
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchable molecular electrocatalysis†
Shifali
Dutt‡
a,
Alagar Raja
Kottaichamy‡
 ad,
Neethu Christudas
Dargily‡
a,
Sanchayita
Mukhopadhyay
a,
Bhojkumar
Nayak
a,
Mruthyunjayachari Chattanhali
Devendrachari
a,
Chatakudhath Prabakaran
Vinod
b,
Harish Makri
Nimbegondi Kotresh
c and
Musthafa
Ottakam Thotiyl
*a
ad,
Neethu Christudas
Dargily‡
a,
Sanchayita
Mukhopadhyay
a,
Bhojkumar
Nayak
a,
Mruthyunjayachari Chattanhali
Devendrachari
a,
Chatakudhath Prabakaran
Vinod
b,
Harish Makri
Nimbegondi Kotresh
c and
Musthafa
Ottakam Thotiyl
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER)-Pune, Dr Homi Bhabha Road, Pashan, Pune 411008, Maharashtra, India. E-mail: musthafa@iiserpune.ac.in
bCatalysis and Inorganic Division, CSIR-NCL, Pune 411008, India
cDepartment of Chemistry, Acharya Institute of Technology, Soldevanahalli, Bangalore 560107, India
dDepartment of Chemistry, Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel
First published on 23rd July 2024
Abstract
We demonstrate a switchable electrocatalysis mechanism modulated by hydrogen bonding interactions in ligand geometries. By manipulating these geometries, specific electrochemical processes at a single catalytic site can be selectively and precisely activated or deactivated. The α geometry enhances dioxygen electroreduction (ORR) while inhibiting protium redox processes, with the opposite effect seen in the β geometry. Intramolecular hydrogen bonding in the α geometry boosts electron density at the catalytic center, facilitating a shift of ORR to a 4-electron pathway. Conversely, the β geometry promotes a 2-electron ORR and facilitates electrocatalytic hydrogen evolution through an extensive proton charge assembly; offering a paradigm shift to conventional electrocatalytic principles. The expectations that ligand geometry induced electron density modulations in the catalytic metal centre would have a comparable impact on both ORR and HER has been questioned due to the contrasting reactivity exhibited by α-geometry and β-geometry molecules. This further emphasizes the complex and intriguing nature of the roles played by ligands in molecular electrocatalysis.
Introduction
Extensive research has been conducted on molecular systems for diverse applications, including fuel cells, batteries, supercapacitors, sensing, and light harvesting.1–9 In these lines, metal phthalocyanines (MPc), such as those based on iron (FePc), cobalt (CoPc), zinc (ZnPc), copper (CuPc), nickel (NiPc), have been the subject of thorough investigation as electrocatalysts for various reactions.10–17 Notably, tetraamino-substituted FePc and CoPc have emerged as widely studied model electrocatalysts for fundamental electrochemical substrates like dioxygen (O2) and proton (H+).18–33 Recent breakthroughs have revealed that these phthalocyanines, particularly when axially coordinated to N atoms, catalyze a 4-electron reduction of oxygen in Zn–air batteries.34–36 In the ongoing electrocatalytic research involving MPc-based organometallic complexes, the selectivity and reaction pathway or mechanism have traditionally been attributed to the central metal ion.37,38. Notably, the role of ligands in facilitating challenging electrochemical transformations has not been unequivocally demonstrated thus far.Here, we illustrate a switchable electrocatalytic effect on the same catalytic metal center toward oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER) through ligand geometry (α geometry and β geometry) assisted hydrogen bonding interactions. A preference for the α geometry ligand for ORR was observed, while the corresponding β geometry ligand showed a specific inclination for HER. In-depth investigations showed that the two geometries exhibit different electrocatalytic behaviours because ligand geometry-driven hydrogen bonding interactions adjust the electron density at the catalytic center with a corresponding modification in interfacial proton charge assembly.
Results and discussion
To demonstrate the ON–OFF effect in electrocatalysis assisted by the ligand geometry, we have synthesized regioisomers of tetraamino-substituted cobalt phthalocyanine (TACoPc) by relocating the –NH2 functional group between the α (α-TACoPc) and β (β-TACoPc) positions on the phthalocyanine ring (Fig. 1a). The presence of parent ion peak for isomeric amino compounds at m/z value of ∼631 in Matrix Assisted Laser Desorption Ionization Time of Flight (MALDI-TOF) (Fig. S1a and b in ESI†) indicate their successful formation. UV-vis spectroscopy revealed the distinctive Q bands in the 600–700 nm range and B band in the 200–350 nm range (Fig. 1b).39 The Q band which is fundamental to ligands, showed a red shift in the α-isomer due to more electronic delocalization compared to the β-isomer.40 ATR-FTIR analysis of isomeric molecules reveals a N–H vibration in the range of 3150–3360 cm−1, C–N vibrations at ∼1149 cm−1, aromatic ring vibration at ∼1635 cm−1, phthalocyanine skeleton vibration at ∼570–850 cm−1, and pyrrole ring vibration at ∼1240 cm−1 (Fig. 1c).41 The Raman spectra exhibits bands at ∼754 cm−1, ∼1187 cm−1, ∼1416 cm−1, ∼1460 cm−1, and ∼1609 cm−1 corresponding to N4-macrocyclic ring, C–H in-plane deformation, C–H in-plane stretching, isoindole ring stretching and C–N in plane stretching respectively, Fig. 1d.42 The morphology of the amine isomeric molecules was examined using high resolution transmission electron microscopy (HRTEM). The TEM image revealed fine nano structural features, and elemental mapping showed a uniform distribution of cobalt, carbon, and nitrogen elements (Fig. 1e and f).43 As cobalt phthalocyanines are paramagnetic, 1D NMR was employed to confirm the successful synthesis of isomeric molecules with zinc-based amino isomeric molecules (Fig. 1g). For aromatic protons, the α-isomer displayed five protons (1H) signals at different chemical shift values. The presence of intramolecular H-bonding between the primary amine group and iminic nitrogen in the Pc ring is indicated by the disappearance of the two protons (labelled d and e in Fig. 1g) that correspond to the aminic proton in α isomer upon D2O addition. On the other hand, the β-isomer for the aminic proton (labelled d in Fig. 1g) only displayed one broad signal, which vanished upon the addition of D2O. It suggests the absence of intramolecular H-bonding interaction in the β isomeric molecule. These molecules are then assembled on nanoporous gold (NPG) electrode because of its well-known three-dimensional nanoporous structure with ultrahigh surface area.44,45 The NPG is prepared as reported in the literature by holding the potential at 2 V vs. Ag/AgCl (3 M KCl), (Fig. S2a, ESI†).44 The enhancement of oxide formation and its reduction response in the treated electrode in comparison to its flat counterpart signal the formation of highly porous NPG architecture. This kind of potential dependent treatment is reported to restructure the Au electrode in such a way that it becomes nanoporous in nature.44 The scanning electron microscopy images (Fig. S2b and c, ESI†) clearly shows a morphological alteration with granular characteristics in the NPG electrode as compared to the bare Au surface, and the X-ray diffraction pattern (Fig. S2d, ESI†) supports the exposure of low index planes, indicating a surface restructuring following electrochemical treatments of the Au electrode.46,47 Following the successful fabrication of the nanoporous gold surface, the NPG electrode was then used as the support for anchoring α-TACoPc and β-TACoPc isomeric molecules via self-assembly approach48,49 (refer Experimental section for more details, ESI†) and subsequently investigated for their ORR and HER activities.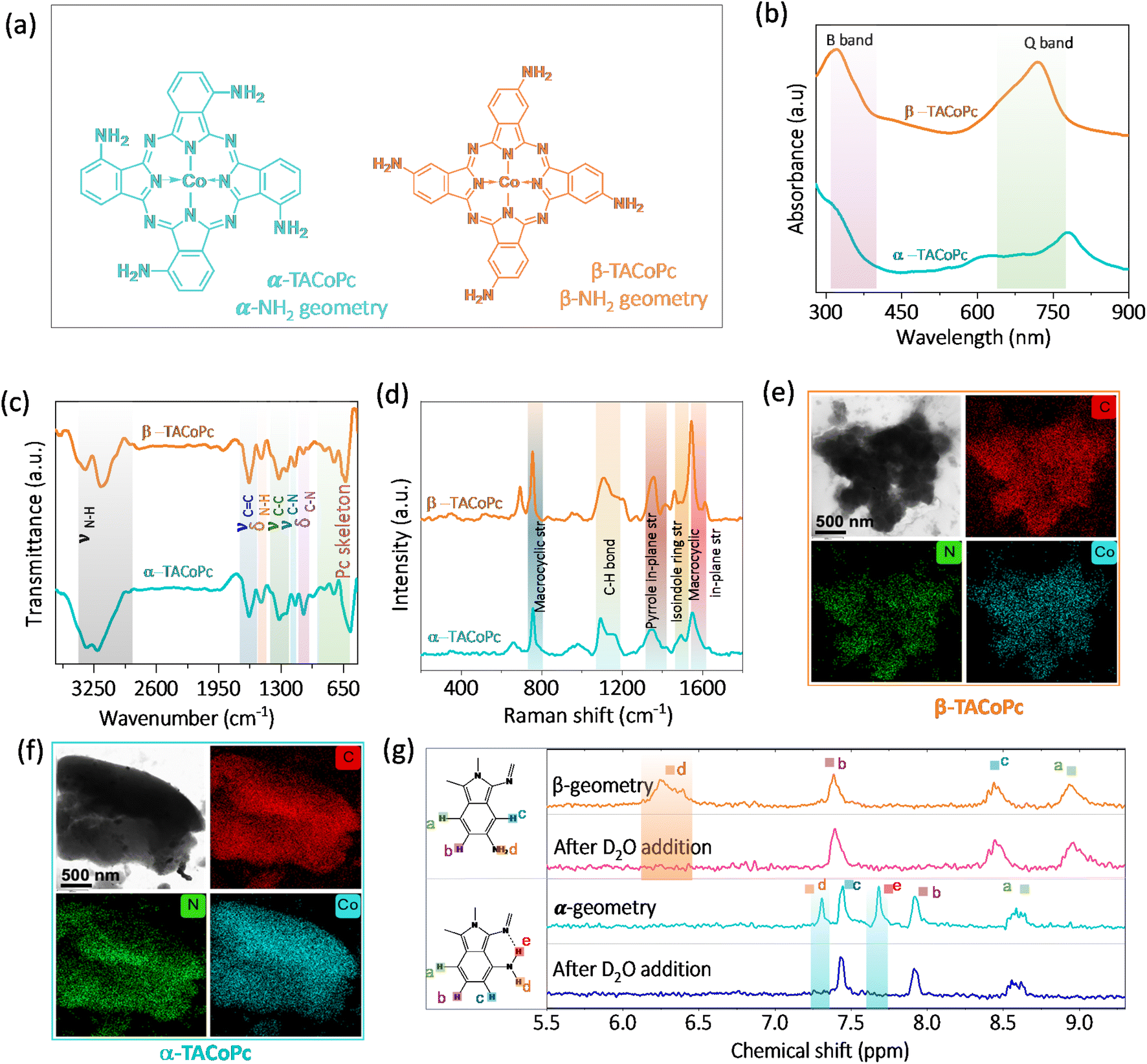 | ||
| Fig. 1 Molecular structures of isomeric (a) α-TACoPc and β-TACoPc molecules. (b) UV-vis spectra. (c) ATR-FTIR spectra, and (d) Raman spectra of isomeric α-TACoPc and β-TACoPc molecules. HRTEM images and elemental mapping of (e) β-TACoPc and (f) α-TACoPc molecules. (g) 1H NMR spectra in DMSO-d6 for the isomeric molecules before and after D2O addition. | ||
Firstly, we have carried out the electrochemical evaluation of NPG modified molecules towards electrochemical ORR and HER simultaneously, Fig. 2a. The simultaneous HER and ORR profiles on α and β-geometry molecules (Fig. 2a) indicate an active ORR with α-geometry molecule and an active HER with β-geometry molecule. The pattern towards the simultaneous ORR and HER remains same even with a planer Au electrode counterpart (Fig. S3, ESI†). This finding is unexpected because both reactions typically involve reduced Co species, therefore one would expect the electron density alterations in the central metal Co due to ligand geometry to influence both ORR and HER to a similar extent. This would suggest that the α-geometry molecule should be equally active for both reactions. However, the observed diametrically opposite electrocatalytic switching behavior challenges this assumption and highlights the complexity of the interactions at play in these reductive electrocatalytic processes.
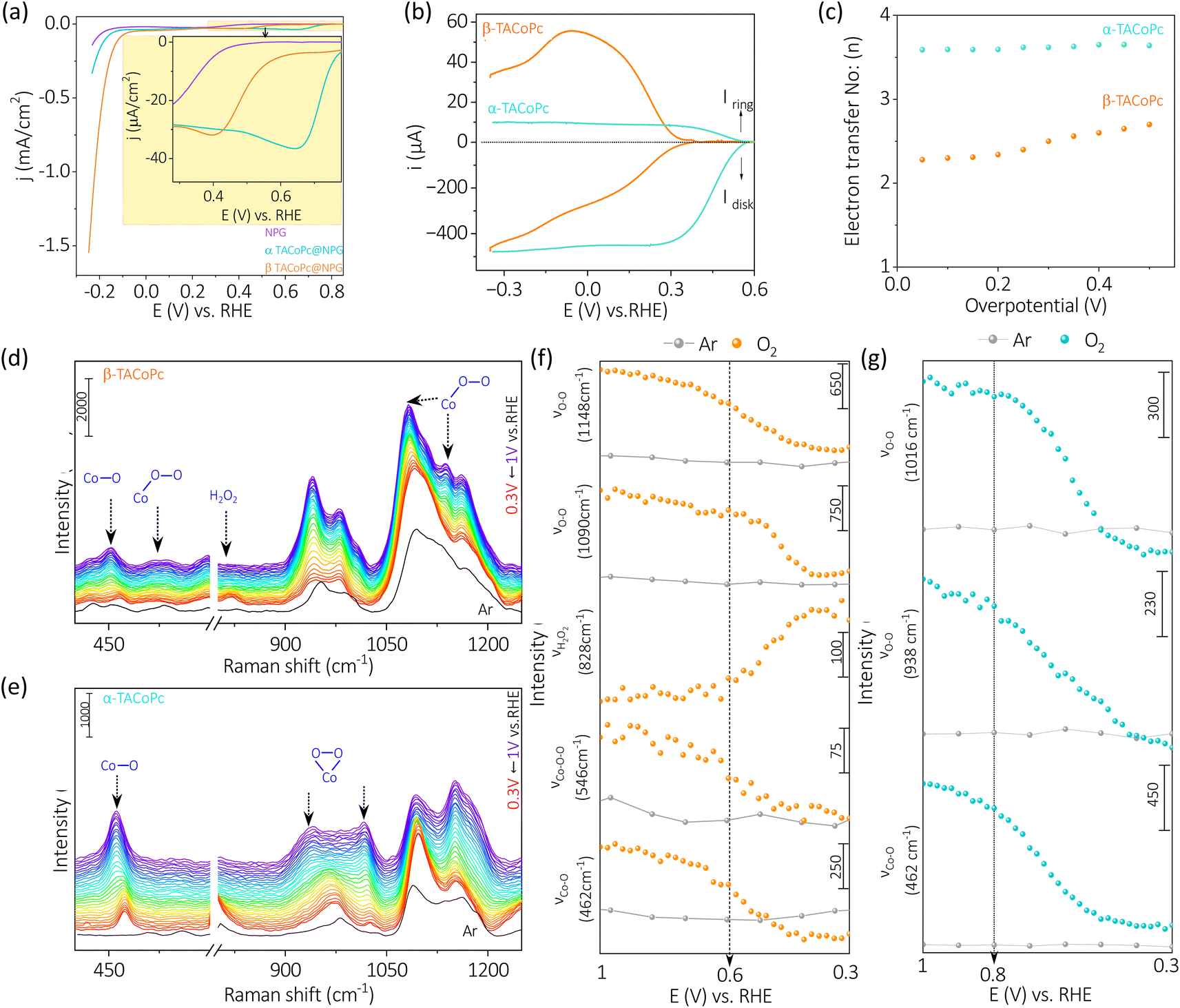 | ||
| Fig. 2 (a) Simultaneous linear sweep voltammograms of α-TACoPc and β-TACoPc modified NPG electrodes in oxygen saturated 0.1 M H2SO4 electrolyte at a scan rate of 5 mV s−1. (b) Rotating ring disk electrode studies (at 5 mV s−1 scan rate) at a rotation rate of 1600 rpm. (c) Number of electrons involved in ORR. In situ electrochemical Raman spectra of (d) β-isomer and (e) α-isomer in 0.1 M H2SO4 solution saturated with oxygen (blue to red trace)/argon (black trace at the open circuit voltage), when the voltage is scanned from 1 V to 0.3 V vs. RHE. The change in intensities for different bands of (f) β-isomer and (g) α-isomer during the reduction scan from 1 V to 0.3 V vs. RHE in 0.1 M H2SO4 solution saturated with oxygen (orange trace for β and blue trace for α)/argon (grey trace). | ||
To shed light on to this intriguing switching in electrocatalysis, α-TACoPc and β-TACoPc modified NPG Au electrodes were separately investigated for ORR and HER. During ORR, a negative shift of nearly 300 mV in ORR onset is observed in favour of the α geometry ligand, indicating an electrocatalytic effect assisted by ligand geometry, (Fig. S4a and Table S1, ESI†). Also, the α geometry molecules maintained a higher steady state potential than the β-geometry molecules, as detailed by the chronopotentiometry data (Fig. S4b†). All these indicate that the same catalytic metal center can be switched ON/OFF by ligand geometry. This is further evident from the parameters reflecting the true rate of electrochemical reactions such as the Tafel slope. The Tafel slope was found to be nearly 81 mV dec−1 on α geometry compared to nearly 122 mV dec−1 on β-geometry indicating an electrocatalytic enhancement in ORR when the ligand adopts α geometry (Fig. S4c and Table S2, ESI†). Hydrodynamic voltammetry using rotating ring-disk electrodes was further performed for oxygen reduction reaction (Fig. 2b) with different ligand geometries. Interestingly, a clear mechanistic change of ORR takes place on the same catalytic Co metal center via ligand geometry. For α geometry, the number of electrons involved in ORR is 4 with a lower peroxide percentile, while for the β counterpart, it is almost 2, with a peroxide proportion of 70–80% (Fig. 2c and S4d†). This trend in electroactivity was found to be same even with a flat Au electrode modified with a monolayer of α-TACoPc and β-TACoPc geometric molecules, suggesting these behaviours are intrinsic to the molecules, (Fig. S5 and Table S3, ESI†). When combined, these findings show that the same electrocatalytic Co center can be turned ON or OFF towards a 4 electron ORR via ligand geometry.
In situ electrochemical Raman spectroscopy was performed on α-TACoPc/β-TACoPc isomers on Au in a 0.1 M H2SO4 solution saturated with oxygen/argon, when the voltage is scanned from 1 V to 0.3 V vs. RHE. The spectra were first collected in argon atmosphere, where no change in spectral features of phthalocyanine were observed while sweeping the potential from 1 to 0.3 V vs. RHE. (Fig. S6a and b, ESI†). Comparative analysis of Raman spectra in an argon environment revealed that the introduction of oxygen not only induced new bands in both α (462, 938, 1016 cm−1) and β (462, 546, 828, 1090, 1148 cm−1) isomers, but also modified their intensity in relation to the applied potentials (refer to Fig. 2d–g). Clear distinctions in bonded oxygen species on the cobalt metal center due to ligand isomerism were validated through in situ Raman spectroelectrochemistry, as shown in Fig. 2d. The appearance of two peaks in the 400–600 cm−1 range in the β isomer, representing Co–O (462 cm−1) and Co–O–O (546 cm−1) species, indicate the presence of linearly bonded dioxygen species (dioxygen end-on binding) on Co catalytic sites (Fig. 2d).50,51 Conversely, such characteristics were notably absent in the α isomer, as indicated in Fig. 2e. The anticipated peak for end-on bonded dioxygen at ∼1125 cm−1 in the β isomer was obscured by closely located phthalocyanine bands within the same range, resulting in coupled bands at 1148 and 1090 cm−1.52,53 The reductive scan-induced consumption of the 1148 cm−1 band, along with an 8 cm−1 shift of the 1090 cm−1 band, back to its original state, supported the concept of coupling and dioxygen binding mode in the β isomer.54 The consumption of these four bands, coupled with the emergence of a band at 828 cm−1 (close to H2O2/O22−, Fig. S6c, ESI†), indicated the end-on binding of dioxygen on the Co sites in the β isomer (Fig. 2f). This implies the occurrence of a two-electron ORR pathway on Co sites when the ligand adopts a β isomerization. In the α isomer molecule, the absence of the Co–O–O band (546 cm−1) and the presence of bands at 938 and 1016 cm−1, approximately 300 cm−1 lower than the end-on bonded dioxygen55 confirmed the side-on binding of dioxygen on the cobalt metal center. Furthermore, all three Raman bands corresponding to dioxygen binding on Co sites in the α isomer (462, 938, 1016 cm−1) exhibited potential-dependent consumption without the emergence of a peak corresponding to peroxide formation at 828 cm−1 (Fig. 2g). These attest the fact that ligand isomerization indeed changes the dioxygen binding mode on the same catalytic Co sites.
In order to prove that the observed catalytic switching effect is not due to the NPG support, a composite of isomeric molecules and multi walled carbon nanotubes (CNT) were prepared (please refer Experimental section for more details, ESI†). The composite electrodes were characterised using various physiochemical techniques such as UV-vis spectroscopy and Raman spectroscopy (Fig. S7, ESI†). The characteristics of CNT in the UV-vis spectra and Raman spectra of the composite catalytic systems point to the integration of CNT with the molecular catalysts (Fig. S7a and b, ESI†). Subsequent rotating ring-disk electrode (RRDE) analysis in an acidic environment (0.1 M H2SO4) suggests a highly active α-TACoPc molecular platform compared to the β-TACoPc system. It is to be noted that, the α-isomerization of the ligand uplifts the activity of the Co catalytic center closer to that of the benchmark Pt/C electrocatalyst, Fig. S7c and Table S4, ESI†.56,57 The higher currents observed in Fig. S7c, ESI† compared to Fig. 2 is attributed to the better dispersion of isomeric molecules on to CNT support. All these suggests that the support materials aid in better dispersion of the electrocatalytic molecules, however the activity trends are intrinsic to the molecules. In order to check the stability of the isomeric molecules (as phthalocyanines are prone to degradation in acidic medium)57 during prolonged electrocatalysis, the Raman spectra were obtained before and after ORR for over 10 hours. It was observed that the Raman bands of the isomeric molecules (in 0.1 M H2SO4 electrolyte) remain almost intact even after prolonged electrocatalysis (Fig. S8, ESI†), suggesting their stability at the electrode/electrolyte interface. As we have recently elucidated, this trend in electrocatalytic ORR is irrespective of the central metal ion and the support material and is majorly dictated by the ligand geometry.58
Though α-TACoPc is found to be active towards ORR than the corresponding β-TACoPc isomer, an opposing effect is observed when HER is probed on the same electrocatalytic systems anchored onto NPG electrodes, Fig. 3a. With α-TACoPc, HER activity was negligible and the LSV profile resembled the trace on the nanoporous support (Fig. 3a), which was intriguing given its superior ORR activity. It's interesting to note that when the ligand geometry is changed to β, there is a noticeable activation in HER, Fig. 3a. This is demonstrated by a large positive shift in the HER onset potential by over 100 mV (Fig. 3a and Table S5, ESI†), suggesting that the molecule with β-geometry, which previously had little ORR activity, is now substantially HER active. This is further clear from the respective chronopotentiometric analysis at a current density of −0.2 mA cm−2 (Fig. 3b). Both the isomeric forms were stable over a period however, with a higher activity on the β-geometry ligand. The Tafel analysis (Fig. 3c and Table S6, ESI†) supports this, showing that the β-geometry demonstrating a lower Tafel slope (∼72 mV dec−1) than the α geometry (∼85 mV dec−1). A flat gold electrode demonstrated a similar trend towards HER (Fig. S9 and Table S7, ESI†) indicating NPG support has only an amplifying effect on this electrocatalytic disparity. Therefore, when the ligand adopts a α-geometry, the catalytic Co center is switched ON towards a 4 electron ORR, while the corresponding β-geometry switched on the same Co center towards electrocatalytic HER.
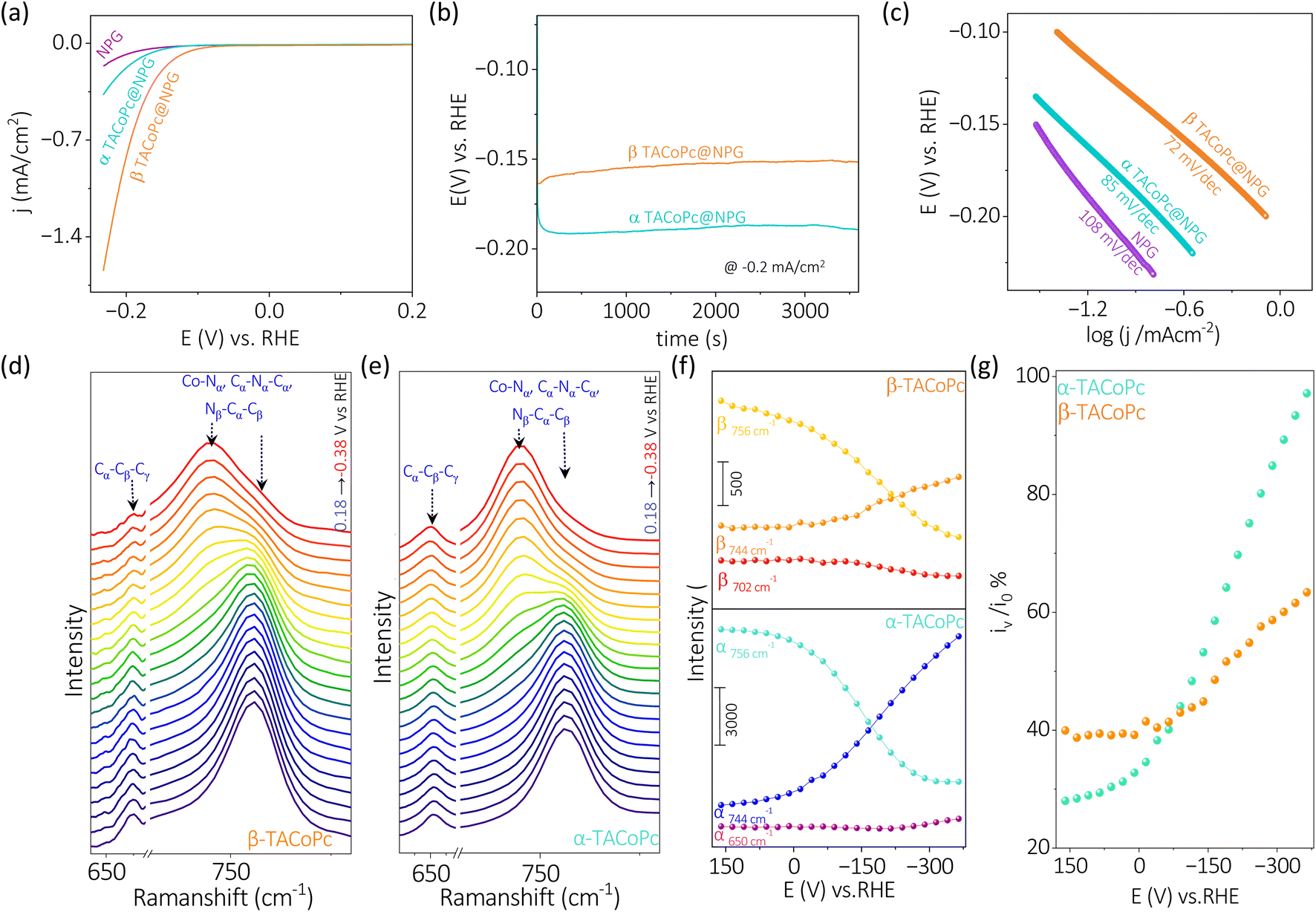 | ||
| Fig. 3 (a) Voltammograms of α-TACoPc and β-TACoPc modified NPG electrodes in 0.1 M H2SO4 at a scan rate of 5 mV s−1, and (b) the corresponding chronopotentiometry profiles at −0.2 mA cm−2. (c) Tafel plots of α-TACoPc and β-TACoPc isomers in 0.1 M H2SO4. In situ electrochemical Raman spectra of (d) β-isomer and (e) α-isomer in 0.1 M H2SO4 solution (blue to red trace) when the voltage is scanned from 0.16 to −0.38 V vs. RHE. The change in intensities for different bands of (f) β-isomer and α-isomer during the reduction scan from 0.16 to −0.38 V vs. RHE in 0.1 M H2SO4 solution. (g) Percentage change in intensity of 744 cm−1 band at different potentials vs. RHE (iv) with respect to the initial intensity of 756 cm−1 band at 0.16 V vs. RHE (i0) (orange trace for β-isomer and blue trace for α-isomer). | ||
The in situ electrochemical Raman spectroscopy of α-TACoPc and β-TACoPc molecules on gold was conducted in 0.1 M H2SO4 solution saturated with argon, by sweeping the potential from 0.16 V to −0.38 V vs. RHE, which corresponds to the hydrogen evolution region (Fig. 3d–g). Alteration in the regime of 600–800 cm−1 was probed which corresponds to coupled signals from metal–nitrogen (M–Nα), bridged nitrogen (Cα–Nα–Cα) and isoindole vibrations (Nβ–Cα–Cβ) of the phthalocyanine skeleton. During HER, Raman bands at 756 cm−1 was consumed with the concomitant generation of a band at 744 cm−1 for both the isomer molecules (Fig. 3d–f). These peaks correspond to coupled vibrations of M–Nα, Cα–Nα–Cα, and Nβ–Cα–Cβ leading to phthalocyanine ring deformation.59–62 The shift of these bands by around 12 cm−1 (in both the isomers) to lower wavelength attest the modification of the phthalocyanine skeleton during the progress of HER and therefore indicates the involvement of ligands in the HER process. On monitoring the ratio of iv to i0 (potential dependent intensity of 744 cm−1 band (iv) vs. initial intensity of 756 cm−1 band (i0) [at 0.16 V vs. RHE] during HER, the α-isomer evidenced an intensity gain of ∼97% as opposed to the β-isomer's 65% intensity gain (Fig. 3g). This suggests that the extent of modification of the phthalocyanine ring is much more pronounced in the α-isomer than the β-isomer during the HER process. Further, the Cα–Cβ–Cγ macrocycle symmetric deformation which is observed at 656 cm−1 for the α-isomer demonstrated a downshift by 4 cm−1 with a commensurate gain in its intensity during the HER. In contrast, the corresponding band for the β-isomer (at 702 cm−1) demonstrated only a depleting trend during the HER. These cumulative observations suggest the involvement of phthalocyanine ring in the HER electrocatalysis via molecular alterations which occurs to a higher extent in the α-isomer than the β-isomer. Therefore, the higher over potential required during HER in the α-isomer may be attributed to the comparatively higher energy required to undergo this significant molecular alterations. Taken together, in situ electrochemical Raman spectroscopy point out the definitive role of ligands in HER molecular electrocatalysis via molecular alterations.
This noticeable difference of the Co metal center's ORR and HER activities just because of the ligand geometry should be due to the types of interactions the molecules are experiencing when –NH2 position is shifted between α and β (Fig. 1a) locations. The proximity of macrocyclic nitrogen atoms to the N atom of –NH2 group in α-TACoPc may favour intramolecular –H bonding interactions as demonstrated in their NMR studies, Fig. 1g. Such interactions may be unfavourable in β-TACoPc due to the large positional separation of the corresponding groups, Fig. 1g. This may affect the electronic density at the catalytic cobalt sites very differently (Scheme S1, ESI†). To verify this electron density alteration, XPS analyses were conducted, Fig. 4a–d. The Co2+/Co3+ ratio extracted from the XPS analysis is significantly improved with a slightly increased satellite feature when the ligand adopts a α geometry (Fig. 4a and b). It is quite noteworthy to mention that the electron density of metal is enriched when the ligand assumes α geometry. From the literature, it is known that the catalytic species of mediated ORR electrocatalysis involving Co complexes are Co2+ (ref. 63 and 64) and its contribution is enriched by the α isomerization of the ligand (Fig. 4a and b). To further verify this, we have carried out EPR spectroscopy for the same concentrations (2 mg ml−1) of isomeric molecules dissolved in DMF solvent. The g value of 2.26 unequivocally indicates the existence of low spin Co2+ paramagnetic species, a feature well-documented in the literature. The EPR spectra of α and β-TACoPc isomers collected at room temperature, (Fig. S10, ESI†) shows a higher intensity in the α isomer (teal trace) illustrating its relatively higher abundance of Co2+.65–68 This enhanced electron density at the catalytic Co center when the ligand assumed a α geometry may indeed change the dioxygen binding mode from end on to side on, leading to a 4 electron ORR as detailed in Fig. 2b–g. It is reported that electron density alteration at the catalytic site can lead to a change in dioxygen binding modes.69,70 Further, intramolecular H bonding in the α geometry molecule, can make the protonation of N atoms difficult and the absence of such H bonding interactions in β geometry may lead to an extensive proton charge assembly over the corresponding ligand. This is investigated by analysing the N 1s XPS spectra and the deconvoluted N 1s peaks, (Co–N (N1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (N2), and C–NH2, (N3))71,72 before and after treating the molecules in acidic medium (Fig. 4c–d). It is observed that N 1s peaks especially those corresponding to N2 and N3 got upshifted in both the geometry molecules on acid treatment however, the upshift is significantly pronounced in the β geometry molecule, Fig. 4c and Table S8 ESI.† A slight difference in the N 1s and Co 2p binding energy assignments are observed especially with respect to 3d metal phthalocyanines non covalently bonded to single walled carbon nanotubes.73 This is attributed to the strong distortion of metal phthalocyanines when it is bonded with carbon nanotubes.73 It is important to note that protonation changes the area ratio of N2 and N3 in both isomeric forms. It is known that area ratio is a measure of the amount of surface species being exposed. Thus, the amplified area ratio of N2 and N3 may suggest their increased surface exposure after protonation.74,75 Nevertheless, N 1s XPS reveals that the extent of protonation is restricted in the case of α geometry due to intramolecular hydrogen bonding, and absence of such interactions leads to an extensive proton charge assembly in the β geometric molecule. This should entail the β geometry molecule's interface more positive than the corresponding α geometry, which is indeed confirmed by the Zeta potential measurements, Fig. 4e. Since protons are the reactant in HER; a preconcentration of protons at the interface of β geometry should lead to enhanced HER kinetics. To further explore the influence of isomerization on the protonation of the ligand, Raman spectra was acquired under open circuit voltage (OCV) conditions by changing the electrolytes.
N (N2), and C–NH2, (N3))71,72 before and after treating the molecules in acidic medium (Fig. 4c–d). It is observed that N 1s peaks especially those corresponding to N2 and N3 got upshifted in both the geometry molecules on acid treatment however, the upshift is significantly pronounced in the β geometry molecule, Fig. 4c and Table S8 ESI.† A slight difference in the N 1s and Co 2p binding energy assignments are observed especially with respect to 3d metal phthalocyanines non covalently bonded to single walled carbon nanotubes.73 This is attributed to the strong distortion of metal phthalocyanines when it is bonded with carbon nanotubes.73 It is important to note that protonation changes the area ratio of N2 and N3 in both isomeric forms. It is known that area ratio is a measure of the amount of surface species being exposed. Thus, the amplified area ratio of N2 and N3 may suggest their increased surface exposure after protonation.74,75 Nevertheless, N 1s XPS reveals that the extent of protonation is restricted in the case of α geometry due to intramolecular hydrogen bonding, and absence of such interactions leads to an extensive proton charge assembly in the β geometric molecule. This should entail the β geometry molecule's interface more positive than the corresponding α geometry, which is indeed confirmed by the Zeta potential measurements, Fig. 4e. Since protons are the reactant in HER; a preconcentration of protons at the interface of β geometry should lead to enhanced HER kinetics. To further explore the influence of isomerization on the protonation of the ligand, Raman spectra was acquired under open circuit voltage (OCV) conditions by changing the electrolytes.
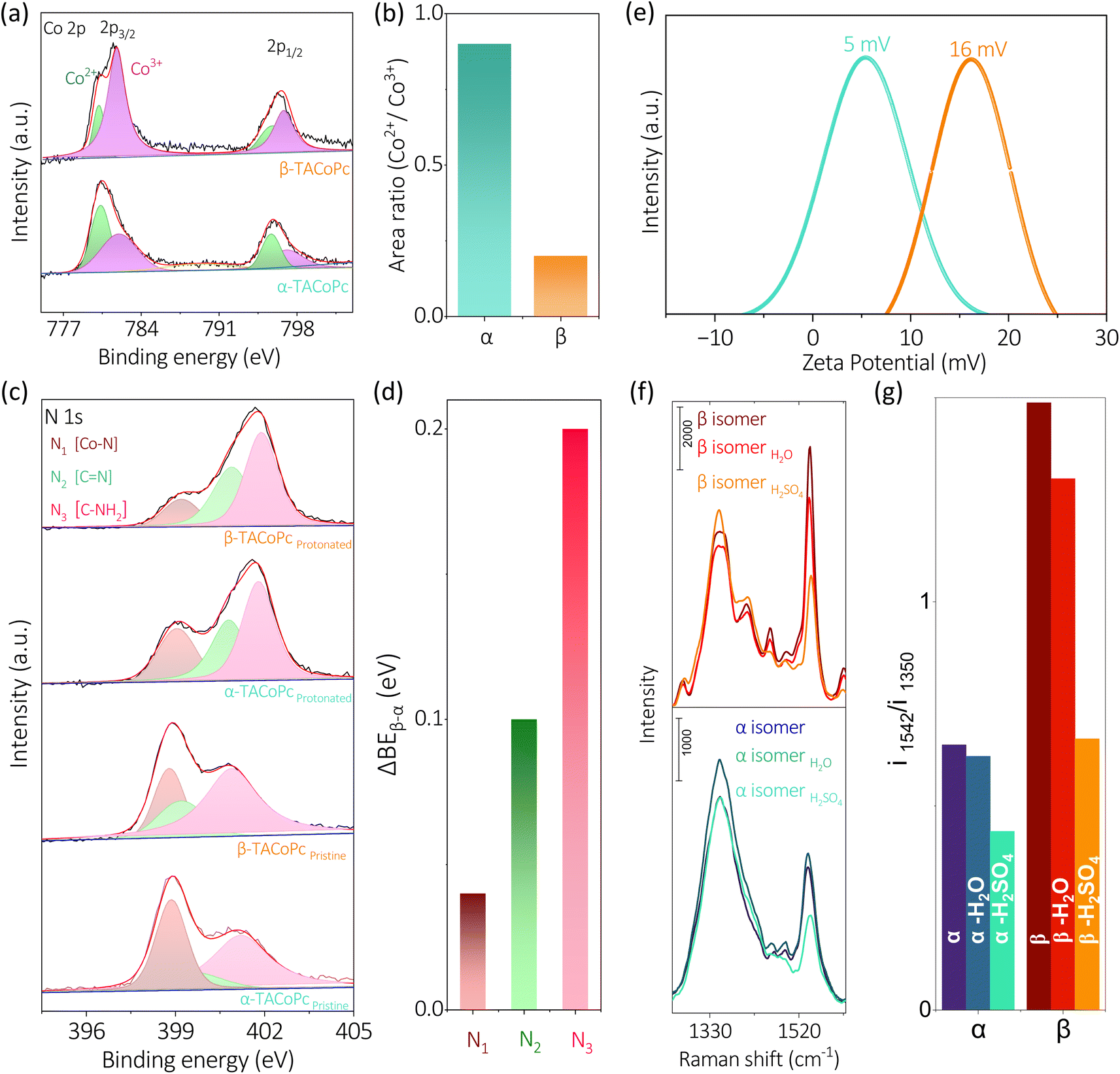 | ||
| Fig. 4 (a) Co 2p XPS spectra of α-TACoPc and β-TACoPc molecules. (b) Co2+/Co3+ area ratio which is extracted from (a). N 1s XPS spectra of (c) pristine and protonated forms of α-TACoPc and β-TACoPc isomers. (d) Bar plot showing the difference of N 1s binding energies between protonated β-TACoPc and protonated α-TACoPc molecules (ΔBEβ–α). (e) Zeta potential profiles of α-TACoPc and β-TACoPc isomeric molecules. (f) Raman spectra of α-TACoPc (bottom panel) and β-TACoPc (top panel) isomers and (g) the intensity ratio of i1542/i1350 peaks in the absence and presence of various solvents. | ||
Alterations in the fingerprint region of the phthalocyanine, in the range from 1260 cm−1 to 1560 cm−1,76 were observed during these experiments (Fig. 4f). The ratio of the most intense peak in this range (i1542/i1350) was analyzed (Fig. 4f). The isomeric molecules exhibited a nearly identical ratio in the absence as well as presence of H2O. However, the presence of protons (as in H2SO4), the ratio of i1542/i1350 was significantly altered for the β isomer, suggesting a pronounced effect of protons in the β isomer compared to the α isomer (Fig. 4g). This observation is consistent with the Zeta potential measurements indicating an extensive proton charge assembly in the β geometry molecule. This explains the higher HER activity of β geometry over the corresponding α geometry. In the previous section discussing Fig. 4, it was observed that the proton charge assembly is more pronounced in the β-isomer compared to the α-isomer as the latter has intramolecular hydrogen bonding interactions. It is believed that the level of proton charge assembly in the isomeric molecules is linked to the extent of molecular alterations shown in Fig. 3 during the process of the HER. The significant proton charge assembly in the β-isomer ligand likely confers an advantage in HER electrocatalysis via minor molecular alterations (Fig. 3). Conversely, the lower degree of proton charge assembly in the α-isomer may necessitate substantial molecular modifications to achieve a comparable reaction rate during the HER. Therefore, this investigation demonstrates that ligand geometry via H bonding interactions can turn ON/OFF the same catalytic center towards ORR and HER and this elucidation of the intricate role of non-covalent interactions is expected to contribute to the rational design of ligands for electrocatalytic applications.
Conclusions
We have demonstrated a ligand-modulated electrocatalytic switching of Co sites in dioxygen electrochemistry and protium redox processes. While the α-geometry precisely turns ON the catalytic centre during dioxygen electrochemistry, the β-geometry switches on the catalytic Co sites during electrocatalytic HER. Detailed investigation indicates that hydrogen bonding interactions triggered by ligand geometry are responsible for this electrocatalytic disparity. When the ligand adopts a α-geometry, the dioxygen binding modes at the Co sites change from the typical end-on mode to an unusual side-on mode, resulting in a 4-electron ORR. Conversely, when the ligand adopts a β-geometry, greater interfacial accumulation of proton charge assembly turns on the Co sites, directing them towards hydrogen evolution processes. The beliefs that ligand geometry-induced electron density alterations in the catalytic central metal would lead to a similar influence on both ORR and HER has been challenged by the observed contrasting reactivity of the α-geometry and β-geometry molecules, that in turn underscores the intricate and multifaceted aspects of ligands in molecular electrocatalysis. Continued research in this area holds promise for advancing the design and optimization of electrocatalysts for diverse applications in sustainable energy technologies.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. D., A. R. K., N. C. D. contributed to this work equally. Their contribution consisted of characterization of molecules, electrochemical experiments, numerous analyses, and writing up results. S. M. and B. N. helped in data curation. H. M. N. K. synthesized the molecules along with M. C. D. C. P. V. helped with the XPS analysis. M. O. T. conceptualized the work and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. O. T. is indebted to DST-SERB (CRG/2020/002549) and DST-WTI (DST/TMD-EWO/WTI/2K19/EWFH/2019/272) for financial support. S. D. and A. R. K. acknowledges financial assistance from UGC-India.References
- Y. Zhu, J. Deng and O. Fontaine, Nat. Energy, 2023, 8, 643–644 CrossRef.
- Y. Li, J. Chen, Y. Ji, Z. Zhao, W. Cui, X. Sang, Y. Cheng, B. Yang, Z. Li, Q. Zhang, L. Lei, Z. Wen, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2023, 62, e202306491 CrossRef CAS PubMed.
- E. Zhang, X. Hu, L. Meng, M. Qiu, J. Chen, Y. Liu, G. Liu, Z. Zhuang, X. Zheng, L. Zheng, Y. Wang, W. Tang, Z. Lu, J. Zhang, Z. Wen, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 18995–19007 CrossRef CAS PubMed.
- J. Lei, Y. Zhang, Y. Yao, Y. Shi, K. L. Leung, J. Fan and Y.-C. Lu, Nat. Energy, 2023, 8, 1355–1364 CrossRef CAS.
- G. M. Peters, J. B. Winegrad, M. R. Gau, G. H. Imler, B. Xu, S. Ren, B. B. Wayland and M. J. Zdilla, Inorg. Chem., 2017, 56, 3377–3385 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed.
- X. Rozhkova, A. Aimukhanov, A. Zeinidenov, V. Paygin, D. Valiev, J. Bisquert, A. Guerrero, A. Alexeev and B. Ilyassov, Synth. Met., 2023, 295, 117347 CrossRef CAS.
- S. Dey, B. B. Wayland and M. J. Zdilla, Inorg. Chem., 2019, 58, 1224–1233 CrossRef CAS PubMed.
- L. Schafzahl, N. Mahne, B. Schafzahl, M. Wilkening, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew Chem. Int. Ed. Engl., 2017, 56, 15728–15732 CrossRef CAS PubMed.
- A. R. Kottaichamy, S. Deebansok, J. Deng, M. A. Nazrulla, Y. Zhu, Z. M. Bhat, M. C. Devendrachari, C. P. Vinod, H. M. N. Kotresh, O. Fontaine and M. O. Thotiyl, Chem. Sci., 2023, 14, 6383–6392 RSC.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef.
- H. Li, H. Zhao, G. Yan, G. Huang, C. Ge, M. Forsyth, P. C. Howlett, X. Wang and J. Fang, Small, 2024, 20, 2304844 CrossRef CAS PubMed.
- G. Zhu, Y. Li, H. Zhu, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2016, 6, 6294–6301 CrossRef CAS.
- C. L. Rooney, M. Lyons, Y. Wu, G. Hu, M. Wang, C. Choi, Y. Gao, C.-W. Chang, G. W. Brudvig, Z. Feng and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202310623 CrossRef CAS PubMed.
- A. Ziani, T. Shinagawa, L. Stegenburga and K. Takanabe, ACS Appl. Mater. Interfaces, 2016, 8, 32376–32384 CrossRef CAS PubMed.
- A. T. Chidembo, K. I. Ozoemena, B. O. Agboola, V. Gupta, G. G. Wildgoose and R. G. Compton, Energy Environ. Sci., 2010, 3, 228–236 RSC.
- C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 5444–5447 CrossRef CAS PubMed.
- G. S. Phun, R. Bhide and S. Ardo, Energy Environ. Sci., 2023, 16, 4593–4611 RSC.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS PubMed.
- J. H. Zagal and M. T. M. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. G. Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- A. N. Oldacre, A. E. Friedman and T. R. Cook, J. Am. Chem. Soc., 2017, 139, 1424–1427 CrossRef CAS PubMed.
- Y. Fu, D. Xu, Y. Wang, X. Li, Z. Chen, K. Li, Z. Li, L. Zheng and X. Zuo, ACS Sustain. Chem. Eng., 2020, 8, 8338–8347 CrossRef CAS.
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS.
- R. Chen, H. Li, D. Chu and G. Wang, J. Phys. Chem. C, 2009, 113, 20689–20697 CrossRef CAS.
- J. Guo, H. Li, H. He, D. Chu and R. Chen, J. Phys. Chem. C, 2011, 115, 8494–8502 CrossRef CAS.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- L. Chen, R. U. R. Sagar, J. Chen, J. Liu, S. Aslam, F. Nosheen, T. Anwar, N. Hussain, X. Hou and T. Liang, Int. J. Hydrogen Energy, 2021, 46, 19338–19346 CrossRef CAS.
- C. P. K. Prabhu, S. Aralekallu, V. A. Sajjan, M. Palanna, S. Kumar and L. K. Sannegowda, Sustain. Energy Fuels, 2021, 5, 1448–1457 RSC.
- I. S. Kwon, I. H. Kwak, J. Y. Kim, H. G. Abbas, T. T. Debela, J. Seo, M. K. Cho, J. P. Ahn, J. Park and H. S. Kang, Nanoscale, 2019, 11, 14266–14275 RSC.
- D. Akyüz, B. Keskin, U. Şahintürk and A. Koca, Appl. Catal., B, 2016, 188, 217–226 CrossRef.
- S. Bhunia, A. Rana, S. Hematian, K. D. Karlin and A. Dey, Inorg. Chem., 2021, 60, 13876–13887 CrossRef CAS PubMed.
- K. Muuli, R. Kumar, M. Mooste, V. Gudkova, A. Treshchalov, H.-M. Piirsoo, A. Kikas, J. Aruväli, V. Kisand, A. Tamm, A. Krumme, P. Moni, M. Wilhelm and K. Tammeveski, Materials, 2023, 16, 4626 CrossRef CAS PubMed.
- Ü. E. Özen, E. Doğan, M. Özer, Ö. Bekaroğlu and A. R. Özkaya, J. Electrochem. Soc., 2016, 163, A2001–A2003 CrossRef.
- H. Xu, S. Ci, Y. Ding, G. Wang and Z. Wen, J. Mater. Chem. A, 2019, 7, 8006–8029 RSC.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y. R. Lu, T. S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- S. Liu, H. Bin Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- A. Cidlina, V. Novakova, M. Miletin and P. Zimcik, Dalton Trans., 2015, 44, 6961–6971 RSC.
- A. Ogunsipe and T. Nyokong, J. Mol. Struct., 2004, 689, 89–97 CrossRef CAS.
- T. Mugadza and T. Nyokong, Electrochim. Acta, 2010, 55, 6049–6057 CrossRef CAS.
- R. Georgescu, C. Boscornea, I. Calinescu and R. State, Rev. Chim., 2015, 66, 1554–1557 CAS.
- Y. Zhang, J. He and R. Yang, J. Mater. Sci., 2022, 57, 16064–16079 CrossRef CAS.
- A. Sukeri, L. P. H. Saravia and M. Bertotti, Phys. Chem. Chem. Phys., 2015, 17, 28510–28514 RSC.
- M. K. Khristosov, S. Dishon, I. Noi, A. Katsman and B. Pokroy, Nanoscale, 2017, 9, 14458–14466 RSC.
- A. A. Farghaly, M. Lam, C. J. Freeman, B. Uppalapati and M. M. Collinson, J. Electrochem. Soc., 2016, 163, H3083–H3087 CrossRef CAS.
- S. Pedireddy, H. K. Lee, W. W. Tjiu, I. Y. Phang, H. R. Tan, S. Q. Chua, C. Troadec and X. Y. Ling, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- A. Maringa, E. Antunes and T. Nyokong, Electrochim. Acta, 2014, 121, 93–101 CrossRef CAS.
- K. Cui, K. S. Mali, D. Wu, X. Feng, K. Müllen, M. Walter, S. De Feyter and S. F. L. Mertens, Angew. Chem., Int. Ed., 2020, 59, 14049–14053 CrossRef CAS PubMed.
- P. Gao, Y. Zeng, P. Tang, Z. Wang, J. Yang, A. Hu and J. Liu, Adv. Funct. Mater., 2022, 32, 1–13 Search PubMed.
- J. Jia, X. Zhao, W. Hu, Y. Wang, J. Huang, J. Huang, H. Li, Y. Peng, H. Ma and C. Xu, J. Mater. Chem. A, 2023, 11, 8141–8149 RSC.
- J. Wei, D. Xia, Y. Wei, X. Zhu, J. Li and L. Gan, ACS Catal., 2022, 12, 7811–7820 CrossRef CAS.
- M. Zhang, M. De Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- D. Nguyen, G. Kang, N. Chiang, X. Chen, T. Seideman, M. C. Hersam, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2018, 140, 5948–5954 CrossRef CAS PubMed.
- H. Nakatsuji and H. Nakai, Chem. Phys. Lett., 1992, 197, 339–345 CrossRef CAS.
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song, J. Zhu, L. Gu, L. Liu, H. Zhao, Y. Li and D. Wang, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed.
- L. Wan, K. Zhao, Y. C. Wang, N. Wei, P. Zhang, J. Yuan, Z. Zhou and S. G. Sun, ACS Catal., 2022, 12, 11097–11107 CrossRef CAS.
- A. R. Kottaichamy, M. A. Nazrulla, M. Parmar, R. Thimmappa, M. C. Devendrachari, C. P. Vinod, M. Volokh, H. M. N. Kotresh, M. Shalom and M. O. Thotiyl, Angew. Chem., Int. Ed., 2024, e202405664 CAS.
- R. Georgescu, C. Boscornea, I. Calinescu and R. State, Rev. Chim., 2015, 66, 1554–1557 CAS.
- S. Harbeck and H. Mack, Experimental and Theoretical Investigations on the IR and Raman Spectra for CuPc and TiOPC, Univ. Tubingen, 2013, pp. 1–19 Search PubMed.
- D. R. Tackley, G. Dent and W. E. Smith, Phys. Chem. Chem. Phys., 2001, 3, 1419–1426 RSC.
- C. A. Melendres and V. A. Maroni, J. Raman Spectrosc., 1984, 15, 319–326 CrossRef CAS.
- E. Jung, H. Shin, B. H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. H. Antink, S. Park, K. S. Lee, S. P. Cho, J. S. Yoo, Y. E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- Q. Zhang, X. Tan, N. M. Bedford, Z. Han, L. Thomsen, S. Smith, R. Amal and X. Lu, Nat. Commun., 2020, 11, 4181 CrossRef PubMed.
- J. M. Assour, J. Am. Chem. Soc., 1965, 87, 4701–4706 CrossRef CAS.
- J. Ding, Z. Wei, F. Li, J. Zhang, Q. Zhang, J. Zhou, W. Wang, Y. Liu, Z. Zhang, X. Su, R. Yang, W. Liu, C. Su, H. Bin Yang, Y. Huang, Y. Zhai and B. Liu, Nat. Commun., 2023, 14, 6550 CrossRef CAS PubMed.
- D. T. Burns and B. D. Flockhart, Philos. Trans. R. Soc. London. Ser. A Phys. Eng. Sci., 1990, 33337–33348 Search PubMed.
- M. Viera, J. Riquelme, C. Aliaga, J. F. Marco, W. Orellana, J. H. Zagal and F. Tasca, Front. Chem., 2020, 8, 1–12 CrossRef PubMed.
- J. Masa, K. Ozoemena, W. Schuhmann and J. H. Zagal, J. Porphyr. Phthalocyanines, 2012, 16, 761–784 CrossRef CAS.
- S. Zheng and Z. Jiujun, J. Phys. Chem. C, 2007, 111, 7084–7090 CrossRef.
- V. Y. Aristov, O. V. Molodtsova, V. V. Maslyuk, D. V. Vyalikh, T. Bredow, I. Mertig, A. B. Preobrajenski and M. Knupfer, Org. Electron., 2010, 11, 1461–1468 CrossRef CAS.
- T. Mthethwa, E. Antunes and T. Nyokong, Dalton Trans., 2014, 43, 8230–8240 RSC.
- E. V Basiuk, L. Huerta and V. A. Basiuk, Appl. Surf. Sci., 2019, 470, 622–630 CrossRef.
- K. Juodkazis, J. Juodkazyte, V. Jasulaitiene, A. Lukinskas and B. Šebeka, Electrochem. Commun., 2000, 2, 503–507 CrossRef CAS.
- A. Kumar, A. Ganguly and P. Papakonstantinou, J. Phys.: Condens.Matter, 2012, 24, 235503 CrossRef PubMed.
- R. R. Cranston and B. H. Lessard, RSC Adv., 2021, 11, 21716–21737 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01284d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |