
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling critical role of metal oxide infiltration in controlling the surface oxygen exchange activity and polarization of SrTi1−xFexO3−δ perovskite oxide electrodes†
Hyunseung Kim‡
 a,
Han Gil Seo‡bc,
Sejong Ahna,
Harry L. Tuller*b and
WooChul Jung*d
a,
Han Gil Seo‡bc,
Sejong Ahna,
Harry L. Tuller*b and
WooChul Jung*d
aDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology (MIT), Cambridge, MA 02139, USA
cDepartment of Materials Science and Engineering, Dankook University, Cheonan 31116, Republic of Korea
dDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: wcjung@snu.ac.kr
First published on 20th February 2025
Abstract
Enhancing the oxygen reduction activity of mixed electronic-ionic conducting oxides holds paramount importance in various energy and fuel conversion technologies. One effective method has involved manipulating surface oxygen kinetics on fluorite-type oxides via controlled acidity of infiltrated binary oxides. This strategy is now extended to the commercially utilized but more complex perovskite-oxide-based electrocatalysts by investigating the impact of surface infiltration of basic CaO or acidic Al2O3 on the oxygen exchange kinetics of perovskite structured SrTi1−x/100Fex/100O3−δ (STFx) mixed conductors. By systematically assessing the degree of activation or deactivation induced by infiltration on STFx as a function of iron concentration x, we validate the applicability of the acid–base approach as well to perovskite oxides. A straightforward infiltration of CaO into STFx with the lowest iron content increased the surface oxygen exchange rate by approximately 40-fold and reduced electrode polarization by 35%. Despite the fact that the surface oxygen exchange rate of uninfiltrated specimens exhibits a divergence of over an order of magnitude, increasing with increases in x, it tends to converge following CaO or Al2O3 infiltration. This highlights the opportunity of utilizing infiltrated STFx with low iron content, offering significantly improved mechanical, thermal, and chemical stability.
Introduction
Perovskite-type oxides that exhibit both electronic and ionic conductivity are currently being extensively investigated as potential cathode materials for solid oxide fuel cells (SOFCs).1–4 The oxygen reduction reaction on the cathode often limits the overall performance of the fuel cell.5,6 The surface oxygen exchange coefficient (kchem) represents the rate at which oxygen exchanges between the solid surface and the surrounding gas atmosphere, and thus serves as a key descriptor in deciding the surface activity of a cathode material.7,8 As such, there is significant interest in developing methods for improving kchem.Recently, we proposed the acidity of infiltrated oxides to be a sensitive descriptor of oxygen exchange kinetics in mixed conducting (Pr,Ce)O2−δ (PCO).7 The relative acidity between two oxides here refers to a measure that indicates the tendency to donate or accept electrons, which is analogous to the relative work function of the materials in question. A relatively basic material has a smaller work function compared to a relatively acidic material, allowing the more basic material to donate electrons to the more acidic material. Basic oxides such as Li2O and CaO were demonstrated, for example, to increase kchem values by orders of magnitude in PCO, reportedly by increasing the surface electron concentration of the mixed conducting PCO, pointing to surface infiltration as a highly promising new strategy for enhancing the activity of SOFC cathodes more generally.7
To date, the infiltration of basic oxides, such as alkaline earth metal oxides, onto the surfaces of perovskite-structured oxides has been largely overlooked, except for very recent, but limited in situ studies within pulsed laser deposition(PLD) chambers.9 Indeed, a long-term controversy exists regarding the impact of excess surface SrO (or other alkaline earth metal oxides) on the surface activity of perovskite-oxide based SOFC cathodes. Most studies including Jung et al. find that excess surface SrO resulting from surface Sr segregation blocks reaction sites and hinders the surface oxygen exchange reaction.4,10–12. In considerably more limited studies, SrO and other alkaline earth metal oxides, intentionally added to the surfaces of perovskite mixed ionic and electronic conductors (MIECs) show improvements, but not at the scale achieved with the acid–base approach.13–15
Extrinsic acidic oxides such as Al2O3, Cr2O3, and SiO2, are additionally recognized for their detrimental impact on the surface activity of MIECs.16–18 The exposure of SOFC electrodes to these acidic oxides is often unavoidable, given the use of Al-based crucibles and Si-based glassware in materials synthesis, and the inevitable presence of Cr from the SOFC interconnects reaching the cathode during operation via vapor phase or surface diffusion. The ability to provide descriptors for the resistance of specific MIECs to poisoning by such acidic oxides is fundamental to the development of high-performance electrochemical devices.
The authors, in a recently published study, examined the surface exchange kinetics and electrode characteristics of a model perovskite oxide system SrTi1−x/100Fex/100O3−δ (STFx), with composition x = 35, designated as STF35.19 In this study, the impact of acidic Cr and basic Ca additives was examined with respect to three different metrics: (i) oxygen exchange coefficient (kchem), (ii) area-specific resistance (ASR), and (iii) peak power density, by utilizing porous STF35 bulk ceramics, symmetric electrochemical cells, and anode-supported single fuel cells, respectively. Acidic Cr-infiltration resulted in a pronounced depression in kchem, an increase in ASR, and a depression in peak power density by approximately 100-fold, 10-fold, and 40%, respectively. Of special note, full recovery of kchem, ASR, and peak power density were achieved by infiltrating basic Ca-species onto initially Cr-poisoned STF35 surfaces. These reversals in surface degradation were in that study largely attributed to the Ca-species induced accumulation of surface electrons, compensating for initial electron depletion induced by the acidic nature of the extrinsic Cr-species catalyst poison. Unexpectedly, the subsequent Ca infiltration simultaneously resulted in measurable decreases in intrinsic Sr segregation, as revealed by X-ray photoelectron spectroscopy(XPS), that was attributed to the reduction of Fe4+ to Fe3+ (driven by electron accumulation) that reduced the strain-induced segregation of the large Sr2+ ion to the surface.
In this study, we extend our investigation of the effects of controlled infiltration on the STFx system by examining how changes in Fe content (x = 35, 50, 80) impact the sensitivity to these treatments. The STFx system represents a group of promising oxygen electrode materials for SOFC as they are Co-free, have a simple composition, are stable in both oxidizing and reducing atmospheres, while possessing comparable activity compared to other popular electrode materials.20–23 Furthermore, this material system is, as we demonstrate, suitable for validating the acid–base approach, given that its surface acidity, which correlates with its work function, can be controlled by adjusting the Fe-to-Ti ratio.24,25 We previously reported that the reactivity of non-surface treated STFx is strongly correlated with the iron concentration.12 The more iron that substitutes for titanium, the smaller the energy distance between the conduction band edge and the Fermi energy (EC − EF), or correspondingly of interest to this study, the smaller the work function. By varying the Fe content of STFx, it thus become possible to investigate the expected impact of oxide infiltrants with well-defined Smith acidities on the host mixed conducting STFx with a given but controllable relative Smith acidity. In this study, basic CaO and acidic Al2O3 were chosen to ensure better experimental control, as they are generally more chemically stable compared to other basic oxides like BaO and Li2O, and acidic oxides like CrO3. The higher basicity of CaO relative to STFx, like for PCO, was indeed found to enhance its surface oxygen exchange kinetics, while acidic Al2O3 instead led to a deterioration in surface activity. In this study we find, however, that the more Fe rich STFx, with correspondingly lower work functions, is less affected by CaO infiltration, while becoming more strongly impacted by Al2O3 infiltration. Indeed, upon reaching a specific level of infiltration, the measured kchem values for various compositions converged to a similar level, despite their un-infiltrated values differing by more than an order of magnitude. These findings indicate that, although CaO and Al2O3 are insulating and, therefore, not inherently reactive, they induce alterations in the effective work function of the STF surface and thus the surface oxygen exchange rate of the perovskite oxides, as demonstrated in our previous study on PCO.7 Furthermore, due to the very straightforward and facile CaO infiltration strategy, we demonstrate that materials with inherently poorer surface exchange performance, but excellent chemical/mechanical stability, especially those with lower Fe content in STFx, have significant potential for achieving comparable levels of surface performance, while coupled with potentially significantly improved mechanical, thermal, and chemical stability.
Experimental
STFx powders (x = 35, 50, 80) were synthesized using the conventional solid-state process. Stoichiometric amounts of SrCO3, TiO2, and Fe2O3 powders were ball-milled in high-purity ethanol (Merck, 99.9%) for 48 hours. After drying, they were calcined in air at 1250 °C for 12 hours following heating and cooling rates of 4 °C min−1. After thorough grinding, they were mixed with a terpinol-based ink vehicle (FuelCellMaterials) and ethanol then ball-milled again for 24 hours. After sufficient time for drying to match the desired viscosity, the STF inks were screen-printed onto single-crystal Al2O3 substrates. To fabricate porous-structured STFx with similar surface area, the annealing time for each composition differed. STF35, STF50, and STF80 were annealed at 1300 °C, 1200 °C, and 1000 °C for 4 hours, respectively. Au paste was applied onto each side of the samples and annealed at 800 °C prior to measurements.Electrical conductivity relaxation (ECR) measurements were performed on specimens supported in a furnace tube with controlled oxygen partial pressure (pO2). A step change in the chamber pO2, from 0.21 to 0.1 atm, was achieved by mixing ultra-high purity nitrogen and oxygen (99.999%) in the appropriate ratios and switching rapidly between the two mixtures with the aid of a 4-way valve. The induced conductivity changes were recorded and fitted to extract the appropriate time constants. The ECR measurements were first performed on STFx specimens without infiltration, followed by measurements on specimens that were subsequently infiltrated with CaO (0.01 M solution of the nitrate) up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm. For the infiltration of Al2O3, the loading amount was limited to 10000 ppm to ensure that the blockage of reaction sites was negligible. Following infiltration, specimens were calcined in situ at 650 °C for 2 hours prior to the relaxation measurements to ensure the decomposition of the nitrate precursor. kchem was calculated using the following equation, as introduced in our previous work:18
000 ppm. For the infiltration of Al2O3, the loading amount was limited to 10000 ppm to ensure that the blockage of reaction sites was negligible. Following infiltration, specimens were calcined in situ at 650 °C for 2 hours prior to the relaxation measurements to ensure the decomposition of the nitrate precursor. kchem was calculated using the following equation, as introduced in our previous work:18
 | (1) |
 corresponds to the surface-area-to-volume ratio of the solid phase.
corresponds to the surface-area-to-volume ratio of the solid phase.
Scanning electron microscopy (SEM, S-4800, Hitachi) was employed to verify the porous structures of the STFx samples. The surface area-to-volume ratio was calculated using quantitative stereology.26 To further investigate the samples after CaO infiltration, scanning transmission electron microscopy (STEM, Talos F200X, Thermo Fisher) combined with energy-dispersive X-ray spectroscopy (EDS) was pursued. Additionally, high-resolution powder X-ray diffraction (XRD) measurements were performed using the SmartLab X-ray diffractometer from Rigaku, utilizing Cu Kα radiation (λ = 1.5406 Å). To confirm the presence of CaO on the surface of the STFx specimens, X-ray photoelectron spectroscopy (XPS) with monochromated Al Kα (hν = 1486.7 eV) radiation was employed, utilizing the Sigma Probe instrument from Thermo VG Scientific.
Results & discussion
As depicted in Fig. 1a, porous STFx specimens were fabricated on single-crystalline Al2O3 substrates. By adjusting the annealing temperature to 1300, 1200, and 1000 °C for STF35, STF50, and STF80, respectively, porous and interconnected specimens with similar area-to-volume ratios were successfully prepared (11181, 12835, and 14488 cm−1 for STF35, STF50, and STF80, respectively), with their surface morphologies illustrated in Fig. 1b. Here, confirming the highly porous structure, ensuring that the physical diffusion length is smaller than the critical thickness, is essential for accurately deriving the surface oxygen exchange coefficient from ECR tests. The critical thickness (LC), defined as LC ≡ D/k, was approximately 50 μm for STF35 and 65 μm for STF50 at 800 °C, and these values become significantly larger at lower temperatures—such as those used in this study—because the activation energy of kchem is greater than that of Dchem. Given that the fabricated highly porous STFx bulk ceramics exhibited grain sizes smaller than 2–3 μm—more than an order or magnitude smaller than the calculated critical thickness—the overall kinetics of the oxygen exchange reaction in this study are governed by the surface exchange reaction. The detailed preparation procedure is described in Fig. S1.† Similar porous morphologies are expected to result in similar distributions of infiltrate particles across the specimens.
were successfully prepared (11181, 12835, and 14488 cm−1 for STF35, STF50, and STF80, respectively), with their surface morphologies illustrated in Fig. 1b. Here, confirming the highly porous structure, ensuring that the physical diffusion length is smaller than the critical thickness, is essential for accurately deriving the surface oxygen exchange coefficient from ECR tests. The critical thickness (LC), defined as LC ≡ D/k, was approximately 50 μm for STF35 and 65 μm for STF50 at 800 °C, and these values become significantly larger at lower temperatures—such as those used in this study—because the activation energy of kchem is greater than that of Dchem. Given that the fabricated highly porous STFx bulk ceramics exhibited grain sizes smaller than 2–3 μm—more than an order or magnitude smaller than the calculated critical thickness—the overall kinetics of the oxygen exchange reaction in this study are governed by the surface exchange reaction. The detailed preparation procedure is described in Fig. S1.† Similar porous morphologies are expected to result in similar distributions of infiltrate particles across the specimens.
