
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in ion-regulated organic room-temperature phosphorescence
Wenbo
Dai
ab,
Yitian
Jiang
a,
Yunxiang
Lei
a,
Xiaobo
Huang
 a,
Peng
Sun
c,
Jianbing
Shi
b,
Bin
Tong
b,
Dongpeng
Yan
*d,
Zhengxu
Cai
*b and
Yuping
Dong
b
a,
Peng
Sun
c,
Jianbing
Shi
b,
Bin
Tong
b,
Dongpeng
Yan
*d,
Zhengxu
Cai
*b and
Yuping
Dong
b
aCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, China
bBeijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing, China. E-mail: caizx@bit.edu.cn
cAdvanced Research Institute of Multidisciplinary Sciences, Beijing Institute of Technology, Beijing, China
dBeijing Key Laboratory of Energy Conversion and Storage Materials, Key Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing, China. E-mail: yandp@bnu.edu.cn
First published on 23rd February 2024
Abstract
Organic room-temperature phosphorescence (RTP) materials have attracted considerable attention for their extended afterglow at ambient conditions, eco-friendliness, and wide-ranging applications in bio-imaging, data storage, security inks, and emergency illumination. Significant advancements have been achieved in recent years in developing highly efficient RTP materials by manipulating the intermolecular interactions. In this perspective, we have summarized recent advances in ion-regulated organic RTP materials based on the roles and interactions of ions, including the ion–π interactions, electrostatic interactions, and coordinate interactions. Subsequently, the current challenges and prospects of utilizing ionic interactions for inducing and modulating the phosphorescent properties are presented. It is anticipated that this perspective will provide basic guidelines for fabricating novel ionic RTP materials and further extend their application potential.
Introduction
Organic room-temperature phosphorescence (RTP), that is persistent luminescence or afterglow emission,1–4 has drawn extensive attention in optical and optoelectronic fields such as data storage,5–7 sensors,8–11 and bioimaging,12–19 owing to its long emission lifetime, large Stokes shifts, and abundant excited states.20–22 Currently, two critical factors are considered indispensable to achieve RTP with high efficiency (ΦP) and long lifetime (τP).23–25 As shown in Scheme 1a, one of the factors is the facilitation of the singlet-to-triplet intersystem crossing (ISC) to populate the triplet state (ΦISC), and the other is the maximum inhibition of the non-radiative relaxation pathways (knr) and oxygen quenching of the excited states (kq).26–29 In the past decades, several effective strategies, such as crystal engineering,30–33 H-aggregation,34–36 supramolecular assembly,37–39 polymerization,12,40–42 host–guest doping43–52 and so on,53–55 have been proposed to develop highly efficient RTP materials. Notably, numerous efforts have been devoted to suppressing the non-radiative transitions by covalent bonding, hydrogen bonding, and van der Waals forces for the realization of long-lived efficiency phosphorescence.28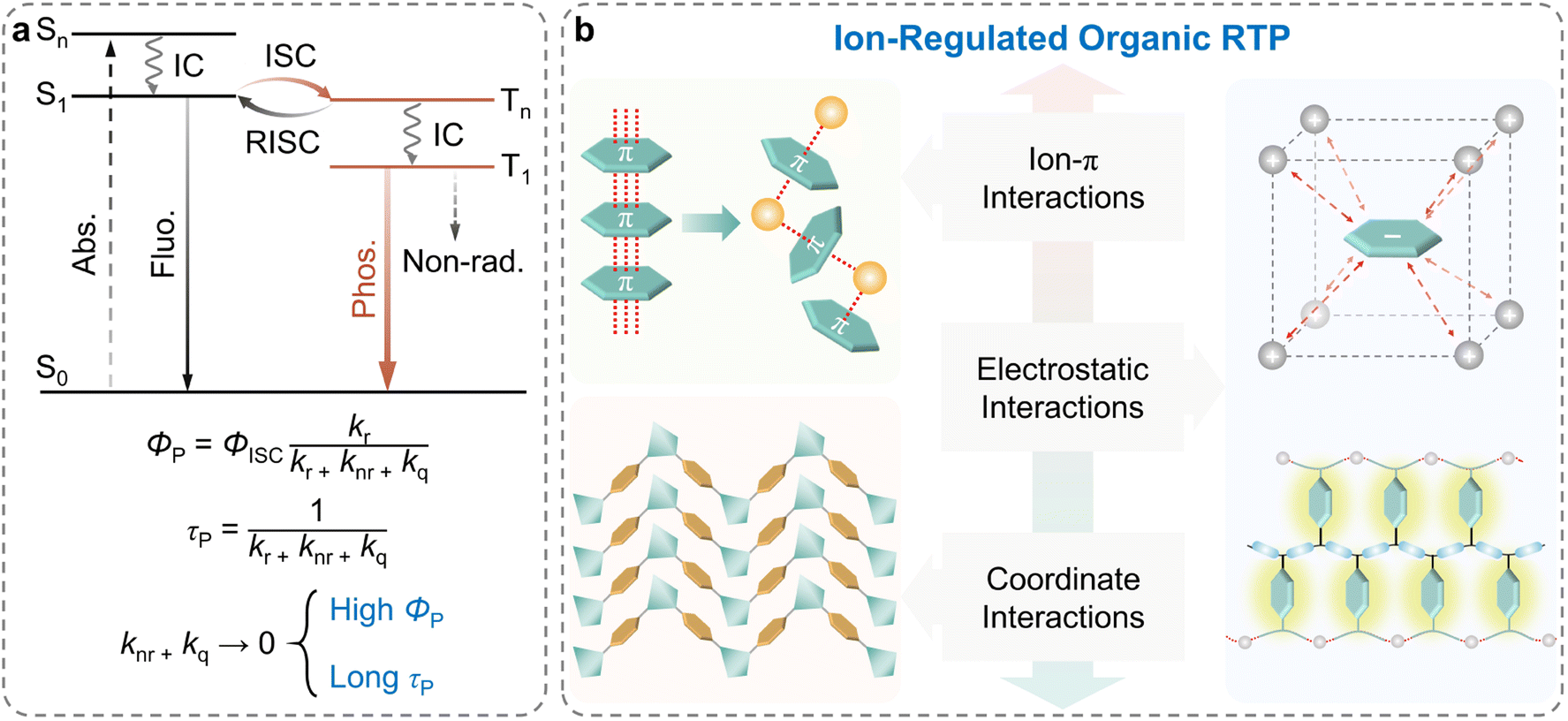 | ||
| Scheme 1 (a) Simplified Jablonski diagram describing the photophysical processes of fluorescence and phosphorescence. (b) Classification of intermolecular interactions in ion-regulated organic RTP materials. Reproduced with permission from ref. 79 (copyright 2022, John Wiley and Sons), ref. 94 (copyright 2019, Springer Nature), and ref. 111 (copyright 2016, John Wiley and Sons). | ||
During the last a few years, many classes of RTP materials particularly organic or metal ions substances have been developed for long lifetime and high quantum yields.56–60 As a novel class of photo-functional materials, ionic systems, compared to ordinary organic neutral systems, could be promising RTP candidates to achieve the aforementioned critical factors due to the following advantages. Firstly, ionic systems could introduce modifiable metal centers and halogen units, enabling the manipulation of the heavy-atom effect to augment spin–orbit coupling (SOC) and, consequently, facilitating the ISC process.61–63 Secondly, the rigid environment established through the ionic or coordinate bonding in the ionic systems could increase molecular conformational rigidity, which could effectively inhibit the non-radiative transitions of the triplet excitons.64–66 Finally, the excited states of ionic systems could be more easily manipulated by modulating the ion species and intermolecular interactions, thereby endowing ionic RTP materials with abundant photophysical characteristics.67–69 Considering these advantages, ionic systems serve as an essential and promising platform for creating novel RTP materials and have attracted wide attention. Despite the large scientific advances in the development of ionic RTP materials, systematic summary for the emission mechanism and rational design strategy still need to be made available.
Herein, we aim to elucidate the significant impact of the different types of ions and intermolecular interactions in ionic systems for achieving and regulating high-performance RTP materials. More specifically, by analyzing the functional role of ions and their phosphorescent properties, we summarize the recent progress in ion-regulated organic RTP materials from the perspective of weak intermolecular interactions (ion–π interactions), electrostatic interactions (in ionic crystals and ionic polymers), and coordinate interactions (Scheme 1b). The representative examples of ionic systems and their RTP properties are listed in Table 1. Please note that due to the introduction of valence and conduction bands in the inorganic parts altering the traditional RTP pathways, metal–organic compounds, such as metal–organic frameworks, metal–organic halides, and hybrid perovskites are not included in this perspective. These materials are, however, discussed in recent review articles which may be referred for detailed insights.70–73 Finally, we illustrate the main issues and challenges for future developments in ionic RTP materials.
| Organic phosphors | Counterions | Wavelengths (nm) | Φ P (%) | Lifetimes (ms) | References |
|---|---|---|---|---|---|
| a Note: ‘/’ refers to the relevant information was not provided in the references. | |||||
| 1,2,3,4-Tetraphenyloxazolium (Fig. 1a) | I− (TPO-I) | 559 | 35.00 | 0.048 | 80 |
| Br− (TPO-Br) | 549 | 36.56 | 0.706 | ||
| Pyromellitic diimide (Fig. 1g) | I− (BrPmDI in 5 wt% laponite) | 470 (3LE) | 5.4 | 0.37 | 84 |
| 560 (3CT) | 0.23 | ||||
| Terephthalic acid (Fig. 2a) | NH4+ (AHT) | 502 | / | 586 | 86 |
| Na+ (DST) | 508 | / | 585 | ||
| K+ (DPT) | 456 | / | 504 | ||
| Tetraphenylphosphonium (Fig. 2e) | F− (TPP F) | 480 | 2 | 25.7 | 88 |
| Cl− (TPP Cl) | 500 | 2 | 108 | ||
| Br− (TPP Br) | 486 | 4 | 6 | ||
| I− (TPP I) | 430 | 42 | 0.0099 | ||
| BF4− (TPP BF4) | 490 | 48 | 43.4 | ||
| ClO4− (TPP ClO4) | 475 | 56 | 399.7 | ||
| Cytosine (Fig. 2f) | PO4− (Cy-PO4) | 430 (365), 495 (395) | / | 126.1, 430.5 | 89 |
| Cl− (Cy-Cl) | 512 (365), 546 (395) | / | 104.8, / | ||
| Terephthalic acid (Fig. 3a) |
|
500 | 9.35 | 862 | 90 |
| 3,4-Methylenedioxyphenylamine (host) | Cl− | 477–679 | 1.40–18.7 | 71.3–1726 | 91 |
| Ammonium salts (guest) (Fig. 3b) | Br− | ||||
| Pyromellitic acid, mellitic acid (Scheme 2) | Na+ | 447 (TSP) | 66.9 | 168.3 | 74 |
| K+ | 420 (TPP) | 28.8 | 132.6 | ||
|
407 (HSM) | 81.6 | 199.1 | ||
| 410 (HPM) | 31.8 | 101.2 | |||
| 454 (TNP) | 96.5 | 184.9 | |||
| Poly(styrene sulfonic acid) | Na+, Li+, K+, Rb+, NH4+, Mg2+, Al3+, Gd3+, Ca2+ | 540 (PSSNa) | 0.79 | 894 | 94 |
| Polyacrylic acid | 480 (PAANa) | 5.79 | 73 (τ1), 222 (τ2) 596 (τ3), 2139 (τ4) | ||
| Polymaleic acid | 523 (PMANa) | 2.77 | 378 | ||
| Poly(maleic acid-co-4-styrenesulfonic acid) (Fig. 4a and d) | 500 (PMANa-co-PSSNa) | 13.08 | 385 | ||
| Poly(4-vinylpyridine) (Fig. 4e) | SO3− (PVP–S) | 524–572 | 6.40 | 80.32–578.3 | 95 |
| Poly(phenylpropenes-co-maleic anhydride) (Fig. 5a) | Na+ (h-PMM-Na) | 498 | / | 401 | 96 |
| K+ (h-PMM-K) | 500 | / | 345 | ||
| Cellulose (Fig. 5b) | Mg2+ | 530 | 5 | 50 | 102 |
| Cellulose 1-cyanomethylimidazolium chloride (Fig. 5c) | Cl− | 550 | 11.81 | 158 | 103 |
| Hemicellulose, lignin, cellulose (Fig. 5d) | Cl− | 545 | 4.7 | 17.5–297 | 105 |
| Chitosan (Fig. 6) | Na+, K+, Mg2+, Ca2+, Zn+, Y3+ | 500 | 0.12–1.5 | 159–1041 | 106 |
| Poly-diallyldimethylammonium (host) | Cl− | 555 (NpA@PAB) | 4.40 | 3.72 | 107 |
| Commercial fluorescent dyes (guest) (Fig. 7a) | Br− | 528 (S-BN@PAB) | 17.2 | 25.9 | |
| 538 (HCM@PAB) | 31.6 | 10.7 | |||
| 510 (QN@PAB) | 13.6 | 13.0 | |||
| 700 (RhB@PAB) | 27.1 | 6.10 | |||
| 581 (PF@PAB) | 1.8 | 4.95 | |||
| Sodium alginate (host) | Na+ | 430–540 | 3.2–25.7 | 310–1200 | 108 |
| Aromatic sodium carboxylates (guest) (Fig. 7c) | |||||
| Terephthalic acid | Zn2+ | 484–533 | 2.2–3.4 | 75–1321 | 112 |
| Isophthalic acid | Cd2+ | ||||
| Trimesic acid (Fig. 8a and b) | |||||
| Chiral L-histidine (Fig. 8g) | Zn2+ | 465–678 | 9.54–21.59 | 141.9–356.7 | 114 |
| 4,5-Dicyanoimidazole (Fig. 9a) | Zn2+ (Zn-DCI) | 536 | 75 | 197.15 | 115 |
| Cd2+ (Cd-DCI) | 441 | 58.4 | 630.15 | ||
| Crown ether (Fig. 10a) | K+ | 490 (B15C5⊃K+) | / | 420 | 119 |
| 490 (DB18C6⊃K+) | / | 330 | |||
| 470 (DB24C8⊃K+) | / | 210 | |||
| 470 (DB30C10⊃K+) | / | 950 | |||
| Phenoxathiine (host) | Zn2+ | 580 | 7.40–20.4 | 56.9–349 | 120 |
| Polyphenylquionline (guest) (Fig. 10d) | |||||
| Benzophenone (host) | Al3+, Cu+/2+, Zn2+, Ga3+, Ag+, Cd2+, In3+ | 512–574 | 1.4–70 | 27–313 | 121 |
| Phenylquinoline (guest) (Fig. 10e) | |||||
Ion–π interactions
Minimizing non-radiative transitions from the molecular motions is the key to obtaining long-lived phosphorescence (Scheme 1a, bottom).1,74 Non-covalent intermolecular interactions, such as π–π interactions, n–π interactions, hydrogen bonds, etc., are pivotal in organic neutral RTP systems, ensuring a rigid molecular environment that effectively impedes the non-radiative transitions of the excitons.28,75–77 In comparison, ion–π interactions, a novel class of non-covalent bonding involving ions and aromatic groups, have attracted increased attention in the development of organic materials exhibiting high luminescence efficiency since their initial application in developing aggregation-induced emission systems in 2017.78 Furthermore, the introduction of heavy halide anions (Cl−, Br−, and I−) can further promote the SOC, thereby enhancing phosphorescence efficiency.79In 2018, Tang et al. integrated the heavy-atom effect with anion–π interactions, proposing a novel approach for constructing purely organic RTP materials.80 A series of highly luminescent organic salts were prepared by simply exchanging the counterions of TPO-P (Fig. 1a). In the solution state, all the compounds exhibited only short-lived fluorescence. However, in the crystalline state, the TPO-I and TPO-Br with heavy halide ions possessed long-lived phosphorescence with the lifetime of 48 μs and 706 μs and the quantum fields of 35.00% and 36.56%, respectively, while the compounds lacking the heavy halide ions only showed fluorescent properties (Fig. 1d and e). Single-crystal analysis and theoretical calculations revealed strong heavy-atom-participated anion–π interactions in the crystalline state for the TPO-I and TPO-Br (Fig. 1b and c). These interactions allowed the heavy atoms to approach the luminescent core closely, enhancing the SOC and charge transfer from the heavy halide ions to the π-conjugated TPO. This feature effectively increased the transition pathways between the singlet and triplet states, while reducing the energy gap between them, thereby boosting the ISC. Meanwhile, the short non-covalent interactions restricted the molecular motion, resulting in the efficient phosphorescence of the TPO-I and TPO-Br. Given the blue fluorescence-yellow phosphorescence dual-emission of the TPO-Br, a balance between the fluorescence and phosphorescence was achieved by incorporating the TPO-Br into various polymers to successfully result in high-performance white-light emission (Fig. 1f).
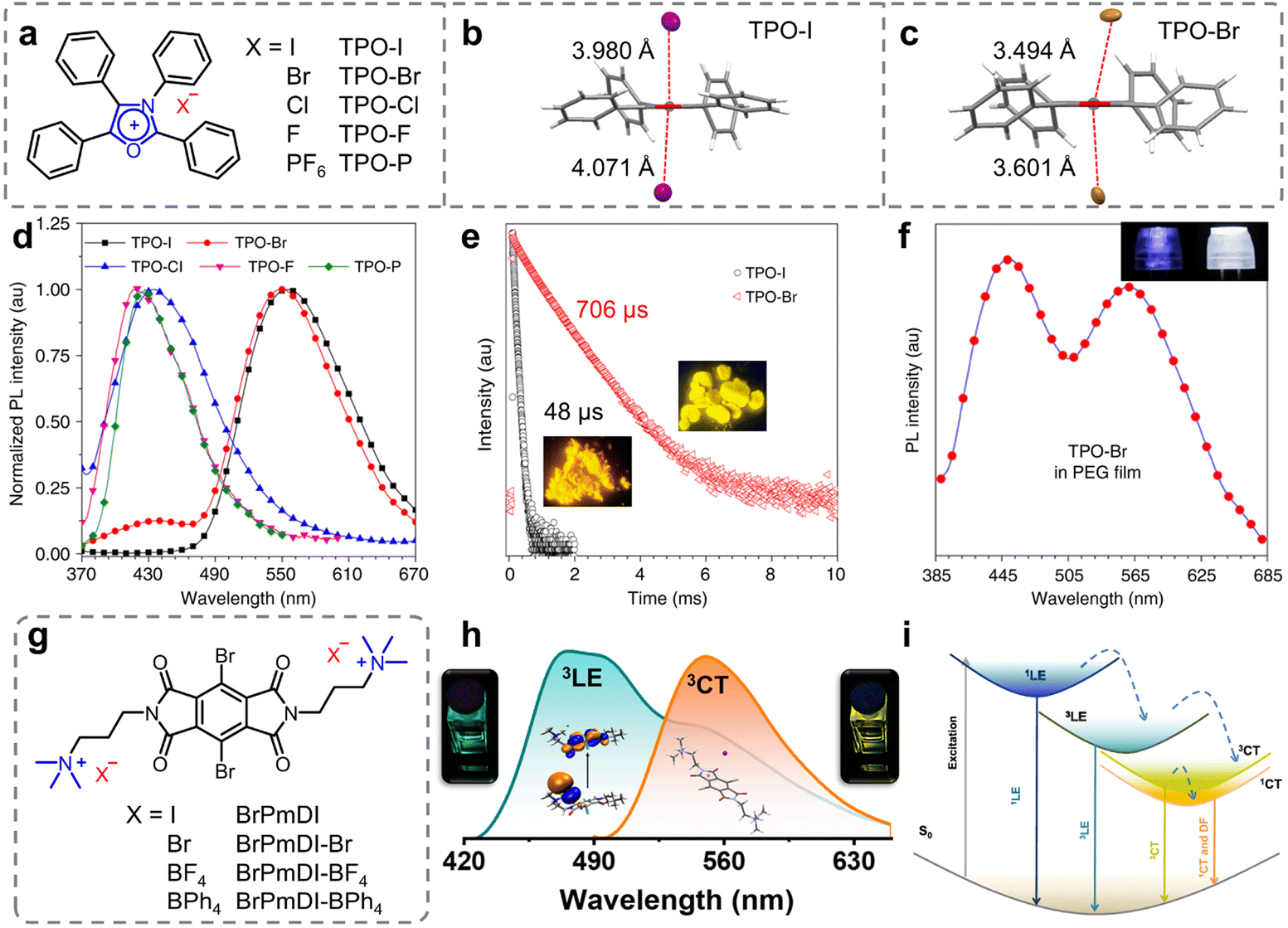 | ||
| Fig. 1 (a) Chemical structures of TPO-I, TPO-Br, TPO-Cl, TPO-F, and TPO-P. Anion–π interactions with distances for (b) TPO-I and (c) TPO-Br. (d) PL spectra of TPO-I, TPO-Br, TPO-Cl, TPO-F, and TPO-P in the solid state. (e) Time-resolved PL decay of d TPO-I and TPO-Br (insets: photographs of TPO-I and TPO-Br were taken under 365 nm UV irradiation). (f) PL spectra of the PEG film containing TPO-Br (2%, m/m) (insets: photographs of 3D printed lampshades without (left) and with (right) TPO-Br taken under UV-LED lamps). (g) Chemical structures of BrPmDI with different counterions. (h) Normalized PL emission spectra at different excitation wavelengths of 370 nm (cyan line) and 470 nm (orange line) showing the contribution both LE and CT emissions. (i) Jablonski diagram showing various emission processes possible from the LE state of the PmDI core and anion–π-induced CT states. Reproduced with permission from (a–f) ref. 80 (copyright 2018, Springer Nature) and (g–i) ref. 84 (copyright 2022, American Chemical Society). | ||
Introducing intramolecular/intermolecular charge transfer (CT) states is another effective approach to achieve high-efficiency phosphorescence.81–83 According to Kasha's exciton model, the CT states can serve as intermediates for minimizing the energy gap between the lowest singlet and triplet states (ΔEST), thereby facilitating the ISC process.1 Based on this strategy, in 2022, George et al. reported anion–π interaction-induced dual RTP emission in pyromellitic diimide (PmDI) derivative, BrPmDI.84 The compound was synthesized through chemical modification of PmDI with bromine atom and iodide counterions (Fig. 1g). Notably, in the BrPmDI single crystals, I− was found stabilized by anion–π interactions of 3.69 Å length within the cationic π-conjugated layers. Meanwhile, the photophysical experiments and theoretical calculations further confirmed the formation of anion–π interaction-induced emissive CT states. Consequently, the researchers effectively embedded the BrPmDI molecules into the supramolecular scaffolding (LAPONITE®) effectively to stabilize the triplet excited states, ultimately achieving dual solution-phase phosphorescence emission from the locally excited (3LE, 470 nm) and the anion–π interaction-induced CT (3CT, 560 nm) triplet states (Fig. 1h and i). This study further demonstrated the feasibility of utilizing ion–π interactions in developing various highly efficient RTP materials and modulating their properties.
Electrostatic interactions
Compared to ordinary neutral systems, organic ionic crystals and polymers are constructed by ionic bonding and ionic cross-linking, which are a kind of electrostatic interaction between an anion and a cation.28 These strong interactions not only construct a rigid environment to restrict molecular motion, but also prevent the quenching effects caused by oxygen and moisture, effectively suppressing the non-radiative relaxation of the triplet excitons. Moreover, the electronic structures of ionic systems are more easily manipulated through charge-state adjustments, allowing for flexible molecular design and optimized RTP performance.Organic ionic crystals
In ionic crystals, in addition to ion–π interactions, there is a widespread presence of strong electrostatic interactions known as ionic bonding. Compared with hydrogen bonding, this type of interaction exhibits distinct properties such as stronger bonds, lack of directional constraints, and non-saturation. The strength of the ionic bonds surpasses that of hydrogen bonds, providing a more rigid environment.85 Such rigidity is crucial in minimizing the non-radiative decay of the triplet excitons. In 2018, Huang et al. introduced a new class of organic ionic crystals, which exhibited intense and ultralong RTP under ambient conditions (Fig. 2a).86 The unique ionic bonding in the organic salts promoted an ordered arrangement to enhance molecular aggregation crucial for the RTP emissions. More importantly, the study demonstrated that changing the cations from NH4+ to Na+, and K+ allowed for tunable phosphorescence colors from yellow-green to sky blue, with emission lifetimes over 586 ms (Fig. 2b–d). Additionally, the research demonstrated the potential for reversible RTP through alternate exposure to ammonia and hydrogen chloride gases, suggesting applications in visual gas sensing. This strategy allowed for the tuning of phosphorescence color and lifetime by altering the counterions, which contributed to expand the range of ultralong RTP materials as well as their applications. Using the same strategy, Qian et al. also reported persistent phosphorescence regulated by anions with tetraphenylphosphonium (TPP+) cations (Fig. 2e).87,88 The strong ionic bonding interactions between the TPP+ and anions were instrumental in facilitating the afterglow generation. Moreover, TPP ClO4 also exhibited size-dependent ultralong afterglow (Fig. 2e).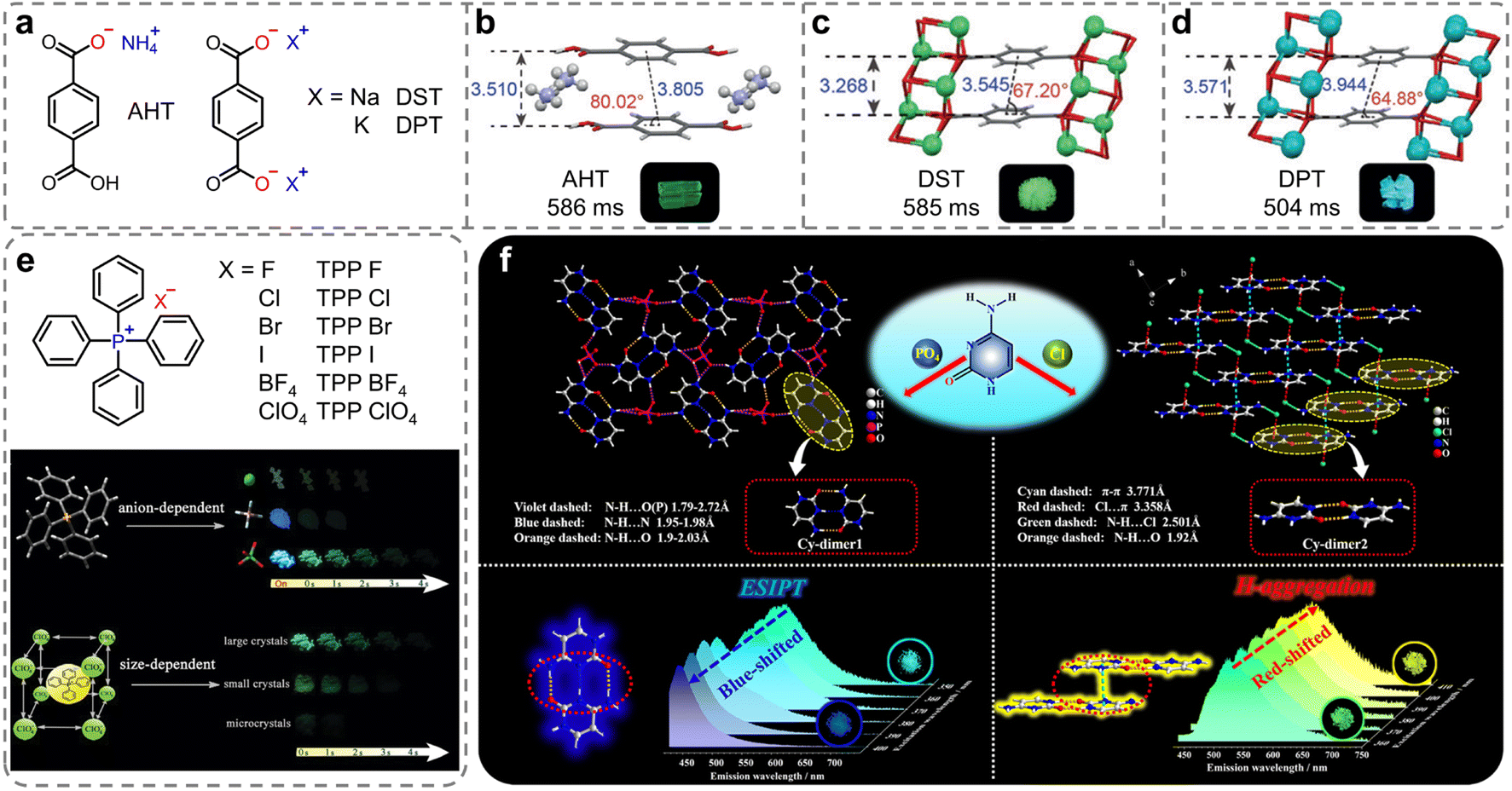 | ||
| Fig. 2 (a) Chemical structures of ionic compounds AHT, DST, and DPT. Molecular packing of (b) AHT, (c) DST and (d) DPT in dimer and the photographs of the ionic crystals under 365 nm lamp off. (e) Chemical structures of ionic compounds TPP X (insets: the schematic illustration of anion-regulated and size-dependent afterglow of tetraphenylphosphonium perchlorate crystals). (f) Schematic representation and single-crystal structures for constructing color-tunable ionic cytosine complexes. Reproduced with permission from (a–d) ref. 86 (copyright 2018, John Wiley and Sons), (e) ref. 88 (copyright 2019, Royal Society of Chemistry), and (f) ref. 89 (copyright 2022, American Chemical Society). | ||
In 2022, Yan et al. introduced a novel category of two-component ionic crystals created through self-assembly of cytosine (Cy) with phosphate (PO4−) and halogens anions (Cl−), named Cy-PO4 and Cy-Cl, respectively.89 The intermolecular interactions of cytosine with the anions led to diverse energy levels and crystal stacking modes, which controlled the molecular aggregation (H-aggregation) and the excited-state intermolecular proton transfer (ESIPT) process. In the Cy-PO4 crystal, cytosine molecules formed a coplanar dimer configuration (Cy-dimer1, Fig. 2f, left) that enabled the ESIPT crucial for the keto–enol transition. For Cy-Cl, face-to-face H-aggregated dimers (Cy-dimer2, Fig. 2f, right), reinforced by π–π interactions, were pivotal for the stabilization of the triplet excitons. Consequently, the H-aggregation-induced green to yellow RTP for Cy-Cl and ESIPT-dominated cyan RTP to deep blue thermally activated delayed fluorescence in Cy-PO4 can be achieved through control of the excitation wavelength, time evolution, and temperature. This work developed a new strategy for color-tunable afterglow from static and dynamic perspectives and has potential applications in information storage and anti-counterfeiting technology.
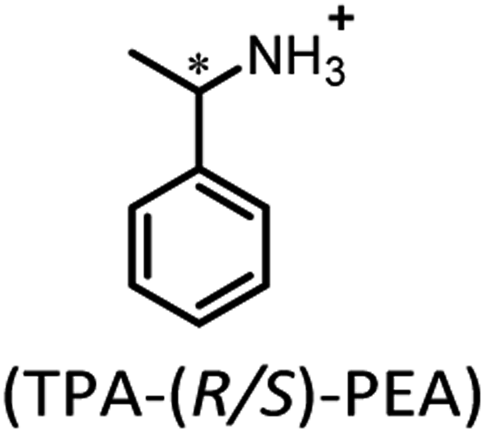
Besides inorganic ammonium cations, organic ammonium cations have been also applied as counterions in constructing efficient RTP materials. In 2018, Duan et al. reported chiral organic ionic crystals derived from terephthalic acid (TPA) and chiral α-phenylethylamines (PEA) exhibiting persistent circularly polarized phosphorescence.90 At room temperature, these ionic crystals exhibited distinct green phosphorescence with lifetimes of up to 862 ms (Fig. 3a). It is noteworthy that the TPA-(R/S)-PEA crystals exhibited significant circularly polarized luminescence signals in both the fluorescence and phosphorescence regions, with the asymmetry factor reaching ≈10−2. Single-crystal analysis revealed that the strong intermolecular interactions between terephthalic acid and chiral amines produced asymmetric torsion angles, which favored the generation of chiral triplet excitons and, thus, exhibited distinct persistent circularly polarized phosphorescence properties. Following that, Lin et al. developed novel metal-free, two-dimensional layered organic ammonium halide crystals (Fig. 3b, HLB and HLC).91 The rigid laminates were composed via various ionic bonding interactions between the ammonium and halide ions. These interactions significantly restricted the molecular motion, which endowed it with significant green RTP. By introducing ammonium halides with diverse triplet energies as guests into phenylethylamine salt hosts, full-color including white persistent phosphorescence emission with ΦP and lifetime up to 18.7% and 1.7 s was achieved, respectively. This phenomenon can be attributed to an efficient Förster energy transfer between the host and guest, and yielding highly efficient RTP of guests (Fig. 3c). Meanwhile, the energy transfer efficiency between the guest and the host in the doped materials strongly depends on the overlapping range of the absorption band of the guest and the emission band of the host. Finally, the authors applied it to various complex anti-counterfeiting and dual-mode fluorescent/phosphorescent chemical sensors.
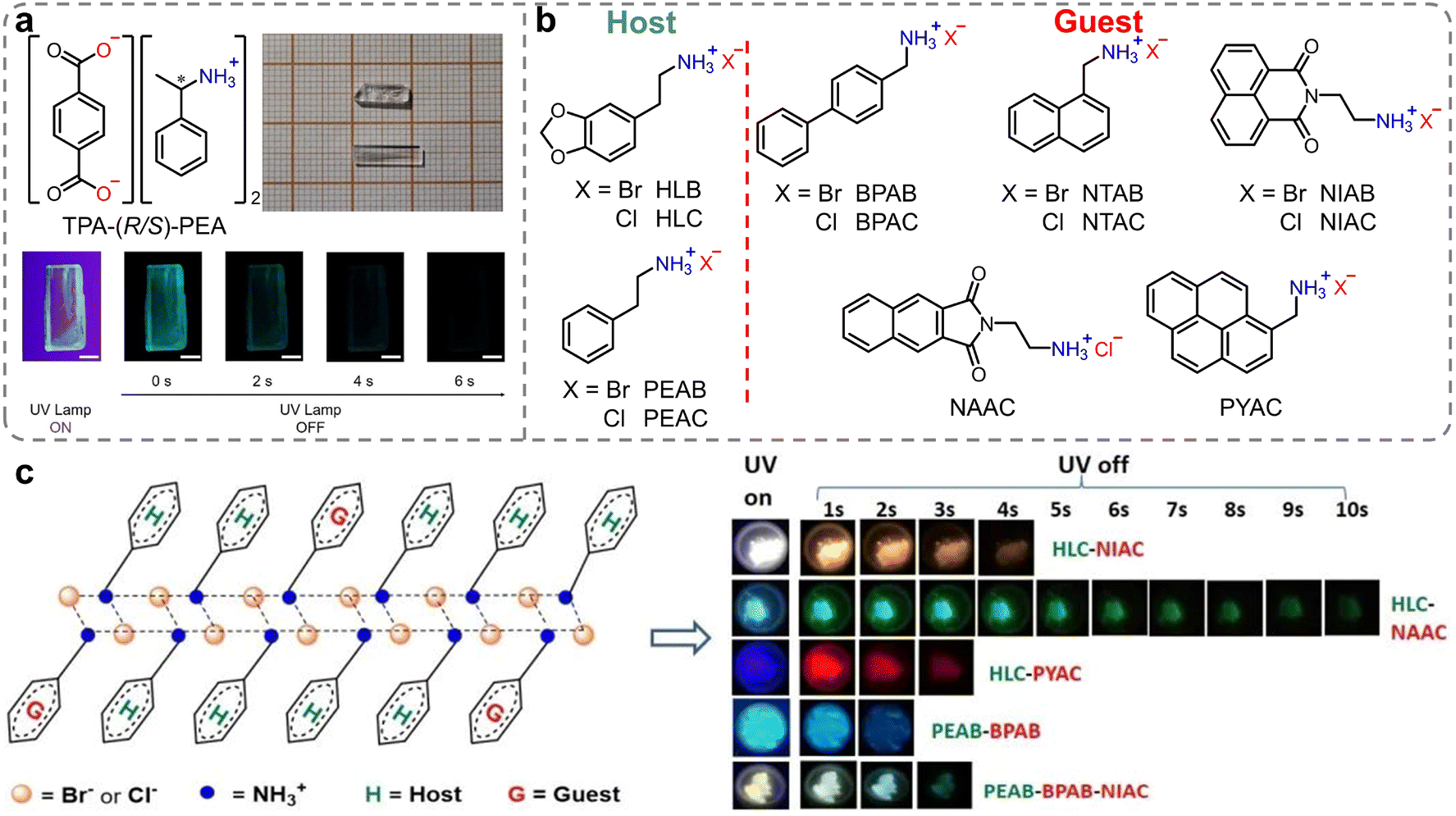 | ||
| Fig. 3 (a) Chemical structures of TPA-(R/S)-PEA organic ionic crystals (insets: images of TPA-(R/S)-PEA crystals in bright field, under UV irradiation and after excitation for different times). (b) Molecular structure of host and guest ammonium halides. (c) Structure diagram of the host–guest system and afterglow photos of doped halides under a UV lamp. Reproduced with permission from (a) ref. 90 (copyright 2018, John Wiley and Sons) and (b and c) ref. 91 (copyright 2021, Royal Society of Chemistry). | ||
It is noteworthy that the H-aggregation strategy in previous works mainly focused on enhancing the RTP performance by strengthening the π–π interactions within the crystal.34,92,93 While the strong π–π interactions between the adjacent phosphorescent chromophores can effectively stabilize the triplet excitons, the substantial dissipation of excitons through the triplet–triplet annihilation channel results in a lower RTP efficiency and a bathochromic shift of phosphorescence.35 This effect seriously impedes to develop deep-blue organic RTP emitters. An et al. presented a strategy for achieving high-efficiency blue phosphorescence by confining isolated chromophores in ionic crystals.74 Pyromellitic acid and mellitic acid with rich carboxyl groups were ionized by sodium hydroxide, potassium hydroxide and alpha-methylbenzylamine to form ionic crystals (Scheme 2). Taking tetrasodium pyromellitate (TSP) crystal as an example, it exhibited intense blue phosphorescence at 450 nm with a quantum efficiency of 66.9%. Single-crystal analysis revealed that the chromophores were isolated by the surrounding Na+ through multiple ionic bonds, forming a cage-like structure. This architecture not only suppressed the molecular motion of the chromophores but also segregated the chromophores with negligible inter-chromophore interactions, thereby achieving high phosphorescence efficiency. Remarkably, tunable phosphorescence colors from blue to deep blue with ΦP up to 96.5% were achieved by altering the charged chromophores and their counterions.
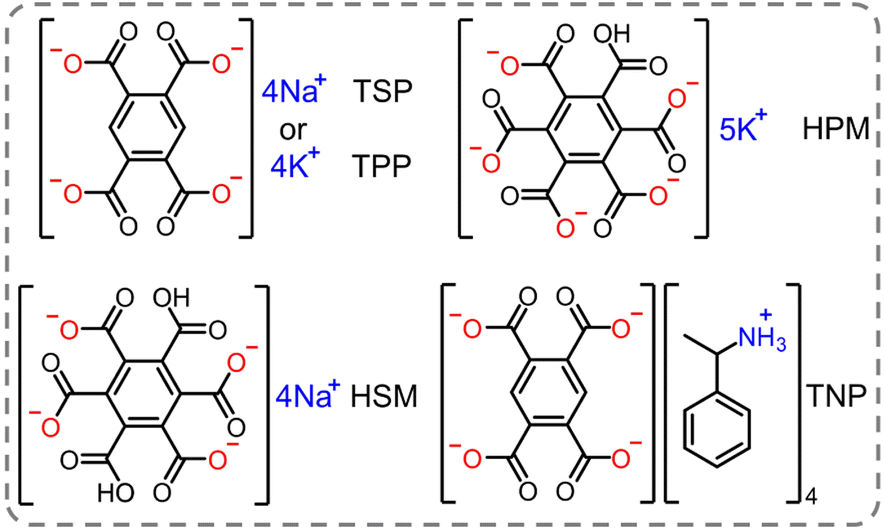 | ||
| Scheme 2 Chemical structure of the ionic crystals (TSP, TPP, HSM, HPM and TNP). | ||
Ionic polymers
Apart from organic ionic crystals, the strong ionic bonding and ionic cross-linking interactions present in ionic polymers can restrict the molecular motion, thus decreasing the non-radiative decay of the triplet excitons. In 2019, Huang et al. proposed a simple strategy to achieve ultralong phosphorescence in polymers via ionic cross-linking (Scheme 1b).94 They initially constructed sodium polystyrene sulfonate (PSSNa) as an ionic model polymer. As shown in Fig. 4a, under ultraviolet (UV) light of 365 nm wavelength, PSSNa film exhibited sky-blue fluorescence and yellow afterglow, closely related to the ionic cross-linking of the sulfonate sodium substituents. Impressively, multicolor long-lived phosphorescent emissions were achieved by tuning the excitation wavelength, with the afterglow colors adjustable from blue to orange at 77 K, originating from different phosphorescent centers (Fig. 4b). Experimental data and theoretical calculations showed that introducing ionic bonding interactions in polymers effectively inhibited the molecular motion of the aromatic luminescent groups, ensuring efficient emission from various phosphorescent centers (Fig. 4c). Moreover, phosphorescence signals of the PSSNa polymer could still be detected even at a temperature of 443 K, owing to strong ionic bonding interactions. This design concept could be applied to both aromatic and non-aromatic polymer systems, namely PAANa, PMANa, and PMANa-co-PSSNa. With the chromophore variations, the RTP emission colors were successfully tuned from yellow to blue (Fig. 4d). Under ambient conditions, the lifetimes of blue RTP emission band at 480 nm for PAANa were reaching up to 2139 ms (τ4, Table 1). The research enhanced the properties of traditional polymer materials and had great application prospects in fields like flexible displays, lighting, data encryption, and biomedicine, due to the wide availability and low cost of polymeric materials. Following this study, to further demonstrate the universality of color-adjustable phosphorescence via ionic bonding cross-linking strategy, they investigated the effects of sulfonation on the photophysical properties of another amorphous polymer poly(4-vinylpyridine) (PVP) (Fig. 4e).95 After ionization with 1,4-butanesultone, the PVP-S phosphor exhibited an ultralong organic phosphorescence lifetime of 578.36 ms, which was 525 times longer than that of the unmodified PVP (Fig. 4f). Additionally, excitation-dependent color-tunable phosphorescence was also observed at both room temperature and 77 K, with emission colors ranging from blue to red. The facile and feasible chemical ionization strategy opened up new avenues for the development of organic smart RTP polymers.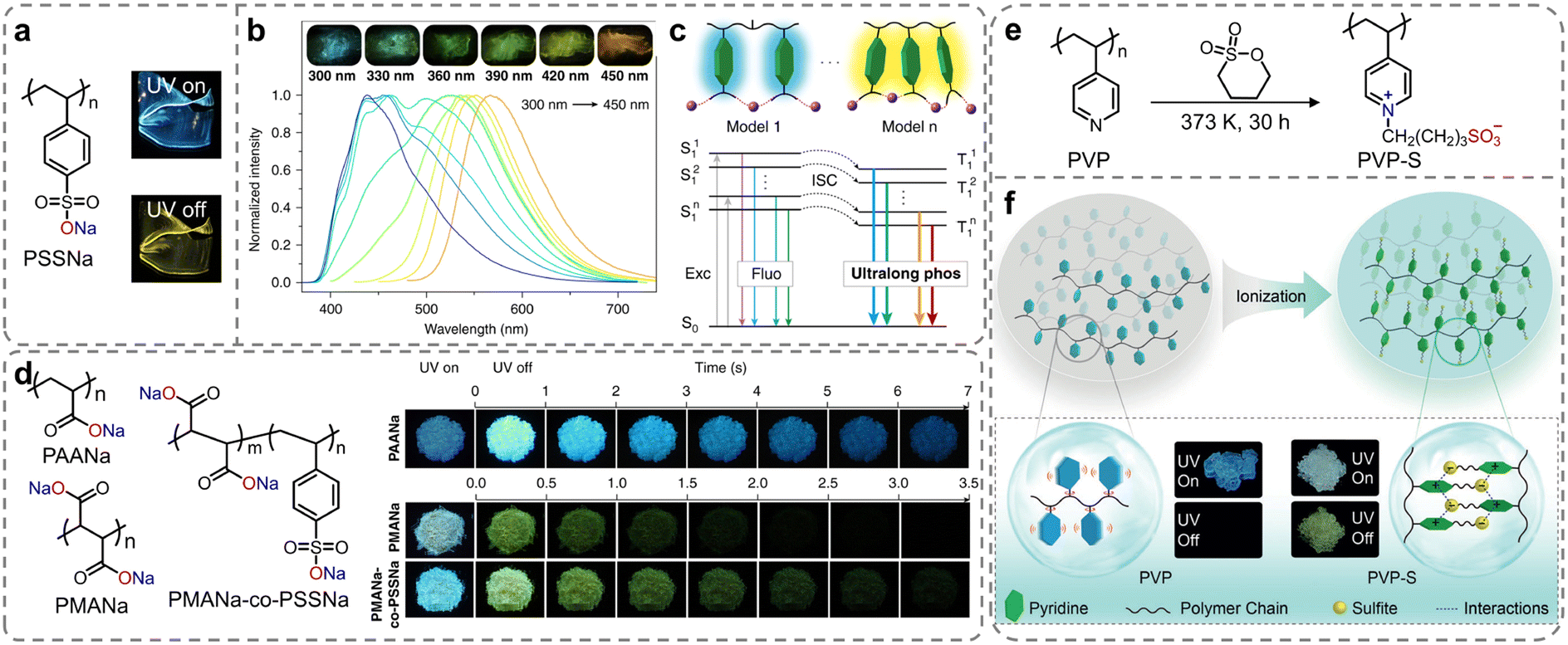 | ||
| Fig. 4 (a) Molecular structure of PSSNa polymer (insets: photographs of PSSNa solids taken under a 365 nm lamp on and off). (b) Excitation dependent phosphorescence spectra of PSSNa polymer at 77 K (insets show photographs taken after PSSNa polymer excited by 300, 330, 360, 390, 420, and 450 nm at 77 K). (c) Proposed mechanism for multicolor ultralong phosphorescence as the excitation wavelength changed. (d) Molecular structures of PAANa, PMANa, and PMANa-co-PSSNa (insets: photographs of PAANa, PMANa, and PMANa-co-PSSNa polymers taken before and after the irradiation of a 310 nm UV lamp). (e) Synthesis of the PVP-S polymer. (f) Ionization enabling ultralong organic phosphorescence in polymers. Reproduced with permission from (a–d) ref. 94 (copyright 2019, Springer Nature) and (e and f) ref. 95 (copyright 2019, John Wiley and Sons). | ||
Similar to the aforementioned strategy, Deng et al. achieved ultralong RTP emission in bio-based non-conjugated polymers through a simple hydrolysis process (Fig. 5a).96 The bio-based polymer was synthesized via free radical solution copolymerization using biomass phenylpropenes and maleic anhydride as monomers. This as-prepared polymer with no traditional luminescent groups could emit blue fluorescence. It is noteworthy that the polymer, after undergoing hydrolysis in a sodium hydroxide solution, exhibited significant enhancement in its fluorescence and afterglow, with a lifetime of 401 ms (Fig. 5a). Furthermore, the type of metal cations in the carboxylate notably affected the phosphorescent properties of the hydrolyzed product.
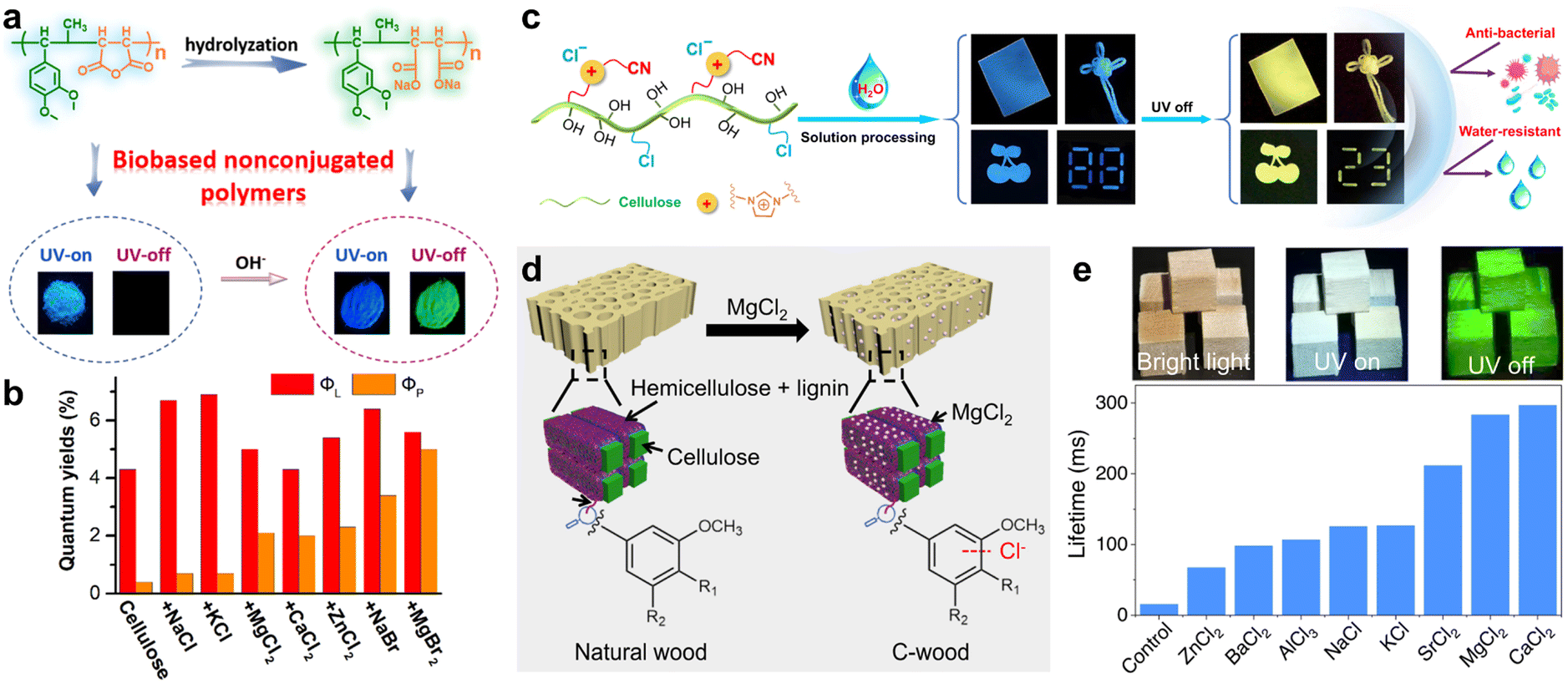 | ||
| Fig. 5 (a) Schematic illustration and the photographs for preparing biomass copolymers. (b) Total luminescence and phosphorescence quantum yields of dried cellulose doped with salts. (c) Chemical structure, processability, and performance of the cationic cellulose derivative with RTP property. (d) Schematic showing the preparation of room temperature phosphorescent C-wood from natural basswood. (e) Photographs of C-wood under bright field (left), UV irradiation (middle), and after switching off the UV light (right), and the lifetime of C-wood prepared using different salts. Reproduced with permission from (a) ref. 96 (copyright 2021, American Chemical Society), (b) ref. 102 (copyright 2022, American Chemical Society), (c) ref. 103 (copyright 2022, Springer Nature), and (d and e) ref. 105 (copyright 2023, Springer Nature). | ||
Unlike the traditional organic conjugated luminescent molecules with extended π-structures, non-traditional luminescent molecules are non-conjugated compounds with electron-rich subunits and have drawn increasing attention in recent years.97 Owing to the enhancement of SOC by heteroatoms, these non-traditional luminescent molecules, characterized by clustering-triggered emission characteristics, are potential materials for realizing RTP.98–100 Among these, cellulose, a naturally occurring renewable resource known for its low toxicity and environmental friendliness, has gained widespread interest.101 However, due to the lack of conjugated groups, the cellulose exhibits weak absorption and low luminescence quantum yields. In 2022, Tian et al. proposed an effective method to enhance the phosphorescence of cellulose (Fig. 5b).102 By doping halide salts into cellulose, the absorption transition from halide anions to polysaccharide chains was induced, along with an increase in the ISC via the external heavy-atom effect. Moreover, the inclusion of divalent metal ions within cellulose effectively minimized the non-radiative relaxations via electrostatic interactions. Consequently, the ΦP of MgBr2-doped dry cellulose could reach up to 5% (Fig. 5b). Zhang et al. developed a cationic phosphorescent cellulose derivative by introducing ionic moieties, such as cyano-methylimidazolium cations and chloride ions into the cellulose chains (Fig. 5c).103 The cyano group and nitrogen atoms in the imidazolium cations facilitated the ISC process. Furthermore, the multiple interactions between the cyano groups, chloride ions, and cellulose's hydroxyl groups, through hydrogen bonding and electrostatic forces, effectively suppressed the non-radiative transitions, leading to excellent RTP performance with a maximum ΦP and lifetime of 11.81% and 158 ms, respectively. Additionally, adding a small amount of glutaraldehyde to the cationic cellulose solution could form a stable dual cross-linked structure, resulting in phosphorescent patterns with superior antimicrobial and water-resistant properties (Fig. 5c). More recently, they also successfully constructed tunable excitation-dependent and visible-light-excited ultralong RTP materials by manipulating the aggregated state of anionic cellulose trimellitates.104
Chen et al. demonstrated a sustainable method to create RTP materials from natural wood using external chloride anion treatment (Fig. 5d).105 Specifically, treating wood with MgCl2 aqueous solution at room temperature resulted in wood containing chloride ions (named C-wood, Fig. 5d), where the halide chloride ions could enhance the SOC and increased the phosphorescence lifetime. The C-wood consequently exhibited intense RTP emission with a lifetime of 297 ms, which was nearly 17 times longer than that of the natural wood (Fig. 5e). Similarly treated with various other chloride anion salts, the treated woods exhibited prolonged phosphorescence lifetimes in comparison to the untreated form (Fig. 5e).
In non-aromatic resources, aside from cellulose, chitosan is a highly biocompatible polymer composed of aminoglucose monomers that was extensively utilized as a hydrophilic matrix due to its easily accessible (Fig. 6a).101 The chemical structure of chitosan facilitates easy physical or chemical modifications. Moreover, the incorporation of functional groups such as amino, hydroxyl, and carbonyl within the chitosan structure promotes the generation of oxygen clusters, which are advantageous for RTP emission. In 2016, Zhang et al. demonstrated that N-substituted naphthalimides conjugated with chitosan exhibited RTP properties and found practical application in time-resolved bioimaging.81 In 2023, Tian et al. also investigated the enhancement and control of chitosan's RTP properties by physically mixing halide salts (Fig. 6b).106 Notably, the RTP characteristics of the chitosan could be precisely adjusted by altering the concentrations and types of halide salts. The presence of cations in the halide salts enhanced RTP by binding with hydroxyl groups, minimizing the distance between halide ions and the emissive clusters, thus boosting the external heavy-atom effect. Moreover, this interaction could also increase the rigidity of the matrix, favorably influencing RTP emissions.
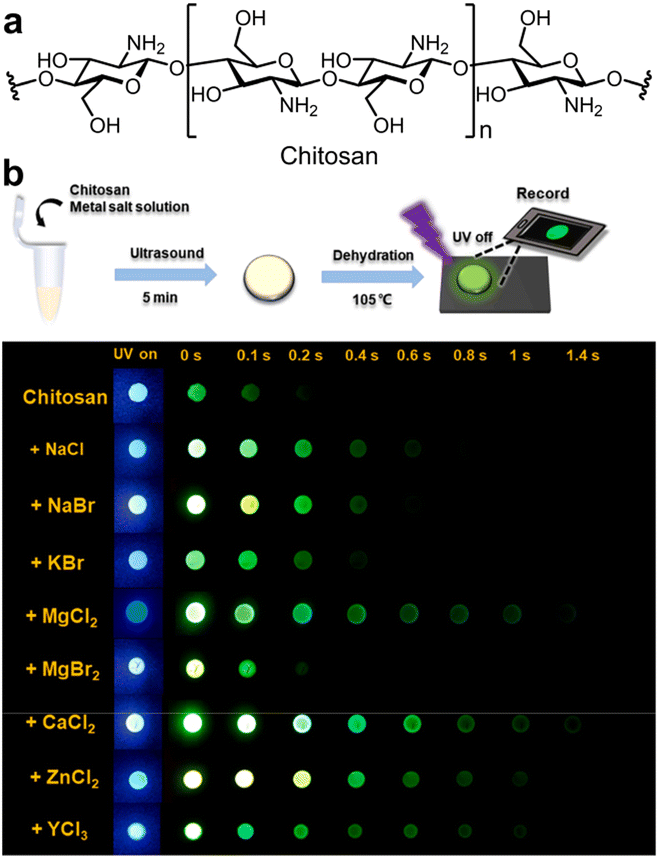 | ||
| Fig. 6 (a) Structure of chitosan. (b) Luminescence imaging of dried chitosan doped with different metal halide under a UV lamp (365 nm) on and off. Reproduced with permission from ref. 106 (copyright 2023, Elsevier). | ||
Compared to chemical modification, direct doping of chromophores into ionic polymers is considered a more straightforward and quantifiable method for preparing RTP materials and modulating their properties.27 The presence of a robust ionic bonding network in polymers is considered highly effective for stabilizing excited triplet excitons, thereby facilitating the generation of RTP emission. Ma et al. constructed a series of organic doped RTP systems by incorporating commercial fluorescent dyes into ionic polymer matrix with external heavy-atom effects and rigid ionic bonding network (Fig. 7a and b).107 This poly-diallyldimethylammonium bromide (PAB) matrix containing bromide ions facilitated the ISC process via an external heavy-atom effect in the doped systems, leading to the generation of excited triplet states. Concurrently, a rigid ionic bonding network formed between the bromide anions and the quaternary ammonium cations in the polymer played a crucial role in reducing the non-radiative deactivation, contributing to the generation of effective RTP emissions. This work offered a practical approach for developing purely organic RTP polymers without the use of complex processes. Based on the same strategy, Yuan et al. developed amorphous sodium alginate (SA) doped with various aromatic sodium carboxylates, which allowed for easy and fast fabrication of flexible, transparent, re-dissolvable polymer films with multi-tunable afterglow colors, ranging from blue to red and even white (Fig. 7c).108 The formation of strong hydrogen and ionic bonds between the SA chains and the doped phosphors impeded the molecular motion. Furthermore, the excitation- and time-dependent afterglows could be achieved and controlled by adjusting the doping concentration, co-doping, and utilizing the triplet to singlet Förster resonance energy transfer (Fig. 7d). This innovative approach demonstrated potential in advanced multi-mode anti-counterfeiting applications, offering a significant advancement in the field of smart luminescent materials.
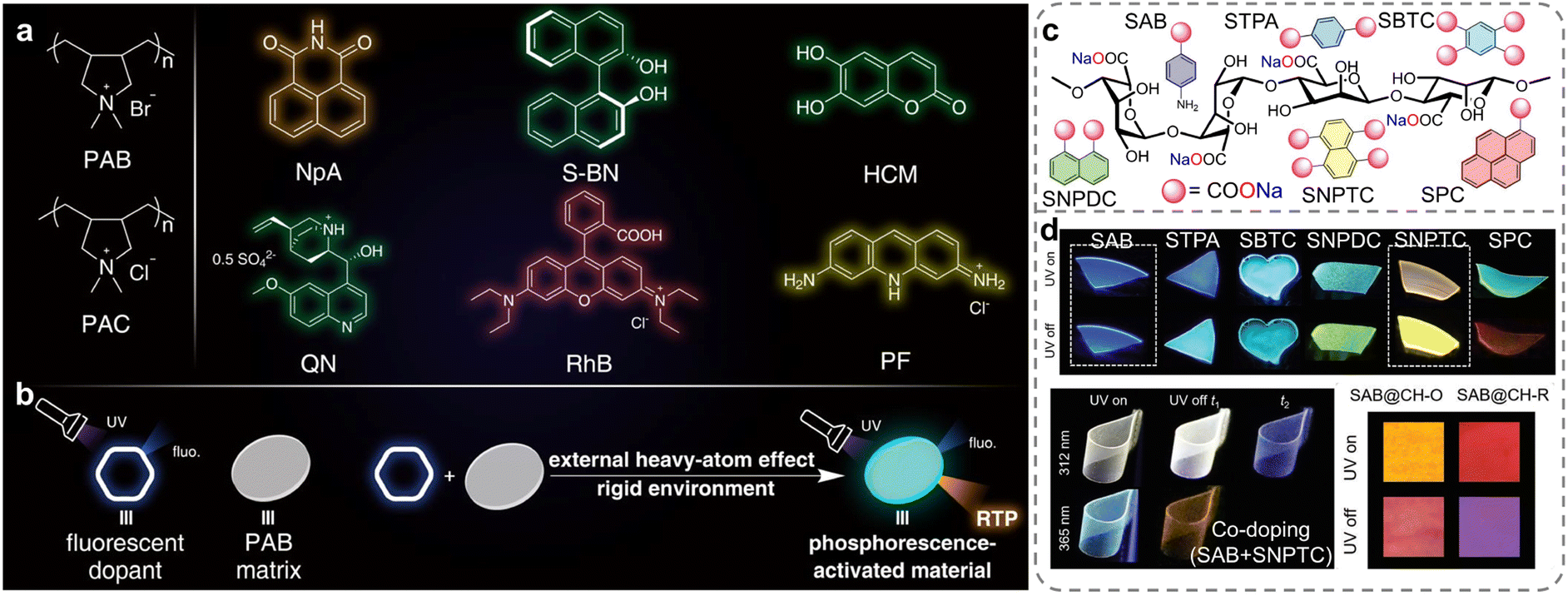 | ||
| Fig. 7 (a) Structures of the polymers (PAB and PAC) and the dopants: NpA, S-BN, HCM, QN, RhB and PF. (b) Illustration of the general strategy. (c) Structures of the aromatic sodium carboxylates and SA. (d) Luminescence photographs of single-component-doped, co-doped, and TS-FRET SA films with multicolor and multi-tunable afterglows. Reproduced with permission from (a and b) ref. 107 (copyright 2021, John Wiley and Sons) and (c and d) ref. 108 (copyright 2022, John Wiley and Sons). | ||
Coordinate interactions
Coordinate bonding, a unique type of covalent bond, where only one atom provides the electron pairs for bonding, is prevalent in organometallic compounds.109 Strong coordinate bonding interactions create rigid architectures for the chromophores, which are crucial for reducing the non-radiative decay of triplet excitons.110 Meanwhile, the coordinate inorganic metals can facilitate the SOC due to the heavy-atom effect, thereby extending the phosphorescence lifetime and enhancing the phosphorescence efficiency.111 Moreover, manipulation of the metal centers and ligands allows tuning of the photophysical properties, including the emission wavelength and lifetime.Coordination polymers, differ from purely organic materials, being formed through coordinate bonding between organic ligands and metal coordination ions. Due to their tunable and stable characteristics, coordination polymers have been used in various applications like sensors, displays, and bioimaging.70 In 2016, Yan et al. demonstrated a significant enhancement in long-lived RTP properties of aromatic acids through coordinate bonding interactions with common metal ion, like Zn2+.112,113 The coordinate bonding formed between Zn2+ and terephthalic acid (TPA) played a crucial role in reducing the non-radiative deactivation. As shown in Fig. 8a, the TPA molecules were tightly encapsulated within the Zn-based rigid framework, effectively inhibiting the non-radiative transitions and boosting the RTP emission.112 Moreover, the organic–metal coordination polymers displayed tunable phosphorescence by varying the metal ions and aromatic ligands, achieving an RTP lifetime of up to 1.3 s (Fig. 8b and c). This work offered a cost-effective way to produce high-performance RTP metal–organic hybrid materials without relying on noble or rare-earth metals, which can be favorable to illumination and sensor applications. Recently, they designed and prepared a series of supramolecular glasses (SGs) based on zinc-L-histidine complexes using an evaporation-induced self-assembly strategy (Fig. 8d).114 The formation of SGs was primarily driven by the synergistic effect of strong coordinate bond and hydrogen bond among Zn2+, L-histidine, and H2O (Fig. 8g). These interactions endowed the SGs with color-tunable circularly polarized afterglow ranging from blue to red with an asymmetry factor as high as 9.5 × 10−3 and a lifetime of up to 356.7 ms (Fig. 8e and f). This work not only presented the SGs with broad tunability in the afterglow and significant circular polarization but also introduced an evaporation-induced self-assembly approach for the macroscopic self-assembly of chiral organic–metal hybrids for photonic applications.
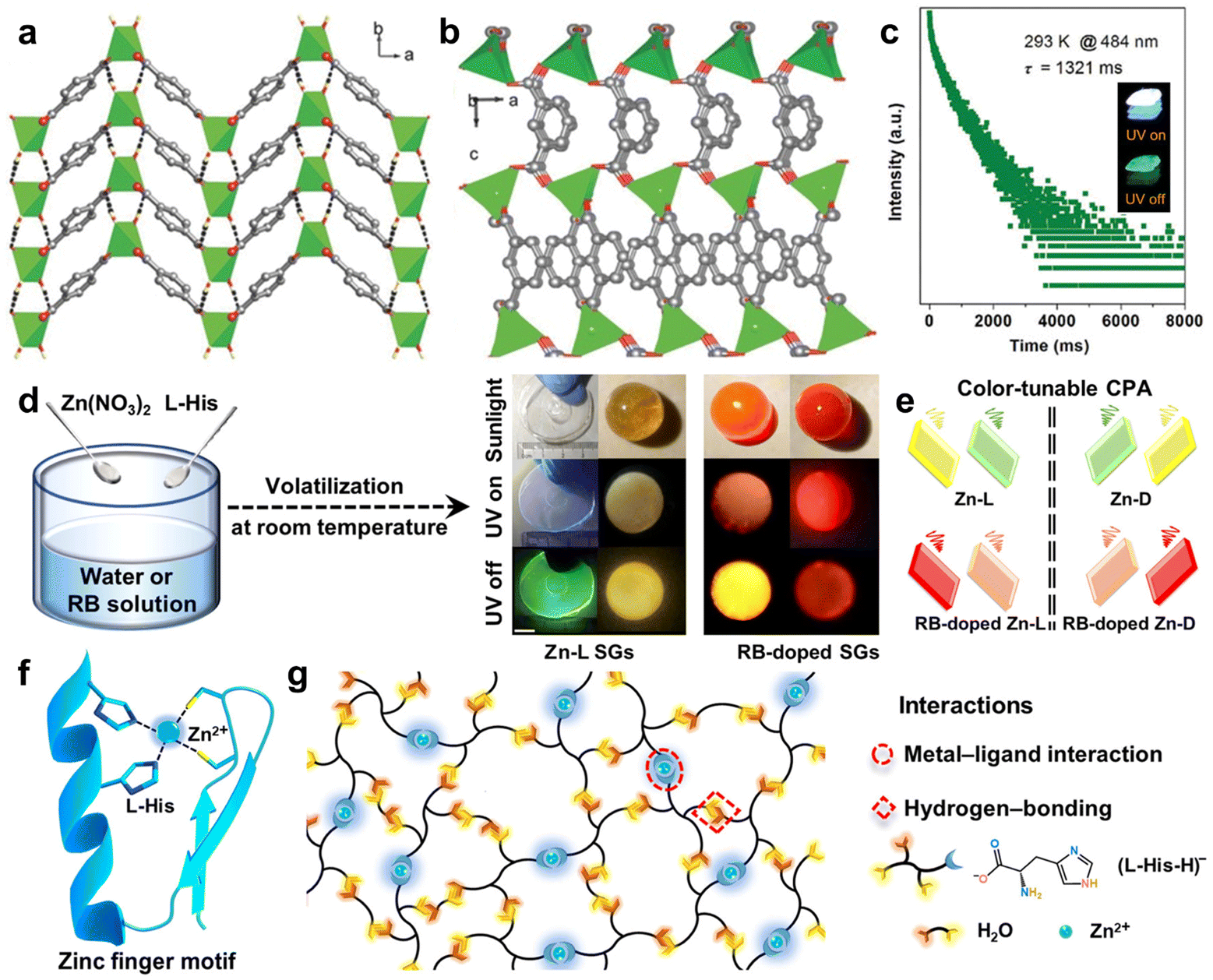 | ||
| Fig. 8 Crystal structures of (a) Zn-TPA and (b) Zn-IPA. (c) Time-resolved emission decay curve (484 nm) for Zn-IPA under ambient conditions. (d) Preparation of Zn-L and RB-doped SGs, and photos of the obtained Zn-L and RB-doped SGs taken under the sunlight, and before and after UV-light turned off. (e) Multicolored CPA emissions from chiral SGs. Spiral lines represent circularly polarized afterglow-active signals. (f) C2H2-type zinc finger motif. (g) Schematic representation of the SGs assembly. Reproduced with permission from (a–c) ref. 112 (copyright 2016, John Wiley and Sons) and (d–g) ref. 114 (copyright 2023, Springer Nature). | ||
In addition to typical aromatic acids, other organic compounds are also viable as organic ligands for constructing coordination polymers with RTP performance. In 2022, Yan et al. developed transparent bulk glasses with an extended phosphorescence lifetime of 630.15 ms and a high photoluminescence quantum yield of up to 75% at ambient conditions via the bottom-up supramolecular coordination self-assembly strategy (Fig. 9a).115 These glasses, comprising metal ions (Zn2+ and Cd2+) and imidazolate-based ligands, featured a structure resembling the random network SiO2 glasses (Fig. 9a). The study revealed that these hybrid glass materials possessed a high Young's moduli and hardness, providing a rigid network that reduced the non-radiative transitions of triplet excitons, thereby enhancing the phosphorescence efficiency (Fig. 9b). Moreover, the luminescence of these hybrid glasses was not a mix of fluorescence and phosphorescence but a full phosphorescent emission directly originating from various triplet state energy levels, demonstrating a high utilization rate of triplet excitons in the materials (Fig. 9c).
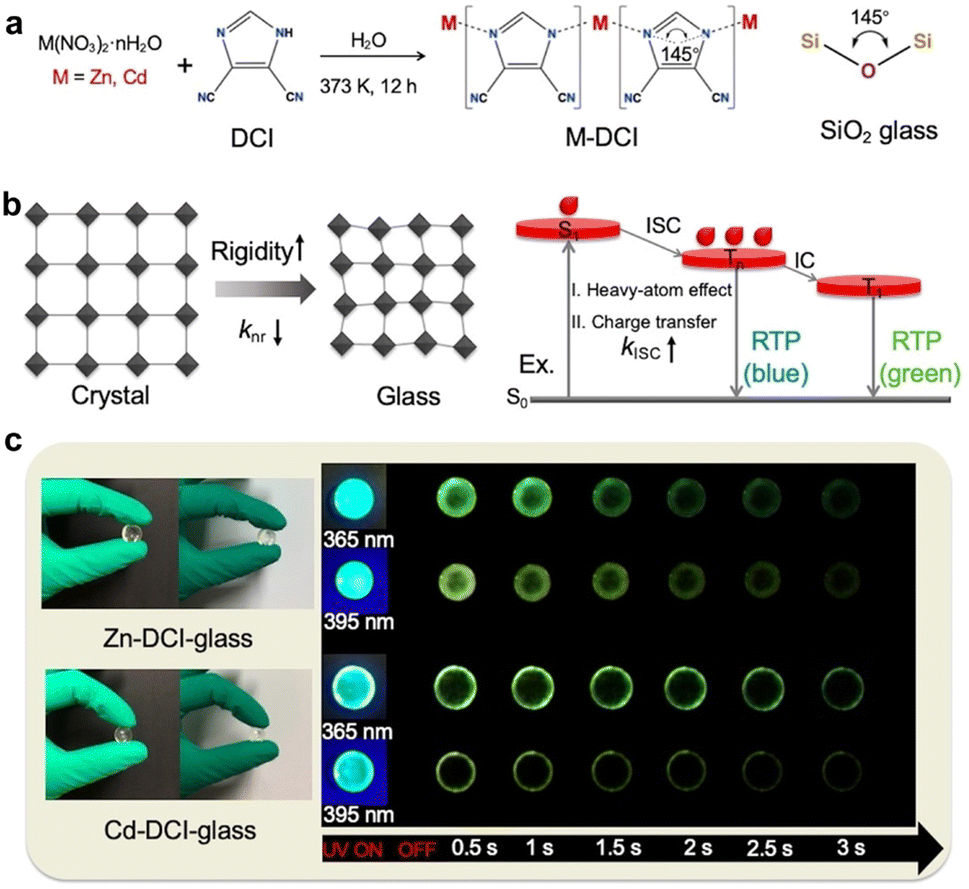 | ||
| Fig. 9 (a) The synthetic procedure of glasses compared to the basic Si–O–Si building unit in classical inorganic zeolites. (b) Schematic mechanism of highly efficient RTP of glasses highlighting the rigid molecular environment for inhibiting non-radiative relaxation and the enhanced ISC process for harvesting triplet excitons. (c) Photographs of the glasses taken before and after turning off the UV light (365 nm and 395 nm). Reproduced with permission from ref. 115 (copyright 2022, John Wiley and Sons). | ||
Dynamic coordinate bonds are a type of bonding used in coordination chemistry.109 These form between metal ions and organic ligands, but unlike traditional coordination bonds, dynamic coordination bonds can form and break rapidly and reversibly. This dynamic nature is crucial in the processes of self-assembly, self-healing, responsive polymers, and the design of smart materials.116–118 In 2019, Tang et al. discovered that a series of crown ethers with different ethylene glycol chains exhibited RTP in the crystalline state (Fig. 10a).119 The flexible glycol chains of crown ethers imparted characteristic ionic selectivity, which combined with the crystallization tendency, allowed for morphological control of the crystalline state, enabling tunable phosphorescence. When K+ was integrated into crown ethers, the phosphorescence lifetime could increase. The mechanism of K+ response was related to the strong coordinate interaction between the multiple oxygen atoms in the crown ether and K+, suppressing non-radiative relaxation (Fig. 10b). Additionally, the phosphorescence lifetime of crown ethers complexed with K+ increased with the length of the glycol chain. Utilizing this “activated” phosphorescence, the authors constructed an advanced multi-level information encryption system in the solid state, where the inherent guest responsiveness of crown ethers allowed for precise design and fabrication of more intelligent luminescent materials (Fig. 10c).
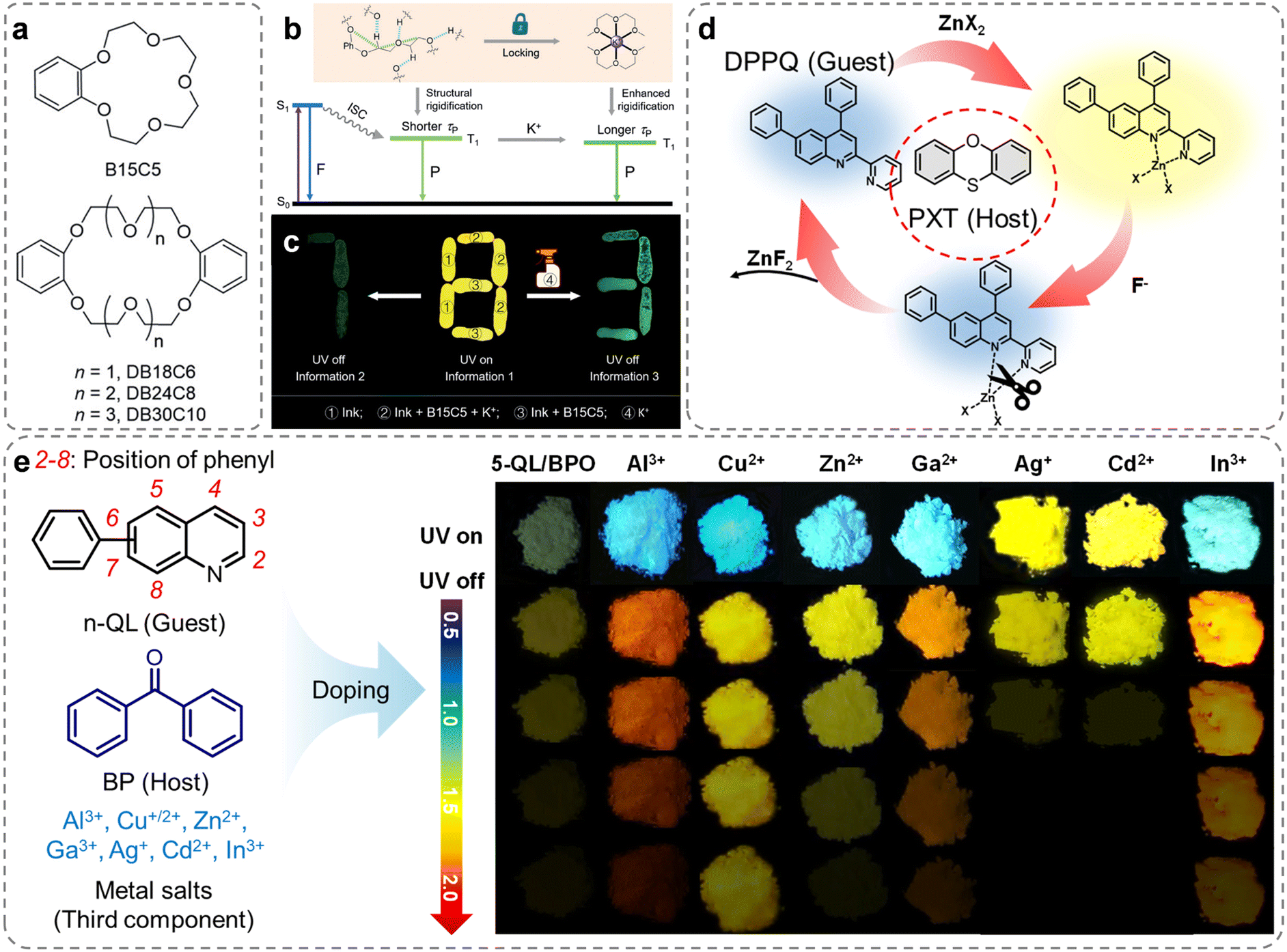 | ||
| Fig. 10 (a) The molecular structures of crown ethers with different ethylene glycol chains. (b) Theoretical models based on Jablonski diagram for understanding the performance of crown ether-based tunable RTP in the solid state. (c) Photograph shows advanced-encryption application based on K+ “activated” persistent phosphorescence strategy. (d) Schematic diagram and molecular structures of host and guest molecule for dynamic manipulation of RTP properties. (e) Molecular structures of the different components and the photographs of three-component doped materials composed of 5-QL/BP and various metal salts under 365 nm UV light on and off. Reproduced with permission from (a–c) ref. 119 (copyright 2019, John Wiley and Sons), (d) ref. 120 (copyright 2023, John Wiley and Sons), and (e) ref. 121 (copyright 2022, American Chemical Society). | ||
In 2022, Dong et al. introduced an effective approach by incorporating dynamic metal–ligand coordination into host–guest doped systems, offering a means to fabricate regulated RTP materials with controlled properties (Fig. 10d).120 The addition of a minimal amount of Zn2+ in the three-component doped system demonstrated remarkable phosphorescence performance. Through single-crystal analysis and theoretical calculations, it was revealed that the coordinate interactions significantly augmented the SOC of the guest, leading to an elevated ISC rate and phosphorescent radiative rate. By manipulating the Zn2+ counterions and adjusting the doping ratio of the guest, facile modulation of luminous color and ΦP could be achieved. Additionally, the reversible nature of metal–ligand coordinate interactions enabled dynamic manipulation of coordination-activated and dissociation-deactivated RTP (Fig. 10d). Employing a similar approach, a series of three-component doped systems were assembled by introducing phenylquinoline isomers (n-QL) as the guest molecules and seven metal ions (Al3+, Cu+/2+, Zn2+, Ga3+, Ag+, Cd2+, and In3+) as the third component into benzophenone (Fig. 10e).121 The coordination between the guest molecules and the metal ions resulted in a comprehensive enhancement of phosphorescence properties, encompassing the phosphorescence efficiency, wavelength, and lifetime, in the doped materials (Fig. 10e). Both studies presented a straightforward, efficient, and universally applicable strategy for fine-tuning the phosphorescent characteristics of doped materials.
Summary and outlook
Numerous efforts have been devoted to suppress the non-radiative transitions and enhance the phosphorescence efficiency by creating ionic interactions. In this perspective, we first illustrated that utilizing ionic compounds as organic RTP materials offers several advantages: enhanced phosphorescence efficiency due to strong SOC facilitated by heavy atoms, increased rigidity in the molecular architecture which can reduce non-radiative decay, and the possibility for tunable emissions by varying the ionic components. Subsequently, we systematically explored the influence of various intermolecular interactions in ionic systems, including the ion–π interactions, electrostatic interactions, and coordinate interactions on the phosphorescence properties at ambient conditions. In summary, ionic compounds provide greater flexibility of their molecular design and control, contributing to the optimization of RTP.For further developments in ion-regulated organic RTP materials, the following issues and challenges need to be considered. Firstly, we need to deeply understand the functions and roles of ionic interactions in the phosphorescence emission, to achieve effective manipulation of the RTP performance including the phosphorescence color, lifetime, and quantum yield. Secondly, compared to classical inorganic systems, the long luminescence lifetimes in the studied ionic systems are significantly shorter. Moreover, high-performance materials of simultaneously possessing both ultra-long phosphorescence lifetimes and high phosphorescence efficiency in ionic systems are rare, and need to be further enriched. Thirdly, the inherent rigidity of ionic crystals results in poor processability, which severely hinders their practical applications in flexible electronics. Thus, considerable attention needs to be paid to the development of amorphous ionic RTP materials for easy processing. Fourthly, most of the reported ionic RTP materials exhibit short absorption and emission wavelengths, which impede their utility in vivo applications due to the limited tissue penetrability. Developing ionic RTP materials with infrared or near-infrared phosphorescence emission will be crucial to extend their applications in vivo. Finally, introducing functional groups to develop stimuli-responsive ionic RTP materials will endow them with more functionalities and support much wider applications.
Author contributions
Prof. Zhengxu Cai and Dongpeng Yan conceived the project. Wenbo Dai and Yitian Jiang conducted the literature review. Wenbo Dai carried out the concepts, design, definition of intellectual content, and manuscript preparation. Prof. Zhengxu Cai and Dongpeng Yan revised and finalized the manuscript. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Key Research and Development Program of China (2018YFA0901800), The National Natural Scientific Foundation of China (Grant number: 22222501, 22175023), and the Natural Science Foundation of Beijing Municipality (2232022). This work was also supported by BIT Research and Innovation Promoting Project (Grant no. 2023YCXZ016).Notes and references
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS
.
- S. Hirata, Appl. Phys. Rev., 2022, 9, 011304 CAS
- M. Fang, J. Yang and Z. Li, Prog. Mater. Sci., 2022, 125, 100914 CrossRef CAS
- J. Gu, Z. Li and Q. Li, Coord. Chem. Rev., 2023, 475, 214872 CrossRef CAS
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed
- S. Guo, W. Dai, X. Chen, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS
- N. Gan, X. Zou, Y. Zhang, L. Gu and Z. An, Appl. Phys. Rev., 2023, 10, 021313 CAS
- X. Wang, H. Shi, H. Ma, W. Ye, L. Song, J. Zan, X. Yao, X. Ou, G. Yang, Z. Zhao, M. Singh, C. Lin, H. Wang, W. Jia, Q. Wang, J. Zhi, C. Dong, X. Jiang, Y. Tang, X. Xie, Y. Yang, J. Wang, Q. Chen, Y. Wang, H. Yang, G. Zhang, Z. An, X. Liu and W. Huang, Nat. Photon., 2021, 15, 187–192 CrossRef CAS
- F. Li, M. Wang, S. Liu and Q. Zhao, Chem. Sci., 2022, 13, 2184–2201 RSC
- H. Sun and L. Zhu, Aggregate, 2022, 4, e253 CrossRef
- Y. Zhang, W. Zhang, J. Xia, C. Xiong, G. Li, X. Li, P. Sun, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2023, 62, e202314273 CrossRef CAS PubMed
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef
- Q. Dang, Y. Jiang, J. Wang, J. Wang, Q. Zhang, M. Zhang, S. Luo, Y. Xie, K. Pu, Q. Li and Z. Li, Adv. Mater., 2020, 32, 2006752 CrossRef PubMed
- W. Dai, Y. Zhang, X. Wu, S. Guo, J. Ma, J. Shi, B. Tong, Z. Cai, H. Xie and Y. Dong, CCS Chem., 2022, 4, 2550–2559 CrossRef CAS
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 186 CrossRef CAS PubMed
- Y. Zhao, J. Yang, C. Liang, Z. Wang, Y. Zhang, G. Li, J. Qu, X. Wang, Y. Zhang, P. Sun, J. Shi, B. Tong, H.-Y. Xie, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2023, e202317431 Search PubMed
- Y. Si, Y. Zhao, W. Dai, S. Cui, P. Sun, J. Shi, B. Tong, Z. Cai and Y. Dong, Chin. J. Chem., 2023, 41, 1575–1582 CrossRef CAS
- Y. Zhang, J. Li, J. Zhao, X. Li, Z. Wang, Y. Huang, H. Zhang, Q. Liu, Y. Lei and D. Ding, Angew. Chem., Int. Ed., 2023, e202313890 Search PubMed
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed
- W. Shao and J. Kim, Acc. Chem. Res., 2022, 55, 1573–1585 CrossRef CAS PubMed
- H. Shi, W. Yao, W. Ye, H. Ma, W. Huang and Z. An, Acc. Chem. Res., 2022, 55, 3445–3459 CrossRef CAS PubMed
- L. Gu, X. Wang, M. Singh, H. Shi, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2020, 11, 6191–6200 CrossRef CAS
- Q. Peng, H. Ma and Z. Shuai, Acc. Chem. Res., 2021, 54, 940–949 CrossRef CAS
- Z. Xu, Y. He, H. Shi and Z. An, SmartMat, 2023, 4, e1139 CrossRef CAS
- C. C. Kenry and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef PubMed
- J. Guo, C. Yang and Y. Zhao, Acc. Chem. Res., 2022, 55, 1160–1170 CrossRef CAS PubMed
- S. Cai, X. Yao, H. Ma, H. Shi and Z. An, Aggregate, 2023, 4, e320 CrossRef CAS
- Y. Zhang, C. Xiong, W. Wang, W. Dai, Y. Ren, J. Xia, G. Li, J. Shi, B. Tong, X. Zheng, X. Shao, Z. Cai and Y. Dong, Aggregate, 2023, 4, e310 CrossRef CAS
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed
- E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977–9981 CrossRef CAS PubMed
- S. Garain, S. S. N. Ansari, A. Kongasseri, B. C. Garain, S. K. Pati and S. J. George, Chem. Sci., 2022, 13, 10011–10019 RSC
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photon., 2019, 13, 406–411 CrossRef CAS
- S. Li, L. Fu, X. Xiao, H. Geng, Q. Liao, Y. Liao and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 18059–18064 CrossRef CAS PubMed
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed
- X. K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS PubMed
- G. Wu, F. Li, B. Tang and X. Zhang, J. Am. Chem. Soc., 2022, 144, 14962–14975 CrossRef CAS PubMed
- H. Gao and X. Ma, Aggregate, 2021, 2, 38 CrossRef
- Z. Huang, Z. He, B. Ding, H. Tian and X. Ma, Nat. Commun., 2022, 13, 7841 CrossRef CAS PubMed
- B. Ding, X. Ma and H. Tian, Acc. Mater. Res., 2023, 4, 827–838 CrossRef CAS
- Y. Lei, W. Dai, Y. Tian, J. Yang, P. Li, J. Shi, B. Tong, Z. Cai and Y. Dong, J. Phys. Chem. Lett., 2019, 10, 6019–6025 CrossRef CAS PubMed
- Y. Wang, J. Yang, Y. Gong, M. Fang, Z. Li and B. Z. Tang, SmartMat, 2020, 1, 1006 CrossRef
- W. Dai, T. Bianconi, E. Ferraguzzi, X. Wu, Y. Lei, J. Shi, B. Tong, B. Carlotti, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 3, 1767–1777 CrossRef CAS
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS PubMed
- X. Chen, W. Dai, X. Wu, H. Su, C. Chao, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, Chem. Eng. J., 2021, 426, 131607 CrossRef CAS
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CrossRef CAS PubMed
- X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Small, 2022, 18, e2104073 CrossRef PubMed
- G. Li, W. Dai, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, ChemPhotoChem, 2022, 7, e202200198 CrossRef
- Y. Ren, W. Dai, S. Guo, L. Dong, S. Huang, J. Shi, B. Tong, N. Hao, L. Li, Z. Cai and Y. Dong, J. Am. Chem. Soc., 2022, 144, 1361–1369 CrossRef CAS PubMed
- Y. Lei, W. Dai, G. Li, Y. Zhang, X. Huang, Z. Cai and Y. Dong, J. Phys. Chem. Lett., 2023, 14, 1794–1807 CrossRef CAS PubMed
- B. Wang, Z. Sun, J. Yu, G. I. N. Waterhouse, S. Lu and B. Yang, SmartMat, 2022, 3, 337–348 CrossRef CAS
- K. Wang, L. Qu and C. Yang, Small, 2023, 19, e2206429 CrossRef PubMed
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C. H. Liu, H. M. Titi and D. F. Perepichka, Nat. Chem., 2023, 15, 83–90 CrossRef CAS PubMed
- X. Liang, X.-F. Luo, Z.-P. Yan, Y.-X. Zheng and J.-L. Zuo, Angew. Chem., Int. Ed., 2021, 60, 24437–24442 CrossRef CAS
- X. Xu, Z. Chen, Y. Lei, X. Sun, M. Liu, H. Wu and X. Huang, Chem. Commun., 2022, 58, 11143–11146 RSC
- I. Partanen, O. Al-Saedy, T. Eskelinen, A. J. Karttunen, J. J. Saarinen, O. Mrozek, A. Steffen, A. Belyaev, P. T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2023, 62, e202305108 CrossRef CAS PubMed
- F. Nie, B. Zhou and D. Yan, Chem. Eng. J., 2023, 453, 139806 CrossRef CAS
- Y. Liu, Y. Ma, X. Fang, T. Chen, F. Nie and D. Yan, J. Mater. Chem. C, 2023, 11, 15855–15860 RSC
- P. She, Y. Yu, Y. Qin, Y. Zhang, F. Li, Y. Ma, S. Liu, W. Huang and Q. Zhao, Adv. Opt. Mater., 2019, 8, 1901437 CrossRef
- S. Liu, Y. Lin and D. Yan, Sci. Bull., 2022, 67, 2076–2084 CrossRef CAS PubMed
- J. Wei, M. Zhu, T. Du, J. Li, P. Dai, C. Liu, J. Duan, S. Liu, X. Zhou, S. Zhang, L. Guo, H. Wang, Y. Ma, W. Huang and Q. Zhao, Nat. Commun., 2023, 14, 4839 CrossRef CAS
- S. Liu, X. Fang, B. Lu and D. Yan, Nat. Commun., 2020, 11, 4649 CrossRef CAS PubMed
- B. Zhou, G. Xiao and D. Yan, Adv. Mater., 2021, 33, e2007571 CrossRef PubMed
- G. Xiao, X. Fang, Y. J. Ma and D. Yan, Adv. Sci., 2022, 9, e2200992 CrossRef
- O. Pajuelo-Corral, A. Rodríguez-Diéguez, G. Beobide, S. Pérez-Yáñez, J. A. García, E. San Sebastian, J. M. Seco and J. Cepeda, J. Mater. Chem. C, 2019, 7, 6997–7012 RSC
- L. J. Xu, A. Plaviak, X. Lin, M. Worku, Q. He, M. Chaaban, B. J. Kim and B. Ma, Angew. Chem., Int. Ed., 2020, 59, 23067–23071 CrossRef CAS
- S. Feng, Y. Ma, S. Wang, S. Gao, Q. Huang, H. Zhen, D. Yan, Q. Ling and Z. Lin, Angew. Chem., Int. Ed., 2022, e202116511 CAS
- R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC
- Z. Qi, B. Zhou and D. Yan, Mater. Chem. Front., 2023, 7, 3475–3493 RSC
- B. Zhou and D. Yan, Adv. Funct. Mater., 2023, 33, 2300735 CrossRef CAS
- X. Li, Y. Wang, Z. Zhang, S. Cai, Z. An and W. Huang, Adv. Mater., 2023, e2308290 Search PubMed
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed
- L. Xiao and H. Fu, Chem.–Eur. J., 2019, 25, 714–723 CrossRef CAS
- F. Xiao, M. Wang, Y. Lei, W. Dai, Y. Zhou, M. Liu, W. Gao, X. Huang and H. Wu, J. Mater. Chem. C, 2020, 8, 17410–17416 RSC
- N. Liu, Y. Pan, Y. Lei, M. Liu, C. Peng, Z. Cai, G. Shen, H. Wu, X. Huang and Y. Dong, Chem. Eng. J., 2022, 433, 133530 CrossRef CAS
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed
- G. Jiang, J. Yu, J. Wang and B. Z. Tang, Aggregate, 2022, 3, e285 CrossRef CAS
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed
- A. Cheng, H. Su, X. Gu, W. Zhang, B. Zhang, M. Zhou, J. Jiang, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202312627 CrossRef CAS PubMed
- S. Garain, S. M. Wagalgave, A. A. Kongasseri, B. C. Garain, S. N. Ansari, G. Sardar, D. Kabra, S. K. Pati and S. J. George, J. Am. Chem. Soc., 2022, 144, 10854–10861 CrossRef CAS
- P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 CAS
- Z. Cheng, H. Shi, H. Ma, L. Bian, Q. Wu, L. Gu, S. Cai, X. Wang, W. W. Xiong, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 678–682 CrossRef CAS PubMed
- G. Chen, H. Feng, F. Feng, P. Xu, J. Xu, S. Pan and Z. Qian, J. Phys. Chem. Lett., 2018, 9, 6305–6311 CrossRef CAS PubMed
- G. Chen, S. Guo, H. Feng and Z. Qian, J. Mater. Chem. C, 2019, 7, 14535–14542 RSC
- G. Xiao, Y. J. Ma, X. Fang and D. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 30246–30255 CrossRef CAS PubMed
- W. Chen, Z. Tian, Y. Li, Y. Jiang, M. Liu and P. Duan, Chem.–Eur. J., 2018, 24, 17444–17448 CrossRef CAS PubMed
- S. Feng, Q. Huang, S. Yang, Z. Lin and Q. Ling, Chem. Sci., 2021, 12, 14451–14458 RSC
- D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375–1406 CrossRef CAS PubMed
- Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS PubMed
- S. Z. Cai, H. L. Ma, H. F. Shi, H. Wang, X. Wang, L. X. Xiao, W. P. Ye, K. W. Huang, X. D. Cao, N. Gan, C. Q. Ma, M. X. Gu, L. L. Song, H. Xu, Y. T. Tao, C. F. Zhang, W. Yao, Z. F. An and W. Huang, Nat. Commun., 2019, 10, 4247 CrossRef PubMed
- H. Wang, H. Shi, W. Ye, X. Yao, Q. Wang, C. Dong, W. Jia, H. Ma, S. Cai, K. Huang, L. Fu, Y. Zhang, J. Zhi, L. Gu, Y. Zhao, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 18776–18782 CrossRef CAS PubMed
- B. Zhao, S. Yang, X. Yong and J. Deng, ACS Appl. Mater. Interfaces, 2021, 13, 59320–59328 CrossRef CAS PubMed
- S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC
- T. Zhu, T. Yang, Q. Zhang and W. Z. Yuan, Nat. Commun., 2022, 13, 2658 CrossRef CAS PubMed
- S. Tang, Z. Zhao, J. Chen, T. Yang, Y. Wang, X. Chen, M. Lv and W. Z. Yuan, Angew. Chem., Int. Ed., 2022, 61, e202117368 CrossRef CAS
- J. Zhang, P. Alam, S. Zhang, H. Shen, L. Hu, H. H. Y. Sung, I. D. Williams, J. Sun, J. W. Y. Lam, H. Zhang and B. Z. Tang, Nat. Commun., 2022, 13, 3492 CrossRef CAS PubMed
- X. Luo, B. Tian, Y. Zhai, H. Guo, S. Liu, J. Li, S. Li, T. D. James and Z. Chen, Nat. Rev. Chem, 2023, 7, 800–812 CrossRef
- Z. Zhu, L. Zeng, W. Li, W. Xu and D. Tian, ACS Sustain. Chem. Eng., 2022, 10, 16752–16759 CrossRef CAS
- X. Zhang, Y. Cheng, J. You, J. Zhang, C. Yin and J. Zhang, Nat. Commun., 2022, 13, 1117 CrossRef CAS PubMed
- J. You, X. Zhang, Q. Nan, K. Jin, J. Zhang, Y. Wang, C. Yin, Z. Yang and J. Zhang, Nat. Commun., 2023, 14, 4163 CrossRef CAS
- Y. Zhai, S. Li, J. Li, S. Liu, T. D. James, J. L. Sessler and Z. Chen, Nat. Commun., 2023, 14, 2614 CrossRef CAS PubMed
- L. Zeng, Z. Zhu, R. Mo, W. Li, W. Xu and D. Tian, Chem. Eng. J., 2023, 460, 141452 CrossRef CAS
- Z.-A. Yan, X. Lin, S. Sun, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 19735–19739 CrossRef CAS PubMed
- Z. Wang, A. Li, Z. Zhao, T. Zhu, Q. Zhang, Y. Zhang, Y. Tan and W. Z. Yuan, Adv. Mater., 2022, 34, e2202182 CrossRef PubMed
- A. Winter and U. S. Schubert, Chem. Soc. Rev., 2016, 45, 5311–5357 RSC
- Y. J. Ma, Z. Qi, G. Xiao, X. Fang and D. Yan, Inorg. Chem., 2022, 61, 16477–16483 CrossRef CAS PubMed
- H. Liu, K. Zhang, P. F. Gao, J. H. Luo, Y. Y. Jiang, M. S. Zhou, T. Li, X. L. Zhu and H. R. Fu, Inorg. Chem., 2022, 61, 1636–1643 CrossRef CAS PubMed
- X. Yang and D. Yan, Adv. Opt. Mater., 2016, 4, 897–905 CrossRef CAS
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC
- F. Nie, K. Z. Wang and D. Yan, Nat. Commun., 2023, 14, 1654 CrossRef CAS
- B. Zhou, Z. Qi and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202208735 CrossRef CAS PubMed
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed
- H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef PubMed
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed
- P. Wei, J. Liu, G. G. Shan, X. Zhang, H. Zhang, J. Qi, W. Zhao, H. H. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 59, 9293–9298 CrossRef
- W. Dai, G. Li, Y. Zhang, Y. Ren, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, Adv. Funct. Mater., 2023, 33, 2210102 CrossRef CAS
- Y. Guo, K. Chen, Z. Hu, Y. Lei, X. Liu, M. Liu, Z. Cai, J. Xiao, H. Wu and X. Huang, J. Phys. Chem. Lett., 2022, 13, 7607–7617 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |