
Chemical and physical properties of orthoconic liquid crystals: 2H NMR spectroscopy and molecular dynamics simulations†
Anna
Drzewicz
 a,
Martina
Rossi
b,
Mario
Cifelli
b,
Giacomo
Saielli
cd,
Marzena
Tykarska
e and
Valentina
Domenici
*b
a,
Martina
Rossi
b,
Mario
Cifelli
b,
Giacomo
Saielli
cd,
Marzena
Tykarska
e and
Valentina
Domenici
*b
aInstitute of Nuclear Physics Polish Academy of Sciences, PL-31342 Krakow, Poland
bDipartimento di Chimica e Chimica Industriale, University of Pisa, via Moruzzi 13, Pisa 56124, Italy. E-mail: valentina.domenici@unipi.it
cCNR, Institute on Membrane Technology Unit of Padova, Via Marzolo, 1 – 35131, Padova, Italy
dDepartment of Chemical Sciences, University of Padova, Via Marzolo, 1 – 35131, Padova, Italy
eInstitute of Chemistry, Military University of Technology, PL-00908 Warszawa, Poland
First published on 20th January 2025
Abstract
In the field of chiral smectic liquid crystals, orthoconic antiferroelectric liquid crystals (OAFLCs) have attracted the interest of the scientific community due to the very high tilt angle, close to 45°, and the consequent optical properties. In the present study, the first 2H NMR investigation is reported on two samples, namely 3F5HPhF9 and 3F7HPhF8, showing the phase sequence isotropic–SmC*–SmCA* and the phase sequence isotropic–SmA–SmC*–SmCA*, respectively, when cooling from the isotropic to the crystalline phases. To this aim, the liquid crystals were doped with a small amount of deuterated probe biphenyl-4,4′-diol-d4. The trend of 2H NMR spectra versus temperature indicates the presence of very high values of the tilt of the deuterated probe in the antiferroelectric phase for both samples. The trend of the local order parameters and that of the tilt angle were compared with the results obtained for the same samples by means of different experimental techniques, namely X-ray diffraction and electrooptical measurements. Moreover, a computational study was performed on the sample labelled 3F5HPhF9 using fully atomistic classical molecular dynamics simulation of the orthoconic phase. The results obtained from the MD simulations show a very large molecular tilt of the molecules (about 42°) when packed in layers and this value is in very good agreement with experimental results. The present research aims to give additional clues about the molecular origin of the very peculiar high tilt angle of orthoconic liquid crystals in the antiferroelectric phase.
1. Introduction
The liquid crystal (LC) chiral phases are a subject of great technological interest because of their chirality, which imparts particular physical properties to these mesophases and thereby undoubtedly impacts their technological applications.1–7 The chirality of LC molecules leads to significant changes at the supramolecular organization level. For example, the chiral nematic phase exhibits a distinct structure compared to the nematic phase, featuring a helical arrangement of microdomains with homogeneous orientation within each microdomain.1,2 In the case of smectic phases, chirality introduces many possible structures almost always associated with the presence of helical distributions giving rise to additional new physical properties.8 The most important chiral smectic mesophase is undoubtedly the SmC* phase having ferroelectric properties,1,2,6 where LC molecules in each smectic layer are inclined at a tilt angle, θ, with respect to the normal to the smectic planes (see the representation in Fig. 1a). Moreover, the molecular chirality determines the reduction in the symmetry of this smectic phase (from C2h for achiral SmC to C2 for chiral SmC) and the formation of helical arrangements:8 although the tilt angle remains constant, as molecules pass from one plane to another, they change the azimuthal angle, ϕ, thus describing a helical pattern, where the axis of the helix is perpendicular to the planes themselves. The chirality of SmC* affects the macroscopic properties of this phase.1,2,6,8 For instance, each SmC* layer possesses a non-zero spontaneous polarization, PS. Moving from one plane to another, the director, n, and the polarization, PS, rotate describing a helical pattern (as sketched in Fig. 1a). This characteristic determines the property of ferroelectricity typical of SmC* phases.1–8 In addition to the ferroelectric phase SmC* there are also numerous variants, such as the so-called antiferroelectric smectic phase, namely SmCA*, discovered in 1989 by Chandani et al.9 SmCA* is a chiral smectic phase with antiferroelectric properties originating from the antiparallel orientation of molecular dipole moments and thus of the net spontaneous polarization cancelled in consecutive layers. This structure is called ‘anticlinic’ since the molecules are inclined at the same tilt angle, but alternately (+θ and −θ) in consecutive layers, so that the supramolecular structure of this mesophase results from the superposition of two helices with a 180° shift of the directors (see the representation in Fig. 1b).5,9 The interest in the SmC* and SmCA* mesophases is related to technological applications such as the liquid crystal displays.1,4–6 The application of an external electric field determines the orientation of molecules of the smectic layers all in the same direction of the electric field; in fact, the application of electric fields perpendicular to the axis of the helix of the phase allows the “unwinding” of the helical structure10 giving rise to a net macroscopic polarization, P, parallel to the applied electric field, E. Moreover, by reversing the polarity of the electric field, the direction in which the helix is unwound changes, so the P vector will consequently change its direction by 180°. The speed at which it is possible to change from one configuration to another and consequently the switching time, τ, is a typical feature of the liquid crystalline chiral mesogen and it is related to the molecular structure and to the supramolecular arrangement of the specific LC molecules within these mesophases.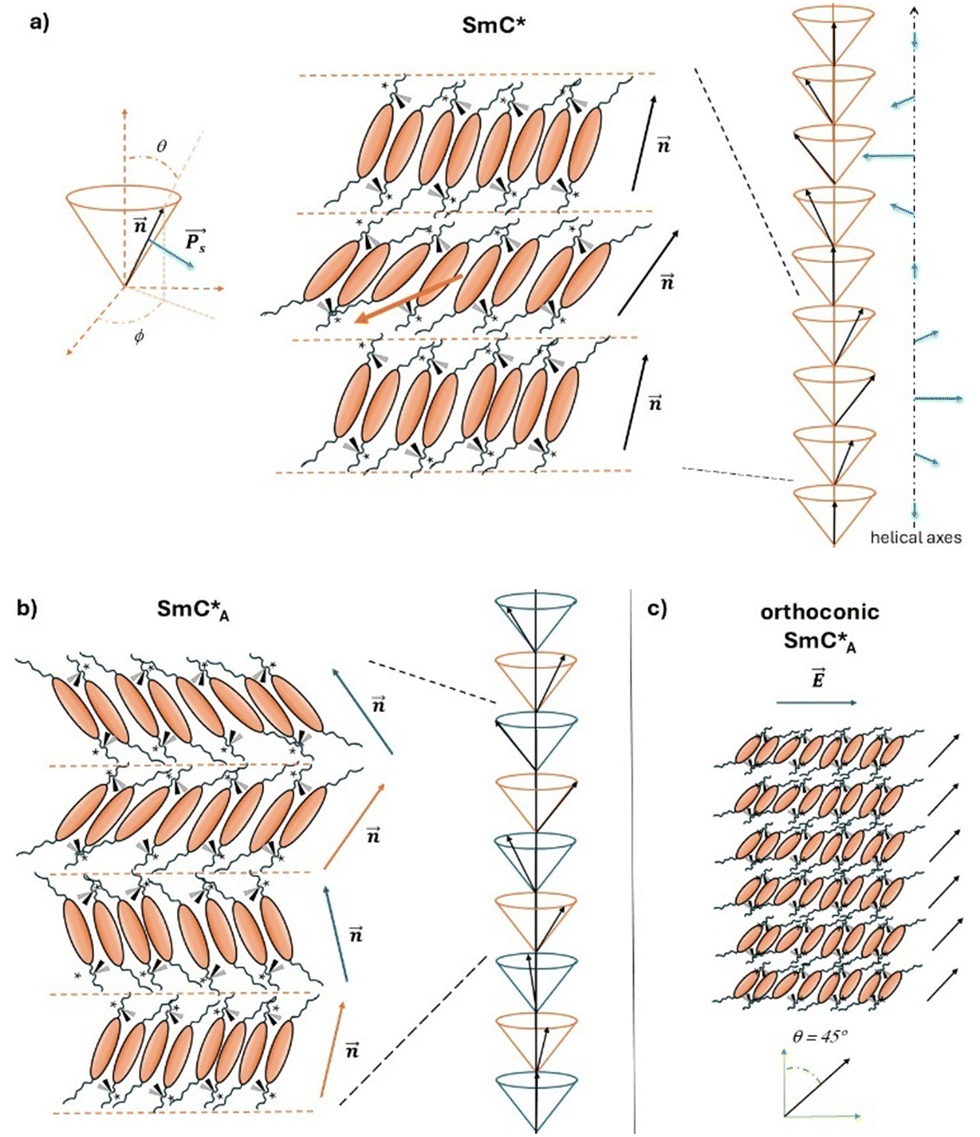 | ||
| Fig. 1 (a) Representation of the ferroelectric SmC* phase. On the left, the orientation of the phase director, n, and the spontaneous polarization, PS, within a smectic layer, and the tilt angle, θ, and the azimuthal angle, ϕ, are shown. In the center, a representation of three consecutive smectic layers is reported where the chiral LC molecules are arranged to better visualize the symmetry properties of the phase (the orange arrow in the center indicates the C2 axis of symmetry). On the right, the helical distribution of the phase director (black arrows) and spontaneous polarization (blue arrows) within a helical pitch of the SmC* phase is drawn. (b) On the left, a representation of four consecutive smectic layers of the antiferroelectric SmCA* phase is reported. The alternate distribution of directors, with the azimuthal angle rotated by 180° passing from one layer to another, is put in evidence by the use of orange and blue arrows. On the right, a portion of the helical distribution of directors in the SmCA* phase is represented. The two helices with a 180° shift of the director vectors through the helical axis are drawn in orange and blue colors. (c) Representation of six consecutive layers of the SmCA* phase formed by orthoconic liquid crystals when an electric field, E, is applied (blue arrow). The orientation of the phase director (black arrow) for each layer and the tilt angle of 45° are shown. | ||
A particular case of chiral smectic liquid crystals, both of ferroelectric and antiferroelectric properties, is represented by the so called orthoconic liquid crystals.5,11 They exhibit peculiar structural characteristics and consequently peculiar optical and electro-optical properties.5 As shown in Fig. 1c, the orthoconic antiferroelectric liquid crystals possess a tilt angle of ∼45° with respect to the normal to the smectic layers. This property results in a variation of the molecular tilt from one plane to another of about 90°. As a consequence, the orientation of the optical axis of the antiferroelectric phase is perpendicular to the plane of molecular tilt.5,12 Moreover, since the inclinations of the director in adjacent layers of the SmCA* phase are directed oppositely, their net polarization becomes close to zero, which could be useful for resolving some limitations of the SmC* and SmCA* phases used in liquid crystal displays.1,3,5,12 In fact, from the discovery of ferroelectric liquid crystals in 1975 and of antiferroelectric liquid crystals in 1989, these systems were indicated as potential candidates to replace the twisted nematic display. First surface-stabilized ferroelectric liquid crystal displays based on ferroelectrics (SSFLCDs) were realized already in 1995,7 based on the discovery of the SSFLC effect,13 which brought to an improvement of 2–3 orders of magnitude in the characteristic switching time, τ, with values of 10–100 μs.1,4,5,7 However, SSFLCDs exhibit several defects due to the helicoidal arrangement of molecules and the packing of smectic layers close to the surface. Among the most studied defects, the zigzag chevron defects on the display surface compromise the optical properties and efficiency of SSFLCDs. The discovery of antiferroelectric liquid crystals, with their typical anticlinic structure, was very promising. However, antiferroelectric liquid crystal displays present several problems related to the reproducibility, as there are no studies demonstrating the stability of these phases after many cycles. Additionally, as with SSFLCDs, the contrast between the ‘on’ and ‘off’ states remains not optimal.5 All these aspects brought to the search for liquid crystals with an optical axis forming 90° moving from one layer to another, in order to solve the contrast problem. The first orthoconic liquid crystal was produced by the group led by R. Dąbrowski more than twenty years ago:14 it was actually not a pure compound, but a mixture of LC compounds and the achieved tilt angle was close to 45°. Several series of pure LC compounds showing an orthoconic antiferroelectric smectic phase have been synthesized later on15–20 and few chemical–physical investigations have been published21–24 to correlate the molecular properties to the macroscopic physical properties of these phases. These research works are mainly based on experimental methods aimed to determine chemical and physical properties, such as the temperature dependence of the helical pitch, helix inversion, spontaneous polarization and optical tilt.25–29 However, several basic questions are still open. For instance, the molecular and structural reasons for the formation of highly tilted phases need more investigations, either experimental or computational ones, and justify the interest in the ferroelectric and antiferroelectric smectic phases formed by orthoconic LCs.
One of the most important features of ferroelectric and antiferroelectric smectic phases is related to the molecular tilt angle: in common calamitic ferroelectrics, the maximum tilt angle is around 22–23°, while in antiferroelectrics, it reaches a maximum value of 25–30°.4 Several techniques can be used to determine the tilt angle, each of them focusing on a particular molecular property: for example, the optical method, which is mostly related to detection of electron density, gives the information from rigid cores; X-ray method, which is mostly related to atomic density, gives the information from whole molecules; and nuclear magnetic resonance (NMR) techniques, whose specificity depends on the observed magnetic nuclei, such as 2H or 13C. NMR spectroscopy is a powerful technique to study liquid crystals30–35 and it was widely applied to the study of ferroelectric liquid crystals.36–43 In particular, the presence of deuterons on specific molecular sites (i.e. on the aromatic rings of the rigid core or on the lateral chains) enables one to get information on different parts of the molecules, such as the local orientational order and the average tilt of different molecular fragments.30–33,36,38,39,43
2H NMR spectroscopy was of help in understanding the ordering properties and the temperature dependence of the tilt angle in ferroelectric liquid crystals38–43 and in understanding the supramolecular properties and the peculiar effect of the external magnetic field in unwinding the helical structure of the SmC* phase.37,39,44,45 In combination with other NMR techniques, such as 1H NMR diffusometry and 13C NMR spectroscopy, 2H NMR spectroscopy gave important contributions to the understanding of the dynamic behaviour at the ferroelectric–antiferroelectric phase transition and of the conformational properties at the SmA–SmC* phase transition.37,39,43–49 To our knowledge, no 2H NMR studies have been performed on orthoconic liquid crystals.
Other than experimental techniques, a few computational studies have been used to investigate the structural and dynamic properties of ferroelectric and antiferroelectric LCs, though the long-range orientational and positional order, and slow dynamics, rendered their simulations not a trivial task.50–52 Early semiempirical quanto-mechanic (QM) calculations and molecular dynamics (MD) simulations of a small box of 4 × 4 × 4 molecules of MHPOBC ((S)-4-[(1-methylheptyloxy)carbonyl]phenyl 4′-octyloxybiphenyl-4′-carboxylate), a topologically similar compound to the ones investigated in this work, except for the presence of fluorine atoms, were reported in 1996 by Toriumi et al.53 focusing on the conformation of the chiral chain with respect to the rigid core. However, to the best of our knowledge, no molecular simulations of the orthoconic liquid crystalline phase have been reported in the literature.
In the present research paper, we are reporting the first 2H NMR investigation of two orthoconic liquid crystals, namely two compounds of the 3FmHPhFr series, in their mesophases by using a small deuterated probe diluted in them. This approach allowed us to obtain the temperature trend of the orientational order parameter Szz of the molecular probe, namely the biphenyl-4,4′-diol-d4, which is likely to follow the trend of the orientational order of the orthoconic molecular aromatic core. For one orthoconic system we could obtain the temperature behaviour of the tilt angle of the molecular probe from 2H NMR experiments. Electrooptic, X-ray and transmission measurements of the two pure compounds were also reported and the electrooptic tilt angle, the layer spacing and the pitch behaviour in the ferroelectric and antiferroelectric phases of these two orthoconic LCs are reported and discussed in view of the literature about orthoconic systems. Moreover, a detailed computational study was performed on one of the two orthoconic LCs by running a series of fully atomistic classical molecular dynamics simulations of the orthoconic phase. The results obtained from the MD simulations show a very large molecular tilt of the molecules (about 42°) when packed in layers and this value is in very good agreement with the experimental results. In this paper, all the obtained results, experimental and computational ones, are discussed giving rise to a more complete picture of conformational, structural and orientational properties of orthoconic liquid crystals.
2. Experimental part
2.1. Materials
The samples analysed in this work are two fluorinated orthoconic liquid crystals of the 3FmHPhFr series (where m and r indicate the number of carbon atoms in the lateral chains), which are a series of fluorinated orthoconic compounds synthesized in 2015 by Milewska et al.19,20 The structure of the two compounds is shown in Fig. 2.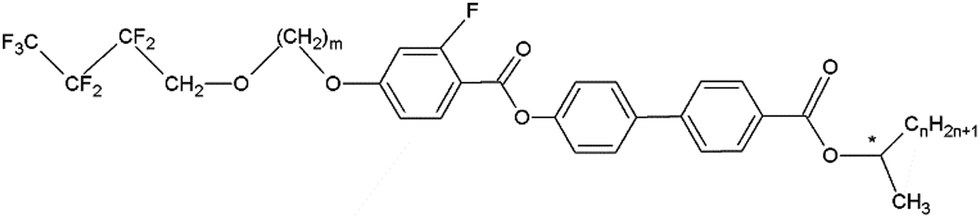 | ||
| Fig. 2 Molecular structure of the compounds investigated in this work, namely 3FmHPhFr. | ||
Both samples present a fluorine atom substituted in the benzene ring close to the lateral achiral fluorinated chain. The chiral chain linked to the biphenyl fragment is an ester derivative. The difference between the two compounds is related to the length of both the achiral and chiral chain, as clearly indicated in their labels: 3F7HPhF8 and 3F5HPhF9. The interest in these compounds specifically concerns the occurrence of the SmC* and SmCA* phases and the temperature dependence of the tilt angle.19,20 Moreover, some compounds of the same series exhibit a reversal of the helical pitch at certain temperatures, typically in the SmCA* phase, making them interesting from an application point of view.23 Among the various fluorinated compounds synthesized by Milewska et al.19,20 we chose these two specific compounds due to their relatively low clearing point (Tc < 100 °C) which is better for NMR instrument requirements.
The characterization of the mesophases of these two pure samples is reported in ref. 19, 20 and 23. In this work, to perform 2H NMR spectroscopy, a deuterated probe, namely biphenyl-4,4′-diol-d4, was added in a small percentage to the LC compounds. The samples were prepared by mixing approximately 100 mg of each liquid crystal with about 1.5 mg of deuterated biphenyl-4,4′-diol-d4 to have a detectable amount of deuterium nuclei and, at the same time, not altering the sequence of liquid crystalline phases of the starting LC samples.
In order to check the effect of the deuterated probe on the mesomorphic properties of the orthoconic LCs, the prepared mixtures were investigated by means of differential scanning calorimetry (DSC) and polarized optical microscopy (POM).
2.2. Experimental methods
2.3. Computational methods
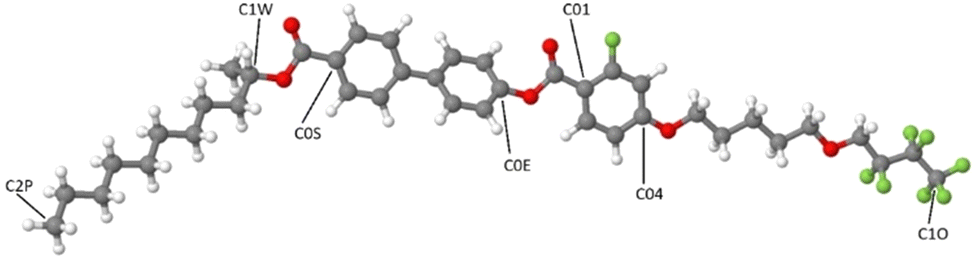
Molecular dynamics simulations were run with the software package GROMACS.56 The force field (FF) and topology files were generated through the web server LigParGen; thus the FF is based on the OPLS-AA parameters (Optimized Potential for Liquid Simulations-All Atom) developed by the Jorgensen group.57–59 The starting box for the simulation was built as follows: the elongated conformer shown in Fig. 3, with the chains in all-trans conformation, was energy minimized at the density functional theory (DFT) level with the popular B3LYP functional (see for instance ref. 60–63) and the 6-31G(d) Gaussian basis set. The molecule was then replicated, approximately oriented with the molecular long axis along the z axis of the box, 80 times with PACKMOL64 in a box of 100 × 100 × 40 Å.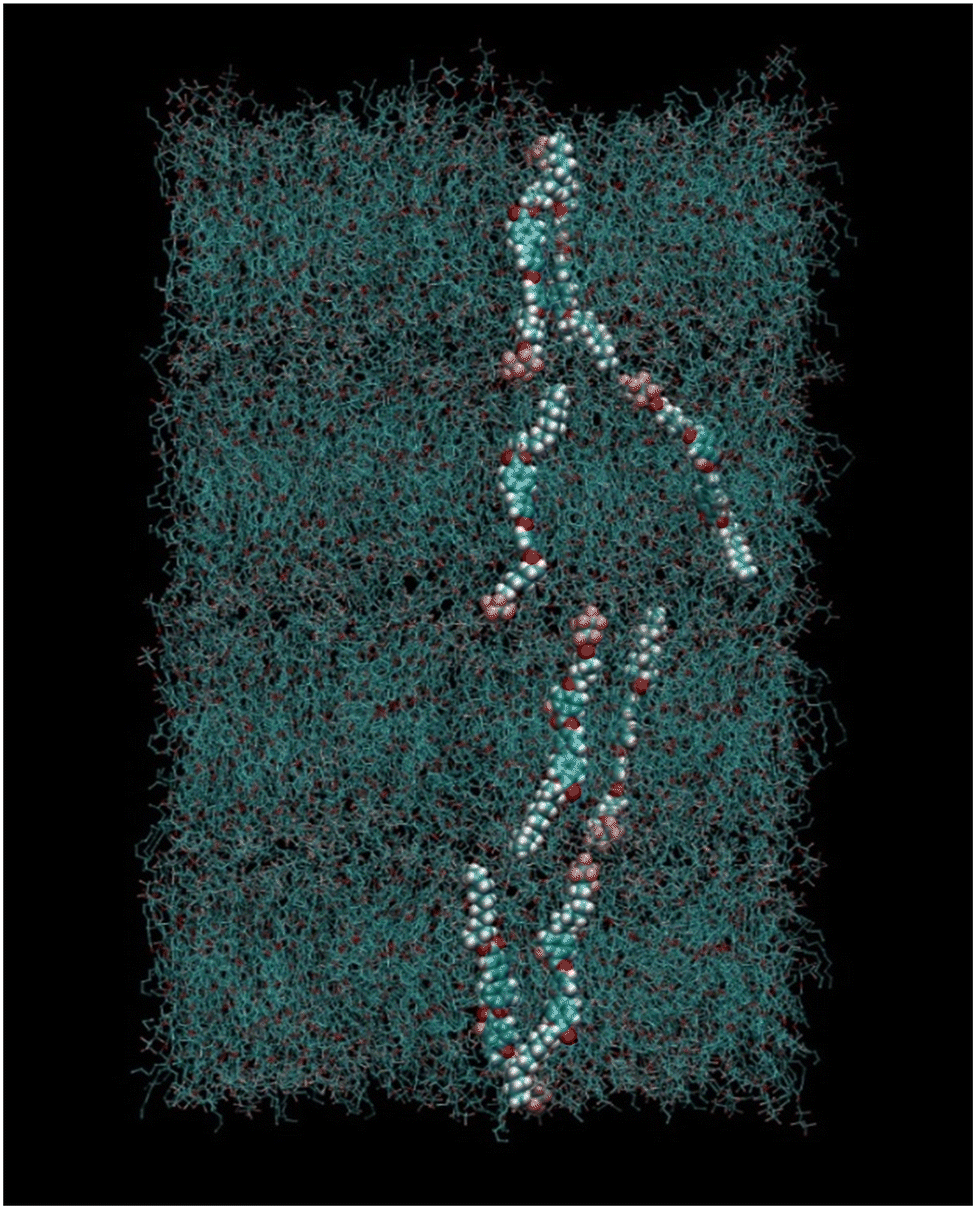 | ||
| Fig. 3 The all-trans conformer of 3F5HPhF9 used to build the starting simulation box. Some representative atoms are labelled and referenced later in the main text. | ||
Half molecules were placed with the fluorinated chain up (+z) and half with the fluorinated chain down (−z). This initial box, having a very low density, was relaxed with the Berendsen barostat65 and semi-isotropic pressure coupling (meaning that the xy axes of the simulation box could fluctuate independently with respect to the z axis) under periodic boundary conditions (PBC) until the density was around 1.2 g mL−1 (box size of about 47.2 × 47.2 × 38.4 Å). This small box was then doubled in the xy direction and replicated 4 times in the z direction in order to obtain a system of 1280 molecules (128![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms) in a SmA arrangement. Such initial configuration is shown in Fig. 4.
000 atoms) in a SmA arrangement. Such initial configuration is shown in Fig. 4.
 | ||
| Fig. 4 Snapshot of the initial simulation box with molecules arranged in a SmA phase, containing four layers. A few selected molecules are highlighted for clarity. | ||
The box was then submitted to 150 ns of simulation in the NpT ensemble, using semi-isotropic pressure coupling, PBC, leap-frog integrator, 1 fs time step, X–H bonds constrained with the LINCS algorithm,66 at a temperature of 400 K and pressure 1 bar using the velocity rescale thermostat and the Berendsen barostat.
We noted that, after 150 ns, each layer of the system spontaneously tilted by a relatively large amount, as can be seen in Fig. S1 (ESI†). The spontaneous tilt suggests that this system can form a stable orthoconic phase; however the small numbers of layers do not allow for a coherent arrangement of the tilt of each layer with respect to the one immediately above or below as in a true orthoconic phase (see Fig. 1c). Therefore, we proceeded to build “by hand” an orthoconic structure by replacing the two bottom layers with a replica of the two top layers of Fig. S1 (ESI†), having a chevron like arrangement. The starting box so obtained was preliminarily relaxed by a short MD run and then it was equilibrated at 400 K for 200 ns in the NpT ensemble with fully anisotropic pressure coupling, that is, each box size was allowed to change independently. The box appeared stable, as can be judged by the time dependence of the length of each axis, which fluctuates around an average value without any apparent drift, as seen in Fig. S2 (ESI†). The 200 ns run with fully anisotropic pressure coupling allowed evaluation of the average box size for the final production run in the NVT ensemble, which is 104.6 × 103.1 × 132.5 A, corresponding to a density of 1.15 g mL−1. In Fig. 5 we show a snapshot of the box used for the NVT simulations exhibiting the orthoconic phase. The box was equilibrated in the NVT ensemble for 500 ns followed by a production run of 50 ns from which configurations were saved every 10 ps for further analysis. The temperature set for the simulations is relatively high since the orthoconic phase is highly ordered; therefore using a simulation temperature slightly higher than the experiments allows speeding up the exploration of the configuration space qualitatively maintaining the microscopic details of the structure.
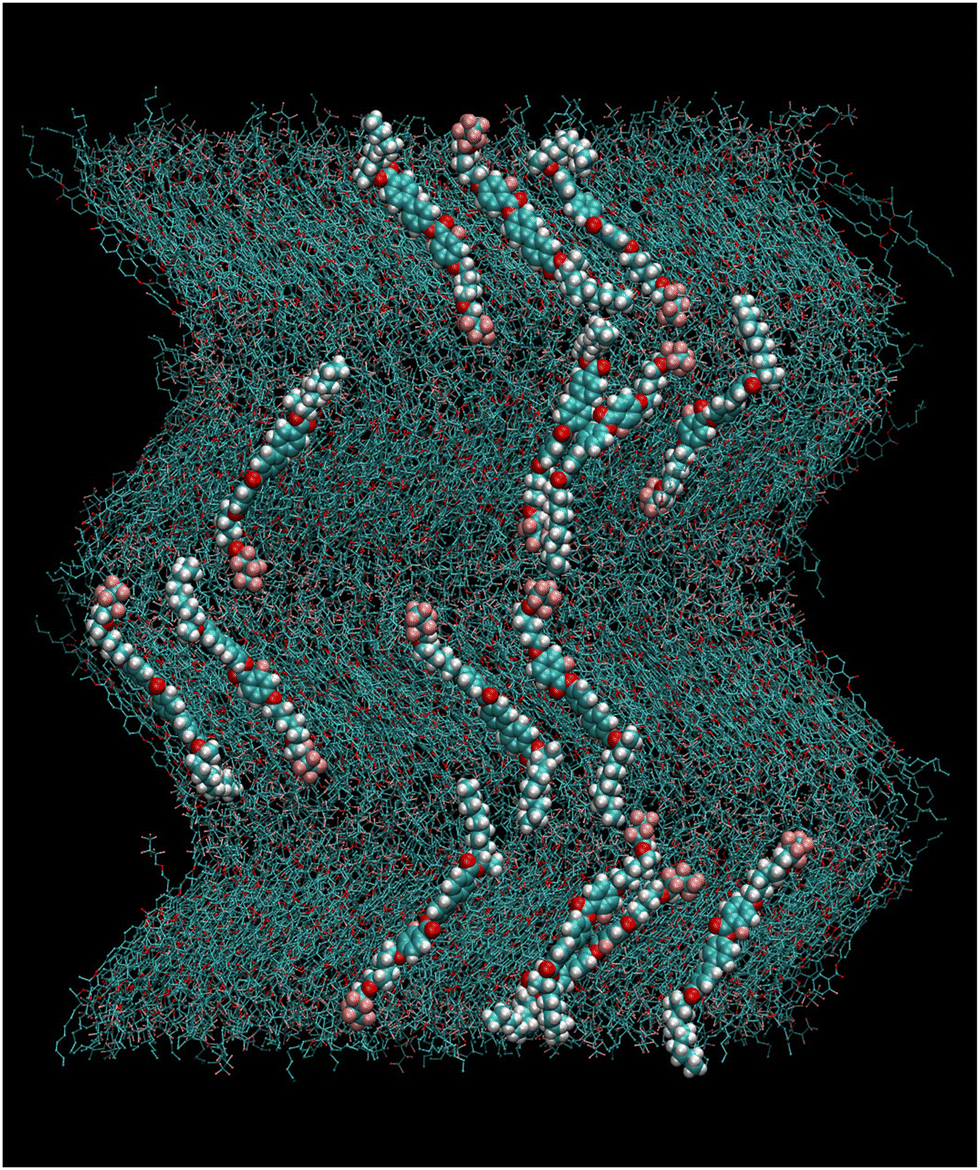 | ||
| Fig. 5 Snapshot of the box with molecules in the orthoconic phase used for the NVT production run at T = 400 K. A few selected molecules are highlighted for clarity. | ||
3. Results and discussion
3.1. Mesophase characterization
The characterization of the mesophase behaviour of the two fluorinated orthoconic LC samples containing a small quantity of biphenyl-4,4′-diol-d4 aims to understand how the addition of a deuterated probe modifies the transition temperatures of the two pure LC samples. The DSC curves of the two mixtures containing the probe are reported in the ESI† (Fig. S3 and S4). Transition temperatures and enthalpy values for the four samples, obtained on cooling and on heating the samples, are reported in Table 1. From these data, it is possible to identify two phenomena common to both samples with the probe, namely the absence of the SmCA* – Cr phase transition in the cooling cycle and the very low sensitivity of the experimental apparatus to observe the transition from SmC* to SmCA* in the cooling cycle. The Cr – SmCA* phase transition is indeed only detectable during the heating cycle and this phenomenon, which is due to the supercooling effect, was observed in the first study on the pure LC samples, too.19,20 Other transitions, namely Iso – SmA, Iso – SmC* and SmA – SmC*, are of the first order and they are well visible in both the heating and the cooling cycle. Concerning the SmCA* – SmC* phase transition, it presents a very small enthalpy value, as it is a weak first order transition: this makes it very difficult to observe it by DSC. A similar behaviour was reported for the pure orthoconic LC samples.19,20| Sample and processs | Phase | T (°C) [ΔH (kJ mol−1)] | Phase | T (°C) [ΔH (kJ mol−1)] | Phase | T (°C) [ΔH (kJ mol−1)] | Phase | T (°C) [ΔH (kJ mol−1)] | Phase |
|---|---|---|---|---|---|---|---|---|---|
| 3F5HPhF9 | Cr | SmCA* | SmC* | Iso | |||||
| Heating | * | 35.7 [24.70] | * | 81.2 [0.07] | * | 85.3 [4.43] | * | ||
| 3F5HPhF9 + probe-d4 | Cr | SmCA* | SmC* | Iso | |||||
| Heating | * | 37.43 [23.54] | * | 80.52 [0.002] | * | 85.64 [4.65] | * | ||
| Cooling | * | * | * | 84.16 [4.56] | * | ||||
| 3F7HPhF8 | Cr | SmCA* | SmC* | SmA | Iso | ||||
| Heating | * | 34.2 [27.72] | * | 85.1 [0.69] | * | 89.9 [0.82] | * | 91.5 [3.33] | * |
| 3F7HPhF8 + probe-d4 | Cr | Sm*CA | SmC* | SmA | Iso | ||||
| Heating | * | 37.96 [24.37] | * | 86.48 [nd] | * | 91.44 [0.67] | * | 93.05 [3.86] | * |
| Cooling | * | * | * | 90.37 [0.67] | * | 91.80 [3.87] | * |
To better distinguish these two phases and identify their transition temperature, an optical microscope with polarized light can be of help. The liquid crystalline phases for both mixtures of orthoconic LCs with the deuterated probe were observed by using POM, and a selection of images of the mesophases of the two samples is shown in Fig. 6.
 | ||
| Fig. 6 Photographs of 3F5HPhF9 added with the deuterated probe obtained on cooling the sample from the isotropic phase: (a) in the SmC* phase at T = 86 °C and (b) in the SmCA* phase at T = 78 °C. Photographs of 3F7HPhF8 added with the deuterated probe obtained on cooling the sample from the isotropic phase: (c) in the SmA phase at T = 91 °C, (d) in the SmC* phase at T = 89 °C, and (e) in the SmCA* phase at T = 60 °C and at T = 30 °C (f). | ||
For simplicity, POM pictures are reported only in the process of cooling the samples; however, they have been captured on heating the samples, too. In Fig. 6(a), small and large domains of the SmC* phase of the mixture of 3F5HPhF9 sample with the formation of typical textures of smectic phases can be seen. The monochrome blue background indicates the presence of homeotropic alignment: in addition to the well-aligned domains of the helicoidal structures of the smectic phase, there are areas where the helix is unwound due to the surface effect. Fig. 6(b) shows the SmCA* phase of the sample 3F5HPhF9 in the presence of the deuterated probe. This phase exhibits the same types of defects as the SmC* phase, so that the SmC* – SmCA* transition can only be noticed through a colour change due to the sudden variation of birefringence at the phase transition. This phenomenon is due to the change of organization of molecules in layers, namely molecules tilted by +θ reorient in every second layer to −θ, either cooling or heating the sample while keeping the polarizers fixed. Fig. 6(c)–(f) show the sequence of mesophases observed on cooling the sample 3F7HPhF8 + probe. The SmA phase is characterized by large domains with typical focal conics with a brilliant green colour. The transition from the SmA to the SmC* phase and from the SmC* to the SmCA* phase is well determined by the rapid change in colour. Fig. 6(f) shows the SmCA* phase of the 3F7HPhF8 liquid crystal observed at T = 30 °C, i.e., below the crystallization temperature. The reason why the crystalline phase was not observable at low temperatures is attributed to the phenomenon of supercooling, which was also observed by DSC. The values of the phase transition temperature for the two samples obtained on cooling and on heating the samples are reported in Table 2.
| Sample and process | Phase | T (°C) | Phase | T (°C) | Phase | T (°C) | Phase | T (°C) | Phase |
|---|---|---|---|---|---|---|---|---|---|
| 3F5HPhF9 | Cr | SmCA* | SmC* | Iso | |||||
| Heating | * | 37.1 | * | 83.3 | * | 87.6 | * | ||
| 3F5HPhF9 + probe-d4 | Cr | SmCA* | SmC* | Iso | |||||
| Heating | * | * | 81.7 | * | 86.3 | * | |||
| Cooling | * | * | 78.3 | * | 85.4 | * | |||
| 3F7HPhF8 | Cr | SmCA* | SmC* | SmA | Iso | ||||
| Heating | * | 36.0 | * | 87.3 | * | 92.2 | * | 94.1 | * |
| 3F7HPhF8 + probe-d4 | Cr | SmCA* | SmC* | SmA | Iso | ||||
| Heating | * | * | 86.3 | * | 91.2 | * | 92.5 | * | |
| Cooling | * | * | 82.0 | * | 90.2 | * | 91.4 | * |
From POM measurements, a decrease of phase transition temperatures is observed in both cooling and heating cycles of samples with probe in comparison to samples without probe. This decrease of the transition temperatures can be explained as the effect of dilution of the LC by the short molecules of the deuterated probe. Moreover, if we compare these results with the data reported in the literature for the pure samples,19,20 it can be stated that the addition of a small amount of deuterated probe (about 1.5% by weight) slightly modifies these temperatures for both 3F5HPhF9 and 3F7HPhF8 samples. These little differences (from about 1 °C up to 4 °C in the case of the SmC*–SmCA* transition of the 3F7HPhF8 sample) are quite reasonable in liquid crystalline samples, since the heating and cooling rates can slightly affect the observed temperatures.67 It should be noted that small differences of about 1–4° of the phase transition temperatures from pure samples to deuterated samples are typically observed in liquid crystals.37,38,41,42 The most important aspect, for the purpose of the present work, is that the addition of a small percentage of deuterated probe does not suppress the mesophases, but only slightly modifies the transition temperatures.
3.2. Orientational order, tilt angle, layer spacing and helical pitch from experimental techniques
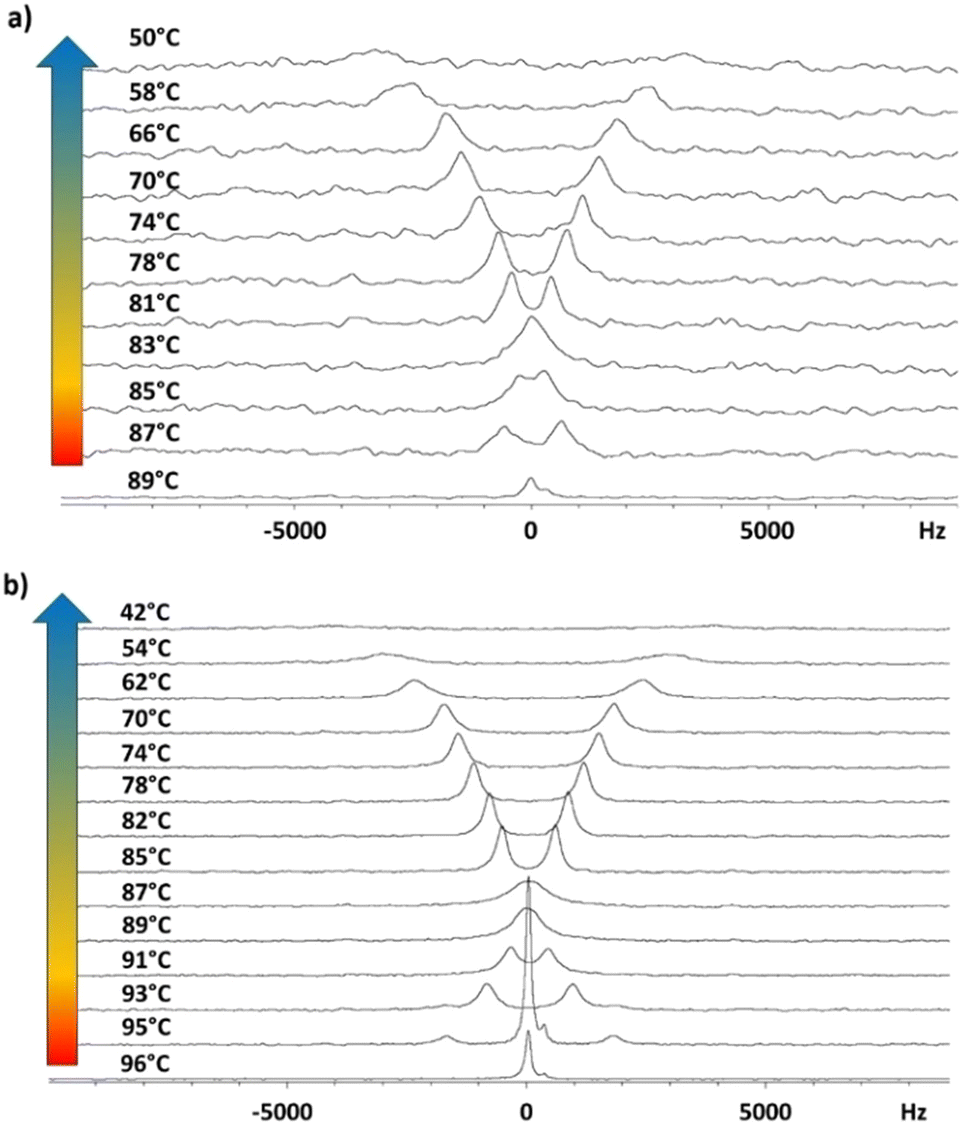 | ||
| Fig. 7 Stackplot of 2H NMR spectra recorded on cooling the sample from the isotropic phase to the crystal phase for the samples (a) 3F5HPhF9 with a small amount of biphenyl-4,4′-diol-d4 and (b) 3F7HPhF8 with a small amount of biphenyl-4,4′-diol-d4. | ||
Spectra recorded with proton decoupling are not reported here. However, 1H decoupling has the only effect of slightly reducing the linewidth, without altering the shape and main features of the spectra. This is due to the fact that in the case of low values of the orientational order, the 1H–2H dipolar coupling is very small. The transition from the isotropic to the liquid crystalline phase is identified by the spectral change from a singlet (typical of the isotropic phase) to a doublet (typical of oriented mesophases). In Fig. 7(b), the presence of a biphasic region (isotropic and smectic A phases) around T = 95 °C is observed in the case of the 3F7HPhF8 sample. The phase transition between different mesophases can be better visualized by the trend of the experimental quadrupolar splitting, Δνq, as a function of temperature, T (see Fig. 8). The vertical solid lines indicate the phase transition temperatures, which are well identified by a discontinuity in the temperature trend of the quadrupolar splitting for both samples. The vertical dashed lines indicate the occurrence of the crystalline phase as observed from 2H NMR measurements, which is typically associated to broad signals, as seen in Fig. 7. Qualitatively, the peculiarity of these two samples with respect to typical ferroelectric and antiferroelectric phases30,36,37,43 is the temperature behaviour of the quadrupolar splitting, Δνq, which changes in sign on decreasing the temperature, passing through zero. This feature, which has never been observed in any other ferroelectric smectic liquid crystals,30,32,36–43 is clearly observed in the stackplot of the 2H NMR spectra reported in Fig. 7, too.
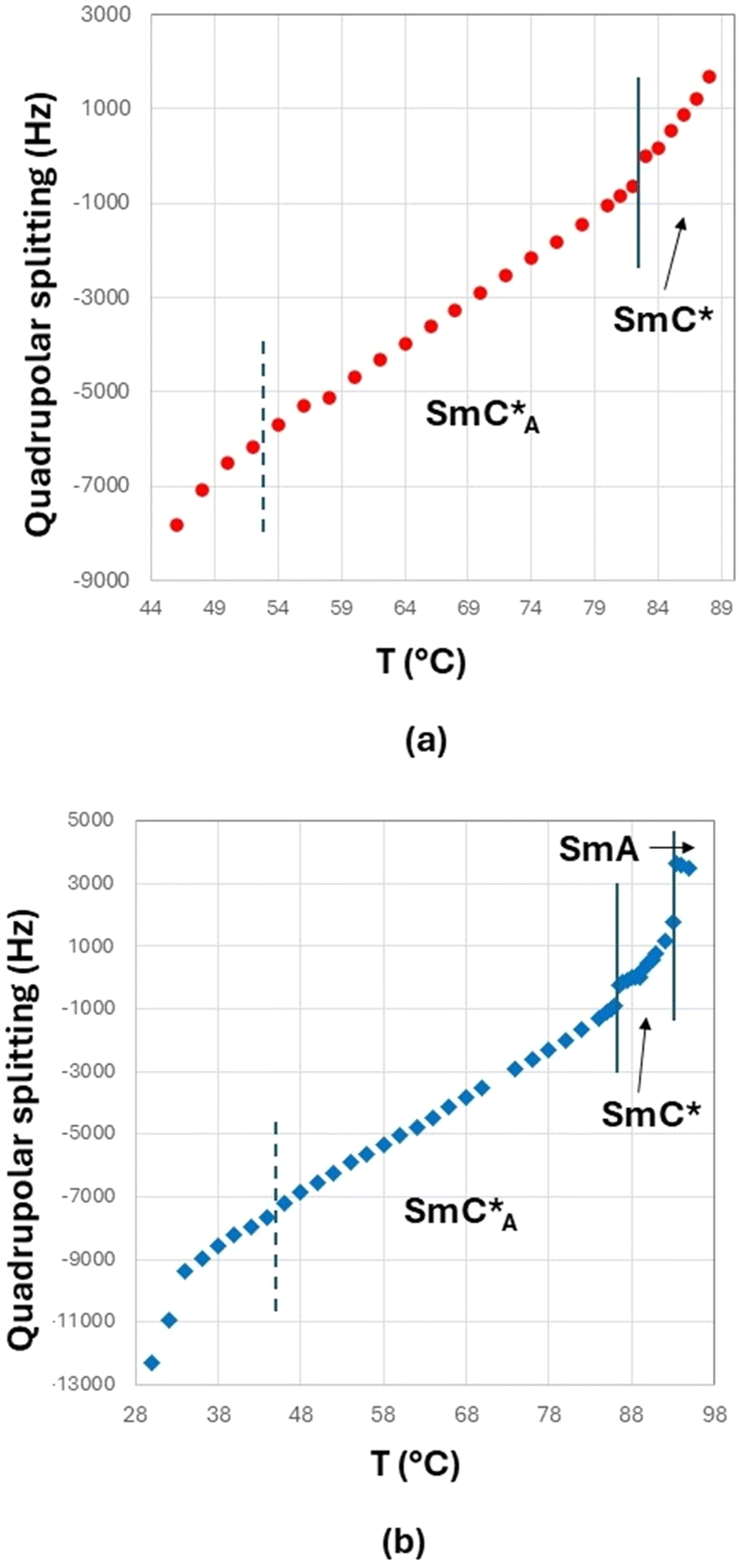 | ||
| Fig. 8 Trend of the quadrupolar splitting (Hz) as a function of temperature (°C), as determined by cooling the sample from the isotropic to the crystal phase, (a) for the 3F5HPhF9 sample with a small amount of biphenyl-4,4′-diol-d4 and (b) for the 3F7HPhF8 sample with a small amount of biphenyl-4,4′-diol-d4. The experimental error is ±100 Hz, which is the applied line broadening of the spectral processing. The vertical lines indicate the phase transitions as determined by 2H NMR. See the text for further explanations. | ||
In the case of the 3F5HPhF9 sample (see Fig. 8(a)), the direct transition from the isotropic to the SmC* phase is followed by a continuous decrease of the quadrupolar splitting with a discontinuity at 82–83 °C corresponding to the transition from the SmC* phase to the SmCA* phase.
In the case of the 3F7HPhF8 sample (see Fig. 8(b)), the value of the quadrupolar splitting increases on decreasing the temperature in the SmA phase, and it starts decreasing at the transition from the SmA phase to the SmC* phase (93–94 °C). Below this temperature, the value of the quadrupolar splitting continues to decrease and a discontinuity at 86–87 °C is observed corresponding to the transition from the SmC* phase to the SmCA* phase.
As reported in the literature for ferroelectric smectic LC phases,30,37,43 the decrease of the quadrupolar splitting on decreasing the temperature is typical of chiral smectic phases with the helical axis aligned parallel to the external magnetic field or, similarly, with the smectic layers’ normal parallel to the external magnetic field. There are a few examples of LC ferroelectric compounds displaying a different trend:39 an increase of the quadrupolar splitting on decreasing the temperature in the SmC* phase. This behaviour, however, is due to the unwinding of the helical axis of the SmC* phase by the external NMR magnetic field. In that case, the layers are tilted with respect to the magnetic field, while the phase director, n, is aligned parallel to the magnetic field, as first observed in a ferroelectric smectic LC.45 This phenomenon is due to the fact that the critical magnetic field for the unwinding of the supramolecular SmC* structure, Hc, is lower than the operating NMR magnetic field, H.39 In the present case, namely for both 3F5HPhF9 and 3F7HPhF8 samples, the critical field Hc is clearly higher than H (=9.4 T); in fact we do not observe any helix unwinding.
As well-known from previous studies,30–35,37,39,43 it is possible to relate the experimental quadrupolar splitting, Δνq, with the local orientational order parameters, the so-called Saupe order parameters, Sii, according to eqn (1):68
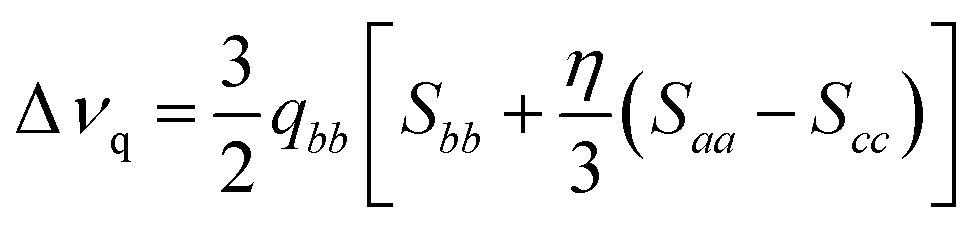 | (1) |
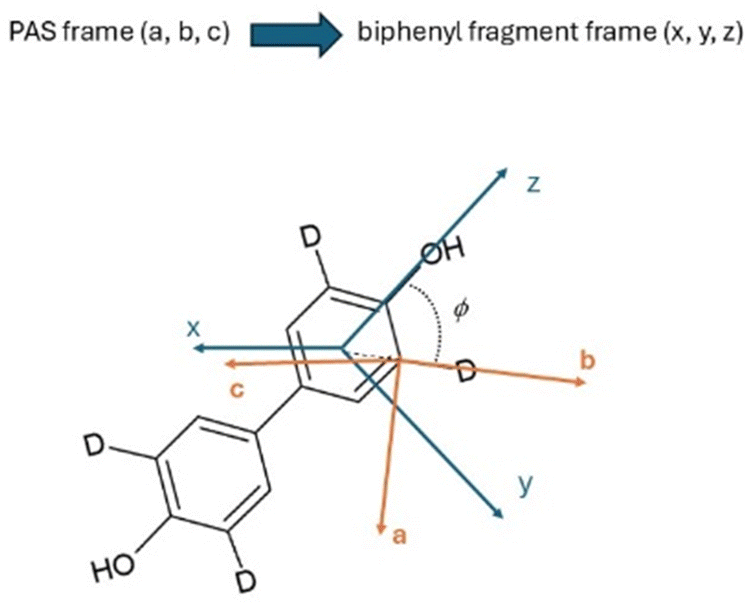 | ||
| Fig. 9 Principal axes system (PAS) versus local biphenyl fragment system, with relative axes orientation, for the deuterated probe biphenyl-4,4′-diol-d4. The angle ϕ between the ‘b’ axis and the ‘z’ axis is shown. | ||
The following equation arises:30
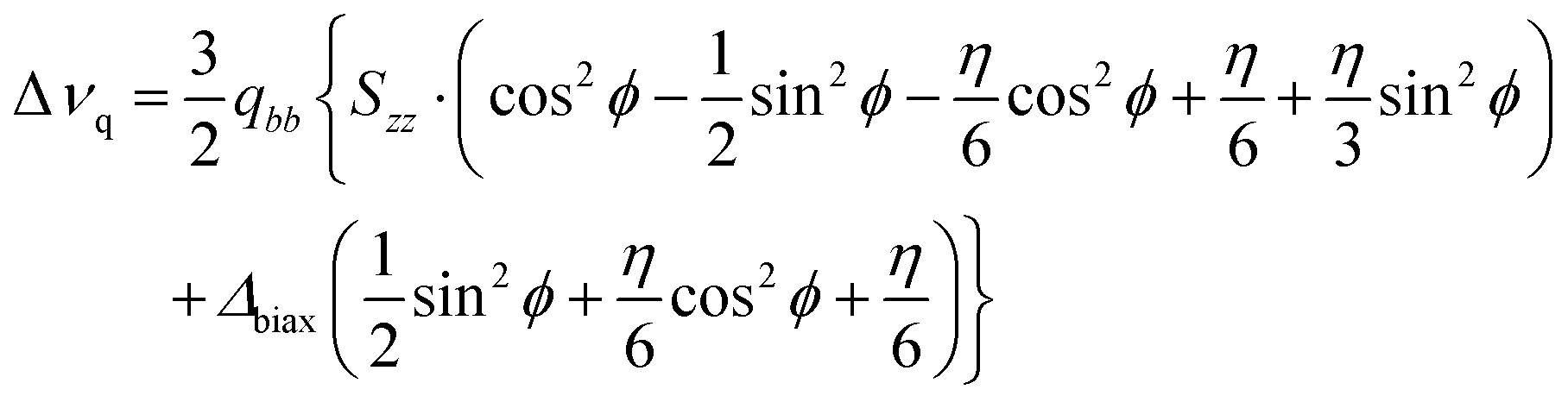 | (2) |
In the absence of additional experimental data (such as 1H–2H or 2H–2H dipolar couplings), a temperature independence of the fragment biaxiality is assumed. Two cases are considered and discussed. The first case is the assumption of uniaxiality of the deuterated probe (Δbiax = 0), which is reasonable in uniaxial phases, but probably not in the case of highly biaxial phases, such as the ferroelectric ones.30–32,37 The second case is the assumption of a relatively high value of the biaxiality for the molecular probe (Δbiax = 0.05). This value of biphenyl biaxiality is the highest value found in the SmC* phase by 2H NMR analysis for LC chiral smesogens,30,37 and it can be considered a reasonable value in highly ordered mesophases, such as the ones investigated in the present work. Note that the exact sign of the quadrupolar splitting used in eqn (2), namely Δνq, is opposite to that reported in Fig. 8, in order to get reliable values of the orientational order parameters.68,69
In Fig. 10, the values of the principal orientational order, Szz, obtained by fixing the biphenyl fragment biaxiality to these two extreme values (Δbiax = 0 − orange symbols and Δbiax = 0.05 − blue symbols) are reported for the two orthoconic samples.
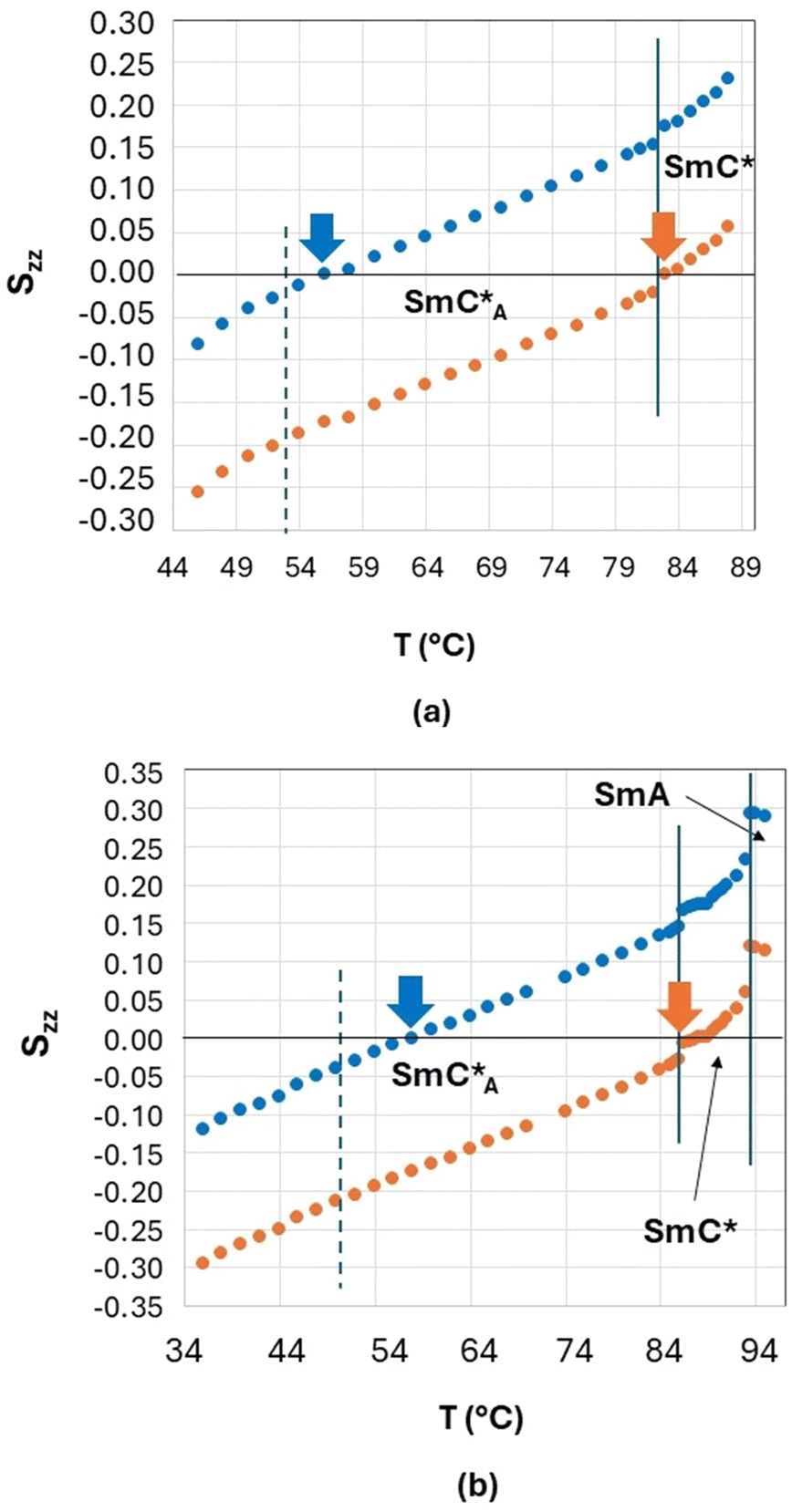 | ||
| Fig. 10 Values of the principal orientational order, Szz, obtained by fixing the fragment biaxiality, versus temperature (a) for the 3F5HPhF9 sample with a small amount of biphenyl-4,4′-diol-d4 and (b) for the 3F7HPhF8 sample with a small amount of biphenyl-4,4′-diol-d4. Values obtained with Δbiax = 0 are represented by orange symbols, while values obtained with Δbiax = 0.05 are represented by blue symbols. The vertical lines indicate the mesophase transitions and the orange and blue arrows indicate the temperature corresponding to zero orientational order in the two cases (Δbiax = 0 and Δbiax = 0.05, respectively). | ||
As shown in Fig. 10, the values of Szz range from 0.3 to −0.3 depending on the sample and based on the fixed value of fragment biaxiality. These values are rather small with respect to typical values of Szz of ferroelectric and antiferroelectric smectic liquid crystals,30–43 but this is reasonable, since we have used a molecular deuterated probe diluted in the LC phases. As known in the literature,70 the orientational order of a deuterated molecular probe is typically scaled with respect of the orientational order of the LC molecules, mainly due to the fast dynamics which characterize small molecular probes (both reorientational and translational ones) thus giving rise to a partial averaging out of the observed spectra. However, it is reasonable to assume that the deuterated probe, being a substituted biphenyl fragment, is quite representative of the aromatic core of the orthoconic LC samples, which contain a biphenyl moiety. This means that the values of Szz obtained for the deuterated probe are probably lower than the values of Szz of the LC molecules, as typically observed in molecular probes, however, the Szz temperature dependence and the peculiar behaviour observed in the antiferroelectric phase is probably the same of the orthoconic LC molecules. In fact, what is unique of the case reported in this paper is the fact that the orientational order passes from positive to negative values, in a gradual way, passing through zero. This aspect can be explained with the very high value of the tilt angle, as will be discussed in the following paragraph.
 which depends on the tilt angle θ(T):
which depends on the tilt angle θ(T):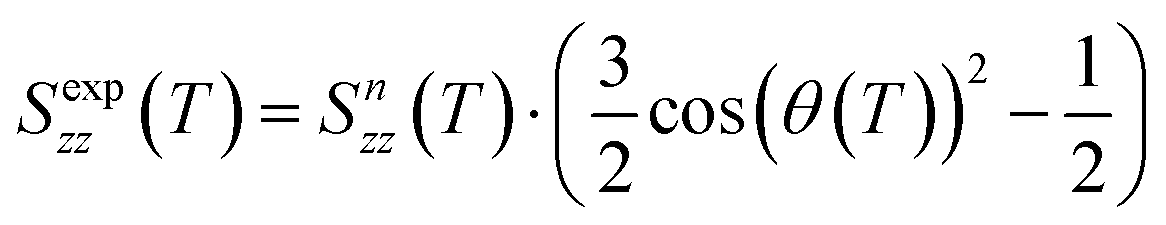 | (3) |
Since the value of Snzz(T) is not accessible from NMR experiments except in the case of helix unwinding,39 it is normally extrapolated by the SmA phase.30–37 In the case of the 3F7HPhF8 sample added with the small deuterated molecular probe, the value of the orientational order of the LC mesophase, Snzz(T), was indeed extrapolated from the SmA phase, where the molecules are on average aligned along the magnetic field. These extrapolated values of Snzz(T) are reported in Fig. 11 together with the experimental orientational order parameter Sexpzz(T) determined by NMR, for the case of biaxiality Δbiax = 0 and Δbiax = 0.05.
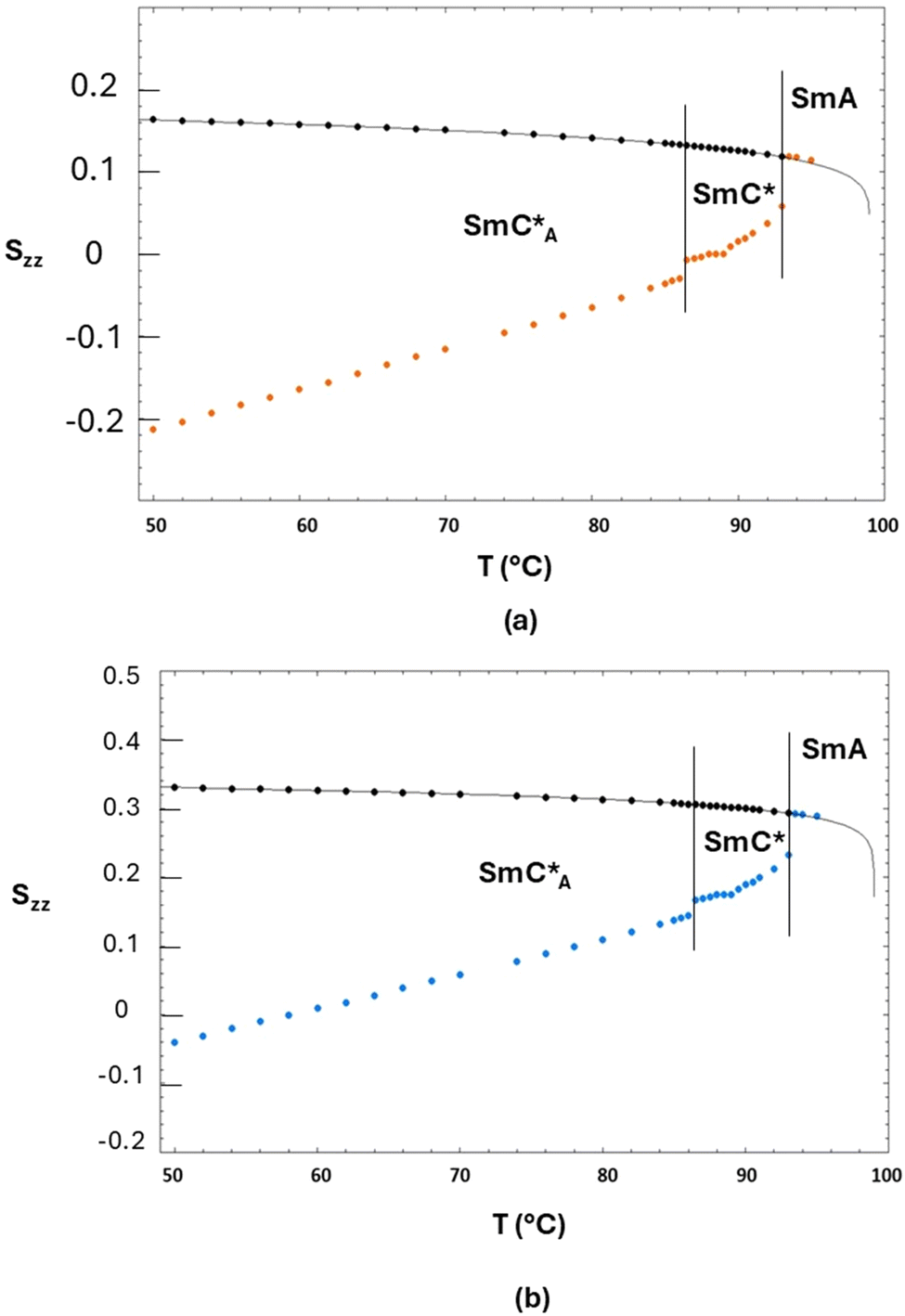 | ||
| Fig. 11 (a) Experimental orientational order parameter Sexpzz(T) determined by NMR with Δbiax = 0 (orange symbols) and (b) experimental orientational order parameter Sexpzz(T) determined by NMR with Δbiax = 0.05 (blue symbols) superimposed with the values (black symbols) of the mesophase orientational order Snzz(T) extrapolated from the SmA phase of the 3F7HPhF8 sample. The black curve is the best fitting curve by using eqn (4). | ||
Fig. 11(a) represents the values of the orientational order determined by fixing the fragment biaxiality to 0 (orange circles) and Fig. 11(b) represents the values of the orientational order determined by fixing the fragment biaxiality to 0.05 (blue circles).
As seen from the fitting of the extrapolated data (black circles in Fig. 9(a) and (b)), the trend of Snzz(T) is well represented by the Landau-de Gennes equation:30
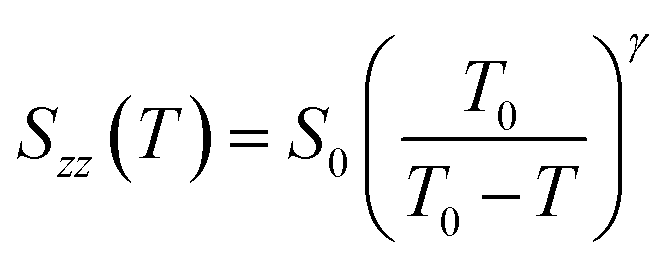 | (4) |
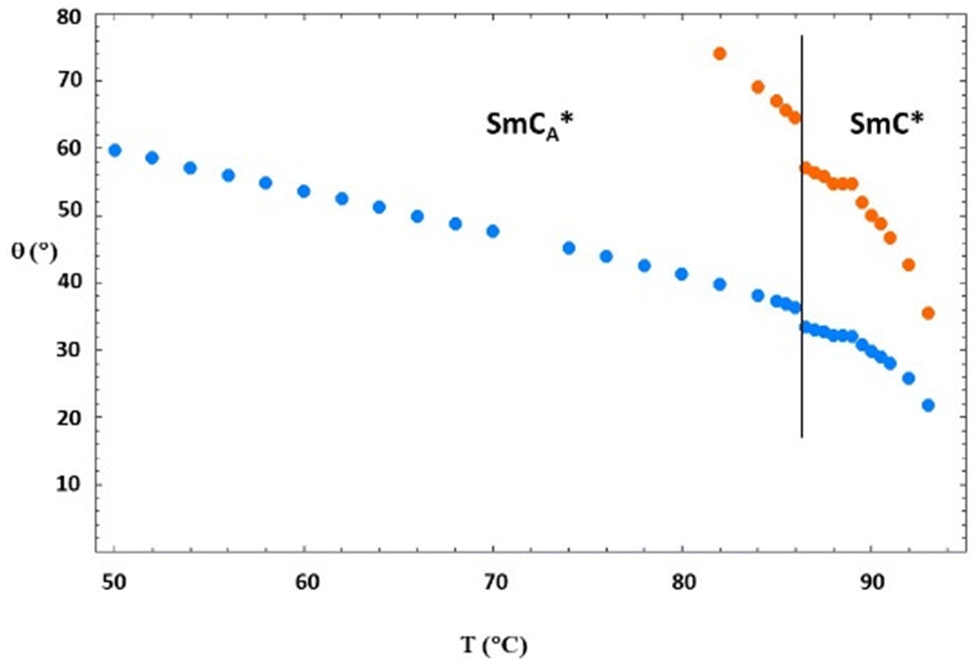 | ||
| Fig. 12 Values of the tilt angle θ(T) obtained by using eqn (3) as described in the text. The blue symbols refer to the case of Δbiax = 0.05 and the orange symbols refer to the case of Δbiax = 0. | ||
It is interesting to note that in the case of Δbiax = 0, the values of the tilt angle are extremely high in the SmC* phase, thus indicating that this case is not reasonable. In contrast, in the case of Δbiax = 0.05, the values of the tilt angle in the SmC* phase are in the range of 22°–33°, which is rather similar to that in the case of normal ferroelectric phases.30–37,39,43 When decreasing the temperature, in the SmCA* phase, the tilt angle increases from 35° at the SmC*–SmCA* phase transition to 60° close to the crystalline phase. However, at this lower temperature, since the system probably undergoes pre-crystallization inside the NMR magnet, data may be affected by a bias. It is interesting to note that the average value of the tilt angle in the antiferroelectric phase from 87 °C to about 50 °C is about 45°, which is the ideal tilt angle of orthoconic LC samples.19,20
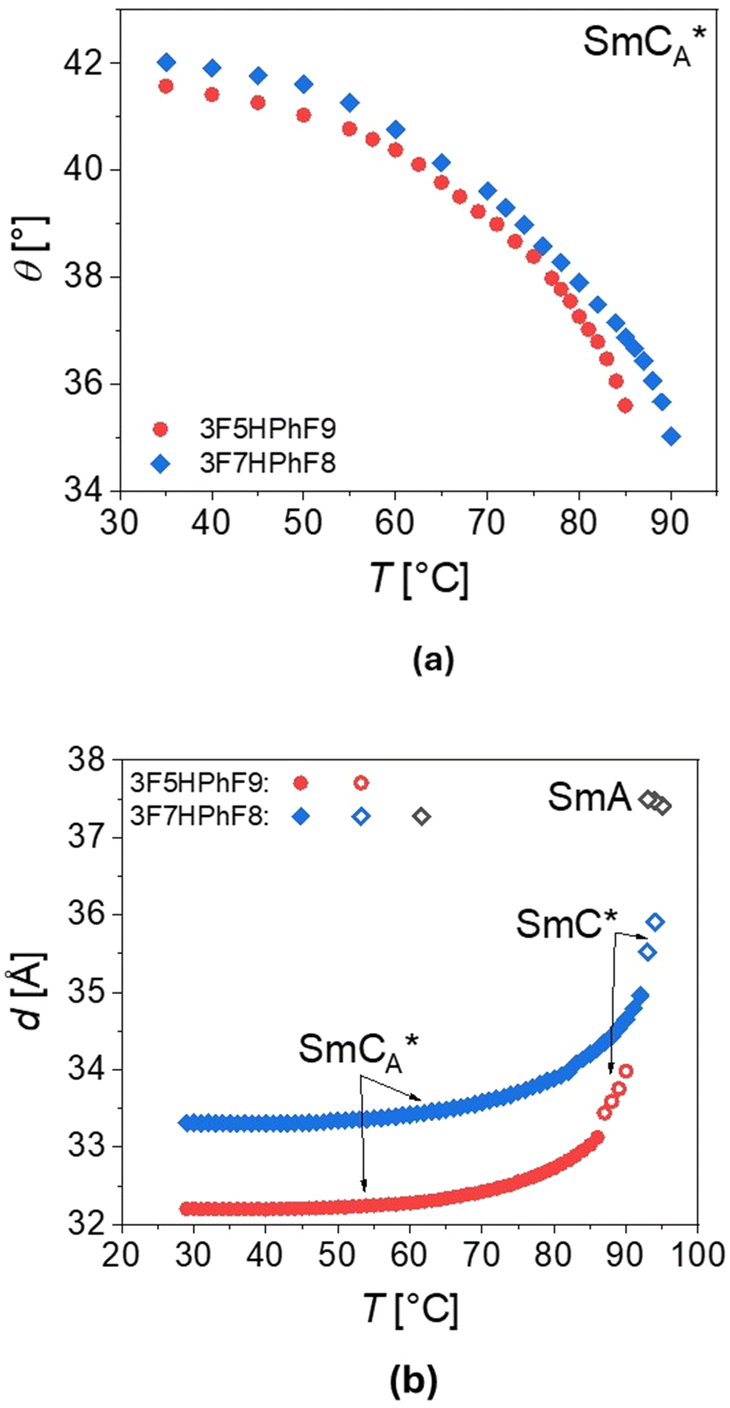 | ||
| Fig. 13 Temperature dependence of the tilt angle θ (a) and layer spacing d (b) for 3F5HPhF9 (red points) and 3F7HPhF8 (blue points), obtained as described in the text. | ||
The thickness of the smectic layers d was determined from the low-angle peak of the X-ray diffraction patterns. For 3F7HPhF8, the thickness is larger in both SmCA* and SmC* phases than for the 3F5HPhF9 compound. For both compounds, the thickness of the layer in the SmCA* phase rapidly increases with temperature before the SmCA*–SmC* transition. Generally, the XRD patterns of the synclinic order in the SmC* phase and the anticlinic order in the SmCA* phase are indistinguishable, although the SmCA*–SmC* transition is sometimes visible as a discontinuity in the location or intensity of the low-angle peak from the smectic layer’ thickness, as we observed. In the SmA phase of the 3F7HPhF8 compound, the layer spacing decreases with increasing temperature. At the SmA–SmC* transition, the layer spacing decreases discontinuously from 37.5 Å to 36.0 Å, which corresponds to the relative layer shrinkage of ca. 4%.
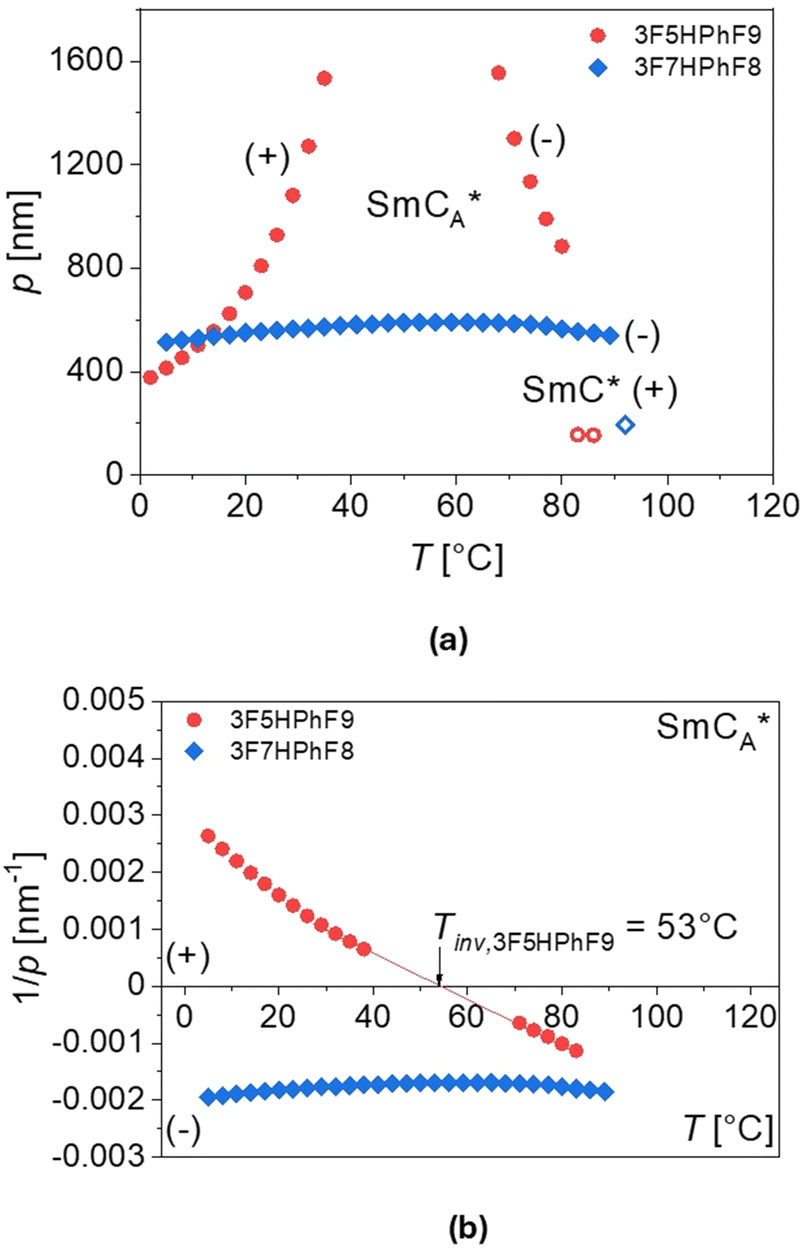 | ||
| Fig. 14 Temperature dependence of the helical pitch p (a) and the inverse helical pitch 1/p (b) for 3F5HPhF9 (red points) and 3F7HPhF8 (blue points). The (+) and (−) signs denote the right- and left-handed helix, respectively. | ||
3.3. Computational investigations
The MD simulations offer a microscopic view of the structure of the orthoconic phase of 3F5HPhF9. In Fig. 15 we show the density profiles along the z axis of various reference atoms in the molecule (see Fig. 3). The density profiles reveal the layered structure of the phase. Some atoms appear to have a single peak per layer while some others have two peaks per layer. This can be easily understood as a result of the presence of the same number of molecules oriented with the fluorinated chain either up or down. This is consistent with the actual model of the antiferroelectric phase in orthoconic LCs, as reported in Fig. 1c. The extreme parts of the rigid core, atoms C0S (and also C1W) or C04, have two peaks per layer corresponding to the molecules oriented up or down within the same layer. Atoms in the central part of the rigid core show a broader single peaked distribution since the central part of the core is at the same level with respect to the z axis for both orientations. The terminal carbon of the two chains should, in principle, also show a doubly peaked density profile; however this is broadened by the flexibility of the chains. The effect is very strong in the alkyl chain (C2P), where the two peaks are completely lost, while for the fluoroalkyl chain terminal carbon (C1O) there is a residual bimodal distribution of the fluorinated ends within each layer. This suggests a larger degree of disorder of the alkyl chain compared to the polyfluorinated chain, a point that will be further discussed below. | ||
| Fig. 15 (top) Density profiles along the director of the phase (z axis) of the atoms labelled C0E, C1O, C2P and C01. (bottom) Density profiles along the director of the phase (z axis) of the atoms C04, C0S and C1W. For labels, see Fig. 3. | ||
The layer thickness can be obtained from the distance of the peaks. We selected the three density profiles showing a single peak per layer, that is, the number density of C2P, C0E and C01 carbon atoms. From these, we obtained a value of the layer thickness of 33.17 ± 0.005 Å. The result is in reasonable agreement with the experiments showing a layer thickness ranging from about 32.2 A at room temperature to 33.2 A at 350 K, just before the transition into the SmA phase. Moreover, we can estimate the average orientation of the molecules in the bulk phase; some selected orientational data are reported in Table S1 (ESI†). As we can see, the rigid core of the molecules is on average oriented at approximately 42° with respect to the director of the phase, which is perpendicular to the layers. Moreover, the orientation in the yz plane points in opposite directions in adjacent layers (see also Fig. 4). This is true for the biphenyl axis, the phenyl axis and the whole rigid core. The result is in qualitative agreement with the experimental observation of a tilt angle ranging from about 42° at room temperature, down to about 36° at 350 K, just before the transition into the SmA phase. Finally, in Fig. 16, we report some selected orientational distribution functions obtained from the simulations.
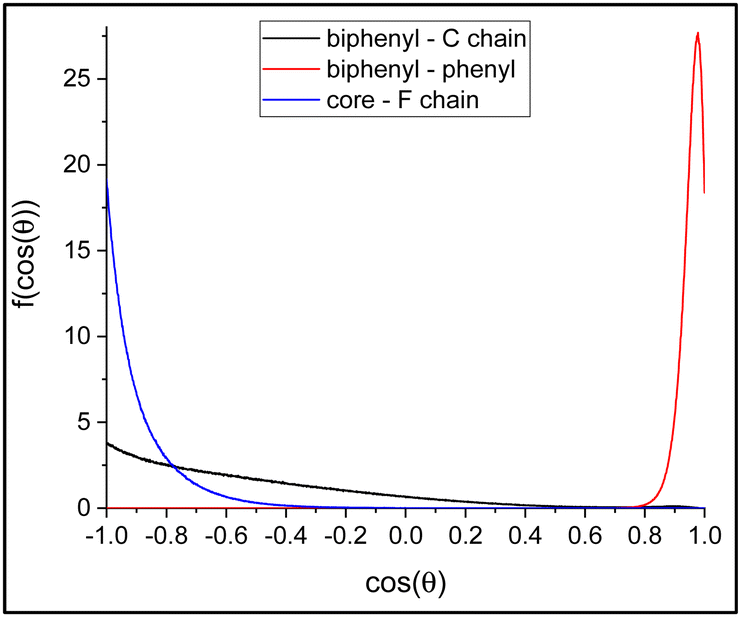 | ||
| Fig. 16 Selected distribution functions f(cosθ), that is, the probability to find a given vector with a cosθ value between cosθ and cosθ + dcosθ. (red line) The distribution of the angle between the biphenyl group (C0S–C0E vector) and the phenyl group (C01–C04 vector); (black line) the distribution of the angle between the biphenyl group (C0S–C0E vector) and the alkyl chiral chain (C1W–C2P vector); and (blue line) the distribution of the angle between the rigid core (C0S–C04 vector) and the polyfluorinated chain (C1O–C04 vector). | ||
As we can see, the distribution of the angle between the phenyl and biphenyl moiety in the bulk is very strongly peaked at cosθ = 0.9759, corresponding to approximately 12.6°, indicating a rigid core with the two aromatic groups well aligned with each other. In contrast, the average relative orientation of the two chains shows interesting differences. The distribution of the angle between the rigid core and the polyfluorinated chain is peaked at 180° and it appears relatively narrow, a result that can be expected and attributed to the orienting mean field of the LC phase favouring an all-trans conformation of the chain. Instead, the probability distribution of the angle between the biphenyl core and the chiral alkyl chain is very broad. Indeed, the maximum probability is still for an orientation at 180°; however, a non-negligible population can be found also at relatively small angles even less than 90°. This suggests a much less ordered arrangement of the alkyl chain compared to the polyfluorinated chains, as already inferred from the density profiles of Fig. 15. In fact, conformers with the alkyl chain at approximately 90° with the rigid core can be clearly spotted in the snapshot of the simulation box in Fig. 4. To confirm this structural arrangement, we ran a scan of the dihedral angle between the carbonyl carbon, the oxygen, the chiral carbon (C1W) and the next carbon in the alkyl chain (see Fig. 3), from 0 to 360° in steps of 10°, using the software Gaussian 1672 at the semiempirical AM1 level of theory.73 The calculations revealed that the conformation shown in Fig. 17, featuring an alkyl chain approximately perpendicular to the rigid core, is the absolute minimum and it lies 0.8 kcal mol−1 lower in energy than the elongated conformer shown in Fig. 4.
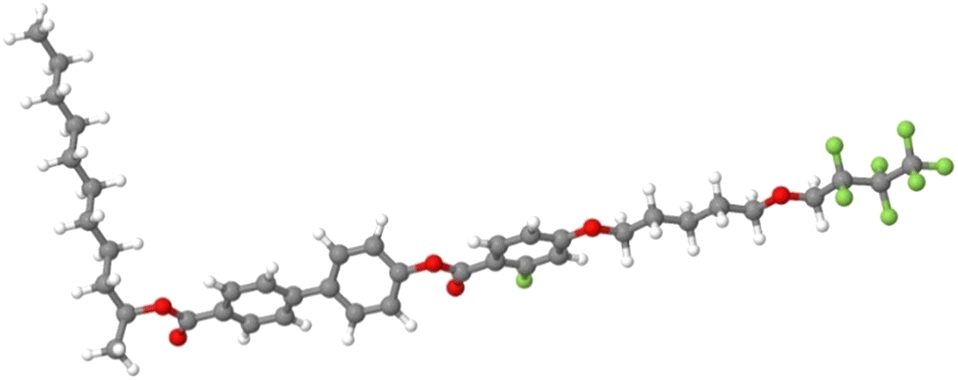 | ||
| Fig. 17 Lowest energy conformation at the AM1 semiempirical level of theory, in gas phase, obtained by scanning the dihedral angle representing the orientation of the alkyl chain with respect to the rigid core (see text for details). | ||
The results discussed here based on the computational study of the 3F5HPhF9 orthoconic liquid crystal are in agreement with the X-ray structure of a structurally similar compound showing the ferroelectric and antiferroelectric phases studied by Toriumi et al.53 The evidence that a ‘bent’ conformation could be favored in highly ordered smectic phases, such as the ferroelectric phase SmC*, with respect to less ordered ones, such as SmA, is supported by previous 13C NMR and 2H NMR investigations on liquid crystals showing the transition from SmA to SmC* phases.38,43,47,49,74
4. Conclusions
In this paper, the first 2H NMR investigation and fully atomistic molecular dynamics study of orthoconic liquid crystals are reported. Two orthoconic LC systems of the series 3FmHPhFr were doped with a small amount of a deuterated probe in order to investigate these two LC molecules by 2H NMR spectroscopy. The mesophase characterization of these mixtures was investigated by POM and DSC, showing that the addition of the probe does not influence the occurrence of the ferroelectric and antiferroelectric phases of the investigated liquid crystals. Only small differences in temperature transitions were observed, as expected due to the addition of the small deuterated probe. As a main result, the trend of the 2H NMR spectra within the mesophases revealed a peculiar phenomenon, not observed in any other ferro-/antiferroelectric LC systems. In particular, the trend of the quadrupolar splitting passes from negative to positive values on decreasing the temperature, and for certain values of the temperature it is zero. This behaviour is a clear indication of very high values of the tilt angle of the longitudinal axis of the deuterated probe with respect to the helical axis of the SmC* and SmCA* phases, which is parallel to the external magnetic field. By comparing the 2H NMR results with those of ferroelectric LC mesogens we can confirm that for both orthoconic LC systems the helical supramolecular structure is not unwound by the operating NMR magnetic field (9.46 T). In the case of one of the two orthoconic systems here investigated, that is, the one showing a SmA phase at high temperatures, the trend of the tilt angle in the tilted LC mesophases was estimated by analysing the 2H NMR spectra and it was compared with that obtained from electrooptic measurements. These values are quite in agreement, confirming the typical properties of orthoconic liquid crystals with a tilt angle close to 45°. Additionally, in this work, a detailed fully atomistic molecular dynamics investigation was performed on one of these two similar LC molecules. Among the obtained results, MD simulations indicate that the rigid core of these molecules is on average oriented at approximately 42° with respect to the director of the phase, which is perpendicular to the layers. This value is in very good agreement with experimental results commented above. In addition, MD simulations show that in the packed organization of the orthoconic LC, which is typical of the antiferroelectric phase, the most favored conformer has the lateral chiral chain quite bent with respect to the aromatic core axis, suggesting a significant effect of the supramolecular organization on the averaged conformer. The evidence of a bent conformation in the chiral smectic mesophases formed by the orthoconic mesogen is in agreement with previous NMR and theoretical studies on ferroelectric liquid crystals. Further investigations on these systems are in progress to clarify the relationship between the molecular structure of these fluorinated molecules and the occurrence of very high tilted smectic phases.Author contributions
Anna Drzewicz: investigation, formal analysis. Mario Cifelli: investigation. Marzena Tykarska: conceptualization, writing – review & editing. Giacomo Saielli: investigation, formal analysis, writing – original draft, writing – review & editing. Martina Rossi: investigation, formal analysis. Valentina Domenici: supervision, conceptualization, investigation, writing – original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. D. and M. R. thank Maria Rita Carosi and Chiara Pelosi for DSC measurements of the LC mixtures. Calculations were run on the Linux clusters of the C3P Community of the Department of Chemical Sciences of the University of Padova which is acknowledged. A. D. thanks Przemysław Morawiak from the Military University of Technology in Warsaw, Poland, for XRD measurements of the pure compounds.References
- S. T. Lagerwall, Ferroelectric and antiferroelectric liquid crystals, Wiley-VCH, New York, 1998 Search PubMed.
- D. Demus, S. Diele, S. Grande and H. Sackmann, Polymorphism in Thermotropic Liquid Crystals, in Advances in Liquid Crystals, ed. G. H. Brown, Liquid Crystal Institute, Kent State University, Kent, Ohio, 1983, vol. 6, pp. 1–107 Search PubMed.
- R. B. Meyer, L. Liebert, L. Strzelecki and P. Keller, J. Physique Lett., 1975, 36, 69 CrossRef.
- I. Musevic, R. Blinc and B. Zeks, The Physics of Ferroelectric and Antiferroelectric Liquid Crystals, World Scientific, 2000 Search PubMed.
- S. T. Lagerwall, Adv. Colloid Interface Sci., 2014, 208, 1 CrossRef CAS PubMed.
- A. Yoshizawa, Crystals, 2024, 14, 350 CrossRef CAS.
- D. M. Walba, Science, 1995, 270, 250 CrossRef CAS.
- J. W. Goodby, Proc. R. Soc. A, 2012, 468, 1521 CrossRef CAS.
- A. D. L. Chandani, E. Gorecka, Y. Ouchi, H. Takezoe and A. Fukuda, Jpn. J. Appl. Phys., 1998, 28, L1265 CrossRef.
- R. Blinc, in Models for Phase Transitions in Ferroelectric Liquid Crystals: Theory and Experimental Results, Phase Transitions in Liquid Crystals, ed. S. Martellucci and A. N. Chester, NATO Science Series B: Physics, Plenum Press, New York, 1992, ch. 22, pp. 343–363 Search PubMed.
- P. Rudquist, Liq. Cryst., 2013, 40, 1678 CrossRef CAS.
- K. D’Have, P. Rudquist, S. T. Lagerwall, H. Pauwels, W. Drzewiński and R. Dabrowski, Appl. Phys. Lett., 2000, 76, 3528 CrossRef.
- N. A. Clark and S. T. Lagerwall, Appl. Phys. Lett., 1980, 36, 899 CrossRef CAS.
- K. D’have, A. Dahlgren, P. Rudquist, J. P. F. Lagerwall, G. Anderson, M. Matuszczyk, S. T. Lagerwall, R. Dabrowski and W. Drzewinski, Ferroelectrics, 2000, 244, 115 CrossRef.
- R. Dąbrowski, J. Gąsowska, J. Otón, W. Piecek, J. Przedmojski and M. Tykarska, Displays, 2004, 25, 9 CrossRef.
- L. Marino, S. Marino, D. Wang, E. Bruno and N. Scaramuzza, Soft Matter, 2014, 10, 3842 RSC.
- M. Żurowska, R. Dąbrowski, J. Dziaduszek, K. Garbat, M. Filipowicz, M. Tykarska, W. Rejmer, K. Czupryński, A. Spadło, N. Bennis and J. M. Otón, J. Mater. Chem., 2011, 21, 2144 RSC.
- R. Dąbrowski, W. Drzewiński, J. Dziaduszek, J. Gąsowska, P. A. Henderson, P. Kula, J. M. Otón and N. Bennis, Opto-Electron. Rev., 2007, 15, 32 Search PubMed.
- K. Milewska, W. Drzewiński, M. Czerwiński and R. Dąbrowski, Liq. Cryst., 2015, 42, 1601 CAS.
- K. Milewska, W. Drzewiński, M. Czerwiński, R. Dąbrowski and W. Piecek, Mater. Chem. Phys., 2016, 171, 33 CrossRef CAS.
- A. Drzewicz, A. Bombalska and M. Tykarska, Liq. Cryst., 2019, 46, 754 CrossRef CAS.
- M. Tykarska, A. Drzewicz, M. Szala and M. Żurowska, Liq. Cryst., 2018, 45, 1385 CrossRef CAS.
- M. Tykarska, M. Czerwiński and A. Drzewicz, J. Mol. Liq., 2019, 292, 110379 CrossRef CAS.
- M. Tykarska and M. Czerwiński, Liq. Cryst., 2016, 43, 462 CrossRef CAS.
- P. Nayek, S. Ghosh, S. Kundu, S. K. Roy, T. P. Majumder, N. Bennis, J. M. Oton and R. Dabrowski, J. Phys. D: Appl. Phys., 2009, 42, 225504 CrossRef.
- P. Nayek, S. Ghosh, S. Roy, T. P. Majumder and R. Dabrowski, J. Mol. Liq., 2012, 175, 91 CrossRef CAS.
- Ł. Kolek, M. Jasiurkowska-Delaporte, M. Massalska-Arodź, W. Szaj and T. Rozwadowski, J. Mol. Liq., 2020, 320, 114338 CrossRef.
- R. Dąbrowski, P. Kula, Z. Raszewski, W. Piecek, J. M. Otón and A. Spadło, Ferroelectrics, 2010, 395, 116 CrossRef.
- J. Fitas, A. Dłubacz, P. Fryń, M. Marzec, T. Jaworska-Gołąb, A. Deptuch, K. Kurp, M. Tykarska and M. Żurowska, Liq. Cryst., 2017, 44, 566 CrossRef CAS.
- M. Cifelli, V. Domenici and C. A. Veracini, Curr. Opin. Colloid Interface Sci., 2013, 18, 190 CrossRef CAS.
- V. Domenici, Pure Appl. Chem., 2011, 83, 67 CAS.
- V. Domenici, Pure Appl. Chem., 2007, 79, 21 CAS.
- V. Domenici, Liq. Cryst. Today, 2017, 26, 2 CrossRef.
- V. Domenici, C. A. Veracini and B. Zalar, Soft Matter, 2005, 1, 408 RSC.
- V. Domenici, Soft Matter, 2011, 7, 1589 RSC.
- V. Domenici, M. Geppi and C. A. Veracini, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 1 CrossRef CAS.
- M. Cifelli, V. Domenici, A. Marini and C. A. Veracini, Liq. Cryst., 2010, 37, 935 CrossRef CAS.
- V. Domenici, M. Cifelli, A. Marchetti, M. Lelli, V. Hamplova, M. Kaspar and C. A. Veracini, Mol. Cryst. Liq. Cryst., 2012, 553, 103 CrossRef CAS.
- V. Domenici, A. Marini, C. A. Veracini, J. Zhang and R. Y. Dong, ChemPhysChem, 2007, 8, 2575 CrossRef CAS PubMed.
- M. Cifelli, V. Domenici, S. V. Dvinskikh, M. Glogarova and C. A. Veracini, Soft Matter, 2010, 6, 5999 RSC.
- D. Catalano, L. Chiezzi, V. Domenici, M. Geppi and C. A. Veracini, J. Phys. Chem. B, 2003, 107, 10104 CrossRef CAS.
- D. Catalano, V. Domenici, A. Marini, C. A. Veracini, A. Bubnov and M. Glogarova, J. Phys. Chem. B, 2006, 110, 16459 CrossRef CAS PubMed.
- V. Domenici, Crystals, 2024, 14, 823 CrossRef CAS.
- V. Domenici, M. Lelli, M. Cifelli, V. Hamplova, A. Marchetti and C. A. Veracini, ChemPhysChem, 2014, 15, 1485 CrossRef CAS PubMed.
- D. Catalano, M. Cifelli, V. Domenici, K. Fodor-Csorba, R. Richardson and C. A. Veracini, Chem. Phys. Lett., 2001, 346, 259 CrossRef CAS.
- M. Cifelli, V. Domenici and C. A. Veracini, Mol. Cryst. Liq. Cryst., 2005, 429, 167 CrossRef CAS.
- A. Marini and V. Domenici, Ferroelectrics, 2010, 395, 46 CrossRef.
- A. Yoshizawa, H. Kikuzaki and M. Fukumasa, Liq. Cryst., 1995, 18, 351 CrossRef CAS.
- A. Marini and V. Domenici, J. Phys. Chem. B, 2010, 114, 10391 CrossRef CAS PubMed.
- S. M. Yayloyan, L. S. Bezhanova and E. B. Abrahamyan, Ferroelectrics, 2000, 245, 157 CrossRef CAS.
- F. Biscarini, C. Zannoni, C. Chiccoli and P. Pasini, Mol. Phys., 1991, 73, 439 CrossRef CAS.
- D. Paschek, S. Y. Yakovenko, A. A. Muravski and A. Geiger, Ferroelectrics, 1998, 212, 45 CrossRef CAS.
- H. Toriumi, M. Yoshida, M. Mikami, M. Takeuchi and A. Mochizuki, J. Phys. Chem., 1996, 100, 15207 CrossRef CAS.
- Z. Raszewski, J. Kędzierski, P. Perkowski, W. Piecek, J. Rutkowska, S. Kłosowicz and J. Zieliński, Ferroelectrics, 2002, 276, 289 CrossRef CAS.
- I. Muševič, M. Škarbot, G. Heppke and H. T. Nguyen, Liq. Cryst., 2002, 29, 1565 CrossRef.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, P19 CrossRef.
- W. L. Jorgensen and J. Tirado-Rives, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6665 CrossRef CAS PubMed.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2017, 121, 3864 CrossRef CAS PubMed.
- L. S. Dodda, I. Cabeza de Vaca, J. Tirado-Rives and W. L. Jorgensen, Nucleic Acids Res., 2017, 45, W331 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463 CrossRef CAS.
- M. E. Neubert, Characterization of mesophase types and transitions, in Liquid Crystals: Experimental Study of Physical Properties and Phase Transitions, ed. S. Kumar, Cambridge University Press, Cambridge, UK, 2001, pp. 29–62 Search PubMed.
- C. A. Veracini, NMR spectra in liquid crystals: the partially averaged spin Hamiltonian, in Nuclear Magnetic Resonance of Liquid Crystals, ed. J. W. Emsley, Reidel, Dordrecht, Netherlands, 1985, ch. 5, vol. 141, pp. 99–118 Search PubMed.
- J. W. Emsley, Measurement of orientational ordering by NMR, in Nuclear Magnetic Resonance of Liquid Crystals, ed. J. W. Emsley, Reidel, Dordrecht, Netherlands, 1985, ch. 15, vol. 141, pp. 379–412 Search PubMed.
- C. A. de Lange, J. G. Snijders and E. E. Burnell, On the orientation of small molecules in anisotropic solvents, in Nuclear Magnetic Resonance of Liquid Crystals, ed. J. W. Emsley, Reidel, Dordrecht, Netherlands, 1985, ch. 9, vol. 141, pp. 181–205 Search PubMed.
- S. Lagerwall, A. Dahlgren, P. Jägemalm, P. Rudquist, K. Dhavé, H. Pauwels, R. Dabrowski and W. Drzewiński, Adv. Funct. Mater., 2001, 11, 87 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef CAS.
- A. Marchetti, V. Domenici, V. Novotna, M. Lelli, M. Cifelli, A. Lesage and C. A. Veracini, ChemPhysChem, 2010, 11, 1641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01321b |
| This journal is © The Royal Society of Chemistry 2025 |