
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe intriguing case of cyclic triimidazole: an emerging scaffold for the preparation of multiemissive, bio-medical and hybrid inorganic–organic materials
Alessandra
Forni
 a,
Daniele
Malpicci
ab,
Daniele
Maver
ab,
Elena
Lucenti
a and
Elena
Cariati
*ab
a,
Daniele
Malpicci
ab,
Daniele
Maver
ab,
Elena
Lucenti
a and
Elena
Cariati
*ab
aInstitute of Chemical Sciences and Technologies “Giulio Natta” (SCITEC) of CNR, via Golgi 19, 20133 Milano, Italy
bDepartment of Chemistry, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy. E-mail: elena.cariati@unimi.it
First published on 20th January 2025
Abstract
Among organic small molecules characterized by multifaceted behaviour, triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine, TT, having a triazinic central ring with three annelated imidazoles, represents a fascinating but still underinvestigated scaffold endowed with intriguing luminescence and coordination properties. Here we comprehensively gather studies, mainly conducted by our research group, on several fully organic or hybrid inorganic–organic TTs revealing their AIE, RTP, mechanochromic and excitation dependent photoluminescence behaviours. Preliminary applications of TTs in sensing and bio-medicine are also reported, opening avenues for further studies in these fields and widening the potentiality of TT and its derivatives. On the whole, the results shown here clearly demonstrate that cyclic triimidazole can be rightfully included in the toolbox of powerful scaffolds inspiring the preparation of multifunctional molecular materials.
1. Introduction
The engineering of organic small molecules characterized by multifaceted properties is certainly a longstanding research goal. In this regard, nitrogen-containing heterocyclic compounds represent an appealing class for different reasons: (i) they are important building blocks to generate bio-probes with significant therapeutic potential;1–3 (ii) they can be used to obtain organic and hybrid inorganic–organic polymeric materials;4–6 and (iii) they present structural features exploitable for the preparation of supramolecular assemblies through intermolecular interactions. In particular, several luminescent systems have been obtained by π–π stacking interactions and hydrogen or halogen bonds.7–11Among different nitrogen-heterocycles, imidazoles and triazines represent versatile scaffolds, which have been used to develop several bioactive compounds with anticancer, antibacterial, antifungal, antitubercular, analgesic, and anti-HIV activities.12–17 Moreover, imidazole and its derivatives have been frequently employed in the synthesis of metal compounds, e.g. numerous luminescent M(I, II) d10 complexes and coordination polymers (CPs), due to the electron-rich nature of the five-membered imidazole ring.18–21 Besides, the triazine moiety with its three N atoms has been exploited to build molecules able to establish multiple supramolecular interactions resulting in rigidified structures with enhanced emissive features.22–25 Importantly, triazine cores have also gained significant attention in the past few decades as building blocks for the preparation of covalent triazine frameworks (CTFs) as promising porous organic materials.26–28
In this regard, triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine or cyclic triimidazole (hereafter TT) represents an intriguing molecule formed by a triazinic central ring with three annelated imidazoles (see Scheme 1a). It possesses C3h molecular symmetry and three nitrogen atoms available for coordination. While TT is known since 1973,29 its properties and reactivity remained mainly unexplored until the recent publication of a convenient synthetic procedure.30 This is the reason why studies on TT are still limited despite its promising potentialities as highlighted in the past few years mainly through investigations performed by our research group. Specifically, TT and a large family of its derivatives (TTs) have been prepared and characterized for their emissive and coordination features. The multicomponent fluorescence and phosphorescence behaviours of most TTs have been discovered but only preliminarily exploited for sensing and bioimaging.
 | ||
| Scheme 1 Synthesis of TT derivatives. | ||
In this perspective, after a brief overview of fused π-extended nitrogen rich cores exploited for the preparation of multifunctional materials, we focus on the TT family detailing the synthetic procedures and the outstanding emissive features of the core itself and its many fully organic or hybrid inorganic–organic derivatives. Specifically, a deep photophysical investigation of TT functionalized with halogens, ethynyl, carboxyl and chromophoric (including pyrene, pyridines, carbazole and thiophene) groups has been conducted in previous works and summarized and rationalized here together with results obtained for selected hybrid metal compounds with TTs as ligands. Finally, an additional paragraph related to the use of TTs as sensors and biomolecules is included.
It is our belief that the present perspective could be a spring of inspiration for other researchers so that the potentiality of the TT scaffold could be more deeply exploited.
2. Overview of selected π-extended fused aza-heterocycles
In the past few decades, polycyclic aromatic hydrocarbons based on fused nitrogen-rich heterocycles have attracted increasing attention from the scientific community for their possible applications in various fields of supramolecular chemistry and materials science. The introduction of the nitrogen heteroatom in combination with the annealing of the aromatic rings has proven to be an efficacious strategy to prepare stable compounds. Fused aza-heterocycles are, in fact, characterized by a lower susceptibility to degradation through oxidation or dimerization in comparison with their nitrogen-free counterparts.31 Consequently, the rigid, planar and electron deficient aromatic scaffolds of heptazine (Hz), quinazoline (Qn), hexaazatriphenylene (HAT) and benzotristriazine (BTT) have been exploited as cores for the preparation of promising functional materials, including covalent organic and metal–organic frameworks, to be used in different areas spanning from liquid crystals, n-type semiconductors, sensors, non-linear optical chromophores, gas absorption, energy storage, and catalysts to photo- and electro-luminescence (Fig. 1).32–38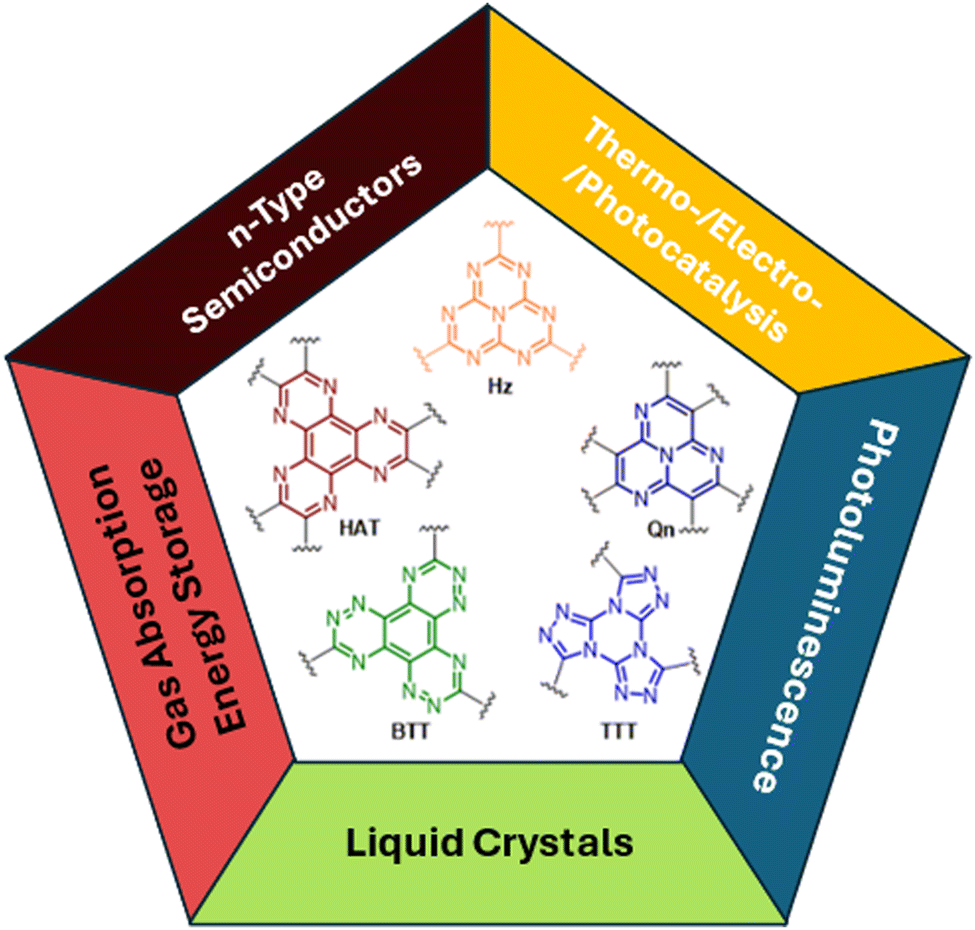 | ||
| Fig. 1 Examples of π-extended fused aza-heterocycles and their main fields of applications. | ||
In particular, among planar and rigid nitrogen-rich aromatic scaffolds, tris[1,2,4]triazolo[1,3,5]triazine (TTT) has recently emerged as a useful tecton for the preparation of liquid crystals and TADF (thermally activated delayed fluorescence) emitters. TTT, characterized by a central triazinic core with three annulated triazoles, with its nine sp2 hybridized nitrogen atoms and C3 symmetry, closely resembles cyclic triimidazole and therefore, in the present work, deserves closer inspection of the results so far obtained with its derivatives.
The first TTT described in the literature was pyroguanazole, synthesized in 1912 by thermal treatment of 3,5-diamino-1,2,4-triazole or guanazole.39 However, since that early report, it remained mainly unexplored until 1961 when a seminal work by Huisgen and coworkers reported the synthesis of triphenyl-substituted TTT by reaction of cyanuric chloride with 1-phenyltetrazole.40 This synthetic protocol was thoroughly explored later on, starting from the 2000s, when the ability of TTTs to self-assemble into columnar superstructures with mono-dimensional conducting properties was foreseen as a great opportunity to be exploited in molecular electronics and in the realization of discotic liquid crystals.41–46 Interestingly, the strong coupling between lateral donor substituents and the central acceptor aromatic core together with the octupolar geometry endowed TTTs with linear and non-linear optical features (i.e. fluorescence and two-photon absorption).47,48
Moreover, the thermal stability and highly ordered structures of discotic materials led to development of compounds characterized by high charge mobility together with light emission capability exploitable in electroluminescent devices.
Through appropriate functionalization of the central electron-acceptor core with donor groups (e.g., phenoxazines, acridines, biacridines or carbazoles) the twisted D–A geometry of TTTs results in blue and green TADF emitters endowed, in some cases, with aggregation-induced enhanced emission (AIEE).49–54 Furthermore, appropriately designed TTT-based dendrimers have been proposed as hot exciton materials to be used for the preparation of efficient and stable OLEDs.55,56
More recently, it has been found that the introduction of thiophene bridges in between bulky donor groups (di-3,6-tert-butyl-carbazole or 9,9-dimethyl-9,10-dihydroacridine) and the TTT acceptor core provides solutions with an impressive emissive behavior comprising delayed fluorescence (associated with triplet–triplet annihilation, TTA) and RTP. The latter has been justified by the positive effect of the bulky donor groups on inhibiting non-radiative decays from the triplet state, as confirmed by the similar lifetimes recorded in solution and in the solid state in the Zeonex matrix.57
Other interesting photoluminescence features have been observed for TTTs with a phenothiazine heterocycle as a donor functionality. Modulation of the donor strength and steric constraints through the incorporation of methyl groups at different positions of the emitter provided multifunctional materials exhibiting AIEE, dual-TADF, aggregation-induced delayed fluorescence (AIDF), and RTP characteristics. In addition, by appropriately choosing the polarity of the host matrix and by controlling the singlet–triplet energy gap, the possibility to switch between RTP and TADF processes has been demonstrated.58
3. The TT family
3.1 Synthetic procedures for TT and TTs
The first report on TT dates back to 1973 when the self-condensation at 373 K of 2-fluoro-imidazole into its cyclic trimer was reported.29 Trimerization reactions were also successfully performed by using as starting materials 2-fluoro- and 2-chloro-imidazoles variously substituted in 4,5-positions or 1-iodo-2-X-4,5-dicyanoimidazoles (X = Cl, Br, I) (Scheme 1a and b).59–62 In a similar way, thermolysis of 2-aryloxybenzimidazoles was reported for the preparation of tris(benzimidazo)[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine (TT-Benzo) (Scheme 1c).63 An alternative and more effective synthetic route for the preparation on a multigram scale of TT and TT-Benzo was further proposed by exploiting solvent-free thermolysis of copper(II) imidazolates. In particular, treatment in vacuum of the blue polymorph of copper(II) diimidazolate [Cu(C3H3N2)2]n at temperatures above 513 K afforded TT together with, as the by-product in a 1 to 5 ratio, isomer Iso-A showing an imidazole ring with 1,5- instead of 1,2-annelation (see Scheme 1d).30,64 Similarly, treatment at 513 K of bis-benzimidazolium hexachlorodicuprate(II) (HBz)2[Cu2Cl6] produced TT-Benzo in fairly good yields (see Scheme 1e).65More recently, a multicomponent approach based on selective multiple Groebke–Blackburn–Bienaymé (GBB) reactions performed on the melamine substrate yielded novel hexasubstituted cyclic triimidazoles.66,67 By appropriately choosing the isocyanide and aldehyde reactants, five different TT-based derivatives were easily isolated in fairly good yields (Scheme 1f). In the case of the halo-aryl substituted products, post-transformation into more complex tripodal scaffolds was further achieved by intramolecular Ullmann-type amination or multiple Suzuki–Miyaura cross-coupling reactions, the latter providing star-shaped compounds with nanometric dimensions.66,67
 | ||
| Scheme 2 Synthesis and chemical structures of 1–3X (X = Cl, Br, I). | ||
Interestingly, the reactive positions in halogenation, as well as in all functionalization reactions of TT further described, are the chemically equivalent 3, 7 and 11 ones. Since halogenation through NXS is not specific and provides mixtures of products, a careful control of reaction conditions (NXS equivalents, solvent and addition of catalytic acid) and purification procedures were necessary to obtain 1-3X in good yields.
1–3Cl were synthetized by mild chlorination of the TT scaffold with N-chlorosuccinimide (NCS).68 Electrophilic chlorination is not a selective reaction and provides a mixture of mono-, di- and trisubstituted compounds in relative quantities related to the amount of NCS employed. To assess the best synthetic conditions, catalysts, solvents and NCS equivalents were screened. The isolation of 1–3Cl in good yields (47%, 61% and 16%, respectively) was obtained by treating TT with NCS in acetonitrile or dioxane at room temperature with the addition of a protic acid such as trifluoroacetic or p-toluenesulfonic acid. The three compounds were isolated using chromatographic techniques and characterized by NMR and MS.
1–3Br were prepared by mild bromination using one, two or three equivalents of N-bromosuccinimide (NBS), respectively.69,70 The synthesis of 3Br required an acetonitrile (ACN)/dichloromethane (DCM) mixture and the addition of trifluoroacetic acid as a catalyst. Products were purified by chromatography affording 1Br and 3Br in good yields (85 and 90%) while 2Br was obtained in 40% yield together with 1Br and 3Br as by-products. NMR structural assignment of 1Br and 2Br, despite an extensive investigation comprising 1D (1H and 13C) and 2D (COSY, 1H–13C HSQC, 1H–13C HMBC, and 1H–15N-HMBC) experiments, was difficult due to the lack of crucial and diagnostic long-range correlations between quaternary carbon and proton signals. Implementation of the experimental results by DFT calculations highlighted 4JH–C correlations about twice the 3JC–H ones, allowing the proper chemical shift assignment for both compounds.72
Finally, mild electrophilic reaction of TT with N-iodosuccinimide (NIS) and catalytic amounts of trifluoroacetic acid in ACN provided 1I and 2I, both compounds being isolated and purified by chromatography and crystallization techniques.71
1–3Br were extensively used in Suzuki–Miyaura, Stille, Sonogashira and Ullmann cross-couplings mediated by Pd(0) or Cu(I) (see Scheme 3) with yields comparable to or slightly lower than those obtained from other brominated aryls (e.g. phenylbromide) using the same catalysts.73–751–3Br react at high temperature, usually above 90 °C, otherwise leading to dehalogenation. The yields decrease, as expected, in polysubstitution reactions (values are in line with those reported for phenylbromide taken as a reference).76,77
 | ||
| Scheme 3 Overview of C–C coupling products. | ||
Suzuki–Miyaura cross-coupling: 1–3Br were reacted with commercially available boronic acids/esters such as pyrid-4-yl-,78 pyren-1-yl-boronic acid79,80 or 9H-carbazolyl-phenyl-,81 2-fluoropyrid-4-yl-,82 2,2′-bithiofen-5-yl-,83 and 9-ethyl-9H-carbazole-3-boronic acid pinacol ester84 in the presence of a Pd(0) precursor and a base, resulting in the isolation of different TTs (Scheme 4).
 | ||
| Scheme 4 Synthesis of TT derivatives by Suzuki–Miyaura cross-coupling. | ||
Stille cross-coupling reaction: in the same way, brominated TTs can react in Stille cross-coupling reaction. In this regard, 1–3Br were reacted with pyrid-2-yl-,78,85 thiophen-2-yl-,83 and 4-hexylthiophen-2-yl86 tributylstannyls in dry refluxing toluene for 16 h under an inert atmosphere (Scheme 5).
 | ||
| Scheme 5 Synthesis of TT derivatives by Stille cross-coupling. | ||
Ullmann-type cross-coupling reaction: moreover, 1Br was reacted according to Ullmann-type cross-coupling reaction mediated by CuI with carbazole, affording TT-(N)-Cz in good yield (Scheme 6).84
 | ||
| Scheme 6 Synthesis of TT derivatives by Ullmann-type cross-coupling. | ||
Sonogashira cross-coupling reaction: halogenated TTs were also tested in Sonogashira type cross-coupling reactions. Recently, functionalization of TT with an ethynyl group was performed by following two different synthetic procedures using either 1Br or 1I as the starting material, both methods converging to TT-CCH (Scheme 7).87
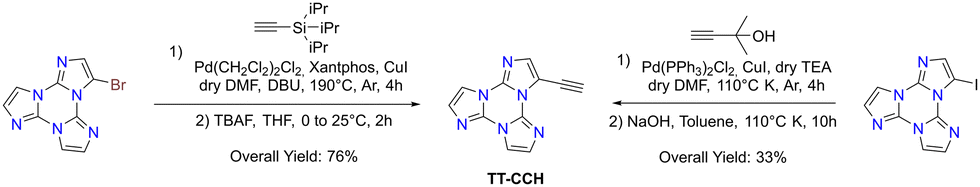 | ||
| Scheme 7 Synthesis of TT derivatives by Sonogashira cross-coupling. | ||
Formylation reaction: formyl-TTs (TT-(CHO)1–3) were prepared by direct formylation of the scaffold via a lithium salt intermediate followed by quenching with DMF.78 The same synthetic route was exploited in the formylation of TTPyr, resulting in the formation of the (CHO)2TT-Pyr derivative, in which the two carbonyl groups are directly connected to the central triazinic core (Scheme 8).88 Moreover, the TT-COOH ligand has been obtained through quenching of the lithium salt using CO2.89
 | ||
| Scheme 8 Synthesis of TT derivatives by formylation. | ||
Direct arylation: recently, a new synthetic approach based on direct and regioselective palladium-catalyzed C–H functionalization of the 3-position of the TT scaffold has been reported.90 Optimization of the reaction conditions allowed achieving high conversion and selectivity with either electron-rich and electron-poor or sterically demanding (hetero)aryl halides (Scheme 9).
 | ||
| Scheme 9 Synthesis of TT derivatives by direct arylation. | ||
3.2 Electrochemical properties
In order to obtain a deeper insight into the structure/property relationship of TTs (Scheme 10), a systematic cyclic voltammetry investigation was performed.83,91TT is characterized by little electrochemical activity, with first oxidation and first reduction requiring extreme potentials, close to the DMF potential window limits, and with the three imidazole units behaving as three nearly independent redox sites. On the other hand, Iso-A is slightly more reactive, due to the electron-rich nature of the 1,5-annelated ring, which undergoes first oxidation at a milder potential, while reduction, centred on the remaining two almost equivalent 1,2-annelated moieties, is similar to that of TT. Methylation of one nitrogen atom of the imidazolic ring in [TTMe][CF3SO3] resulted in a significant positive shift of the first reduction potential, due to the electron poor nature of the alkylated N site. In 1–3Br, the electrochemical cleavage of the C–X bond is only slightly influenced by the number of halogens. These findings further strengthened the hypothesis that each imidazole unit in the cyclic trimer acts as an almost independent redox site, with very poor heteroannular aromaticity.91 | ||
| Scheme 10 Chemical structures of [TTMe][CF3SO3], TT-4Py1–3, TT-Thio1–3 and TT-biThio1–3. | ||
Comparison of the electrochemical activity of TT substituted with a pyridine moiety attached to the ortho (TT-2Py) or para (TT-4Py) position revealed that the effect of the linking position of the pyridyl group is more effective on the HOMO or oxidation process (mostly centred on TT) than the LUMO or reduction (mostly pyridine-centred). In the case of TT-2Py, a slight negative shift of the first oxidation potential was observed, probably due to a conjugation, rather than the inductive effect, between the TT core and the 2Py moiety.
On the other hand, a remarkable positive shift of both reduction and oxidation processes was observed for TT-4Py. As a result, both derivatives display a HOMO–LUMO gap dramatically decreased with respect to TT, although significantly smaller for TT-2Py than for TT-4Py. The introduction of multiple 4Py moieties in TT-4Py2 and TT-4Py3 favoured the reduction process, as pointed out by the less negative values with increasing number of pyridine substituents (TT-4Pyn −2.64/−2.60/−2.54 V for n = 1/2/3, respectively).
In the case of a more electron rich substituent, as the thiophene moiety in the TT-Thio1–3 series, the TT core is endowed with more favourable first oxidation and first reduction potentials and a narrower HOMO–LUMO gap. The derivatives are not only significantly electron richer than the pyridine-substituted ones, but display also a more effective conjugation efficiency, as confirmed by DFT calculations disclosing the significant involvement of the thiophene terminals in both HOMOs and LUMOs. However, no electrochemical coupling reactions have been observed for TT-Thio1–3, probably due to the fact that the thiophene pendants are too small in comparison with the TT core.
In order to efficiently promote coupling and formation of an electroactive film on the electrode surface, the bithiophene TT-biThio1–3 series was investigated, revealing smaller energy gaps than their thiophene counterparts, with the HOMO and the LUMO mainly located on the bithiophene moiety. Electrodeposition experiments in monofunctionalized TT-biThio solutions, despite the formation of an electroactive product on the electrode surface, did not provide evidence of active film growth, probably due to the formation of a dimeric layer on the electrode surface. On the other hand, di- and trisubstituted monomers TT-biThio2–3 produced fast and regular growth of electroactive films on the electrode surface upon potential cycling around their first oxidation peak (Fig. 2). These promising results on the fast and reproducible oxidative electrodeposition of oligo/polymer films based on alternate TT and tetrathiophene units constitute an attractive springboard for the development of highly electroactive TT-based materials.83
 | ||
| Fig. 2 First and fourth stability cycles in monomer-free solution and electrochromism of [TT-biThio2]n films electrochemically formed on the ITO electrode. Reproduced from ref. 83 under the terms of the Creative Commons CC BY license. Copyright 2023, Published by Elsevier Ltd. | ||
3.3 Photophysical and structural properties
Single component materials characterized by rich emissive behaviour including synergic aggregation induced emissive (AIE) and room temperature long lived (room temperature phosphorescence, RTP) features are receiving growing attention from the scientific community due to the benefits they offer in different fields (e.g. bioimaging,92–95 anti-counterfeiting,96–100 and displays101). Long lived RTP can be achieved through organic compounds but phosphorescence is traditionally considered as the exclusive realm of heavy metal complexes. In fact, inter-system crossing (ISC) and, consequently, phosphorescence lifetime, depend on spin–orbit coupling (SOC), which is large in the presence of heavy atoms. However, through appropriate molecular design and supramolecular engineering, pure organic long-lived phosphors have become numerous and have been already tested for different applications.102–104Many TTs revealed a rich photophysical behaviour comprising multiple fluorescence and phosphorescence of molecular and supramolecular origins, anti-Kasha emissions and excitation dependent photoluminescence (see the following). Multiple emissions derived from molecular electronic levels can originate from the anti-Kasha mechanism, that is, radiative deactivation from high energy Sn (or Tn) states, or from the presence of different conformers either in the ground or in the excited states. While anti-Kasha behaviour is frequently associated with a large S1–S2 energy gap and a high S0–S2 oscillator strength (f), it can also occur in systems having a smaller S1–S2 gap, but Sn–S1 internal conversion (IC) prohibited on symmetry grounds.105 Examples of both anti-Kasha mechanisms have been observed for TTs (see simplified mechanisms in Fig. 3).
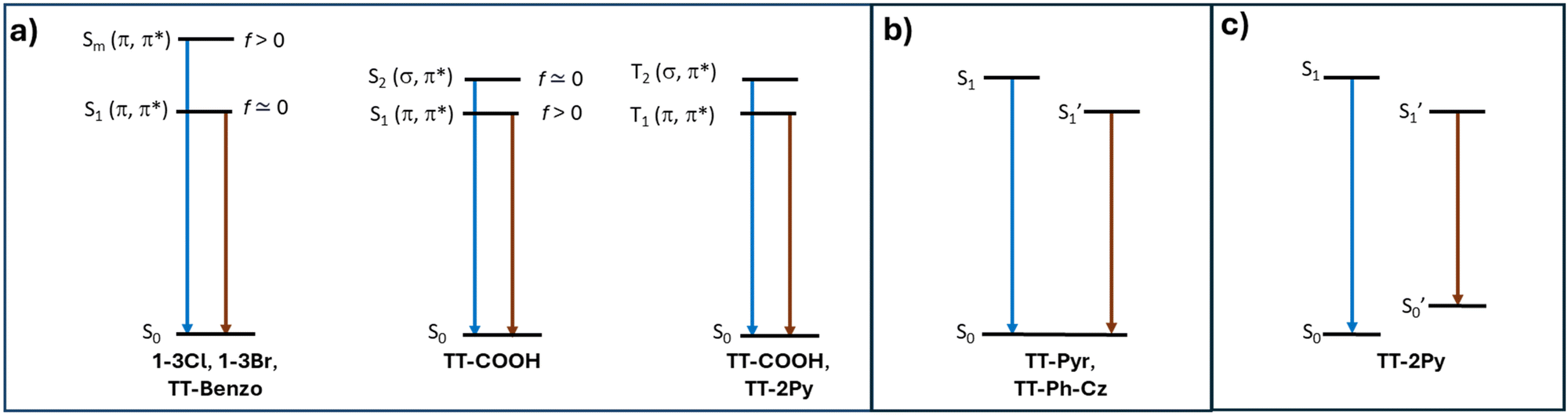 | ||
| Fig. 3 Simplified energy level for organic TTs displaying anti-Kasha behavior (a) and multiple emissions from different excited (b) or ground (c) state conformers. | ||
The reported excitation dependent behavior of many TTs can be explained as the selective suppression of stronger signals (usually fast components at high energy) allowing the monitoring of the weaker ones (typically red shifted phosphorescence) by low energy excitation to singlets of aggregated species or of different conformers or, less frequently, by direct triplet population. Moreover, halogenated derivatives of TT represent remarkable examples of excitation dependent deactivation paths. These systems, in fact, display phosphorescence from T1 only when excited at sufficiently high energy to populate an Sn from which ISC to Tn is favorable. Subsequent IC to T1 results in the observed phosphorescence (see later).
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with significant distortion from the expected C3h symmetry, a result of the establishment of strong intermolecular π–π stacking interactions.30TT molecules, in fact, pack in an alternating AB face-to-face antiparallel fashion forming columnar aggregates where relatively electron-rich and electron-poor regions approach each other (Fig. 4). This structural pattern represents a distinctive feature of the TT scaffold being observed, with obvious variations in relative distances and orientations, in almost all TTs, as shown below.
space group with significant distortion from the expected C3h symmetry, a result of the establishment of strong intermolecular π–π stacking interactions.30TT molecules, in fact, pack in an alternating AB face-to-face antiparallel fashion forming columnar aggregates where relatively electron-rich and electron-poor regions approach each other (Fig. 4). This structural pattern represents a distinctive feature of the TT scaffold being observed, with obvious variations in relative distances and orientations, in almost all TTs, as shown below.
 | ||
| Fig. 4 Crystal packing of TT exhibiting AB stacking arrangement with distances between molecular planes given in Å. | ||
Crystals of TT display at 298 K an intense vibrationally resolved fluorescence at 400 and 424 nm with a RTUP (lifetime, τ, ≅1 s) low energy tail (overall quantum yield, Φ, equal to 30%), which was well disclosed in the delayed spectrum where vibrationally resolved peaks at 525 and 570 nm become visible (Fig. 5). After grinding in a mortar, a loss of the vibronic components of both prompt and RTUP emissions was observed together with a quantum efficiency reduction from 30% to 22%.
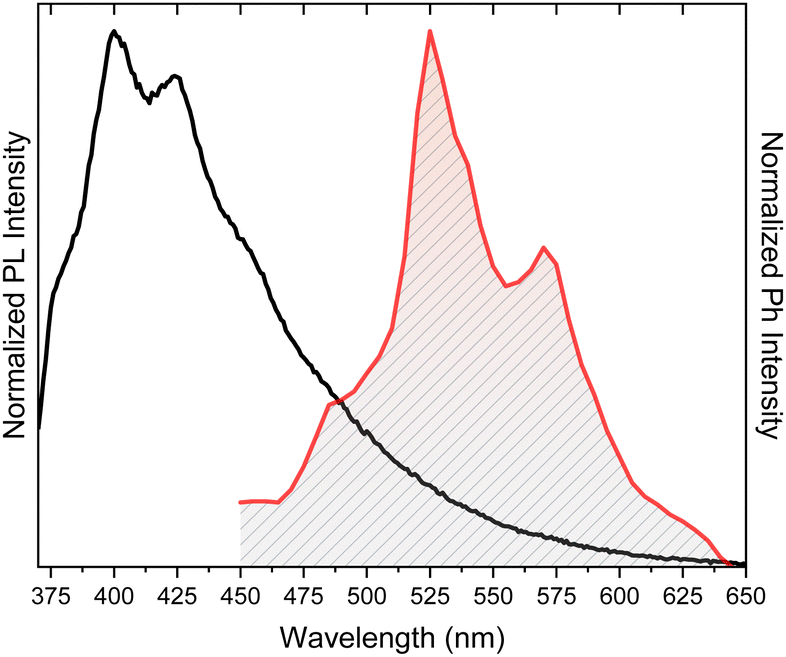 | ||
| Fig. 5 Photoluminescence (PL, black line, λexc = 350 nm) and phosphorescence (red line, time delay 472 ms, λexc = 374 nm) of TT crystals at 298 K. | ||
Geometry optimizations based on either DFT calculations on dimeric and tetrameric aggregates106 or hybrid QM/MM calculations107 highlighted the role of π–π interactions in distorting TT from the C3h symmetry, as experimentally observed. It was also evidenced107 that low- and middle-frequency normal modes, that is, the triazine/imidazole twisting and stretching motions, respectively, are effective only for the isolated (in vacuo or DCM solution) molecule, while they are suppressed for a molecule restricted in a cluster. These restrictions result in a strong increase of the quantum yield and are therefore responsible for the observed AIE behaviour of TT. The phosphorescence efficiency in the solid state was explained by the strong decrease in the S1–T1 energy gap when going from the isolated molecule to aggregates106,107 and by a remarkable intersystem crossing (kISC) and negligible reverse intersystem crossing (krISC) rates,107 in agreement with the lack of delayed fluorescence.
The π–π interactions observed in many TTs are therefore expected to be relevant in affecting their photophysics. As a further proof, many of them, including TT itself, display mechanochromic features (see the following and Table 3).
These results, in agreement with previous reports on the active role played by H-aggregation in activating RTUP,108,109 were further confirmed by investigation of the photophysics of Iso-A, the isomer of TT having one imidazole ring with 1,5- instead of 1,2-annelation. This compound, crystallizing in the P21/c space group, is characterized by CIE behaviour, being slightly emissive in solution at 298 K (in DCM, Φ = 2.8%) but quite so in crystals due to a strong fluorescence at about 415 nm (Φ = 13%).106 The absence of a long-lived component was confirmed also at 77 K. Its single crystal XRD analysis revealed aggregation of π–π stacked ribbons formed by dimeric units interconnected through cyclic C–H⋯N hydrogen bonds (HBs) and joined by co-crystallized water molecules. Weaker π–π interactions are suggested by the greater slippage of the parallel-packed columns and longer distance between centroids of the triazinic rings when compared to TT, in agreement with the lack of the ultralong emission component.
Owing to its inherent asymmetry associated with the presence of nitrogen atoms, TT is a prochiral molecule and its deposition on a surface generates two enantiomers.110 The two are stabilized by π–metal interaction and their interconversion is not possible since it would require flipping the molecule out of plane by 180°. Along the surface plane, TT self-assembles through symmetric HB pairing of its left- (S) or right-turning (R) enantiomers, giving rise to spontaneously resolved two-dimensional hexameric networks consisting exclusively of S- (or R-) enantiomers (Fig. 6 top), as experimentally observed for TT deposited on a Ag(111) substrate.110 The heterochiral assembly, though possible, was predicted by DFT calculations to be less stable than the homochiral one. STM images revealed the presence of kidney-shaped enantiomeric islands, consisting of hexagonal porous networks of TT on the metal surface separated by a dark region, which was associated with a defective zone where RR islands ‘capture’ S enantiomers (and vice versa) during the deposition process (Fig. 6 bottom).
 | ||
| Fig. 6 Top: fragment of a model for the energetically most favorable homochiral 2D assembly of TT (the S-enantiomers are shown). Bottom: STM images of the nanoporous 2D networks formed by deposition of TT molecules on Ag(111) showing the kidney-shaped islands formed by TT (left, 400 × 400 nm, 1 V, 20 pA image) and one specific interface zone between two different domains of the hexagonal network (right, 40 × 40 nm, 1 V, 40 pA image). Adapted with permission from ref. 110 under the terms of the Creative Commons CC BY license. Copyright 2023, American Chemical Society. | ||
 | ||
| Scheme 11 Chemical structure of nX. | ||
In fact, 1–3X display a quite complex, excitation dependent photoluminescence with emissions including dual fluorescence, molecular phosphorescence and supramolecular RTP and RTUP covering a wide portion of the visible region.68–71
1–3Cl crystallize in the P, C2/c and P21/c space groups, where molecules form hydrogen bonded corrugated layers, which stack in a slipped manner, evidencing the presence of π–π stacking interactions between TT units and, only for 3Cl, very short (rCl⋯N = 3.014 and 3.151 Å) halogen bonds (XB) corresponding to 8.7 and 4.5% shortening with respect to the sum of the vdW (van der Waals) radii.
Crystals of 1–3Cl are characterized by multiple, excitation dependent emissive features (Φ of about 24, 12 and 10% respectively) comprising dual fluorescence (a high energy component, HEF, in the 330–350 interval, and a low energy one, LEF, in the 350–400 nm portion of the spectrum) and triple phosphorescence (HEP 405–440 nm, MEP 480–550 nm and LEP 550–560 nm) with the lowest energy contribution (LEP) visible only at 77 K (see Fig. 7 and Table 3).
 | ||
| Fig. 7 Normalized PL emission spectra of 1–3Cl crystals at 298 K (full lines) and 77 K (dashed lines). Left 1Cl, 298 K: λexc = 300 nm (black), λexc = 340 nm (blue) and λexc = 440 nm (red). 77 K: λexc = 280 nm (black), λexc = 300 nm (red), λexc = 370 nm (blue) and λexc = 440 nm (green). Middle 2Cl, 298 K: λexc = 280 nm (black), λexc = 308 nm (blue), λexc = 375 nm (red) and λexc = 413 nm (green). 77 K: λexc = 280 nm (black), λexc = 308 nm (red), λexc = 400 nm (blue) and λexc = 434 nm (green). Right 3Cl, 298 K: λexc = 300 nm (black), λexc = 330 nm (red), λexc = 374 nm (blue) and λexc = 440 nm (green). 77 K: λexc = 280 nm (black), λexc = 330 nm (red), λexc = 373 nm (blue) and λexc = 440 nm (green). | ||
Through spectroscopic, structural and computational analyses, HEF, LEF, HEP and LEP were assigned to excited states of molecular origin while MEP was interpreted as derived from aggregated species. More specifically, HEF corresponds to radiative deactivation from a high energy singlet level (Sn) of (π,π*) character and large oscillator strength, while an S1 state of (π,π*) character and much lower energy and oscillator strength is responsible for LEF. Altogether these features result in the observed anti-Kasha behaviour. Regarding the three phosphorescence types, HEP was correlated to T1 of (π,π*) character, while LEP to high energy triplets of (π,σ*) character (Tσ, Fig. 8). Aggregation induced MEP, whose intensity increases by increasing the crystalline grade of the samples and the strength of π–π interactions in the crystal structure (intensity increasing in the order: 1Cl < 3Cl ≅ 2Cl), was assigned to TT stacking interactions.
 | ||
| Fig. 8 Main molecular orbitals involved in the (π,σ*) Tσ state of 2Cl (isosurface value: 0.02). | ||
 | ||
| Fig. 9 1Br in DCM (10−4 M): top: absorption (black dotted line) and PL emission (λexc = 280 nm; blue solid line) at 298 K. Bottom: excitation profile (λem = 580 nm; black dotted line) and PL emission (λexc = 280 nm; red solid line) at 77 K. Adapted with permission from ref. 70. Copyright 2017, John Wiley and Sons. | ||
On the basis of DFT and TDDFT calculations, the RT emission was assigned, according to an anti-Kasha mechanism, to a high energy Sn state of (π,π*) character, while the phosphorescence observed at 77 K was explained by the presence of high energy 3(σ,σ*) and, only for 3Br, 3(σ,π*) levels, which facilitate an efficient ISC (by both El Sayed and heavy atom effects) from the closest  level, followed by IC to T1. By exciting at longer wavelengths, in fact, the appropriate high energy
level, followed by IC to T1. By exciting at longer wavelengths, in fact, the appropriate high energy  level cannot be populated and only fluorescence from Sn was observed.
level cannot be populated and only fluorescence from Sn was observed.
1–3Br crystallize in the P21/c, P and P21/c space groups respectively, forming π-stacked arrangements of dimeric (3Br) or columnar (1Br and 2Br) TT units (see Fig. 10) with π–π stacking interactions’ strength in the order 2Br > 3Br > 1Br.
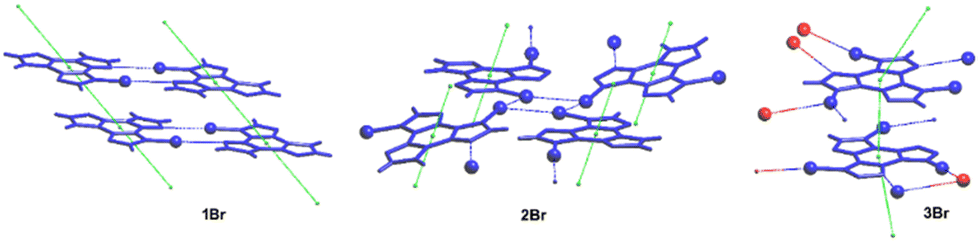 | ||
| Fig. 10 Views of the π–π stacking in 1–3Br; Br atoms are shown as spheres and XB interactions are highlighted by blue dashed lines. Red spheres in 3Br refer to atoms belonging to different layers. Reproduced with permission from ref. 69. Copyright 2018, John Wiley and Sons. | ||
The structures of 2Br and 3Br highlight the presence of Br⋯Br XBs with formation of tetrameric Br4 (2Br, see Fig. 10 centre) and trimeric Br3 (3Br) units. The centrosymmetric Br4 synthon (2Br) is characterized by high rigidity, as evidenced by both its approximate coplanarity and Br⋯Br distances 4 and 1% shorter than two times the bromine vdW radius. In the Br3 unit (3Br), instead, only two molecules have close Br⋯Br contact (3% shortening), while the third one is rather far from the other two and significantly out from their ls (least squares) plane, indicating a less stable XB supramolecular aggregation.
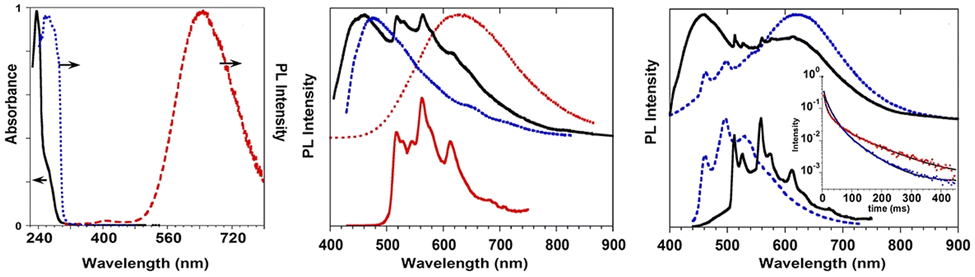
Crystals of 1–3Br display excitation dependent multicomponent spectra (Φ < 0.1 for 1Br and 3Br and about 14% for 2Br) including the Sn–S0 emission, HEF, only observed in solution at RT, with maxima in the 330–440 nm interval. In addition, in 1Br and 3Br, the S1–S0 fluorescence LEF (with maxima in the 430–530 nm interval) is activated owing to distorting packing forces that reduce the symmetry of the molecular π-electron system (see Fig. 11 for 3Br), resulting in dual fluorescence. The broad long-lived component detected at 470 nm for 2Br at 298 K and at 490 nm for 3Br at 77 K was ascribed to the extrinsic heavy atom effect caused by the bromine atom in the Br⋯Br XB motifs (the Br4 cyclic units in 2Br and the Br3 cyclic units in 3Br). The structured RTUP (at about 550–650 nm), observed in the emission spectra of powders of 2Br and 3Br (see Fig. 11 for 3Br), was associated with the presence of stacking interactions among TT units. This RTUP cannot be totally excluded even for 1Br since the crystallinity degree and the crystal size of the examined samples may play a role in the relative intensity of phosphorescence emissions. Finally, the three compounds display at 77 K and upon excitation at short wavelengths (280 nm) the strong phosphorescence (at 573, 558 and 590 nm for 1Br, 2Br and 3Br, respectively) observed also in the frozen solutions and therefore assigned to molecular features.
 | ||
| Fig. 11 Powders of 3Br left: at 298 K. Top: Prompt emission (λexc = 280 nm, red solid line; λexc = 340 nm, blue solid line) and excitation profiles (λem = 420 nm, dashed blue line). Bottom: Delayed emission (λexc = 340 nm, 1 ms delay, window 50 ms; black solid line) and excitation profiles (λem = 550 nm, dotted black line). Right: at 77 K. Top: prompt emission (λexc = 280 nm, red solid line; λexc = 340 nm, blue solid line; λexc = 385 nm, green solid line) and excitation profiles (λem = 420 nm, dotted blue line). Bottom: delayed emission λexc = 360 nm, 100 μs delay, window 500 μs, red solid line; λexc = 385 nm, 100 μs delay, window 500 μs, green solid line; λexc = 385 nm, 5 ms delay, window 10 ms, black solid line) and excitation profiles (λem = 523 nm, dotted green line; λem = 600 nm, dotted red line). Adapted with permission from ref. 69. Copyright 2018, John Wiley and Sons. | ||
 | ||
| Fig. 12 Left: 1I in DCM: absorption spectrum at 298 K (black solid); PL emission and excitation spectra at 77 K (λexc = 280 nm, red dashed; λem = 648 nm, blue dotted); centre: emission spectra of 1I crystals at 298 K: top: PL at λexc = 300 nm (red dotted), λexc = 370 nm (black solid), λexc = 415 nm (blue dashed); bottom: phosphorescence spectrum (λexc = 370 nm, delay 50 ms, window 200 ms, red solid); right: emission spectra of 1I crystals at 77 K. Top: PL at λexc = 320 nm (blue dotted), λexc = 370 nm (black solid); bottom: phosphorescence spectra at λexc = 320 nm (delay 10 ms, window 50 ms, blue dotted) and λexc = 370 nm (delay 50 ms, window 200 ms, black solid). Phosphorescence decays at λem = 460 nm (λexc = 320 nm, blue points) and λem = 558 nm (λexc = 370 nm, red points) with their three-exponential fits (black lines) are shown in the inset. Adapted with permission from ref. 71. Copyright 2019, John Wiley and Sons. | ||
1I and 2I crystallize in the C2/c and P space groups, respectively, where XB and π–π stacking interactions act in a cooperative way. In 1I, non-equivalent I⋯N XB bonds (rI⋯N = 2.878 and 3.020 Å, corresponding to 18 and 14% shortening with respect to the sum of the vdW radii) are established on both sides of the molecule, giving rise to helicoidal chains (see Fig. 13). Four halogen bonded chains are intervolved along the helix axis, so that the pitch of the helix, 16.388 Å long, comprises four molecules with the interplanar distance (3.309 Å) facilitating strong π–π interactions. The crystal structure of 2I is isomorphous with that of 2Br, where molecules form tetrameric I⋯I XB cyclic units, I4, slightly more rigid than the Br4 ones. The I⋯I distances, 3.717 and 3.780 Å, are in fact 6 and 5% shorter than two times the iodine vdW radius, to be compared with 4 and 1% shortening observed in the Br4 unit.
 | ||
| Fig. 13 Partial views along b- (left) and c-axes (right) of 1I crystal structure showing columnar π–π stacks (centroids of the triazinic rings shown as red circles) interconnected through I⋯N XB (light blue dashed lines) to form intervolved quadruple helices along the b-axis. Reproduced with permission from ref. 71. Copyright 2019, John Wiley and Sons. | ||
Crystals of 1I and 2I show at room temperature quite similar excitation dependent emissive features (Φ < 0.1 and 7%, respectively). In particular, upon low energy excitation, 1I and 2I display a broad fluorescence (at 476 and 443 nm, respectively) (see Fig. 12 for 1I) similar to that observed in solution at 77 K and analogously attributed to radiative deactivation from S1. At higher excitation energy, RTUP becomes visible (at 517, 563, 612 nm and 625 nm, respectively). By exciting at very shorter wavelengths (<300 nm) the molecular phosphorescence (630 and 680 nm, respectively) present also in solution at 77 K is activated. Investigation of crystals of 1I at 77 K (Fig. 12) revealed an additional phosphorescence contribution at 460, 495 and 530 nm. Even in this case, RTUP was assigned to π–π aggregates while the new 77 K phosphorescence of ms order observed at 460 nm, firstly ascribed to the presence of I⋯N XB, was, after investigation of [MI(TT)]n coordination polymers (see later), interpreted as due to I⋯C intermolecular electronic coupling with partial orbital overlapping111 between the heavy iodine atom and the TT unit. Finally, the molecular phosphorescence of 1I and 2I, visible also at 298 K, is much more intense than that observed at 77 K for nBr, in agreement with the presence of the heavier iodine atom on the molecule.
Comparison of the photophysical behaviour of halogenated compounds is reported in Fig. 14. The effect of the heavy halogen atoms can be accurately evaluated only through lifetimes of LEP, decreasing from 1Cl to 1I. In fact, LEP is associated with molecular electronic levels and it is observed for all 1X under the same conditions (solid state, 77 K) allowing a reliable comparison.
 | ||
| Fig. 14 Comparison through a simplified Jablonski diagram of solid 1–3X. Lifetimes of LEP for 1X are reported.a Even at RT for 1–2I. | ||
To evaluate the extrinsic iodine heavy atom effect on the photoluminescence of TT isolated from the intrinsic one, a structural and spectroscopic investigation was performed on TT·DITFB, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal self-assembled through I⋯N XB between TT and 1,4-diiodotetrafluorobenzene (see Fig. 15), DITFB.
:1 cocrystal self-assembled through I⋯N XB between TT and 1,4-diiodotetrafluorobenzene (see Fig. 15), DITFB.
 | ||
| Fig. 15 Left: chemical structures of TT·DITFB. Right: partial view of its crystal structure showing columnar π–π stacks of TT (centroids of the triazinic rings shown as red circles) interconnected through I⋯N XB (light blue dashed lines) with DITFB to form infinite 1D zig-zag chains. Adapted with permission from ref. 71. Copyright 2019, John Wiley and Sons. | ||
Its crystal structure (P21/n space group) consists of heteromeric zig-zag infinite 1D chains self-assembled through I⋯N XB, where TT acts as double XB acceptor and DITFB as a double XB donor (Fig. 15) with I⋯N distances, rI⋯N = 3.031 and 3.006 Å, shorter by 14 and 15%, respectively, than the sum of vdW radii. Adjacent chains are connected through strong π–π stacking interactions among TT units.
Crystals of TT·DITFB show at 298 K (overall Φ equal to 5%) only broad fluorescence (at about 410 nm) and structured phosphorescence (at 496, 528 and 566 nm), while at 77 K broad, red phosphorescence (at 720 nm) dominates the spectrum comprising additional bands (Fig. 16). π–π stacking interactions among TT units were recognized as responsible for the structured phosphorescence, while the low energy contribution was associated through DFT/TDDFT calculations to the iodine extrinsic heavy atom effect, resulting in the presence of 3(σ,σ*) and 3(π,σ*) levels allowing SOC from close singlet states of different characters. This ‘extrinsic-molecular phosphorescence’, observed only at 77 K, is less efficient than the intrinsic one observed also at 298 K for 1I and 2I. Among the other bands observed at 77 K, additional phosphorescence was recognized through delayed spectra (at 463, 497, and 537 nm) and associated with the I⋯C intermolecular electronic coupling (TI–S0), similarly to what concluded for 1I.
 | ||
| Fig. 16 Left: Emission spectra of TT·DITFB at 298 K: top: PL at λexc = 300 nm (blue) and λexc = 350 nm (black); bottom: phosphorescence (λexc = 350 nm, delay 0.5 ms, window 1 ms, red dashed); right: emission spectra of TT·DITFB crystals at 77 K: top: PL at λexc = 340 nm (black), λexc = 300 nm (red); bottom: phosphorescence at λexc = 320 nm (delay 10 ms, window 50 ms, blue dashed) and λexc = 370 nm (delay 10 ms, window 50 ms, red dotted). Adapted with permission from ref. 71. Copyright 2019, John Wiley and Sons. | ||
Based on these results, it was established that the molecular phosphorescence is better activated through an intrinsic heavy atom effect, being observed at 298 K only in 1I and 2I. Moreover, the I⋯C induced phosphorescence was confirmed as an extrinsic heavy atom effect, being observed in both 1I and TT·DITFB.