 | ||
| Fig. 1 Experimental set-up for measuring surface oxygen exchange coefficient, kchem of porous specimens. (a) Schematic of DC conductivity relaxation measurement set-up and (b) surface morphology of SrTi1−x/100Fex/100O3−δ samples (x = 35, 50, 80). | ||
Infiltration of CaO and Al2O3 was carried out on STFx samples, with the analysis focusing on STF50 samples using STEM-EDS, XRD, and XPS for confirmation of successful infiltration. As illustrated in Fig. 2a and 2b, CaO and Al2O3 were observed to deposit as particles, rather than covering the entire surface that could potentially limit the surface exchange reaction. Despite annealing at 700 °C for 10 hours, conditions exceeding those of ECR measurements, the XRD patterns of the specimens exhibited no visible shift, indicating that Ca and Al species did not incorporate into the lattice, within the limits of sensitivity, but exist as second-phases. The XRD peaks of CaO and Al2O3 were not prominently displayed due to their low content of 10000 ppm (see Fig. 2c). Note that the diffraction peaks observed at 2 theta values of 28.4°, 47.3°, and 56.1° correspond to the (111), (220), and (311) planes of silicon, respectively.27 These peaks originate from the silicon powder employed as a calibration standard in the X-ray diffraction analysis. Judging from the Ca 2p core-level spectrum of CaO-infiltrated STF50 (Fig. 2d), STF50 infiltrated with CaO exhibited a single Ca 2p doublet with a Ca 2p3/2 peak at approximately 346 eV. Considering that Ca incorporated into the perovskite lattice would exhibit a higher binding energy as seen in CaTiO3 of Fig. S2a,† the presence of only one doublet at Ca 2p3/2 peak around 346 eV indicates the exclusive existence of CaO on STF50. Similarly, the Al 2p spectrum of Al2O3-infiltrated STF50 in Fig. 2e also shows a single peak at approximately 74.5 eV, corresponding to the signal from Al2O3 shown in Fig. S2b,† which is distinct from the Al signal associated with SrAl2O4 species (73.6 eV).
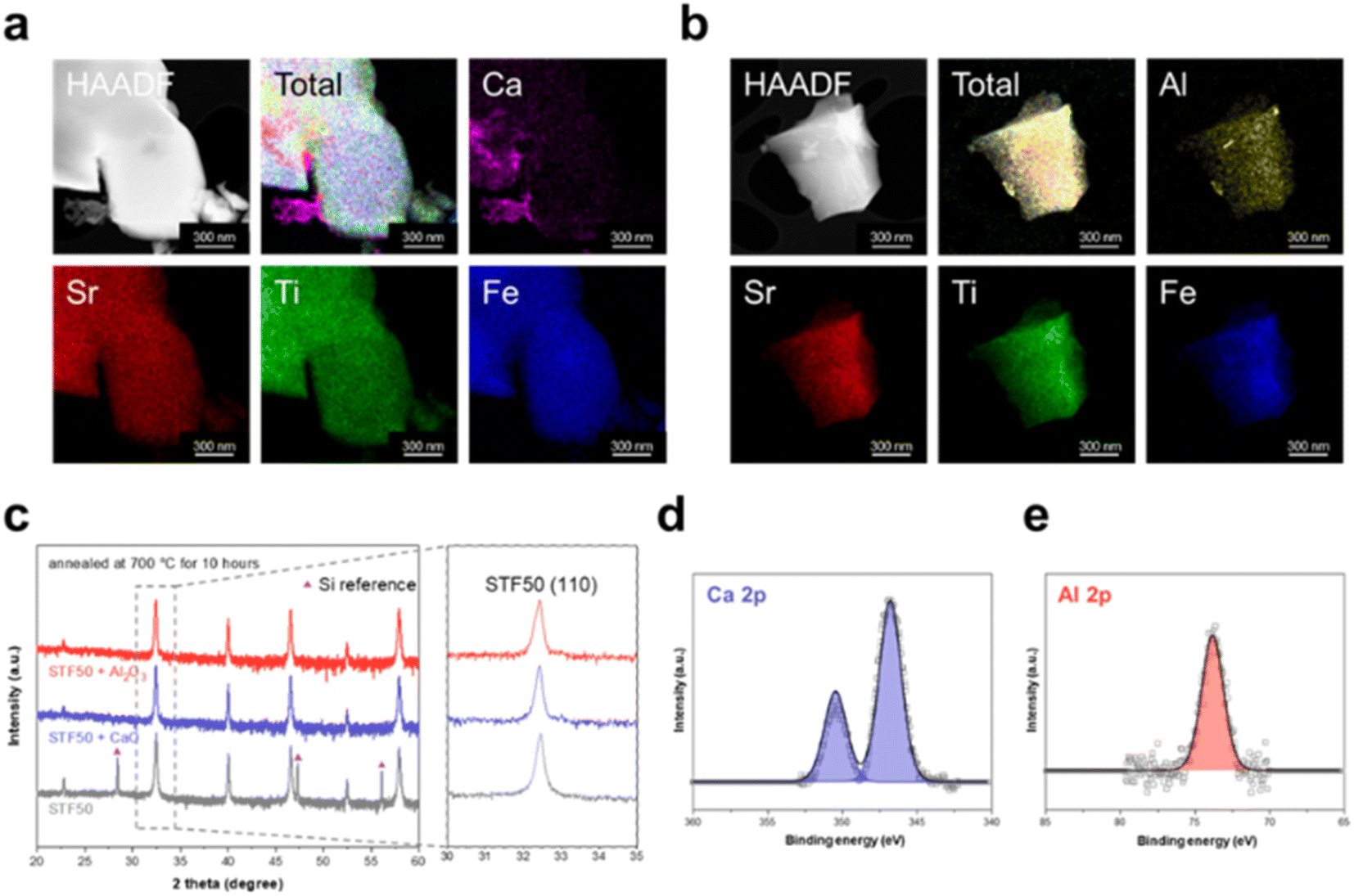 | ||
| Fig. 2 Verification of CaO and Al2O3 infiltration of SrTi0.5Fe0.5O3−δ (STF50). Scanning transmission electron microscopy (STEM) and energy-dispersive X-ray spectroscopy (EDS) imaging of the STF50 specimen infiltrated with (a) CaO and (b) Al2O3 following annealing at 650 °C. (c) X-ray diffraction patterns of pristine, CaO-, and Al2O3-infiltrated samples, along with the magnified (110) peak. X-ray photoelectron spectroscopy (XPS) analysis on (d) Ca 2p for CaO-infiltrated and (e) Al 2p for Al2O3-infiltrated specimens. | ||
