
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon-conducting polymer thin films via chemical vapor deposition polymerization
Kwang-Won
Park†
 a,
Christina H
Yu†
b,
Shuaicheng
Fu†
a and
Rong
Yang
*a
a,
Christina H
Yu†
b,
Shuaicheng
Fu†
a and
Rong
Yang
*a
aRobert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: ryang@cornell.edu
bDepartment of Materials Science and Engineering, Cornell University, Ithaca, NY 14853, USA
First published on 7th February 2025
Abstract
Ion-conducting polymers (ICPs), benefiting from the movement of ions instead of electrons, have attracted significant interest in various scientific and technological fields, including drug delivery, water purification, and electrochemical devices. This review aims to highlight recent advances in the synthesis of ICP thin films, with a particular focus on chemical vapor deposition (CVD) technologies. Traditional solution-based methods for ICP thin film deposition face challenges, including non-uniformity, low-throughput manufacturing, and the generation of hazardous wastes. In comparison, CVD eliminates the drawbacks associated with solution-based processes. They offer precise control film properties, including high purity, conformal coating, delicate control over thickness, etc. This review organizes the latest developments in CVD-based ICP synthesis, based on material properties and the synthesis strategy, into direct deposition and post-polymerization modification, ionogels, hydrogels, and ultrathin siloxane or silazane-based polymer films. By providing an up-to-date review of the materials and synthesis, we aim to position CVD polymerization as an effective strategy for future materials development/production and device fabrication in energy, sustainability, and healthcare where ion conductivity is desired.
1. Introduction
Ion-conducting polymers (ICPs) offer a combination of ionic conductivity and mechanical flexibility, making them highly versatile for a wide array of technological applications, including energy storage devices, fuel cells, sensors, membranes, wearable electronics, and biomedical devices.1 ICP films are particularly crucial in the context of energy storage devices, such as lithium-ion batteries (LIBs), as these films serve as solid electrolytes, separator coatings, and artificial solid–electrolyte interphases (SEIs), addressing several critical challenges. For example, ICP coatings on anode surfaces can prevent dendrite formation, which is a major safety concern in LIBs, and accommodate the significant volume changes in high-capacity anode materials like Si. There have been more than 4000 publications each year during the past decades regarding ICP films and their applications. Among them, perfluorinated sulfonic acid (PFSA) has been most widely applied, ever since Nafion® was developed by Dupont, due to its excellent ionic conductive performance.2Traditional methods for fabricating ICP thin films often rely on solution processing like spin casting, spray coating, and layer-by-layer assembly. However, these methods face limitations, such as poor coating conformality on nano- and micro-structured substrates due to the dominating capillary effects at this length scale. In addition, the ICP film quality may be limited due to the undesirable effects of solvents and their surface tension, which can cause issues like pinhole defects due to dewetting. Further challenges revolve around bench-to-manufacturing scale-up and environmental concerns associated with the generation of large quantities of hazardous liquid waste. These limitations become more significant as there is a growing demand for ICP coatings on 3D structures, e.g., in energy storage and CO2 reduction, and scalable manufacturing processes.
One potential synthesis technique to address these challenges and produce high-quality conformal functional thin films is chemical vapor deposition (CVD) polymerization. CVD is a technique for depositing thin films from gaseous precursors, and it offers significant advantages for precise control over film properties. It involves one or more monomer species delivered in the vapor phase.2 These species activate in a vacuum chamber (e.g., by plasma or hot wires), encounter a substrate, and create a high-purity and conformal thin film, achieving polymerization and film formation in a single step. Due to their advantageous film quality and process scalability, there is an emerging interest in applying CVD techniques to create ICP thin films. Significantly, because CVD techniques offer a solvent-free and relatively low-temperature (temperature of the substrate to be coated is often kept at <30 °C) reaction environment that preserves the integrity of chemical functional groups and sensitive or delicate substrates (e.g., reactive metal anode), CVD-deposited ICP films have been leveraged for efficient ion transport and enhanced electrochemical stability of energy storage devices.3–6
Moreover, the precise control over synthesis conditions afforded by CVD processes allows for the tuning of polymer composition and film structure. This capability is particularly important for creating polymer thin films to satisfy multiple material design targets, e.g., high ionic conductivity combined with hydrophobicity to simultaneously suppress hydrogen-evolution reactions (HER) and dendrite formation in batteries.6 Recent studies have demonstrated the successful application of various monomers, including acrylates, methacrylates, styrenes, and siloxanes, to produce ICP films with good ionic conductivities.7–11 While a majority of these materials have been used in LIBs, the impact of CVD-deposited ICP films extends beyond energy storage. These materials are also being explored for use in flexible and wearable electronics, where their ability to form conformal coatings on complex geometries is highly advantageous. For example, composition-tunable composite hydrogels produced via initiated CVD (iCVD) have shown promise in wearable applications by offering adjustable ion conductivities and robust performance in flexible and rugged substrates like textiles.12,13
While various CVD processes have emerged as novel techniques for synthesizing functional polymer films, including iCVD, oxidative CVD (oCVD), and plasma-enhanced CVD (PECVD), each offering unique advantages, iCVD has been the most common method for depositing ICP films, hence chosen as the focus of this review. iCVD enables chain-growth polymerization using monomers with one or more vinyl moieties. Although the resulting polymers are electrically insulating, they could be made ionically conductive through the selection of suitable monomers, substrate materials, and/or modification of functional groups, as discussed in this review. A general experimental setup for both iCVD and photoinitiated CVD (piCVD) can be seen in Fig. 1.14,15 In contrast, oCVD enables step-growth polymerization, producing electrically conductive conjugated polymers with ionic species as dopants. Since the electrical conductivity of oCVD polymers leads to different conduction mechanisms and applications, it will not be explored in this review. Readers can refer to other comprehensive reviews on oCVD polymers for more information.16–18 Additionally, there have been no reports of ICPs produced through PECVD or parylene CVD, to the best of our knowledge.
 | ||
| Fig. 1 (A) The schematic of an initiated chemical vapor deposition reactor. The custom-built cylindrical chamber is connected to a vacuum pump, thereby creating a natural vapor flow from the source of precursors (i.e., initiators and monomers) to the throttle valve that regulates the flow to the vacuum pump. The pressure within the vacuum chamber is controlled by a feedback loop between the throttle valve and the pressure gauge. Substrates (e.g., Si wafers) are positioned on a cooled stage equipped with a chiller, maintaining a controlled substrate temperature, commonly ranging from 5 to 50 °C. The initiators and monomers are introduced by heating them to create vaporized molecules. An array of filaments made of resistance wire (i.e., nichrome) is heated to approximately 200 to 300 °C to initiate the polymerization by decomposing the initiators into reactive radicals. Reprinted from ref. 14 with permission from Frontiers, Copyright 2021. (B) Schematic depiction of the piCVD setup. piCVD operates similarly to iCVD, but the radicals are generated by UV-induced decomposition of photoinitiators. Reprinted from ref. 15 with permission from Wiley, Copyright 2024. (C). Reaction mechanism of iCVD. | ||
This review aims to explore recent advancements in ICP films synthesized using vapor-phase precursors, mainly via iCVD, and their distinct features and applications. While other CVD polymerization methods for ICP film synthesis have been rare in recent years, we introduce the few examples identified in the corresponding sections based on the class of ICP they enable. This review is organized based on the types of ICP materials, including ion-containing polymers, polymer–ionic liquids (ILs) composites, hydrogels, and ultrathin ion-conducting films, highlighting their potential to impact a wide cross-section of industries ranging from sustainability to healthcare.
2. iCVD polymerization techniques to synthesize ICP thin films
iCVD is a polymer coating technique invented in the early 2000s.7 A typical iCVD setup and reaction mechanism is shown in Fig. 1. Distinct from most other CVD polymerization techniques, iCVD leverages chain-growth polymerization to synthesize chemically well-defined polymer thin films. Compared to traditional solution-based coating techniques, iCVD enables free radical polymerization in the vapor phase, which allows a uniform coating and control over the coating thickness with nanometer-scale precision.19 Copolymers of chemically distinct monomers and cross-linked polymers can easily be achieved by feeding the monomer vapors with a targeted stoichiometry in the vapor-phase. The temperature of the substrate is kept at around 20–40 °C to promote monomer adsorption and to preserve substrates from potential thermal damage. In addition, iCVD typically achieves 100% functional group retention from the monomers thanks to its room-temperature reaction environment.A typical iCVD process involves several steps. Firstly, two precursors, monomer(s) and an initiator (typically a peroxide like tert-butyl peroxide), are vaporized and delivered into the vacuum reactor. The monomer molecules subsequently absorb onto the cooled substrate. The filament array suspended over the substrate is heated to activate the initiator vapor to generate free radicals and initiate polymerization at the substrate surface through the reaction between radicals and adsorbed monomers. The coating process is terminated by turning off the filament heating and removing the unreacted vapor-phase precursors.
Similar free-radical polymerizations can be initiated by UV radiation instead, and is termed as photoinitiated chemical vapor deposition (piCVD). Instead of using thermal energy produced from a filament heater for the formation of free radicals like in iCVD, piCVD utilizes photoinitiators to create reactive radicals upon UV radiation (Fig. 1B). The main advantages of piCVD compared to iCVD include not requiring a filament heater (hence reduced energy consumption), and the ability to leverage line-of-sight effects to achieve non-conformal coatings if desired.20 It was reported that some monomers (e.g., 2-hydroxyethyl methacrylate (HEMA)) spontaneously cross-link upon UV radiation. So, insoluble polymers can be synthesized in piCVD without the need for cross-linking agents.15 However, the precursors must be photosensitive for initiation, which limits the range of materials suitable for the piCVD process. More critically, achieving ICP films through iCVD using ionic conductive monomers presents a significant challenge, as most of these monomers are difficult to vaporize due to the strong electrostatic intermolecular interactions. Volatility remains a major limiting factor for ICP film deposition via the iCVD technique. Nevertheless, these challenges have been addressed through the use of volatile and pH-sensitive monomers or post-deposition modification strategies, which will be discussed in the later section.
3. Types of ICPs and their synthesis via iCVD
In this section, we will review studies published within the past 10 years about iCVD-processed ICP films. We organize the discussion based on the class of ion-conducting materials, namely: ionomers bearing ion-conducting moieties (Section 3.1), composites comprising polymer thin films impregnated with ionic liquids (Section 3.2), hydrogels with hydration-facilitated ion conduction (Section 3.3), and siloxane films that are not ionized but present flexible chains that readily interact with ions (Section 3.4). For each study, the specific iCVD method, ion conductivity, and application are the main focuses. This section also aims to provide a comparison of ion conductivity values between novel iCVD-deposited ICP films and traditional solution-based conductive films. From the studies, iCVD synthesized ICPs can be comparable against traditionally used polymers such as Nafion® and Sustainion®.3.1. Ion-containing polymers (ionomers)
Ionomers are a class of polymers containing ionic groups covalently bonded to the polymer backbone. They can conduct anions (e.g., OH−, (CF3SO2)2N− (TFSI)) or cations (e.g., H+, Li+, Na+) with ionic conductivities on the order of 1.0 × 10−8 to 10−4 S cm−1.21,22 Cation or proton(H+)-conducting ionomers like Nafion® are commonly employed in electrochemical fuel cells, electrolyzers, batteries, and membranes.2 Nafion® often serves as a proton exchange membrane to facilitate proton conduction while blocking electrons and gas molecules, thereby improving the efficiency and durability of fuel cells.2,23 In batteries such as LIBs, zinc batteries, and all-solid-state batteries (ASSBs), anionic sulfonic acid polymers, such as Nafion®, and sulfonated polyether ether ketone (SPEEK), are employed to create artificial SEI to improve ionic conductivity and stability at the electrode–electrolyte interface, which critically improves the overall battery performance and longevity.2,5,24,25 Additionally, ionomers with zwitterionic functional groups such as poly[1-vinyl-3-(3-sulfopropyl)imidazolium-co-DVB] (pVSpIm-co-DVB) and poly[1-(3-sulfopropyl)-2-vinyl-pyridinium-co-DVB] (pSPV-co-DVB) are utilized in biomaterial coatings for their anti-bacterial properties, where the permanent charge along the polymer backbone is thought to promote material–cell interactions (e.g., the cation–π interaction between polyimidazolium and spike proteins in human coronaviruses).26,27 Traditionally, most ionomer coating techniques are solution-based, including solvent casting, dip coating, and spin coating.28–30 However, solution-based polymerization faces challenges such as poor control over conformality and film thickness and limitations regarding scalability and waste generation.31 In iCVD, ionomer coatings have been made through two methods: direct deposition and post-deposition modification. Using direct deposition, the ionomers are synthesized and coated in a single step. This enables their direct integration into various devices via iCVD polymerization.9 Most cation-conducting ionomers, such as polyacids, have been synthesized via direct deposition.9 In the post-deposition modification method, a non-conductive polymer coating is deposited via iCVD, followed by the introduction of a secondary reagent that reacts with the polymer (usually via a nitrogen quaternization reaction), thus transforming it into an ion-conducting material.| Method | Polymers | Chemical structure | Types of ions | Temperature (°C) | Thickness (nm) | Conductivity (S cm−1) | Other values | Application | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Obtained after cell-assembly. b Electrolyte-swollen state. | |||||||||
| iCVD | pPFDA-co-MAA |

