
Integrating electrocatalysis with biocatalysis for asymmetric synthesis
Yuqi
Lin
 *a,
Jiage
Yu
a and
Ke-Yin
Ye
*ab
*a,
Jiage
Yu
a and
Ke-Yin
Ye
*ab
aKey Laboratory of Molecule Synthesis and Function Discovery (Fujian Province University), College of Chemistry, Fuzhou University, Fuzhou 350108, China. E-mail: Linyuqi@fzu.edu.cn; kyye@fzu.edu.cn
bSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 6th November 2024
Abstract
The integration of electrocatalysis with biocatalysis has recently been developed for the stereoselective synthesis of valuable compounds. This integration leverages the high efficiency and exquisite selectivity of enzymes along with the tunability of electrochemical reactions, providing a versatile approach for asymmetric synthesis with broad prospects. In this highlight, recent advances and future development directions in asymmetric synthesis that integrate electrocatalysis with biocatalysis are presented and discussed. We foresee that more valuable stereoselective transformations using this integrated strategy will be developed in the future.
Enzyme-based biocatalysis has become a powerful tool for generating enantiomerically pure chemicals in organic synthesis due to high enantioselectivity, gentle reaction conditions, and minimal by-product formation.1 Enantioselective oxidoreductases, which account for the majority of known enzymes, play an essential role in redox reactions.2 They have been extensively exploited to facilitate the efficient production of a variety of chiral compounds in asymmetric synthesis.3 However, the inability to efficiently regenerate expensive reduced cofactors has restricted their application.4 Electrochemistry offers an attractive and promising alternative for cofactor regeneration with the advantages of harnessing electricity as a clean energy source, getting rid of by-product generation, and simplifying the product separation process.5 Integrating biocatalysis with electrocatalysis brings broader benefits, such as improved reaction efficiency, enhanced selectivity, and the environmentally benign and sustainable production of enantiopure chemicals.
Electrochemistry, a green and sustainable synthetic tool for organic transformations, has attracted considerable interest in synthetic chemistry.6 For instance, electrochemical approaches have recently emerged as mild and efficient alternatives for the synthesis of organofluorines.7 The application of electrons as cheap and renewable reagents avoids stoichiometric external oxidants or reductants and eliminates undesired by-product generation.8 Furthermore, the reactivity and selectivity of electrochemical reactions can be regulated by precisely controlling the applied potential and electric current.9 However, asymmetric electrosynthesis remains a challenging field of development and typically requires costly transition metals and chiral catalysts.10 Additionally, the high reactivity of oxidatively or reductively generated radicals or reactive intermediates poses a challenge for asymmetric electrosynthesis.11 Integrating electrocatalysis with biocatalysis provides a versatile approach for achieving stereo-control in electrochemistry.
The integration of biocatalysis and electrocatalysis is intriguing due to the combined merits of the high efficiency and exquisite selectivity of enzymes along with the tunability of electrochemical reactions.12 This emerging strategy provides a powerful way to tune substrate and product selectivity, further enriching catalytic strategies to explore more valuable stereoselective transformations for asymmetric synthesis. There are two main modes involved in this integration (Scheme 1). In the first mode, (modified) electrodes are used to regenerate cofactors for the cofactor-dependent oxidoreductases to achieve their natural redox activity. The second mode involves the electroenzymatic cascade reaction, where the electrocatalytic redox reaction initially converts the substrate or co-substrate into an intermediate, which is subsequently catalyzed by the enzymes to generate the chiral product. Although the application of this combined strategy in asymmetric organic synthesis is still in its infancy, recent breakthroughs indicate that it holds great promise for the exploration of novel asymmetric reactions. Herein, we highlight recent advances in asymmetric synthesis that integrate electrocatalysis with biocatalysis and discuss future directions in this field.
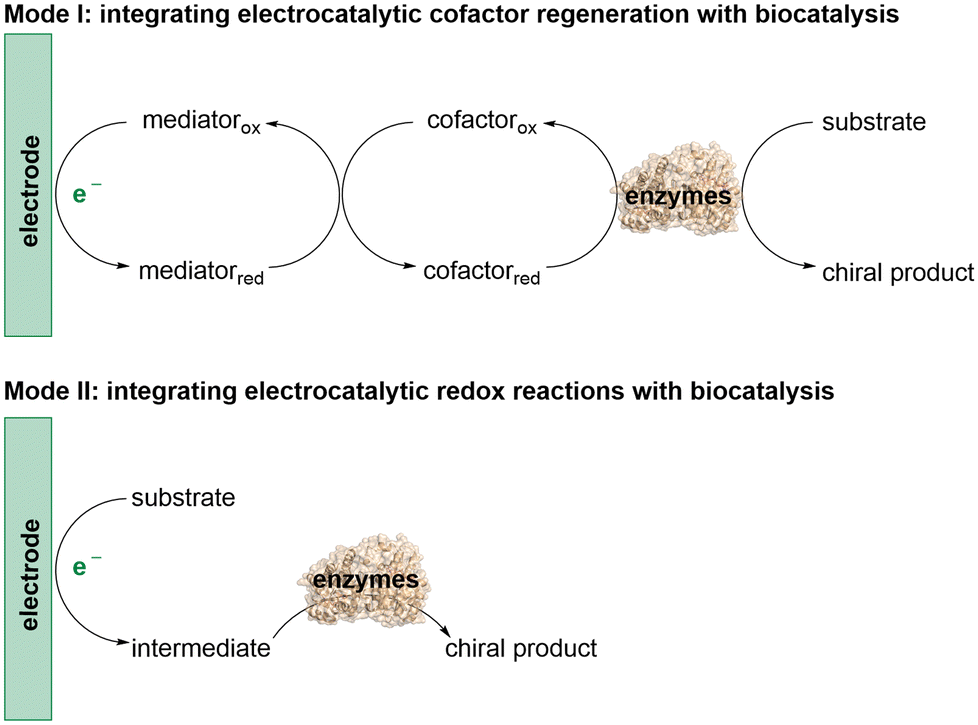 | ||
| Scheme 1 Modes in the integration of electrocatalysis and biocatalysis. | ||
The Minteer group constructed an enhanced bioelectrocatalytic platform for N2 fixation that combines nitrogenase, diaphorase (DI), L-alanine dehydrogenase (AlaDH), and ω-transaminase (ω-TA) with electrodes to convert N2 into chiral amine intermediates in one-pot under mild conditions (Scheme 2).13 This bioelectrocatalytic system used an H-shaped dual-chamber reactor with oxidized methyl viologen (MV2+) as an electron mediator. The cathode directly provides electrons for reduced MV+, realizing the efficient regeneration of MV+. The reduced MV+ then donates electrons to nitrogenase to facilitate the reduction of N2 to NH3. DI consumes reduced MV+ to regenerate NADH, while MV+ is recycled by the cathode. AlaDH, using NADH as a coenzyme, captures NH3 and transfers it to the acceptor pyruvate to generate alanine. Then the amino group of alanine is transferred by ω-TA to various ketone substrates, ultimately generating enantiomerically pure chiral amine intermediates. Notably, AlaDH consumes the by-product pyruvate to reproduce alanine, shifting the reaction equilibrium towards the amine product side. Employing this system, different prochiral ketones could be aminated to produce the corresponding chiral amines with perfect stereoselectivity (>99% ee).
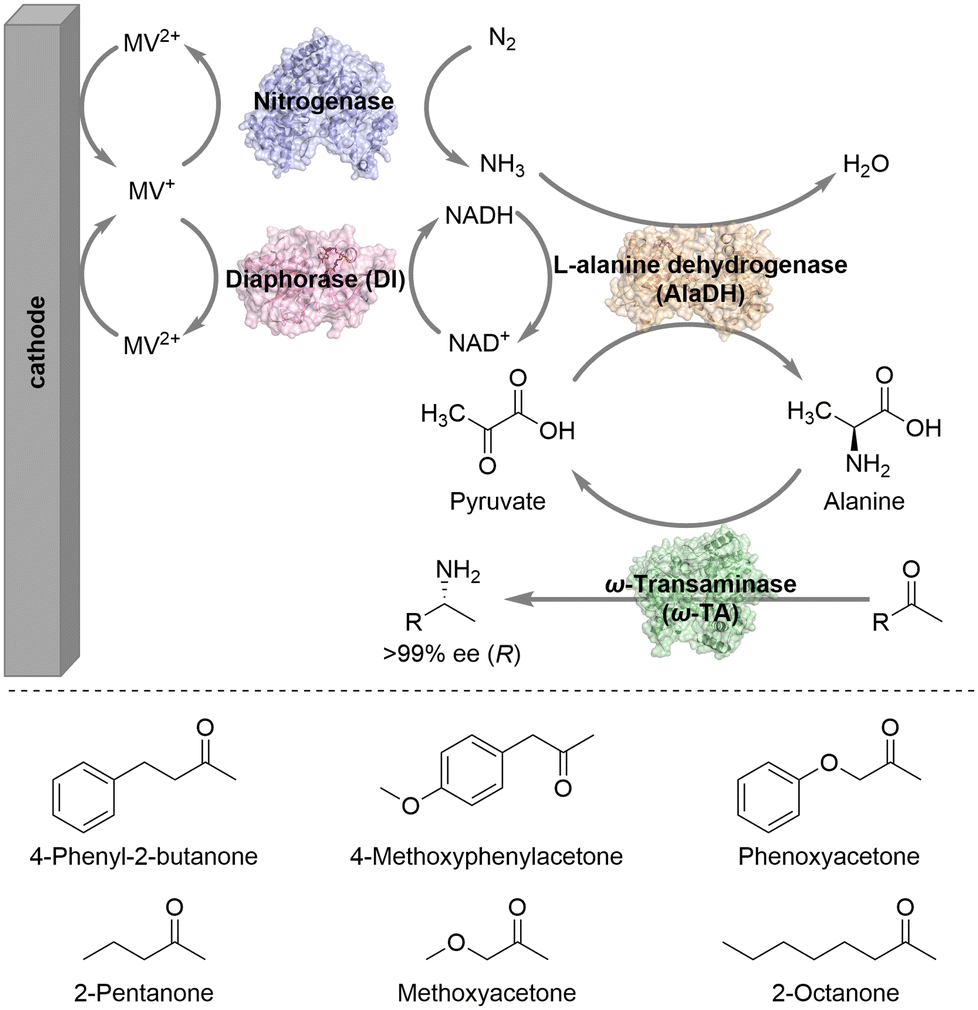 | ||
| Scheme 2 A bioelectrocatalytic platform converts N2 into chiral amine intermediates (mode I). | ||
To overcome the low efficiency of cofactor regeneration and substrate solubility problems in oxidoreductase-based asymmetric synthesis, Minteer and colleagues developed a novel two-phase bioelectrocatalytic system. This system enables the efficient synthesis of chiral β-hydroxy nitriles by integrating alcohol dehydrogenase (AdhS), halohydrin dehalogenase (HHDH) with diaphorase (DH)-modified electrodes (Scheme 3).14 Diaphorase (DH) was immobilized on the electrode surface with a cobaltocene-modified poly(allylamine) redox polymer to form DH/Cc-PAA bioelectrode. In this bioelectrode, Cc-PAA polymer mediates the transfer of electrons from the electrode to DH, which further utilizes the electrons to catalyze the reduction of NAD+ to efficiently regenerate the reduced cofactor NADH. NADH-dependent AdhS then catalyzes the reduction of the model substrate COBE to (S)-CHBE by consuming NADH. HHDH catalyzes the dehalogenation and cyanation of (S)-CHBE to (R)-CHCN. Notably, the use of methyl tert-butyl ether (MTBE) as the second organic phase enhances substrate loading, prevents spontaneous substrate hydrolysis, alleviates substrate inhibition of HHDH activity, improves product yields, and prolongs the lifetime of DH/Cc-PAA bioelectrode. By applying this two-phase bioelectrocatalytic system, different chiral β-hydroxy nitriles could be produced with high enantioselectivity (up to 96% ee).
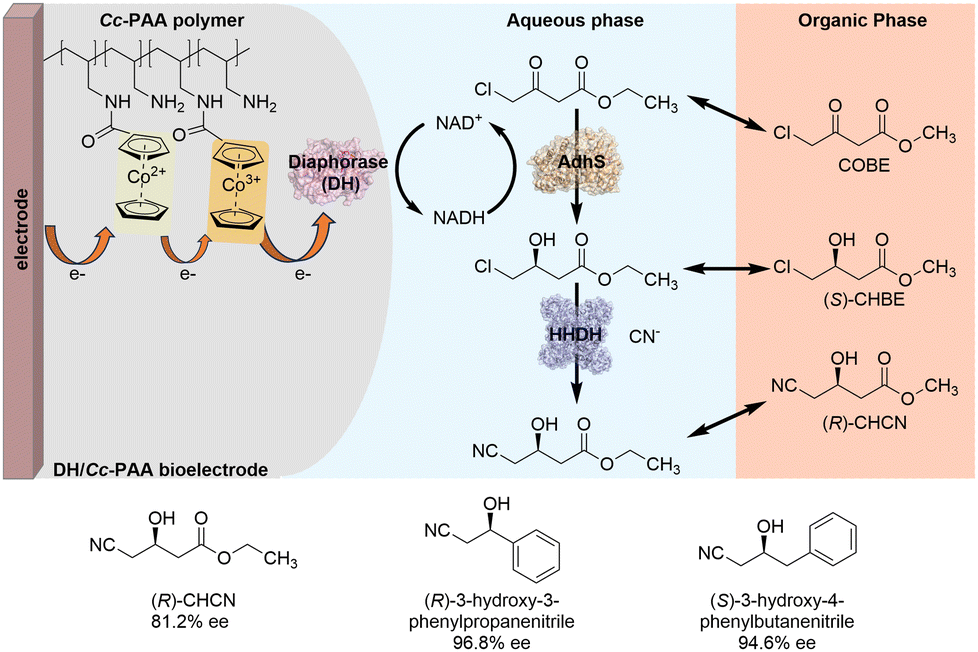 | ||
| Scheme 3 Two-phase bioelectrocatalytic system for the efficient synthesis of chiral β-hydroxy nitriles (mode I). | ||
In recent years, the integration of electrochemistry and biocatalysis for the conversion of CO2 into value-added chemicals has garnered significant attention.15 The Plumere group presented a bioelectrocatalytic approach that combines electricity-driven cofactor NADPH regeneration with enzymatic CO2 fixation for the stereoselective C–C bond formation (Scheme 4).16 They employed 2,2′-viologen modified polyvinyl alcohol (V2+-PVA) hydrogel as the redox polymer to co-immobilize ferredoxin NADP+ reductase (FNR) with NADPH-dependent crotonyl-CoA carboxylase/reductase (Ccr) on electrode. The V2+-PVA/FNR films showed excellent electrocatalytic activity and stability, providing efficient electron transfer from the electrode to FNR for reducing NADP+ to NADPH. Sufficient supply of NADPH enabled the Ccr-catalyzed incorporation of CO2 into crotonyl-CoA for the stereoselective generation of (2S)-ethylmalonyl-CoA. After optimization of conditions, the system was successfully scaled up to achieve a maximum yield of 57% and a faradaic efficiency of 91%.
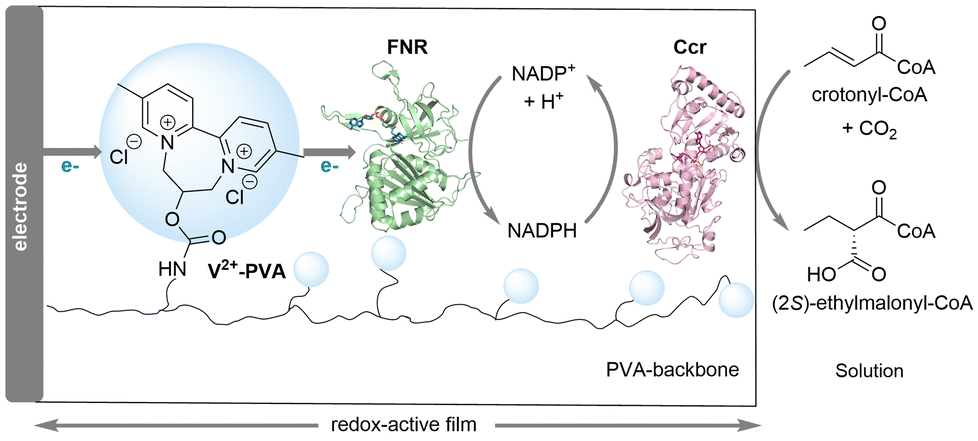 | ||
| Scheme 4 A bioelectrocatalytic approach for CO2 fixation and the stereoselective C–C bond formation (mode I). | ||
The free diffusion of Ccr in solution generally leads to a significant decrease in reaction yields. Therefore, by co-immobilizing the enzymes within a smaller volume, the proximity of the biocatalysts facilitates rapid diffusive transport of intermediates and cofactors among the active sites of the enzymes, resulting in increased reaction rates and enhanced electrocatalytic performance.
Armstrong and co-workers reported a unique approach for driving and controlling enantioselective conversions by coupling nanoconfined enzyme cascades with bidirectional electrocatalysis.17 They co-confined ferredoxin NADP+ reductase (FNR) and alcohol dehydrogenase (ADH) variants within the nanopores of an indium tin oxide (ITO) layer to form (FNR + ADH)@ITO electrode. FNR catalyzed the rapid and reversible NADP+/NADPH interconversion in a bidirectional manner controlled by the applied electrode potential. At the same time, ADH immediately recycled NADP+ or NADPH to catalyze redox interconversions between ketone and secondary alcohol enantiomers. The deracemization of alcohols could be efficiently achieved through a two-stage process in one pot with two (FNR + ADH)@ITO electrodes in sequence. In the first stage, an electrode containing an enantioselective ADH variant selectively oxidized the undesired alcohol enantiomer to ketone. After a quick switch to the electrode containing another ADH variant with opposing enantioselectivity and a pH adjustment, the ketone was irreversibly reduced to the desired alcohol enantiomer with >90% ee in the second stage.
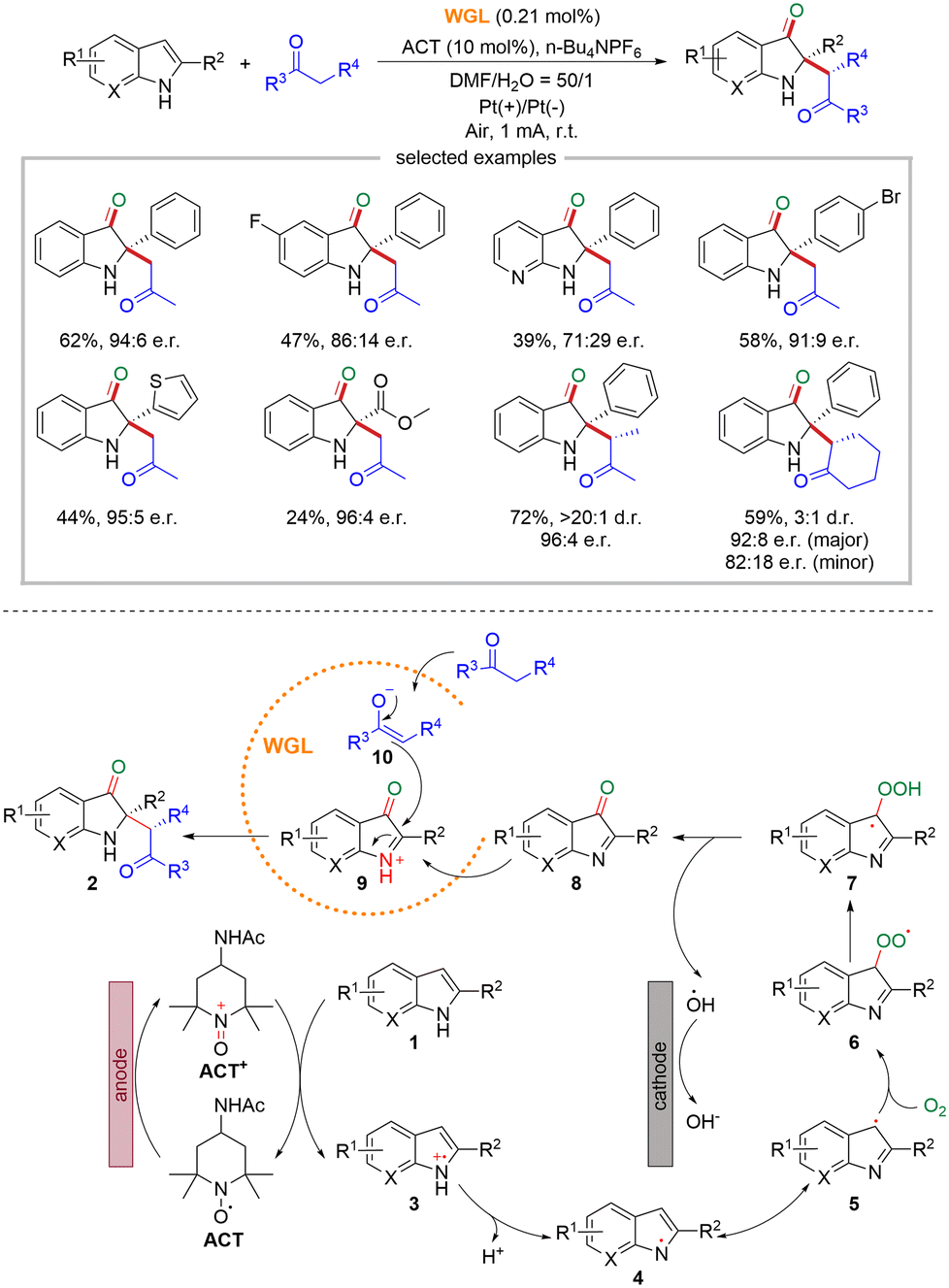
The majority of the established strategies that integrate biocatalysis and electrocatalysis were limited to oxidoreductases, while electrochemistry is used for cofactors regeneration.18 In 2022, Guan and co-workers reported an innovative approach that combined enzymes other than oxidoreductases with electrosynthesis, going beyond cofactor regeneration and expanding the scope and synthetic potential of the integrated strategy.19 They take advantage of the catalytic promiscuity of hydrolase with electrosynthesis for the asymmetric synthesis of complex chiral compounds (Scheme 5). Specifically, they carried out the enantioselective oxidative cross-coupling of 2-substituted indoles and simple ketones in an undivided electrochemical cell with wheat germ lipase (WGL) as the biocatalyst, platinum sheets as electrodes, 4-acetamido 2,2,6,6-tetramethyl-1-piperideoxy (ACT) as the redox mediator, n-Bu4NPF6 as electrolyte, and DMF/H2O as the solvent. A wide range of 2,2-disubstituted 3-carbonyl indoles featuring a stereogenic quaternary carbon center could be successfully obtained with good yields (up to 78%), excellent enantioselectivity (up to 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 e.r.) and diastereoselectivity (>20:1 d.r.).
:4 e.r.) and diastereoselectivity (>20:1 d.r.).
 | ||
| Scheme 5 Integration of the catalytic promiscuity of lipase (WGL) with electrosynthesis for asymmetric oxidative cross-coupling reaction (mode II). | ||
With regard to the mechanism, ACT is initially oxidized at the anode to generate ACT+, which subsequently oxidizes substrate 1 to the N-centered radical cation 3via single electron transfer. Then N-centered radical cation 3 undergoes deprotonation to generate N-centered radical 4. Its resonance structure, the C-centered radical 5, reacts with O2 to produce radical 6, which further undergoes hydrogen transfer to afford radical 7. Subsequently, radical 7 cleaves a hydroxyl radical to produce intermediate 8. In the active site of WGL, intermediate 8 is protonated to form the electrophilic cationic intermediate 9, while ketone loses a proton to generate enolate 10, which then nucleophilically attacks intermediate 9 to yield the final product 2.
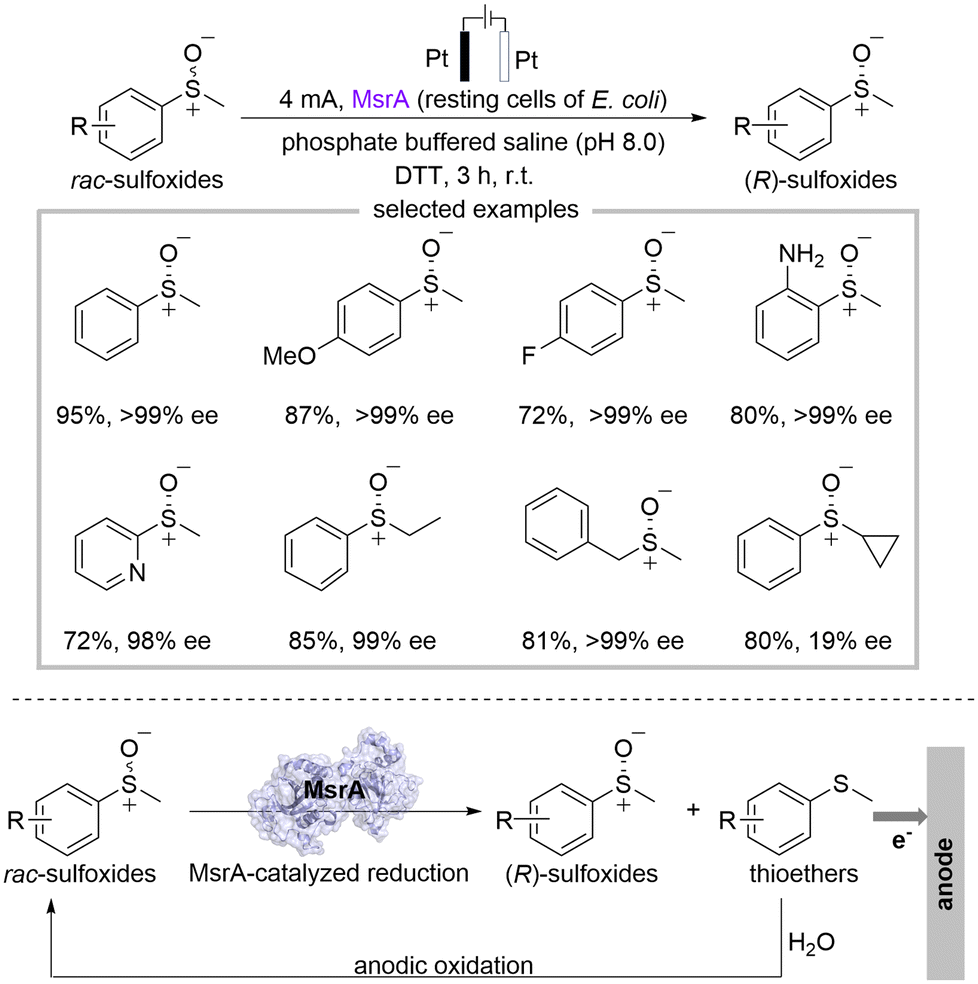
Xu's group reported a deracemization strategy for achieving chiral sulfoxides through the combination of electrocatalysis and biocatalysis (Scheme 6).20 The deracemization process was conducted in an undivided cell with platinum electrodes, methionine sulfoxide reductase A (MsrA) as the biocatalyst, dithiothreitol (DTT) as terminal reductants, and the phosphate buffered saline (PBS) as the electrolyte solution under 4 mA constant current in the atmosphere. Chiral sulfoxides can be achieved up to 95% yield with 99% ee by integrating MsrA-catalyzed reduction for kinetic resolution of racemic sulfoxides with the non-stereoselective anodic oxidation of thioethers. This strategy can be successfully applied to a wide range of substituted sulfoxides, showing good functional group tolerance. Remarkably, the reaction can be easily scaled up. Mechanistic experiments revealed that the anodic oxidation of thioethers proceeded via a radical pathway, and H2O served as the oxygen atom source during the anodic oxidation process.
 | ||
| Scheme 6 Combination of electrocatalysis and biocatalysis for chiral sulfoxides (mode II). | ||
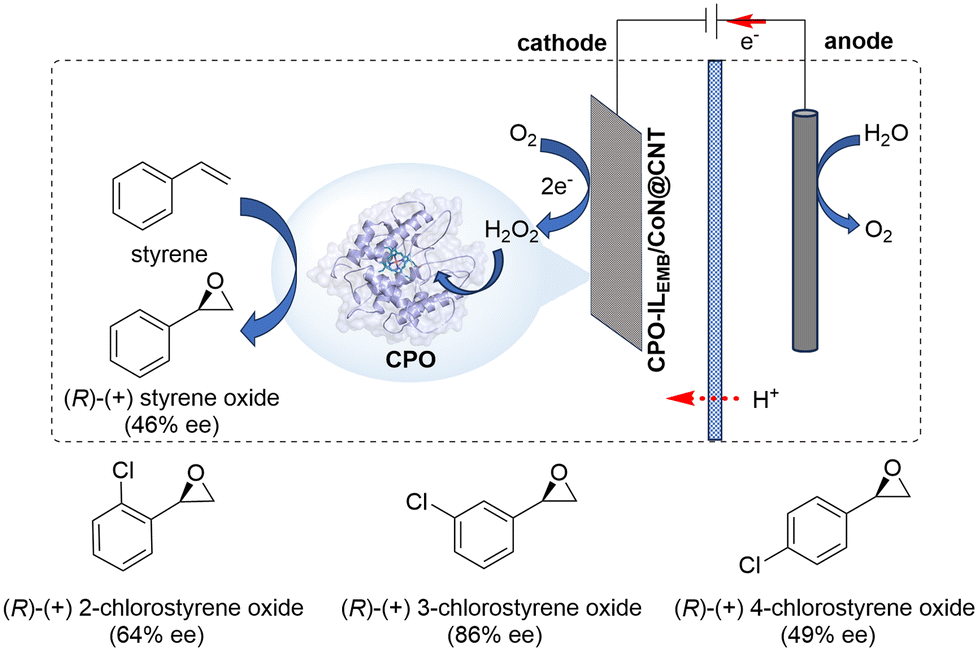
Recently, Chen and co-workers developed a one-pot electroenzymatic cascade platform that integrated the advantages of enzymatic catalysis and electrocatalysis for efficient asymmetric epoxidation of styrene derivatives (Scheme 7).21 Chloroperoxidase (CPO), a heme-containing peroxidase, was functionalized using 1-ethyl-3-methylimidazolium bromide (ILEMB) and immobilized on cobalt nitrogen-doped carbon nanotubes (CoN@CNT) to form CPO-ILEMB/CoN@CNT biohybrid. This biohybrid was then dropped on electrode surfaces. The electrochemical measurements showed that CoN@CNT has excellent electrocatalytic properties for oxygen reduction, producing H2O2 with high Faradaic efficiency and rate. In situ electrogenerated H2O2 was directly utilized by CPO to catalyze the asymmetric epoxidation of styrene, with the merit of maintaining the high catalytic activity of CPO. In a sealed H-type electrolytic cell, CPO-ILEMB/CoN@CNT achieved 27% styrene conversion within 30 min and yielded 46% ee of chiral styrene oxide with a cascade catalytic efficiency 17 times higher than that of free CPO-ILEMB in solution. The universality of this electroenzymatic cascade platform was further validated by the successful epoxidation of various chloro-substituted styrene derivatives with high enantioselectivity (49–86% ee).
 | ||
| Scheme 7 A one-pot electroenzymatic cascade platform for asymmetric epoxidation of styrenes (mode II). | ||
In this highlight, the latest advances in asymmetric synthesis that integrate electrocatalysis and biocatalysis have been demonstrated. This versatile approach has gained prominence as it combines the advantages of both electrocatalysis and biocatalysis. By utilizing electricity as a clean energy source and employing enzymes with high stereoselectivity and even with catalytic promiscuity as biocatalysts, the green and efficient production of chiral chemicals can be achieved without the need for stochiometric redox reagents while minimizing the production of by-products. Based on these inspiring breakthroughs, future development direction should focus on the following aspects.
(1) Exploring new types of enzymes for integration with electrocatalysis, which will greatly diversify the reaction types and broaden the application range.
(2) Further design and construction of novel and efficient electroenzymatic cascade reaction systems with good compatibility between enzymes and electrodes, which is crucial for realizing more enantioselective transformations.
(3) Applications of protein engineering such as directed evolution to enhance the activity and stability of enzymes under electrochemical and non-aqueous conditions to achieve new and interesting reactivity.
(4) Design and application of aqueous–organic biphasic media and improved enzyme immobilization techniques as pertinent strategies to improve reaction productivity.
(5) Exploring the non-natural activities of enzymes driven by electricity to achieve novel-to-nature reactivity,22 which will enhance the versatility of enzymes and open up exciting possibilities for new synthetic pathways and the discovery of novel reactions.
We envisage that the integration of biocatalysis and electrocatalysis will be a versatile approach for asymmetric synthesis, leading to more valuable stereoselective transformations and promising for a multitude of synthetic applications.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Educational Research Project for Young and Middle-aged Teachers in Fujian Province (JAT231005), Fuzhou University (511311), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2024Z08) is gratefully acknowledged.References
- (a) F. H. Arnold, Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture), Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed; (b) G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, The Crucial Role of Methodology Development in Directed Evolution of Selective Enzymes, Angew. Chem., Int. Ed., 2020, 59, 13204–13231 CrossRef CAS PubMed; (c) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Engineering the third wave of biocatalysis, Nature, 2012, 485, 185–194 CrossRef CAS PubMed; (d) P. de María, G. de Gonzalo and A. Alcántara, Biocatalysis as Useful Tool in Asymmetric Synthesis: An Assessment of Recently Granted Patents (2014–2019), Catalysts, 2019, 9, 802 CrossRef; (e) C. K. Winkler, J. H. Schrittwieser and W. Kroutil, Power of Biocatalysis for Organic Synthesis, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed; (f) Y. Lin, W. Xue, H. Li, B. Su, J. Lin and K. Y. Ye, Advances in Enzymatic Incorporation of Small Fluorine Modules, Eur. J. Org. Chem., 2024, 27, e202400003 CrossRef CAS.
- (a) X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways, Chem, 2017, 2, 621–654 CrossRef CAS; (b) M. Yuan, M. J. Kummer, R. D. Milton, T. Quah and S. D. Minteer, Efficient NADH Regeneration by a Redox Polymer-Immobilized Enzymatic System, ACS Catal., 2019, 9, 5486–5495 CrossRef CAS.
- (a) J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Biocatalytic Oxidation Reactions: A Chemist's Perspective, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed; (b) F. Hollmann, D. J. Opperman and C. E. Paul, Biocatalytic Reduction Reactions from a Chemist's Perspective, Angew. Chem., Int. Ed., 2020, 60, 5644–5665 CrossRef PubMed.
- (a) R. A. Sheldon and D. Brady, The limits to biocatalysis: pushing the envelope, Chem. Commun., 2018, 54, 6088–6104 RSC; (b) H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Methods for the regeneration of nicotinamide coenzymes, Green Chem., 2013, 15, 1773 RSC.
- (a) C. Zhang, H. Zhang, J. Pi, L. Zhang and A. Kuhn, Bulk Electrocatalytic NADH Cofactor Regeneration with Bipolar Electrochemistry, Angew. Chem., Int. Ed., 2022, 61, e202111804 CrossRef CAS PubMed; (b) Y. Li, G. Liu, W. Kong, S. Zhang, Y. Bao, H. Zhao, L. Wang, L. Zhou and Y. Jiang, Electrocatalytic NAD(P)H regeneration for biosynthesis, Green Chem. Eng., 2024, 5, 1–15 CrossRef; (c) H. K. Song, S. H. Lee, K. Won, J. H. Park, J. K. Kim, H. Lee, S. J. Moon, D. K. Kim and C. B. Park, Electrochemical Regeneration of NADH Enhanced by Platinum Nanoparticles, Angew. Chem., 2008, 120, 1773–1776 CrossRef; (d) L. Zhang, N. Vilà, G.-W. Kohring, A. Walcarius and M. Etienne, Covalent Immobilization of (2,2′-Bipyridyl) (Pentamethylcyclopentadienyl)-Rhodium Complex on a Porous Carbon Electrode for Efficient Electrocatalytic NADH Regeneration, ACS Catal., 2017, 7, 4386–4394 CrossRef CAS.
- (a) J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls, Acc. Chem. Res., 2019, 53, 45–61 CrossRef PubMed; (b) E. J. Horn, B. R. Rosen and P. S. Baran, Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed; (c) S. R. Waldvogel and B. Janza, Renaissance of Electrosynthetic Methods for the Construction of Complex Molecules, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef CAS PubMed; (d) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Modern Electrochemical Aspects for the Synthesis of Value–Added Organic Products, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- (a) Y. Zheng, W. Lu, T. Ma and S. Huang, Recent advances in photochemical and electrochemical strategies for the synthesis of sulfonyl fluorides, Org. Chem. Front., 2024, 11, 217–235 RSC; (b) R. Shaw, N. Sihag, H. Bhartiya and M. R. Yadav, Harnessing photocatalytic and electrochemical approaches for C–H bond trifluoromethylation and fluoroalkylation, Org. Chem. Front., 2024, 11, 954–1014 RSC.
- (a) J. C. Siu, N. Fu and S. Lin, Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (b) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, A Survival Guide for the “Electro-curious”, Acc. Chem. Res., 2019, 53, 72–83 CrossRef PubMed.
- (a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, Organic Electrochemistry: Molecular Syntheses with Potential, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (b) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- (a) X. Chang, Q. Zhang and C. Guo, Asymmetric Electrochemical Transformations, Angew. Chem., Int. Ed., 2020, 59, 12612–12622 CrossRef CAS PubMed; (b) K. Yamamoto, M. Kuriyama and O. Onomura, Anodic Oxidation for the Stereoselective Synthesis of Heterocycles, Acc. Chem. Res., 2019, 53, 105–120 CrossRef PubMed; (c) L. Li, Y. Li, N. Fu, L. Zhang and S. Luo, Catalytic Asymmetric Electrochemical α–Arylation of Cyclic β–Ketocarbonyls with Anodic Benzyne Intermediates, Angew. Chem., Int. Ed., 2020, 59, 14347–14351 CrossRef CAS PubMed.
- X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Electricity-driven asymmetric Lewis acid catalysis, Nat. Catal., 2018, 2, 34–40 CrossRef.
- (a) H. Chen, F. Dong and S. D. Minteer, The progress and outlook of bioelectrocatalysis for the production of chemicals, fuels and materials, Nat. Catal., 2020, 3, 225–244 CrossRef; (b) D. G. Boucher, E. Carroll, Z. A. Nguyen, R. G. Jadhav, O. Simoska, K. Beaver and S. D. Minteer, Bioelectrocatalytic Synthesis: Concepts and Applications, Angew. Chem., Int. Ed., 2023, 62, e202307780 CrossRef CAS PubMed; (c) C. J. Long, Y. H. He and Z. Guan, Emerging Strategies for Asymmetric Synthesis: Combining Enzyme Promiscuity and Photo–/Electro–redox Catalysis, Asian J. Org. Chem., 2023, 12, e202200685 CrossRef CAS.
- H. Chen, R. Cai, J. Patel, F. Dong, H. Chen and S. D. Minteer, Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates, J. Am. Chem. Soc., 2019, 141, 4963–4971 CrossRef CAS PubMed.
- F. Dong, H. Chen, C. A. Malapit, M. B. Prater, M. Li, M. Yuan, K. Lim and S. D. Minteer, Biphasic Bioelectrocatalytic Synthesis of Chiral β-Hydroxy Nitriles, J. Am. Chem. Soc., 2020, 142, 8374–8382 CrossRef CAS PubMed.
- (a) R. Cai, R. D. Milton, S. Abdellaoui, T. Park, J. Patel, B. Alkotaini and S. D. Minteer, Electroenzymatic C–C Bond Formation from CO2, J. Am. Chem. Soc., 2018, 140, 5041–5044 CrossRef CAS PubMed; (b) M. Yuan, S. Sahin, R. Cai, S. Abdellaoui, D. P. Hickey, S. D. Minteer and R. D. Milton, Creating a Low–Potential Redox Polymer for Efficient Electroenzymatic CO2 Reduction, Angew. Chem., Int. Ed., 2018, 57, 6582–6586 CrossRef CAS PubMed; (c) S. K. Kuk, K. Gopinath, R. K. Singh, T.-D. Kim, Y. Lee, W. S. Choi, J.-K. Lee and C. B. Park, NADH-Free Electroenzymatic Reduction of CO2 by Conductive Hydrogel-Conjugated Formate Dehydrogenase, ACS Catal., 2019, 9, 5584–5589 CrossRef CAS; (d) G. Morello, B. Siritanaratkul, C. F. Megarity and F. A. Armstrong, Efficient Electrocatalytic CO2 Fixation by Nanoconfined Enzymes via a C3-to-C4 Reaction That Is Favored over H2 Production, ACS Catal., 2019, 9, 11255–11262 CrossRef CAS; (e) Y. Guo, X. Hong, Z. Chen and Y. Lv, Electro-enzyme coupling systems for selective reduction of CO2, J. Energy Chem., 2023, 80, 140–162 CrossRef CAS; (f) X. Tan and J. Nielsen, The integration of bio-catalysis and electrocatalysis to produce fuels and chemicals from carbon dioxide, Chem. Soc. Rev., 2022, 51, 4763–4785 RSC.
- L. Castañeda-Losada, D. Adam, N. Paczia, D. Buesen, F. Steffler, V. Sieber, T. J. Erb, M. Richter and N. Plumeré, Bioelectrocatalytic Cofactor Regeneration Coupled to CO2 Fixation in a Redox–Active Hydrogel for Stereoselective C−C Bond Formation, Angew. Chem., Int. Ed., 2021, 60, 21056–21061 CrossRef PubMed.
- L. Wan, R. S. Heath, C. F. Megarity, A. J. Sills, R. A. Herold, N. J. Turner and F. A. Armstrong, Exploiting Bidirectional Electrocatalysis by a Nanoconfined Enzyme Cascade to Drive and Control Enantioselective Reactions, ACS Catal., 2021, 11, 6526–6533 CrossRef CAS.
- (a) F. Hollmann, K. Hofstetter, T. Habicher, B. Hauer and A. Schmid, Direct electrochemical regeneration of monooxygenase subunits for biocatalytic asymmetric epoxidation, J. Am. Chem. Soc., 2005, 127, 6540–6541 CrossRef CAS PubMed; (b) R. Wu, C. Ma and Z. Zhu, Enzymatic electrosynthesis as an emerging electrochemical synthesis platform, Curr. Opin. Electrochem., 2020, 19, 1–7 CrossRef CAS; (c) H. Chen, T. Tang, C. A. Malapit, Y. S. Lee, M. B. Prater, N. S. Weliwatte and S. D. Minteer, One-Pot Bioelectrocatalytic Conversion of Chemically Inert Hydrocarbons to Imines, J. Am. Chem. Soc., 2022, 144, 4047–4056 CrossRef CAS PubMed; (d) R. Wu, F. Li, X. Cui, Z. Li, C. Ma, H. Jiang, L. Zhang, Y. H. P. J. Zhang, T. Zhao, Y. Zhang, Y. Li, H. Chen and Z. Zhu, Enzymatic Electrosynthesis of Glycine from CO2 and NH3, Angew. Chem., Int. Ed., 2023, 62, e202218387 CrossRef CAS PubMed.
- C. J. Long, H. Cao, B. K. Zhao, Y. F. Tan, Y. H. He, C. S. Huang and Z. Guan, Merging the Non–Natural Catalytic Activity of Lipase and Electrosynthesis: Asymmetric Oxidative Cross–Coupling of Secondary Amines with Ketones, Angew. Chem., Int. Ed., 2022, 61, e202203666 CrossRef CAS PubMed.
- X. Duan, D. Cui, M. Wang, Y. Tian, J. Shi, Y. Wu, S. Liu, Z. Zhu and J. Xu, Deracemization of Sulfoxides Combining Electrocatalysis and Biocatalysis, Chin. J. Chem., 2024, 42, 3107–3112 CrossRef CAS.
- X. Zhu, Y. Ding, S. Li, Y. Jiang and Y. Chen, Electroenzymatic cascade reaction on a biohybrid boosts the chiral epoxidation reaction, Sci. Bull., 2024, 69, 483–491 CrossRef CAS PubMed.
- (a) D. C. Miller, S. V. Athavale and F. H. Arnold, Combining chemistry and protein engineering for new-to-nature biocatalysis, Nat. Synth., 2022, 1, 18–23 CrossRef PubMed; (b) Z. Liu and F. H. Arnold, New-to-nature chemistry from old protein machinery: carbene and nitrene transferases, Curr. Opin. Biotechnol., 2021, 69, 43–51 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2024 |