Surface oxygen exchange coefficients, kchem, for STFx specimens were determined through ECR measurements both before and after infiltration with CaO and Al2O3 at various temperatures, as illustrated in Fig. 3. The kchem of pristine STFx increased with increasing iron content, resulting in values of approximately 1.83 × 10−6, 6.63 × 10−6, and 32.23 × 10−6 cm s−1 at 600 °C for STF35, STF50, and STF80, respectively. The activation energy, Ea, calculated using eqn (2) was found to be 1.48, 1.44, and 1.37 eV for STF35, STF50, and STF80, respectively, where A is the pre-exponential, k is the Boltzmann constant and T is the temperature.
| kchem = A·exp(−Ea/kT) | (2) |
 | ||
| Fig. 3 Surface oxygen exchange coefficient, kchem, measured with respect to temperature before and after infiltration with (a) CaO and (b) Al2O3. Colored arrows indicate range of enhancement with CaO activation or depression with Al2O3 deactivation in kchem for SrTi0.65Fe0.35O3−δ (STF35) (orange), SrTi0.5Fe0.5O3−δ (STF50) (green), and SrTi0.2Fe0.8O3−δ (STF80) (blue), respectively, at selected temperatures. Error bars account for the standard deviation from average kchem values obtained from two samples. | ||
This trend, of increasing kchem and decreasing Ea with increasing Fe content, aligns well with findings in earlier reports, albeit in terms of electrochemical surface oxygen exchange coefficients, kq, rather than kchem.12 Detailed comparisons of the surface oxygen exchange coefficients and activation energies are provided in Fig. S3.†
Following infiltration of CaO, kchem for STFx specimens exhibited a significant increase in magnitude as illustrated in Fig. 3a. As observed with basic oxide infiltration in previous studies, kchem increased with the amount of infiltrate until it reached saturation. (Fig. S4†) Notably, Ea did not exhibit noticeable changes, consistent with existing literature indicating that only the pre-exponential factor (A) increases upon infiltration with basic oxides.7,17,28 Of note, STF35, of the three compositions studied, shows the most substantial enhancement in kchem with the degree of improvement diminishing as iron content increases.
In contrast, Fig. 3b reveals that the infiltration of Al2O3 led to a severe reduction in the surface activity of STFx, with the least deterioration observed for STF35 and an increase in degradation degree with increasing iron content. Like that found for CaO infiltration, Ea remained relatively unchanged upon Al2O3 infiltration. Although the spread in kchem values for the un-infiltrated STFx materials varied by more than an order of magnitude with Fe content at a given temperature, the values of the infiltrated specimens tended to converge, overcoming the effect of Fe content. We return later to this feature and point out how this may be beneficial in terms of improving the long-term stability of STF-based SOFC cathodes.
The Smith acidity α of the three respective STFx specimens was determined using eqn (3), referenced from our previous work.29
 | (3) |
Due to the higher basicity of iron oxide compared to titanium oxide, STFx exhibits increasing basicity with increasing iron concentration (see Table 1). The acidity of STFx falls between that of more basic CaO (αCaO = −7.5) and more acidic Al2O3 (αAl2O3 = – 2.0).30 As depicted in Fig. 4a, illustrating the relative Smith acidity of STFx specimens and infiltrates, the infiltration of basic CaO would be expected to have the most significant impact on the surface of STF35, which is the most acidic among the STFx specimens. Conversely, the detimental effect of acidic Al2O3 infiltration is expected to be minimal for STF35, that has the highest acidity among the STFx specimens.
| STF35 | STF50 | STF80 | |
|---|---|---|---|
| α | −4.6 | −4.7 | −5.0 |
 | ||
| Fig. 4 Relative Smith acidity of SrTi1−x/100Fex/100O3−δ (STFx, x = 35, 50, 80) specimens and their infiltrates, along with trends in degree of activation. (a) Qualitative relative Smith acidity of the STFx specimens, along with that of CaO, and Al2O3. (b) Activation factor in case of CaO infiltration (c) Deactivation factor in case of Al2O3 infiltration. | ||
We define the degree of impact that the infiltrants have on kchem by considering the ratio of kchem (following to infiltration) vs. kchem (prior to infiltration) as in eqn (4) below, that we call the activation or deactivation factor depending on whether kchem is enhanced or depressed due to the infiltration process.
| (de)activation factor ≡ kchem(after infiltration)/kchem(prior to infiltration) | (4) |
In Fig. 4b and c, the calculated activation and deactivation factors for the CaO and Al2O3 infiltration cases are presented respectively for measurements performed at 575 °C. The surface activity enhancement by CaO is most pronounced for the more highly acidic STF35, which exhibits the greatest acidity difference with CaO. Conversely, the deactivation effect by Al2O3 infiltration is most noticeable for the most basic STF80, demonstrating the greatest acidity difference with Al2O3.
The correlation observed in the activation or deactivation factor influenced by infiltrates, consistent with the relative acidity of the specimens, provides compelling evidence supporting the hypothesis that the acidity of infiltrates acts as a critical descriptor for the surface oxygen exchange rate in mixed-conducting perovskite oxides. It is suggested that the upper and lower limits of kchem depend on the simultaneous optimization of the concentration of electrons, oxygen vacancies, and adsorbed oxygen atoms on the surface. By introducing additional basic materials onto the surface whose activity is constrained either by the availability of electrons,7 or adsorbed oxygen,9 kchem of perovskite oxides can be significantly enhanced. These observations in the ternary STFx perovskite system extends the material scope beyond PCO, a binary fluorite oxide, as outlined in our earlier studies.7
The enhanced surface oxygen exchange rate likewise led to increased electrode activity, as measured by electrochemical impedance spectroscopy on symmetrical cells (STFx|(Sm,Ce)O2−δ|STFx), as shown in Fig. S5.† While the electrochemical performance of CaO-infiltrated STF35 does not exactly match that of Fe-rich samples, it should be remembered that the electrode resistance plotted in Fig. S5† is also influenced by the morphology (surface area) and current collection characteristics of the specific electrode under study. By appropriately engineering the electrode structure and improving current collection to resemble that of the STF80 example, the electrochemical performance (electrode activity) of STF35 would be expected to be nearly equivalent to that of the STF80 example. Nevertheless, the trend in the activation factor of electrode activity aligns well with the trend observed in kchem.
With the established link between surface oxide acidity and the oxygen exchange kinetics of perovskite oxides, there is potential for gaining deeper insights into poisoning effects in solid oxide fuel cells. This understanding can contribute to the formulation of strategies for activity enhancement through binary oxide infiltration. The acid–base approach further underscores the significant influence of surface properties on the oxygen exchange kinetics of STFx.
Furthermore, given that the oxygen exchange rates of all the CaO-activated STFx specimens, irrespective of Fe content, fall within a similar range, suggests the potential for greater flexibility in identifying new MIEC electrocatalysts for use in SOFC systems. While the higher Fe content STF80 without CaO activation has over an order of magnitude higher oxygen exchange rate than STF35, increased Fe content comes with two detrimental features. First, with the higher concentration of redox variable Fe content comes an expected higher chemical expansion coefficient.31 Thus, by enhancing kchem of STF35 by CaO infiltration to values comparable to that of STF80, lower Fe content electrodes with reduced chemical expansion coefficients could be applied without loss of performance. Furthermore, we previously reported that with the higher Fe/Ti ratio in STF comes a higher degree of Sr segregation, given the lower ionic radius of Fe versus that of Ti.10 Since increasing levels of Sr segregation to the surface of Sr containing MIEC perovskite cathodes has been associated with enhanced area specific resistances (ASR), this would point to another benefit of being able to operate STF with lower Fe contents.8,10,32 Furthermore, after annealing the CaO- and Al2O3-infiltrated specimens at 700 °C for 10 hours, the CaO-infiltrated specimen exhibited a reduced amount of non-lattice Sr species, interpreted as a result of surface Sr segregation, whereas the Al2O3-infiltraed specimen showed an increased degree of surface Sr segregation (Fig. S6†). The acid–base approach not only enables STF compositions with lower iron content, which are intrinsically more resistant to Sr segregation, to achieve activity levels comparable to those of compositions with higher iron content, but also it significantly suppresses the rate of surface Sr segregation, as demonstrated above. Lastly, we note that STF, in general, does not rely on the use of Co, well known to be a critical element given its high demand in lithium or metal–air battery cathodes33–37 and its limited accessibility geographically.38,39 As this study demonstrates, MIECs such as STF composed of readily available and non-critical elements, but initially exhibiting non-optimum oxygen exchange kinetics can be activated through the infiltration of affordable and abundant basic oxides, such as CaO, thereby pointing the way to replace commonly utilized SOFC cathodes such as (La,Sr)(Co,Fe)O3−δ (LSCF) with Co-free materials.
Conclusions
The acid–base approach was expanded to the model mixed conducting perovskite oxide SrTi1−x/100Fex/100O3−δ (STFx) electrocatalysts by examining how the surface infiltration of basic CaO or acidic Al2O3 and Fe content influence the surface oxygen exchange kinetics. Through a systematic evaluation of the degree of (de)activation induced by binary oxide infiltration on STFx as a function of iron concentration x, we were also able to demonstrate the importance of the acidity of the host electrode material relative to that of the infiltrants in impacting their ability to influence the exchange kinetics. Interestingly, despite significant divergence in the surface oxygen exchange rate of uninfiltrated specimens with variations in x, kchem tended to converge to a specific value following activation with CaO or deactivation with Al2O3 infiltration. This result not only demonstrates applicability to a variety of multi-component oxide systems where reactivity is determined by acid/base infiltration, but simultaneously highlights the benefits of using infiltrated STFx with low iron, and more generally, variable valent cation content, with improved mechanical, thermal, and chemical stability.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. L. T. and W. J. conceived the idea and guided the whole project. They, along with H. K., H. G. S. and S. A., conceptualized the idea, designed the experiments, analyzed the data, created the figures, and prepared the manuscript. The devices were fabricated, electric measurements were conducted by H. K. and H. G. S. All authors have read the paper, concur with its contents, and approve the submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2022-NR066812, No. RS-2023-00236572). H. G. Seo and H. L. Tuller received partial support from the R.P. Simmons Chair of Ceramics and Electronic Materials, Department of Materials Science and Engineering, Massachusetts Institute of Technology. It was also supported by Research Institute of Advanced Materials (RIAM). H. Kim also thanks the BK21-Four program through National Research Foundation of Korea (NRF) for providing support to him as an MIT visiting PhD student.Notes and references
- Y. Zhang, B. Chen, D. Guan, M. Xu, R. Ran, M. Ni, W. Zhou, R. O'Hayre and Z. Shao, Nature, 2021, 591, 246–251 CrossRef CAS PubMed.
- P. Boldrin and N. P. Brandon, Nat. Catal., 2019, 2, 571–577 CrossRef CAS.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 15014 CrossRef CAS.
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Nat. Mater., 2016, 15, 1010–1016 CrossRef CAS PubMed.
- J. Liu, J. K. Kim, Y. Wang, H. Kim, A. Belotti, B. Koo, Z. Wang, W. C. Jung and F. Ciucci, Energy Environ. Sci., 2022, 15, 4069–4082 RSC.
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H. Il Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS.
- C. Nicollet, C. Toparli, G. F. Harrington, T. Defferriere, B. Yildiz and H. L. Tuller, Nat. Catal., 2020, 3, 913–920 CrossRef CAS.
- B. Koo, H. Kwon, Y. Kim, H. G. Seo, J. W. Han and W. Jung, Energy Environ. Sci., 2018, 11, 71–77 RSC.
- A. Merieau, M. Siebenhofer, C. Böhme, M. Kubicek, O. Joubert, J. Fleig and C. Nicollet, J. Mater. Chem. A, 2024, 12, 13960–13969 RSC.
- W. Jung and H. L. Tuller, Energy Environ. Sci., 2012, 5, 5370–5378 RSC.
- H. Wang and S. A. Barnett, J. Electrochem. Soc., 2018, 165, F564–F570 CrossRef CAS.
- W. C. Jung and H. L. Tuller, Adv. Energy Mater., 2011, 1, 1184–1191 CrossRef CAS.
- C. Argirusis, S. Wagner, W. Menesklou, C. Warnke, T. Damjanovic, G. Borchardt and E. Ivers-Tiffée, Phys. Chem. Chem. Phys., 2005, 7, 3523–3525 RSC.
- S. F. Wagner, C. Warnke, W. Menesklou, C. Argirusis, T. Damjanović, G. Borchardt and E. Ivers-Tiffée, Solid State Ionics, 2006, 177, 1607–1612 CrossRef CAS.
- Y. Zhu, D. Liu, H. Jing, F. Zhang, X. Zhang, S. Hu, L. Zhang, J. Wang, L. Zhang, W. Zhang, B. Pang, P. Zhang, F. Fan, J. Xiao, W. Liu, X. Zhu and W. Yang, Sci. Adv., 2022, 8, eabn4072 CrossRef CAS PubMed.
- A. Staerz, H. G. Seo, T. Defferriere and H. L. Tuller, J. Mater. Chem. A, 2022, 10, 2618–2636 RSC.
- H. G. Seo, A. Staerz, G. Dimitrakopoulos, D. Kim, B. Yildiz and H. L. Tuller, J. Power Sources, 2023, 558, 232589 CrossRef CAS.
- H. G. Seo, A. Staerz, D. S. Kim, D. Klotz, C. Nicollet, M. Xu, J. M. LeBeau and H. L. Tuller, Energy Environ. Sci., 2022, 15, 4038–4047 RSC.
- H. G. Seo, H. Kim, W. Jung and H. L. Tuller, Appl. Catal., B, 2024, 355, 124172 CrossRef CAS.
- S. L. Zhang, H. Wang, M. Y. Lu, A. P. Zhang, L. V. Mogni, Q. Liu, C. X. Li, C. J. Li and S. A. Barnett, Energy Environ. Sci., 2018, 11, 1870–1879 RSC.
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Joule, 2018, 2, 478–496 CrossRef CAS.
- S. Molin, W. Lewandowska-Iwaniak, B. Kusz, M. Gazda and P. Jasinski, J. Electroceram., 2012, 28, 80–87 CrossRef CAS.
- A. Mroziński, S. Molin and P. Jasiński, J. Solid State Electrochem., 2020, 24, 873–882 CrossRef.
- M. Ghaffari, M. Shannon, H. Hui, O. K. Tan and A. Irannejad, Surf. Sci., 2012, 606, 670–677 CrossRef CAS.
- A. Rothschild, W. Menesklou, H. L. Tuller and E. Ivers-Tiffée, Chem. Mater., 2006, 18, 3651–3659 CrossRef CAS.
- E. E. Underwood, in Microstructural Analysis, ed. J. L. McCall and W. M. Mueller, Springer US, Boston, MA, 1973, pp. 35–66 Search PubMed.
- C. R. Hubbard, H. E. Swanson and F. A. Mauer, J. Appl. Crystallogr., 1975, 8, 45–48 CrossRef.
- H. G. Seo, A. Staerz, D. S. Kim, J. M. LeBeau and H. L. Tuller, Adv. Mater., 2023, 35, e2208182 CrossRef PubMed.
- C. Nicollet and H. L. Tuller, Chem. Mater., 2022, 34, 991–997 CrossRef CAS.
- D. W. Smith, J. Chem. Educ., 1987, 64, 480–481 CrossRef CAS.
- N. H. Perry, J. J. Kim, S. R. Bishop and H. L. Tuller, J. Mater. Chem. A, 2015, 3, 3602–3611 RSC.
- B. Koo, J. Seo, J. K. Kim and W. Jung, J. Mater. Chem. A, 2020, 8, 13763–13769 RSC.
- S. Lee and A. Manthiram, ACS Energy Lett., 2022, 7, 3058–3063 CrossRef CAS.
- H. H. Ryu, H. H. Sun, S. T. Myung, C. S. Yoon and Y. K. Sun, Energy Environ. Sci., 2021, 14, 844–852 RSC.
- M. Li and J. Lu, Science, 2020, 367, 979–980 CrossRef CAS PubMed.
- J. Ko and Y. S. Yoon, J. Korean Ceram. Soc., 2023, 60, 591–613 CrossRef CAS.
- J. Kim, J. Lee, S. Kim and W. Jung, Electron. Mater. Lett., 2024, 20, 450–458 CrossRef CAS.
- S. van den Brink, R. Kleijn, B. Sprecher and A. Tukker, Resour. Conserv. Recycl., 2020, 156, 104743 CrossRef.
- E. Savinova, C. Evans, É. Lèbre, M. Stringer, M. Azadi and R. K. Valenta, Resour. Conserv. Recycl., 2023, 190, 106855 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08529a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |