h under mild reaction conditions. The catalyst recovery and reusability studies performed on a representative reaction demonstrated efficient recycling of the catalyst. We also present a model in which a non-covalent interaction, namely the C–H⋯O hydrogen bond, between the carbonyl group of ε-CL and a methylene CH2 group of dicationic ammonium salts triggers monomer activation to initiate the polymerization.
Results and discussion
Synthesis of salt
The salt N,N′-dibutyl-N,N,N′,N′-tetramethylethane-1,2-diammonium bromide (1) was synthesized using a procedure reported in the literature.55Ring-opening polymerisation of ε-CL
The catalytic activity of 1 was first verified for the ROP of 100 equivalents of ε-CL at 60 °C using toluene as the solvent (Table 2). It was intriguing to note that a 37.6% conversion in the first two hours was observed without any external initiator. With this encouraging finding, a first screening was conducted to find the optimal time for the complete conversion without any external initiator. Upon further proceeding with the reaction, 58.8, 79.9, and 98.3% conversions were attained in 4, 6, and 8 h, respectively. After quenching with BnOH, 99% of the polymer product was isolated by precipitating the crude reaction mixture in water. The solvent toluene was recovered from water using a separating funnel. The turnover frequency (TOF) calculated for the catalyst was 12.2 h−1, substantially higher than the value calculated for the same reaction reported using an ionic liquid-based catalyst (Table 1). Even though the highest TOF of 13.3 h−1 could be achieved in the first 6 h, the polymerization was continued for 8 h to ensure >95% monomer conversion. The other green chemistry metrics evaluated for this reaction such as E-factor, atom economy (AE), atom efficiency, carbon efficiency (CE), product mass intensity (PMI), effective mass yield (EMI) and reaction mass efficiency (RME) were also improved significantly compared to the reported IL catalyst (Table 1). Fig. FS10 in the ESI† shows the 1H NMR spectrum of the prepared PCL showing the typical peaks as reported in the literature.25 The chemical shifts of 4.07, 2.29, 1.64, and 1.34 ppm are due to PCL, and the peak due to the methylene protons adjacent to the ω-chain end of the hydroxyl group is observed at 3.65 ppm. The chemical shifts of 7.32 and 5.10 ppm are due to the aromatic methylene protons of the BnO− moiety attached to an ester linkage, respectively. Additionally, the number-average molecular weight of the polymer estimated from 1H NMR agreed with that calculated from the feed monomer to initiator ratio and monomer conversions (Table 2, entry 4).|
|
||||
|---|---|---|---|---|
| Green chemistry metric | Acronym formula | Ref. | Previous approach | Current work |
| E-factor |
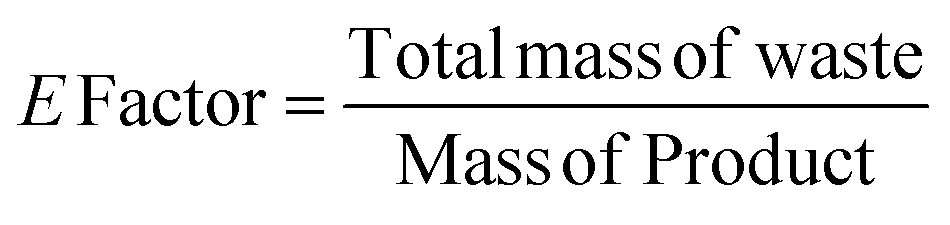
|
67 and 68 | 0.5 | 0.01 |
| Atom economy (AE) |
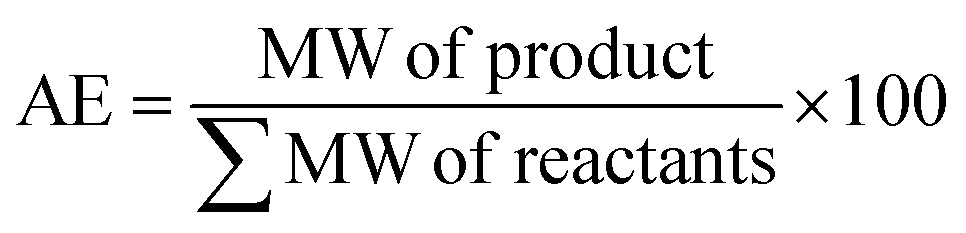
|
67 and 68 | 98% | 100% |
| Atom efficiency (AE) |
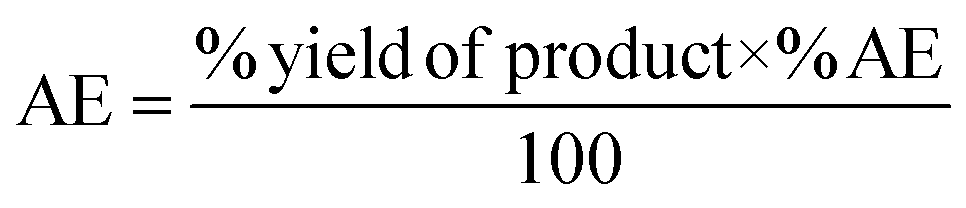
|
67 and 68 | 98% | 99% |
| Carbon efficiency (CE) |
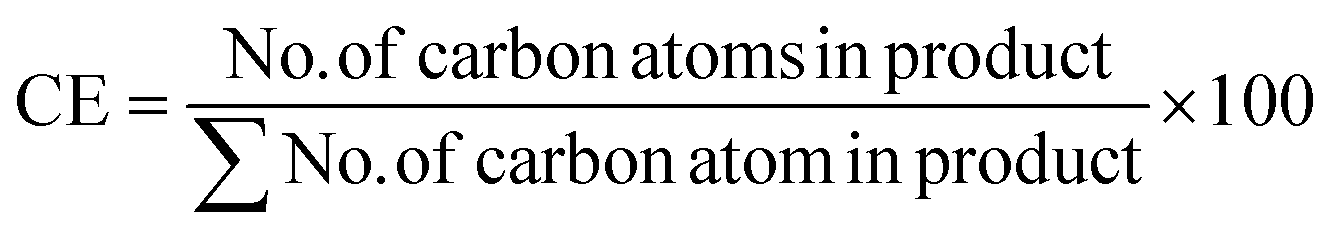
|
67 and 68 | 100% | 100% |
| Product mass intensity (PMI) |
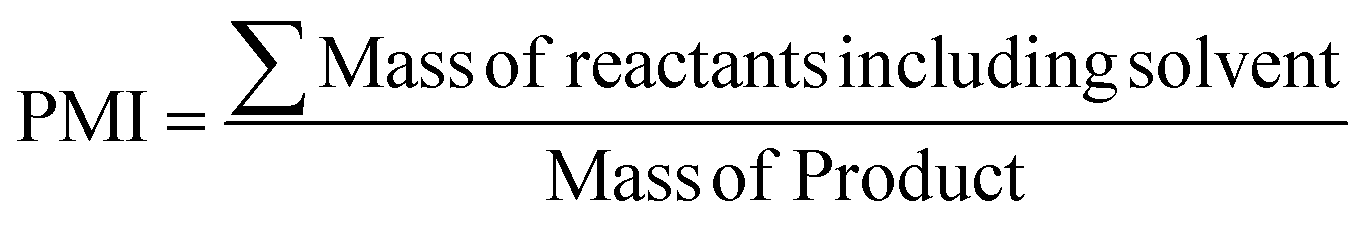
|
67 and 68 | 1.5 | 1.02 |
| Reaction mass efficiency (RME) |
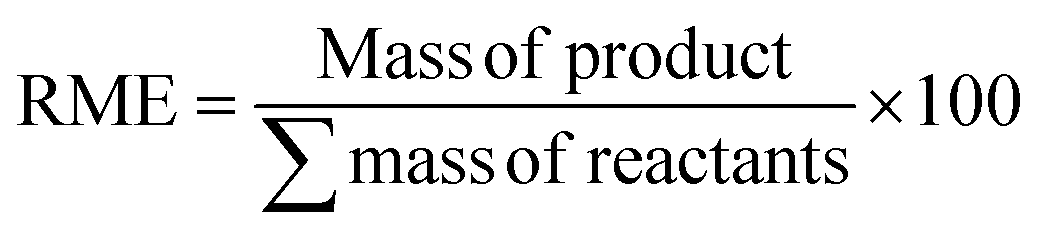
|
67 and 68 | 66% | 98.9% |
|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M :M |
Cat (mol L−1) | Monomer (mol L−1) | Time (h) | Temp. (°C) | Conv.b | Yieldc | TONd | TOF (h−1)e |
M
n theof (kDa) |
M n exp (kDa) | PDI |
| a In toluene, [catalyst] = 0.026 mmol. b Conversions were determined by crude mixture 1H NMR spectroscopy. c Yield = (weight of the polymer obtained/weight of the monomer used) × 100. d TON = [(mol epoxide/mol catalyst) × isolated polymer yield (%)]. e TOF = TON/time (h). f M n (theo) = molecular weight of chain-end + 114 gmol−1 × (salt: M) × conversion. g In THF (2 mg mL−1) and molecular weights were determined by GPC-LLS (flow rate = 0.5 mL min−1). Universal calibration was carried out with polystyrene standards, laser light scattering detector data, and a concentration detector. Each experiment is duplicated to ensure precision. h In THF as the solvent. | ||||||||||||
| 1 | 1:100 |
0.006 | 0.64 | 2 | 60 | 37.6 | 21 | 20.7 | 10.3 | 4.4 | 3.1 | 1.6 |
| 2 | 1:100 |
0.006 | 0.64 | 4 | 60 | 58.8 | 48 | 47.3 | 11.8 | 6.8 | 5.9 | 1.4 |
| 3 | 1:100 |
0.006 | 0.64 | 6 | 60 | 79.9 | 81 | 79.8 | 13.3 | 9.2 | 8.8 | 1.3 |
| 4 | 1:100 |
0.006 | 0.64 | 8 | 60 | 98.3 | 99 | 97.5 | 12.2 | 11.3 | 11.0 | 1.1 |
| 5 | 1:200 |
0.006 | 1.28 | 8 | 60 | 98.1 | 99 | 198 | 24.8 | 22.5 | 21.9 | 1.1 |
| 6 | 1:300 |
0.006 | 1.95 | 8 | 60 | 97.8 | 98 | 294 | 36.8 | 33.6 | 33.1 | 1.2 |
| 7 | 1:400 |
0.006 | 2.6 | 8 | 60 | 96.7 | 97 | 388 | 48.5 | 44.3 | 43.5 | 1.2 |
| 8 | 1:500 |
0.006 | 3.25 | 8 | 60 | 95.5 | 96 | 480 | 60 | 54.6 | 51.6 | 1.4 |
| 9 | 0:100 |
0 | 0.64 | 8 | 60 | 0 | 0 | — | — | — | ||
| 10h | 1:100 |
0.006 | 0.64 | 8 | 60 | 22.5 | 14 | 13.7 | 1.7 | Nd | nd | nd |
The monomer-to-catalyst ratio plays a crucial role in the ROP of cyclic esters. Next, we carried out the polymerization by successively increasing the monomer loading to 200, 300, 400, and 500 equivalents at a given catalyst concentration. High conversions of the monomer were achieved for all the reactions to give PCLs with precisely controlled molecular weights. Effective utilization of the catalytic species at the molecular level was confirmed for all reactions by their high turnover number (TON = 97 to 488). The GPC analysis confirmed a narrow molecular weight distribution (PDI = 1.1 to 1.4), indicating side reactions; for example, the transesterification, which became more predominant at high temperatures and low monomer concentrations, is restricted. As shown in Fig. FS9 in the ESI,† the average molecular weight (Mn) obtained for the polymers showed a linear dependency on the monomer-to-catalyst ratio, indicating the single-site, living behavior of the catalytic species. The influence of solvent on the polymerization was also examined (Table 2). Good control of the polymerization was observed in toluene. Polymerization in the presence of THF as a solvent was extremely sluggish, where a conversion of up to 22.5 percent could be obtained in 8 hours (Table 2, entry 10). The competitive interactions of THF with the catalyst molecule could cause a sluggish reaction in the presence of the THF solvent.
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) have often been helpful to gain valuable structural insights into PCL and determine its purity and stability. The DSC thermogram of a representative polymer (Table 2, entry 4) showed the glass transition temperature (Tg) to be −58.06 °C and the melting temperature (Tm) to be 58.01 °C, which is typical behavior of conventional pure PCL (Fig. 2). Under a nitrogen atmosphere, the TGA curve of the PCL sample displayed a one-step degradation, a single weight loss phenomenon, corresponding to pyrolysis of pure commercial grade PCL.58 The initiation of degradation was found to occur at 365.5 °C, and the final degradation temperature (Tf) was 428.5 °C with a maximum decomposition peak at 410.2 °C (Tmax) (Fig. 2).
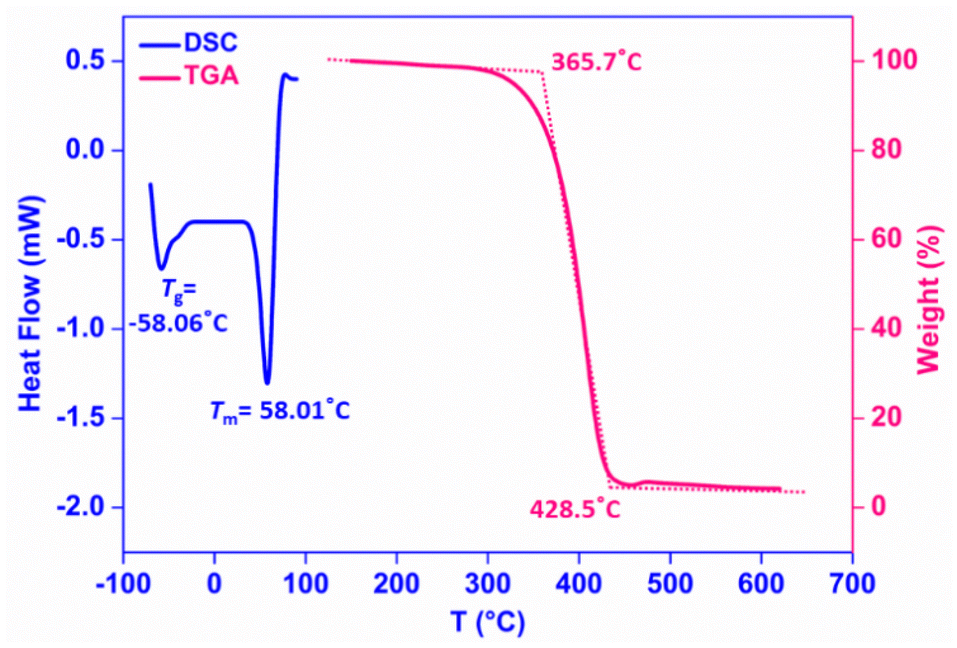 | ||
| Fig. 2 DSC and TGA curves of PCL samples (entry 4, Table 1). | ||
Having established the catalytic activity of 1 in ROP of ε-CL, we further investigated the polymerization behavior to understand the kinetics. The kinetic plots of [CL]0/[CL] versus the catalyst (1) were found to be linear, indicating first-order dependence on the ε-CL concentration (Fig. FS3 in the ESI†). The first-order dependence of the polymerization reactions substantiates the presence of only one initiator and, consequently, follows the second-order rate law, which can be expressed as rate = −d[CL]/dt = kp[cat]1[CL]1. The activation parameters for the ROP of ε-CL in CDCl3 were found to be ΔH‡ = 10.8 kJ mol−1 K, ΔS‡ = −0.14 kJ (mol K)−1, and ΔEa‡ = 13.5 kJ mol−1. The ΔG‡ value for the ROP of ε-CL catalyzed by 1 at 60 °C was calculated to be 57.4 kJ mol−1.
Therefore, the rate expression can be summarized as
| −d[CL]/dt = kapp[Salt]1[CL]1 | (1) |
| −d[CL]/dt = kobs[CL]1 | (2) |
The mechanism for cyclic ester ROP of various organocatalysts has been extensively investigated.59–66 Our NMR results demonstrate the presence of the benzyl ester end group in the polymer chain when benzyl alcohol is used as a quenching agent, indicating that the ring-opening of ε-CL takes place through cleavage of the C(acyl)–O bond (Fig. FS10 in ESI†). From the 1H NMR spectra of a 1:10 catalyst to CL mixture, the resonance signals of CH3 (1), CH2 (2), and CH2 (3) were noted to be deshielded to slightly higher chemical shift positions. In contrast, the other two signals corresponding to CH3 (6) and CH2 (5) remained unchanged (Fig. 3).
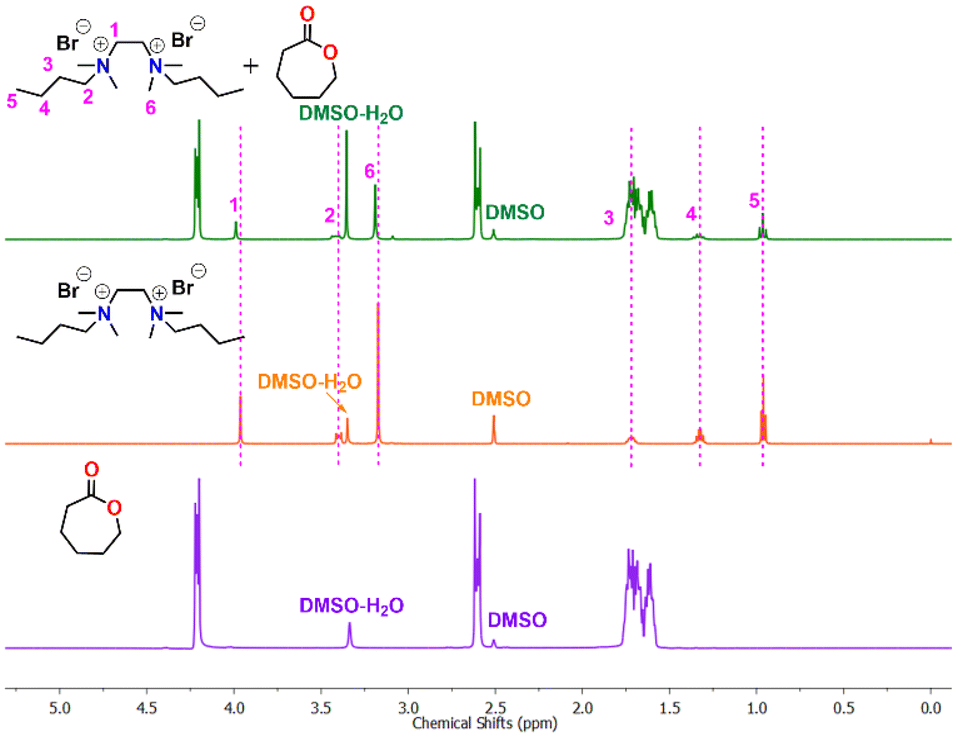 | ||
| Fig. 3 1H NMR spectra of complex formed in situ in the mixture of salt 1 and ε-CL in DMSO. | ||
The observed deshielding of these specific sets of CH3 and CH2 protons is significant and caused by C–H⋯O hydrogen bonding with the carbonyl group of the ε-CL monomer. The chemical shift of CH2 (4) could not be determined due to its overlap with other CH signals from active polymer chains. Inspired by the above observation, we anticipated that the methyl/methylene groups directly attached to quaternary ammonium groups are involved in CH3⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C type interactions with the carbonyl group of the ε-CL monomer. The formation of such non-covalent interactions in a similar system containing quaternary ammonium salt and a cyclic ester is also reported in the literature.51 These non-covalent interactions increase the electrophilicity of the ε-CL monomer and it subsequently becomes susceptible to nucleophilic attack. The bromide anion, which remains as an ion pair close to the quaternary ammonium group, is considered to initiate the polymerization by the nucleophilic attack on the carbonyl-carbon of ε-CL followed by acyl–oxygen bond scission. This leads to the formation of an acyl bromide at one end of the polymer chain and active oxyanion species at the other end, which reacts with the second monomer for further propagation. Upon adding a quenching agent such as BnOH, the ester is formed as one of the end groups by nucleophilic displacement of the bromide anion from the acyl bromide. Hence, the proposed activation mechanism for ε-CL ring opening catalyzed by (DBTMEDA)Br2 is shown in Fig. 4.
C type interactions with the carbonyl group of the ε-CL monomer. The formation of such non-covalent interactions in a similar system containing quaternary ammonium salt and a cyclic ester is also reported in the literature.51 These non-covalent interactions increase the electrophilicity of the ε-CL monomer and it subsequently becomes susceptible to nucleophilic attack. The bromide anion, which remains as an ion pair close to the quaternary ammonium group, is considered to initiate the polymerization by the nucleophilic attack on the carbonyl-carbon of ε-CL followed by acyl–oxygen bond scission. This leads to the formation of an acyl bromide at one end of the polymer chain and active oxyanion species at the other end, which reacts with the second monomer for further propagation. Upon adding a quenching agent such as BnOH, the ester is formed as one of the end groups by nucleophilic displacement of the bromide anion from the acyl bromide. Hence, the proposed activation mechanism for ε-CL ring opening catalyzed by (DBTMEDA)Br2 is shown in Fig. 4.
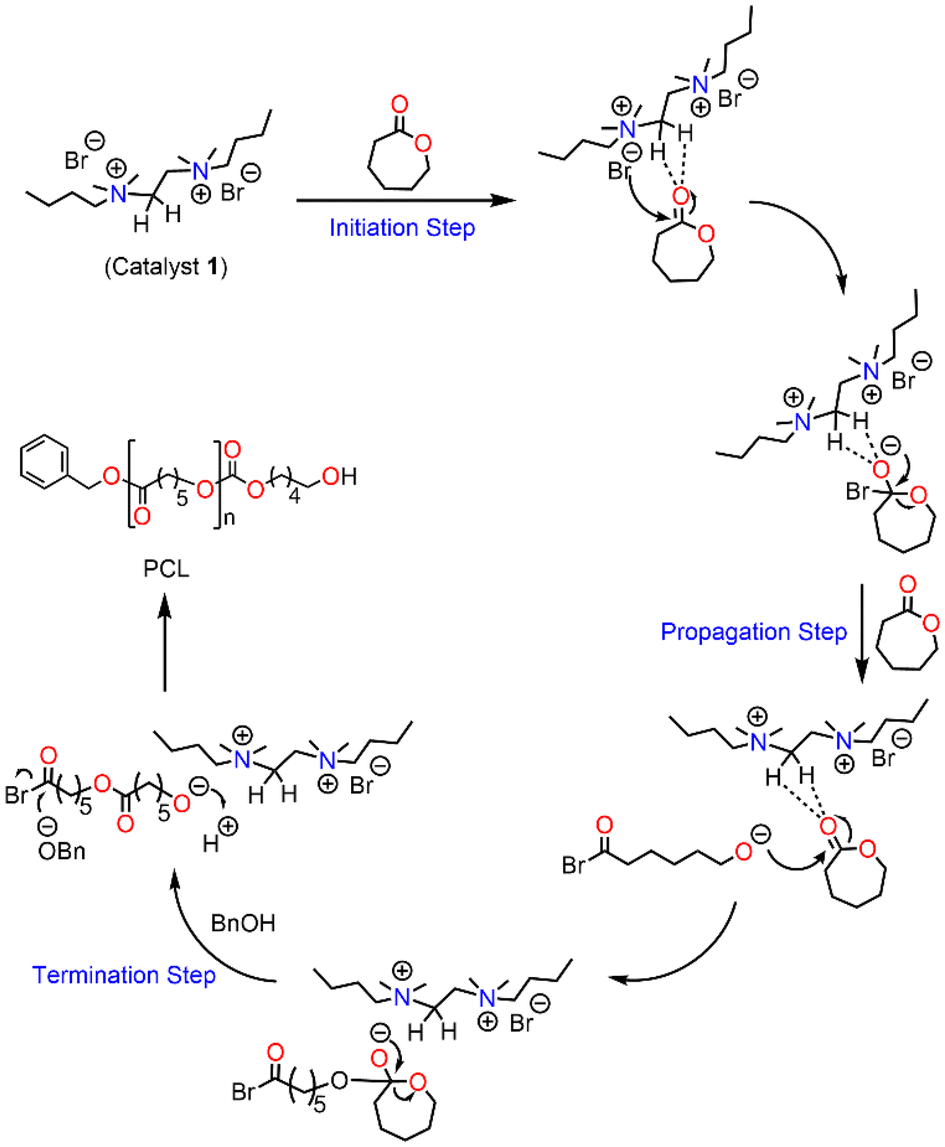 | ||
| Fig. 4 Proposed mechanism of N,N′-dibutyl-N,N,N′,N′-tetramethylethane-1,2-diammonium bromide catalysed ROP of ε-CL. | ||
DFT studies of the ROP process
Previously, the role of CH⋯OC interactions between an imidazole salt and cyclic esters has been substantiated computationally for the ROP of cyclic esters.51 We have carried out geometry optimization at the B3LYP/6-31G**69–72 levels on mixtures of ε-CL and a cationic catalyst in the absence of the counter ion. Here, we have analyzed two possible mixtures of ε-CL and a cationic catalyst, which possess different orientations of these species (A and B). Although these calculations are not directly related to the experimental observation of the ROP, the number of possible interactions present in the DFT-optimized mixtures can directly relate to the activation of ε-CL. In species A, we have observed that the O atom (CO) of ε-CL is involved in the hydrogen bonding interactions with quaternary ammonium dicationic salt, where we observed one strong hydrogen bond (O1–H1) = 1.98 Å and three additional weak interactions, i.e. (O1–H2 = 3.08 Å, O1–H3 = 2.33 Å, and O1–H4 = 2.46 Å Fig. 5a). In contrast, we do not observe any possible hydrogen bonding interaction in species B, which is optimized by keeping ε-CL and quaternary ammonium dicationic salt side by side. Next, we analyzed the hydrogen bonding interaction using the non-covalent interaction (NCI) plot in A and B, where high-density NCI regions represent the hydrogen bonds while the weak interactions extend over large regions of intermolecular contacts (Fig. 5b). In species A, we observed round, highly localized blue color NCI72,73 domains in the region where oxygen and hydrogen interact (O1—H1), followed by a vastly scattered region representing weak van der Waals interactions between O1–H2, O1–H3, and O1–H4. In species A, the partial charges on the hydrogen atoms involved in H-bonds ranged from +0.25 to +0.29 au, while a large positive charge of +0.82 au on the C atom of the (CO) of ε-CL indicates higher electrophilicity.
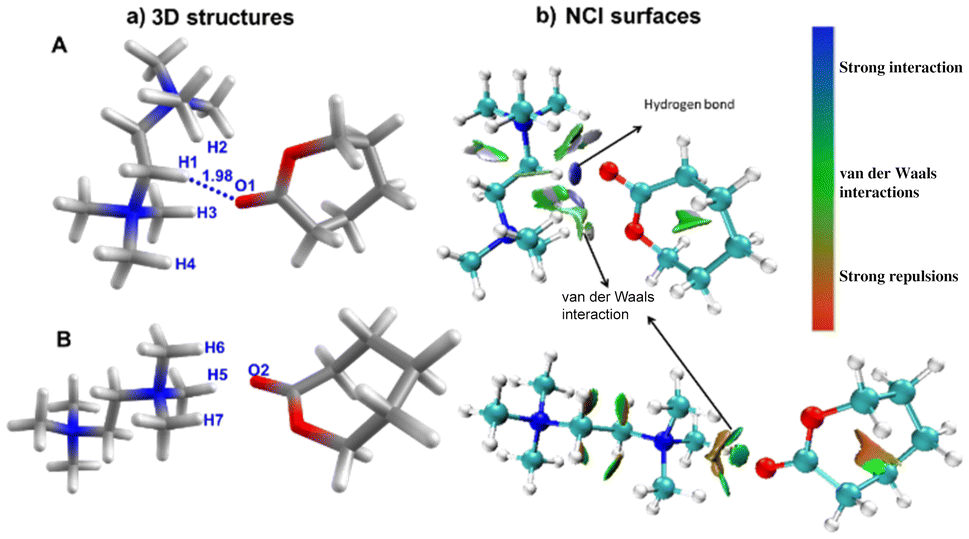 | ||
| Fig. 5 (a) Molecular modelling of complexes between the cationic catalyst and ε-CL in different orientations of these species (A and B) (b) NCI surfaces (iso value = 0.5). Color code: C (black), O (red), H (grey), and N (blue). | ||
On the other hand, the NCI plot of species B does not reveal any hydrogen bonding interaction, which agrees with the structural topology. In addition, we have performed energy decomposition analysis (EDA)74 on species A and B by fragmenting them in quarternary ammonium dicationic salt and ε-CL (see the ESI† for details). The total intermolecular interaction computed total interaction energies (ΔEint) are −38.3 and −31.1 kcal mol−1 for species A and B, respectively, indicating that the former species is stabilized by ∼7 kcal mol−1 compared to species B. The decomposed energy shows that the significant contribution emerges from electrostatic interactions, followed by Pauli (repulsive in nature) and orbital interactions. Due to hydrogen bonding interactions, we observed relatively higher electrostatic and orbital interaction energies in species A than in species B. The steric repulsion (Pauli) is relatively large in species A, due to the large number of protons close to ε-CL. Moreover, the dispersion interactions are more robust in species A than in species B.
The recyclability and reusability features of the catalyst were also assessed in terms of its possible application in the industry. It was studied for the ROP of ε-CL (100 equivalent) under the standard reaction conditions (Table 3). After the polymerization (8 h), the entire reaction mixture was added dropwise to water, where PCL was precipitated as a solid, and the catalyst was dissolved in water. The polymer was then isolated from water by a simple filtration technique. The filtrate containing the catalyst was evaporated to recover and reuse the catalyst for the next cycle. The process was repeated for up to five cycles while keeping the initial catalyst-to-monomer ratio at 1:100, and the results are summarized in Table 3 and Fig. 6. Remarkably, about 95% of the used catalyst could be recovered in the first cycle and reused up to six successive cycles of reaction without substantial loss in catalytic efficiency (Table TS3 in the ESI†). The gradual drop in catalyst recovery in the subsequent cycles is considered mainly due to the difficulty associated with handling of smaller mass of the catalyst in the lab setup. The solvent recovery study carried out using a higher-scaled reaction (∼10×) indicated about 95% recovery of toluene after the reaction (Table TS2 in the ESI†).
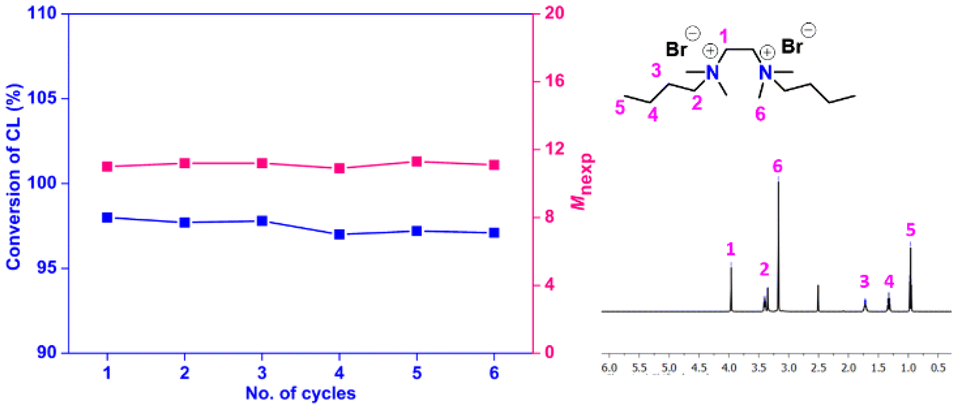 | ||
| Fig. 6 (a) Recycling studies of N,N′-dibutyl-N,N,N′,N′-tetramethylethane-1,2-diammonium bromide as the catalyst for the ROP of ε-CL and (b) 1H NMR spectra of the catalyst after 6th cycle. | ||
The catalytic performance of the supported organocatalysts generally shows a noticeable drop after a few cycles because of their intrinsic defects, such as the loss of attached organocatalysts from the supports or the mass transfer of the reactants to the supports.75,76 However, the polymerization was under outstanding control in the present case, affording PCL with the expected molecular weight and narrow dispersity in each cycle (Table 3). To further substantiate the result, the recycled catalyst obtained after the 6th cycle was analyzed by 1H NMR (Fig. 6b) and the spectra revealed no change before and after the recyclability experiment, indicating that the catalyst remains reasonably stable even after six cycles.
Conclusions
In summary, N,N′-dibutyl-N,N,N′,N′-tetramethylethane-1,2-diammonium bromide was investigated to exhibit its catalytic performance in the ROP of ε-CL at 60 °C. Efficient ROP was demonstrated with very good control over the polymerization without an external initiator. To the best of our knowledge, this is the first report on organocatalyzed cyclic ester ROP accomplished without using an external initiator. Various well-defined PCLs with precisely controlled Mn were successfully synthesized and characterized with NMR, GPC, TGA, and DSC. The fact that the catalyst can be easily recovered and reused without the loss of catalytic activity makes this catalyst an attractive recyclable organocatalyst for preparing biodegradable polyesters, avoiding the tedious and uneconomical heterogeneous catalyst fabrication process.Author contributions
Shweta Sagar: methodology, data curation, formal analysis, software, and writing – original draft. Shiva Lall Sunar: catalyst synthesis and analysis. Priyanku Nath: data curation and formal analysis. Aranya Ray: data curation and formal analysis. Mridula Choudhary: DFT calculation and data analysis. Saurabh K. Singh: DFT calculation and data analysis. Alok Sarkar: supervision and writing – review & editing. Tarun K. Panda: conceptualization, supervision, project administration, resources, and writing – review & editing.Data availability
All the data used in this work are available in the ESI and the references cited in the manuscript.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from SERB, India under project no. CRG/2022/001941. The Department of Chemistry at IIT Hyderabad is acknowledged for its instrumental facilities. S. S. acknowledges the Prime Minister's Research Fellowship (2000829) for financial assistance. P. N. and S. L. S. thank CSIR (09/1001(15565)/2022-EMR-I and 09/1001(0090)/2021-EMR-I) for their PhD fellowships. A. R. and M.C. thank MHRD and UGC for their PhD fellowships. Part of the graphical abstract images is designed by brgfx / Freepik.References
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed
.
-
H. J. Böhm and G. Schneider, Protein-ligand interactions from molecular recognition to drug design, Wiley-VCH, 2003 Search PubMed
-
J. W. Steed, J. L. Atwood and P. A. Gale, in Supramolecular Chemistry, Wiley, 2012 Search PubMed
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS
-
M. Shahinpoor, Polymer Sensors and Actuators, Springer Berlin Heidelberg, 2000 Search PubMed
- J. J. Shea, IEEE Electr. Insul. Mag., 2005, 15, 50–51 Search PubMed
- S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061–1069 CrossRef CAS PubMed
-
G. A. Jeffrey and G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, New York, 1997, vol. 12 Search PubMed
-
J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, 2011 Search PubMed
- E. A. Meyer, R. K. Castellano, F. Diederich, R. K. Castellano, F. Diederich and E. A. Meyer, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS
- J. C. Ma, D. A. Dougherty and M. Beckman, Chem. Rev., 1997, 97(5), 1303–1324 CrossRef CAS PubMed
- C. Tanford, Science, 1978, 200, 1012–1018 CrossRef CAS PubMed
- L. Pauling, Chem. Eng. News, 1946, 24(10), 1375–1377 CrossRef CAS
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed
- W. Zhao, Y. Lv, J. Li, Z. Feng, Y. Ni and N. Hadjichristidis, Nat. Commun., 2019, 10(1), 3590 CrossRef PubMed
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC
- Data Bridge Market Research, https://markets.businessinsider.com/news/stocks/polycaprolactone-pcl-market-destine-to-reach-usd-909-81-billion-with-an-excellent-cagr-of-10-50-by-2029-1032246582 (accessed on 30th March 2024).
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef PubMed
- H. Zhang, Y. Dong, K. Huang, J. Liu, B. Dong and F. Wang, Eur. Polym. J., 2019, 118, 633–641 CrossRef CAS
- H. C. Huang, B. Wang, Y. P. Zhang and Y. S. Li, Polym. Chem., 2016, 7, 5819–5827 RSC
- R. Olejník, M. Bílek, Z. Růžičková, Z. Hoštálek, J. Merna and A. Růžička, J. Organomet. Chem., 2015, 794, 237–246 CrossRef
- D. J. Darensbourg and O. Karroonnirun, Macromolecules, 2010, 43, 8880–8886 CrossRef CAS
- V. Paradiso, V. Capaccio, D. H. Lamparelli and C. Capacchione, Coord. Chem. Rev., 2021, 429, 213644 CrossRef CAS
- J. Chen, X. Wu, L. Zhang, Z. Duan and B. Liu, Polym. Chem., 2022, 13, 2971–2979 RSC
- S. Sagar, H. Karmakar, P. Nath, A. Sarkar, V. Chandrasekhar and T. K. Panda, Chem. Commun., 2023, 59(56), 8727–8730 RSC
- S. Sagar, K. Bano, A. Sarkar, K. Pal and T. K. Panda, Eur. J. Inorg. Chem., 2022, 34, e202200494 CrossRef
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC
- R. A. Qazi, M. S. Khan, L. A. Shah, R. Ullah, A. Kausar and R. Khattak, Polym.-Plast. Technol. Mater., 2020, 59, 928–951 CAS
- B. Guo and P. X. Ma, Sci. China: Chem., 2014, 57, 490–500 CrossRef CAS
- K. Fukushima and K. Nozaki, Macromolecules, 2020, 53, 5018–5022 CrossRef CAS
- C. J. Zhang and X. H. Zhang, Sci. China: Chem., 2019, 62, 1087–1089 CrossRef CAS
- M. Nikulin and V. Švedas, Molecules, 2021, 26(09), 2750 CrossRef CAS PubMed
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC
- S. I. Shoda, H. Uyama, J. I. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS PubMed
- C. Thomas, F. Peruch, A. Deffieux, A. Milet, J. P. Desvergne and B. Bibal, Adv. Synth. Catal., 2011, 353, 1049–1054 CrossRef CAS
- J. Liu, C. Chen, Z. Li, W. Wu, X. Zhi, Q. Zhang, H. Wu, X. Wang, S. Cui and K. Guo, Polym. Chem., 2015, 6, 3754–3757 RSC
- A. Alba, A. Schopp, A. P. De Sousa Delgado, R. Cherif-Cheikh, B. Martín-Vaca and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 959–965 CrossRef CAS
- S. Koeller, J. Kadota, A. Deffieux, F. Peruch, S. Massip, J. M. Léger, J. P. Desvergne and B. Bibal, J. Am. Chem. Soc., 2009, 131, 15088–15089 CrossRef CAS PubMed
- G. Gontard, A. Amgoune and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3253–3256 CrossRef CAS
- O. I. Kazakov and M. K. Kiesewetter, Macromolecules, 2015, 48, 6121–6126 CrossRef CAS
- O. I. Kazakov, P. P. Datta, M. Isajani, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2014, 47, 7463–7468 CrossRef CAS PubMed
- F. Shi, Y. Gu, Q. Zhang and Y. Deng, Catal. Surv. Asia, 2004, 8(3), 179–186 CrossRef CAS
- Y. Li, S. Hu, J. Cheng and W. Lou, Chin. J. Catal., 2014, 35, 396–406 CrossRef CAS
- L. Zhang, M. Xian, Y. He, L. Li, J. Yang, S. Yu and X. Xu, Bioresour. Technol., 2009, 100, 4368–4373 CrossRef CAS PubMed
- P. Naert, K. Rabaey and C. V. Stevens, Green Chem., 2018, 20, 4277–4286 RSC
- M. Vafaeezadeh and M. M. Hashemi, J. Chem. Eng., 2014, 250, 35–41 CrossRef CAS
- R. Kore and R. Srivastava, J. Mol. Catal. A: Chem., 2013, 376, 90–97 CrossRef CAS
- A. Kaoukabi, F. Guillen, H. Qayouh, A. Bouyahya, S. Balieu, L. Belachemi, G. Gouhier and M. Lahcini, Ind. Crops Prod., 2015, 72, 16–23 CrossRef CAS
- N. Cheechana, W. Benchaphanthawee, N. Akkravijitkul, P. Rithchumpon, T. Junpirom, W. Limwanich, W. Punyodom, N. Kungwan, C. Ngaojampa, P. Thavornyutikarn and P. Meepowpan, Polymers, 2021, 13, 4290 CrossRef CAS
- M. C. Bubalo, K. Radošević, I. R. Redovniković, I. Slivac and V. G. Srček, Arh. Hig. Rada Toksikol., 2017, 68, 171–179 CrossRef CAS PubMed
- S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038 CrossRef CAS
- B. Jastorff, R. Störmann, J. Ranke, K. Mölter, F. Stock, B. Oberheitmann, W. Hoffmann, J. Hoffmann, M. Nüchter and B. Ondruschka, Green Chem., 2003, 5, 136–142 RSC
- R. Kawai, S. Yada and T. Yoshimura, ACS Omega, 2019, 4, 14242–14250 CrossRef CAS PubMed
- R. Zana, J. Colloid Interface Sci., 2002, 248, 203–220 CrossRef CAS
- R. Zana, Curr. Opin. Colloid Interface Sci., 1996, 1, 566–571 CrossRef CAS
- K. Fukushima, D. Tabuani, C. Abbate, M. Arena and P. Rizzarelli, Eur. Polym. J., 2011, 47, 139–152 CrossRef CAS
- K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772–1782 CrossRef CAS
- N. Zhu, Y. Liu, J. Liu, J. Ling, X. Hu, W. Huang, W. Feng and K. Guo, Sci. Rep., 2018, 8, 3734 CrossRef PubMed
- Y. Zhao, X. Lim, Y. Pan, L. Zong, W. Feng, C. H. Tan and K. W. Huang, Chem. Commun., 2012, 48, 5479–5481 RSC
- D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249–2256 RSC
- X. Zhi, J. Liu, Z. Li, H. Wang, X. Wang, S. Cui, C. Chen, C. Zhao, X. Li and K. Guo, Polym. Chem., 2016, 7, 339–349 RSC
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef CAS PubMed
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS
- P. Li, Y. Xi, L. Li, H. Li, W. H. Sun and M. Lei, Inorg. Chim. Acta, 2018, 477, 34–39 CrossRef CAS
- J. Martínez, J. F. Cortés and R. Miranda, Processes, 2022, 10, 1274 CrossRef
- S. Mandal, R. Narvariya, S. L. Sunar, I. Paul, A. Jain and T. K. Panda, Green Chem., 2023, 25, 8266–8272 RSC
- C. Lee, E. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- W. J. Hehre, K. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed
- O. Gleeson, G. L. Davies, A. Peschiulli, R. Tekoriute, Y. K. Gun'Ko and S. J. Connon, Org. Biomol. Chem., 2011, 9, 7929–7940 RSC
- U. Yolsal, T. A. R. Horton, M. Wang and M. P. Shaver, Prog. Polym. Sci., 2020, 111, 101313 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04411h |
| This journal is © The Royal Society of Chemistry 2025 |