|
H+ | RT | 1000 | 7 × 10−1 | — | Fuel cell (PEM) | 9 |
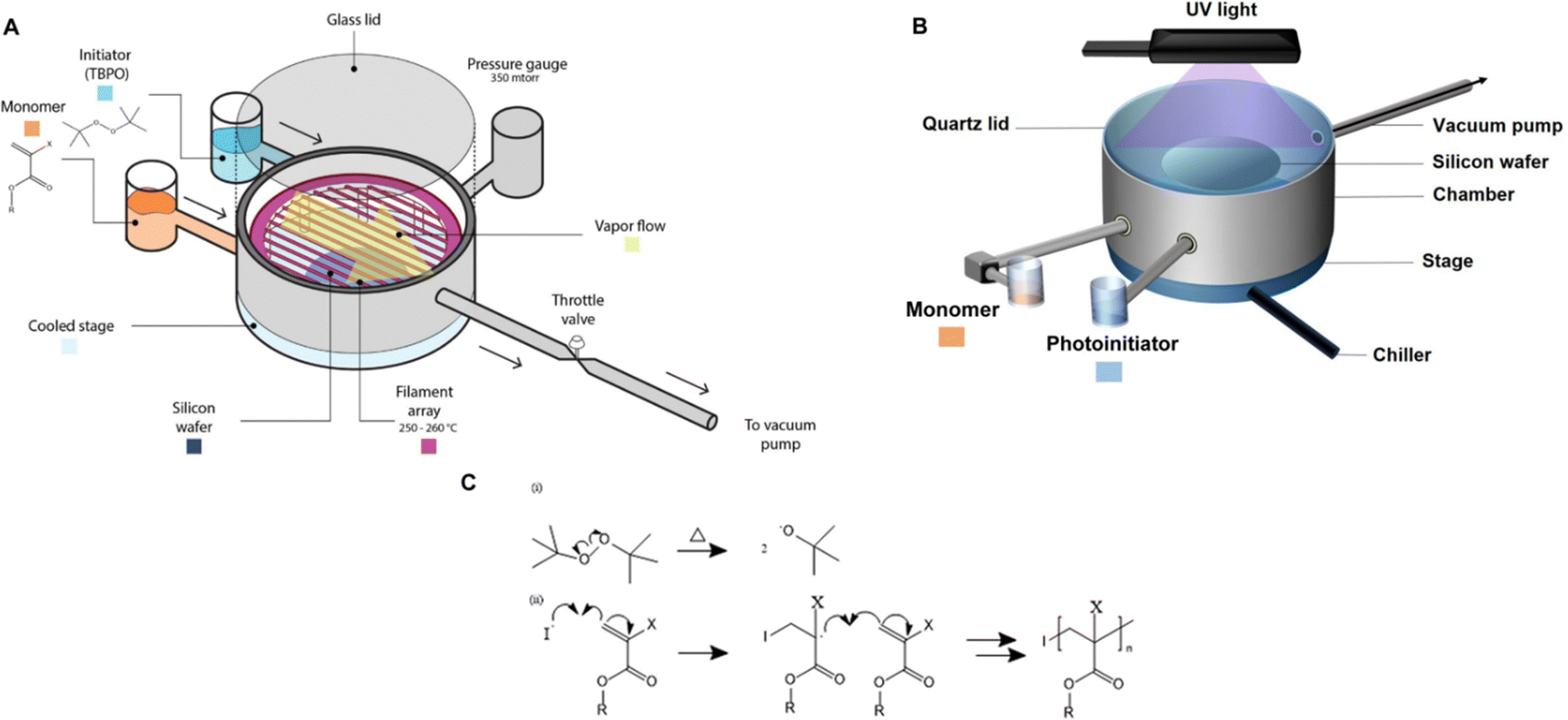
| iCVD | pAA |
|
Li+ | — | 100 | 2.95 × 10−3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
— | ASSBs (SEI) | 5 |
| iCVD | pAA |

|
— | — | 40 | — | — | Synaptic transistor | 41 |
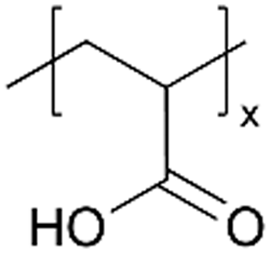
| iCVD | pHEMA |
|
I−/I3− | — | — | — | — | DSSC (electrolyte) | 45 |
| pGMA |

|
I−/I3− | — | — | — | — | |||
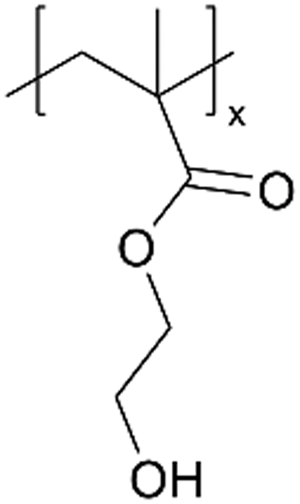
| p4VP |
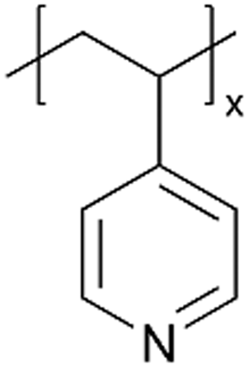
|
I−/I3− | — | — | — | — | |||
| iCVD | pVIm |
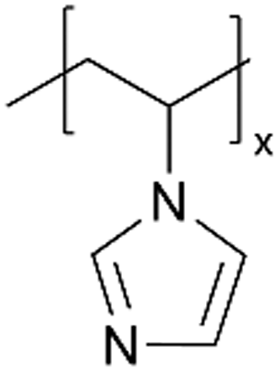
|
— | — | — | — | — | DSSC (electrolyte) | 46 |
| iCVD | pDMAMS |
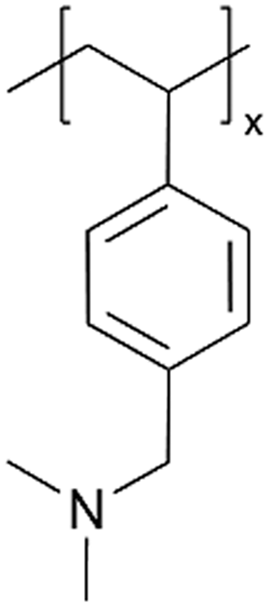
|
Li+ | RT | 100 | 6.54 × 10−3b |
190 ohms | LIB (SEI) | 8 |
| iCVD | pDMAEMA-co-VBC |
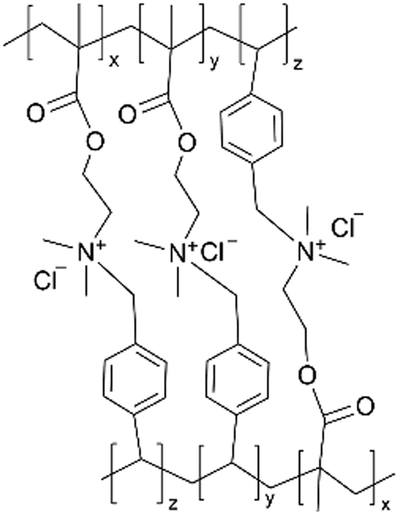
|
— | — | 500 | — | — | Oil/water separate membrane | 40 |
| iCVD | pEGDMA |
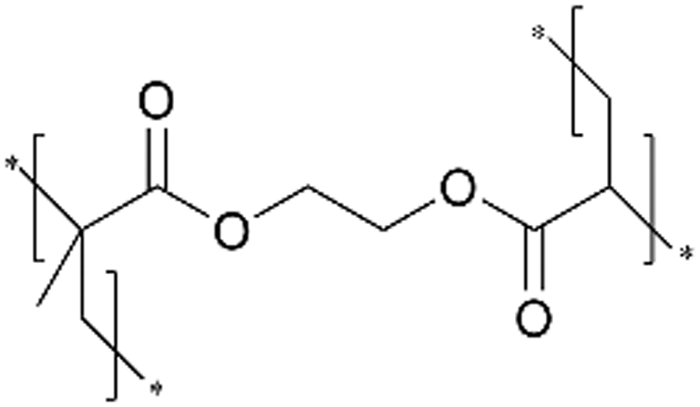
|
— | — | 20 | — | — | Polymer electrolyte-gated synaptic transistor (PEGST) | 49 |
For example, copolymers of 1H,1H,2H,2H-perfluorodecyl acrylate (PFDA) and methacrylic acid (MAA) (pPFDA-co-MAA) have been synthesized via iCVD as proton exchange membranes for fuel cell applications.9 Hydrophobicity and ion-conductivity are required simultaneously for fuel cell applications.23 Hydrophobicity prevents flooding in the cell and enhances cell performance, while ion conductivity is critical for ion transport and minimizing resistance.23,32 A study in 2013 highlighted iCVD's capability to synthesize copolymers of hydrophobic and hydrophilic monomers.9 This is generally difficult to achieve with conventional solution-based techniques due to the lack of a common solvent for the two monomer classes. MAA served as the proton exchange group due to its proton conductive –COOH groups, while PFDA increased the hydrophobicity of the polymer due to its fluorinated carbon groups.33 The copolymer composition was varied systematically to prevent water uptake and increase proton conductivity by altering the monomer feed ratio between MAA and PFDA during the iCVD process. A 1 μm thick pPFDA80-co-MAA20 film exhibited ionic conductivity comparable to commercial Nafion® membranes, with a value as high as 7.0 × 10−1 S cm−1.
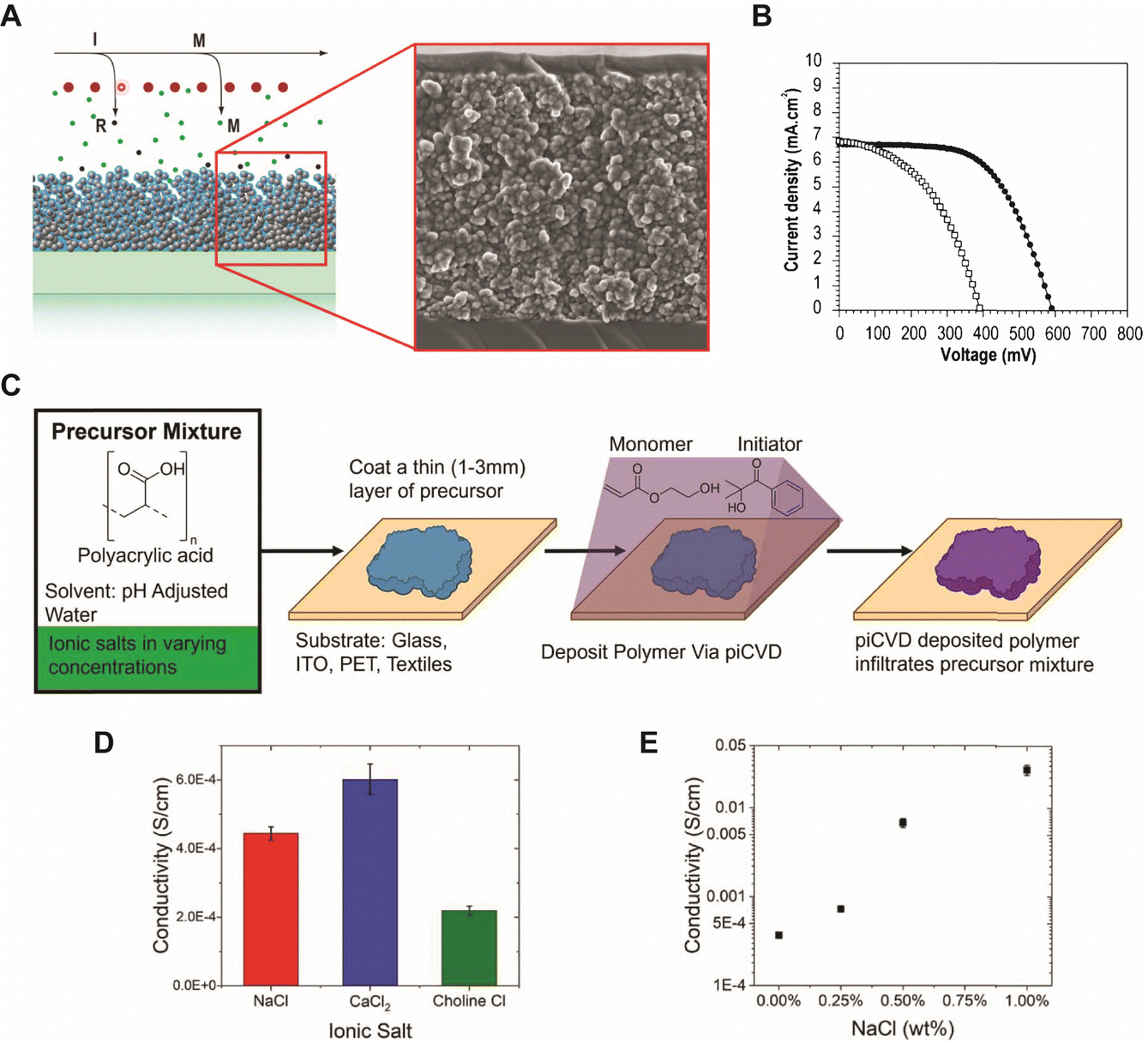
Polymer coatings have been extensively used to mitigate safety issues, such as short circuits between the anode and cathode in batteries.34,35 Since iCVD can apply polymer films without requiring solvent or high temperatures, it avoids potential damage to surface or electrochemical cells, making it ideal for various functional polymers. Notably, cross-linked polymers effectively prevent dendrite formation during lithiation and accommodate the significant volume changes in anode materials like silicon.36–38 Recently, poly(acrylic acid) (pAA) films were applied to stabilize the interface between sulfide solid electrolytes (SSEs) and indium (In) anodes in ASSBs.5 ASSBs, despite reducing the risk of explosion and broadening material selection by replacing liquid electrolytes with solid electrolytes, are known to be unstable due to complex interfacial phenomena at the electrode material and electrolyte interface. The electrochemical reaction between anode material and electrolyte often leads to the formation of a heterogeneous corrosion layer, known as SEI, causing irreversible electrolytes and lithium consumption and the formation of dendrites while posing significant safety concerns and limitations to long-term performance.39 The challenges of an unstable SSE/In interface and charge transfer at the anode surface were addressed by incorporating iCVD polymers at the interface. Fig. 2A highlights the iCVD synthesis process for the polymer layer on the In metal anode.5Fig. 2B presents the surface roughness analysis obtained through AFM, revealing that the root-mean-square roughness (Rq) values remain consistent. This indicates that the iCVD process results in a uniform and well-controlled coating. Among different iCVD polymers, pAA-coated In (In@pAA) displayed good long-term half-cell cycle performance with Li6PS5Cl (LPSCl), a promising SSE known for its high ionic conductivity of 3.4 × 10−3 S cm−1 (obtained using the half-cell assembly). This performance is attributable to the higher affinity of pAA for Li+ ions and its contribution to forming a stable SEI. It was hypothesized that spontaneous chemical reactions occurred between the polar bonds of pAA and highly reactive LPSCl during cell assembly, leading to the formation of native SEI, although the precise reaction steps are not well understood. XPS confirmed the formation of Li2CO3 as a result of the interactions between the –COOH groups of In@pAA and LPSCl. The high ionic conductivity of the SEI layer significantly reduced the internal resistance of all-solid-state full cells. Consequently, the full cells with In@pAA demonstrated robust performance with a capacity retention of 64.8% after 100 cycles.
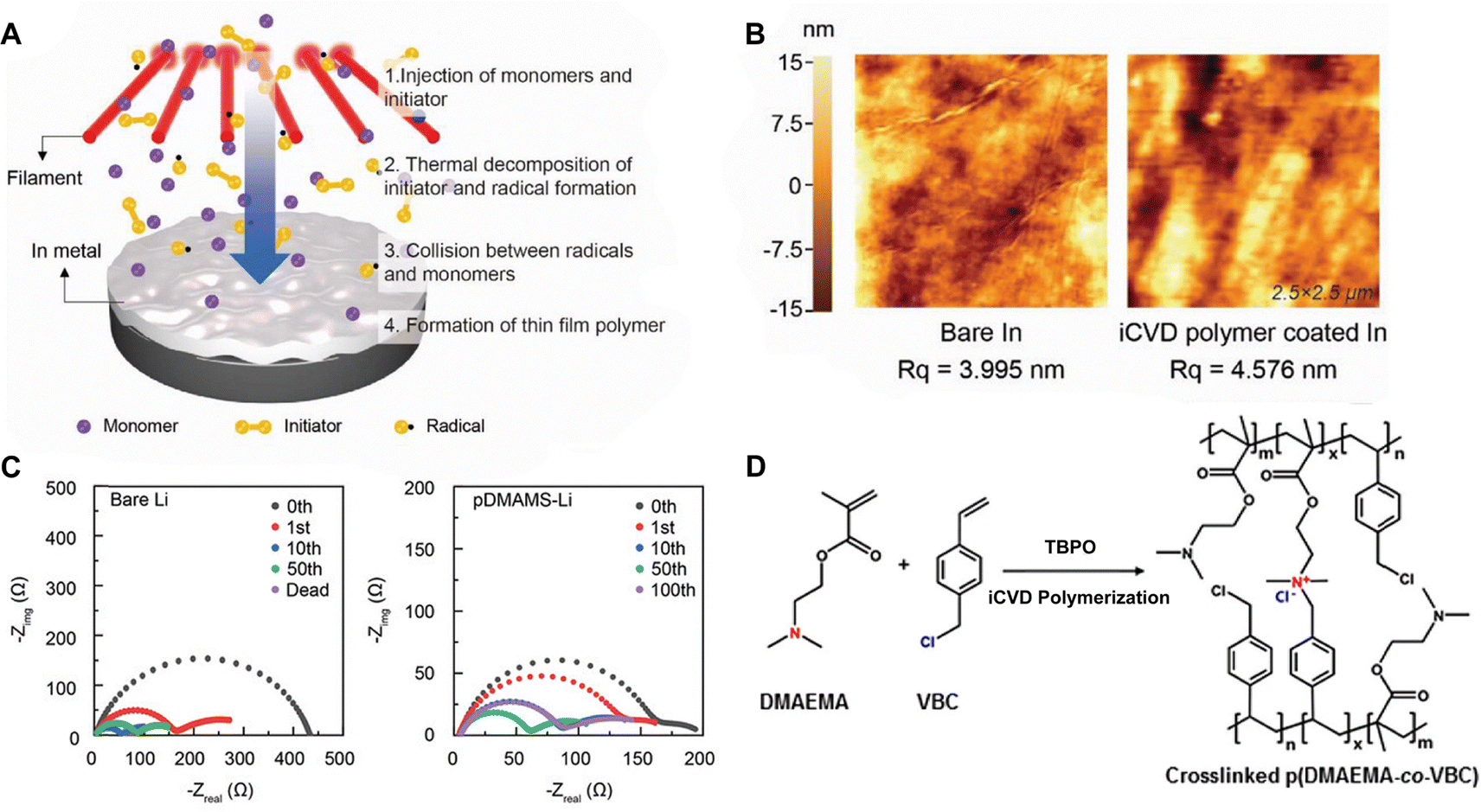 | ||
| Fig. 2 (A) Schematic illustration representing the iCVD process for artificial SEI layer formation on the In metal anode. (B) Comparison of surface morphology of In metal foil before and after the iCVD process characterized by AFM. Reprinted from ref. 5 with permission from Wiley, Copyright 2024. (C) Nyquist plots of symmetric bare-Li and 100 nm pDMAMS-Li cells at 0th to 100th cycles. Reprinted from ref. 8 with permission from Wiley, Copyright 2023. (D) Synthetic scheme and chemical structure of cross-linked pDMAEMA-co-VBC copolymer. The m and n represent the unreacted contents of DMAEMA and VBC, respectively. The x is the ionic cross-linked part generated through the Menshutkin reaction. These m, n, and x values are changed by feed ratios of each monomer into the iCVD chamber. Reprinted from ref. 40 with permission, Copyright 2017 American Chemical Society. | ||
Moreover, a bio-inspired organic synaptic transistor was developed that mimics biological synapses for neuromorphic computing applications.41 Large-scale integration on a flexible or stretchable substrate with uniformity and precise control of film thickness was achieved. The device is composed of a thin layer of ion-doped polyelectrolyte, created through the iCVD deposition of pAA followed by in situ ion doping with tert-amine cation, 2-(dimethylamino)ethyl acetate, which mimics acetylcholine, one of the key neurotransmitters. While the precise ion conductivity was not reported, the formation of ionic complexes imparted ionic conductivity to the films, which exhibited the behavior predicted by the simplified Randle's model, whereas pristine pAA showed typical capacitance behavior in electrochemical impedance spectroscopy (EIS) measurements. This highlights the critical importance of the ionic functional groups in facilitating ion transport. Consequently, the resulting devices demonstrated low power consumption, synaptic plasticity, and compatibility with stretchable substrates, making them promising candidates for future artificial nervous systems and wearable systems.
In addition to polymers with acid groups, those with basic groups, such as tertiary amine groups, have also been explored. iCVD polymers have demonstrated significant potential as polymer electrolytes in energy conversion devices. Polymer electrolytes offer advantages over conventional liquid electrolytes, including leak prevention, flexibility, and relatively high ionic conductivity, enhancing the operability and durability of electrochemical cells.42 In dye-sensitized solar cells (DSSCs), the mesoscopic features of TiO2 electrodes are crucial for increasing cell efficiency by providing photosensitizers with a larger surface area for enhanced light harvesting. While infiltrating electrolyte materials into the mesoporous TiO2 has been a significant challenge, iCVD has been leveraged to form uniform, conformal coatings and effectively fill nanometer-sized pores in mesoporous TiO2 photoanodes.43 It was demonstrated that iCVD allows excellent pore filling for photoanode layer thickness up to 12 μm.44 Furthermore, poly(2-hydroxyethyl methacrylate) (pHEMA), poly(glycidyl methacrylate) (pGMA), and poly(4-vinyl pyridine) (p4VP) have been deposited via iCVD for DSSCs45 to address the major challenges of liquid electrolyte leakage and evaporation, which limits the long-term operation of DSSCs. The three polymers were chosen to understand the influence of distinct chemical functional groups on DSSC performance. Theoretical and experimental studies have shown that the formation of complexes between lithium iodide and polymer side groups affects open circuit voltage (Voc). Specifically, pHEMA exhibited the largest Voc due to the strong affinity of hydroxyl and carbonyl groups toward Li+ ions compared to epoxide and pyridine moieties of pGMA and p4VP, respectively. Moreover, the basic pyridine group in p4VP could create a better blocking layer between TiO2 and triiodide, compared to pGMA and pHEMA, leading to lower charge transfer resistance and an increase in current density (Jsc). This finding indicates that DSSC device characteristics, including Jsc, Voc, and fill factor (FF), can be tuned by varying polymer electrolyte chemistry.
Poly(1-vinylimidazole) (pVIm) has also been synthesized via iCVD as a solid-state electrolyte for mesoporous TiO2 photoanodes in DSSCs.46 It has been hypothesized to enhance the ion conduction of the iodide-triiodide redox couple (I−/I3−), because pVIm has been shown to bind strongly to Li+ ions via Lewis acid–base interaction.47 Indeed, the DSSCs with pVIm conformally covering the TiO2 photoanode outperformed those with liquid electrolytes in terms of efficiency, Jsc, and Voc. This improved performance can be attributed to two potential reasons. Firstly, the basic character of the pVIm electrolyte results in strong acid–base interaction with the electrode, creating a blocking layer that prevents electron recombination at the interface, thereby leading to a higher Jsc in DSSCs. Secondly, the strong binding between pVIm and Li+ reduces the surface concentration of Li+ on the electrode, improving the Voc of the DSSC iCVD-synthesized poly(1-vinyl-2-pyrrolidone) pVP has also been explored as a solid-state electrolyte for DSSCs.48 Due to the solubility of pVP, the thin films were cross-linked with ethylene glycol diacrylate (EGDA) at a cross-linking density of 20%. First principles modeling showed that cross-linked pVP-co-EGDA electrolytes could increase the recombination rate constant, improving the shunt resistance and affecting interfacial processes significantly by shifting the conduction band of TiO2.
A “swollen-soft-scaffold (3S)” strategy has been developed using iCVD to enable ion conductivity in a bifunctional polymer layer, namely solvogel, which conducts metal ions via its permeability to liquid electrolytes.8 As such, the degree of swelling is crucial for high ion conductivity. Among the polymers tested, including poly(1H,1H,2H,2H-perfluorooctyl acrylate) (pPFOA), poly(ethylene glycol dimethacrylate) (pEGDMA), pAA, poly(divinylbenzene) (pDVB), and poly(dimethylaminomethyl styrene) (pDMAMS), pDMAMS showed the largest swelling capability. EIS and cyclic voltammetry (CV) revealed that the iCVD coatings are electrochemically stable, except for pPFDA. The electrolyte-swollen pDMAMS matrix exhibited an ionic conductivity of 6.54 × 10−3 S cm−1. Fig. 2C shows the Nyquist impedance of pDMAMS–Li cells compare to bare-Li cells, as well as the Nyquist impedance of pDMAMS–Li across different cycles. The results demonstrate that pDMAMS effectively reduces interfacial resistance (≈190 Ω for pDMAMS–Li, compared with ≈440 Ω for bare-Li), leading to improved performance compared to bare-Li cells.8 XPS revealed that the ion conductivity may have partially benefitted from a potential electrochemical reaction with diethyl carbonate (a component of the electrolyte), which converted the amine to quaternary ammonium cations. The solvogel has been shown to reduce Li dendrite formation when applied to the Li anode in LIBs during full cell operations by forming a miscible, homogeneous layer with the native SEI. pDMAMS coatings thicker than 100 nm have been shown to hinder Li+ ion transport and increase the interfacial resistance.
In addition, the in situ reactions between functional moieties in a CVD-synthesized copolymer have been designed to imbue ion conductivity into polymer thin films.40 For example, the reaction between tertiary amine (nucleophile) and alkyl halide (electrophile) has been leveraged to create anion-conducting quaternary ammonium side chains. Fig. 2D shows the mechanism of the single-step reaction to create cross-linked ionic copolymer.40 Using iCVD, a copolymer of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and 4-vinylbenzyl chloride (VBC) has been deposited, resulting in a cross-linked network via the amine–alkyl halide reaction.
Furthermore, ultrathin (∼20 nm) pEGDMA has been successfully demonstrated as an all-solid-state polymer electrolyte in polymer electrolyte-gated synaptic transistor (PEGST), which is a potential alternative to electrolyte-gated transistors (EGTs) for artificial and deep neural networks for neuromorphic hardware implementation.49 The iCVD deposition of pEGDMA allowed wafer-scale fabrication with nanoscale thickness and uniformity, making the solid-state PEGST more stable and CMOS-compatible. The dynamic behaviour of the pEGDMA as an electrolyte gate dielectric was investigated based on the distribution of protons as mobile ions that migrated in response to external electric fields. The resulting PEGST showed potential for neuromorphic computing due to its scalability, low power consumption, and excellent linear and symmetric synaptic behaviour.
The direct deposition of ICP thin films via CVD represents an innovative fabrication technique that offers numerous advantages for creating uniformed and conformal ionomer coatings. By carefully selecting monomers or monomer pairs, it is possible to achieve reasonable ion conductivity in as-deposited thin films. Innovative reaction schemes, such as those between the side chains of comonomers to create a permanent change, have advanced the all-dry synthesis of ion-conducting polymers. Although the selection of monomers is still limited due to the low volatility of ionic species, these novel approaches highlight the critical role of direct deposition in advancing the performance and durability of electrochemical devices through precise control over polymer composition and structure.
| Method | Polymers | Chemical structure before modification | Final chemical structure | Types of ions | Temperature (°C) | Thickness (nm) | Conductivity (S cm−1) | Other values | Application | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Mixed ionic–electronic conductivity. | ||||||||||
| iCVD | p4VP·ICl |
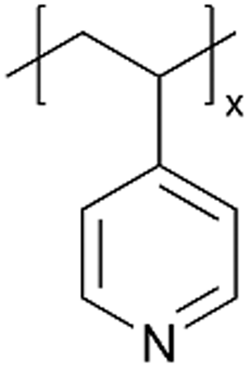
|

|
Li+ | 20 | 1600 | 1.7 × 10−6a |
— | LIB (Solid-state cathodes) | 58 |
| iCVD | pDMAMS+ |
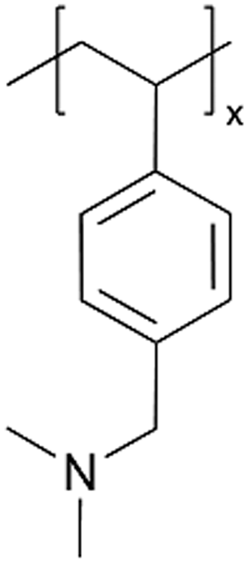
|
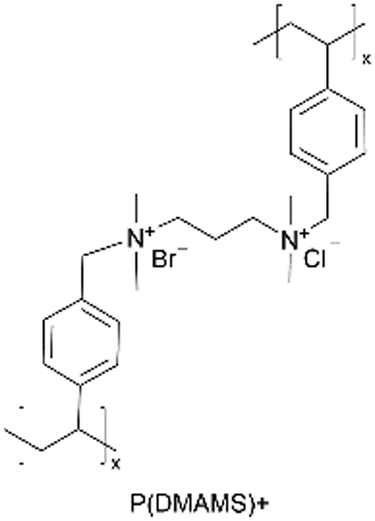
|
Br− | RT | Dry: 580 ± 59 | Dry: 2.5 × 10−8 ± 6.0 × 10−9 | 20 MΩ | 3D SSB (SSE) | 60 |
| Wet: 1000 ± 500 | Wet: 3.3 × 10−6 ± 2.0 × 10−6 | |||||||||
| HCO3− | Dry: 502 ± 83 | Dry: 3.5 × 10−7 ± 1.3 × 10−7 | 2 MΩ | |||||||
| Wet: 2500 ± 500 | Wet: 2.8 × 10−5 ± 8.4 × 10−6 | |||||||||
| OH− | Dry: 502 ± 83 | Dry: 5.3 × 10−7 ± 1.7 × 10−7 | 5 MΩ | |||||||
| Wet: 2500 ± 500 | Wet: 2.0 × 10−5 ± 8.6 × 10−6 | |||||||||
| iCVD | p4VP-co-DVB |
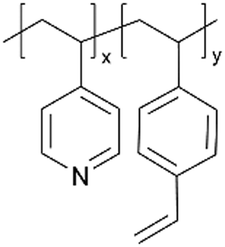
|

|
Li+ | RT | 10–500 | — | — | LIB (SEI) | 50 |
| pVIm-co-DVB |

|
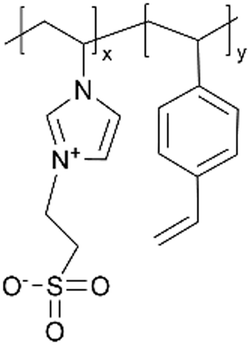
|
— | — | ||||||
| pDMAEMA-co-DVB |
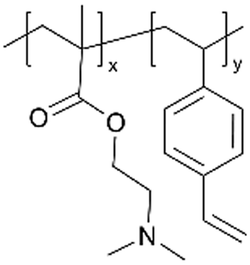
|

|
— | — | ||||||
| iCVD | p4VP-co-DVB |

|
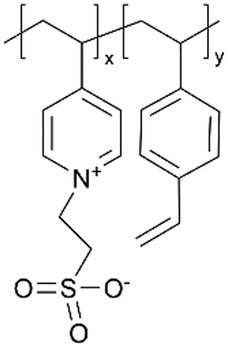
|
Zn+ | — | <1000 | — | 10 ± 4 Ω | Zinc batteries (SEI) | 25 |
| iCVD | pPFDA |

|
|
— | — | <300 | — | — | Zinc batteries (SEI) | 6 |
| iCVD | pMAA-co-EGDA |

|
|
Li+ | 20 | 1000 | 6 × 10−9 | — | Solid-state lithium microbatteries (Electrolyte) | 61 |
| 100 | 1 × 10−5 | |||||||||
Anion-conducting ICPs have been synthesized by reacting iCVD-synthesized p4VP films with gas-phase ICl, leading to protonated pyridinium side groups.58 The p4VP-iodine monochloride (p4VP·ICl) was subsequently used as a polymeric charge transfer complex cathode in LIBs and significantly improved the ionic conductivity compared to traditional organic cathodes. Fig. 3A proposes the reaction between p4VP and ICl in gas phase. FT-IR confirmed that thermal heterolysis of ICl resulted in α-H substitution in the p4VP backbone, creating halogenated p4VP.58 The resulting p4VP·ICl exhibited a mixed ionic–electronic conductivity of 1.7 × 10−6 S cm−1 with an electronic conductivity of 4.1 × 10−7 S cm−1 (22% of the total conductivity) at room temperature. Moreover, due to the substrate-independent nature of the iCVD technique, flexible batteries could be fabricated using those organic cathodes.
 | ||
| Fig. 3 (A) Proposed reactions between p4VP and ICl during gas phase incorporation. Thermal heterolysis of ICl may result in α-H substitution in p4VP and further generate pyridinium, which is detected by FTIR. Reprinted from ref. 58 with permission from The Royal Society of Chemistry, Copyright 2022. (B) AFM and SEM analyses of pDMAMS films as a function of processing step. The SEM image includes contamination particles to show image is in focus. Reprinted from ref. 59 with permission from The Royal Society of Chemistry, Copyright 2024. (C) Schematic illustration of the fabrication of zwitterionic polymeric interphases. Step 1: iCVD precursor polymer film (25 °C for p4VP-co-DVB and 30 °C for pVIm-co-DVB and pDMAEMA-co-DVB). Step 2: derivatization. Reprinted from ref. 50 with permission from American Chemical Society, Copyright 2024. (D) Schematic illustration of the ultrathin and semicrystalline pPFDA interphases of interest in this study. Reprinted from ref. 6 with permission from American Chemical Society, Copyright 2024. | ||
A similar approach has been used to synthesize ICPs by post-polymerization modification using a di-halogen alkylating reagent.60 In this process, iCVD was used to synthesize conformal pDMAMS coatings on 2D and 3D substrates, which were subsequently exposed to di-halogen alkylating reagents to yield pDMAMS+ (containing quaternary ammonium moieties for anion conductivity, Table 2). The successful conversion was proven by NMR, FTIR, and XPS.59 The quaternary ammonium was combined with anions, including Br−, HCO3−, and OH−, the electrochemical performance of which was characterized under dry and wet states. The AFM was conducted at each processing step during the functionalization of polymer films, including annealing, quaternization, and ion exchange (Fig. 3B), demonstrating that a smooth morphology (RMS roughness ≤1 nm) is retained throughout the functionalization process steps.59 The three dry films (pDMAMS+ (Br−), pDMAMS+ (HCO3−), and pDMAMS+ (OH−)) displayed total electrical resistances of 20 MΩ, 2 MΩ, and 5 MΩ, respectively. In contrast, EIS indicated that the wet pDMAMS+ films exhibited higher ionic conductivity than their dry counterparts. For example, the ionic conductivity of the pDMAMS+ (Br−) film was 2.5 × 10−8 ± 6.0 × 10−9 S cm−1 when dry and 3.3 × 10−6 ± 2.0 × 10−6 S cm−1 when wet. Detailed ionic conductivity data for all materials tested can be found in Table 2. Redox-probe voltammetry results further demonstrated that pDMAMS+ allows negatively charged redox probes to reach the electrode surface while blocking positively charged redox probes. This behavior resembles commercial ionomers such as Fumion®.
Despite their charge neutrality, zwitterionic polymers have recently been used in batteries and supercapacitors, which points to their potential ion conductivity.62–64 While there have been limited reports that directly measure the ion conductivity of cross-linked zwitterionic thin films, their application in lithium metal batteries without increasing interfacial resistance hints at reasonable Li+ conductivities in the sulfobetaine zwitterionic polymer thin films. In such applications, these thin films were used to enhance the reversibility (retention of Coulombic efficiency (CE)) and electrochemical stability (cycle life and retention of capacity) of lithium metal batteries.5Fig. 3C shows the iCVD mechanism, as well as the modification mechanism of the zwitterionic polymer.50 Specifically, during the iCVD process, polymer films were fabricated through free radical polymerization by introducing monomers, cross-linkers, and initiators into the vacuum chamber. After the completion of iCVD process, the resulting polymer underwent a post-deposition modification, in which the tertiary nitrogen groups were converted into quaternized sulfobetaine moieties, forming a zwitterionic polymer. When comparing sulfobetaine zwitterionic polymers bearing different cationic groups (e.g., ammonium, pyridinium, and imidazolium), electrochemical characterizations revealed that the ammonium-based zwitterionic polymer outperformed other zwitterions in terms of interfacial impedance, CE, and battery cycle life, which was attributed to the enhanced Li+ solvation and reduced cation activity.
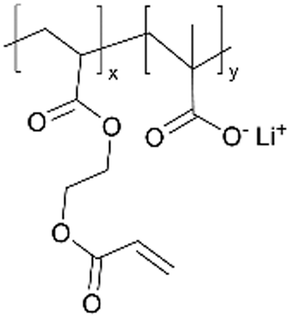
Similar effects of the zwitterionic polymer coatings have been captured in aqueous Zn batteries, hinting at their general cation conductivity.25 It is worth noting that in order to suppress the HER, a gradient coating (zwitterionic gradient (ZG)) was synthesized with p4VP placed at the coating-anode interface and pyridinium sulfobetaine polymer at the coating-electrolyte interface. While the interfacial ion transportation at the polymer–electrolyte interphase was enhanced by the zwitterionic polymer, the fact that the battery performance (capacity, efficiency, cycling stability, prevention of dendrite formation) was enhanced and not reduced from the p4VP layer implies that p4VP may exhibit cation conductivity as well. EIS spectra confirmed the protection of the ZG interphase against electrochemical corrosion on the Zn anode in an alkaline aqueous environment. Consequently, high-performance Zn‖NiOOH batteries with the ZG interphase demonstrated long-term durability.
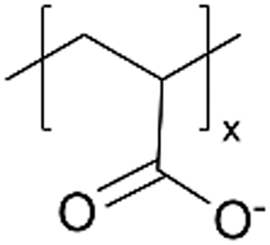
While the aforementioned examples invariably require an additional step to convert a polymer of presumed low ion conductivity to an ion-conducting one, the post-polymerization modification could occur spontaneously in operando to enhance ion conductivity. One such example is iCVD-synthesized pPFDA coatings.6 pPFDA bears a labile fluorocarbon side group that is prone to hydrolysis in alkaline environments, giving rise to a carboxylic acid side group with demonstrated cation conductivity.65Fig. 3D demonstrates the hydrolysis reaction of pPFDA in alkaline environment, where the ester group in pPFDA at the interface is hydrolyzed, creating a carboxylic acid group (COO−).6 That hydrolysis reaction is faster in amorphous pPFDA regions and slower in crystalline regions, thus giving rise to the adaptive formation of cation-conducting channels (concentrated in amorphous regions) embedded in stable and hydrophobic crystalline regions to maintain film stability and hydrophobicity. This design of adaptive ion channel formation has been demonstrated in a Zn aqueous battery.6 The spontaneous hydrolysis of amorphous pPFDA is shown to facilitate cation transportation and enhance interfacial stability, as shown by a decrease in interfacial impedance of EIS measurement using a 150-nm thick semi-crystalline pPFDA coating. The crystalline domains are shown to protect the electrode, inhibit side reactions like HER, and improve the overall cycling stability. The pPFDA interphases, with adaptive ion channel formation during battery cycling, can extend the cycle life of full Zn‖MnO2 batteries to over 11000 cycles.
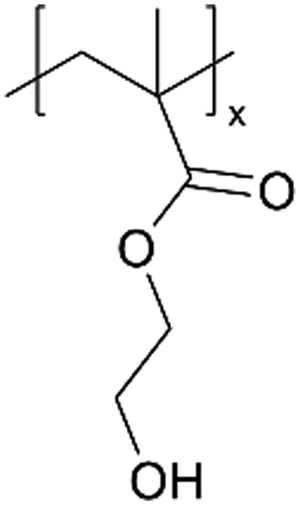
Recently, a new pathway for fabricating ICPs has been developed via ion exchange, as demonstrated by Li+-conducting polyelectrolytes in 3D micro batteries,61 where a pMAA-co-EDGA thin film is synthesized using iCVD as solid-state thin film electrolytes These films gained Li+-conductivity through a H+/Li+ ion exchange. The cross-linking provided by EDGA rendered the polymer films insoluble, preventing dissolution during ion exchange while enhancing the mechanical properties. The highest ion conductivity was observed in pMAA91-co-EDGA9 films, reaching a value of 9.59 × 10−7 S cm−1. Notably, the ion exchange strategy led to a transference number of 1, thus eliminating transport limitations associated with conventional salt-loaded polymer electrolytes. Its simultaneous improvement in ion conductivity, stability, and mechanical robustness addresses critical challenges in 3D micro batteries.61
In summary, post-deposition modification methods have emerged as a powerful strategy to overcome the limitations of direct deposition in the synthesis of ICPs via iCVD. These methods enable the transformation of non-conductive polymers into ionically conductive materials through simple nucleophilic substitution reactions, spontaneous side-chain hydrolysis, or ion exchange, thereby expanding the range of potential precursors and the palette of ion-conducting polymers. The ability to fine-tune the chemical composition and structure of these polymer films post-deposition allows for the simultaneous yet independent optimization of key performance indicators, including mechanical strength, ion conductivity, and electrochemical stability, making post-polymerization modification a promising technique in the advancement of high-performance ICPs.
3.2. Stable ionogels
Ionogels are a class of gel material comprised of Ionic liquids (ILs) dispersed in a solid network like polymers.70,71 These composite materials merge the beneficial properties of both polymers and ILs. ILs are renowned for their use as solvents due to their remarkable chemical and thermal stability, wide electrochemical window, and exceptional ionic conductivity.72 However, their liquid state presents practical and environmental challenges, such as processability, leakage, and toxicity.73 Consequently, ionogels, especially polymer–IL (PIL) composites, have attracted intensive research interest. Traditional methods for producing PIL composites include solution-casting, electrospinning, and printing. For example, the combination of HEMA monomer and a polymerizable ionic liquid, 1,4-di(vinylimidazolium)butane bisbromide (DVIMBr) have been polymerized in 1-butyl-3 methylimidazolium hexafluorophosphate ([bmim][PF6]) solvent, resulting in the in situ entrapment of the IL in the gel during polymerization and cross-linking.74 Electrochemical characterizations, including CV and EIS measurements, have shown that these composite materials have conductivity as high as 3.0 × 10−4 S cm−1.ILs are known for their low volatility, which makes them unique liquid substrates that are compatible with CVD techniques. For example, iCVD polymers have been discovered to form polymer films on top of an IL layer or polymeric particles within an IL layer (Table 3), depending on the spreading coefficient.66,67,75 The spreading coefficient indicates whether it is energetically favorable for the polymer to spread over a liquid.75 Its common expression is given in eqn (1):
| S = γLVS = γLV × (1 + cosθ) − 2γPV | (1) |
| Method | Polymers | Chemical structure | IL | IL structure | Types of ions | Temperature (°C) | Thickness (nm) | Conductivity (S cm−1) | Application | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| ILiCVD | pHEMA |
|
[bmim][PF6] |
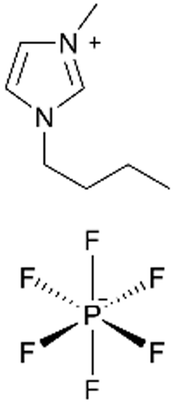
|
— | — | 200–350 | — | Fuel cells (electrolyte) | 66 |
| pPFDA |
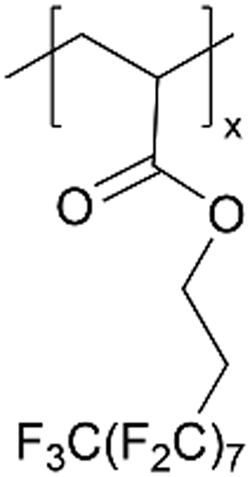
|
|||||||||
| iCVD | pHEMA |

|
[emim][BF4] |

|
— | 25 | Total: 3000–20000 (IL: ∼3000) |
1.0 × 10−2 | Fuel cell membranes, thin-film transistors | 67 |
| iCVD | pHEMA-co-EGDMA |

|
[amim][DCA] |
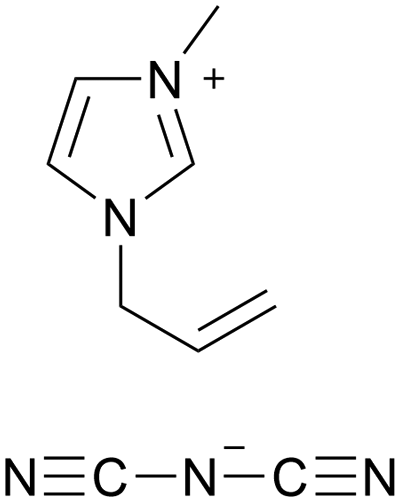
|
— | RT | 450–1000 | 1.8 × 10−2 | Fuel cell membranes | 68 |
| CW-IR | Polyurea |
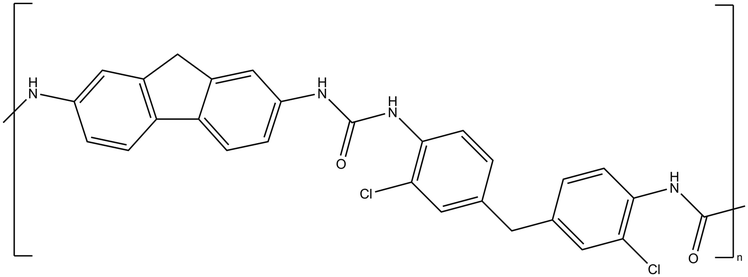
|
[emim][TFSA] |
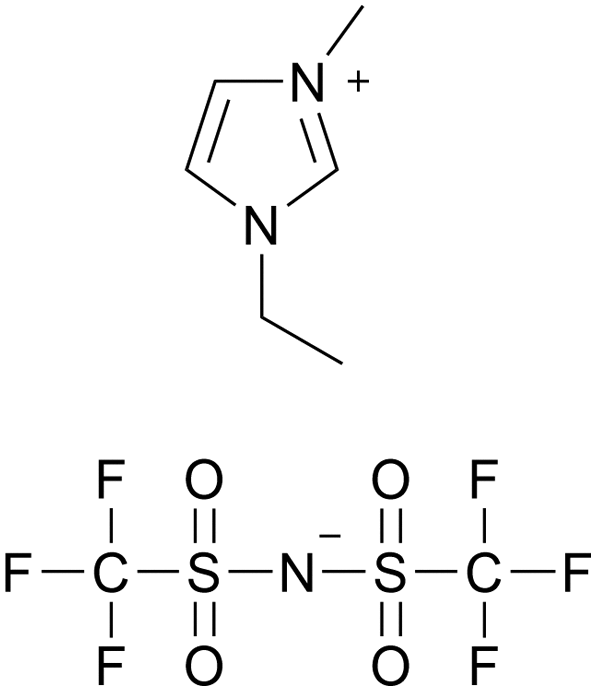
|
— | 20 | — | Electric double-layer transistors | 69 |
Since its first demonstration in 2011, 4 polymers with varying surface energy have been deposited on/in IL using iCVD, a method termed ILiCVD (Fig. 4A).66 Beyond the spreading coefficient, the temperature at which polymerization occurs also plays a critical role in determining the morphology of the CVD-enabled ionogels. For example, pHEMA has been deposited in the presence of the IL, [bmim][PF6] using iCVD. A free-standing film formed after 2 minutes of deposition, and a skin layer completely encapsulating the IL formed after 15 minutes (Fig. 4B). When varying stage temperatures for the 15-minute deposition of pHEMA, 35 °C resulted in a skin layer, 55 °C resulted in a film with polymer pieces concentrated at the edge of the IL droplet, and 75 °C led to polymer pieces floating throughout the droplet (Fig. 4C).
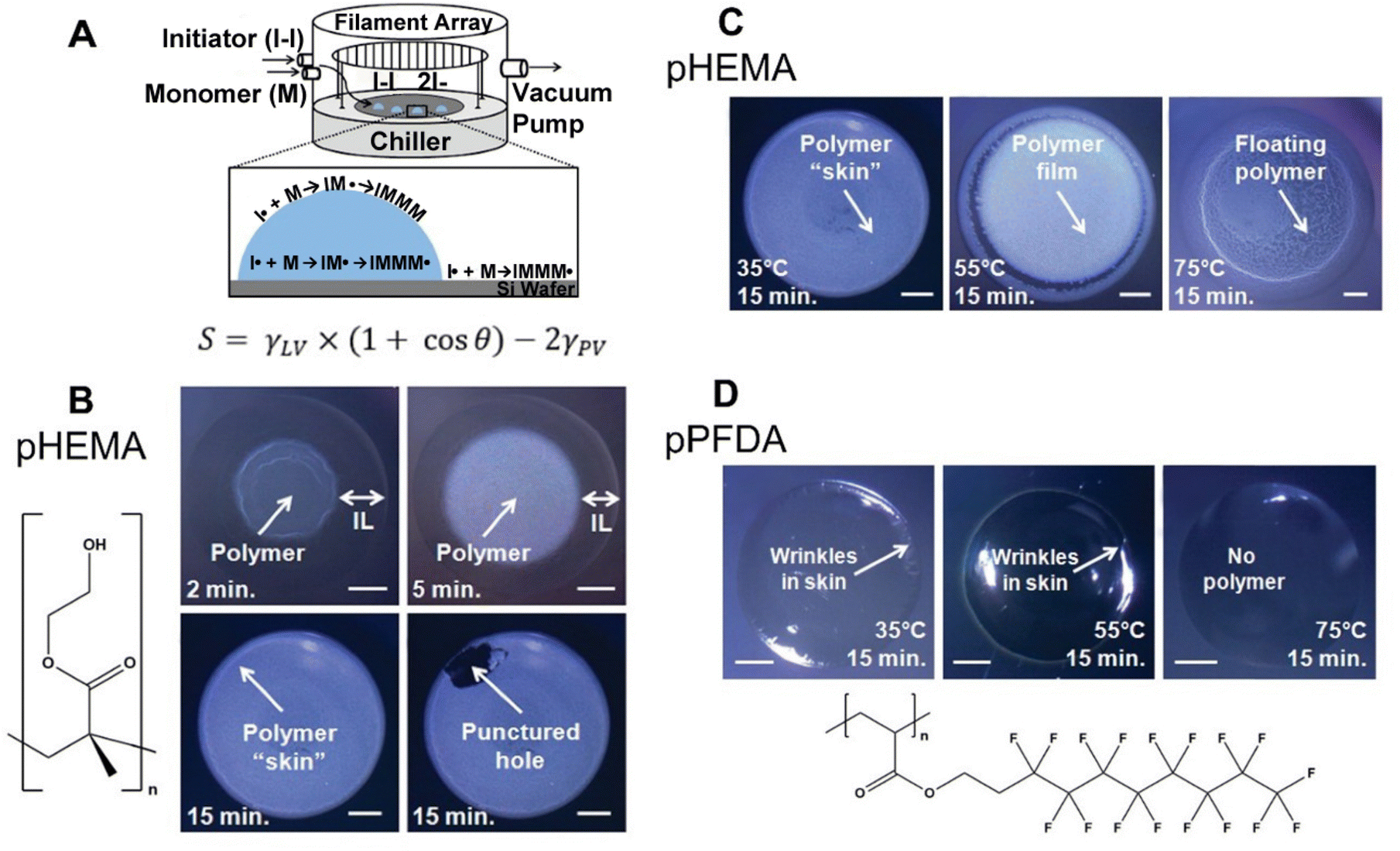 | ||
| Fig. 4 (A) ILiCVD schematic with spreading coefficient expression. (B) pHEMA depositions on [bmim][PF6] with time as the variable changed. Scale bars are 1 mm. (C) Depositions with stage temperature as the variable changed. Scale bars are 1 mm. (D) pPFDA depositions on [bmim][PF6] for 15 minutes at different temperature conditions. Scale bars are 1 mm. Reprinted from ref. 66 with permission from American Chemical Society, Copyright 2011. | ||
Changing the polymer from pHEMA to pPFDA led to skin formation at 35 °C and 55 °C, but not at 75 °C since the monomer's vapor/IL interface concentration was low with a PM/Psat of 0.03 (0.1–0.7 are considered desirable PM/Psat values) along with the low solubility of PFDA (Fig. 4D).66 The different solubilities of monomers in ILs can be utilized to create heterogeneous polymer films through simultaneous or sequential depositions using iCVD. Combining insoluble monomers like PFDA and soluble monomers such as EGDA leads to a pEGDA ionogel at the polymer–IL interface and a copolymer film on top of the ionogel.76
In subsequent studies, ionogels comprising high-molecular-weight polymer networks infiltrated by ILs have been produced via iCVD on thin IL layers.67 The solubility of HEMA in 1-ethyl-3-methylimidazolium tetrafluoroborate ([emim][BF6]) enabled polymerization at the IL–vapor interface as well as within the IL layer. Polymerization in ILs resulted in higher propagation rates and lower termination rates, leading to the formation of longer polymer chains within the IL layer. Increasing the chamber pressure further increased the molecular weight. The as-synthesized ionogels achieved an ionic conductivity of 1.0 × 10−2 S cm−1 at 25 °C, comparable to pure IL counterpart (2.2 × 10−2 S cm−1) and pHEMA-[emim][BF4] gels made via in situ polymerization (2.0 × 10−2 S cm−1).77 Longer deposition times increased the polymer concentration in the iCVD-enabled ionogel, transitioning it from a viscous liquid to a solid gel, demonstrating the ability to manipulate ionogel properties by adjusting synthesis conditions.
A copolymer of HEMA and EGDMA (pHEMA-co-EGDMA) on IL droplets of 1-allyl-3-methylimidazolium dicyanamide ([Amim][DCA]) has recently been made via iCVD, with ionic conductivity values as high as 1.8 × 10−2 S cm−1 in ambient air, higher than that for the neat [Amim][DCA] (i.e., 1.7 × 10−2 S cm−1).68 This result is in contrast with previous studies where high conductivity in pMAA–HEMA–EGDMA ionogels was only observed in high humidity (≥95%) after prolonged equilibration times of up to 400 minutes.68 Precoating substrates with a pHEMA-co-EGDMA could improve the spreading and uniformity of the solidified ionogel, but decrease overall conductivity. In addition, varying deposition conditions influenced the homogeneity and conductivity of the resulting ionogel membranes (Fig. 5A).68 An increase in pre-flow time, IL:HEMA ratio, and top-coating thickness all seemed to decrease the conductivity.
 | ||
| Fig. 5 (A) Effect of various deposition conditions on the resultant conductivity σ. “Pre-flow time” refers to the time HEMA was pre-flowed into the system. “IL:HEMA ratio” refers to the ratio of IL and HEMA when drop casted onto the substrate instead of pure IL. “Pre-coating” refers to the influence of precoating the samples with pHEMA-EGDMA before drop-casting. “Top-coating” refers to the influence of the thickness of the pHEMA-EGDMA top coating on the conductivity. Reprinted from ref. 68 with permission from American Chemical Society, Copyright 2020. (B) Schematic of porous PU-IL film procedure. (C) Impedance measurements of PU-IL film based on annealing at 100 °C or 200 °C in air or vacuum. Reference impedance of the substrate and pure IL was also given. (D) Schematic of all production stages represented in D. (E) Impedance data of the rewriteability experiment at different production stages. Reprinted from ref. 69 with permission from American Chemical Society, Copyright 2020. | ||
Another liquid substrate similarly used is liquid crystal, which has a similar structural order of a crystal while being a liquid.78 Previously, nematic liquid crystals were used to template iCVD-produced pDVB microspheres and chemical vapor polymerized (CVP) nanofibers due to its ordered structure.79,80
Besides iCVD, a recent study employed an “all-in-vacuum” process to fabricate an IL-containing porous PU nanogel electrolyte for electric double-layer transistors (EDLTs).69 4,4′-Methylene bis(2-chlorophenyl isocyanate) (MBCI) and 2,7-diaminofluorene (DAF) monomers were co-deposited at room temperature via continuous-wave IR (CW-IR) laser deposition (Fig. 5B). CW-IR was first introduced in 2007,81 and organic sources in crucibles are ablated by an IR laser (λ = 808 nm), showing equivalent quality to that fabricated by thermal evaporation and similar controllability to pulsed laser deposition (PLD). Following monomer deposition, IL 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)-amide ([emim][TFSA]) was deposited using the same method. Subsequent annealing of the bilayer thin film led to the formation of a porous PU-IL composite nanogel film. The composite film annealed in air at 100 °C showed comparable ionic conductivity to a neat IL film (Fig. 5C), indicating a uniform distribution of the IL within the porous PU matrix. In contrast, annealing of the composite film at 200 °C in vacuum increased the impedance to levels comparable to those of bare substrate, attributable to the almost complete evaporation of IL molecules from the composite thin film. Additionally, the rewriteability of the composite films was demonstrated, as the IL can easily be removed and reintroduced into the PU matrix to make the film insulating or highly conductive, respectively (Fig. 5D). Annealing in vacuum at 100 °C for 10 min can remove the IL, while depositing the IL and then vacuum annealing at 60 °C for 10 min can reintroduce IL into the material. The rewriteability was further demonstrated through impedance measurements (Fig. 5E). The impedance data indicate that the PU-IL composite nanogel film retained consistent impedance ranges with and without the incorporation of IL into the film, respectively.
Nevertheless, a potential issue with CW-IR deposition is that the target selection is largely limited to those that can absorb IR.82 An example of a substance that does not absorb IR light is IL, such as [bmim][NTf2]. To resolve this issue, an IR absorbent like Si powder can be added to the target. Recently, binary mixtures of IL droplets of [bmim][NTf2] and 1-butyl-3-methylimidazolium hyxafluorophosphate ([bmim][PF6]) mixed with Si powder have been deposited onto a sapphire (0001) substrate via CW-IR.82
In summary, by integrating the exceptional ion conductivity of ILs with the structural benefits of polymers, such as mechanical properties and solidity, researchers have created composites that exhibit high ionic conductivity, stability, and versatility. Innovations such as the ILiCVD system and the creation of high molecular weight PIL gels have demonstrated the potential of these composites for various applications, including energy storage and biomedical devices. In developing an ionogel, one must consider conditions such as the IL concentration, stage temperature, deposition time, and IL and monomer solubility. Still, the current understanding of PILs is not complete. For example, conflicting reports exist for ionogels’ conductivity relative to the base IL.67,68 Solution-based methods have attributed a decreased ionic conductivity in ionogels to a decrease in ion mobility, which could occur at greater ion size, polymer concentration (which lowers ion concentration), viscosity, or upon changes in morphology (e.g., from a porous to dense architecture).83–85 While those factors have not been as extensively studied for iCVD-produced ionogels, this represents a great opportunity to gain a deeper understanding of the structure–property relationships.86 Another property to consider for the future is the dielectric constant, as it has been demonstrated to be directly correlated to the ionic conductivity of solution-based ionogels,87 as shown in eqn (2):
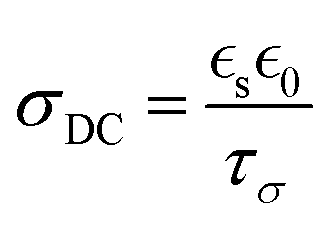 | (2) |
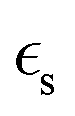 is the static dielectric constant,
is the static dielectric constant,  is the vacuum permittivity constant, and τσ is the time scale when the counter ion motion is diffusive.87 Despite the gaps in knowledge, these advancements highlight the promising future of CVD processes for PIL composites in overcoming the limitations of pure ILs.
is the vacuum permittivity constant, and τσ is the time scale when the counter ion motion is diffusive.87 Despite the gaps in knowledge, these advancements highlight the promising future of CVD processes for PIL composites in overcoming the limitations of pure ILs.
3.3. Hydrogels
Charge-neutral hydrogels have proven effective in facilitating ion transportation.88,89 These hydrogels often retain water molecules within their polymer network (mesh size of ∼10 nm), demonstrating physicochemical properties that resemble liquid water. This feature makes hydrogels elastic solids and ionic conductors.90 Several hydrogels, e.g., pHEMA,91 poly(hydroxypropyl methacrylate) (pHPMA), and pHEA,92,93 that have been synthesized using iCVD (Table 4) have demonstrated rapid swelling with water content as high as 35%.94 Notably, pHEMA is not sufficiently hydrophilic to dissolve in water;92 therefore, incorporating a cross-linker is required for gel properties.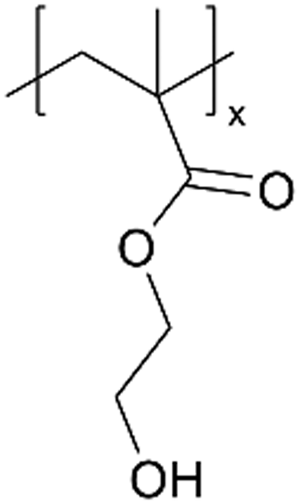

Based on that ion conductivity, iCVD-synthesized pHEMA films have been integrated into DSSCs as quasi-solid-state polymer electrolytes (Fig. 6A), resulting in enhanced device performance compared to cells with conventional liquid electrolytes.44,95 By engineering the synthesis conditions, the pHEMA films were shown to infiltrate mesoporous TiO2 electrodes, leading to an increase in Voc, while Jsc does not change significantly (Fig. 6B).44,95 This suggests that quasi-solid-state polymer electrolytes can maintain ion transport properties similar to those of liquid electrolytes. EIS analysis revealed an increased electron lifetime of ∼50 ms in the polymer electrolyte due to reduced recombination, compared to ∼12 ms in the liquid electrolyte.
 | ||
| Fig. 6 (A) Schematic of the pore filling process during iCVD, and corresponding cross sectional SEM image of polymer coated TiO2 electrode. (B) Effect of redox solvent on the performance of DSSCs fabricated with the pHEMA polymer electrolyte (solid circle) and the corresponding liquid electrolyte (hollow square) containing pure propylene carbonate. Reprinted from ref. 44 with permission from American Chemical Society, Copyright 2011. (C) Schematic of piCVD gel process. (D) Conductivity of the polymer gels with identical concentrations of 75 mmol with 3 different salts. (E) Conductivity of samples with increasing concentrations of NaCl showing a trend of increasing conductivity. Reprinted from ref. 12 with permission from IOP Publishing, Copyright 2023. | ||
An interesting class of composition-tunable composite hydrogels has been developed using piCVD recently.12 A mixture of pAA and NaCl dissolved in water was doctor-bladed onto a substrate as a precursor layer, which was subsequently coated by pHEA using piCVD (Fig. 6C).12 The precursor layer likely underwent dehydration during the piCVD step, giving rise to interconnected polymer networks of pAA and pHEA and preventing their dissolution in water in subsequent processing. Additionally, as mentioned in Section 2, highly cross-linked polymer thin films can be fabricated via piCVD without additional cross-linkers.15 The ionic conductivity of the composite hydrogel films was tunable from 1 × 10−5 to 3 × 10−2 S cm−1 by adjusting the salts (Fig. 6D) and salt concentrations (Fig. 6E), and/or the piCVD deposition conditions (e.g., concentration of initiator and reaction time).12
While the ion conductivity of hydrogels is often coupled with the content of water and/or salt, hydrogel-based ICPs have the distinct advantage of biocompatibility. As such, they are particularly promising for biosensing, actuation, and human–machine interactions, e.g., in developing soft robotics, neurosensing, and next-generation wearable devices. The distinct conformality and substrate-independence of CVD-synthesized thin films render such ICPs compatible with a wide range of substrates and devices.
3.4. Ultrathin siloxane and silazane polymer films
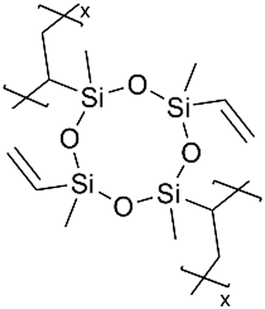
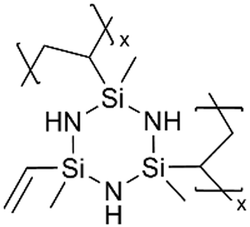
Although dielectric polymers like polysiloxanes were initially utilized as dielectric materials and insulators, they were found to conduct Li+ ions effectively after lithiation. Several ultra-thin dielectric polymer films have been successfully applied to LIBs with considerable Li+ ion conductivity using monomers like 1,3,5-trimethyl-1,3,5-trivinyl cyclotrisiloxane (V3D3) and 2,4,6,8-tetravinyl-2,4,6,8-tetramethyl cyclosiloxane (V4D4). Homopolymers of cyclic siloxane (Si–O) and silazanes (Si–N) can be described as organic covalent networks due to their multiple reactive vinyl groups (three and four vinyl groups for V3D3 and V4D4, respectively), potentially forming permeation channels for metal ions like Li+.71 The deposition of conformal thin films at the nanometer scale at room temperature has been leveraged in many battery applications, as summarized in Table 5 and detailed below.| Method | Polymer | Chemical structure | Types of ions | Temperature (°C) | Thickness (nm) | Conductivity (S cm−1) | Other values | Application | Ref. |
|---|---|---|---|---|---|---|---|---|---|
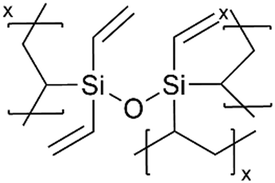
| iCVD | pHVDS |
|
Li+ | RT | ∼3.9 | 8.7 × 10−6 | — | LIB (Separator) | 11 |
| iCVD | pV4D4 |

|
Li+ | RT | 25 | 5.0 × 10−8 | — | LIB (Electrolyte) | 10 |
| iCVD | pV4D4 |

|
Li+ | RT | 25 | 7.5 × 10−8 | — | LIB (Electrolyte) | 96 |
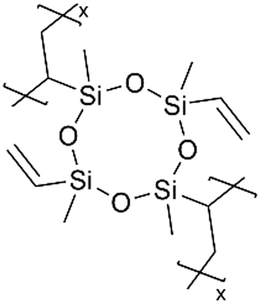
| iCVD | pV4D4 |
|
Li+ | RT | 25 | 6.0 × 10−8 | — | LIB (SEI) | 97 |
| iCVD | pV3D3 |

|
Li+ | RT | 20 | 8.0 × 10−9 | — | LIB (Electrolyte) | 98 |
| pV4D4 |
|
Li+ | RT | 25 | 6.0 × 10−8 | ||||
| pV3N3 |
|
Li+ | RT | 25 | 2.0 × 10−9 | ||||
| pV4N4 |

|
Li+ | RT | 10 | 2.0 × 10−9 | ||||
| iCVD | pV3D3 |

|
Li+ | — | 10–40 | — | — | LIB (SEI) | 99 |
| iCVD | pV4D4 |

|
Li+ | — | 25 | — | — | AFLB (SEI) | 100 |
A ∼3.9 nm thick cross-linked polyhexavinyldisiloxane (pHVDS) coating has been synthesized via iCVD, which exhibited a Li+ ionic conductivity of 8.7 × 10−6 S cm−1.11 When applied to poly ethylene (PE) separators, the coating led to a modest improvement of ion conductivity from 6.6 × 10−4 S cm−1 to 8.4 × 10−4 S cm−1, which was attributed to a greater wettability of the LIB electrolyte (i.e., LiPF6 in ethylene carbonate and ethyl methyl carbonate) on the pHVDS coating than uncoated PE. Half coin-type cells using a pHVDS-coated PE separator showed enhanced electrochemical performance in terms of capacity (by almost two times), rate capability, and stability after 70 charge–discharge cycles.
Siloxane-based polymer films have also been used for solid-state electrolytes in LIBs due to their Li+ ion conductive characteristics. In 3D battery applications, ultrathin and conformal ICP films that uniformly cover 3D electrodes with complex geometries serve as solid-state electrolytes, whose nano-scale ion diffusion lengths compensate for modest ionic conductivities. Lithiation of a 25-nm thick pV4D4 film exhibited a Li+ ion conductivity of 7.5 × 10−8 S cm−1 at room temperature (Fig. 7A).96 The ultra-thin pV4D4 films (∼10 nm-thick) were deposited via iCVD as smooth and conformal coatings that uniformly cover Ag nanowires (Fig. 7B), demonstrating minimal morphological or thickness changes during lithiation and, thus, reasonable stability.
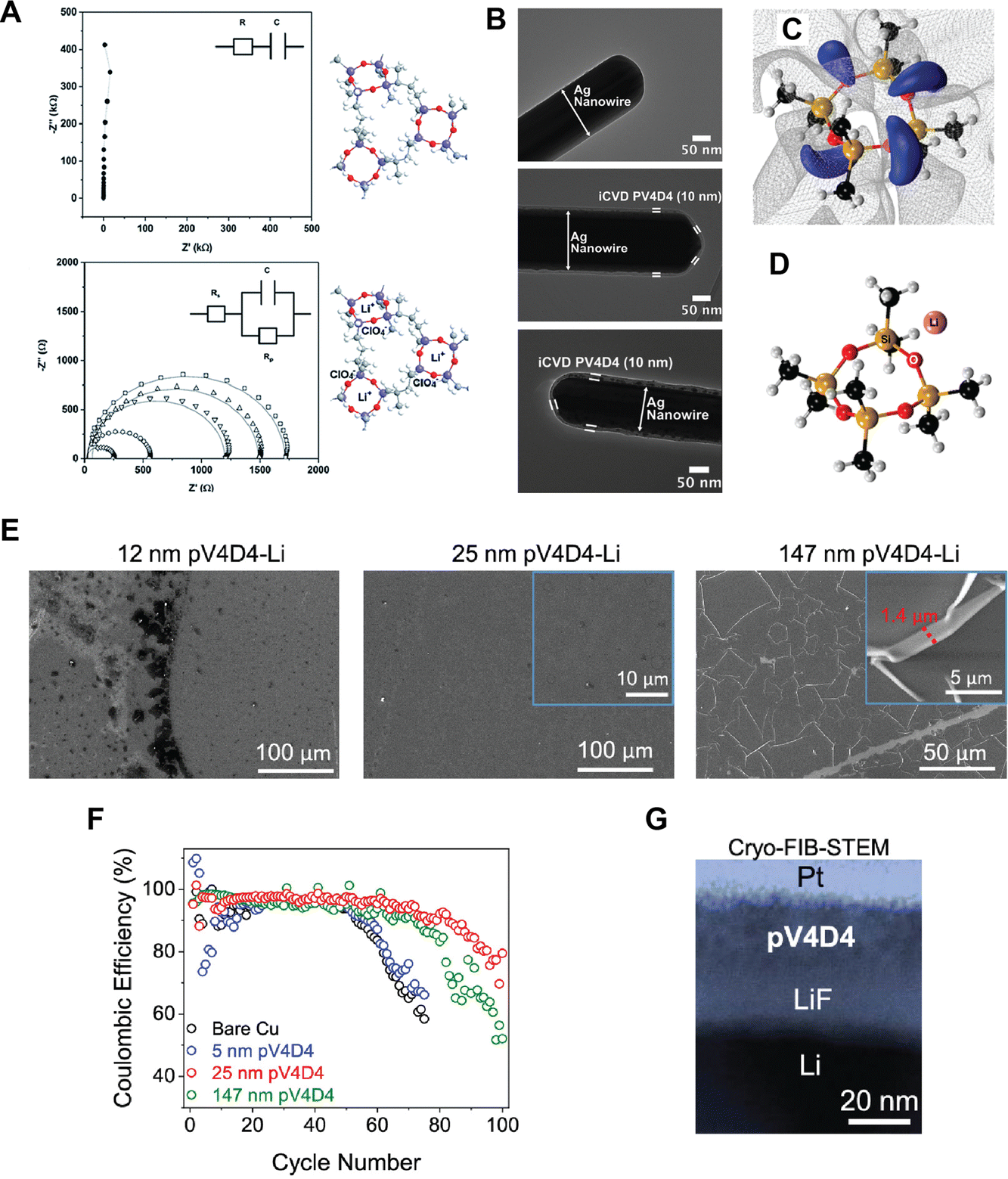 | ||
| Fig. 7 (A) Impedance spectra of 25-nm-thick PV4D4 films that are unlithiated (top) and lithiated (bottom), and their corresponding molecular structures (blue: Si; red: O). The data were taken in the frequency range from 100 kHz to 100 Hz using a 10-mV (RMS) potential. The circuits used for equivalent circuit fitting are shown as insets. (B) TEM images of (top) uncoated Ag nanowire, (middle) 10 nm pV4D4 coated Ag nanowire, and (bottom) 10 nm pV4D4 coated Ag nanowire after lithiation. Reprinted from ref. 96 with permission from The Royal Society of Chemistry, copyright 2015. (C) The electrostatic potential map for the ground state configuration of the V4D4 monomer in which regions of the negative potential are colored blue and the isosurface of the zero potential is indicated by the gray mesh. The colored spheres indicate the positions of the silicon (gold), oxygen (red), carbon (black), and hydrogen (white) atoms. (D) illustrate the Li+ coordination environment in the vicinity of the ring structures in V4D4. Reprinted from ref. 10 with permission from The American Chemical Society, copyright 2015. (E) SEM images of pV4D4 films deposited on Si wafers after soaking in the electrolyte. (F) Coulombic efficiency for each pV4D4 coating thickness. Each data point represents the average of two cells. (G) Cross-sectional cryo-FIB-STEM image of pV4D4-coated Li anode that created LiF SEI layer. Reprinted from ref. 100 with permission from The American Chemical Society, copyright 2023. | ||
Density functional theory (DFT) calculations revealed that the Li+–siloxane ring interactions are stable near the electronegative oxygen atoms. For example, the electrostatic potential of the ground state configuration of a single V4D4 shows a particularly negative electrostatic potential, which is localized near the oxygen atoms, highlighted in blue (Fig. 7C).10 Optimized geometries of single Li+ ion-coordinated V4D4 (Fig. 7D) demonstrate that Li+ is stable within the blue-highlighted regions in Fig. 7C. Furthermore, based on the investigation on the Li+–siloxane ring binding energies, it was asserted that the ring structure in pV4D4 films would lead to higher Li+ ion conductivity than pV3D3 or polysilazane, attributed to the more energetically favorable Li+ binding sites and lower ring strain in V4D4.
Cyclic polysilazanes, such as poly(1,3,5-trivinyl-1,3,5-trimethylcyclotrisilazane) (pV3N3) and poly(1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasilazane) (pV4N4), have also been synthesized and characterized for their ion conductivity.98 By replacing the O atoms in polysiloxanes with N atoms, polysilazane films, including pV3N3 and pV4N4, showed lower ionic conductivities at room temperature, averaging around 2.0 × 10−9 S cm−1, compared to pV4D4, which exhibited an ion conductivity of 6.0 × 10−8 S cm−1. That has been attributed to the polarity and flexibility of the Si–O–Si bonds, which facilitate ion transport through the molecular motion of the cross-linked network.
Those ultra-thin siloxane and silazane films have been applied as artificial SEI layers in LIBs to restrict the liquid electrolytes from accessing the anode, hence preventing electrolyte reduction, while allowing Li+ transfer. They have been shown to maintain the passivation of Si electrodes, even with their volumetric evolutions during cycling. In one example, a 25-nm-thick pV4D4 film on a Si electrode improved the initial CE by 12.9% and capacity retention by 64.9% over 100 cycles relative to untreated Si electrodes.97 By comparing the film thicknesses of 0, 15, 25, 35, and 45 nm, it was shown that thicknesses greater than 25 nm lead to reduced ion conductivity, slower ion transport, and poor lithiation of the silicon anode.
Anode materials, including graphite and Cu, have also been modified by polysiloxane films. LIBs with pV3D3-coated graphite anodes exhibited greater cycling stability, reduced fade of capacity over 100 charge/discharge cycles, and better performance under high loads and overcharge conditions.99 With 50-nm pV3D3 coating on the graphite anode, the cells remained well-behaved in EIS measurements and demonstrated Nyquist responses, indicating good Li+ ion transfer into the graphite. Post-mortem XPS measurements revealed that the pV3D3 layer led to SEIs with desirable compounds (LiF) and improved uniformity while reducing undesirable side reactions compared to the cells with uncoated graphite anodes.
Recently, a Cu current collector coated with a 25-nm-thick pV4D4 layer enabled robust Li plating and stripping by serving as an artificial SEI that mitigates electrolyte decomposition and facilitates a uniform permeation flux of Li+, pointing to their potential applications in anode-free Li metal batteries (AFLBs).100 While a 25-nm-thick pV4D4 layer as an artificial SEI was shown to enable Li deposition underneath the film, increasing the thickness to 147 nm led to Li nucleation and deposition atop the pV4D4 layer, likely caused by the permeation resistance associated with a thicker layer. In addition, exposure to a liquid electrolyte significantly altered the thickness and morphology of the 147-nm-thick pV4D4 films (Fig. 7E). The wrinkle-like features were attributed to the compressive stress upon deswelling. The ultrathin pV4D4 films remained stable after the exposure to electrolytes.100 EIS also indicated that the lithiation and delithiation of the 25 nm pV4D4 were stable over 100 cycles, pointing to high cycling efficiency (retaining a CE > 80%), especially compared to cells with 5, 25, or 147-nm pV4D4 films (Fig. 7F).100 Post-mortem analysis demonstrated that the 25-nm pV4D4 effectively blocked electrolytes from accessing the anode and the associated decomposition while promoting an artificial SEI comprising LiF (Fig. 7G).100 These effects were partially attributed to the Li+ ionic conductivity of the 25-nm-thick pV4D4, although the intrinsic ionic conductivity value was omitted.
The siloxane- and silazane-based ICPs have the distinct advantages of tunable cross-linking density, (electro)chemical stability, and deformation tolerance compared to other ICP thin films synthesized via CVD polymerization techniques, making such ICPs excellent candidates for artificial SEI layers in energy storage devices. In addition, the CVD technologies afford nanometer-scale precision in thickness control, which has been leveraged to form ultrathin siloxane- and silazane-based ICP coatings that deliver targeted interfacial properties (e.g., mechanical stability) without introducing additional ion-conducting resistance.
4. Conclusions and perspectives
This review summarizes the recent updates on ICP films synthesized via CVD polymerization techniques (predominantly iCVD and its variants). The ICPs discussed include ionomer thin films that have been directly deposited or synthesized via post-deposition modifications, ionogels, hydrogels, and ultrathin siloxane-based polymers. The uniform, conformal, high-purity, and thickness-controlled ICP films, synthesized in a solvent-free and at mild temperatures using various CVD techniques, are becoming increasingly prevalent in energy storage devices. They present a unique advantage when combined with complex nano- and micro-structures, such as 3D electrodes in batteries, due to the conformality and substrate independence, which points to device miniaturization and, in some cases, improved ion conductivity.Many CVD-enabled ICP thin films have been successfully integrated into batteries as artificial SEIs or solid-state electrolytes, exhibiting reasonable ionic conductivity and electrochemical stability. The all-dry synthesis and processing conditions allow ICP coatings to be applied directly on highly reactive alkali metals or transition metals, thus bypassing common side reactions caused by impurities or solvents that are introduced during conventional solution-based coating methods. Although most CVD-synthesized ICPs have shown lower ionic conductivities than liquid electrolytes, recent advancements, such as ionogel and hydrogel composites, have narrowed that gap. Furthermore, the nanometer-scale thickness control afforded by CVD technologies has been leveraged to synthesize ultrathin ICP films to compensate for the lower ionic conductivity by shortening the ion diffusion length. That is particularly effective in 3D battery electrodes or electrolytes, where CVD could enable the conformal growth of ICP nanolayers.
Iontronic devices, including artificial muscles, skins, touchpads, sensors, and wearable electronics, could also benefit from CVD-synthesized ICPs. With the combination of ion conductivity and biocompatibility, CVD-synthesized hydrogels have the potential to mimic living tissues in terms of mechanical, chemical, and electrical properties, bridging the human–machine interface and facilitating their integration. While most devices reported to date employ planar interfacial structures, CVD technologies will allow the facile integration of ion-conducting hydrogels into 3D interfaces/devices with complex nano/micro-structures, pointing to enhanced sensitivity and responsiveness, e.g., in artificial skins and neuromorphic devices.
A third potentially fruitful direction of future research we would like to highlight is the application of CVD-synthesized ICPs in electrochemistry, e.g., the electro-reduction of CO2. Such electrochemical processes often pose multi-faceted challenges to the interface design, which could be addressed by leveraging the precision of synthesis enabled by CVD polymerization. For example, the design of gas-diffusion layers in a CO2-reduction flow cell simultaneously requires high surface area (for electrocatalysis), high ionic conductivity, and hydrophobicity (to improve the selectivity and efficiency), which could be met by synthesizing conformal ultra-thin ICPs, their copolymers, or composites using CVD polymerization. Nevertheless, achieving ionic conductivities that are on par with current gold standards such as Nafion®, Sustainion®, or Aquivion® could be challenging for CVD polymerization.
While ion conductivity, film quality, and device integration/fabrication have attracted the most attention in recent research, this review also aims to highlight several key knowledge gaps in the fundamental structure–property relationships, which, once addressed, could guide the design of next-generation ICPs. For example, in the development of ionogels, a better understanding of the effects of ion mobility, polymer concentration, dielectric constant, viscosity, and morphology on ion conductivity could lead to ICP composites with designer properties to meet the aforementioned multi-faceted requirements. Furthermore, the exploration of novel ICP chemistries that are compatible with CVD polymerization remains limited, with a particular unmet need in proton or cation exchange polymers beyond the well-established (meth)acrylic acid chemistry. The widely accepted best-in-class ICPs, such as the PFSAs family, remain to be achieved using all-dry synthesis methods. That breakthrough will likely require major innovations in the CVD process and/or the development of new monomers.
This review showcases the transformative potential of CVD-synthesized ICP films in advancing energy storage, iontronic devices, and electrochemical processes, where recent breakthroughs are paving the way for enhanced performance and device miniaturization. While challenges remain in achieving higher ionic conductivities, the path forward is full of promise, with exciting opportunities for novel chemistries and innovative designs to shape the next generation of advanced materials.
Author contributions
CY, SF, and KWP conducted the literature search and composed the manuscript. RY directed the study and revised the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Cornell Atkinson Center for Sustainability. The project was also sponsored by the Department of the Navy, Office of Naval Research under ONR award N00014-23-1-2189, and the National Science Foundation Faculty Early Career Development Program [grant number CMMI-2144171].Notes and references
- J. Owen, Comprehensive Polymer Science and Supplements, Elsevier, 1989, pp. 669–686 Search PubMed.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- in CVD Polymers: Fabrication of Organic Surfaces and Devices, ed. K. K. Gleason, Wiley, 1st edn, 2015 Search PubMed.
- Y. Cheng, A. Khlyustova, P. Chen and R. Yang, Macromolecules, 2020, 53, 10699–10710 CrossRef CAS.
- S. Cho, Y. Kim, Y. Song, J. Ryu, K. Choi, J. Yang, S. Lee, S. G. Im and S. Park, Adv. Funct. Mater., 2024, 2314710 CrossRef CAS.
- P. Chen, S. Jin, S. Hong, Y. Qiu, Z. Zhang, Y. Xu, Y. L. Joo, L. A. Archer and R. Yang, J. Am. Chem. Soc., 2024, 146, 3136–3146 CrossRef CAS PubMed.
- S. K. Murthy, B. D. Olsen and K. K. Gleason, Langmuir, 2002, 18, 6424–6428 CrossRef CAS.
- J. Bae, K. Choi, H. Song, D. H. Kim, D. Y. Youn, S. Cho, D. Jeon, J. Lee, J. Lee, W. Jang, C. Lee, Y. Kim, C. Kim, J. Jung, S. G. Im and I. Kim, Adv. Energy Mater., 2023, 13, 2203818 CrossRef CAS.
- A. M. Coclite, P. Lund, R. Di Mundo and F. Palumbo, Polymer, 2013, 54, 24–30 CrossRef CAS.
- B. Reeja-Jayan, N. Chen, J. Lau, J. A. Kattirtzi, P. Moni, A. Liu, I. G. Miller, R. Kayser, A. P. Willard, B. Dunn and K. K. Gleason, Macromolecules, 2015, 48, 5222–5229 CrossRef CAS.
- Y. Yoo, B. G. Kim, K. Pak, S. J. Han, H.-S. Song, J. W. Choi and S. G. Im, ACS Appl. Mater. Interfaces, 2015, 7, 18849–18855 CrossRef CAS.
- E. D. Patamia and T. L. Andrew, Flex. Print. Electron., 2023, 8, 045003 CrossRef CAS.
- S. Z. Homayounfar, S. Rostaminia, A. Kiaghadi, X. Chen, E. T. Alexander, D. Ganesan and T. L. Andrew, Matter, 2020, 3, 1275–1293 CrossRef.
- A. Khlyustova and R. Yang, Front. Bioeng. Biotechnol., 2021, 9, 670541 CrossRef PubMed.
- E. Güney, J. Karimzadeh Khoei, B. S. Sengul, F. Can and G. Ozaydin Ince, J. Appl. Polym. Sci., 2024, 141, e55210 CrossRef.
- A. Dianatdar and R. K. Bose, J. Mater. Chem. C, 2023, 11, 11776–11802 RSC.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1801564 CrossRef.
- D. Bhattacharyya, R. M. Howden, D. C. Borrelli and K. K. Gleason, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1329–1351 CrossRef CAS.
- M. E. Alf, A. Asatekin, M. C. Barr, S. H. Baxamusa, H. Chelawat, G. Ozaydin-Ince, C. D. Petruczok, R. Sreenivasan, W. E. Tenhaeff, N. J. Trujillo, S. Vaddiraju, J. Xu and K. K. Gleason, Adv. Mater., 2010, 22, 1993–2027 CrossRef CAS PubMed.
- C. A. Dorval Dion and J. R. Tavares, Powder Technol., 2013, 239, 484–491 CrossRef CAS.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- S. Favero, I. E. L. Stephens and M. Titirci, Adv. Mater., 2024, 36, 2308238 CrossRef CAS PubMed.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- W. Li, L. C. Bradley and J. J. Watkins, ACS Appl. Mater. Interfaces, 2019, 11, 5668–5674 CrossRef CAS.
- S. Jin, P.-Y. Chen, Y. Qiu, Z. Zhang, S. Hong, Y. L. Joo, R. Yang and L. A. Archer, J. Am. Chem. Soc., 2022, 144, 19344–19352 CrossRef CAS.
- P. Chen, J. Lang, Y. Zhou, A. Khlyustova, Z. Zhang, X. Ma, S. Liu, Y. Cheng and R. Yang, Sci. Adv., 2022, 8, eabl8812 CrossRef CAS.
- A. Khlyustova, M. Kirsch, X. Ma, Y. Cheng and R. Yang, J. Mater. Chem. B, 2022, 10, 2728–2739 RSC.
- E. Yu. Safronova, D. Yu. Voropaeva, A. A. Lysova, O. V. Korchagin, V. A. Bogdanovskaya and A. B. Yaroslavtsev, Polymers, 2022, 14, 5275 CrossRef PubMed.
- A. Olejnik, J. Karczewski, A. Dołęga, K. Siuzdak and K. Grochowska, Coatings, 2020, 10, 810 CrossRef CAS.
- S. Farzin, T. J. Johnson, S. Chatterjee, E. Zamani and S. K. Dishari, Front. Chem., 2020, 8, 690 CrossRef CAS PubMed.
- W. E. Tenhaeff and K. K. Gleason, Adv. Funct. Mater., 2008, 18, 979–992 CrossRef CAS.
- F. P. García De Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- J. R. Robalo, S. Huhmann, B. Koksch and A. Vila Verde, Chem, 2017, 3, 881–897 CAS.
- X. Kong, P. E. Rudnicki, S. Choudhury, Z. Bao and J. Qin, Adv. Funct. Mater., 2020, 30, 1910138 CrossRef CAS.
- T. Kasuga, H. Yagyu, K. Uetani, H. Koga and M. Nogi, ACS Appl. Nano Mater., 2021, 4, 3861–3868 CrossRef CAS.
- G. Zheng, C. Wang, A. Pei, J. Lopez, F. Shi, Z. Chen, A. D. Sendek, H.-W. Lee, Z. Lu, H. Schneider, M. M. Safont-Sempere, S. Chu, Z. Bao and Y. Cui, ACS Energy Lett., 2016, 1, 1247–1255 CrossRef CAS.
- Q. Pan, D. M. Smith, H. Qi, S. Wang and C. Y. Li, Adv. Mater., 2015, 27, 5995–6001 CrossRef CAS PubMed.
- P. Ye, X. Li, K. He, A. Dou, X. Wang, A. Naveed, Y. Zhou, M. Su, P. Zhang and Y. Liu, J. Power Sources, 2023, 558, 232622 CrossRef CAS.
- J. Pu, C. Zhong, J. Liu, Z. Wang and D. Chao, Energy Environ. Sci., 2021, 14, 3872–3911 RSC.
- M. Joo, J. Shin, J. Kim, J. B. You, Y. Yoo, M. J. Kwak, M. S. Oh and S. G. Im, J. Am. Chem. Soc., 2017, 139, 2329–2337 CrossRef CAS PubMed.
- J. Yu, Y. Kim, C. Lee, B. Jeong, J. Kim, J. Han, J. Yang, S. Yun, S. G. Im and Y. Choi, Small, 2024, 20, 2312283 CrossRef CAS PubMed.
- B. Li, L. Wang, B. Kang, P. Wang and Y. Qiu, Sol. Energy Mater. Sol. Cells, 2006, 90, 549–573 CrossRef CAS.
- C. Hsieh and K. K. S. Lau, Adv. Mater. Interfaces, 2015, 2, 1500341 CrossRef.
- S. Nejati and K. K. S. Lau, Nano Lett., 2011, 11, 419–423 CrossRef CAS PubMed.
- Y. Y. Smolin, S. Nejati, M. Bavarian, D. Lee, K. K. S. Lau and M. Soroush, J. Power Sources, 2015, 274, 156–164 CrossRef CAS.
- A. G. Kuba, Y. Y. Smolin, M. Soroush and K. K. S. Lau, Chem. Eng. Sci., 2016, 154, 136–142 CrossRef CAS.
- N. Pekel, H. Savaş and O. Güven, Colloid Polym. Sci., 2002, 280, 46–51 CrossRef CAS.
- Y. Y. Smolin, S. Janakiraman, M. Soroush and K. K. S. Lau, Thin Solid Films, 2017, 635, 9–16 CrossRef CAS.
- J. Yu, C. Lee, D. Kim, H. Park, J. Han, J. Hur, J. Kim, M. Kim, M. Seo, S. G. Im and Y. Choi, Adv. Funct. Mater., 2021, 31, 2010971 CrossRef CAS.
- S. Stalin, P. Chen, G. Li, Y. Deng, Z. Rouse, Y. Cheng, Z. Zhang, P. Biswal, S. Jin, S. P. Baker, R. Yang and L. A. Archer, Matter, 2021, 4, 3753–3773 CrossRef CAS.
- R. Yang and K. K. Gleason, Langmuir, 2012, 28, 12266–12274 CrossRef CAS PubMed.
- H. Z. Shafi, A. Matin, Z. Khan, A. Khalil and K. K. Gleason, Surf. Coat. Technol., 2015, 279, 171–179 CrossRef CAS.
- H. Shu, P. Chen and R. Yang, Chem. Bio Eng., 2024, 1, 516–534 CrossRef CAS.
- A. Khlyustova, Y. Cheng and R. Yang, J. Mater. Chem. B, 2020, 8, 6588–6609 RSC.
- T. Franklin and R. Yang, ACS Biomater. Sci. Eng., 2020, 6, 182–197 CrossRef CAS PubMed.
- K. K. Gleason, J. Vac. Sci. Technol., A, 2020, 38, 020801 CrossRef CAS.
- C. Falcón García, M. Kretschmer, C. N. Lozano-Andrade, M. Schönleitner, A. Dragoŝ, Á. T. Kovács and O. Lieleg, npj Biofilms Microbiomes, 2020, 6, 1 CrossRef PubMed.
- Z. Li, F. Hu, N. Huo and W. E. Tenhaeff, J. Mater. Chem. A, 2022, 10, 8390–8400 RSC.
- H. O. Ford, B. L. Chaloux, B. Jugdersuren, X. Liu, C. A. Klug, J. B. Miller, X. Zuo, M. W. Swift, M. D. Johannes, J. W. Long, D. R. Rolison and M. B. Sassin, RSC Appl. Interfaces, 2024, 1, 531–543 RSC.
- H. O. Ford, B. L. Chaloux, Y. Kim, J. W. Long, D. R. Rolison and M. B. Sassin, RSC Appl. Interfaces, 2024, 1, 522–530 RSC.
- Y. Gao and W. E. Tenhaeff, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2019, 37, 051401 Search PubMed.
- K. Aissou, M. Coronas, J. Richard, C. Bakkali-Hassani, S. Vishwakarma, E. Petit, A. Van Der Lee and S. Roualdes, J. Polym. Sci., 2024, 62, 4567–4575 CrossRef CAS.
- H. Kwon, S. Kim, H. Park, J. S. Park, J. Park, Y. Lee, S. J. Yang and U. H. Choi, Adv. Funct. Mater., 2024, 2406727 CrossRef CAS.
- L. Chai, Z. Zou, Z. Yang and G. Yang, J. Mater. Chem. A, 2024, 12, 20288–20299 RSC.
- S. Shindler and R. Yang, Langmuir, 2023, 39, 1215–1226 CrossRef CAS PubMed.
- P. D. Haller, R. J. Frank-Finney and M. Gupta, Macromolecules, 2011, 44, 2653–2659 CrossRef CAS.
- R. J. Frank-Finney, L. C. Bradley and M. Gupta, Macromolecules, 2013, 46, 6852–6857 CrossRef CAS.
- M. Kräuter, M. Tazreiter, A. Perrotta and A. M. Coclite, Macromolecules, 2020, 53, 7962–7969 CrossRef PubMed.
- H. Watanabe, R. Takazawa, R. Takahashi, S. Maruyama and Y. Matsumoto, ACS Appl. Nano Mater., 2020, 3, 9610–9615 CrossRef CAS.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- C. Yan, W. Li, Z. Liu, S. Zheng, Y. Hu, Y. Zhou, J. Guo, X. Ou, Q. Li, J. Yu, L. Li, M. Yang, Q. Liu and F. Yan, Adv. Funct. Mater., 2024, 34, 2314408 CrossRef CAS.
- S. P. M. Ventura, F. A. E. Silva, M. V. Quental, D. Mondal, M. G. Freire and J. A. P. Coutinho, Chem. Rev., 2017, 117, 6984–7052 CrossRef CAS PubMed.
- O. Lebedeva, D. Kultin and L. Kustov, Eur. Polym. J., 2024, 203, 112657 CrossRef CAS.
- M. Taghavikish, S. Subianto, Y. Gu, X. Sun, X. S. Zhao and N. R. Choudhury, Sci. Rep., 2018, 8, 10918 CrossRef CAS PubMed.
- P. D. Haller, L. C. Bradley and M. Gupta, Langmuir, 2013, 29, 11640–11645 CrossRef CAS PubMed.
- L. C. Bradley and M. Gupta, Langmuir, 2013, 29, 10448–10454 CrossRef CAS PubMed.
- A. Noda and M. Watanabe, Electrochim. Acta, 2000, 45, 1265–1270 CrossRef CAS.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926 CrossRef CAS PubMed.
- K. C. K. Cheng, M. A. Bedolla-Pantoja, Y.-K. Kim, J. V. Gregory, F. Xie, A. De France, C. Hussal, K. Sun, N. L. Abbott and J. Lahann, Science, 2018, 362, 804–808 CrossRef CAS PubMed.
- A. Jain, S. Pal, N. L. Abbott and R. Yang, arXiv, 2023, preprint, arXiv:2303.05662, 10.48550/ARXIV.2303.05662.
- S. Yaginuma, J. Yamaguchi, M. Haemori, K. Itaka, Y. Matsumoto, M. Kondo and H. Koinuma, J. Phys.: Conf. Ser., 2007, 59, 520–525 CrossRef CAS.
- Y. Takeyama, S. Maruyama and Y. Matsumoto, Sci. Technol. Adv. Mater., 2011, 12, 054210 CrossRef PubMed.
- K. Zhao, H. Song, X. Duan, Z. Wang, J. Liu and X. Ba, Polymers, 2019, 11, 444 CrossRef PubMed.
- M. Wang, J. Hu and M. D. Dickey, JACS Au, 2022, 2, 2645–2657 CrossRef CAS PubMed.
- A. V. Agafonov, E. P. Grishina, N. O. Kudryakova, L. M. Ramenskaya, A. S. Kraev and V. D. Shibaeva, Arab. J. Chem., 2022, 15, 103470 CrossRef CAS.
- M. Wang, J. Hu and M. D. Dickey, NPG Asia Mater., 2023, 15, 66 CrossRef.
- P. Kuray, W. Mei, S. Sheffield, J. Sengeh, C. Pulido, C. Capparelli, R. Hickey and H. Hickner, Front. Energy Res., 2020, 8, 569387 CrossRef.
- X. Li, D. Wang and F. Ran, Energy Storage Mater., 2023, 56, 351–393 CrossRef.
- J. Han, K. Wang, W. Liu, C. Li, X. Sun, X. Zhang, Y. An, S. Yi and Y. Ma, Nanoscale, 2018, 10, 13083–13091 RSC.
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- E. Sevgili and M. Karaman, Thin Solid Films, 2019, 687, 137446 CrossRef.
- J. Choi, J. Yoon, M. J. Kim, K. Pak, C. Lee, H. Lee, K. Jeong, K. Ihm, S. Yoo, B. J. Cho, H. Lee and S. G. Im, ACS Appl. Mater. Interfaces, 2019, 11, 29113–29123 CrossRef CAS PubMed.
- K. Chan and K. K. Gleason, Langmuir, 2005, 21, 8930–8939 CrossRef CAS PubMed.
- S. Nejati and K. K. S. Lau, Thin Solid Films, 2011, 519, 4551–4554 CrossRef CAS.
- N. Chen, B. Reeja-Jayan, J. Lau, P. Moni, A. Liu, B. Dunn and K. K. Gleason, Mater. Horiz., 2015, 2, 309–314 RSC.
- B. H. Shen, S. Wang and W. E. Tenhaeff, Sci. Adv., 2019, 5, eaaw4856 CrossRef CAS PubMed.
- N. Chen, B. Reeja-Jayan, A. Liu, J. Lau, B. Dunn and K. K. Gleason, Macromol. Rapid Commun., 2016, 37, 446–452 CrossRef CAS PubMed.
- R. Carter, J. F. Parker, M. B. Sassin, E. J. Klein, M. A. Wolak, C. T. Love and J. W. Long, J. Electrochem. Soc., 2020, 167, 060510 CrossRef CAS.
- R. B. Nuwayhid, J. Yeom, J. Watt, H. O. Ford, J. W. Long, R. Carter and C. T. Love, ACS Appl. Energy Mater., 2023, 6, 12072–12083 CrossRef CAS.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |