
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical solid state single electron transfer from reduced organic hydrocarbon for catalytic aryl-halide bond activation†
Amit
Biswas
,
Anup
Bhunia
 * and
Swadhin K.
Mandal
*
* and
Swadhin K.
Mandal
*
Department of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata, Mohanpur-741246, India. E-mail: swadhin.mandal@iiserkol.ac.in; bhunia1988@gmail.com
First published on 2nd February 2023
Abstract
Solid-state radical generation is an attractive but underutilized methodology in the catalytic strong bond activation process, such as the aryl-halide bond. Traditionally, such a process of strong bond activation relied upon the use of transition metal complexes or strongly reducing photocatalysts in organic solvents. The generation of the aryl radical from aryl halides in the absence of transition-metal or external stimuli, such as light or cathodic current, remains an elusive process. In this study, we describe a reduced organic hydrocarbon, which can act as a super reductant in the solid state to activate strong bonds by solid-state single electron transfer (SSSET) under the influence of mechanical energy leading to a catalytic strategy based on the mechano-SSSET or mechanoredox process. Here, we investigate the solid-state synthesis of the super electron donor phenalenyl anion in a ball mill and its application as an active catalyst in strong bond (aryl halide) activation. Aryl radicals generated from aryl halides by employing this strategy are competent for various carbon–carbon bond-forming reactions under solvent-free and transition metal-free conditions. We illustrate this approach for partially soluble or insoluble polyaromatic arenes in accomplishing solid–solid C–C cross-coupling catalysis, which is otherwise difficult to achieve by traditional methods using solvents.
Introduction
Recent advances in synthetic methods based on mechanochemistry received significant attention.1–5 In the last few decades, new synthetic methods have been developed to address the efficiency, selectivity, and atom economy issues. More importantly, mechanochemistry has helped the chemical community to perform organic reactions in a nonfluid medium. Besides sustainability, mechanochemical reactions using ball milling have provided access to large chemical spaces with complementary reactivities from conventional homogeneous processes. In 2021, IUPAC recognized the mechanochemical process as one of the top 10 emerging technologies in chemistry.6 The potential advantages of mechanochemical reactions using ball milling are that they can minimize the use of corrosive and harmful solvents, avoid external heating, and have less reaction time. In this context, it may be recalled that the application of mechanical energy with the assistance of ball mills in accomplishing C–C cross-coupling catalysis is considered an emerging strategy, although in the presence of transition metal-based catalysts.7–14 The success of mechanochemical organic synthesis is mainly limited to ionic reactions.15,16 In fact, the application of mechanochemical processes in radical chemistry is quite limited due to the intrinsic unpredictable nature of highly reactive radical intermediates.17,18 A limited application of mechanical forces has been reported to form radicals by the degradation of generic polymers or by homolytic cleavage of covalent bonds.19–28 More recently, the use of piezoelectric materials as reusable charge transfer catalysts has shown a promising impact in this growing field.29–36 Seminal work from Kubota, Ito, and co-workers provided the first evidence of the mechanoredox process for C–C cross-coupling catalysis using a piezoelectric material (BaTiO3).29 However, this method could not achieve the goal of a transition metal-free process as BaTiO3 was used as the redox reagent. It was not catalytic since it used a super stoichiometric amount of the piezoelectric material (4–5 equiv. BaTiO3). Moreover, such a method is limited to non-step- and non-atom-economic aryl diazonium salts as coupling partners. Additionally, the reaction of piezoelectric materials has been limited to low-reduction potential substrates and failed to activate aryl halides. In this context, catalytic C–C cross-coupling reactions activating aryl halides that avoid transition metals, light, and solvents are yet to be realized. In parallel efforts, to avoid transition metals, small organic molecules have been used as electron transfer catalysts.37,38 From organocatalysis to radical chemistry, new types of catalysis have been propelled using small organic molecules at a breathtaking speed which was unthinkable a few years ago.39 A series of organic compounds were utilized as redox catalysts for the transition-metal-free bond breaking/forging process.40–43 Among such approaches, the single electron transfer (SET) process by small organic molecules is particularly limited to photochemical or electrochemical transformations. However, purely chemical SET using small organic molecules was accomplished recently in C–C cross-coupling reactions in assembling complex aromatic compounds,44,45 which relied mainly on Pd-based organometallic catalysts over the past several decades. The base-promoted homolytic aromatic substitution (BHAS) is one such attempt that has been widely recognized as a chemically driven SET process.46 However, these reactions have several limitations, such as the necessity of using semi-stoichiometric catalyst loading, harsh reaction conditions, and hazardous solvents. Alternatively, we introduce herein a small organic odd alternant hydrocarbon such as a phenalenyl (PLY) based molecule that would be an ideal candidate for solid-state single electron transfer (SSSET). PLY-based radicals are long known in the construction of organic magnetic semiconductors,47–52 molecular switches,53–55 molecular batteries,56,57 quantum spin simulators,58 and molecular spin memory devices.59 All these intriguing material properties of PLY radicals originate from their facile solid-state electron transfer ability.60,61 Such facile electron transfer in the solid state is attributed to the fact that the conduction molecular orbital of the PLY radical is formally non-bonding. Thus, the electron occupancy in this orbital does not contribute to the overall bond order resulting in minimal reorganization energy.62 This plays a favourable role in the intramolecular phonon contribution to the polaron binding energy resulting in facile electron transfer in the solid state.62,63 In 2013, it was demonstrated that solid-state electron transfer from PLY-based radicals under external stimuli such as the magnetic field plays a vital role in constructing a molecular-scale quantum spin memory device (Fig. 1a).59 In such a design, solid-state electron transfer from the surface of a ferromagnet was engineered to generate a PLY-based radical, which results in an unexpected interfacial magnetoresistance of more than 20% as it creates a spin-based bias (Fig. 1a). In the present work, it was argued that reduced PLY-based molecules could perform solid-state single electron transfer (SSSET) under external mechanical force to accomplish C–C cross-coupling catalysis leading to the development of the transition metal-free mechano-SSSET or mechanoredox process. Our experiments exploit the cationic PLY I (orange-coloured solid) as the standard redox system. Cyclic voltammetry revealed two consecutive reduction potentials generating mono and doubly reduced PLY species at E11/2 = −0.85 V and E21/2 = −1.88 V.64 It may be noted that the second reduction potential (E21/2 = −1.88 V) is significantly higher than the flat band potential of polarized BaTiO3 (−0.59 V);65 thus, it might offer activation of a strong bond (aryl-halide). In the solid state, PLY cation I (12πe− species) can accept two electrons stepwise from an organic electron donor generating the mono-reduced green-coloured radical PLY II (13πe− species), which upon further 1e− reduction generates the brown-coloured double reduced PLY III (14πe− anionic species). The double-reduced PLY III can successfully initiate solid-state electron transfer to aryl/heteroaryl halide substrates generating aryl/heteroaryl radicals under ball-milling conditions. Such solid-state single electron transfer and activation of aryl halides were integrated into a catalytic cycle which affords the construction of various C–C cross-coupled products under solvent-free and transition metal-free conditions. This strategy based on solid-state single electron transfer under the influence of mechanical energy (Fig. 1c) develops an efficient method for various types of C–C cross-coupling reactions (such as direct C–H arylation and Heck-coupling) using aryl halides (Fig. 1c). In particular, this approach works very well for partially soluble or insoluble polyaromatic arenes in accomplishing solid–solid C–C cross-coupling catalysis under solvent-free and transition metal-free conditions for the first time.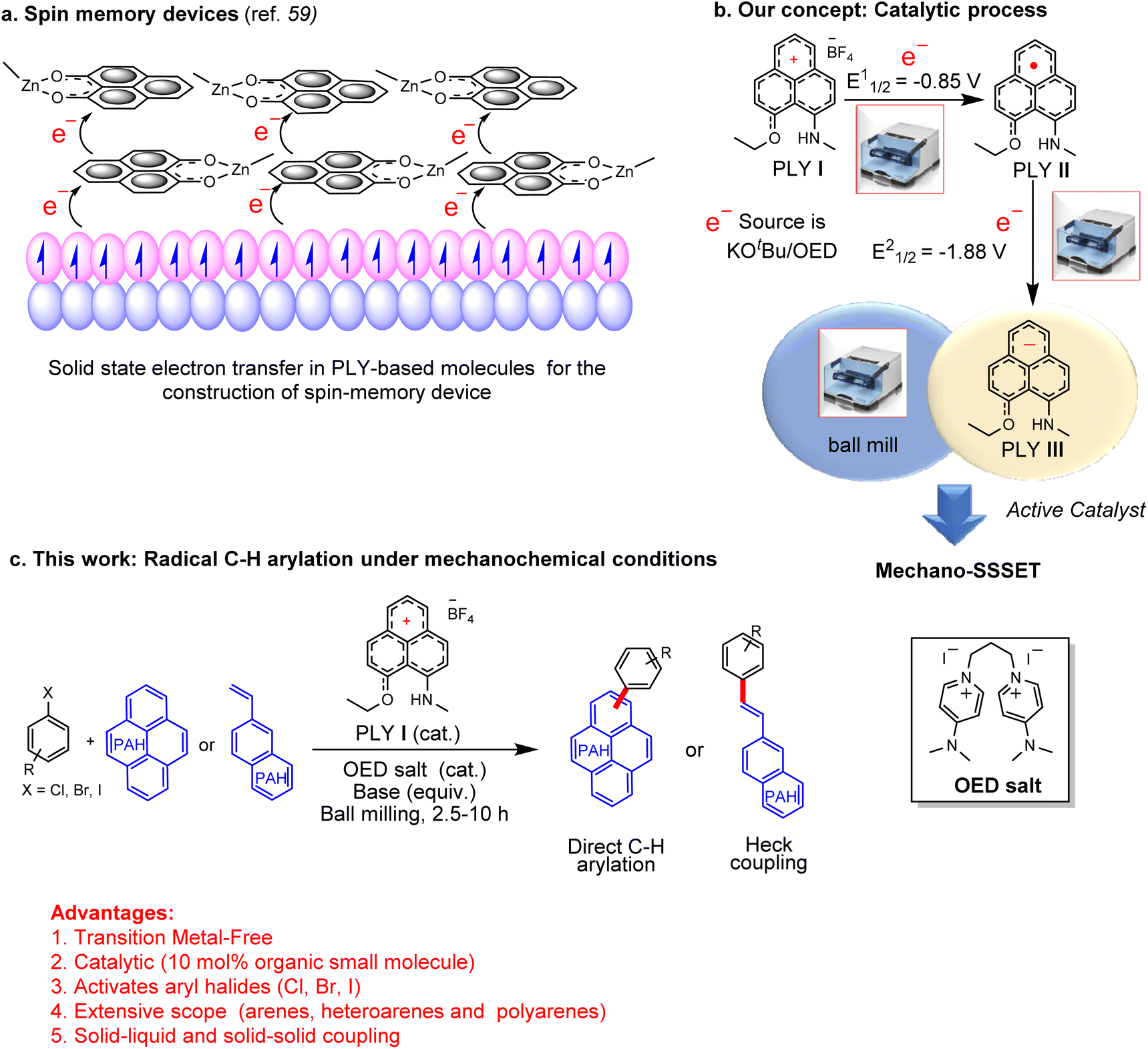 | ||
| Fig. 1 (a) Construction of a spin molecular device relying upon solid-state electron transfer to the phenalenyl (PLY) moiety; (b) our strategy based on solid-state electron transfer avoiding transition metal under mechanical energy: “mechano-SSSET” (c) this work: aryl halide activation under solvent-free and transition metal-free conditions accomplishing direct C–H arylation and Heck-coupling for various substrates including partially soluble or insoluble polyaromatic arenes. Organic electron donor (OED). | ||
Results and discussion
Continuing our program with phenalenyl-based radicals,60,61 we sought to explore and expand the synthetic utility of PLY-based catalysts for transition-metal-free solid-state SET using mechanical energy generated by ball mills (Fig. 1b). We have chosen the PLY (N, O) cation (PLY I) as the standard redox system, which was synthesized according to the previously reported literature method.64 In our earlier study, we noticed that the Zn-PLY complex was reduced to a Zn-PLY radical complex by interfacial solid-state electron transfer from a ferromagnetic Co-metal surface.59 Inspired by this solid-state electron transfer to the PLY moiety, we envisaged that cationic PLY could be reduced through solid-state electron transfer by an organic electron donor.Initially, we treated PLY I with various electron donors by changing the stoichiometry of reducing agents (TDAE or KOtBu or in situ generated organic electron donor (OED) or metallic K, see ESI, Table S5†) in the solid state under mechanical force within a ball mill for 15 min. The addition of one equivalent amount of reducing agent generated a green compound, which was identified as a mono-reduced PLY radical species (PLY II). The radical PLY II was characterized by EPR and solid-state UV-visible spectroscopy. The green compound showed EPR activity with a g value of 2.004 (see the inset of the solid-state UV-visible spectrum of PLY II, Fig. 2a). The UV absorbance maximum of PLY I in the solid state was observed at 471 nm, while the mono-reduced green solid PLY II showed maximum absorbance at 476 nm and 682 nm. The absorbance in the 682 nm region is identified as the signature of PLY radical species66 (Fig. 2a). Next, we studied the catalytic activity of PLY II in the solid state. However, using a ball mill, we failed to activate aryl halides with this mono-reduced green solid compound (PLY II) generated under mechanical force. Mono-reduced PLY II could not activate aryl halides in ball milling, indicating that the reduction potential of PLY II is not sufficiently high to activate aryl halides (Fig. 2a). Cyclic voltammetry (CV) of the PLY (N, O) cation (PLY I) has earlier shown64 that it can take up to two electrons (at E11/2 = −0.85 V and E21/2 = −1.88 V) successively to form double reduced PLY (N, O) species (PLY III).
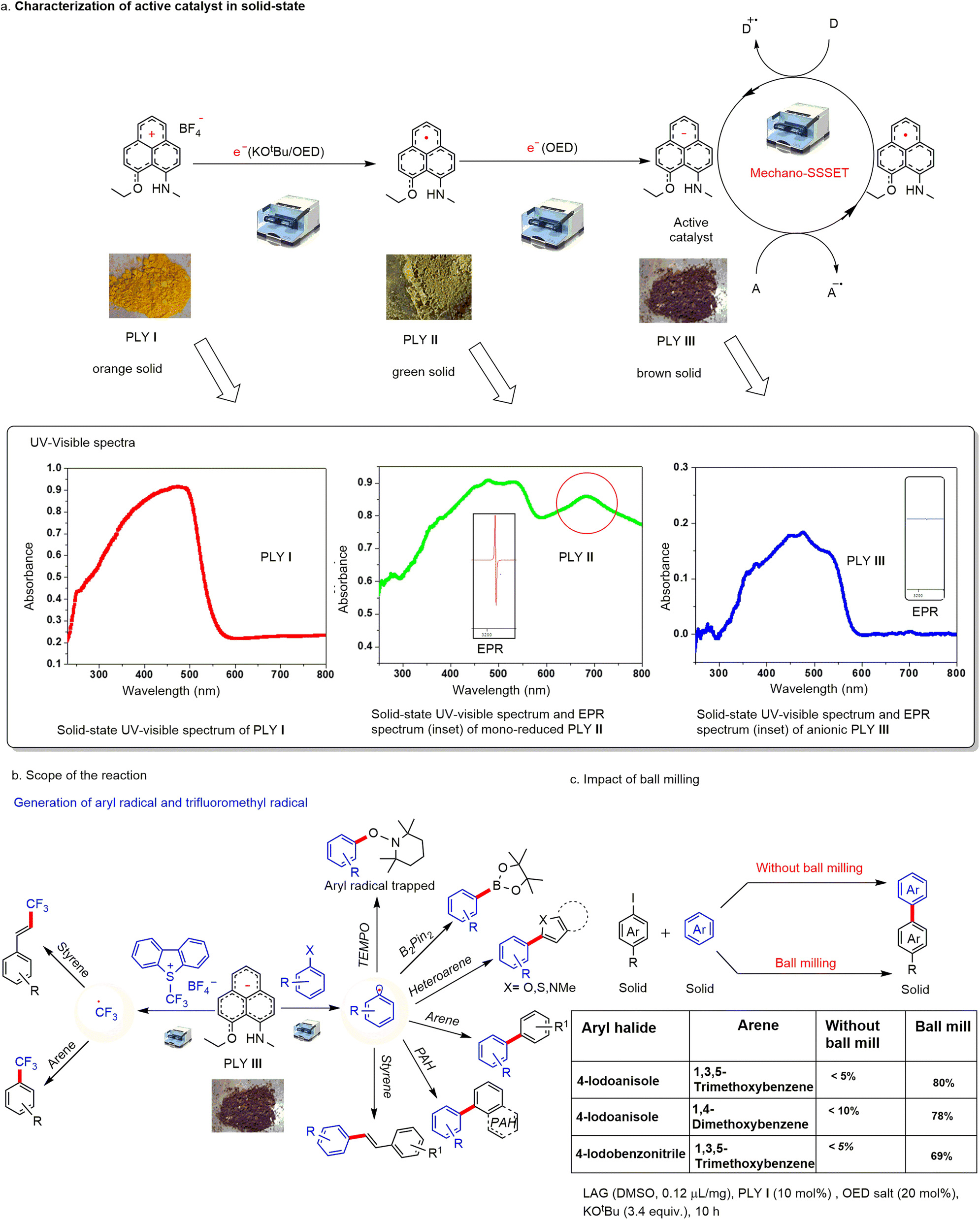 | ||
| Fig. 2 (a) Generation and characterization of reduced PLY species (PLY II and PLY III) from solid-state UV-visible and EPR spectra; (b) generation of an aryl radical and trifluoromethyl radical and scope of reaction (c) impact of ball milling in the solid-state electron transfer process. Liquid assisted grinding (LAG). | ||
It may be noted that such stepwise reduction and characterization of PLY II and PLY III were established earlier either by single crystal X-ray crystallography, NMR or EPR spectroscopy as well as DFT studies when metallic K was used as a reductant.68 Furthermore, such PLY II and PLY III were characterised by UV-vis spectroscopy.69 Very recently, we have reported the generation of PLY III in solution by stepwise reduction of PLY I with OED following a similar mechanism.70 Therefore, we tried further to reduce the green solid PLY II with another equivalent of organic electron donor (OED)67 under ball milling for 30 min, and we observed the formation of brown solid PLY III which was characterized by EPR and UV experiments. The brown solid showed EPR inactivity at 25 °C (inset of solid-state UV-visible spectrum of PLY III, Fig. 2a) and showed maximum absorbance at 474 nm, while the peak at 682 nm vanished as revealed from its solid-state UV-visible spectrum (Fig. 2a). This result indicates the formation of the double reduced anionic PLY III. To our delight, PLY III was found to be an excellent reductant to undergo solid-state single electron transfer (SSSET), generating aryl radicals by activating aryl halides. Such an aryl radical was trapped when treated with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and characterized by mass spectroscopy (Fig. 2b). Due to high reactivity, the double-reduced PLY III decomposes upon exposure to air. However, it can be stored in a glovebox for a week at −20 °C without losing its reactivity. Upon exposure to air, PLY III changes its colour immediately from brown to black and decomposes. We used this brown solid (PLY III) for aryl halide bond activation and subsequent trapping of the aryl radical to form various carbon–carbon bonds. This observation suggests that in situ generated PLY III may be able to reduce an acceptor, and subsequent reductive quenching by a donor can regenerate the active catalyst. Therefore, it may open the possibility of integrating such phenomena into a catalytic cycle (Fig. 2a).
For the optimization, we used the MM400 mixer mill with a jar of 5 mL volume and two stainless steel balls with a 10 mm diameter. The frequency of the vibration was 30 Hz, and the time of the reaction varied from the substrate to substrate. Initially, we tested the redox coupling process with two liquid reactants (aryl halide and arene/styrene) under solvent-free conditions to realize the catalytic application of the reduced hydrocarbon (PLY III). The cross-coupling reaction worked efficiently to form a variety of aryl–aryl and hetero-biaryl products with a broad substrate scope (see ESI, Scheme S11† for details). The reaction time varies from 2.5–10 h. The reaction was compatible with various electronically substituted liquid haloarenes (iodo, chloro, and bromo). Also, a range of heteroarene derivatives such as N-methyl pyrrole, furan, thiazole, benzofuran, and arene derivatives with diverse functionalization (e.g., fluoro, trifluoromethyl, t-Butyl, methoxy) worked well to derive the products with good yields (47–78%). Moreover, aryl radicals were also engaged with styrenes to derive Heck-coupled products. The reaction in a solvent-free medium was superior to the homogeneous process reported earlier68,69 as it does not need the metallic K as a reductant or DMF/DMSO as a solvent. Furthermore, we observed that the dehalohydrogenation of aryl halides is minimized in the ball milling process, which is a major side reaction that significantly reduces the isolated yield of the desired C-C cross-coupled product when performed in an organic solvent (see ESI, Scheme S30†).
With this initial success under solvent-free conditions in C–C cross-coupling between two liquid substrates, we became interested in performing C–C cross-coupling reactions between solid and liquid or solid and solid substrates. Note that the cross-coupling reaction of (partially)insoluble materials under homogeneous conditions is a formidable challenge. Hence, we assumed that the application of mechanical energy on PLY III with the assistance of ball mills could be utilized to couple solid–solid and liquid–solid substrates under neat conditions. We optimized the coupling of solid and liquid substrates to demonstrate the proposed methodology in a solvent-free medium. After extensive screening (see ESI, Table S1† for further details), we found that 4-cyanoiodobenzene (1.0 equiv., solid) and N-methylpyrrole (10.0 equiv., liquid) in the presence of 10 mol% PLY (N, O) cation PLY I, KOtBu (3.4 equiv.) and 20 mol% OED salt [1,1′-(propane-1,3-diyl)bis(4-(dimethylamino)pyridin-1-ium) iodide] afforded the bi(hetero)aryl [4-(1-methyl-1H-pyrrol-2-yl)benzonitrile] product (1) in 72% yield (Fig. 3) under an inert atmosphere. In a control experiment, we noticed that in the absence of PLY I, it only afforded 18% of the desired product, which is most likely mediated by direct electron transfer from OED. The reaction only afforded a trace amount (∼5%) of the desired product in the absence of OED salt. Note that the active catalyst PLY III was generated in situ during the ball milling process in the presence of OED. The PLY III catalyst decomposes in the presence of oxygen. Hence the reaction under open-air conditions worked with less efficiency (yield 40–54%; see ESI, Table S12†) compared to inert conditions. Notably, temperature monitoring with an infrared thermometer inside the milling jar after the reaction was recorded at a maximum 40 °C, which suggests that the temperature did not significantly increase under the optimized reaction conditions (see ESI, Fig. S18†). The ICP OES measurements of PLY I, OED salt, KOtBu, and 4-iodoanisole revealed the palladium (Pd) level < 1 parts per billion (ppb) (see ESI, Table S16†). Such observations exclude alternative Pd-catalyzed reaction pathways.
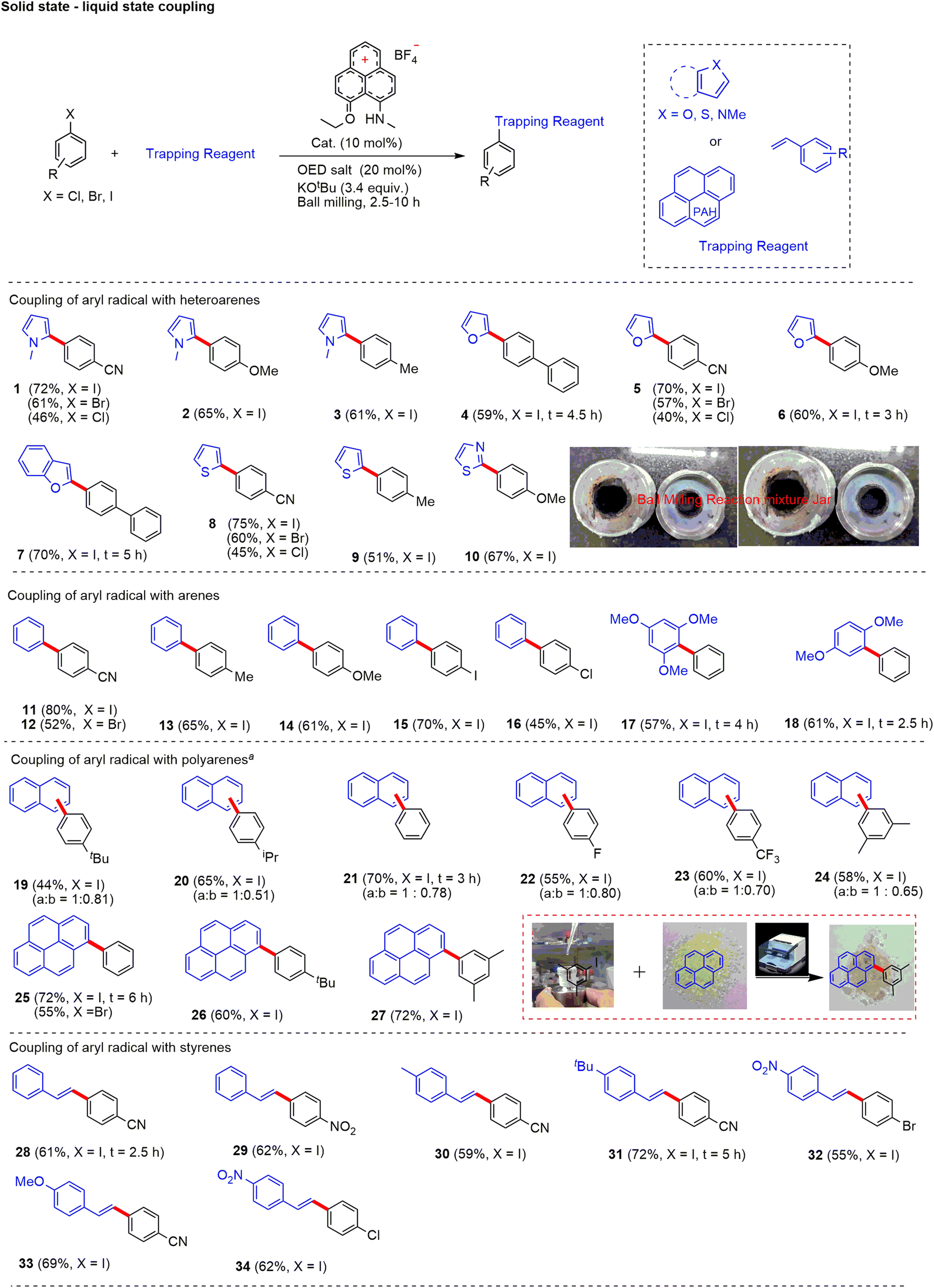 | ||
| Fig. 3 Substrate scope of direct C–H arylation with a variation of heteroarenes, arenes, and styrenes on the liquid–solid interphase under transition-metal-free and solvent-free conditions. Standard conditions: aryl halide (0.3 mmol), arene/heteroarene (10 equiv.), PLY I (10 mol%), OED salt 20 mol%, KOtBu 3.4 equiv., ball milling, aryl halide (0.4 mmol). Unless otherwise specified, the reaction time is 10 h. Reaction conditions for polyarene (5–7 equiv.), aryl halide (0.4 mmol), PLY I (10 mol%), OED salt (20 mol%), KOtBu (3.4 equiv.). The average yield of two reactions is reported. | ||
Further, to our delight, we successfully employed this protocol to develop various cross-coupling reactions such as C–H arylation with heteroarene, arene and polyarene, borylation reaction with B2Pin2 resulting in C–B bond formation, and Heck-type coupling reactions (Fig. 2b) under transition metal-free and solvent-free conditions. Furthermore, we observed that the brown-coloured PLY III species can generate the trifluoromethyl radical from the Umemoto reagent, which was used successfully to accomplish catalytic arene trifluoromethylation of styrene or arene (Fig. 2b). To investigate the role of mechanical energy during this catalytic process, we have performed a series of control experiments keeping all other parameters unchanged under ball milling conditions and normal conditions without using ball milling. In every case, the ball milling conditions outperformed, suggesting that mechanical stimulation is essential for such catalytic reactions under solvent-free conditions (Fig. 2c, also see ESI, Table S10†). The solid-state reaction with pre-generated PLY III in simple magnetic stirring under neat conditions at 40 °C afforded only a negligible amount of product (∼5%); however, the reaction under the ball milling conditions resulted in 80% yield (see ESI, Scheme S31†), which further emphasizes the importance of the mechanical energy. Furthermore, the reaction of solid substrates at higher temperatures (40 °C) in various organic solvents also failed to achieve the desired products, which establishes the advantages of the ball milling method (see ESI, Tables S14 and S15†).
Next, we explored the substrate scope for direct C–H arylation reactions with the variation of arene and polyarene coupling partners (Fig. 3). Delightfully, partially soluble polyaromatic arenes/styrenes/aryl halides were successfully engaged in the coupling sequence, including various solid–solid coupling reactions (see below). The direct coupling of aryl halides (iodo, chloro and bromo) with heteroarenes worked efficiently to deliver the desired product with good to excellent yields. Heteroarenes such as N-methyl pyrrole (1–3), furan (4–6), benzofuran (7), thiophene (8 and 9), and thiazole (10) were engaged efficiently to afford the desired C–H arylated products (up to 75% isolated yield). Notably, all heterocycles produced a single regioisomer. Moreover, the variation of electronics of aryl halides with electron-deficient to electron-rich substituents at the aryl ring was well tolerated. This method delivered a wide variety of unsymmetrical biaryl derivatives. Haloarenes (iodo, chloro, and bromo) equipped with various electron-donating and withdrawing substituents such as cyano, phenyl, alkyl, and methoxy worked well to provide the biaryl derivatives. The products (11–18) were obtained from the coupling of benzene (11–16), 1,3,5-trimethoxybenzene (17), and 1,4-dimethoxybenzene (18) to afford C–H arylated products efficiently (up to 80% isolated yield). Importantly, in all of the cases, only mono-selective arylation was observed. Moreover, dihaloarenes (1,4-diiodo or 1,4-dichloro or 1,4-diiodo arene) were engaged in the cross-coupling process to obtain mono halogen containing arylated products (15 and 16) with up to 70% isolated yields, which can be further functionalized to value-added products. Next, we tested the efficacy of this method for polyarene functionalization. Generally, polyarene activation is considered challenging due to high conjugation and less solubility in organic solvents. Mechanochemical processes are quite efficient for dealing with these poorly soluble substrates and have been reported only with Pd-based catalysts.71 However, such activation was never reported under transition metal-free and solvent-free conditions. Herein, for the first time, we arylated naphthalene (19–24) and pyrene (25–27) under transition metal-free conditions.
Next, we focused on the development of solvent-free Heck-coupling methods. The Heck reaction is a Pd-catalyzed coupling process forming C–C bonds between an aryl halide and an alkene. In the mechanochemical method, efforts were made toward solvent-free Heck-coupling using Pd-catalysts.7 However, transition metal-free Heck coupling using the mechanochemical method has not been reported. Our investigations on the scope of this methodology were continued by studying the Heck-coupling reaction of the resultant aryl radical with styrene derivatives. Delightfully, the metal-free radical mediated Heck-coupling reaction worked effectively under ball milling conditions between the liquid and solid substrates. Arylation of styrene proceeds smoothly with various electronically substituted arenes (28–34) to afford the desired products with up to 72% isolated yield. The variations of aryl halides with substitutions such as nitro (29), dihalo (32 and 34), and cyano (28, 30, 31 and 33) tolerated the reaction conditions very well (Fig. 3).
Following this, we focused on coupling between two solid substrates (Fig. 4). To facilitate the reaction, we added a minimal amount of DMSO/DMF (0.12 μL mg−1) for liquid-assisted grinding (LAG). LAG helped to mix the reactants in the ball mill for an efficient solid-state reaction.72 Solid arenes such as 1,3,5-trimethoxy and 1,4-dimethoxy coupled efficiently with the aryl radical generated from solid haloarenes to afford the desired products (35–44) with a yield up to 79%. Moreover, polyarenes such as pyrene (45 and 46), anthracene (47), phenanthrene (48), and naphthalene (49 and 50) worked well to offer arylated products with up to 68% yield. For some substrates (36, 38, 41, 44, 48 and 52), reactions were completed faster (2.5–6 h) than the optimized time (10 h). Additionally, we noticed that without LAG, solid–solid coupling worked with moderate efficiency to afford the expected products with lower yield (by 10–15%) in comparison to the LAG-method. Next, we tried trifluoromethylation of solid styrene and arene using the Umemoto reagent. The reaction worked efficiently with these solid substrates affording the fluoroalkylated alkene derivative (54, 61%). Note that a similar outcome has only been reported through a Pd(0) catalyst.73 Further, the reaction of electron-rich solid arene provided trifluoromethyl arene (55) in 52% yield. Moreover, the aryl radical generated from trifluoromethyl substituted iodoarene reacted successfully with B2Pin2 to deliver the corresponding borylated product 56 with a 65% yield.
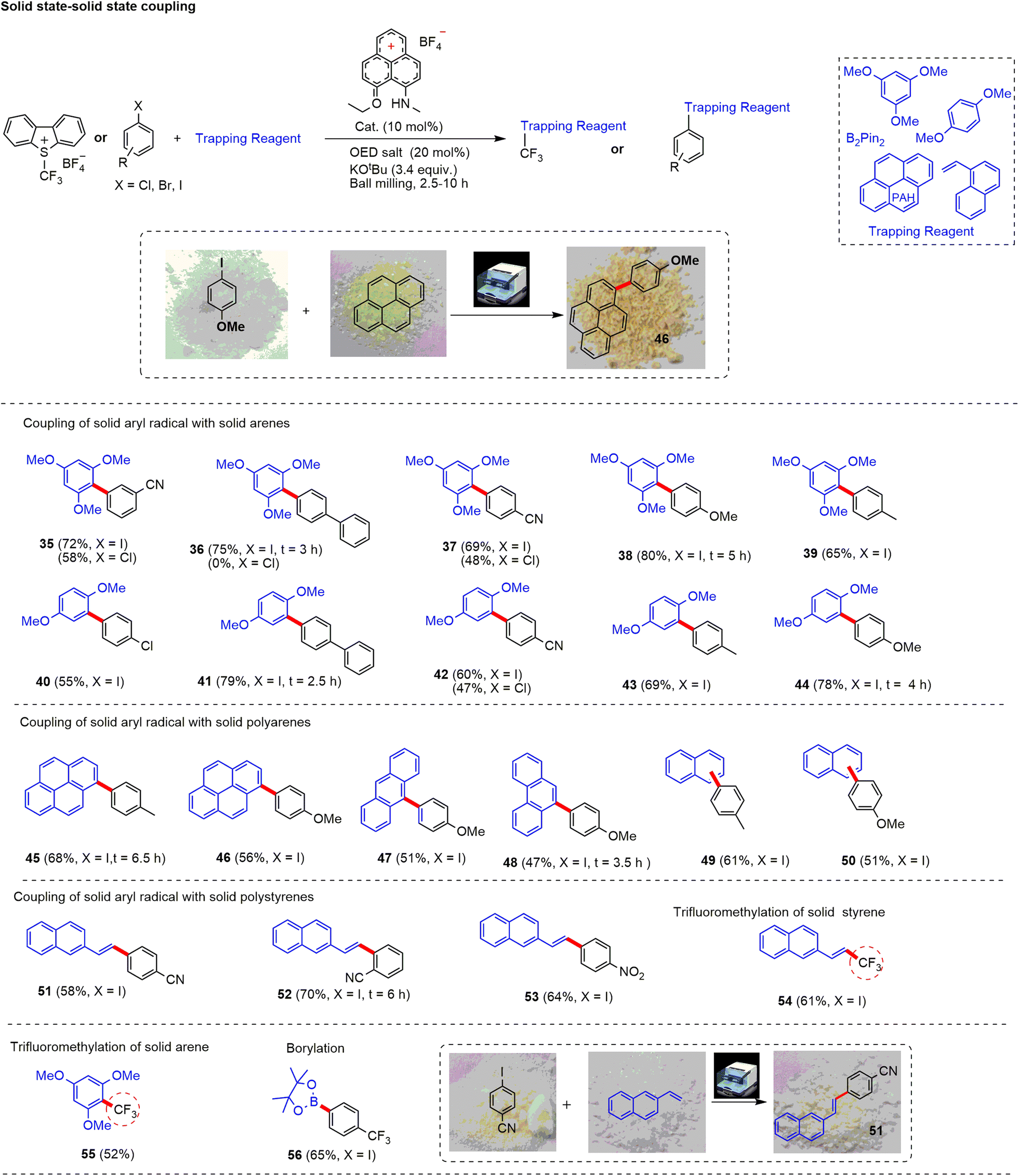 | ||
| Fig. 4 Substrate scope of solid–solid coupling reactions. Reaction conditions: aryl halide (0.3 mmol), styrene (3.5–5 equiv.), PLY I (10 mol%), OED salt 20 mol%, KOtBu 3.4 equiv., LAG (DMSO, 0.12 μL mg−1), ball milling, unless otherwise specified, the reaction time is 10 h. | ||
Conclusion
The outcome of various C–C cross-coupling reactions described above reveals for the first time that such reactions can be accomplished under solvent-free and transition metal-free conditions with broad substrate scopes. This mechanochemical method is suitable for coupling between liquid–liquid, liquid–solid, and solid–solid substrates. The diverse scope of the process covers an array of organic transformations such as C–H arylation, Heck-coupling, borylation, and trifluoromethylation reactions. In addition, the C–H functionalization of partially soluble or insoluble materials is quite difficult. This method can overcome these limitations under mechanochemical conditions. The efficacy of this process was attributed to the ability of the solid-state electron transfer process to generate the doubly reduced phenalenyl-species (PLY III) under ball milling conditions. Historically such ability of solid-state electron transfer in phenalenyl-based molecules was known over several decades in designing various spin-based molecular materials. This study establishes for the first time that such a property can be integrated into a catalytic cycle in the solid state. This result is anticipated to open up a new domain of applications of a small organic molecule as a catalyst using this mechano-SSSET or the mechanoredox concept in catalysis.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
S. K. M. conceived the original approach of this work. S. K. M., A. B., and Am. B. (Amit) designed the experiments. Am. B. performed all experiments. S. K. M., A. B., and Am. B. analyzed the results and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the generous financial support of the CSIR, India (Grant No. 01/3090/21/EMR-II). Anup Bhunia thanks DST SERB for the Ramanujan Fellowship Grant (RJF/2020/000099). Amit Biswas thanks CSIR-New Delhi for a senior research fellowship.Notes and references
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- I. Halasz, A. Puškarić, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. E. Dinnebier, B. Patel, W. Jones, V. Strukil and T. Friščič, Angew. Chem., Int. Ed., 2013, 52, 11538–11541 CrossRef CAS PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef.
- F. Gomollon-Bel, Ten Chemical Innovations That Will Change Our World, Chem. Int., 2019, 41, 12–17 CAS.
- A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- K. Kubota and H. Ito, Trends Chem., 2020, 2, 1066–1081 CrossRef CAS.
- D. Tan, C. Mottillo, A. D. Katsenis, V. Štrukil and T. Friščič, Angew. Chem., Int. Ed., 2014, 53, 9321–9324 CrossRef CAS PubMed.
- K. Kubota, T. Seo, K. Koide, Y. Hasegawa and H. Ito, Nat. Commun., 2019, 10, 111 CrossRef PubMed.
- R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed.
- V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed.
- A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763–775 CrossRef.
- P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202207118 CAS.
- K. J. Ardila-Fierro, S. Lukin, M. Etter, K. Užarević, I. Halasz, C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2020, 59, 13458–13462 CrossRef CAS PubMed.
- C. Bolm and J. G. Hernández, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- A. Studer and D. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- B. G. Fiss, A. J. Richard, G. Douglas, M. Kojic, T. Friščič and A. Moores, Chem. Soc. Rev., 2021, 50, 8279–8318 RSC.
- K. Budny-Godlewski, I. Justyniak, M. K. Leszczyński and J. Lewiński, Chem. Sci., 2019, 10, 7149–7155 RSC.
- K. Kubota, N. Toyoshima, D. Miura, J. Jiang, S. Maeda, M. Jin and H. Ito, Angew. Chem., Int. Ed., 2021, 60, 16003–16008 CrossRef CAS PubMed.
- J. M. McBride, Acc. Chem. Res., 1983, 16, 304–312 CrossRef CAS.
- M. Kuzuya, J. Niwa and S.-I. Kondo, Mol. Cryst. Liq. Cryst., 2006, 277, 343–349 CrossRef.
- H. Sharma, N. Kaur, N. Singh and D. O. Jang, Green Chem., 2015, 17, 4263–4270 RSC.
- M. J. Muñoz-Batista, D. Rodriguez-Padron, A. R. Puente-Santiago and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 9530–9544 CrossRef.
- C. Espro and D. Rodríguez-Padrón, Curr. Opin. Green Sustain. Chem., 2021, 30, 100478 CrossRef CAS.
- A. I. Martín-Perales, A. M. Balu, I. Malpartida and R. Luque, Curr. Opin. Green Sustain. Chem., 2022, 38, 100714 CrossRef.
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed.
- Y. Pang, J. W. Lee, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 22570–22576 CrossRef CAS PubMed.
- C. Schumacher, J. G. Hernández and C. Bolm, Angew. Chem., Int. Ed., 2020, 59, 16357–16360 CrossRef CAS PubMed.
- M. M. Amer, R. Hommelshiem, C. Schumacher, D. Kong and C. Bolm, Faraday Discuss., 2023, 241, 79–90, 10.1039/d2fd00075j.
- J. A. Leitch and D. L. Browne, Eur. J. Chem., 2021, 27, 9721–9726 CrossRef CAS PubMed.
- H. Lv, X. Xu, J. Li, X. Huang, G. Fang and L. Zheng, Angew. Chem., Int. Ed., 2022, 61, e202206420 CAS.
- P. Chakma, S. M. Zeitler, F. Baum, J. Yu, W. Shindy, L. D. Pozzo and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202215733 CrossRef CAS PubMed.
- Z. Ren, Y. Peng, H. He, C. Ding, J. Wang, Z. Wang and Z. Zhang, Chin. J. Chem., 2023, 41, 111–128 CrossRef.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- J. M. Lassaletta, Nat. Commun., 2020, 11, 3787 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- S. Reischauer and B. Pieber, iScience, 2021, 24, 102209 CrossRef CAS PubMed.
- A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. L. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- W. Liu, H. Cao, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed.
- C. L. Sun, H. Li, D. G. Yu, M. Yu, X. Zhou, X. Y. Lu, K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed.
- X. Chi, M. E. Itkis, B. O. Patrick, T. M. Barclay, R. W. Reed, R. T. Oakley, A. W. Cordes and R. C. Haddon, J. Am. Chem. Soc., 1999, 121, 10395–10402 CrossRef CAS.
- S. K. Mandal, M. E. Itkis, X. Chi, S. Samanta, D. Lidsky, R. W. Reed, R. T. Oakley, F. S. Tham and R. C. Haddon, J. Am. Chem. Soc., 2005, 127, 8185–8196 CrossRef CAS PubMed.
- S. K. Mandal, S. Samanta, M. E. Itkis, D. W. Jensen, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 1982–1994 CrossRef CAS PubMed.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281–284 CrossRef CAS PubMed.
- S. K. Pal, P. Bag, A. Sarkar, X. Chi, M. E. Itkis, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2010, 132, 17258–17264 CrossRef CAS PubMed.
- P. Bag, S. K. Pal, M. E. Itakis, A. Sarkar, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2013, 135, 12936–12939 CrossRef CAS PubMed.
- M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443–1445 CrossRef CAS PubMed.
- J. S. Miller, Angew. Chem., Int. Ed., 2003, 42, 27–29 CrossRef CAS PubMed.
- R. G. Hicks, Nat. Chem., 2011, 3, 189–191 CrossRef CAS PubMed.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS PubMed.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- A. Ueda, S. Suzuki, K. Yoshida, K. Fukui, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2013, 52, 4795–4799 CrossRef CAS PubMed.
- K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, P. Lazic, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blugel, M. Munzenberg and J. S. Moodera, Nature, 2013, 493, 509–513 CrossRef CAS PubMed.
- J. Ahmed and S. K. Mandal, Chem. Rev., 2022, 122, 11369–11431 CrossRef CAS PubMed.
- A. Mukherjee, S. C. Sau and S. K. Mandal, Acc. Chem. Res., 2017, 50, 1679–1691 CrossRef CAS PubMed.
- R. C. Haddon, Nature, 1975, 256, 394–396 CrossRef CAS.
- R. C. Haddon, Aust. J. Chem., 1975, 28, 2343–2351 CrossRef CAS.
- S. R. Roy, A. Nijamudheen, A. Pariyar, A. Ghosh, P. K. Vardhanapu, P. K. Mandal, A. Datta and S. K. Mandal, ACS Catal., 2014, 4, 4307–4319 CrossRef CAS.
- H. Guo, A. Jaworski, Z. Chen, C. Lu, A. Slabon and U. Häassermann, Inorganics, 2021, 9, 62 CrossRef CAS.
- K. Uchida and T. Kubo, J. Synth. Org. Chem., Jpn., 2016, 74, 1067–1077 CrossRef.
- J. A. Murphy, J. Garnier, S. R. Park, F. Schoenebeck, S. Z. Zhou and A. T. Turner, Org. Lett., 2008, 10, 1227–1230 CrossRef CAS PubMed.
- J. Ahmed, P. Datta, A. Das, S. Jomy and S. K. Mandal, Chem. Sci., 2021, 12, 3039–3049 RSC.
- A. Banik and S. K. Mandal, ACS Catal., 2022, 12, 5000–5012 CrossRef CAS.
- S. Sil, A. S. Bhaskaran, S. Chakraborty, B. Singh, R. Kuniyil and S. K. Mandal, J. Am. Chem. Soc., 2022, 144, 22611–22621 CrossRef CAS PubMed.
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270–1274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06119h |
| This journal is © The Royal Society of Chemistry 2023 |