On the whole, studies on 1-3X revealed 1-3Cl as the best performing among the full series, preserving the solid state multifaceted emissive behaviour but with enhanced AIE features (highest quantum yield) with respect to the bromine and iodine analogues. Moreover, the role of the extended network of intermolecular interactions in activating multiple radiative deactivation channels comprising fast and long-lived components was fully disclosed. In particular, strong π–π interactions dominating the crystal structures of all halo derivatives were deemed responsible for the RTUP, while extrinsic heavy atom effect was recognized as efficaceous in activating green phosphorescence only in the case of Br and I derivatives, owing to the low heavy-atom effect played by chlorine.
 | ||
| Scheme 12 Chemical structure of TT-CCH. | ||
 | ||
| Fig. 17 Absorption (dotted line), excitation (dashed line, λem: 374 nm) and PL emission (continuous line, λexc: 290 nm) spectra of TT-CCH in DCM. | ||
For polymethylmethacrylate (PMMA)-blended films with low fluorophore loading (dye/matrix w/w% equal to 0.1) a single, unstructured fluorescence, HEF (at 330 nm), and a broad phosphorescence (at 442 nm), HEP, were observed at an appropriate excitation wavelength (Fig. 18). Blended films with higher loadings (w/w% equal to 5) display dual fluorescence (HEF at about 342 nm and LEF at about 383 nm) and dual phosphorescence (HEP at 442 nm and LEP at about 522 nm).
 | ||
| Fig. 18 PL emission spectra of TT-CCH in a PMMA matrix (0.1 w/w%, dotted line; 5 w/w%, full line) at 298 K. λexc: 290 nm (black lines), 330 nm (blue line), 380 nm (red lines) and 445 nm (green line). Inset: The delayed spectra are shown for the 5 w/w% concentration at short (40 μs delay, 200 μs window; red line) and long (4 ms delay, 20 ms window; green line) delay times. | ||
The compound crystallizes in the monoclinic P21/c space group forming infinite ribbons with TT-CCH molecules connected through quite short CH⋯N HBs forming cyclic patterns (rH⋯N = 2.33 and 2.48 Å, the former involving the acidic C(sp)–H bond). The ribbons are overlapped giving rise to columnar π–π aggregates.
Crystals of TT-CCH show excitation dependent behaviour (Φ = 16%) comprising dual fluorescence (HEF at 314, 326, 338 nm and LEF at 354, 367, 377 nm) and dual phosphorescence (HEP at 434 nm and LEP at 538 nm). The relative intensity of the two fluorescent signals is temperature and grinding dependent, with LEF being attenuated through grinding and by increasing the temperature (Fig. 19). Of the four emissions, HEF and HEP were interpreted as molecular phenomena, while LEF and LEP as due to the supramolecular ones. In agreement with this hypothesis, in PMMA films, LEF and LEP appear only by increasing the dye-loading and HEF/LEF relative intensity shows mechanochromic dependence.
 | ||
| Fig. 19 Normalized PL emission spectra of TT-CCH crystals. Left at 298 K: Prompt (full line) and delayed (dashed line) spectra; λexc.: 275 nm (black); 330 nm (blue) and 486 nm (green); λexc.: 310 nm (dashed red, delay 15 μs, window 400 μs; dashed green, delay 1 ms, window 15 ms). Middle at 77 K: prompt spectra; λexc.: 275 nm (black); 300 nm (blue); λexc.: 340 nm (red); λexc.: 450 nm (green). Right at 298 K: before (black) and after (blue) manual grinding in a mortar; λexc.: 275 nm. | ||
The origin of LEF was clarified through DFT/TDDFT calculations on both the isolated molecule and different small aggregates, comprising CH⋯N and/or π⋯π stacked dimers and tetramers. The computed molecular S1 level possessing (π,π*) character is localized on the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond and the portion of the TT moiety directly bonded to it and lies very close to a triplet state from which HEP is originated. For the CH⋯N aggregates, S1 gradually red shifts and acquires an impressive increase of its oscillator strength, from 0.25 (monomer) to 0.74 (dimer) and then 1.64 (tetramer). For π–π stacked dimeric and tetrameric species a much lower increase in oscillator strength was computed, indicating that appearance of LEF is due to the strong HB characterizing the TT-CCH crystal structure and not to π–π stacking interactions, which are instead responsible for LEP as often observed among TTs.
C bond and the portion of the TT moiety directly bonded to it and lies very close to a triplet state from which HEP is originated. For the CH⋯N aggregates, S1 gradually red shifts and acquires an impressive increase of its oscillator strength, from 0.25 (monomer) to 0.74 (dimer) and then 1.64 (tetramer). For π–π stacked dimeric and tetrameric species a much lower increase in oscillator strength was computed, indicating that appearance of LEF is due to the strong HB characterizing the TT-CCH crystal structure and not to π–π stacking interactions, which are instead responsible for LEP as often observed among TTs.
space group with the formation of intramolecular OH⋯N hydrogen bonding and π–π stacking interactions involving the TT units. Crystals of TT-COOH display excitation dependent behaviour (Φ = 26%, Fig. 20) comprising dual fluorescence (HEF at about 342 nm and LEF at 386, 408 and 432 nm) and triple phosphorescence (HEP at 445 nm, MEP at 487 nm, LEP at 549, 590 and 642 nm).
 | ||
| Scheme 13 Chemical structure of TT-COOH. | ||
 | ||
| Fig. 20 Normalized PL emission spectra of TT-COOH crystals at RT (continuous lines) and 77 K (dotted lines). λexc = 300 nm (black); 340 nm (blue); 390 nm (green); 440 nm (red) and 500 nm (grey). Inset: Normalized emission (continuous, λexc = 340 nm) and excitation profiles (dotted, λem = 409 nm) at 77 K. | ||
Based on spectroscopic, structural and theoretical investigations, the emissions were recognized as molecular (HEF, LEF, HEP and MEP) or π–π aggregate (LEP) features (Fig. 20). The molecular contributions were associated with the presence of low energy excited states of (π,π*) and (σ,π*) symmetries, justifying separate deactivation radiative paths. Moreover, calculations on H-bonded dimers did not result in a remarkable increase of the S1 oscillator strength as instead obtained for TT-CCH, as a consequence of the weaker interaction in TT-COOH. Therefore, the latter was classified as an anti-Kasha emitter differently from the former.
 | ||
| Scheme 14 Chemical structure of TT-Benzo. | ||
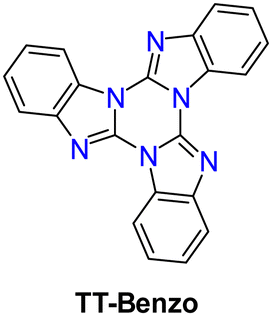
TT-Benzo crystallizes in the P space group112 in an arrangement very similar to that of TT but with a higher intermolecular distance along its π–π columnar aggregates. At 298 K, microcrystalline powders of TT-Benzo show excitation dependent properties comprising both dual fluorescence (at 335, 350, 366 nm and 407 nm, the first being quite similar to the one observed in solution) and RTUP (at 530 nm); the latter is assignable to π–π stacking interactions (Fig. 21). The dual fluorescence was interpreted through DFT/TDDFT calculations, which revealed the presence of an S0–S1 transition of (π,π*) character and zero oscillator strength for symmetry reasons, and two almost degenerate transitions at slightly higher energy with large oscillator strength (S0–S2 and S0–S3). These electronic conditions are effective in producing anti-Kasha behaviour105,113,114 and, on this basis, the high energy fluorescence observed in solution and in the solid state was associated with S2–S0 radiative deactivation. In the solid state, the S0–S1 transition acquires intensity due to the partial loss of molecular symmetry through intermolecular interactions, resulting in the S1–S0 fluorescence.
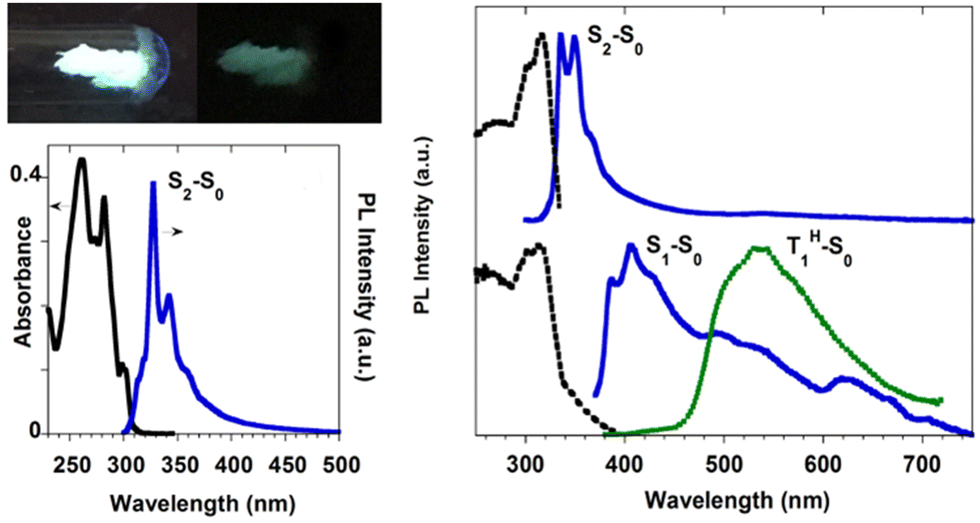 | ||
| Fig. 21 (left) Top: powders of TT-Benzo at 77 K with UV irradiation on (left) and off (right). Bottom: DCM solutions at 298 K: absorption (black) and PL emission (blue, λexc = 260 nm). Right: Powders at 298 K: top: excitation (dashed black, λexc = 348 nm) and PL emission (blue, λexc = 260 nm). Bottom: excitation (dashed black, λem = 408 nm), PL emission (blue, λexc = 370 nm), and phosphorescence (green dotted, 10 ms delay, window 50 ms, λexc = 358 nm). Reproduced with permission from ref. 70. Copyright 2017, John Wiley and Sons. | ||
 | ||
| Scheme 15 Chemical structure of TTPyr1–3. | ||
 | ||
| Fig. 22 Normalized PL emission (solid lines) and excitation (dashed lines) spectra at 298 K. Left: DMSO solutions of TTPyr1 (blue line), TTPyr2 (red) and TTPyr3 (black), λexc = 350 nm, λem = 420 nm. Right: TTPyr1 in PMMA (5% w/w), λexc = 345 nm; λem = 397 nm. | ||
Only for TTPyr1 single crystals suitable for XRD analysis were obtained by various methods. In particular, three crystalline forms, namely TTPyr(RT) (P21/c space group, Fig. 23), TTPyr(Et) (C2/c space group, Fig. 24) and TTPyr(HT) (Pna21 space group, Fig. 25), were isolated. The asymmetric unit of TTPyr(Et) contains one TTPyr and half ethanol molecule interacting via hydrogen bonds (Fig. 24).
 | ||
| Fig. 23 X-Ray crystal structure of TTPyr(RT). Left: View of a single TTPyr molecule (ellipsoid drawn at 30%); right: view of molecular packing (hydrogen atoms are omitted for clarity). Reproduced with permission from ref. 80. Copyright 2021, John Wiley and Sons. | ||
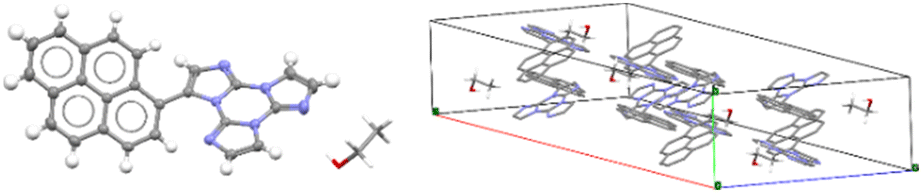 | ||
| Fig. 24 X-Ray crystal structure of TTPyr(Et). Left: View of a single TTPyr molecule interacting through hydrogen bonds with an ethanol molecule (ellipsoid drawn at 30%, only one model of disordered ethanol molecule is shown); right: view of the molecular packing (hydrogen atoms are omitted for clarity and only one model is shown for ethanol molecules). Reproduced with permission from ref. 80. Copyright 2021, John Wiley and Sons. | ||
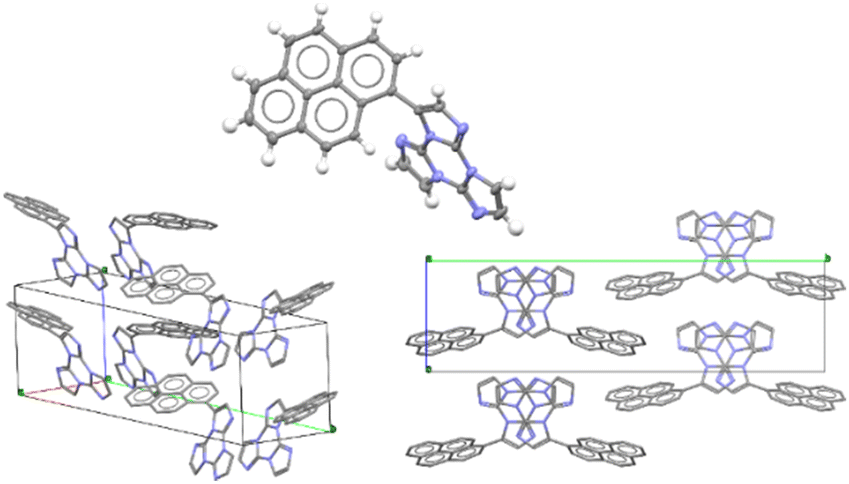 | ||
| Fig. 25 X-Ray crystal structure of TTPyr(HT). Top center: View of a single TTPyr molecule (ellipsoids are drawn at 30%); bottom: two views of the molecular packing (hydrogen atoms are omitted for clarity). Reproduced with permission from ref. 80. Copyright 2021, John Wiley and Sons. | ||
In TTPyr(RT) and TTPyr(Et), the TT unit of one molecule stacks in between two Pyr units of neighbouring molecules, forming infinite columns of π–π stacking fragments (Fig. 23 and 24). On the other hand, in TTPyr(HT) the molecules form columns where only the TT fragments are facing each other and the pyrene units of adjacent molecules protrude in opposite directions (Fig. 25).
Crystals of TTPyr(RT) and TTPyr(Et) display a similar excitation dependent photophysical behaviour (Φ about 40%), comprising one fluorescence band (at 490 and 493 nm, respectively) and one phosphorescence band (at 550 and 555 nm, respectively) (Fig. 26) with relative intensity varying with crystallinity of the examined sample.
 | ||
| Fig. 26 Normalized PL emission (full line) and excitation (dashed line) spectra at RT. Left: TTPyr(Et) crystals. λexc = 300 nm (black line), λexc = 405 nm (red line), λexc = 495 nm (blue line); λem = 494 nm (black line); λem = 610 nm (blue line). Middle: TTPyr(RT) crystals. λexc = 300 nm (black line), λexc = 405 nm (red line), λexc = 490 nm (blue line); λem = 466 nm (red line), λem = 555 nm (blue line). Right: TTPyr(HT) crystals. λexc = 300 nm (black line), λexc = 440 nm (red line), λexc = 480 nm (blue line); λem = 445 nm (black line), λem = 483 nm (red line), λem = 524 nm (blue line). | ||
In agreement with its different disposition of the chromophore inside the structure with respect to the other crystalline forms, TTPyr(HT) displays a macroscopically different photophysics with dual fluorescence (HEF and LEF at 420, 450 nm and 480 nm, respectively) and single phosphorescence (at 550 nm) with relative intensity depending on the excitation energy. For all phases, the phosphorescence feature was assigned to π–π (TT-TT or TT-Pyr interactions) aggregated species. For what concerns fluorescence, DFT/TDDFT calculations on dimeric prototypes of TTPyr(RT) evidenced a blocked conformation with reduced twisting with respect to the isolated molecule and therefore optimized lower energy S1 minimum, resulting in the observed red shifted single fluorescence (monitored in solution). On the other hand, the TTPyr(HT) dimer, where the pyrene moiety is still free to get the two S0 minima, produces dual fluorescence.
Relatively to TTPyr2 and TTPyr3, showing in powder only one fluorescence band (at 490 and 476 nm, respectively) and one phosphorescence band (at 528 and 522 nm, respectively), the lack of structural details allowed drawing only qualitative conclusion. In particular, aggregated species were deemed responsible for the long-lived emission appearing at the same wavelength as that of TTPyr LEP.
 | ||
| Scheme 16 Chemical structures of TT-HThio and TT-HThio3. | ||
TT-HThio crystallizes in the P space group with head-to-head π-stacked aggregates. No single crystals were obtained for TT-(HThio)3.
Crystals of the former and powder of the latter display crystallization enhanced emission (CEE) through an excitation dependent behaviour comprising one fluorescence band (at 376 and 382 nm, and 400 nm, respectively) and two phosphorescence bands (HEP at 425 and 451 nm, and 428 and 453 nm, respectively; LEP at 497, 530, and 578 nm, and 514 and 550 nm, respectively) (overall Φ = 26% and 22%, respectively, Fig. 27). CEE behaviour was confirmed by a decrease in Φ (18%) through grinding crystals of TT-HThio in a mortar. From DFT/TDDFT and X-ray studies, HEP and LEP observed in both solid compounds were assigned to molecular and aggregated π–π (TT-TT) species.
 | ||
| Fig. 27 Normalized PL emission of crystals of TT-HThio: (continuous lines) and of crystalline powders of TT-(HThio)3 (dashed dotted lines) spectra. Left 298 K at λexc = 300 nm (black), ≅380 nm (blue), 450 nm (red); right 77 K, emission at λexc = 300 nm (black), 360 nm (blue), ≅440 nm (red). | ||
 | ||
| Scheme 17 Chemical structures of TT-(N)-Cz, TT-(C)-Cz and TT-Ph-Cz. | ||
3.3.5d.1 TT-(N)-Cz . In dilute DCM solution TT-(N)-Cz shows at room temperature two sets of absorption bands at 280 and 288 nm and 313 and 326 nm and a broad fluorescence band at 345 nm (Φ = 27%). At 77 K, a strong fluorescence band at 339 nm and a much weaker vibrationally resolved phosphorescence band at 411, 439, and 466 nm are observed. Blended PMMA films (TT-(N)-Cz 0.5 wt%, Fig. 28) display at RT in vacuum one fluorescence band at 345 nm (Φ = 28%) and one vibrationally resolved phosphorescence band (maxima at 412, 440 and 462 nm).
 | ||
| Fig. 28 TT-(N)-Cz normalized emission spectra (λexc = 300 nm). PL of DCM solutions (10−6 M, red) at 298 K (left) and 77 K (right) in air. PL of PMMA films (0.5 wt%) in vacuum (black) and delayed spectra (blue dashed; delay 200 μs, window 500 μs at 298 K (left); delay 5 ms, window 10 ms at 95 K (right). | ||
TT-(N)-Cz crystallizes in the monoclinic P21/c (TT-(N)-CzM) and in the triclinic P (TT-(N)-CzT) space groups. In both TT-(N)-Cz polymorphs, TT and Cz are almost orthogonal, with structures dominated by π–π stacking interactions between TT moieties (see Fig. 29).
 | ||
| Fig. 29 Crystal packing of TT-(N)-CzM (left) and TT-(N)-CzT (right), showing the shorter distances between triazinic geometrical centroids (green spheres) and intermolecular contacts shorter than the sum of vdW radii (light grey dashed lines). Ellipsoids at 30% probability. Reproduced with permission from ref. 84. Copyright 2023, by Elsevier Ltd. | ||
Crystals of both polymorphs (Fig. 30) revealed quite similar excitation dependent emissive features (Φ = 13 and 16% for TT-(N)-CzT and TT-(N)-CzM, respectively) comprising HEF (349 and 357 nm for TT-(N)-CzT and TT-(N)-CzM, respectively), LEF (in the 380–402 nm interval), HEP (442, 464, and 437 nm, respectively) and LEP (509 and 517 nm, respectively).
 | ||
| Fig. 30 Normalized emission at 298 K (left) and 77 K (right). Top: TT-(N)-CzT crystals: PL spectra (continuous lines): λexc = 300 nm (black) and λexc = 400 nm (red); delayed spectra (blue dashed): delay 1 ms, window 5 ms. Bottom: PL emission spectra of: TT-(N)-CzM crystals, λexc = 300 nm (red continuous); λexc = 355 nm (red dotted); TT-(N)-CzT crystals, λexc = 300 nm, before (black) and after (blue) grinding. | ||
The origin of this multicomponent emissive behaviour was disclosed through spectroscopic, structural and computational studies revealing the molecular or the aggregated nature of each contribution. LEF and LEP possess aggregated origin as proven by both their absence in diluted solutions and blended films and their quenching through crystal grinding. In addition, LEP appears in the same spectral region where RTP is observed for TT emitters having columnar or dimeric π–π aggregates. Based on theoretical studies, the LEF origin was as well attributed to strong π–π interactions involving the TT scaffold. In fact, TDDFT calculations performed on the molecule resulted, among the others, in an S1 state of Cz character and an S3 state of TT character, both having (π,π*) symmetry. For π–π dimeric prototypes a clear stabilization accompanied by a noteworthy increase in the oscillator strength was calculated only for S3, which has been therefore identified as the lowest energy excited state in the crystal (SH).
3.3.5d.2 TT-(C)-Cz . In dilute DCM solution, TT-(C)-Cz shows at RT two absorption bands at 239 and 290 nm with a tail at about 330 nm and a broad fluorescence at 380 nm (Φ = 20%). At 77 K, a broad long-lived component at about 500 nm (LEP) is clearly visible in the PL spectrum. PMMA films (TT-(C)-Cz 0.5 wt%, Fig. 31) in vacuum display one fluorescence band at 377 nm and two phosphorescence bands at about 417 nm (HEP) and 515 nm (LEP), which appear to be vibronically resolved at 90 K (at 412, 442, and 500 nm, respectively).
 | ||
| Fig. 31 TT-(C)-Cz normalized emission spectra (λexc = 300 nm). PL of DCM solutions (2 × 10−6 M, red continuous) at 298 K (left) and 77 K (right). PL of PMMA films (0.5 wt%, at 298 K, left, and 90 K, right) in vacuum (black) and delayed spectra (blue dashed, delay 0.2 ms, window 0.5 ms; red dashed, delay 50 ms, window 10 ms at 95 K. | ||
TT-(C)-Cz crystallizes in the Pna21 space group with two independent molecules in its a.u., i.e., A and B, having the dihedral angle between TT and Cz units equal to 34.2° (A) and 37.8° (B), indicating partial conjugation. Moreover, differently from the TT-(N)-Cz polymorphs, in TT-(C)-Cz the π–π stacking interactions between TT units are replaced by analogous interactions between TT and Cz.
TT-(C)-Cz crystals (Φ = 28%, Fig. 32) display excitation dependent PL spectra comprising one fluorescence band at 402 nm with shoulder at 420 nm and one phosphorescence band at 460 nm with shoulder at 486 nm (HEP). Moreover, an additional phosphorescence band (LEP at 531 and 580 nm and at 500 and 540 nm at RT and 77 K, respectively) was disclosed in delayed spectra.
 | ||
| Fig. 32 Normalized emission spectra of TT-(C)-Cz crystals at 298 K (left) and 77 K (right). PL (continuous line) λexc = 300 nm (black) and λexc = 415 nm (red), delayed spectra (dashed line) λexc = 385 nm (298 K, delay 1 ms, window 5 ms; 90 K, blue: delay 0.5 ms, window 0.5 ms; red: delay 10 ms, window 20 ms). | ||
Supported by DFT/TDDFT calculations, fluorescence and HEP were associated with molecular excited states. They are red shifted with respect to the corresponding ones in TT-(N)-Cz owing to the reduced molecular twisting and the consequently increased electronic communication between TT and Cz. Again, LEP was attributed to a triplet of aggregated origin and its appearance in solutions or blended films was justified by the presence of aggregated forms even at very low concentrations. However, in the present case, less efficacious π–π stacking interactions between TT and Cz moieties result in RTP visible only in delayed experiments and in the absence of LEF.
3.3.5d.3 TT-Ph-Cz . The compound shows at 298 K in dilute DCM solutions four absorption maxima at 236, 293, 310 and 340 nm and a broad, structureless fluorescence band at 370 nm with a shoulder at 350 nm (Φ = 63%). At 77 K, a narrowing of the band (maximum at 353 nm) was observed together with the appearance of a broad phosphorescence band centered at 512 nm. The molecular or aggregate origin of the latter could not be established since, even in dilute solutions, the presence of small aggregates in frozen DCM is possible.68,85 PMMA films (TT-Ph-Cz 0.5 wt%, Fig. 33) in vacuo display at 298 and 77 K a photophysical behaviour strongly resembling that of frozen solution. In particular, one narrow fluorescence band with vibronic replicas at 347 and 360 nm (Φ = 60.2%) and a broad phosphorescence band (490 or 508 nm, at 298 and 90 K, respectively) were observed. Speculating rigidification effects, spectra of dilute glycerol solution were collected. At 298 K, a broad band centered at 380 nm and a narrow peak at 350 nm were visible. Importantly, disappearance of the 380 nm component was observed by increasing the viscosity of the solution at low temperature. These results suggested dual emission from a Franck–Condon (FC) and a relaxed emitting state of molecular TT-Ph-Cz, as confirmed by theoretical calculations. Intriguingly, using solvent/non-solvent (DMSO/water equal to 20/80%) mixtures, RTP (430 nm) nanoaggregates were prepared.
 | ||
| Fig. 33 TT-Ph-Cz. Normalized emission spectra (λexc = 300 nm). Top: PL in DCM (2 × 10−6 M, red lines) at 298 K (left) and 77 K (right) in air. PL of PMMA films (0.5 wt%, at 298 K, left, and 90 K, right) in vacuum (black) and delayed spectra (blue dashed, delay 0.2 ms, window 0.5 ms). Bottom: PL in glycerol (2 × 10−6 M, right) at 248 K (dashed line) and 298 K (continuous line). PL in DMSO (10−6 M) at 298 K (left) with increasing H2O volume. 0% (black continuous), 20% (red continuous), 50% (black dashed), 70% (red dashed-dotted), 80% (black dashed-dotted). Delayed spectrum (blue dashed, delay 0.2 ms, window 0.5 ms) of the 80% water fraction solution. | ||
TT-Ph-Cz crystallizes in three phases: TT-Ph-CzM (monoclinic C2/c space group), TT-Ph-CzT (triclinic P) and TT-Ph-CzO (orthorhombic Pbca), all characterized by the same TT-Cz columnar aggregates. TT-Ph-CzM includes, in its a.u., one MeOH molecule, which is lost at 393 K, resulting in a single-crystal-to-single-crystal transition to TT-Ph-CzT and, at 443 K, to TT-Ph-CzO (Fig. 34). Transformation of TT-Ph-CzM into TT-Ph-CzT was found to be reversibly accomplished through grinding and MeOH exposure.
 | ||
| Fig. 34 Crystal structures of TT-Ph-CzM (top), TT-Ph-CzT (middle, showing the two disordered forms by different color gradation) and TT-Ph-CzO (bottom). In a and c, the shorter distances between triazinic and pyrrolic geometrical centroids (green spheres) and intermolecular contacts shorter than the sum of vdW radii (light grey dashed lines) are reported. Ellipsoids at 30% probability. Reproduced from ref. 81 under the terms of the Creative Commons CC BY license. Copyright 2023, John Wiley and Sons. | ||
The interconversion among the three phases was followed through thermal analysis, photoluminescence investigations and XRPD measurements.
TT-Ph-CzM crystals display multiple emissions (Fig. 35) comprising at 298 K, HEF (375 and 408 nm), LEF overlapped with HEP (425 nm) and LEP (540 nm, overall Φ = 43%) and, at 77 K, only HEF (373 and 388 nm) and LEP (523 nm). The absence of LEF and HEP at 77 K was interpreted with two possible excited state conformations, with the relaxed one being not accessible at low temperature, in agreement with results obtained in solution. In addition, LEP was associated with π–π stacking interactions among TT and Cz units.
 | ||
| Fig. 35 Normalized PL (full lines) and delayed (dashed lines) spectra. Left: TT-Ph-CzM crystals (λexc = 300 nm) at 298 K (blue, blue dashed: delay 0.2 ms, window 0.5 ms) and 77 K (red, red dashed: delay 1 ms, window 5 ms). Middle: TT-Ph-CzO crystals at 298 K (black: λexc = 300 nm; blue: λexc = 370 nm; red: λexc = 360 nm, delay 1 ms, window 5 ms). Right: TT-Ph-CzT crystals at 298 K (black: λexc = 300 nm; blue full: λexc = 375 nm; blue dashed: λexc = 300 nm, delay 0.2 ms, window 0.5 ms. | ||
T-Ph-CzT and TT-Ph-CzO display, as well, multiple emissions but with reduced contribution from LEF/HEP at 298 K. This difference was related to the co-crystallized MeOH in TT-Ph-CzM, forming strong HBs with the TT unit. These, on one hand, provide conformational freedom to the Ph-Cz fragment resulting in LEF and, on the other hand, contribute to suppressing non-radiative deactivation channels highly competitive with HEP.
These studies revealed how synergistic and different combinations of TT and a chromophoric fragment (Cz) can result in quite different emissive behaviours due to both molecular reasons and aggregation modes. Crystals of TT-(N)-Cz (both polymorphs), TT-(C)-Cz and TT-Ph-Cz, with or without co-crystallized MeOH, display one LEP band associated with π–π stacking interactions involving only the TT unit or TT and Cz units. Moreover, TT-(N)-Cz and TT-Ph-Cz are characterized by HEF of molecular origin and LEF of different nature in the two compounds: in TT-(N)-Cz LEF derives from the TT-based π–π aggregated species, while in TT-Ph-Cz it is associated with conformational freedom resulting in rigidochromic and multistimuli responsive behaviour.
 | ||
| Scheme 18 Chemical structures of TT-2Py and TT-4PyF derivatives. | ||
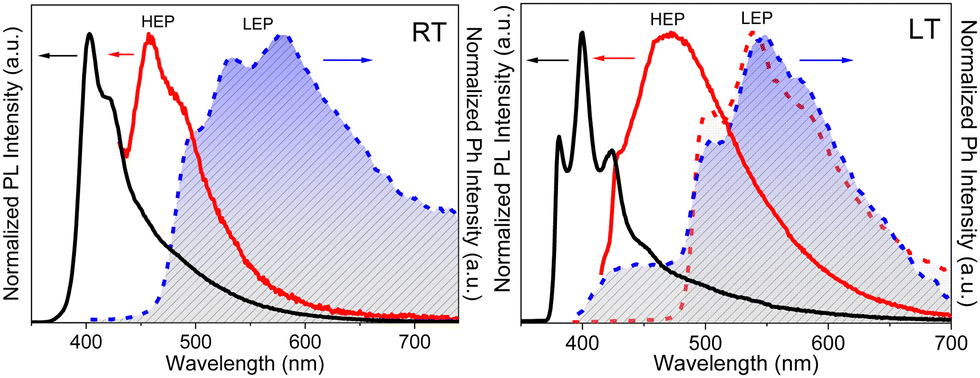
In dilute DCM and ACN solutions at 298 K, TT-2Py displays absorption bands at about 235 and 290 nm and one emission band at 350 nm (Φ ≅ 17%). Detailed investigation of delayed spectra of deareated solutions and solvent–non-solvent mixtures indicated the presence of different long-lived emissions. In particular, HEP and MEP at 345 and 380–420 nm, respectively, and two LEPs in the 490–560 nm interval, were observed.
TT-2Py crystallizes as three different polymorphs (A, Pbcn; H, P21/c and X, P21/c space group) depending on the recrystallization solvent (CH2Cl2/CH3OH, CH3CN/H2O or CH3CN, respectively). All of them display, in their crystal structure, π–π stacking interactions among TT moieties, characterized by slightly different intermolecular distances and slippage features. The TT units are further anchored to each other by several short C–H⋯N HBs in the plane roughly perpendicular to the stacking axis. On the other hand, the pyridinic ring is involved only in weak interactions, resulting in slightly different tilting with respect to TT (41.47, 43.7 and 37.6/39.6° in A, H, and the two independent molecules of X, respectively).
According to an extensive spectroscopic, structural and theoretical investigation, LEP and DRP were attributed to dimeric or columnar π–π interactions among TT units, respectively. Noteworthily, while DRP was observed only for crystals of the polymorph with the strongest π–π interaction, the LEP components are present in all phases, including dilute solutions, where the presence of aggregated species cannot be totally excluded. The remaining emissions, namely HEF, HEP, MEP and LEF, were associated with molecular electronic states: HEF to S1, HEP to a high energy Tn, MEP to T1 and LEF to S1 of a different conformer. In more detail, HEP anti-Kasha emission is due to the difficult IC from Tn to T1 having different characters ((σ/π,π*) and (π,π*), respectively). LEF is associated with the presence, in the TT-2Py ground state (Fig. 36 left), of a local minimum (C) where the pyridinic N atom faces the TT unit rather than on the opposite side, as found in the minimum energy geometry (A). Although conformer C is not observed in any of the TT-2Py polymorphs, its minority presence cannot be excluded in any of the examined phases (solution, blended films and, as defects, in the crystal phase), with the two minima being separated by a low (≅2 kcal mol−1) barrier (B). The two observed fluorescence components are therefore associated with radiative decay from S1 of relaxed conformers A (HEF) and C (LEF), with the latter emission being detectable only by excluding the stronger HEF. The mechanism proposed to explain LEF, requiring population of a triplet Tm of low energy followed by ISC to S1, has been supported by pump–probe experiments.
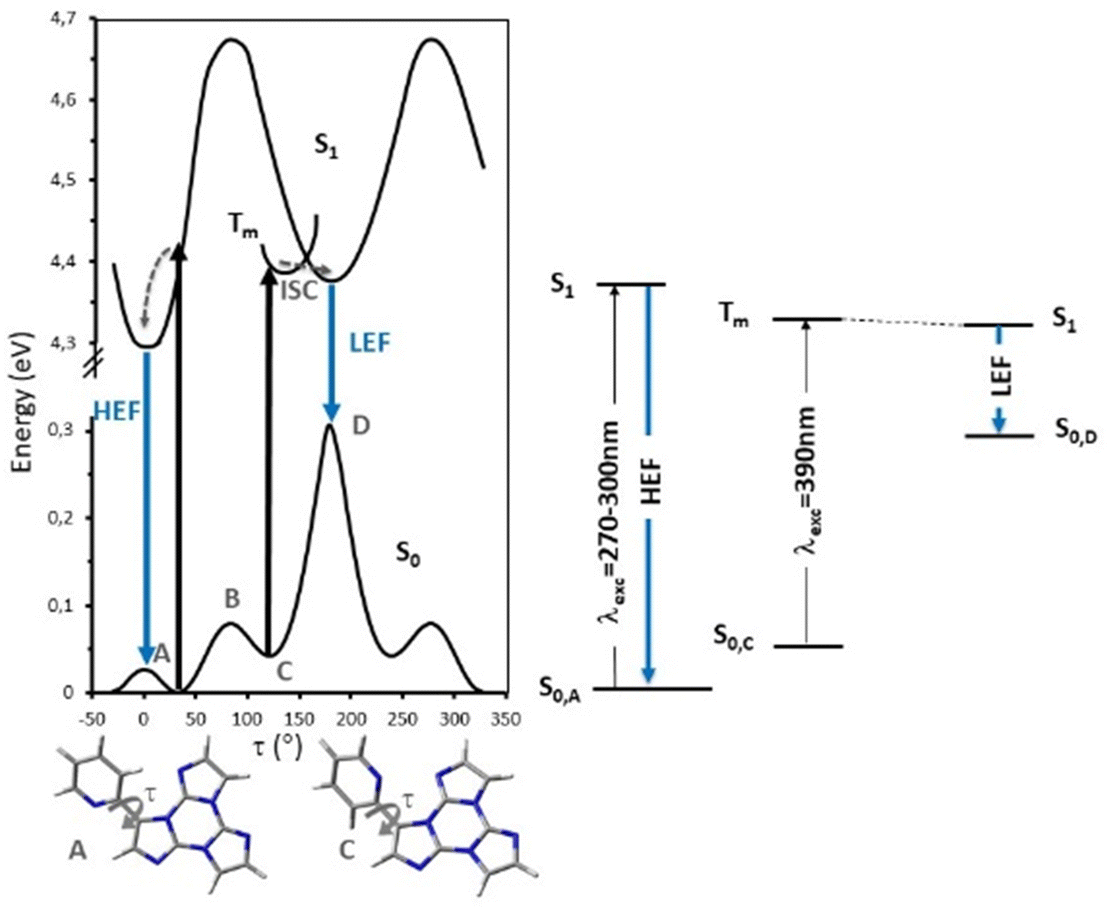 | ||
| Fig. 36 Left: scans of the relaxed potential energy surfaces of S1 and S0 of TT-2Py along the Npy–Cpy–CTT–CTT torsion angle, τ, at the (TD)-ωB97X/6-311++G(d,p) level of theory. Tm represents a generic triplet level, A and C denote minima on S0 and B is the barrier between the two minima. Energies are relative to the S0 state equilibrium geometry. Right: simplified Jablonski diagram of the fluorescence components of TT-2Py. | ||
To deepen the comprehension of the mechanisms involved in TT-2Py photophysical behaviour, blended PMMA films were exposed to acidic vapors to give TT-2PyH+, where a proton is added to the pyridinic nitrogen atom. According to DFT scan calculations, the conformation of the protonated species was predicted to be locked due to the formation of a strong N–H+⋯N intramolecular hydrogen bond, accessible after rotation around the TT-pyridine bond by about 180° with respect to the neutral form. Such blocked structure predicted the suppression of LEF. Moreover, all computed transitions (both singlet and triplet states) were found to be red shifted with respect to the neutral form and to possess (π,π*) character, therefore excluding the possible presence of anti-Kasha HEP. In agreement with this finding, for TT-2PyH+/PMMA blended films, only one fluorescent band (at 412 nm) was observed (Fig. 37), together with a weak phosphorescent band, both at lower energy with respect to the corresponding ones of TT-2Py.
 | ||
| Fig. 37 Photophysical properties of TT-2Py in PMMA (TT-2Py/PMMA 10% wt) before (solid lines) and after (dashed line) HCl exposure. PL emission spectra λexc = 300 nm (black lines), λexc = 350 nm (violet), λexc = 390 nm (blue), and λexc = 450 nm (green). | ||
3-(2-Fluoropyridin-4-yl)triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine, TT-4PyF (Scheme 18), displays in dilute ACN solution at 298 K two absorption maxima at 227 and 291 nm and an intense fluorescence band at 358 nm (Φ = 50%).82 At 77 K, an additional long-lived contribution is present at 454 nm. Similarly, fluorescence (at 348 nm) and green phosphorescence (at 415 and 436 nm) are observed at 298 K in PMMA blended films (w/w 6%).
The compound crystallizes in the P space group with columnar π–π (TT-TT) aggregates. Crystals of TT-4PyF are characterized by fluorescence (at 373 nm) and dual phosphorescence (HEP at 403, 424 and 446 nm, and LEP at 547 nm, Fig. 38) with overall Φ = 25%. Based on structural and computational studies, HEP has been assigned to a molecular triplet while LEP has been ascribed to π–π aggregates. The presence of a single high energy fluorescence in TT-4PyF having only one conformer further supports the hypothesis of a different conformer at the origin of LEF in TT-2Py.
 | ||
| Fig. 38 Crystals of TT-4PyF at 298 K. Upper panel: normalized PL emission (λexc = 300 nm, black solid line; λexc = 360 nm, blue solid line; λexc = 480 nm, red solid line) and excitation (λem = 373 nm, black dashed line; λem = 425 nm, blue dashed line; λem = 570 nm, red dashed line) spectra. Bottom panel: Normalized phosphorescence spectra (λexc = 300 nm; delay 200 μs, window 1 ms, blue dashed line; delay 5 ms, window 20 ms, red solid line). Reproduced from ref. 82 under the terms of the Creative Commons CC BY license. Copyright 2019 by the authors. Licensee MDPI, Basel, Switzerland. | ||
In this regard, an additional interesting feature of TT derivatives is the presence, at the vertexes of a regular triangle, of three nitrogen atoms potentially available for coordination, leading to various metal complexes and coordination polymers especially with filled-shell d10 systems, which lack low lying ligand-field excited states and offer an opportunity to observe other excited states.
In particular, four coordination compounds, namely [Zn3(CH3COO)6(H2O)2](TT)2, [Cd(H2O)6](ClO4)2(TT)2, [Cd(H2O)6](BF4)2(TT)2 and [Zn(H2O)6](BF4)2(TT)2, accommodating TT as a guest in their crystal lattice, were isolated and investigated to clarify the extrinsic heavy metal effect on the chromophore's photophysics.
In crystals of [Zn3(CH3COO)6(H2O)2](TT)2 the TT moieties are organized in π–π stacked columns similar to those found in TT itself (Fig. 39).123
 | ||
| Fig. 39 Motifs of chromophore's aggregation in TT (left); [Zn3(CH3COO)6(H2O)2](TT)2 (centre); [Cd(H2O)6](ClO4)2(TT)2, [Cd(H2O)6](BF4)2(TT)2 and [Zn(H2O)6](BF4)2(TT)2 (right) with distances between centroids equal to 3.450 Å for the former and to 3.423 Å for the latter two. Reproduced with permission from ref. 123. Copyright 2019, John Wiley and Sons. | ||
In agreement with this finding, crystals of [Zn3(CH3COO)6(H2O)2](TT)2 display at 298 K both fluorescence (at about 400 nm) and RTUP (at about 555 nm, Fig. 40); the latter is associated with the presence of the observed stacking interactions. By comparison between the metal complex and TT, the external heavy atom effect in the intensification of the RTUP emission was disclosed.
 | ||
| Fig. 40 Normalized PL spectra. Left: crystals of [Zn3(CH3COO)6(H2O)2](TT)2. Emission (full lines) at λexc = 300 nm (black), λexc = 340 nm (red), λexc = 450 nm (green), and excitation (dashed lines) at λem = 416 nm (red) and λem = 600 nm (green). Right: Emission spectra of [Zn3(CH3COO)6(H2O)2](TT)2 (λexc = 340 nm, red) and TT (λexc = 350 nm, blue). | ||
In the three isostructural compounds, [Cd(H2O)6](ClO4)2(TT)2, [Cd(H2O)6](BF4)2(TT)2 and [Zn(H2O)6](BF4)2(TT)2, TT molecules form stacked dimers instead of columns.
Crystals of [Cd(H2O)6](ClO4)2(TT)2 display at 298 K a broad, featureless emission band comprising one fluorescence band (at 421 nm) and one RTUP band (at about 550 nm), which were associated with the π–π dimers. The fluorescence/RTUP intensity ratio is higher in [Cd(H2O)6](ClO4)2(TT)2 than [Zn3(CH3COO)6(H2O)2](TT)2, indicating that RTUP is more sensitive to the supramolecular organization of the chromophores themselves (columns vs. dimers) rather than to the extrinsic heavy atom effect.
The different heavy atom effect exerted by Cd vs. Zn was highlighted by comparing [Cd(H2O)6](BF4)2(TT)2 and [Zn(H2O)6](BF4)2(TT)2 both showing one fluorescence band (at 383 and 370 nm, respectively) and one RTUP band (at 441 and 469 nm and 394 and 422 nm, respectively). The latter possesses a shorter triplet lifetime in the cadmium complex compared to zinc (τ = 125 ms and 542 ms, respectively), indicating higher SOC in the former.
The observed emission red-shift of [Cd(H2O)6](ClO4)2(TT)2 with respect to the two other isostructural compounds was associated with the different counter ions through DFT and TDDFT calculations on TT·ClO4− and TT·BF4− interacting units. In fact, while HOMOs of TT·BF4− are essentially localized on TT, those of TT·ClO4− are delocalized on both interacting units, suggesting in the latter case charge transfer character for the S0–S1 transition. The CT character of emissive levels of [Cd(H2O)6](ClO4)2(TT)2 was therefore deemed responsible for the observed red shift of its emissions.
Zn(II) and Cd(II) coordination compounds with TT have also been obtained, in particular the cationic [Zn(TT)(NO3)(H2O)3](NO3) and the neutral [Cd(TT)2(NO3)2(H2O)2] complexes where TT ligands form infinite stacking columns with TT-TT distances much shorter in the former (the distance between triazine geometric centroids equal to 3.85 Å vs. 4.88–4.94 Å).124
Crystals of the zinc complex display an intense fluorescence band (at 439 nm) with a long-lived tail (at about 550 nm) (overall Φ = 1%), which was assigned, due to crystallographic considerations, to π–π aggregates of the ligand. Comparison of the photophysical parameters (quantum yields and lifetimes) of [Zn(TT)(NO3)(H2O)3](NO3) with those of TT itself and the [Zn3(CH3COO)6(H2O)2](TT)2 cocrystal, having a similar organization of TT inside the structure, allowed us to conclude that the presence of Zn(II) facilitates depopulation of S1 through ISC to a triplet level with kr < knr, with the intrinsic (coordination) effect being more pronounced than the extrinsic (cocrystal) one.
Crystals of [Cd(TT)2(NO3)2(H2O)2] at 298 K show one fluorescence band (at 430 nm) and one phosphorescence band (at 505 nm), which, again based on crystallographic considerations, were identified as ligand-centered emissions.
The versatility of TT as a coordinating ligand is also manifested in the isolation of several Cu(I) and Ag(I) coordination polymers (CPs), ranging from 1D chains to 3D coordination networks, in which TT is either a monodentate or bi- and tri-dentate bridging ligand.125,126 Among these, two Cu and three Ag CPs were obtained with the required high purity grade for reliable photophysical measurements. In particular, 1D double-stranded stair [MI(TT)]n and 3D [MCl(TT)]n (M = Cu, Ag) isostructural polymers and a 3D Ag(I) derivative, namely [Ag3(TT)4]n(NO3)3n·6nH2O, were isolated and characterized. [MI(TT)]n crystallized in the monoclinic P21/c space group forming double-stranded stairs of [MI]n composition, decorated on both sides with monodentate TT ligands. The TT ligands on both sides of the chains are all parallel and superimposed allowing π–π stacking interactions.
Crystals of [MI(TT)]n display an excitation dependent emissive behaviour. For crystals of [AgI(TT)]n at 298 K one fluorescence band at 385 nm and three phosphorescence bands (HEP at 411 and 445 nm, MEP at 446, 476 and 509 nm, and LEP at 494, 530, 575, 620 and 680 nm) were visible (overall Φ equal to 19%). For [CuI(TT)]n at an appropriate excitation wavelength three phosphorescence bands were observed (Φ = 18%). Two of them (MEP at 431, 460 and 487 nm and LEP at 536, 582 and 623 nm) fall in the same spectral range as that of [AgI(TT)]n, while the third one (at 568 nm), activated only by exciting at high energy (λexc < 390 nm), is specific for [CuI(TT)]n and assigned to a 3XMLCT state. Based on structural considerations, LEP was associated with π–π stacking interactions between TT units and MEP was associated with I⋯C intermolecular electronic coupling, also in view of its similarity, in both position and shape, with that observed in 1I and TT·DITFB.71 The structures of these four compounds are in fact characterized by comparable I⋯C contacts between adjacent stairs (in [MI(TT)]n), helices (in 1I) and chains (in TT·DITFB). The similarity of LEP and MEP in the two isostructural [MI(TT)]n polymers was a clear indication that the metal plays a secondary role in these emissions and can be therefore referred to as an “external” perturber. Ag(I) HEP was interpreted as due to deactivation of a ligand centered triplet state as supported by the energy spacing (about 1300–1550 cm−1) of its vibrational components, which can be associated with a vibronic progression involving imidazole ring modes. The absence of fluorescence and HEP in [CuI(TT)]n was justified, through theoretical studies, with the greater metal contribution to the intramolecular emissive behaviour of copper resulting in the suppression of fluorescence through easy ISC to close triplets and activation of the XMLCT phosphorescence.127
3D coordination polymers [MCl(TT)]n crystallize in the cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . Unlike [MI(TT)]n polymorphs, the TT ligand uses the nitrogen atoms of all of its three imidazole moieties to coordinate the metal atoms. Moreover, the ligands are also involved in π–π stacking interactions. At 298 K, crystals of [AgCl(TT)]n display an excitation-dependent emission spectrum comprising one fluorescence band (at 448 nm) and two superimposed phosphorescence bands (in the 530–615 nm range), one of molecular origin and the other assigned to TT-TT π–π dimers. For [CuCl(TT)]n only two long-lived emissions (at 515 and 560 nm) were observed, with the lowest energy one assigned as well to π–π dimers and the other to a molecular triplet. Again, the presence of fluorescence for the Ag(I) polymorph was interpreted as due to a reduced metal contribution of Ag(I) with respect to Cu(I) in the emitting states.127
. Unlike [MI(TT)]n polymorphs, the TT ligand uses the nitrogen atoms of all of its three imidazole moieties to coordinate the metal atoms. Moreover, the ligands are also involved in π–π stacking interactions. At 298 K, crystals of [AgCl(TT)]n display an excitation-dependent emission spectrum comprising one fluorescence band (at 448 nm) and two superimposed phosphorescence bands (in the 530–615 nm range), one of molecular origin and the other assigned to TT-TT π–π dimers. For [CuCl(TT)]n only two long-lived emissions (at 515 and 560 nm) were observed, with the lowest energy one assigned as well to π–π dimers and the other to a molecular triplet. Again, the presence of fluorescence for the Ag(I) polymorph was interpreted as due to a reduced metal contribution of Ag(I) with respect to Cu(I) in the emitting states.127
The 3D coordination polymer [Ag3(TT)4]n(NO3)3n·6nH2O crystallizes in the cubic space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d with each silver atom coordinated to four TT molecules in a distorted tetrahedral environment and no stacking interactions between TT molecules. Its crystals display at 298 K (Φ equal to 7.5%) excitation-dependent emissions comprising a broad fluorescence band (at 404 nm) and two phosphorescence bands (at 454 and 556 nm), which were related, according to DFT/TDDFT calculations, to triplet states of different nature indicating anti-Kasha behaviour.
3d with each silver atom coordinated to four TT molecules in a distorted tetrahedral environment and no stacking interactions between TT molecules. Its crystals display at 298 K (Φ equal to 7.5%) excitation-dependent emissions comprising a broad fluorescence band (at 404 nm) and two phosphorescence bands (at 454 and 556 nm), which were related, according to DFT/TDDFT calculations, to triplet states of different nature indicating anti-Kasha behaviour.
space group where the deprotonated TT-COO− residue coordinates in an N,O-bidentate chelate coordination mode forming a rare-met seven-membered metallochelate ring giving an extended H-bonded layer. Adjacent layers stack in a zip-like mode with partial overlap of coordinated luminophores.
Crystals of zinc and cadmium complexes, the only emissive in this series (Φ = 9 and 1.4%, respectively), display a photophysical behaviour quite similar to that of free TT-COOH. This suggests an external role of the metal (whose evidence is limited to a lower quantum yield in metal containing derivatives) as supported by DFT-TDDFT calculations. In fact, similarly to TT-COOH, two molecular fluorescence components (HEF at 338 and 352 nm for Zn and Cd, respectively; LEF at 382, 405 and 430 for both compounds) and phosphorescence components (HEP at 452 nm, MEP at 490 and 523 nm, respectively) with anti-Kasha behaviour were observed for the two complexes (Fig. 41). Moreover, an additional LEP (at 556 and 577 nm, respectively) of π–π stacked origin was present in the PL spectrum of the compounds.
 | ||
| Fig. 41 Normalized PL emission spectra at RT. Left: crystals of [Zn(TT-COO)2(H2O)2]λexc = 280 nm (black); 340 nm (blue); 390 nm (green); 435 nm (red) and 493 nm (grey). Right: crystals of [Cd(TT-COO)2(H2O)2]λexc = 300 nm (black); 340 nm (blue); 390 nm (green); 454 nm (red) and 523 nm (grey). | ||
A series of Cu(II) coordination compounds with pyridine-substituted derivatives, namely 3-(pyridin-2-yl)triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine (TT-2Py) and 3-(pyridin-4-yl)triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine (TT-4Py), were reported.128 In particular, a mononuclear complex [Cu(NO3)2(H2O)2(TT-4Py)2]·2H2O, a binuclear one [Cu2(CH3COO)4(TT-2Py)2], and a 1D coordination polymer [Cu(NO3)2(TT-2Py)]n, where luminophores coordinate to the metal ion as either monodentate terminal ligands or bidentate-chelate ones, were obtained. In spite of the rich variety of structural patterns, accommodating both hydrogen bonds and π⋯π stacking interactions with different overlapping areas of the stacking ligands, the compounds are scarcely emissive in agreement with the paramagnetic character of the Cu(II) ion.
The ability of TT-2Py to act as a chelating ligand giving a seven-membered metallacycle through the nitrogen atom of the pyridinic ring and the other on the imidazolic unit, as observed in [Cu(NO3)2(TT-2Py)]n, was exploited for the preparation of other coordination compounds. In particular, TT-2Py was reacted with Re(I) and Cu(I) further evidencing its versatility in adopting different coordination modes.
With Re(I), a mononuclear ([ReCl(CO)3(TT-2Py)]) and a hexanuclear species ([{Re(μ-Cl)(CO)3(μ-TT-2Py)}{Re2(μ-Cl)2(CO)6}]2) were isolated.129 In the mononuclear complex the rhenium atom attains an octahedral geometry and bears three terminal carbonyl ligands in a facial arrangement, one terminal chloro ligand and two nitrogen atoms of the chelating TT-2Py ligand (Fig. 42), namely the nitrogen atom of the pyridyl substituent and the nitrogen atom of the triimidazotriazine moiety, similarly to what is observed in [Cu(NO3)2(TT-2Py)]n. The hexanuclear species contains two [ReCl(CO)3(TT-2Py)] moieties with geometry quite similar to the one of the mononuclear complexes, which bridge two additional [Re2(μ-Cl)2(CO)6] fragments through the chloro ligand and a nitrogen atom of the triimidazotriazine moiety (Fig. 43).
 | ||
| Fig. 42 View of the rhenium complex [ReCl(CO)3(TT-2Py)] as found in its crystal structure, with a partial labelling scheme. Ellipsoids are drawn at the 50% probability level. Reproduced from ref. 129 under the terms of the Creative Commons license and with permission from the Royal Society of Chemistry. Copyright 2023. | ||
 | ||
| Fig. 43 View of the rhenium complex [{Re(μ-Cl)(CO)3(μ-TT-2Py)}{Re2(μ-Cl)2(CO)6}]2 as found in the crystal structure of its n-hexane solvate, with a partial labelling scheme. Ellipsoids are drawn at the 50% probability level. Reproduced from ref. 129 under the terms of the Creative Commons license and with permission from the Royal Society of Chemistry. Copyright 2023. | ||
The absorption and emission spectra of the two complexes were recorded in dilute DCM and toluene solutions, respectively. The mononuclear derivative shows two broad absorption bands at 247 and 301 nm both of which, based on DFT/TDDFT calculations, are assigned to MLCT + XLCT states. Moreover, for the lower energy transition, an additional ILCT contribution was predicted from calculations. The hexanuclear species displays absorption bands at 234, 264 and 320 nm, with a shoulder at 346 nm with not clearly established origin.
Upon excitation at 365 nm, the two compounds display a broad unstructured phosphorescence band (at 583 and 597 nm, Φ = 0.4 and 0.01%, for the mono- and the hexanuclear species, respectively). In the solid state, both derivatives maintain the structureless profile with a concomitant important hypsochromic shift of the phosphorescence maxima (532 and 563 nm, respectively) and increase in the photoluminescence quantum yield (12 and 2.5%, respectively). Such AIE features were attributed to restriction of the vibrational motions.
By mixing copper iodide with TT-2Py in ACN at 298 K in a 2:1 molar ratio, an orange emissive solid, namely [Cu2I2(TT-2Py)]n, was obtained.130 This Cu(I) CP, crystallizing in the P21/n space group, is characterized by a quite rare crystal structural motif, comprising a double-stranded (CuI)2 stair and a single CuI zig-zag chain (Fig. 44), which, among copper(I) halide based compounds, was reported in the literature only once previously (CSD Refcode GAJBEB131).
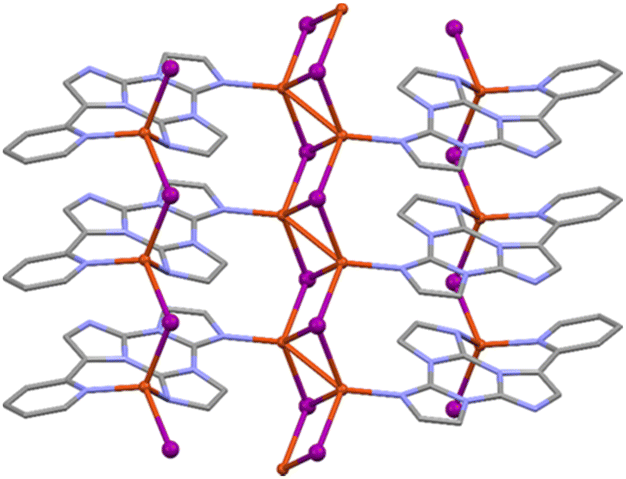 | ||
| Fig. 44 View of a single monodimensional chain of [Cu2I2(TT-2Py)]n showing the central double stranded stair and the two lateral single zig-zag structural motifs bridged by the ligand. Reproduced from ref. 130 under the terms of the Creative Commons CC BY license. Copyright 2023 by the authors. Licensee MDPI, Basel, Switzerland. | ||
Powders of [Cu2I2(TT-2Py)]n display at 298 K an orange broad excitation independent emission of XMLCT character centered at 588 nm (quantum yield, Φ, equal to 7%; τ = 1.33 μs) shifted to 580 nm (τ = 17.89 μs) at 77 K (Fig. 45). Based on the one order of magnitude increase of lifetime upon cooling (from 298 to 77 K) and on the presence of a triplet almost isoenergetic with S1, a TADF mechanism at the origin of the single emission of [Cu2I2(TT-2Py)]n was not fully excluded.
 | ||
| Fig. 45 Normalized excitation (dashed) and PL emission (continuous) spectra of crystalline powders of [Cu2I2(TT-2Py)]n at 298 K (black, λem = 588 nm; λexc = 300 nm) and 77 K (red, λem = 580 nm; λexc = 300 nm). Reproduced from ref. 130 under the terms of the Creative Commons CC BY license. Copyright 2023 by the authors. Licensee MDPI, Basel, Switzerland. | ||
To further investigate the coordination ability of the TT ligands, the tri-pyridinic substituted derivative, namely 3,7,11-tri(pyridin-4-yl)triimidazo[1,2-a:1′,2′-c:1′′,2′′-e][1,3,5]triazine, hereafter TT-4Py3, having three pyridyl groups with nitrogen atoms in the para-position with respect to TT and oriented with a trigonal symmetry, was synthesized and reacted with Ag(I), Cd(II) and Cu(I).132 Three layered CPs, [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2, [Cd{TT-4Py3}(NO3)2(EtOH)]n and [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) were obtained and characterized by SCXRD, XRPD, IR and photoluminescence spectroscopies and their emissive properties were interpreted with the aid of DFT/TDDFT calculations.
[Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 and [Cd{TT-4Py3}(NO3)2(EtOH)]n crystallize, respectively, in P21/c and P space groups to give very similar 2D coordination motifs, where the ligand acts as a trigonal donor through the pyridyl nitrogen atoms bridging three different metal ions, so that each metal is coordinated to three different ligands (Fig. 46). [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) crystallizes in the P21/c space group without significant inter-layer interactions between aromatic fragments except for a short distance (3.735 Å) between the triazinic centroids of two TT-4Py3 ligands belonging to two parallel neighboring layers (Fig. 46), according to quite strong I⋯π halogen bonds.133–138 Powders of TT-4Py3 and crystals of [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 and [Cd{TT-4Py3}(NO3)2(EtOH)]n display excitation dependent emissive behaviour while those of [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) are characterized by a single, broad, excitation independent emission. In more detail, TT-4Py3 shows at 298 K and an appropriate excitation wavelength, one fluorescence band (at 365 nm) and two phosphorescence bands (at 458 and 549 nm). Similarly, crystals of [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 show at 298 K one fluorescence band (at 390 nm) and two phosphorescence bands (at 460 and 530 nm) (overall Φ ≅ 1%, Fig. 47). At 77 K the three emissions are still visible in slightly different positions (at 395 nm; 450 nm; 510, 548, 592 and 655 nm, respectively) but with different relative intensities with respect to that observed at room temperature suggesting different deactivation paths at the two temperatures.
 | ||
| Fig. 46 Crystal structures of [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2, [Cd{TT-4Py3}(NO3)2(EtOH)]n and [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH): views of a single molecular layer down the [1, 1, −1], [1, 0, −2] and [1, 0, 1] directions for [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 (top), [Cd{TT-4Py3}(NO3)2(EtOH)]n (middle) and [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) (bottom). Adapted with permission from ref. 132. Copyright 2024, John Wiley and Sons. | ||
 | ||
| Fig. 47 Normalized PL emission spectra of [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 crystals at 298 K (full lines) and 77 K (dashed lines); λexc: 300 nm (black lines); 358 nm (red full line); 400 nm (red dashed line) and 460 nm (blue line). Adapted with permission from ref. 132. Copyright 2024, John Wiley and Sons. | ||
Crystals of [Cd{TT-4Py3}(NO3)2(EtOH)]n display at 298 K and an appropriate excitation wavelength (Fig. 48), one fluorescence band (at about 405 nm) and two phosphorescence bands (at about 455 and 550 nm, overall Φ ≅ 15%), slightly shifted and better vibronically resolved at 77 K (at 346, 365, 381, 403 and 421 nm; 450 nm; 498 and 536 nm, respectively).
 | ||
| Fig. 48 Normalized PL emission spectra of [Cd{TT-4Py3}(NO3)2(EtOH)]n crystals at 298 K (full lines) and 77 K (dashed lines); λexc: 300 nm (black lines); 370 nm (red full line); 380 nm (red dashed line) and 450 nm (blue lines). Adapted with permission from ref. 132. Copyright 2024, John Wiley and Sons. | ||
Interestingly, in both [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 and [Cd{TT-4Py3}(NO3)2(EtOH)]n, the presence of the metal results in a red shift of the fluorescence with respect to that observed for the free ligand, while it does not affect the phosphorescence emission maxima. The latter were assigned to molecular (HEP) and supramolecular (LEP) contributions (π–π stacking arrangement of the ligands).
DFT/TDDFT calculations on both the isolated ligand and discrete models of the two compounds, comprising the metal ion and its full coordinating sphere, revealed that the set of singlet and triplet excited states display an alternation of pure (π,π*) and mixed (σ/π,π*) levels, with the σ contribution mainly located on the pyridine moieties. While for TT-4Py3 the lower energy singlet states have mixed 1(σ/π,π*) (S1) or pure 1(σ,π*) character (S2 ÷ S4), and T1 is a 3(π,π*) state, coordination to the Ag(I) and Cd(II) ions results in stabilization of the (π,π*) singlets, which are located at lower energy with respect to the almost unvaried 1(σ/π,π*) and 1(σ,π*) ones. The low energy triplets, instead, remain almost unperturbed. This explains the observed red shift of the fluorescence emission, differently from HEP which remains energetically unaffected by complexation. Moreover, inspection of the orbitals mainly involved in the transitions revealed that in both compounds LUMOs and HOMOs are essentially ligand-centered. Only for [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2, however, a partial, non-negligible delocalization on the metal was calculated, resulting in a modest MLCT contribution. The absence of a metal contribution in the transitions of [Cd{TT-4Py3}(NO3)2(EtOH)]n explained its much higher quantum yield compared to that of [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2, due to the reduction, in the former, of the ‘detrimental’ (from this point of view) path towards the phosphorescence emission.
The single broad phosphorescence (at about 650 and 606 nm at RT and 77 K, respectively) observed for crystals of [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) was associated with multiple contributions mainly related to the ligand itself on the basis of its long lifetime (τ = 6.36 and 34.47 ms at 298 and 77 K, respectively, Fig. 49) and DFT/TDDFT calculations, providing low energy triplet states with prevalent ILCT character.
 | ||
| Fig. 49 Normalized PL emission (λexc: 360 nm) and excitation spectra of [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) crystals at 298 K (full line) and 77 K (dashed lines); (λem: 645 nm full line, 602 nm dashed line). Reproduced with permission from ref. 132. Copyright 2024, John Wiley and Sons. | ||
Reaction of TT-2Py, TT-4Py and TT-4Py3 with europium nitrate afforded two salts, namely (TT-2PyH)[Eu(NO3)4(H2O)2] and (TT-4PyHTT-4Py)2[Eu(NO3)5(H2O)]·(CH3CN)2, where the protonated ligands are accommodated as guests in the crystal lattices, and a neutral complex, [Eu(NO3)3(H2O)2(TT-4Py3)].139
(TT-2PyH)[Eu(NO3)4(H2O)2] shows a significant planarization of the (TT-2PyH)+ cation provided by intramolecular NH⋯N HBs resulting in supramolecular chains with an alternation of organic and inorganic motifs within the HB layers (Fig. 50 left). The twisted protonated TT-4Py ligands in (TT-4PyHTT-4Py)2[Eu(NO3)5(H2O)]·(CH3CN)2 form packed HB layers with channels accommodating the bulky inorganic anions (Fig. 50 right).
 | ||
| Fig. 50 Left: crystal structure of (TT-2PyH)[Eu(NO3)4(H2O)2]: fragment of the H-bonded layer. Right: crystal structure of (TT-4PyHTT-4Py)2[Eu(NO3)5(H2O)]·(CH3CN)2: fragment of crystal packing with indication of stabilising short contacts. Adapted with permission from ref. 139. Copyright 2024, American Chemical Society. | ||
Differently, in [Eu(NO3)3(H2O)2(TT-4Py3)], a direct coordination between one of the N-pyridine binding sites of TT-4Py3 and the Eu(III) metal centre is present (Fig. 51).
 | ||
| Fig. 51 Crystal structure of [Eu(NO3)3(H2O)2(TT-4Py3)]. Left: The molecule of [Eu(NO3)3(H2O)2(TT-4Py3)] with the partial numbering scheme. The thermal ellipsoids are shown at the 50% probability level. Right: fragment of the H-bonded layer. Reprinted with permission from ref. 139. Copyright 2024, American Chemical Society. | ||
Photoluminescence investigation of (TT-4PyHTT-4Py)2[Eu(NO3)5(H2O)]·(CH3CN)2 and [Eu(NO3)3(H2O)2(TT-4Py3)] revealed a broad unstructured triazinic ligand-based emission with very small Eu(III) contribution for the former and an efficient ligand–metal energy transfer for the latter, as manifested by its bright red emission comprising only the characteristic emissive features of Eu(III).
3.4 Applications of TTs
In this scenario, two emissive pyrene derivatives of TT, namely TTPyr and (CHO)2TTPyr (Scheme 19), were proposed as single-molecule fluorescent probes for the detection of various nitroaromatic energetic hazardous materials.88 The two sensors were investigated by means of fluorescence-quenching titrations in solution through the incremental addition of analytes, comprising a large number of highly energetic molecules and explosives (Scheme 20). A strong attenuation of the emission intensity was observed for both compounds in the presence of nitroaromatics (phenols and toluenes) while negligible effects were recorded with aliphatic or inorganic energetic materials.
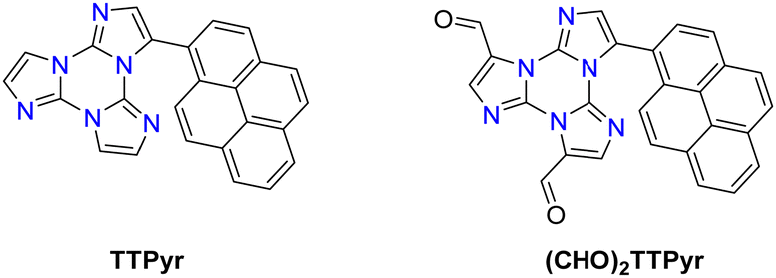 | ||
| Scheme 19 Chemical structures of TTPyr and (CHO)2TTPyr. | ||
 | ||
| Scheme 20 List of explosives tested with TTPyr and (CHO)2TTPyr probes. | ||
Fluorescence quenching titrations were analysed through the Stern–Volmer (SV) equation: I0/I = 1 + KSV[Q], where I0 and I are the emission intensities before and after addition of the quencher, respectively, [Q] is the quencher concentration and KSV is the SV constant. For those analytes absorbing in the blue region (e.g. nitrophenols) and thus overlapping and affecting the 420 nm TTPyr emission according to the secondary inner filter effect, the red-shifted fluorescence of (CHO)2TTPyr at 565 nm represented a valid alternative. Evaluation of the interaction strength between probes and analytes, by means of KSV and limit of detection (LOD) calculations, provided values that are quite comparable for both TT probes (Table 1), according to the following order: aliphatic and inorganic explosives < nitrotoluenes < nitrophenols, with the strongest response in the case of picric acid (PA).
| Explosive | TTPyr | (CHO)2TTPyr | ||
|---|---|---|---|---|
| K SV (M−1) | LOD (μM) | K SV (M−1) | LOD (μM) | |
| RDX | 2.30 × 10+2 | 113 | 1.66 × 10+2 | 164 |
| PETN | 1.74 × 10+2 | 114 | −1.15 × 10+1 | — |
| NG (dynamite) | 2.00 × 10+2 | 126 | 2.17 × 10+1 | 880 |
| AN | 1.89 × 10+2 | 120 | 1.54 × 10+2 | 146 |
| 2NT | 1.71 × 10+3 | 18.15 | 4.70 × 10+2 | 41.62 |
| 3NT | 2.57 × 10+3 | 17 | 5.34 × 10+2 | 30.5 |
| 4NT | 9.75 × 10+2 | 28.41 | 5.79 × 10+2 | 29.57 |
| DNT | 1.31 × 10+3 | 18.46 | 1.06 × 10+3 | 31.76 |
| TNT | 2.17 × 10+3 | 8.2 | 1.45 × 10+3 | 10.27 |
| 2NP | 3.48 × 10+3 | 12.56 | 4.86 × 10+3 | 3.66 |
| 3NP | 1.81 × 10+3 | 24.13 | 4.90 × 10+3 | 7.58 |
| 4NP | 8.99 × 10+3 | 4.87 | 1.12 × 10+4 | 2.53 |
| PA | 1.46 × 10+4 | 2.85 | 1.25 × 10+4 | 2.76 |
Moreover, time-resolved measurements in the absence and in the presence of increasing concentration of PA or TNT were performed on TTPyr with the aim to discriminate between static or dynamic quenching. The results showed constant excited state lifetimes even at very high quencher concentration, suggesting a static quenching with formation of a non-emissive dark-complex between the probe and the analyte in the ground state. Such evidence was further supported by the isolation and characterization by SCXRD of a TTPyr/PA adduct with 2:1 stoichiometry.
In the panorama of highly energetic molecules, TT was used as a stabilizer of all-carbon-nitrated azoles, usually suffering from drawbacks such as poor thermal stability, frictional and impact sensitivities, deliquescence and hydrolysis.142,143 By exploiting the donor properties of the electron-rich TT scaffold, three cocrystals with electron-poor acceptors, namely 5-nitrotetrazole (HNT), 3,5-dinitro-1,2,4 triazole (HDNT) and 3,4,5-trinitropyrazole (TNP), were prepared (Fig. 52).144 All cocrystals exhibited better performances, in terms of thermal stability and mechanical sensitivity, with respect to the pure azole components. By means of SCXRD and theoretical calculations it was suggested that the combination of strong hydrogen bonds (N–H⋯N, C–H⋯N, and C–H⋯O) and weak NO2⋯π and π-stacking interactions encountered in the highly energetic adducts could be responsible for their significantly improved performances.
 | ||
| Fig. 52 Crystal structures of: (left) 1:1 TT/HNT cocrystals, (middle) 2:1 TT/HDNT cocrystals and (right) 1:1 TT/TNP cocrystals. Displacement ellipsoids are shown at the 50% probability level. Adapted with permission from ref. 144. Copyright 2022, American Chemical Society. | ||
To this scope, TTPyr was selected as the test compound due to its luminescence properties such as long-lived emission, tunable emission range and high quantum yields both in solution and as a solid phase.80 Evaluation of TTPyr emissive behaviour in DMSO by adding increasing amounts of a non-solvent (water) showed a gradual increase and a concomitant blue shift of the emission from 420 to 400 nm by increasing the water content up to 70% in volume (Fig. 53 left). Further water addition resulted in an attenuation of this emission with appearance of an additional low energy fluorescence component (associated with aggregated species) at 480 nm, the wavelength suitable for bacteria and cell imaging studies. Staining experiments with eukaryotic (HeLa and HLF) and prokaryotic (S. aureus and E. coli) cells revealed, through confocal laser scanning microscopy, that TTPyr can enter both type of cells at low concentration in an appropriate time window, locating at cytoplasm level (Fig. 53 right). In addition, MTT assays pointed out the good biocompatibility properties of TTPyr, characterized by distinct cytotoxicity towards cancer cells rather than normal cells. These findings provide useful insights and indications to further expand the TT family aiming at the development of probes for the intracellular environment sensing to be potentially applied as luminescent drugs for anticancer and phototheranostic studies.
 | ||
| Fig. 53 Left: PL emission spectra of TTPyr (10−5 M) in DMSO/H2O mixtures with different water fractions (fwater) at RT (λexc= 340 nm). Right: HeLa cell co-staining experiment (TTPyr 10 μM). Adapted with permission from ref. 80. Copyright 2021, John Wiley and Sons. | ||
G-Quadruplexes (G4) are non-canonical secondary structures of DNA or RNA nucleic acids that self-folds in regions rich in guanine bases organized in planar layers of four guanines (G-quartets), which are repeated inside the biopolymer sequence.149,150 Intriguingly, it has been reported that putative G4-forming sequences are present in genomic regions and they are involved in several pathologies, such as cancer or viral infections, where they can promote or rule the genetic information transfer. Additionally, they are found to have a role in DNA repair and maintenance,151,152 and hence there is interest in targeting G4s as an appealing and novel promising antitumor strategy.150,153,154 There is rich literature on G4 ligands, which are characterized by some common structural features such as the presence of large planar aromatic systems, able to stack with the planar G-quartets, and the presence of polar or positively charged tails, which interact with the negatively charged phosphate DNA backbone and the grooves and loops of G4s.155–157 Based on these observations, the flat aromatic TT was appropriately functionalized by the introduction of polar and hydrophilic groups. In particular, TT-2Pyn and TT-4Pyn series were methylated on the pyridinic nitrogen, resulting in water soluble salts with the positive charge at different distances from the central core (Scheme 21).78
 | ||
| Scheme 21 Preparation of TT-(2PyMeI)n and TT-(4PyMeI)n. | ||
In parallel, a family of polar guanyl hydrazones TT-Gnx (x = 1–3) were synthesized through imination of the corresponding formyl-TT-(CHO)x with aminoguanidine hydrochloride (Scheme 22).158,159
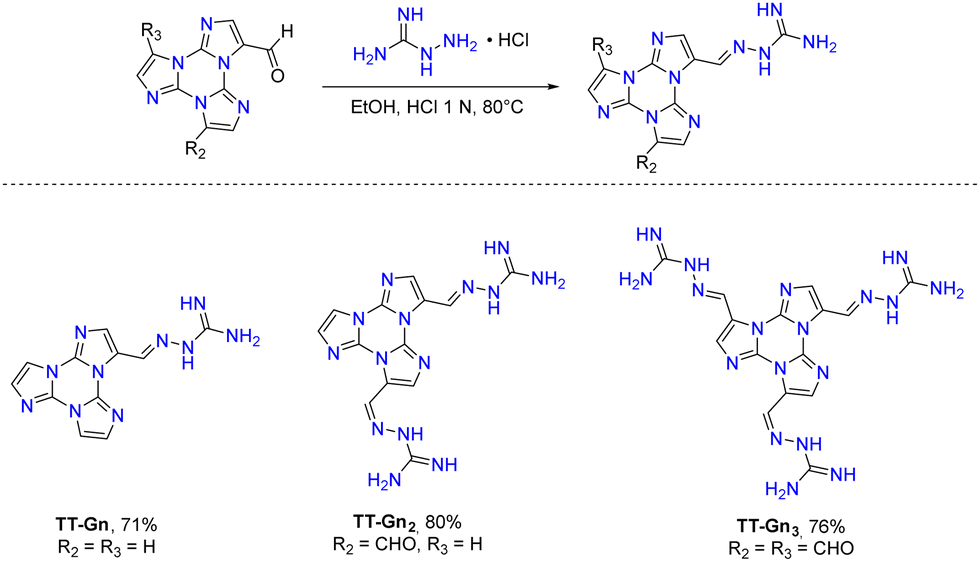 | ||
| Scheme 22 Preparation of TT-Gnx (x = 1–3). | ||
The methyl pyridinium and guanyl hydrazone TTs were tested as G4 stabilizers by using sequences from the oncogene promoter regions of BCL-2, c-KIT, c-MYC (BCL2 G4, c-KIT2 G4 and c-MYC G4, respectively) and a 26-mer truncation of the human telomeric DNA sequence (Tel26 G4). A biophysical in vitro screening of the ligands was carried out by circular dichroism (CD) analysis and CD-melting experiments, to evaluate the ability of the synthesized compounds to interact and stabilize the G4 targets. The results revealed a noteworthy G4-stabilizing effect for most of the tested compounds even at low concentrations, with stabilization increasing with the number of substituents on the TT core (Table 2). Moreover, a comparison of ligand-G4s affinity for the TT-(2PyMeI)n and TT-(4PyMeI)n series revealed better interaction for the para-substituted one, highlighting the importance of the positive charge distance from the central scaffold.
| Compounds | ΔTm (°C) | |||
|---|---|---|---|---|
| BCL2 | c-KIT2 | c-MYC | Tel26 | |
| TT-(2PyMeI) | −1.6 | 1.9 | 0.8 | 0.9 |
| TT-(2PyMeI)2 | 0.2 | 1.0 | 4.7 | 2.1 |
| TT-(2PyMeI)3 | 6.4 | 4.3 | 8.1 | 5.0 |
| TT-(4PyMeI) | 3.2 | 7.0 | 2.1 | 6.6 |
| TT-(4PyMeI)2 | 13.7 | 15.3 | 3.8 | 16.6 |
| TT-(4PyMeI)3 | >30 | 22.0 | 2.1 | 22.0 |
| TT-Gn | 7.2 | 11.0 | 6.2 | 2.3 |
| TT-Gn2 | >30 | 27.8 | 10.8 | 8.4 |
| TT-Gn3 | >30 | 27.7 | 16.9 | 11.7 |
| Compound | 298 K | 77 K | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Φ (%) | Origin | λ em (nm) | τ av | Origin | λ em (nm) | τ av | ||||
Tσ: triplet of (π,σ*) symmetry, TBr: triplet located on the Br halogen bond; Tn: high energy triplet; Sn: high energy singlet; TI: triplet associated with the extrinsic I effect, 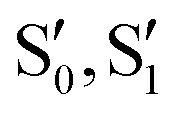 : S0, S1 from a second conformer; SH: singlet from π–π aggregates; TH: triplet from π–π aggregates; TH′: triplet from a π–π dimer. : S0, S1 from a second conformer; SH: singlet from π–π aggregates; TH: triplet from π–π aggregates; TH′: triplet from a π–π dimer. |
||||||||||
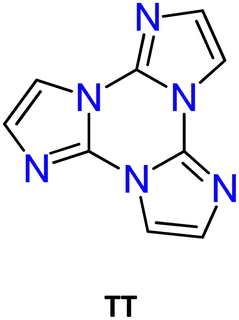
|
DCM | 2 | S1–S0 | 400 | 7.09 ns | S1–S0 | 435 | 14.31 ns | 106 | |
| TH–S0 | 500 | 0.927 s | ||||||||
| pwd | 18 | S1–S0 | 425 | 9.89 ns | S1–S0 | 430 | 12.32 ns | |||
| TH–S0 | 520 | 0.555 s | TH–S0 | 520 | 0.963 s | |||||
| cryst | 30 | S1–S0 | 400 | 7.67 ns | S1–S0 | 403 | 8.96 ns | |||
| TH–S0 | 525 | 0.970 s | TH–S0 | 510 | 1.075 s | |||||

|
cryst | 13 | S1–S0 | 415 | 2.98 ns | S1–S0 | 420 | 14.41 ns | 106 | |

|
cryst | 24 | Sn–S0 | 333, 348 | 0.77 ns | Sn–S0 | 333, 349 | 1.22 ns | 68 | |
| S1–S0 | 350 | 1.02 ns | S1–S0 | 367, 378, 383 | 4.76 ns | |||||
| T1–S0 | 405, 428 | 269.74 μs | T1–S0 | 406, 429, 450 | 1.66 ms | |||||
| TH–S0 | 510 | 25.99 ms | TH–S0 | 487, 515 | 778.44 ms | |||||
| Tσ–S0 | 558 | 11.92 ms | ||||||||

|
cryst | 12 | Sn–S0 | 329 | n.d. | Sn–S0 | 329, 339, 348 | n.d. | 68 | |
| S1–S0 | 397 | 1.41 ns | S1–S0 | 362, 380, 401 | 1.48 ns | |||||
| T1–S0 | 434 | 1.33 ms | T1–S0 | 436 | 4.38 ms | |||||
| TH–S0 | 481, 512, 552 | 49.13 ms | TH–S0 | 483, 515, 554 | 530.56 ms | |||||
| Tσ–S0 | 549 | 12.68 ms | ||||||||

|
cryst | 9 | Sn–S0 | 327, 341, 349 | n.d. | Sn–S0 | 318, 332 | n.d. | 68 | |
| S1–S0 | 373 | 0.65 ns | S1–S0 | 358, 380 | 0.96 ns | |||||
| T1–S0 | 416, 438 | 13.66 ms | T1–S0 | 416, 439 | 516.60 ms | |||||
| TH–S0 | 492, 523 | 142.45 ms | TH–S0 | 490, 521 | 700.58 ms | |||||
| Tσ–S0 | 550 | 11.22 ms | ||||||||

|
DCM | 3 | Sn–S0 | 328, 342, 358 | 0.68 ns | T1–S0 | 580 | 0.276 ms | 69,70 | |
| pwd | <0.1 | Sn–S0 | 326, 345, 365, 382 | 0.89 ns | Sn–S0 | 344, 365, 378 | 0.86 ns | |||
| S1–S0 | 426, 530 | 5.52 ns | S1–S0 | 457, 492, 530 | 2.54 ns | |||||
| T1–S0 | 573 | 0.274 ms | ||||||||

|
DCM | <0.1 | Sn–S0 | 380 | 5.22 ns | T1–S0 | 575 | 0.288 ms | 69,70 | |
| pwd | 14 | Sn–S0 | 395, 419, 443 | 0.91 ns | Sn–S0 | 409, 434, 462 | 1.96 ns | |||
| TBr–S0 | 470 | 1.07 ms | TBr–S0 | 433, 461, 484 | 4.05 ms | |||||
| TH–S0 | 553, 600, 646 | 28.85 ms | T1–S0 | 558 | 0.302 ms | |||||

|
DCM | <0.1 | Sn–S0 | 370 | 10.91 ns | T1–S0 | 585 | 0.263 ms | 69 | |
| pwd | <0.1 | Sn–S0 | 394, 418, 444 | 1.02 ns | Sn–S0 | 392, 417, 440 | 1.51 ns | |||
| S1–S0 | 415, 437 | n.d. | S1–S0 | 414, 440 | n.d. | |||||
| TH–S0 | 555, 605, 656 | 18.42 ms | TH–S0 | 545, 596, 650 | n.d. | |||||
| TBr–S0 | 490 | 18.11 ms | ||||||||
| T1–S0 | 590 | 0.200 ms | ||||||||

|
DCM | T1–S0 | 630 | 27.27 μs | 71 | |||||
| cryst | <0.1 | S1–S0 | 476 | 1.37 ns | S1–S0 | 458 | 2.77 ns | |||
| TI–S0 | 460, 495, 530 | 34.85 ms | ||||||||
| TH–S0 | 517, 563, 612 | 63.69 ms | TH–S0 | 511, 526, 558, 573, 610 | 66.47 ms | |||||
| T1–S0 | 630 | 0.53 μs | T1–S0 | 640 | 23.66 μs | |||||

|
cryst | 7 | S1–S0 | 443 | 1.23 ns | 71 | ||||
| TH–S0 | 625 | 9.47 ms | ||||||||
| T1–S0 | 680 | 3.47 ms | ||||||||

|
cryst | 5 | S1–S0 | 410 | 2.56 ns | S1–S0 | 440 | 3.41 ns | 71 | |
| TI–S0 | 463, 497, 537 | 14.65 ms | ||||||||
| TH–S0 | 496, 528, 566 | 21.48 ms | TH–S0 | 490, 527, 560 | 20.01 ms | |||||
| T1–S0 | 720 | 6.83 μs | ||||||||
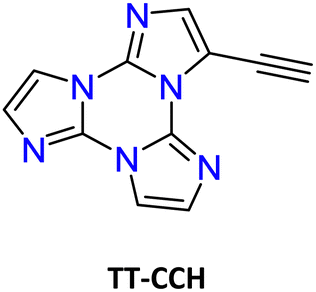
|
DCM | 2 | S1–S0 | 363 | n.d. | 87 | ||||
| PMMA | 3 | S1–S0 | 342 | 0.99 ns | ||||||
| SH–S0 | 383 | 1.46 ns | ||||||||
| T1–S0 | 439 | 0.16 ms | ||||||||
| TH–S0 | 522 | 6.9 ms | ||||||||
| cryst | 16 | S1–S0 | 314, 326, 338 | n.d. | S1–S0 | 310, 327, 337 | n.d. | |||
| SH–S0 | 354, 367, 377 | 1.05 ns | SH–S0 | 354, 364, 383 | 1.47 ns | |||||
| T1–S0 | 396, 420 | 0.25 ms | T1–S0 | 395, 419 | 6.39 ms | |||||
| TH–S0 | 545 | 4.66 ms | TH–S0 | 489, 523 | 44.84 ms | |||||
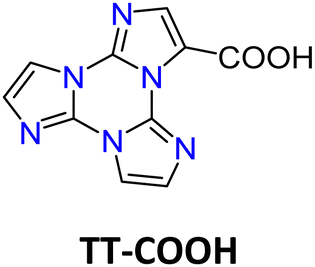
|
DMSO | 3.8 | S2–S0 | 327, 343 | n.d. | 89 | ||||
| S1–S0 | 404 | 3.55 ns | ||||||||
| cryst | 26 | S2–S0 | 342 | 2.32 ns | S2–S0 | 326, 341 | 2,51 | |||
| S1–S0 | 386, 408, 432 | 2.75 ns | S1–S0 | 385, 408, 435 | 5,51 | |||||
| T2–S0 | 445 | 3.64 ms | T2–S0 | 457 | 117 ms | |||||
| T1–S0 | 487 | 5.58 ms | T1–S0 | 505 | 526 ms | |||||
| TH–S0 | 549, 590, 642 | 117 ms | TH–S0 | 549, 590, 642 | 1.1 s | |||||

|
DCM | 17 | S2–S0 | 327, 341, 359 | 3.24 ns | S2–S0 | 338, 352, 373 | 5.06 ns | 70 | |
| pwd | 18 | S2–S0 | 335, 350, 366 | 2.61 ns | S2–S0 | 339, 352, 370 | 1.93 ns | |||
| S1–S0 | 387, 407, 429 | n.d. | S1–S0 | 394, 413, 437 | n.d. | |||||
| TH–S0 | 497, 536, 623 | 0.339 s | TH–S0 | 504, 537, 621 | 0.573 s | |||||

|
DMSO | 92 | S1–S0 | 420 | 2.76 ns | 79,80 | ||||
| cryst | TT-Pyr(Et) | 53 | S1–S0 | 493 | 2.97 ns | S1–S0 | 463, 493, 520 | 4.17 ns | ||
| TH–S0 | 555 | 4.95 ms | TH–S0 | 553, 596 | 5.37 ms | |||||
| TT-Pyr(RT) | 20 | S1–S0 | 490 | 1.85 ns | S1–S0 | 463, 492, 520 | 2.66 ns | |||
| TH–S0 | 550 | 4.62 ms | TH–S0 | 556, 599 | 5.61 ms | |||||
| TT-Pyr(HT) | 21 |
|
422, 443 | 0.62 ns |

|
413, 450 | 1.56 ns | |||
| S1–S0 | 483 | 1.99 ns | S1–S0 | 478 | 2.91 ns | |||||
| TH–S0 | 524, 563 | 23.4 ms | TH–S0 | 553, 590 | 26.2 ms | |||||

|
DMSO | 78 | S1–S0 | 419 | 9.22 ns | 79 | ||||
| pwd | 40.2 | S1–S0 | 490 | 4.64 ns | ||||||
| TH–S0 | 528 | 20.54 ms | ||||||||

|
DMSO | 74.4 | S1–S0 | 422 | 11.16 ns | 79 | ||||
| pwd | 36.9 | S1–S0 | 476 | 5.12 ns | ||||||
| TH–S0 | 522 | 40.62 ms | ||||||||

|
DMSO | 3 | S1–S0 | 565 | 2.76 ns | 88 | ||||

|
DCM | 11 | S1–S0 | 370 | 0.81 ns | S1–S0 | 365 | 1.71 ns | 86 | |
| PMMA | 15 | S1–S0 | 365 | 0.72 ns | ||||||
| cryst | 26 | S1–S0 | 376 | 1.15 ns | S1–S0 | 368 | 1.48 ns | |||
| T1–S0 | 425, 451 | 5.91 ms | T1–S0 | 420, 448 | 10.61 ms | |||||
| TH–S0 | 497, 530, 578 | 50.22 ms | TH–S0 | 494, 553, 577 | 132.01 ms | |||||
| Ground cryst | 18 | S1–S0 | 376 | 1.11 ns | S1–S0 | 375 | 1.91 ns | |||
| T1–S0 | 428, 441 | 5.20 ms | T1–S0 | 423, 443 | 9.56 ms | |||||
| TH–S0 | 500 | 29.81 ms | TH–S0 | 492, 513 | 60.84 ms | |||||

|
DCM | 5 | S1–S0 | 380 | 1.17 ns | S1–S0 | 370 | 1.42 ns | 86 | |
| PMMA | 17 | S1–S0 | 372 | 0.90 ns | S1–S0 | |||||
| pwd | 22 | S1–S0 | 382, 400 | 0.42 ns | S1–S0 | 364, 381 | 1.23 ns | |||
| T1–S0 | 428, 453 | 15.10 ms | T1–S0 | 423, 447 | 153.42 ms | |||||
| TH–S0 | 514, 550 | 41.51 ms | TH–S0 | 478, 512, 544 | 327.16 ms | |||||

|
DCM | 27 | S1–S0 | 337, 345 | 3.47 ns | S1–S0 | 339 | 5.69 ns | 84 | |
| T1–S0 | 411, 439, 466 | 2.75 s | ||||||||
| PMMA | 28 | S1–S0 | 333, 345 | 9.58 ns | S1–S0 | 345 | 10.25 ns | |||
| T1–S0 | 412, 440, 462 | 319.3 ms | T1–S0 | 411, 439, 466b | 82.87 ms | |||||
| cryst | TT-(N)-CzT | 13 | S1–S0 | 350 | 2.37 ns | S1–S0 | 329, 343, 352 | 4.30 ns | ||
| SH–S0 | 379, 390, 398 | 28.02 ns | SH–S0 | 377, 385, 394 | 52.13 ns | |||||
| T1–S0 | 442, 464 | 23.36 ms | T1–S0 | 440, 465 | 82.87 ms | |||||
| TH–S0 | 509 | 247.78 ms | TH–S0 | 495, 516 | 1.14 s | |||||
| TT-(N)-CzM | 16 | S1–S0 | 357, 367 | 3.66 ns | S1–S0 | 344, 358 | 6.34 ns | |||
| SH–S0 | 386, 391, 402 | 15.39 ns | SH–S0 | 381, 394, 402 | 27.31 ns | |||||
| T1–S0 | 437 | 20.04 ms | T1–S0 | 436 | 71.35 ms | |||||
| TH–S0 | 517 | 0.17 s | TH–S0 | 519 | 0.13 s | |||||
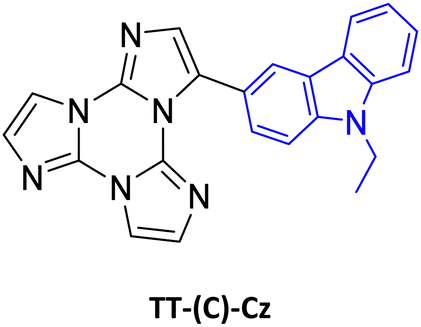
|
DCM | 20 | S1–S0 | 380 | 5.55 ns | S1–S0 | 365, 381 | 5.80 ns | 84 | |
| TH–S0 | 500 | 0.88 s | ||||||||
| PMMA | 33 | S1–S0 | 377 | 9.98 ns | S1–S0 | 361, 379 | 10.49 ns | |||
| T1–S0 | 417 | 46.51 ms | T1–S0 | 412, 442 | 1.65 s | |||||
| TH–S0 | 515 | 134.6 ms | TH–S0 | 500 | 0.24 s | |||||
| cryst | 28 | S1–S0 | 402, 420 | 6.74 ns | S1–S0 | 380, 400, 422 | 8.45 ns | |||
| T1–S0 | 460, 486 | 57.80 ms | T1–S0 | 471 | 0.12 s | |||||
| TH–S0 | 531, 580 | 0.12 s | TH–S0 | 500, 540 | 1.17 s | |||||

|
DCM | 63 | S1–S0 | 350, 370 | 2.86 ns | S1–S0 | 353 | 5.60 ns | 81 | |
| TH–S0 | 512 | 935 ms | ||||||||
| PMMA | 60 | S1–S0 | 347, 360 | 7.70 ns | S1–S0 | 350 (90 K) | 8.79 nsb | |||
| TH–S0 | 490 | 246.8 ms | TH–S0 | 508 | 601.0 ms | |||||
| cryst | TT-Ph-CzM | 43 | S1–S0 | 375, 408 | 2.16 ns | S1–S0 | 373, 388 | 3.45 ns | ||
| T1–S0 | 425 | 20.72 ms | ||||||||
| TH–S0 | 540 | 60.94 ms | TH–S0 | 523 | 684.47 ms | |||||
| TT-Ph-CzT | 22 | S1–S0 | 374, 394 | 3.41 ns | ||||||
| T1–S0 | 432 | 3.03 ms | ||||||||
| TH–S0 | 510 | 10.61 ms | ||||||||
| TT-Ph-CzO | 23 | S1–S0 | 373 | 3.49 ns | ||||||
| T1–S0 | 416 | 15.04 ms | ||||||||
| TH–S0 | 550 | 62.16 ms | ||||||||

|
DCM | 17 | S1–S0 | 351 | 0.62 ns | S1–S0 | 325, 345 | 2.32 ns | 85 | |
| Tn–S0 | 10.2 ms | Tn–S0 | 375 | 13.16 ms | ||||||
| T1–S0 | 408 | 11.01 ms | T1–S0 | 393 | — | |||||
| PMMA | S1–S0 | 350 | 1.18 ns | |||||||
| T1–S0 | 394 | 13.73 ms | ||||||||

|
440 | 3.47 ns | ||||||||
| TH′-S0 | 530 | 15.70 ms | ||||||||
| cryst | TT-2Py-A | 52 | Tn–S0 | 370 | 698 ms | |||||
| T1–S0 | 418 | 0.29 ms | ||||||||

|
450 | — | ||||||||
| TH′–S0 | 510, 570, 608 | 2.09 ms | ||||||||
| TT-2Py-H | Tn–S0 | 374 | 20.38 ms | |||||||
| T1–S0 | 408 | 0.47 ms | ||||||||

|
450 | 1.91 ns | ||||||||
| TH′–S0 | 500 | 18.25 ms | ||||||||
| 528, 562, 610 | 5.98 ms | |||||||||
| TT-2Py-X | Tn–S0 | 362 | 91.83 ms | |||||||
| T1–S0 | 402 | 1.31 ms | ||||||||
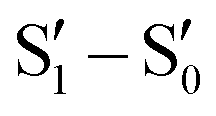
|
466 | 4.13 ns | ||||||||
| TH′–S0 | 524 | 9.38 ms | ||||||||
| TH–S0 | 636, 695, 767 | 111.1 ms | ||||||||

|
pwd | — | S1–S0 | 365 | 1.25 ns | 132 | ||||
| T1–S0 | 458 | 6.31 ms | ||||||||
| TH–S0 | 549 | 66.41 ms | ||||||||
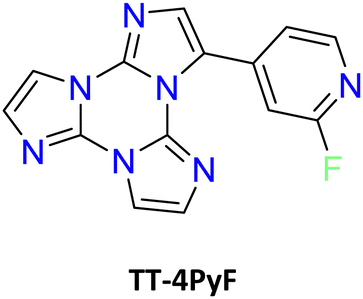
|
ACN | 50 | S1–S0 | 358 | 4.26 ns | S1–S0 | 344 | 4.04 ns | 82 | |
| T1–S0 | 454 | 1.63 s | ||||||||
| PMMA | S1–S0 | 348 | 1.89 ns | |||||||
| T1–S0 | 415, 436 | 3.09 ms | ||||||||
| cryst | 25 | S1–S0 | 373 | 4.77 ns | S1–S0 | 385 | 16.42 ns | |||
| T1–S0 | 403, 424, 446 | 11.64 ms | T1–S0 | 401, 425, 457 | 12.95 ms | |||||
| TH–S0 | 546, 592 | 419.08 ms | TH–S0 | 549 | 1.93 s | |||||
| Hybrid inorganic/organic derivatives | ||||||||||
| [Zn3(CH3COO)6(H2O)2](TT)2 | cryst | 7 | S1–S0 | 375, 395, 418 | 6.75 ns | S1–S0 | 416 | 8.45 ns | 123 | |
| TH–S0 | 555 | 0.650 s | TH–S0 | 554 | 1.303 s | |||||
| [Cd(H2O)6](ClO4)2(TT)2 | cryst | 10 | S1–S0 | 421 | 5.17 ns | S1–S0 | 440 | 5.50 ns | 123 | |
| TH–S0 | 549 | 0.110 s | TH–S0 | 550 | 0.585 s | |||||
| [Cd(H2O)6](BF4)2(TT)2 | cryst | 16 | S1–S0 | 383 | 17.16 ns | S1–S0 | 390 | 18.63 ns | 123 | |
| TH–S0 | 441, 469 | 0.293 s | TH–S0 | 441, 471 | 0.753 ms | |||||
| [Zn(H2O)6](BF4)2(TT)2 | cryst | 9 | S1–S0 | 370 | 3.52 ns | S1–S0 | 371 | 5.66 ns | 123 | |
| TH–S0 | 394, 422 | 0.542 s | TH–S0 | 397, 422 | 1.29 s | |||||
| [Zn(TT)(NO3)(H2O)3](NO3) | cryst | 1 | S1–S0 | 439 | 4.36 ns | S1–S0 | 430 | 5.93 ns | 124 | |
| TH–S0 | 550 | 67.70 ms | TH–S0 | 498 | 534.63 ms | |||||
| [Cd(TT)2(NO3)2(H2O)2] | cryst | S1–S0 | 430 | 0.61 ns | S1–S0 | 430 | 2.15 ns | 124 | ||
| TH–S0 | 505 | 32.69 ms | TH–S0 | 500 | 240.96 ms | |||||
| [Ag(TT)I]n | cryst | 19 | S1–S0 | 385, 400 | 2.15 ns | S1–S0 | 386, 395 | 2.31 ns | 126 | |
| T1–S0 | 411, 445 | <50 μs | T1–S0 | 425, 446 | 421.03 μs | |||||
| TI–S0 | 446, 476, 509 | 2.84 ms | TI–S0 | 440, 470, 506, 541 | 5.67 ms | |||||
| TH–S0 | 494, 530, 575, 620, 680 | 39.76 ms | TH–S0 | 490, 528, 576, 631, 701 | 44.51 ms | |||||
| [Cu(TT)I]n | cryst | 18 | T1–S0 | 568 | 32 μs | T1–S0 | 568 | 47.14 μs | 125,126 | |
| TI–S0 | 431, 460, 487 | — | TI–S0 | 430, 460, 494, 538 | 41.85 μs | |||||
| TH–S0 | 536, 582, 623 | 302.12 μs | TH–S0 | 529, 580, 627 | 1.03 s | |||||
| [Ag(TT)Cl]n | cryst | S1–S0 | 448 | 4.45 ns | 126 | |||||
| T1–S0 | 520 | — | ||||||||
| TH–S0 | 526, 565, 615 | 47.7 ms | ||||||||
| [Cu(TT)Cl]n | cryst | 4 | T1–S0 | 515 | 6.24 μs | 125,126 | ||||
| TH–S0 | 560 | 3.56 ms | ||||||||
| [Ag3(TT)4]n(NO3)3n·6nH2O | cryst | 7.5 | S1–S0 | 404 | 1.49 ns | S1–S0 | 391 | 1.76 ns | 126 | |
| Tn–S0 | 454 | 3.41 ms | Tn–S0 | 471 | 32.69 ms | |||||
| T1–S0 | 556 | 1.04 ms | T1–S0 | 565 | 33.36 ms | |||||
| [Zn(TT-COO)2(H2O)2] | cryst | 9 | S2–S0 | 338 | 0.88 ns | 89 | ||||
| S1–S0 | 382, 405, 430 | 1.94 ns | ||||||||
| T2–S0 | 452 | 1.42 ms | ||||||||
| T1–S0 | 490 | 4.3 ms | ||||||||
| TH–S0 | 566 | 478 ms | ||||||||
| [Cd(TT-COO)2(H2O)2] | cryst | 1.4 | S2–S0 | 352 | 0.77 ns | 89 | ||||
| S1–S0 | 382, 405, 430 | 1.59 ns | ||||||||
| T2–S0 | 452 | 2.04 ms | ||||||||
| T1–S0 | 523 | 10.4 ms | ||||||||
| TH–S0 | 577 | 116 ms | ||||||||
| [ReCl(CO)3(TT-2Py)] | Tol | 0.4 | T1–S0 | 583 | 483 ns | 129 | ||||
| pwd | 12 | T1–S0 | 532 | 100 ms | ||||||
| [Re3Cl3(CO)9(TT-2Py)]2 | Tol | 0.01 | T1–S0 | 597 | 500 ns | 129 | ||||
| pwd | 2.5 | T1–S0 | 563 | 40 ms | ||||||
| [Cu2I2(TT-2Py)]n | cryst | 7.3 | T1–S0 | 588 | 1.33 ms | T1–S0 | 580 | 17.89 ms | 130 | |
| [Cu2I2(TT-2Py)]2 | cryst | 44 | TI–S0 | 515 | 9.35 μs | TI–S0 | 513 | 27.93 μs | ||
| TH–S0 | 598 | 19.07 μs | TH–S0 | 596, 645 | 1.73 ms | |||||
| [Ag{(TT-4Py3}(SO3CF3)]n·½nCH2Cl2 | cryst | 1 | S1–S0 | 390 | 1.66 ns | S1–S0 | 395 | 1.96 ns | 132 | |
| T1–S0 | 460 | 0.836 ms | T1–S0 | 450 | 1.35 ms | |||||
| TH–S0 | 530 | 4.912 ms | TH–S0 | 510, 548, 592, 655 | 262 ms | |||||
| [Cd{TT-4Py3}(NO3)2(EtOH)]n | cryst | 15 | S1–S0 | 405 | 2.54 ns | S1–S0 | 346, 365, 381, 403, 421 | 2.69 ns | 132 | |
| T1–S0 | 455 | 12.62 ms | T1–S0 | 450 | 73.88 ms | |||||
| TH–S0 | 550 | 74.63 ms | TH–S0 | 498, 536 | 1.86 s | |||||
| [(Cu2I2)3{TT-4Py3}4]n·xn(H2O,EtOH) | cryst | ≈1 | TH–S0 | 650 | 6.36 ms | TH–S0 | 606 | 34.47 ms | 132 | |
| (TT-4PyHTT-4Py)2[Eu(NO3)5(H2O)]·(CH3CN)2 | cryst | S1–S0 | 450 | 139 | ||||||
| 5D0–7F2 | 618 | |||||||||
| [Eu(NO3)3(H2O)2(TT-4Py3)] | cryst | 5D0–7F2 | 618 | 283 μs | 139 | |||||
Förster resonance energy transfer (FRET) melting assays on the three best performing ligands, namely TT(4PyMeI)3, TT-Gn2 and TT-Gn3, supported the remarkable stabilizing ability of the three candidates and provided evidence for better performances of guanyl hydrazone derivatives (rather than the pyridinium ones) probably due to higher flexibility of the peripheral fragments allowing more effective interactions with the grooves/loops of G4s. On the other hand, in order to estimate the ligands’ selectivity for G4 over duplex DNA, FRET melting and microscale thermophoresis (MST) experiments were carried out,160,161 overall suggesting the need for improving their selectivity toward G4 structures by an appropriate molecular design.
4. Conclusions
The present perspective comprehensively gathers studies on a promising family based on the cyclic triimidazole scaffold, a small, rigid, planar, N-rich system with excellent π–π stacking ability and with moderate electron withdrawing character. Fascinating observations have already been made by inspection of multiemissive properties and coordination potentialities of different members of the family, including the TT prototype, and the results are here analytically reported according to different classes.Excitation dependent short and long-lived photoluminescence of molecular and supramolecular origin resulting in AIE features has been frequently disclosed. TT's multifaceted emissive behaviour, including dual fluorescence and phosphorescence, RTP from aqueous aggregates, mechanochromism and vapochromism, confirms TT as a powerful building block for designing high-performance single-component luminescent materials.
Intriguingly, some compounds possess anti-Kasha fluorescence (namely 1-3X (X = Cl, Br) and TT-Benzo) or phosphorescence (TT-2Py and [Ag3(TT)4]n(NO3)3n·6nH2O) or both (TT-COOH, [Zn(TT-COO)2(H2O)2] and [Cd(TT-COO)2(H2O)2]). Some others possess dual fluorescence due to emission from different conformers (TT-2Py, TTPyr and TT-Ph-Cz). Finally, all compounds, except Iso-A, display π–π stacking induced phosphorescence. Interpretation of the mechanisms involved in the photophysical behaviour has been achieved through experimental, structural and computational studies. Discrimination between molecular and supramolecular features has been provided by the comparison of photoluminescence in solution and low loaded blended film with respect to crystalline phases. Further proof has been given by mechanochromism associated with supramolecular components and, when possible, comparison among polymorphs.
Preliminary applications of TTs in sensing and bio-medicine open avenues for further studies in these fields and exciting results in a plethora of applications are expected by using TT as a building block for molecular, macromolecular and supramolecular systems.
A close control of the excited-state energy, which is essential for the development of triplet exciton-harvesting organic emitters, such as those with TADF and RTP, is far from being reached and needs further investigations through appropriate functionalization of the TT core with donor groups.57,58 When compared with other fused nitrogen-rich heterocycles, TT enables the preparation of asymmetric scaffolds: brominated derivatives have been revealed as convenient starting materials in C–C and C–N cross-coupling reactions, and therefore, donor–acceptor groups can be introduced on the core by properly reacting 1–2Br.
The presence of three exo-oriented nitrogen atoms on the TT scaffold, its relatively simple functionalization with coordinating groups and the possibility to prepare mono-, di- and tri-substituted systems make TTs particularly attracting in view of their further exploitation in coordination chemistry and isolation of porous metal–organic frameworks (MOF). In addition, tri-substituted TTs with C3 symmetry represent appealing building blocks for the production of covalent organic frameworks (COF), as reported for other π-extended conjugated multiazine or H-bonded organic frameworks (HOF), by fine tuning pendant groups around the central core. Moreover, because of their octupolar character, appropriately functionalized tri-substituted TTs represent suitable candidates for NLO applications and as chromophores for multiphoton absorption.
Due to their planar and prochiral nature, TTs can be exploited for the development of self-organized homochiral two-dimensional nanostructures on different surfaces to be used, for example, in the preparation of graphene-like structures.
A further challenge regarding this family is the application of iso-A, the TT isomer, in the preparation and characterization of molecular and supramolecular materials analogous to those of TT. Moreover, iso-A utilization in the preparation of carbene compounds (through salification of the asymmetric nitrogen and subsequent reaction with metals) as preliminarily reported in the literature64 deserves further investigation.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Chiara Botta, Lucia Carlucci, Clelia Giannini and Daniele Marinotto for their contribution to most of the work cited in this perspective.References
- A. A. Bekhit and A. M. Baraka, Eur. J. Med. Chem., 2005, 40, 1405–1413 CrossRef CAS PubMed.
- M. A. Gouda, M. A. Berghot, G. E. Abd El-Ghani and A. E.-G. M. Khalil, J. Heterocycl. Chem., 2016, 53, 1241–1250 CrossRef CAS.
- E. M. Morsy, E. R. Kotb, H. A. Soliman, H. H. Sayyed and N. A. Abdelwahed, Acta Pol. Pharm., 2015, 72, 465–474 CAS.
- A. Kumar, S. Rana, G. Sharma, P. Dhiman, M. I. Shekh and F. J. Stadler, J. Environ. Chem. Eng., 2023, 11, 110770 CrossRef CAS.
- X. Wei, N. Li, Y. Wang, Z. Xie, H. Huang, G. Yang, T. Li, X. Qin, S. Li, H. Yang, J. Zhu, F. You, C. Wu and Y. Liu, Appl. Mater. Today, 2021, 23, 100995 CrossRef.
- Q. Zhang, J. Zhou, H. Zhang, C. Qi, Q. Zhou, R. Guo, H. Yang, T. Xing, M. Wang, M. Wu and W. Wu, Adv. Funct. Mater., 2024, 34, 2401579 CrossRef CAS.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W.-W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- L. Gu, H. Shi, C. Miao, Q. Wu, Z. Cheng, S. Cai, M. Gu, C. Ma, W. Yao, Y. Gao, Z. An and W. Huang, J. Mater. Chem. C, 2018, 6, 226–233 RSC.
- Y. Liu, G. Zhan, P. Fang, Z. Liu, Z. Bian and C. Huang, J. Mater. Chem. C, 2017, 5, 12547–12552 RSC.
- G.-P. Yong, Y.-M. Zhang, W.-L. She and Y.-Z. Li, J. Mater. Chem., 2011, 21, 18520 RSC.
- W. Akbar, S. Ehsan, S. A. Siddique, M. Sarfraz, F. Shaheen, A. Shafqat, Shahnaz, M. B. A. Siddique, A. Saeed, R. Al-Salahi and Y. El Bakri, ACS Omega, 2024, 9, 34428–34444 CrossRef CAS PubMed.
- S. H. Lee, S. Kim, M. H. Yun, Y. S. Lee, S. N. Cho, T. Oh and P. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 1515–1518 CrossRef CAS PubMed.
- X. Lu, X. Liu, B. Wan, S. G. Franzblau, L. Chen, C. Zhou and Q. You, Eur. J. Med. Chem., 2012, 49, 164–171 CrossRef CAS PubMed.
- Y. Ozkay, I. Isikdag, Z. Incesu and G. Akalin, Eur. J. Med. Chem., 2010, 45, 3320–3328 CrossRef PubMed.
- B. Rathod, S. Pawar, S. Puri, A. Diwan and K. Kumar, ChemistrySelect, 2024, 9, e202303655 CrossRef CAS.
- M. Tapera, E. Doğan, K. Şahin, G. A. Gözkamane, H. Kekeçmuhammed, S. Sandal, A. C. Gurkan, R. E. Bora, A. Anber, S. Durdagi, Y. Zorlu and E. Sarıpınar, J. Mol. Struct., 2024, 1318 Search PubMed.
- Y. A. Belousov, A. A. Drozdov, I. V. Taydakov, F. Marchetti, R. Pettinari and C. Pettinari, Coord. Chem. Rev., 2021, 445, 214084 CrossRef CAS.
- X.-Q. Liang, H.-P. Xiao, B.-L. Liu, Y.-Z. Li, J.-L. Zuo and X.-Z. You, Polyhedron, 2008, 27, 2494–2500 CrossRef CAS.
- X.-Q. Liang, X.-H. Zhou, C. Chen, H.-P. Xiao, Y.-Z. Li, J.-L. Zuo and X.-Z. You, Cryst. Growth Des., 2009, 9, 1041–1053 CrossRef CAS.
- G. Yuan, Z. Chao, D.-J. Xu, K.-Z. Shao, X.-M. Li, X.-R. Hao and Z.-M. Su, Polyhedron, 2020, 180, 114430 CrossRef CAS.
- A. Barakat, A. El-Faham, M. Haukka, A. M. Al-Majid and S. M. Soliman, Appl. Organomet. Chem., 2021, 35, e6317 CrossRef CAS.
- K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3392–3395 CrossRef CAS PubMed.
- B. Therrien, J. Organomet. Chem., 2011, 696, 637–651 CrossRef CAS.
- M.-H. Yu, X.-T. Liu, B. Space, Z. Chang and X.-H. Bu, Coord. Chem. Rev., 2021, 427, 213518 CrossRef CAS.
- C. Krishnaraj, H. S. Jena, K. Leus and P. Van Der Voort, Green Chem., 2020, 22, 1038–1071 RSC.
- P. Puthiaraj, Y.-R. Lee, S. Zhang and W.-S. Ahn, J. Mater. Chem. A, 2016, 4, 16288–16311 RSC.
- P. Xiong, S. Zhang, R. Wang, L. Zhang, Q. Ma, X. Ren, Y. Gao, Z. Wang, Z. Guo and C. Zhang, Energy Environ. Sci., 2023, 16, 3181–3213 RSC.
- K. L. Kirk, W. Nagai and L. A. Cohen, J. Am. Chem. Soc., 1973, 95, 8389–8392 CrossRef CAS PubMed.
- D. M. Schubert, D. T. Natan, D. C. Wilson and K. I. Hardcastle, Cryst. Growth Des., 2011, 11, 843–850 CrossRef CAS.
- J. L. Segura, R. Juárez, M. Ramos and C. Seoane, Chem. Soc. Rev., 2015, 44, 6850–6885 RSC.
- K. Bergmann and Z. M. Hudson, Faraday Discuss., 2024, 250, 181–191 RSC.
- O. Buyukcakir, R. Yuksel, Y. Jiang, S. H. Lee, W. K. Seong, X. Chen and R. S. Ruoff, Angew. Chem., Int. Ed., 2019, 58, 872–876 CrossRef CAS PubMed.
- H. Chen, X. Suo, Z. Yang and S. Dai, Adv. Mater., 2022, 34, 2107947 CrossRef CAS PubMed.
- J. Li, L. Tao, Y. Wang, Y. Yao and Q. Guo, Front. Chem., 2021, 9, 717569 CrossRef CAS PubMed.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- S. Vijayakumar, A. P. Mohanachandran, R. B. Rakhi, S. Shankar, R. S. Pillai and A. Ajayaghosh, Small, 2024, 20, 2405701 CrossRef CAS PubMed.
- X.-Y. Yan, M.-D. Lin, S.-T. Zheng, T.-G. Zhan, X. Zhang, K.-D. Zhang and X. Zhao, Tetrahedron Lett., 2018, 59, 592–604 CrossRef CAS.
- K. A. Hofmann and O. Ehrhart, Chem. Ber., 1912, 45, 2731–2740 CrossRef.
- R. Huisgen, H. J. Sturm and M. Seidel, Chem. Ber., 1961, 94, 1555–1562 CrossRef CAS.
- R. Cristiano, J. Eccher, I. H. Bechtold, C. N. Tironi, A. A. Vieira, F. Molin and H. Gallardo, Langmuir, 2012, 28, 11590–11598 CrossRef CAS PubMed.
- R. Cristiano, H. Gallardo, A. J. Bortoluzzi, I. H. Bechtold, C. E. M. Campos and R. L. Longo, Chem. Commun., 2008, 5134–5136, 10.1039/B810680K.
- A. G. Dal-Bó, G. G. L. Cisneros, R. Cercena, J. Mendes, L. M. da Silveira, E. Zapp, K. G. Domiciano, R. da Costa Duarte, F. S. Rodembusch and T. E. A. Frizon, Dyes Pigm., 2016, 135, 49–56 CrossRef.
- M. D. S. Kutz, L. A. Suassuna e Bega, C. Francener, G. Farias, F. A. de Campos, H. Bock, I. H. Bechtold, F. Molin and E. Westphal, J. Mol. Struct., 2025, 1321, 139996 CrossRef CAS.
- M. Sperner, N. Tober and H. Detert, Eur. J. Org. Chem., 2019, 4688–4693 CrossRef CAS.
- V. A. Tartakovsky, A. E. Frumkin, A. M. Churakov and Y. A. Strelenko, Russ. Chem. Bull., 2005, 54, 719–725 CrossRef CAS.
- S. Glang, T. Rieth, D. Borchmann, I. Fortunati, R. Signorini and H. Detert, Eur. J. Org. Chem., 2014, 3116–3126 CrossRef CAS.
- H. Zhao, Y. Wang, Z. Liu and B. Dai, RSC Adv., 2014, 4, 13161–13166 RSC.
- X. Chen, S. Wang, H. L. Lee, J. Y. Lee, X. Liao, L. Li, W. Zhu and Y. Wang, Adv. Opt. Mater., 2021, 9, 2101518 CrossRef CAS.
- Z. Fang, S. Wang, J. Liao, X. Chen, Y. Zhu, W. Zhu and Y. Wang, J. Mater. Chem. C, 2022, 10, 4837–4844 RSC.
- R. Hojo, D. M. Mayder and Z. M. Hudson, J. Mater. Chem. C, 2021, 9, 14342–14350 RSC.
- F. Hundemer, E. Crovini, Y. Wada, H. Kaji, S. Bräse and E. Zysman-Colman, Mater. Adv., 2020, 1, 2862–2871 RSC.
- S. K. Pathak, Y. Xiang, M. Huang, T. Huang, X. Cao, H. Liu, G. Xie and C. Yang, RSC Adv., 2020, 10, 15523–15529 RSC.
- S. Wang, X. Wang, K. H. Lee, S. Liu, J. Y. Lee, W. Zhu and Y. Wang, Dyes Pigm., 2020, 182, 108589 CrossRef CAS.
- Y. Yin, S. Zeng, C. Xiao, P. Fan, D. J. Shin, K. J. Kim, H. Nam, Q. Ma, H. Ma, W. Zhu, T. Kim, J. Y. Lee and Y. Wang, Mater. Horiz., 2024, 11, 1741–1751 RSC.
- S. Zeng, C. Xiao, J. Zhou, Q. Dong, Q. Li, J. Lim, H. Ma, J. Y. Lee, W. Zhu and Y. Wang, Adv. Funct. Mater., 2022, 32, 2113183 CrossRef CAS.
- H. Marchi Luciano, G. Farias, C. M. Salla, L. G. Franca, S. Kuila, A. P. Monkman, F. Durola, I. H. Bechtold, H. Bock and H. Gallardo, Chem. – Eur. J., 2023, 29, e202203800 CrossRef CAS PubMed.
- M. Ferreira, N. O. Decarli, A. Nyga, K. Erfurt, J. Lingagouder, L. E. de Sousa, L. de Thieulloy, P. de Silva and P. Data, J. Mater. Chem. C, 2024, 12, 13651–13664 RSC.
- E. C. Coad, P. G. Apen and P. G. Rasmussen, J. Am. Chem. Soc., 1994, 116, 391–392 CrossRef CAS.
- E. C. Coad, J. Kampf and P. G. Rasmussen, J. Org. Chem., 1996, 61, 6666–6672 CrossRef CAS PubMed.
- J. Heredia-Moya and K. L. Kirk, J. Fluorine Chem., 2007, 128, 674–678 CrossRef CAS PubMed.
- Y. Takeuchi, K. L. Kirk and L. A. Cohen, J. Org. Chem., 1979, 44, 4243–4246 CrossRef CAS.
- I. Sadahiro, F. Yoshiaki, S. Shizen and M. Kohji, Bull. Chem. Soc. Jpn., 1975, 48, 956–959 CrossRef.
- D. M. Buck and D. Kunz, Organometallics, 2015, 34, 5335–5340 CrossRef CAS.
- R. Bhattacharya, S. Ray, J. Ray and A. Ghosh, Cent. Eur. J. Chem., 2003, 1, 427 CAS.
- O. Ghashghaei, S. Caputo, M. Sintes, M. Revés, N. Kielland, C. Estarellas, F. J. Luque, A. Aviñó, R. Eritja, A. Serna-Gallego, J. A. Marrugal-Lorenzo, J. Pachón, J. Sánchez-Céspedes, R. Treadwell, F. de Moliner, M. Vendrell and R. Lavilla, Chem. – Eur. J., 2018, 24, 14513–14521 CrossRef CAS PubMed.
- O. Ghashghaei, P. N. Rodríguez and R. Lavilla, Synlett, 2022, 822–835 CAS.
- C. Giannini, A. Forni, D. Malpicci, E. Lucenti, D. Marinotto, A. Previtali, L. Carlucci and E. Cariati, Eur. J. Org. Chem., 2021, 2041–2049 CrossRef CAS.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, A. Colombo, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, ChemPhotoChem, 2018, 2, 801–805 CrossRef CAS.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Pavanello, A. Previtali, S. Righetto and E. Cariati, Angew. Chem., Int. Ed., 2017, 56, 16302–16307 CrossRef CAS PubMed.
- E. Lucenti, A. Forni, C. Botta, C. Giannini, D. Malpicci, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, Chem. – Eur. J., 2019, 25, 2452–2456 CrossRef CAS PubMed.
- S. Di Micco, C. Giannini, A. Previtali, E. Lucenti and G. Bifulco, Magn. Reson. Chem., 2019, 57, 82–92 CrossRef CAS PubMed.
- M. Beinhoff, W. Weigel, M. Jurczok, W. Rettig, C. Modrakowski, I. Brüdgam, H. Hartl and A. D. Schlüter, Eur. J. Org. Chem., 2001, 3819–3829 CrossRef CAS.
- A. Pal, R. Ghosh, N. N. Adarsh and A. Sarkar, Tetrahedron, 2010, 66, 5451–5458 CrossRef CAS.
- M. Mao, M.-G. Ren and Q.-H. Song, Chem. – Eur. J., 2012, 18, 15512–15522 CrossRef CAS PubMed.
- S. Telitel, F. Dumur, T. Faury, B. Graff, M.-A. Tehfe, D. Gigmes, J.-P. Fouassier and J. Lalevée, Beilstein J. Org. Chem., 2013, 9, 877–890 CrossRef CAS PubMed.
- M. Drev, U. Grošelj, B. Ledinek, F. Perdih, J. Svete, B. Štefane and F. Požgan, Org. Lett., 2018, 20, 5268–5273 CrossRef CAS PubMed.
- D. Malpicci, S. Andolina, S. Di Ciolo, E. Lucenti, E. Cariati, S. Marzano, B. Pagano, J. Amato, A. Randazzo and C. Giannini, Eur. J. Org. Chem., 2022, e202200718 CrossRef CAS.
- D. Malpicci, C. Giannini, E. Lucenti, A. Forni, D. Marinotto and E. Cariati, Photochem, 2021, 1, 477–487 CrossRef.
- A. Previtali, W. He, A. Forni, D. Malpicci, E. Lucenti, D. Marinotto, L. Carlucci, P. Mercandelli, M. A. Ortenzi, G. Terraneo, C. Botta, R. T. K. Kwok, J. W. Y. Lam, B. Z. Tang and E. Cariati, Chem. – Eur. J., 2021, 27, 16690–16700 CrossRef CAS PubMed.
- D. Malpicci, A. Forni, C. Botta, C. Giannini, E. Lucenti, D. Marinotto, D. Maver, L. Carlucci and E. Cariati, Chem. – Eur. J., 2023, 29, e202300930 CrossRef CAS PubMed.
- A. Previtali, E. Lucenti, A. Forni, L. Mauri, C. Botta, C. Giannini, D. Malpicci, D. Marinotto, S. Righetto and E. Cariati, Molecules, 2019, 24, 2552 CrossRef CAS PubMed.
- D. Malpicci, S. R. Araneo, S. Arnaboldi, E. Cariati, A. Forni, S. Grecchi, E. Lucenti, D. Marinotto, D. Maver and P. R. Mussini, Electrochim. Acta, 2023, 469, 143117 CrossRef CAS.
- D. Malpicci, A. Forni, C. Botta, C. Giannini, E. Lucenti, D. Marinotto, D. Maver, L. Carlucci and E. Cariati, Dyes Pigm., 2023, 215, 111274 CrossRef CAS.
- E. Lucenti, A. Forni, A. Previtali, D. Marinotto, D. Malpicci, S. Righetto, C. Giannini, T. Virgili, P. Kabacinski, L. Ganzer, U. Giovanella, C. Botta and E. Cariati, Chem. Sci., 2020, 11, 7599–7608 RSC.
- D. Malpicci, A. Forni, E. Cariati, R. Inoguchi, D. Marinotto, D. Maver, F. Turco and E. Lucenti, Molecules, 2023, 28, 140 CrossRef CAS PubMed.
- D. Malpicci, D. Maver, E. Rosadoni, A. Colombo, E. Lucenti, D. Marinotto, C. Botta, F. Bellina, E. Cariati and A. Forni, Molecules, 2024, 29, 1967 CrossRef CAS PubMed.
- M. Formenti, D. Blasi, E. Cariati, L. Carlucci, A. Forni, C. Giannini, M. Guidotti, S. Econdi, D. Malpicci, D. Marinotto and E. Lucenti, Dyes Pigm., 2022, 206, 110637 CrossRef CAS.
- D. Malpicci, A. Forni, E. Lucenti, D. Marinotto, D. Maver, V. Lozovan, V. C. Kravtsov, A. Siminel, M. S. Fonari and E. Cariati, Eur. J. Inorg. Chem., 2024, e202400338 CrossRef CAS.
- E. Rosadoni, L. Ballerini, A. Del Vecchio and F. Bellina, Asian J. Org. Chem., 2024, 13, e202400082 CrossRef CAS.
- M. Magni, E. Lucenti, A. Previtali, P. R. Mussini and E. Cariati, Electrochim. Acta, 2019, 317, 272–280 CrossRef CAS.
- Q. Dang, Y. Jiang, J. Wang, J. Wang, Q. Zhang, M. Zhang, S. Luo, Y. Xie, K. Pu, Q. Li and Z. Li, Adv. Mater., 2020, 32, 2006752 CrossRef PubMed.
- W. Qin, P. Zhang, H. Li, J. W. Y. Lam, Y. Cai, R. T. K. Kwok, J. Qian, W. Zheng and B. Z. Tang, Chem. Sci., 2018, 9, 2705–2710 RSC.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- J. Zhi, Q. Zhou, H. Shi, Z. An and W. Huang, Chem. – Asian J., 2020, 15, 947–957 CrossRef CAS PubMed.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed.
- Y. Li and P. Gao, Chemosensors, 2023, 11, 489 CrossRef CAS.
- B. Sk and S. Hirata, Adv. Sci., 2024, 11, 2308897 CrossRef CAS PubMed.
- J. Tan, Q. Li, S. Meng, Y. Li, J. Yang, Y. Ye, Z. Tang, S. Qu and X. Ren, Adv. Mater., 2021, 33, 2006781 CrossRef CAS PubMed.
- S. Hirata, K. Totani, H. Kaji, M. Vacha, T. Watanabe and C. Adachi, Adv. Opt. Mater., 2013, 1, 438–442 CrossRef.
- S. Datta and J. Xu, ACS Appl. Bio Mater., 2023, 6, 4572–4585 CrossRef CAS PubMed.
- X. Yang, G. I. N. Waterhouse, S. Lu and J. Yu, Chem. Soc. Rev., 2023, 52, 8005–8058 RSC.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- D. Malpicci, E. Lucenti, C. Giannini, A. Forni, C. Botta and E. Cariati, Molecules, 2021, 26, 6999 CrossRef CAS PubMed.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS PubMed.
- H. Li, L. Lv, K. Yuan, S. Pan and Z. Li, Sci. Rep., 2023, 13, 12357 CrossRef CAS PubMed.
- S. Cai, X. Yao, H. Ma, H. Shi and Z. An, Aggregate, 2023, 4, e320 CrossRef CAS.
- S. J. N. Dixit, R. Ghosh and N. Agarwal, Phys. Chem. Chem. Phys., 2025, 27, 175–181 RSC.
- A. Ahsan, X. Wang, R. Sk, M. Heydari, L. Buimaga-Iarinca, C. Wäckerlin, E. Lucenti, S. Decurtins, E. Cariati, T. A. Jung, U. Aschauer and S.-X. Liu, J. Phys. Chem. C, 2023, 127, 23000–23009 CrossRef CAS PubMed.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- D. S. Cati and H. Stoeckli-Evans, CCDC 227635: CSD Communication, 2004 DOI:10.5517/cc7mw2s.
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- M. Kasha, Faraday Discuss. R. Soc. Chem., 1950, 9, 14–19 RSC.
- S. H. Lee, S. H. Kim, S. K. Kim, J. H. Jung and J. S. Kim, J. Org. Chem., 2005, 70, 9288–9295 CrossRef CAS PubMed.
- I. Suzuki, M. Ui and A. Yamauchi, J. Am. Chem. Soc., 2006, 128, 4498–4499 CrossRef CAS PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS PubMed.
- C. Chen, K. C. Chong, Y. Pan, G. Qi, S. Xu and B. Liu, ACS Mater. Lett., 2021, 3, 1081–1087 CrossRef CAS.
- J. Song, Y. Wang, L. Qu, L. Fang, X. Zhou, Z.-X. Xu, C. Yang, P. Wu and H. Xiang, J. Phys. Chem. Lett., 2022, 13, 5838–5844 CrossRef CAS PubMed.
- S. Yuan, Y. Zhang, J. Chen, Y. Yu, L. Yue, Q. Sun, H. Zhang, S. Xue and W. Yang, Adv. Opt. Mater., 2022, 10, 2200090 CrossRef CAS.
- Y. Zhang, Q. Sun, J. Chen, S. Cui, H. Zhang, S. Xue and W. Yang, Chem. Eng. J., 2022, 447, 137458 CrossRef CAS.
- E. Cariati, A. Forni, E. Lucenti, D. Marinotto, A. Previtali, S. Righetto, C. Botta, V. Bold, V. Kravtsov and M. S. Fonari, Chem. – Asian J., 2019, 14, 853–858 CrossRef CAS PubMed.
- M. S. Fonari, V. C. Kravtsov, V. Bold, E. Lucenti, E. Cariati, D. Marinotto and A. Forni, Cryst. Growth Des., 2021, 21, 4184–4200 CrossRef CAS.
- E. Lucenti, E. Cariati, A. Previtali, D. Marinotto, A. Forni, V. Bold, V. C. Kravtsov, M. S. Fonari, S. Galli and L. Carlucci, Cryst. Growth Des., 2019, 19, 1567–1575 CrossRef CAS.
- D. Malpicci, E. Lucenti, A. Forni, D. Marinotto, A. Previtali, L. Carlucci, P. Mercandelli, C. Botta, S. Righetto and E. Cariati, Inorg. Chem. Front., 2021, 8, 1312–1323 RSC.
- A. Forni, E. Cariati, L. Carlucci, E. Lucenti, D. Marinotto, S. Pieraccini and M. Sironi, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2021, 77, 865–870 CrossRef CAS.
- E. Melnic, V. C. Kravtsov, E. Lucenti, E. Cariati, A. Forni, N. Siminel and M. S. Fonari, New J. Chem., 2021, 45, 9040–9052 RSC.
- D. Malpicci, D. Maver, D. Maggioni, P. Mercandelli, L. Carlucci, E. Cariati, P. Mussini and M. Panigati, New J. Chem., 2023, 47, 21463–21474 RSC.
- D. Malpicci, D. Blasi, D. Marinotto, A. Forni, E. Cariati, E. Lucenti and L. Carlucci, Crystals, 2023, 13, 149 CrossRef CAS.
- J.-C. Li, H.-X. Li, H.-Y. Li, W.-J. Gong and J.-P. Lang, Cryst. Growth Des., 2016, 16, 1617–1625 CrossRef CAS.
- D. Malpicci, D. Blasi, A. Forni, E. Lucenti, D. Marinotto, M. Formenti, D. Maver, P. Mercandelli, E. Cariati and L. Carlucci, Eur. J. Inorg. Chem., 2024, e202400185 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- A. Forni, S. Pieraccini, D. Franchini and M. Sironi, J. Phys. Chem. A, 2016, 120, 9071–9080 CrossRef CAS PubMed.
- A. Forni, S. Pieraccini, S. Rendine, F. Gabas and M. Sironi, ChemPhysChem, 2012, 13, 4224–4234 CrossRef CAS PubMed.
- A. Forni, S. Pieraccini, S. Rendine and M. Sironi, J. Comput. Chem., 2014, 35, 386–394 CrossRef CAS PubMed.
- Q. J. Shen, X. Pang, X. R. Zhao, H. Y. Gao, H.-L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 5027–5034 RSC.
- P. Smart, Á. Bejarano-Villafuerte, R. M. Hendry and L. Brammer, CrystEngComm, 2013, 15, 3160–3167 RSC.
- V. C. Kravtsov, V. Bold, N. Siminel, Y. Chumakov, M. Gdaniec, E. Lucenti, D. Malpicci, D. Maver and M. S. Fonari, Cryst. Growth Des., 2024, 24, 4875–4883 CrossRef CAS.
- V. Kumar, H. Kim, B. Pandey, T. D. James, J. Yoon and E. V. Anslyn, Chem. Soc. Rev., 2023, 52, 663–704 RSC.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- R. Haiges, G. Bélanger-Chabot, S. M. Kaplan and K. O. Christe, Dalton Trans., 2015, 44, 7586–7594 RSC.
- G. Hervé, C. Roussel and H. Graindorge, Angew. Chem., Int. Ed., 2010, 49, 3177–3181 CrossRef PubMed.
- P. Peng, N. Ding, C. Zhao, Y. Li, J. Liu, S. Li and S. Pang, Cryst. Growth Des., 2022, 22, 2158–2167 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- W. Xu, M. M. S. Lee, Z. Zhang, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, D. Wang and B. Z. Tang, Chem. Sci., 2019, 10, 3494–3501 RSC.
- K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012–11042 CrossRef CAS PubMed.
- F. P. L. Lim and A. V. Dolzhenko, Eur. J. Med. Chem., 2014, 85, 371–390 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- S. Marzano, B. Pagano, N. Iaccarino, A. Di Porzio, S. De Tito, E. Vertecchi, E. Salvati, A. Randazzo and J. Amato, Int. J. Mol. Sci., 2021, 22, 10315 CrossRef CAS PubMed.
- P. Zizza, C. Cingolani, S. Artuso, E. Salvati, A. Rizzo, C. D'Angelo, M. Porru, B. Pagano, J. Amato, A. Randazzo, E. Novellino, A. Stoppacciaro, E. Gilson, G. Stassi, C. Leonetti and A. Biroccio, Nucleic Acids Res., 2016, 44, 1579–1590 CrossRef PubMed.
- R. Hänsel-Hertsch, J. Spiegel, G. Marsico, D. Tannahill and S. Balasubramanian, Nat. Protoc., 2018, 13, 551–564 CrossRef PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- M. Allegretti, A. Fabi, E. Giordani, C. Ercolani, P. Romania, C. Nisticò, S. Gasparro, V. Barberi, M. Ciolina, E. Pescarmona, D. Giannarelli, G. Ciliberto, F. Cognetti and P. Giacomini, Mol. Cancer, 2021, 20, 151 CrossRef CAS PubMed.
- A. R. Duarte, E. Cadoni, A. S. Ressurreição, R. Moreira and A. Paulo, ChemMedChem, 2018, 13, 869–893 CrossRef CAS PubMed.
- T. Santos, G. F. Salgado, E. J. Cabrita and C. Cruz, Pharmaceuticals, 2021, 14, 769 CrossRef CAS PubMed.
- D. Musumeci, J. Amato, P. Zizza, C. Platella, S. Cosconati, C. Cingolani, A. Biroccio, E. Novellino, A. Randazzo, C. Giancola, B. Pagano and D. Montesarchio, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1341–1352 CrossRef CAS PubMed.
- J. Amato, G. Miglietta, R. Morigi, N. Iaccarino, A. Locatelli, A. Leoni, E. Novellino, B. Pagano, G. Capranico and A. Randazzo, J. Med. Chem., 2020, 63, 3090–3103 CrossRef CAS PubMed.
- J. Amato, R. Morigi, B. Pagano, A. Pagano, S. Ohnmacht, A. De Magis, Y.-P. Tiang, G. Capranico, A. Locatelli, A. Graziadio, A. Leoni, M. Rambaldi, E. Novellino, S. Neidle and A. Randazzo, J. Med. Chem., 2016, 59, 5706–5720 CrossRef CAS PubMed.
- J. Amato, C. Platella, S. Iachettini, P. Zizza, D. Musumeci, S. Cosconati, A. Pagano, E. Novellino, A. Biroccio, A. Randazzo, B. Pagano and D. Montesarchio, Eur. J. Med. Chem., 2019, 163, 295–306 CrossRef CAS PubMed.
- J. Amato, T. W. Madanayake, N. Iaccarino, E. Novellino, A. Randazzo, L. H. Hurley and B. Pagano, Chem. Commun., 2018, 54, 9442–9445 RSC.
| This journal is © The Royal Society of Chemistry 2025 |